

Abstract
Postsynaptic density (PSD) proteins include scaffold, cytoskeletal, and signaling proteins that structurally and functionally interact with glutamate receptors and other postsynaptic membrane proteins. The molecular mechanisms regulating the assembly of PSD proteins and their associations with synapses are still widely unknown. We investigated the molecular mechanisms of Shank1 targeting and synapse assembly by looking at the function of guanylate kinase-associated protein (GKAP) and PSD-95 interactions. Shank1 when it is not associated to GKAP, which binds to the Shank PSD-95-Discs Large-zona occludens-1 domain, forms filamentous and fusiform structures in which the Src homology 3 domain specifically interacts with the ankyrin repeat domain, thus allowing its multimerization via a novel form of intermolecular interaction. Surprisingly, in both COS-7 cells and hippocampal neurons, GKAP forms insoluble aggregates with Shank that colocalize with heat shock protein 70 and neurofilaments, two markers of the aggresomes in which misfolded proteins accumulate. However, the two proteins are organized in clusters in COS cells and synaptic clusters in neurons when both are overexpressed and associated with wild-type PSD-95, but not with palmitoylation-deficient PSD-95. Synaptic activity in neurons induces the formation of Shank and GKAP intracellular aggregation and degradation. Similarly, the overexpression of a GKAP mutant that is incapable of binding PSD-95 induces Shank aggregation and degradation in neurons. Our data suggest a possible functional and structural role of the PSD-95-GKAP complex in Shank and PSD protein assembly and stability to synapses.
Keywords: postsynapstic density, aggresomes, cytoskeleton, PDZ domain, Homer, intermediate filaments
Introduction
The postsynaptic density (PSD) consists of a network of interacting proteins that form an electron-dense organelle right beneath the postsynaptic membrane. Most PSD proteins function as scaffolds that anchor and link glutamate receptors and other postsynaptic membrane proteins to cytoskeletal elements and signaling pathways (Garner et al., 2000; Kennedy, 2000; Scannevin and Huganir, 2000; Sheng and Pak, 2000). Shank and GKAP (guanylate kinase-associated protein) are probably two of the major scaffold proteins organizing the PSD.
Shank1-3 proteins (also called ProSAP, SSTRIP, cortBP, Synamon, and Spank) contain ankyrin repeats, an Src homology 3 (SH3) domain, a PSD-95-Discs Large-zona occludens-1 (PDZ) domain, a proline-rich domain, and a sterile alpha motif (SAM) domain. They are associated with the NMDA receptor-PSD-95 complex by their binding to C-terminal GKAP, and with type I metabotropic glutamate receptors (mGluRs) via an interaction with Homer in the proline-rich domain (Boeckers et al., 1999; Naisbitt et al., 1999; Tu et al., 1999; Zitzer et al., 1999; Sheng and Kim, 2000). A number of actin regulatory molecules bind to Shank in the proline-rich domain, cortactin (Naisbitt et al., 1999; Hering and Sheng, 2003), IRSp53 (Bockmann et al., 2002; Soltau et al., 2002), and AbP1 (Qualmann et al., 2004) or at the PDZ domains, β-PIX (Park et al., 2003). These data suggest that Shank acts as a major scaffold for postsynaptic proteins and as a molecular bridge linking multiple glutamate receptor subtypes to the postsynaptic cytoskeleton. It is also known that the overexpression of Shank1 in hippocampal cultures induces synapse maturation associated with the enlargement of dendritic spines (Sala et al., 2001). Furthermore, a form of mild mental retardation associated with severe expressive language delay and minor facial dysmorphisms has been found in several patients with a 22q13.3 distal deletion that causes ProSAP2/Shank3 haploinsufficiency (Bonaglia et al., 2001; Boeckers et al., 2002; Wilson et al., 2003), thus suggesting that Shank plays a central role in the organization and function of excitatory synapses.
GKAP [also called SAPAP (synapse-associated protein 90-postsynaptic density-95-associated protein) and DAP (Discs large-associated protein)] is a less characterized protein. The four members of the family were originally identified as proteins interacting with the GK domain of PSD-95 (Kim et al., 1997; Satoh et al., 1997; Takeuchi et al., 1997). GKAP has five repeats of 14 amino acids involved in the interaction of PSD-95 and binds to S-SCAM (Hirao et al., 1998), nArgBP2 (Kawabe et al., 1999), Dynein light chain (Naisbitt et al., 2000), and Shank (Boeckers et al., 1999; Naisbitt et al., 1999). GKAP may therefore function as scaffolding proteins that link PSD protein complexes to motor proteins (Naisbitt et al., 2000).
We studied the molecular mechanisms underlying Shank1 and GKAP assembly and targeting to synapses, and found that: (1) Shank and GKAP form aggresomes and are degraded by proteasomes in the absence of PSD-95; (2) in the absence of GKAP, Shank1 multimerizes by means of a new form of multimerization because of an interaction between the ankyrin repeat and SH3 domains; and (3) association of Shank to a PSD-95-GKAP complex seems to be required to ensure correct Shank1 targeting to synapses in developing and mature neurons.
Materials and Methods
Hippocampal neuron cultures and COS-7 cells. The hippocampal neuron cultures were prepared from embryonic day 18 (E18)-E19 rat hippocampi (Charles River, Milan, Italy). Medium-density neurons (150-200 cells/mm2) were prepared as previously described (Brewer et al., 1993) with minor modifications, plated on 18-mm-diameter coverslips, and grown on 12 well plastic tissue culture plates (Iwaki; Bibby Sterilin, Staffordshire, UK). The neurons were transfected using calcium phosphate precipitation (transfection efficiency ∼1%) or infected with lentivirus vectors. The COS-7 cells were cultured and transfected as previous described (Kim et al., 1996); the experiments were performed 48 hr after transfection.
Recombinant DNA and vectors. Full-length hemagglutinin (HA)-tagged Shank1B (HA-Sankh1B) cDNA (Sala et al., 2001) was used as a template for all of the Shank constructs. N-terminal green fluorescent protein (GFP)-tagged versions of the Shank1B deletion constructs were made by replacing the HA tag with a HindIII-AscI insert coding for enhanced GFP (EGFP). Myc-tagged Homer1b, Homer1a, and Homer1aW24A were prepared as previously described (Tu et al., 1998; Xiao et al., 1998). Full-length GKAP1A and GKAP1B expression constructs were prepared as previously described (Kim et al., 1997; Naisbitt et al., 1997). HA-tagged GKAP249L was prepared by means of the PCR amplification of a KpnI-EcoRI fragment coding for the 249-666 region of GKAP1A, and then in-frame subcloned into a GW1-cytomegalovirus expression vector (British Biotechnology, Oxford, UK) with a built-in N-terminal HA tag (N-HA-GW1 vector). For the GKAP249A construct, the C-terminal leucine 666 was mutated in alanine using the Quick-Change Site-Directed Mutagenesis kit (Stratagene, Cedar Creek, TX). All of the constructs were verified by means of sequencing and the expression of proteins of the expected molecular weight in COS-7 cells.
GKAP249L and GKAP249A were subcloned in FUW vector (Lois et al., 2002) containing the ubiquitin promoter. The lentivirus vectors were prepared as previously described (Naldini et al., 1996; Lois et al., 2002). Neurons were infected after 5 d in vitro (DIV5) and solubilized in sample buffer on DIV14.
Immunostaining and antibodies. The neurons and COS-7 cells were fixed in 4% paraformaldehyde and 4% sucrose at room temperature, or in 100% methanol at -20°C for 10 min. Primary and secondary antibodies were applied in GDB buffer (30 mm phosphate buffer, pH 7.4, containing 0.2% gelatin, 0.5% Triton X-100, and 0.8 m NaCl) for 2 hr at room temperature, or overnight at 4°C. The following antibodies were used (source in parentheses): mouse monoclonal anti-HA antibodies (Hoffmann-La Roche, Basel, Switzerland); mouse monoclonal anti-Myc (Santa Cruz Biotechnology, Santa Cruz, CA); GKAP rabbit N1546 and C9589 (Kim et al., 1997; Naisbitt et al., 1997); PSD-95 mouse monoclonal K28/43.1 (a gift from J. Trimmer, State University of New York, Stony Brook, NY); Shank guinea pig number 1123, Homer rabbit 1133 (gifts from E. Kim, Korean Advanced Institute of Science and Technology, Taejon, Korea); tubulin mouse and vimentin mouse (both from Sigma, St. Louis, MO); heat shock protein (Hsp) 70 mouse monoclonal (Stressgen, San Diego, CA); neurofilament mouse monoclonal (Hoffmann-La Roche); GFP mouse monoclonal (Chemicon, Temecula, CA); and FITC-, Cy3- and Cy5-conjugated secondary antibodies (Jackson ImmunoResearch, West Grove, PA). Texas Red-conjugated phalloidin was purchased from Molecular Probes (Eugene, OR).
Glutathione S-transferase fusion protein pull-down assays. Human embryonic kidney (HEK) 293 cells were maintained in DMEM supplemented with 10% fetal bovine serum (Invitrogen, Carlsbad, CA) and penicillin-streptomycin. For transient transfection, the cells were plated on 10 cm Petri dishes (Falcon, Lincoln, NJ) until reaching 70-80% confluency, and were then transfected with an appropriate amount of DNA using a lipofectAMINE transfection kit (lipofectAMINE PLUS; Invitrogen) according to the manufacturer's instructions. Transient expression was allowed to continue for 36-48 hr, after which the cells were washed once with ice-cold PBS, scraped into microcentrifuge tubes, and lysed with 1% Triton X-100 in 50 mm HEPES, pH 7.6, containing 150 mm NaCl, 10 mm Na4P2O7, 2 mm Na3VO4, 100 mm NaF, 10 mm EDTA, and protease inhibitors. The cell lysates were centrifuged at 20,000 × g for 30 min, and the supernatants were transferred to a fresh tube and stored at -20°C until used.
For each pull-down assay, 15 μl of a 75% slurry of glutathione Sepharose-4B was first washed three times with 0.5 ml of PBS for purposes of equilibration. Approximately 30 μg of glutathione S-transferase (GST) fusion protein was added, and the suspension was agitated at 4°C for 30 min. The beads loaded with GST fusion protein were then washed three times with 0.5 ml of PBS to remove any unbound protein. Cell lysate (∼100 μg of total protein) or 0.6 nmol of purified recombinant protein was added to the GST fusion protein-loaded beads, and the suspension was agitated at 4°C for 1 hr. The beads then washed with 0.5 ml of PBS four times to remove any unbound proteins and subsequently boiled with 25 μl of 2× SDS-polyacrylamide gel sample buffer. The proteins eluted off the beads underwent SDS-PAGE, and were then stained with Coomassie Blue R-250 or processed for Western blot analysis.
Image acquisition and quantification. The confocal images were acquired with a Bio-Rad (Hercules, CA) MRC1024 confocal microscope, using a Nikon (Tokyo, Japan) 60× objective with a sequential-acquisition setting at a resolution of 1280 × 1024 pixels. Each image was a z-series projection taken at 0.75 μm deep intervals. The morphometric measurements were made using MetaMorph image analysis software (Universal Imaging, West Chester, PA). The ratio of average immunofluorescence intensity between the spine head and the dendritic shaft was measured in manually selected spine head and dendritic-shaft areas. The cluster area and number were automatically measured and counted by computer after thresholding the cluster area in randomly selected dendrites. The fluorescence intensity of the staining of endogenous or GFP-tag transfected proteins on the cell body was measured as the mean intensity of manually selected areas of transfected neurons. Neurons were randomly selected, and confocal setting was maintained constant during the acquisition of different coverslips.
COS extract solubilization, immunoprecipitation, and Western blotting. The COS cells were washed in PBS buffer with protease inhibitor (Complete EDTA; Hoffmann-La Roche) and extracted in radioimmunoprecipitation assay buffer without SDS buffer (50 mm Tris-HCl, pH 7.6, 150 mm NaCl, 1 mm EDTA, 1% NP-40, and 0.5% deoxycholate). After being agitated for 1 hr at 4°C, the cell extract was spun at 16,000 × g for 15 min and divided into the supernatant (soluble fraction) and pellet (insoluble fraction). The pellet was resuspended in Laemmli buffer one time and the supernatant in Laemmli buffer two times, and then loaded onto 6 or 10% SDS-PAGE gels. Immunoprecipitation was performed as previously described (Hsueh et al., 1997). Infected neurons were resuspended in Laemmli buffer two times and then loaded onto 6 or 10% SDS-PAGE gels. Primary antibodies (mouse anti-HA 1:1000, rabbit anti-GKAP 1:500, mouse anti-PSD-95 1:500, mouse anti-Myc 1:1000, mouse anti-GFP 1:1000, and mouse anti-tubulin 1:1000) were applied overnight in blocking buffer (20 mm Tris, pH 7.4, 150 mm NaCl, 0.1% Tween 20, and 3% dried nonfat milk); the secondary antibodies (HRP-conjugated anti-mouse or anti-rabbit) (Amersham) were used in a ratio of 1:2000. The signal was detected using an ECL detection system (PerkinElmer Life Sciences, Emeryville, CA) and quantified by means of ImageQuant software (Bio-Rad).
Time-lapse imaging. The neurons were plated on glass-bottomed Petri dishes (Iwaki; Bibby Sterilin), transfected using calcium phosphate precipitation on DIV3-4, and observed on DIV5-7. The images were acquired using a Zeiss (Oberkochen, Germany) Axiovert 200 equipped for live cell imaging, including temperature and CO2 concentration controllers, and a Roper MicroMax CCD 512 × 512 camera controlled by Meta-Morph program. The images were acquired every 12-24 min using a 50% neutral filter to reduce light damage.
Results
Shank1 without the PDZ domain forms fusiform and filament aggregates in neurons
It has previously been suggested that Shank1 targeting to synapses depends on its interaction with GKAP (Naisbitt et al., 1999) and the presence of the PDZ domain (Sala et al., 2001); the interaction with Homer1 can also regulate the synapse localization of Shank1 (Sala et al., 2003).
To better investigate the mechanisms of Shank1 targeting to synapses and the related function of the PDZ and other domains, we made a number of GFP-tagged Shank1 constructs devoid of various individual domains and studied their distribution in transfected hippocampal neurons.
All of the constructs were tagged with EGFP at the N terminal (Fig. 1A). The hippocampal neurons were transfected on DIV10-11 and fixed on DIV17-18. The ability of each construct to localize at the synapse and dendritic spine was quantified by the calculation of the GFP intensity ratio between the spine and shaft compartments and the mean cluster size when the transfected protein was capable of forming synaptic clusters (Fig. 1A).
Figure 1.
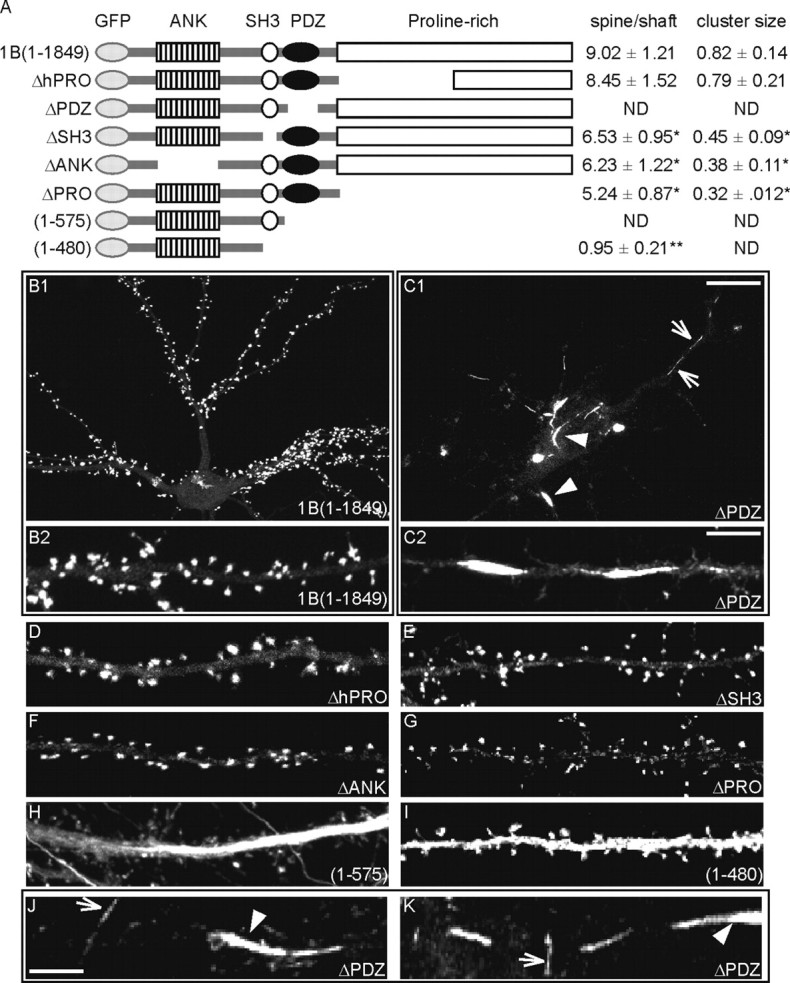
The PDZ domain is necessary for Shank1B targeting to spine. A, Diagram of the wild-type and mutant constructs of Shank1B used in this study (all GFP-tagged at the N terminus). The various domains are indicated. ANK, Ankyrin repeats. For each construct, the spine-shaft signal intensity and cluster size ratios (value ± SEM) were calculated as indicated on the right. ND, Not detected. *p < 0.05 versus GFPShank1B; **p < 0.01 versus GFPShank1B. B-K, Neurons were transfected with GFP-tagged wild-type Shank1B or mutant Shank constructs (as indicated in each panel) on DIV10-11 and fixed on DIV17-18; the images were acquired by means of confocal microscope in the GFP channel. B2 shows enlargements of a neuron dendrite transfected with GFPShank1B; C2, J, and K show enlargements of dendrites from neurons transfected with GFPShank1BΔPDZ. The constructs deleted from the PDZ domain, but with the SH3 and ANK domains, form fusiform (arrowheads) or filiform (arrows) aggregates in dendrites (C1, C2, H, J, K). Scale bars: (in C1) B1, C1, 10 μm; (in C2) B2, C2, D-I, 5 μm; (in J) J, K, 2.5 μm.
As previously reported (Sala et al., 2003), Shank1B, a Shank1 splice variant lacking the SAM domain, is highly enriched in the synapse with a spine-shaft ratio of 9.02 ± 1.21 (mean ± SEM) (Fig. 1A,B1,B2). Similarly, the full-length Shank1 isoform, GFP-Shank1A (Sala et al., 2001), is specifically targeted to synapses and dendritic spines (data not shown).
The result was completely different when only the PDZ domain was deleted. Shank1ΔPDZ was essentially absent from the synapses and became mainly distributed as fusiform (Fig. 1C1, arrowheads) or filamentous aggregates (Fig. 1C1, arrows) localized along the dendrites. Similar aggregates were also formed by the Shank1(1-575) construct, which contains only the ankyrin repeat domain (ANK) and SH3 domain (Fig. 1H). The aggregates formed by Shank1(1-575) were also often localized in the axon (data not shown). The deletion of the PDZ domain therefore induces a dramatic change in the distribution of GFP-tag Shank1 transfected protein. Figure 1, J and K, shows the distribution of GFPShank1ΔPDZ at high magnification; as mentioned above, we classified the aggregates as fusiform (Fig. 1 C1,C2; J,K, arrowheads) or filament-shaped (Fig. 1C1,C2; J,K, arrows).
Deletion of the first half (Shank1BΔhPRO) or the whole of the proline-rich domain (Shank1ΔPRO) does not abolish Shank1 targeting to synapses, although Shank1ΔPRO protein is quantitatively less enriched than Shank1B and Shank1BΔhPRO (spine-shaft ratio 5.24.02 ± 0.87) (Fig. 1A; B2, compare D, G). The clusters formed by Shank1ΔPRO were smaller than those formed by Shank1B and Shank1BΔhPRO (0.32 ± 0.12 vs 0.82 ± 0.14 for Shank1B and 0.79 ± 0.21 for Shank1BΔhPRO) (Fig. 1A). Deletion of the SH3 domain (Shank1ΔSH3) or the ankyrin repeat domain (ANK) (Shank1ΔANK) did not substantially compromise the protein localization at synapses (Fig. 1A,E,F), but the mean cluster size of both constructs was statistically smaller (p < 0.05) than Shank1B (0.45 ± 0.09 for Shank1ΔSH3 and 0.38 ± 0.11 for Shank1ΔANK vs 0.82 ± 0.14 for Shank1B) (Fig. 1A).
Aggregates and synaptic localization were absent from the construct Shank1(1-480), which only contains the ANK domain and some flanking regions (Fig. 1I). Shank1(1-480) is diffuse throughout the dendritic compartment (Fig. 1A,I).
These data suggest that the PDZ domain of Shank1 is essential for protein targeting to synapses as its deletion leads to protein mistargeting and aggregation. They also suggest that the ANK and SH3 domains are involved in the formation of fusiform and filament shaped GFPShank1ΔPDZ aggregates in neurons.
Shank1 in COS cells has a filamentous distribution
To investigate further the nature of the elongated aggregates in neurons shown in Figure 1C, we transfected COS cells with the same set of constructs and found that GFP-Shank1B forms a filamentous structure (Fig. 2A). Similar distributions were observed for the full-length Shank1, GFPShank1A, and the HA-tagged or Myc-tagged Shank1A and Shank1B constructs (data not shown). We then tested all of the deletion constructs, and found that only the constructs lacking the ANK (Fig. 2E) or SH3 domain (Fig. 2D,H) do not form filamentous structures: Shank1ΔANK and Shank1ΔSH3 form small aggregates, and Shank1(1-480) is generally diffuse.
Figure 2.
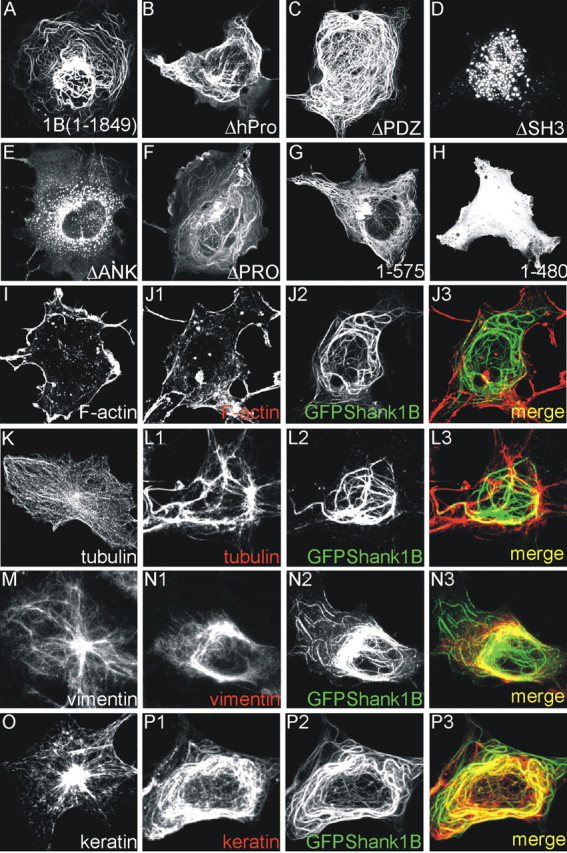
GFP-Shank forms filaments in COS cells. A-H, COS cells were transfected with the same constructs as those shown in Figure 1 and fixed 2 d after transfection. All of the constructs formed filaments in COS cells except the mutants deleted of the ankyrin or SH3 domains (D, E, H). I-P, COS cells were transfected or not with GFP-Shank1B and stained for F-actin (I, J1-J3), tubulin (K,L1-L3), vimentin (M,N1-N3), or keratin (O,P1-P3). The distribution of tubulin, vimentin, and keratin was modified by GFP-Shank1B: see K versus L1 for tubulin, M versus N1 for vimentin, and O versus P1-P3 for keratin.
The results indicate that, as in neurons, the ANK and the SH3 domains are required by COS cells for the filamentous aggregation of Shank1. Interestingly, the deletion of the PDZ domain does not change the filamentous structure of Shank1 in COS cells, thus suggesting that the interaction of the PDZ domain with GKAP (or other proteins) in neurons can block the formation of the dendritic filamentous aggregates by targeting Shank1 to synapses (Fig. 1B2,D,G, compare 2A,B,F).
We then investigated possible connections between GFPShank1B filament and cytoskeletal elements in COS cells. COS cells were transfected with GFPShank1B and stained for F-actin, tubulin, vimentin, and keratin. There was no obvious colocalization of Shank1 with F-actin, tubulin, and vimentin (Fig. 2J1-J3, L1-L3, N1-N3), but GFPShank1B filaments colocalized almost completely with keratin filaments (Fig. 2P1-P3). More surprisingly, we found that tubulin, vimentin, and keratin filaments were reorganized in GFPShank1B-transfected COS cells (Fig. 2, compare K with L1, M with N1, and O with P1). Only the distribution of F-actin was not modified (Fig. 2, compare I,J1).
Shank1 therefore forms a filamentous structure in COS cells that is at least partially localized with keratin and modifies the distribution of endogenous cytoskeletal elements tubulin, vimentin, and keratin.
The ankyrin repeats domain interacts with the SH3 domain
The fact that both the ANK and SH3 domains are required for filamentous aggregation in COS and fusiform or filament aggregates in neurons suggests the presence of an intermolecular interaction between the ANK and SH3 domains in Shank1.
To verify this type of interaction biochemically, we performed pull-down experiments by loading extracts of HEK 293 cells transfected with HA-tagged Shank1 deletion constructs (Fig. 3A) to a glutathione column coupled with GST-SH3. We first found that full-length Shank1B can be pulled down by the SH3 domain (Fig. 3B). We then used the Shank1 construct deleted of various domains (Fig. 3A). Interestingly, all the constructs containing the ANK domain robustly bound to GST-SH3 (Fig. 3A,B). A Shank1 deletion mutant containing only the ANK region (construct 106-340) also bound to the GST-SH3 domain (Fig. 3A,B, lanes 25-27).
Figure 3.

The Shank1 SH3 domain binds to the ankyrin repeat domain (ANK). A, Diagram of the wild-type and mutant constructs of Shank1B (all HA-tagged at the N terminus) used in this study. B, Extracts of HEK cells transfected with wild-type Shank1B or mutant Shank constructs (as indicated above the Western blot) were incubated with GST or GST-SH3, and the pull-down proteins were revealed using HA antibody. In the input lane, ∼10% of the total extract used for the pull-down was loaded. C, GST-SH3, but not GST alone, can pull down purified His6-ANK domains.
To demonstrate the direct interaction between ANK and SH3 domains, we tested the interaction between purified GST-SH3 and His6-ANK proteins in vitro. As shown in Figure 3C, GST-SH3 (but not GST alone) could pull down the purified His6-ANK domain.
The role of the ANK and SH3 domains in the formation of this filament was investigated by transfecting Shank1ΔANK and Shank1ΔSH3 constructs in COS cells together with full-length Shank1B. Neither the Shank1ΔANK nor the Shank1ΔSH3 construct formed filaments when transfected alone (Figs. 2D,E, 4A1-A3,C1-C3) or cotransfected in the same cell (GFPShank1ΔSH3 plus HA-Shank1ΔANK) (Fig. 4E1,E2), but they did form filaments when cotransfected with full-length Myc-Shank1B (Fig. 4B1-B3,D1-D3), thus suggesting that an intermolecular interaction between the two transfected proteins may underlie the formation of the filaments. Thus, apparently both GFPShank1ΔSH3 and GFPShank1ΔANK bound to a template formed by GFPShank1B filaments.
Figure 4.

GFPSh1BΔSH3 and GFPSh1BΔANK form filaments in COS cells only when coexpressed with full-length Shank1B. A, B, COS cells were transfected with GFPSh1BΔSH3 or GFPSh1BΔANK plus or minus Myc-Shank1B and stained for Myc in red; GFPSh1BΔSH3 and GFPSh1BΔANK was visualized on the GFP channel. E, COS cells were transfected with GFPSh1BΔDANK plus HA-Sh1BΔSH3 and stained for HA in red; GFPSh1BΔANK and HA-Sh1BΔSH3 did not colocalize when coexpressed in COS cells. F, COS cells were transfected with Myc-Shank1B alone (lanes 1,5), Myc-Shank1B plus HA-Shank1B (lanes 2, 3, 6, 7), and HA-Shank1B plus Myc-PSD-95 (lanes 4, 8). Extracts immunoprecipitated with HA antibodies were stained with HA and Myc antibodies, as indicated on the right. G, COS cells were transfected with Myc-Shank1B plus GFPShank1ΔSH3 (lanes 1,4), Myc-Shank1B plus GFPShank1ΔANK (lanes 2,5) or HA-Shank1ΔSH3 plus GFPShank1ΔANK (lanes 3,6). Extracts immunoprecipitated with Myc (lanes 4, 5) or HA (lane 6) antibodies were stained with Myc, GFP, or HA antibodies, as indicated on the right.
To further test this possibility, we first looked at the ability of HA-Shank1B to coimmunoprecipitate with Myc-Shank1B when cotransfected in COS cells (Fig. 4F). Although the HA-Shank1B splice variant is devoid of the SAM domain capable of forming a homodimer (Tu et al., 1999), it did specifically immunoprecipitate Myc-Shank1B (Fig. 4F, lanes 6, 7) but not Myc-PSD-95 (Fig. 4F, lane 8). It is therefore not surprising that Myc-Shank1B can associate and precipitate with either GFPShank1ΔSH3 or GFPShank1ΔANK when coexpressed in COS cells (Fig. 4G, lanes 1,2,4,5), whereas HA-Shank1BΔSH3 did not coimmunoprecipitate with GFPShank1BΔANK (Fig. 4G, lanes 3,6).
Taken together, these data suggest that the ANK and SH3 domains play a role in the formation of filamentous aggregates in COS. Our biochemical data show that the SH3 and ANK domains can directly interact with each other, and this interaction leads to the multimerization of Shank1.
The function of ANK and SH3 domains interaction in neurons remains at the moment not clear, although as shown in Figure 1, A, E, and F, deletion of either the SH3 or the ANK domain reduced the cluster size of GFP-tagged proteins, which suggests that the ANK and SH3 domains play a role in the correct accumulation of Shank1 in synapses.
Role of GKAP and PSD-95 in Shank1 synapse assembly
Shank1B forms filaments in COS cells, but is localized to the synapses of mature neurons. COS cells do not express Shank1 binding partners (S. Romorini and G. Piccoli, unpublished observations) such as GKAP and Homer, so we investigated what happens to filament distribution in COS cells when the other Shank1 protein partners are coexpressed.
GFP-Shank1B was cotransfected with GKAP1A, GKAP1B (a splice variant of GKAP that does not bind to Shank) (Naisbitt et al., 1999), Homer1b, Homer1a, and Homer1aW24A (a Homer1a mutant that does not bind to Shank1) (Beneken et al., 2000). Surprisingly, GKAP1A completely disassembled the Shank1B filaments and induced the formation of perinuclear aggregates of GFP-Shank1B (Fig. 5A1-A3,B1-B3). GKAP1B did not induce the formation of GFP-Shank1B aggregates, thus indicating that the binding of Shank1B to GKAP is responsible for the formation of the Shank1B aggregates (Fig. 5C1-C3). Consistent with this observation, all the Shank1 constructs containing the PDZ domain formed aggregates with GKAP (data not shown). Homer1b did not change the filament distribution of Shank, but colocalized with the filaments (Fig. 5D1-D3), whereas Homer1a completely destroyed the filaments and induced the diffused cytosolic distribution of GFP-Shank1B (Fig. 5E1-E3); this localization pattern of Shank1B requires physical binding between Homer1a and Shank because the Homer1aW24A mutant did not have any effect on the organization of Shank1B filaments (Fig. 5F1-F3).
Figure 5.
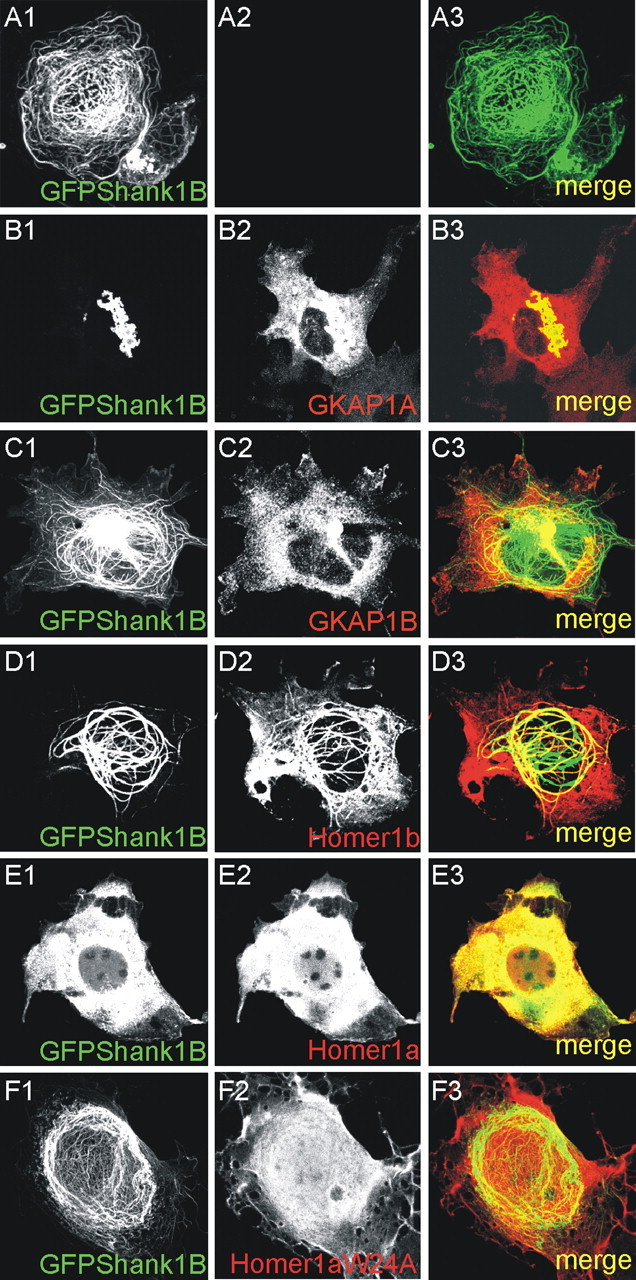
GKAP and Homer1a interfere with Shank1B filament distribution in COS cells. A-F, COS cells were transfected with GFP-Shank1B, alone or in combination with GKAP1A (a splice variant of GKAP that binds to Shank), GKAP1B (a splice variant of GKAP that does not bind to Shank), Homer1b, Homer1a, or Homer1aW24A, as indicated in each panel.
As previously reported (Naisbitt et al., 1999), GFP-Shank1B did not form aggregates with GKAP1A, but was recruited in planar clusters when PSD-95 was also cotransfected (Fig. 6A1-A4). GFP-Shank1B remained in filaments when GKAP1B (which binds PSD-95 but not Shank) was expressed (Fig. 6B1-B4). Only PSD-95-GKAP binding eliminated the Shank1B aggregates because neither Homer1b nor Homer1a could change the Shank1B aggregates induced by GKAP1A (Fig. 6C1-C4,D1-D4). Interestingly, the ability of PSD-95 to recruit Shank1B and GKAP1A in clusters depends on the ability of PSD-95 to form clusters and to be palmitoylated, because PSD-95C3,5S mutated on the two N-terminal cysteines (Hsueh et al., 1997; El-Husseini et al., 2000) is unable to form planar clusters with Shank and GKAP but remain aggregated together with both (Fig. 6E1-E3).
Figure 6.
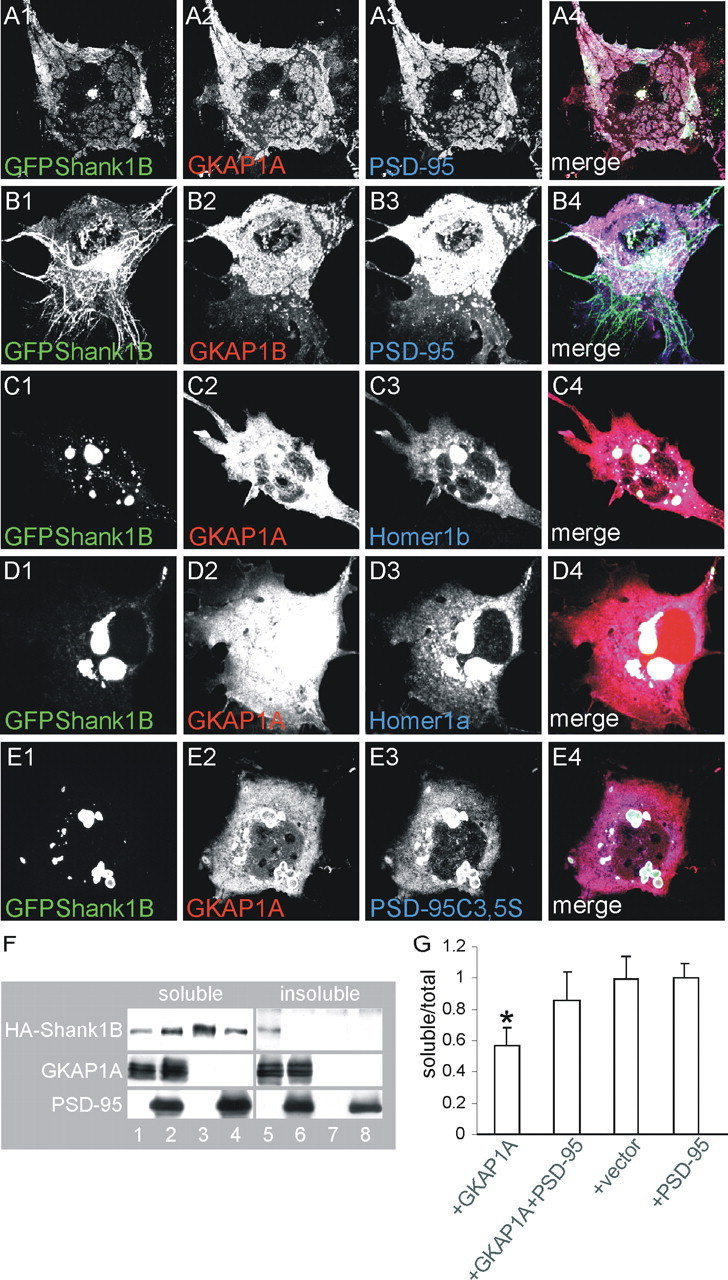
Only GFP-Shank1B, GKAP1A, and wild-type PSD-95 are distributed in clusters when coexpressed in COS cells. A, B, COS cells were triple-transfected with GFP-Shank1B plus GKAP1A or GKAP1B and PSD-95, as indicated in each panel. C-E, COS cells were triple-transfected with GFP-Shank1B plus GKAP1A or GKAP1B and Homer1b or Homer1a or PSD-95C3,5S, as indicated in each panel. The individual channels are shown in grayscale for better resolution; merge is shown in the panels on the right. F, G, HA-Shank1B solubility to detergent was tested in COS cells in the presence of GKAP1A alone or GKAP1A plus PSD-95. HA-Shank1B was soluble when transfected alone (F, lanes 3, 7, G) but became at least 50% insoluble when cotransfected with GKAP1A (F, lanes 1, 5, G). Shank1B solubility was recovered when GKAP1A was cotransfected with PSD-95 (F, lanes 2, 6, G). The graph bars indicate the mean values (±SEM) obtained from at least two independent experiments. *p < 0.05 versus all the other transfection combinations.
It has been previously shown that GKAP makes PSD-95 insoluble to detergent (Hirao et al., 2000). To see whether Shank also becomes insoluble in the presence of GKAP, we transfected COS cells with HA-Shank1B plus GKAP1A, and divided the cell extracts into soluble and insoluble proteins by treating them with 0.5% Triton X-100. Interestingly, although Shank1B alone was almost 100% soluble (Fig. 6F, lanes 3,7), it became ∼50% insoluble when transfected with GKAP1A (Fig. 6F, lanes 1,5). We then investigated what happens to Shank1B solubility when PSD-95 is coexpressed with GKAP1A, and found that it again became almost 100% soluble (Fig. 6F, lanes 2,6). Quantification of the soluble-insoluble ratio showed a statistically significant reduction in Shank1B solubility in the presence of GKAP1A alone, but not with GKAP1A plus PSD-95 (Fig. 6G).
In mature hippocampal neurons, GFP-Shank1B do not form aggregates when transfected alone or together with full-length GKAP1A (Fig. 1B1) (data not shown). We therefore investigated whether GKAP1A deleted of the PSD-95 binding domain induces the formation of endogenous or transfected Shank aggregates. Neurons were transfected with GFP-Shank1B plus GKAP1A truncated of the first 248 aa (GKAP-249L) containing the PSD-95 binding domain. The control was the same GKAP-deleted construct with a mutation at the last C-terminal amino acid (GKAP-249A) that is unable to bind the Shank PDZ domain (Kim et al., 1997). We found that GKAP-249L induced GFP-Shank1B aggregates in the neuronal cell bodies (Fig. 7A1-A3) and almost completely abolished GFP-Shank1B synapse targeting (Fig. 7B1-B3). GKAP-249L almost completely abolished endogenous Shank synaptic staining (Fig. 7F1-F3) and induced the formation of small Shank aggregates in the cell bodies of >50% of the transfected neurons (Fig. 7E1-E3). As expected, GKAP-249A did not form aggregates in the cell bodies or interfere with the synaptic localization of transfected GFP-Shank1B or endogenous Shank (Fig. 7C1-C3,D1-D3,G1-G3,H1-H3).
Figure 7.
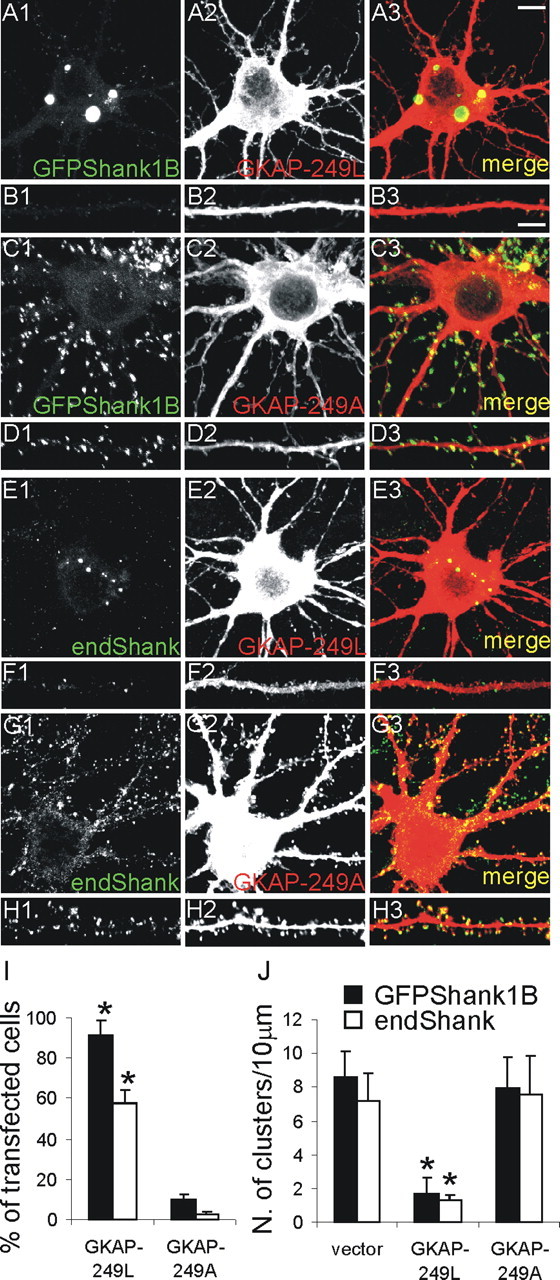
The GKAP1A-C-terminal (deleted of the N-terminal PSD-95 binding domain, GKAP-249L) induces aggregation in the cell body of transfected GFP-Shank1B and endogenous Shank. A-D, Neurons were transfected on DIV11 with Shank1B plus GKAP-249L or GKAP-249A (with a point mutation that abolishes binding to the Shank PDZ domain) as indicated in each panel, and stained on DIV16; B1-B3 and D1-D3, respectively, show higher magnifications of neuron dendrites from A1-A3 and C1-C3. E-H, Neurons were transfected on DIV11 with GKAP-249L or GKAP-249A, as indicated in the panel, and stained for GKAP and endogenous Shank on DIV16; F1-F3 and H1-H3, respectively, show higher magnifications of neuron dendrites from E1-E3 and G1-G3. I, Quantification of the numbers of neurons with intracellular aggregates formed by GFP-Shank1B and endogenous Shank after transfection with the indicated constructs (mean values ± SEM). J, Density of clusters of GFP-Shank1B and endogenous Shank per 10 μm of dendrite length (mean values ± SEM) in neurons transfected with the indicated constructs. At least 15 transfected neurons were quantified for each GKAP mutant construct. Scale bars: (in A3) A1-A3, C1-C3, E1-E3, G1-G3, 10 μm; (in B3) B1-B3, D1-D3, F1-F3, H1-H3, 5 μm. *p < 0.01 versus vector or GKAP-249A.
We measured the number of neurons with GFPShank1B or endogenous Shank aggregates after transfection with GKAP-249L or GKAP-249A. Approximately half of the transfected neurons (57.3 ± 7.2%) had endogenous Shank aggregates and ∼90% (91.5 ± 7.3%) GFP-Shank1B aggregates after GKAP-249L transfection, whereas there were aggregates in only a few cell bodies of neurons transfected with GKAP-249A (2.4 ± 1.3% for endogenous Shank, and 10.2 ± 2.2% for GFPShank1B) (Fig. 7I). GKAP-249L also greatly reduced the number of endogenous and GFP-Shank1B synaptic clusters to 1.7 ± 0.9 and 1.3 ± 0.3 cluster/10 μm, respectively, in comparison with the neurons transfected with vector (8.6 ± 2.0 and 7.1 ± 1.7) or GKAP-249A (8.0 ± 1.8 and 7.6 ± 2.0) (Fig. 7J).
GKAP-Shank aggregates are aggresomes
As GKAP induces Shank1 aggregation in COS and neurons, we wondered whether Shank1 aggregates can be considered aggresomes (Kopito, 2000). COS cells were transfected with GFP-Shank1B, GKAP1A plus or minus PSD-95, and stained for GFP, PSD-95, and Hsp70 (a marker of aggresomes or intracellular protein aggregates) (Kopito, 2000). As shown in Figure 8A1-A4, GFP-Shank1B-GKAP aggregates can be specifically stained with Hsp70 antibodies, whereas Hsp70 staining is diffuse in COS cells triple-transfected with GFP-Shank1B, GKAP1A, and PSD-95 (Fig. 8B1-B4). As shown in Figure 2, O and P1-P3, GFP-Shank1B modifies keratin distribution in COS cells, and the same is true in the case of GFP-Shank1B plus GKAP1A, in which keratin becomes concentrated in the intracellular aggregate (Fig. 8C1-C4), whereas the distribution of keratin is similar to that observed in untransfected COS cells when PSD-95 is cotransfected with GFP-Shank1B plus GKAP1A (Fig. 8D1-D4).
Figure 8.
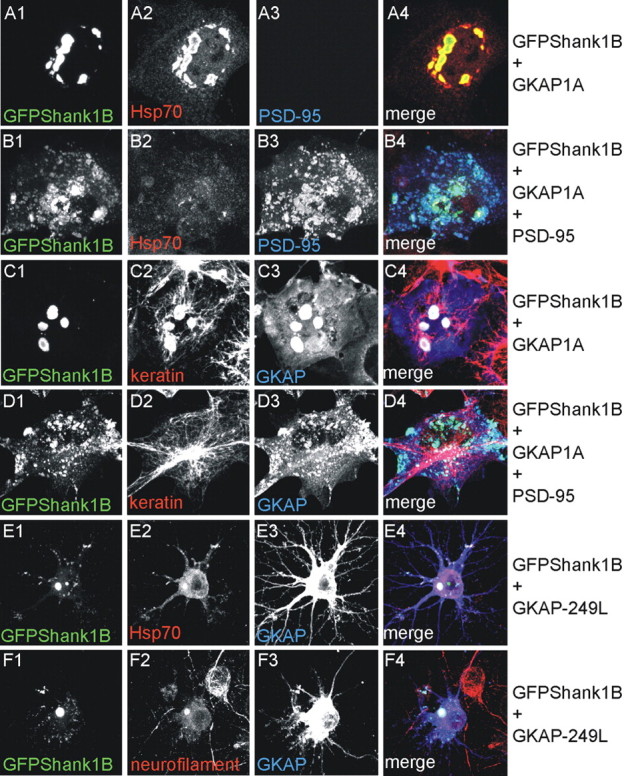
GKAP-Shank aggregates can be considered aggresomes. A-D, As indicated on the right, COS cells were transfected with GFPShank1B and GKAP1A (A, C) or GFPShank1B, GKAP1A, and PSD-95 (B, D) and stained for GFP (A1-D1), Hsp70 (A2, B2), PSD-95 (A3, B3), keratin (C2, D2), and GKAP (C3, D3). As shown in A1-A4, intracellular GFPShank1B-GKAP aggregates can be specifically stained with Hsp70 antibodies. The merge is shown in the panels on the right (A-D4). E, F, As indicated on the right, neurons were transfected on DIV10 with GFPShank1B plus GKAP-249L and stained on DIV15 for GFP (E1, F1), Hsp70 (E2), neurofilament (F2), and GKAP (E3, F3). The merge is shown in the panels on the right (E4, F4). Also in neurons, intracellular GFPShank1B-GKAP aggregates can be specifically stained with Hsp70 or neurofilament antibodies.
We then stained GFP-Shank1B aggregates in neurons for Hsp70 and neurofilaments (another neuron specific marker of aggresomes or intracellular protein aggregates) (Rao et al., 1998; Kopito, 2000; Kopito and Sitia, 2000). In both cases, we found concentrations of these proteins in the GFP-Shank1B aggregates cotransfected with GKAP249L (Fig. 8E1-E4,F1-F4).
The ultimate fate of a misfolded protein is determined by kinetic partitioning between proteasomal degradation and the aggregation of high molecular weight oligomers to form aggresomes. To demonstrate further that GKAP-249L induces both GFP-Shank1B and endogenous Shank misfolding and aggresome relocalization, we treated neurons transfected with GKAP-249L or GKAP-249A with the proteasome inhibitor MG132 for 12 hr, and then stained the transfected neurons for GFP or Shank. Interestingly, we found that MG132 induced the accumulation of both GFP-Shank1B and endogenous Shank in neurons transfected with GKAP-249L (Fig. 9A1,A2,B1,B2,D1,D2,E1,E2) but not in those transfected with GKAP-249A (Fig. 9C1,C2,F1,F2). Quantification of the mean cell body fluorescence intensity showed that MG132 increased staining for both GFPShank1B and endogenous Shank by >100% (from 50.3 ± 13.9 to 116.5 ± 3.8 arbitrary units for GFP-Shank1B, and from 26.8 ± 8.7 to 58.3 ± 7.0 for endogenous Shank) (Fig. 9G). GKAP-249L therefore also at least partially induces Shank degradation by means of a proteasome-dependent mechanism.
Figure 9.
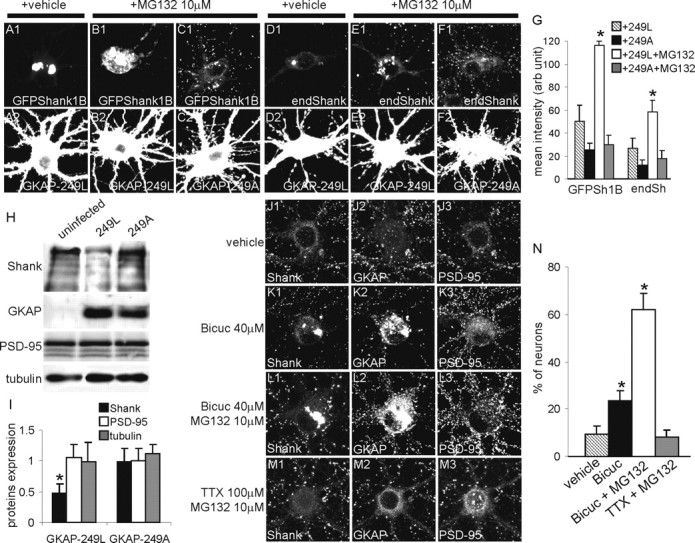
GKAP-249L induces partial degradation of GFPShank1B and endogenous Shank in neurons by means of proteasome degradation. A-F, DIV10 neurons were transfected with GFPShank1B plus GKAP-249L (A, B), GFPShank1B plus GKAP-249A (C), GKAP-249L alone (D, E), or GKAP-249A alone (F) and stained on DIV15 for GFP (A-C1), Shank (D-F1), and GKAP (A-F2). To test whether proteasomes are involved in this process, we treated the neurons with 10 μm of the MG132 proteasome inhibitor for 12 hr. G, The graph indicates the mean fluorescence intensity (arbitrary units ± SE) of the cell bodies of MG132-treated or untreated neurons. At least 10 neurons were measured at each experimental point; *p < 0.01 versus GKAP-249L without MG132. H, Neurons were infected at DIV5 with a lentivirus vector expressing GKAP-249L or GKAP-249A and lysated on DIV14. The expression of endogenous Shank, PSD-95, and tubulin was measured by Western blotting and quantified as shown in I. I, The graph indicates the mean value ± SE of the band intensity ratios between infected and uninfected neurons. The values were obtained from three independent experiments; *p < 0.05 versus GKAP249A. J-M, Bicuculline stimulation induces the formation of endogenous Shank and GKAP aggregates in cell bodies. The neurons were treated for 24 hr as indicated on the left of each panel and then stained for endogenous Shank (J1-M1), GKAP (J2-M2), and PSD-95 (J3-M3). Intracellular aggregates formed by endogenous Shank and GKAP (but not PSD-95) are visible in the neurons treated with bicuculline or bicuculline plus MG132. N, The graph indicates the mean ± SE percentage of neurons with intracellular aggregates. The data were obtained by counting at least 100 neurons from six coverslips obtained from three independent experiments for each treatment; *p < 0.01 versus vehicle and TTX plus MG132.
As a further demonstration that GKAP-249L induces Shank1 degradation, we infected neurons with retrovirus vectors expressing GKAP-249L or GKAP-249A and then used Western blotting to measure the expression of endogenous Shank protein in comparison with endogenous PSD-95 and tubulin proteins. The neurons were infected on DIV5 and lysated on DIV14. Equal amounts of proteins were loaded on SDS-PAGE gel and blotted with Shank, PSD-95, and tubulin.
As shown in Figure 9H, we found that only GKAP-249L reduced the expression of Shank by ∼50% (mean ± SE: 0.48 ± 0.15-fold in comparison with uninfected neurons) (Fig. 9I), whereas GKAP-249A had no effect on Shank expression (mean ± SE: 0.99 ± 0.20-fold in comparison with uninfected neurons) (Fig. 9I). Neither GKAP-249L or GKAP-249A had any effect on PSD-95 and tubulin expression. Taken together, the results of these experiments suggest that GKAP-249L induces specific Shank1 degradation.
It has recently been shown that synaptic activity induces Shank and GKAP (but not PSD-95) degradation by ubiquitination and proteasome degradation (Ehlers, 2003). Our data suggest the possibility that the Shank-GKAP complex is induced to aggregate and degrade in the absence of PSD-95 associated with GKAP. As previously done by Ehlers, we treated neurons with bicuculline or TTX in the presence or absence of MG132, and then stained them with Shank, GKAP, and PSD-95 antibodies looking for cell body aggregates of endogenous proteins.
As shown in Figure 9J-M, we found several cell body intracellular aggregates of Shank and GKAP in neurons treated with bicuculline with or without MG132, but very few aggregates in untreated neurons or neurons treated with TTX and MG132 (mean ± SE: 62.1% ± 6.9 of the neurons treated with 40 μm bicuculline plus 10 μm MG132 and 23.7% ± 3.9 of the neurons treated with 40 μm bicuculline alone, compared with 9.5% ± 3.3 of untreated neurons and 8.2% ± 9.2 of neurons treated with 100 μm TTX plus 10 μm MG132) (Fig. 9N). Interestingly, PSD-95 was almost always absent from these aggregates.
PSD-95—GKAP-Shank complex during synapse development
GKAP therefore induces Shank aggregation in neurons if it cannot associate with PSD-95. To test this further, we investigated whether Shank overexpression forms filaments or aggregates in young neurons (up to DIV7) when both GKAP and PSD-95 expression is low (C. Sala, unpublished observation) that can be rescued by the coexpression of GKAP and PSD-95. DIV4-5 neurons were transfected with GFP-Shank1B, alone or together with GKAP1A or GKAP1A plus PSD-95, and fixed on DIV6-7. Interestingly, GFP-Shank1B alone formed elongated intracellular aggregates (Fig. 10A1-A4,B1-B4), similar to the distribution observed with GFP-Shank1BΔPDZ (Fig. 1C1,C2,J,K), but formed intracellular aggregates when cotransfected with GKAP1A (Fig. 10E1-E4). These aggregates were redistributed in small clusters when PSD-95 was also coexpressed (Fig. 10C1-C4,D1-D4). As expected, GKAP1B did not modify Shank1B distribution in these neurons (Fig. 10F1-F4). These data suggest that GKAP and PSD-95 play a role in correct Shank targeting to, and association with synapses. Also in this case, PSD-95C3,5S was unable to recruit GKAP and Shank in clusters (Fig. 10G1-G4), thus suggesting that PSD-95 binding to GKAP is not enough but the ability of PSD-95 to localize at synapses is required.
Figure 10.
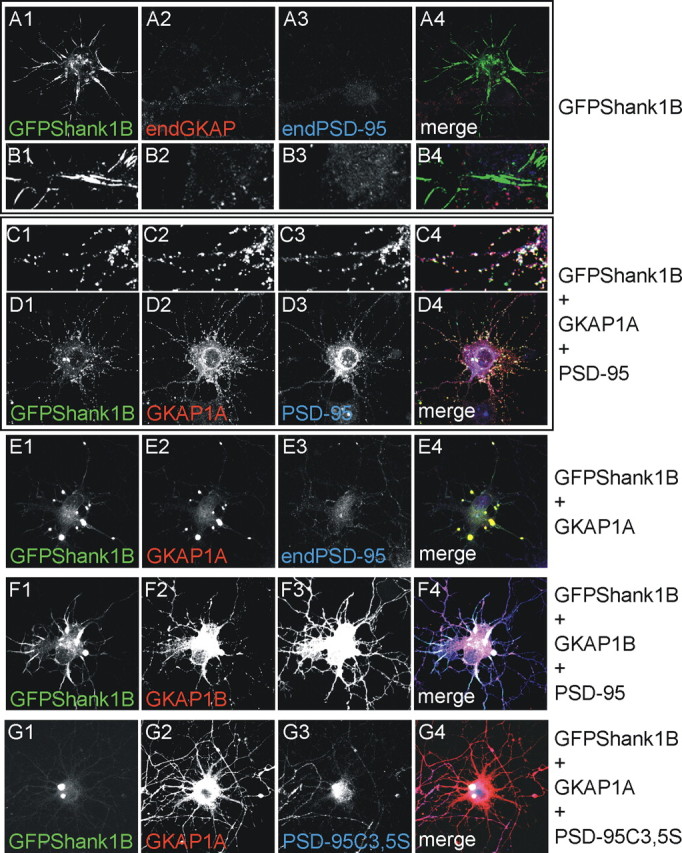
In young neurons, GFPShank1B is localized in synaptic-like clusters only if cotransfected with GKAP and PSD-95. A-D, As indicated on the right, the neurons were transfected on DIV4-5 with GFPShank1B alone (A1-A4, B1-B4), GFPShank1B and GKAP1A (E1-E4), GFPShank1B, GKAP1A, and PSD-95 (C1-C4, D1-D4), GFPShank1B, GKAP1B, and PSD-95 (F1-F4) or GFPShank1B, GKAP1B, and PSD-95C3,5S (G1-G4), and fixed on DIV6-7. The transfected neurons were labeled for GFP (A-G1), GKAP (A-G2), and PSD-95 (A-G3); the merge is shown in the panels on the right (A-G4). B1-B4 and C1-C4, respectively, show enlargements of A1-A4 and D1-D4.
We then investigated whether clusters of PSD-95—GKAP-Shank are localized to synaptic contacts by staining transfected neurons with presynaptic markers, such as bassoon and synaptophysin (Fig. 11A1-A3,B1-B3). We found that ∼65 and 60% of the clusters colocalized with the presynaptic markers, thus indicating that most of the clusters are localized at synapses (Fig. 11C).
Figure 11.

In young neurons, the majority of the clusters formed by GFPShank1B, GKAP, and PSD-95 colocalize with presynaptic markers. A, B, The neurons were transfected on DIV4-5 with GFPShank1B, GKAP1A, and PSD-95 and fixed on DIV6-7. The transfected neurons were labeled for GFP (A1, B1), bassoon (A2), or synaptophysin (B2); the merge is shown in the panels on the right (A3, B3). C, The graph shows the mean ± SE percentage of GFPShank1B clusters that colocalize with bassoon or synaptophysin. The data were obtained by counting at least 20 neurons from six coverslips obtained from three independent experiments for each antibody. D, DIV5 neurons were transfected with GFPShank1B and imaged on DIV8-10 using a CCD camera on an inverted microscope for 2-4 hr at 37°C in a 5% CO2 atmosphere. The images show the pictures acquired after 0, 24, 48, 72, 96, and 120 min. The first frame of the figure shows two small filaments formed by GFPShank1B (arrowheads). These filaments progressively became reorganized in small aggregates, as also indicated by the two arrows in the last frame acquired 120 min after the first.
We finally asked whether neuronal Shank1B filaments are stable or can be reorganized in small cluster, and how rapid this event may be. DIV5 neurons were transfected with GFP-Shank1B and imaged on DIV8-10 using a CCD camera for 2-4 hr. In some cells, we observed a disaggregation of the filaments into small clusters after ∼2 hr (Fig. 11D). The first frame of Figure 11D shows two small filament of GFP-Shank1B (arrowheads), which were progressively reorganized into small aggregates (see the two arrows in the last frame acquired 120 min later). We have performed similar time-lapse imaging experiments using GFP-Shank1B transfected COS cells, but never observed any Shank filament reorganization into small clusters (data not shown). These data suggest that Shank filaments can be reorganized into clusters at least in neurons, but it is still not known whether this is caused by the interaction between GKAP and PSD-95.
Discussion
In this study, we found that GKAP and Shank form intracellular aggregates when GKAP is not associated with synaptic PSD-95. These aggregates are inclusion bodies that can be defined as aggresomes because they colocalize with markers such as Hsp70 and neurofilaments. Furthermore, only the three-protein complex PSD-95—GKAP-Shank is organized in clusters in COS cells, and in synaptic clusters in developing neurons. We also found that Shank1 can multimerize as a result of an intermolecular interaction between the ankyrin repeat and SH3 domains. This multimerization induces the formation of filaments or fusiform aggregates in COS cells and neurons in the absence of GKAP interaction. Our data suggest that PSD-95-GKAP complex regulates Shank targeting and stability to synapses.
Shank1 multimerization
Two possible direct mechanisms of Shank multimerization have been previously described: Naisbitt et al. (1999) showed that Shank3 can multimerize through the SAM-SAM domain interaction, and a PDZ-PDZ interaction has recently been found for Shank1 (Im et al., 2003). CC-Homer (a Homer1b isoform with the coil-coil domain) can also indirectly cross-link two Shank molecules (Tu et al., 1999). We now provide biochemical and morphological evidence of another molecular mechanism of Shank1 multimerization: the binding of the ankyrin repeat domain to the SH3 domain. Classically, the SH3 domain recognizes the PXXP core motif preceded or followed by positively charged residue or residues (Musacchio, 2002), although it has more recently been shown that it can bind other sequences even if they do not contain proline-based sequences but usually contain hydrophobic amino acids (Kang et al., 2000). One characteristic of the ankyrin repeat domain is its predicted central hydrophobic α-helix structure (Bork, 1993) and so, although not totally unexpected, this is the first reported interaction between the SH3 and ankyrin repeat domains. Additionally, the interaction between the SH3 domain and the ankyrin repeats requires all of the ankyrin repeats, because further deletion of any ankyrin repeat abolished the interaction (data not shown). Therefore, the interaction between the SH3 domain and the ankyrin repeats of Shank represents a novel mode of SH3 domain mediated protein binding. It should be noted that also the SH3 domain of the PSD-95 family appears to have an atypical binding specificity to the guanylate kinase-like domain allowing an intramolecular or intermolecular interaction that regulate PSD-95 multimerization (Shin et al., 2000; McGee et al., 2001).
Interestingly, no ligand has been previously described for the Shank1 SH3 domain. In a pull-down experiments on brain extracts using the purified recombinant SH3 domain, we found that Shank itself is a strong interactor (M. Jiang and M. Zhang, unpublished observation), thus suggesting that the ankyrin repeat domain is the major binding partner of SH3.
The ability of Shank1 to multimerize by binding of the SH3 to ankyrin repeat domains correlates with the formation of filaments and fusiform aggregates in COS cells and neurons (Figs. 1, 2,4, 10), although it is difficult to determine how the macromolecular filaments are formed and why this requires the multimerization of Shank1.
We do not know whether endogenous Shank1 forms such filamentous aggregates in hippocampal neurons. We observed transfected GFP tag Shank1 filament and fusiform aggregates under two conditions: (1) when the PDZ domain was deleted and (2) when GFPShank1B (i.e., the full-length Shank1) was overexpressed in immature neurons, a condition in which GKAP (and probably other GKAP-SAPAP) are little expressed. These conditions can be created only if the proteins are overexpressed, thus making it almost impossible to do the experiment looking at endogenous proteins. However, when GFPShank1B is overexpressed in mature neurons, it localizes very well to synapses like the endogenous protein, thus suggesting that overexpression alone does not change the properties of the proteins. It is probable that GKAP and the other GKAP-SAPAP proteins are sufficiently highly expressed in mature neurons to inhibit filament formation. For all of these reasons, we believe that the surprising distribution of GFPShankΔPDZ in mature neurons, and GFPShank1B in immature neurons, is not an overexpression artifact but a consequence of protein-folding properties when PDZ binding partners are unbound to the domain. The function of Shank1 multimerization in neurons needs further investigation.
GFPShank1B modifies the distribution of tubulin, vimentin, and keratin (but not that of F-actin) in COS cells, whereas Shank1 increases F-actin in neuronal dendritic spines (Sala et al., 2001; Usui et al., 2003). These results suggest that other components associated with Shank and/or Homer are necessary for the synaptic recruitment of F-actin in neurons, such as IRSp53, which binds to activated Cdc42 (Bockmann et al., 2002; Soltau et al., 2002), βPIX, a guanine nucleotide exchange factor for Rac1 and Cdc42 small GTPases (Park et al., 2003), Abp1 and cortactin, two actin-binding protein (Naisbitt et al., 1999; Hering and Sheng, 2003; Qualmann et al., 2004).
PSD-95—GKAP-Shank complex assembling to synapses
It has been previously reported that the Shank1 PDZ domain plays a role in Shank1 targeting to synapses (Naisbitt et al., 1999; Sala et al., 2001; Yao et al., 2003), and here we show that the deletion of only the PDZ domain abolishes Shank1 synaptic targeting in neurons. Surprisingly, when Shank interacts with GKAP at the PDZ domain (full-length GKAP in developing neurons and COS cells, or GKAP-249L, which is unable to bind PSD-95, in mature neurons) forms intracellular aggregates that can be defined as aggresomes on the basis of Hsp70 and neurofilament staining (Kopito, 2000). Aggresomes usually accumulate proteins that cells recognize as being incorrectly folded, such as mutated transmembrane proteins or uncompleted protein complexes (Johnston et al., 1998; Kopito and Sitia, 2000).
Our data suggest that the GKAP-Shank complex is accumulated as an aggregate of unfolded proteins in COS cells and neuronal cell bodies. We also show that Shank and GKAP aggregates can be induced by increasing basal synaptic activity by means of bicuculline. It has been previously shown by Ehlers (2003) that synaptic activity induces the elimination of Shank and GKAP (but not PSD-95) from the PSD, and degradation by proteasomes. Our data suggest that Shank and GKAP may form a misfolded and unstable complex when removed from synapses, which may explain why, by competing with endogenous GKAP, GKAP-249L induces Shank aggregation and degradation in neurons. Finally, the fact that these aggregates are more abundant in the presence of a proteasome inhibitor suggests that the ubiquitination-proteasome pathway plays a role in their elimination.
A completely different distribution of Shank1 can be seen when GKAP and PSD-95 are coexpressed. In this case, the three proteins together form planar-like clusters in COS cells (Naisbitt et al., 1999; Tu et al., 1999) and synaptic clusters in developing young neurons. These data suggest at least that Shank1 needs to be bound to GKAP, and GKAP needs to be bound to PSD-95, to ensure the correct synaptic targeting of Shank1.
We have thus found that Shank1 has at least three different morphological conformations: (1) filamentous structure when it is essentially not bound to GKAP, (2) intracellular aggregates when it binds only GKAP, and (3) clusters (planar clusters in COS cells or synaptic clusters in neurons) when it binds the GKAP-PSD-95 complex. It is still unclear whether these different morphological conformations reflect a real molecular and structural change. The fact that the presence of PSD-95 is required to avoid GKAP-Shank aggregates suggests the possibility that the association of PSD-95 with GKAP and Shank acts like a chaperone protein, or that PSD-95 recruits some chaperone proteins to the GKAP-Shank complex to avoid the formation of Shank-GKAP aggregates. Interestingly this PSD-95 property is lost when the N-terminal palmitoylation is abolished.
It is still unknown whether the PSD-95—GKAP-Shank complex represents the minimal protein complex required for PSD assembly: e.g., it is not known when the complex is formed or whether its recruitment to synapses is regulated by synaptic activity, phosphorylation, or lipid binding (El-Husseini Ael et al., 2002). In in vivo and cultured neurons, the expression of the three proteins during development has a similar time course (Kawashima et al., 1997; Lim et al., 1999). Immunolocalization experiments using cultured neurons have shown that PSD-95 is almost always associated with GKAP (and probably Shank) in developing neurons (Rao et al., 1998).
There is evidence suggesting that PSD proteins are recruited to the synapses from the cytosol (Okabe et al., 1999, 2001; Friedman et al., 2000; Bresler et al., 2001; Prange and Murphy, 2001; McGee and Bredt, 2003), unlike the presynaptic compartment, which seems to travel to the synapses preassembled in large transport vesicles (Friedman et al., 2000; Bresler et al., 2004). Although our data (like those from most of the above studies) are mainly based on overexpression studies of Shank, GKAP, and PSD-95 wild-type and mutants in COS cells and neurons, they suggest that the assembly of PSD proteins at synapses may also require a controlled protein-protein interaction assembly and folding mechanism. Furthermore, at least in the case of GKAP and Shank, the association with PSD-95 is required for their complex assembly and stability. Finally, the possibility that a preassembled PSD-95—GKAP-Shank complex is targeted to synapses is intriguing but needs to be more directly tested.
Footnotes
C.S. was supported by the Giovanni Armenise-Harvard Foundation Career Development Program and MIUR 40%. M.P. is a DTI-Telethon assistant scientist. M.Z. was supported by the Research Grants Council of Hong Kong. We thank Morgan Sheng and Alaa El Din El-Husseini for their comments and suggestions and Pavel Osten for producing the lentivirus vectors.
Correspondence should be addressed to Carlo Sala, Consiglio Nazionale delle Ricerche, Institute of Neuroscience, Cellular and Molecular Pharmacology, Department of Pharmacology, University of Milan, Via Vanvitelli 32, 20129 Milan, Italy. E-mail: c.sala@in.cnr.it.
Copyright © 2004 Society for Neuroscience 0270-6474/04/249391-14$15.00/0
References
- Beneken J, Tu JC, Xiao B, Nuriya M, Yuan JP, Worley PF, Leahy DJ (2000) Structure of the Homer EVH1 domain-peptide complex reveals a new twist in polyproline recognition. Neuron 26: 143-154. [DOI] [PubMed] [Google Scholar]
- Bockmann J, Kreutz MR, Gundelfinger ED, Bockers TM (2002) ProSAP/Shank postsynaptic density proteins interact with insulin receptor tyrosine kinase substrate IRSp53. J Neurochem 83: 1013-1017. [DOI] [PubMed] [Google Scholar]
- Boeckers TM, winter C, Smalla KH, Kreutz MR, Bockmann J, Seidenbecher C, Garner CC, Gundelfinger ED (1999) Proline-rich synapse-associated proteins ProSAP1 and ProSAP2 interact with synaptic proteins of the SAPAP/GKAP family. Biochem Biophys Res Commun 264: 247-252. [DOI] [PubMed] [Google Scholar]
- Boeckers TM, Bockmann J, Kreutz MR, Gundelfinger ED (2002) ProSAP/Shank proteins - a family of higher order organizing molecules of the postsynaptic density with an emerging role in human neurological disease. J Neurochem 81: 903-910. [DOI] [PubMed] [Google Scholar]
- Bonaglia MC, Giorda R, Borgatti R, Felisari G, Gagliardi C, Selicorni A, Zuffardi O (2001) Disruption of the ProSAP2 gene in a t(12;22)(q24.1; q13.3) is associated with the 22q13.3 deletion syndrome. Am J Hum Genet 69: 261-268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bork P (1993) Hundreds of ankyrin-like repeats in functionally diverse proteins: mobile modules that cross phyla horizontally? Proteins 17: 363-374. [DOI] [PubMed] [Google Scholar]
- Bresler T, Ramati Y, Zamorano PL, Zhai R, Garner CC, Ziv NE (2001) The dynamics of SAP90/PSD-95 recruitment to new synaptic junctions. Mol Cell Neurosci 18: 149-167. [DOI] [PubMed] [Google Scholar]
- Bresler T, Shapira M, Boeckers T, Dresbach T, Futter M, Garner CC, Rosenblum K, Gundelfinger ED, Ziv NE (2004) Postsynaptic density assembly is fundamentally different from presynaptic active zone assembly. J Neurosci 24: 1507-1520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brewer GJ, Torricelli JR, Evege EK, Price PJ (1993) Optimized survival of hippocampal neurons in B27-supplemented Neurobasal, a new serum-free medium combination. J Neurosci Res 35: 567-576. [DOI] [PubMed] [Google Scholar]
- Ehlers MD (2003) Activity level controls postsynaptic composition and signaling via the ubiquitin-proteasome system. Nat Neurosci 6: 231-242. [DOI] [PubMed] [Google Scholar]
- El-Husseini AE, Craven SE, Chetkovich DM, Firestein BL, Schnell E, Aoki C, Bredt DS (2000) Dual palmitoylation of PSD-95 mediates its vesiculotubular sorting, postsynaptic targeting, and ion channel clustering. J Cell Biol 148: 159-172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El-Husseini Ael D, Schnell E, Dakoji S, Sweeney N, Zhou Q, Prange O, Gauthier-Campbell C, Aguilera-Moreno A, Nicoll RA, Bredt DS (2002) Synaptic strength regulated by palmitate cycling on PSD-95. Cell 108: 849-863. [DOI] [PubMed] [Google Scholar]
- Friedman HV, Bresler T, Garner CC, Ziv NE (2000) Assembly of new individual excitatory synapses: time course and temporal order of synaptic molecule recruitment. Neuron 27: 57-69. [DOI] [PubMed] [Google Scholar]
- Garner CC, Nash J, Huganir RL (2000) PDZ domains in synapse assembly and signalling. Trends Cell Biol 10: 274-280. [DOI] [PubMed] [Google Scholar]
- Hering H, Sheng M (2003) Activity-dependent redistribution and essential role of cortactin in dendritic spine morphogenesis. J Neurosci 23: 11759-11769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirao K, Hata Y, Ide N, Takeuchi M, Irie M, Yao I, Deguchi M, Toyoda A, Sudhof TC, Takai Y (1998) A novel multiple PDZ domain-containing molecule interacting with N-methyl-d-aspartate receptors and neuronal cell adhesion proteins. J Biol Chem 273: 21105-21110. [DOI] [PubMed] [Google Scholar]
- Hirao K, Hata Y, Deguchi M, Yao I, Ogura M, Rokukawa C, Kawabe H, Mizoguchi A, Takai Y (2000) Association of synapse-associated protein 90/postsynaptic density-95-associated protein (SAPAP) with neurofilaments. Genes Cells 5: 203-210. [DOI] [PubMed] [Google Scholar]
- Hsueh Y-P, Kim E, Sheng M (1997) Disulfide-linked head-to-head multimerization in the mechanism of ion channel clustering by PSD-95. Neuron 18: 803-814. [DOI] [PubMed] [Google Scholar]
- Im YJ, Lee JH, Park SH, Park SJ, Rho SH, Kang GB, Kim E, Eom SH (2003) Crystal structure of the shank PDZ-ligand complex reveals a class I PDZ interaction and a novel PDZ-PDZ dimerization. J Biol Chem 278: 48099-48104. [DOI] [PubMed] [Google Scholar]
- Johnston JA, Ward CL, Kopito RR (1998) Aggresomes: a cellular response to misfolded proteins. J Cell Biol 143: 1883-1898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang H, Freund C, Duke-Cohan JS, Musacchio A, Wagner G, Rudd CE (2000) SH3 domain recognition of a proline-independent tyrosine-based RKxxYxxY motif in immune cell adaptor SKAP55. EMBO J 19: 2889-2899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawabe H, Hata Y, Takeuchi M, Ide N, Mizoguchi A, Takai Y (1999) nArgBP2, a novel neural member of ponsin/ArgBP2/vinexin family that interacts with synapse-associated protein 90/postsynaptic density-95-associated protein (SAPAP). J Biol Chem 274: 30914-30918. [DOI] [PubMed] [Google Scholar]
- Kawashima N, Takamiya K, Sun J, Kitabatake A, Sobue K (1997) Differential expression of isoforms of PSD-95 binding protein (GKAP/SAPAP1) during rat brain development. FEBS Lett 418: 301-304. [DOI] [PubMed] [Google Scholar]
- Kennedy MB (2000) Signal-processing machines at the postsynaptic density. Science 290: 750-754. [DOI] [PubMed] [Google Scholar]
- Kim E, Cho K-O, Rothschild A, Sheng M (1996) Heteromultimerization and NMDA receptor-clustering activity of chapsyn-110, a member of the PSD-95 family of proteins. Neuron 17: 103-113. [DOI] [PubMed] [Google Scholar]
- Kim E, Naisbitt S, Hsueh Y-P, Rao A, Rothschild A, Craig AM, Sheng M (1997) GKAP, a novel synaptic protein that interacts with the guanylate kinase-like domain of the PSD-95/SAP90 family of channel clustering molecules. J Cell Biol 136: 669-678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kopito RR (2000) Aggresomes, inclusion bodies and protein aggregation. Trends Cell Biol 10: 524-530. [DOI] [PubMed] [Google Scholar]
- Kopito RR, Sitia R (2000) Aggresomes and Russell bodies. Symptoms of cellular indigestion? EMBO Rep 1: 225-231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim S, Naisbitt S, Yoon J, Hwang JI, Suh PG, Sheng M, Kim E (1999) Characterization of the Shank family of synaptic proteins. Multiple genes, alternative splicing, and differential expression in brain and development. J Biol Chem 274: 29510-29518. [DOI] [PubMed] [Google Scholar]
- Lois C, Hong EJ, Pease S, Brown EJ, Baltimore D (2002) Germline transmission and tissue-specific expression of transgenes delivered by lentiviral vectors. Science 295: 868-872. [DOI] [PubMed] [Google Scholar]
- McGee AW, Bredt DS (2003) Assembly and plasticity of the glutamatergic postsynaptic specialization. Curr Opin Neurobiol 13: 111-118. [DOI] [PubMed] [Google Scholar]
- McGee AW, Dakoji SR, Olsen O, Bredt DS, Lim WA, Prehoda KE (2001) Structure of the SH3-guanylate kinase module from PSD-95 suggests a mechanism for regulated assembly of MAGUK scaffolding proteins. Mol Cell 8: 1291-1301. [DOI] [PubMed] [Google Scholar]
- Musacchio A (2002) How SH3 domains recognize proline. Adv Protein Chem 61: 211-268. [DOI] [PubMed] [Google Scholar]
- Naisbitt S, Kim E, Weinberg RJ, Rao A, Yang F-C, Craig AM, Sheng M (1997) Characterization of guanylate kinase-associated protein, a postsynaptic density protein at excitatory synapses that interacts directly with postysynaptic density-95/synapse-associated protein 90. J Neurosci 17: 5687-5696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naisbitt S, Kim E, Tu JC, Xiao B, Sala C, Valtschanoff J, Weinberg RJ, Worley PF, Sheng M (1999) Shank, a novel family of postsynaptic density proteins that binds to the NMDA receptor/PSD-95/GKAP complex and cortactin. Neuron 23: 569-582. [DOI] [PubMed] [Google Scholar]
- Naisbitt S, Valtschanoff J, Allison D, Sala C, Kim E, Craig A, Weinberg R, M S (2000) Interaction of the postsynaptic density-95/guanylate kinase domain-associated protein complex with a light chain of myosin-V and dynein. J Neurosci 20: 4524-4534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naldini L, Blomer U, Gallay P, Ory D, Mulligan R, Gage FH, Verma IM, Trono D (1996) In vivo gene delivery and stable transduction of nondividing cells by a lentiviral vector. Science 272: 263-267. [DOI] [PubMed] [Google Scholar]
- Okabe S, Kim HD, Miwa A, Kuriu T, Okado H (1999) Continual remodeling of postsynaptic density and its regulation by synaptic activity. Nat Neurosci 2: 804-811. [DOI] [PubMed] [Google Scholar]
- Okabe S, Urushido T, Konno D, Okado H, Sobue K (2001) Rapid redistribution of the postsynaptic density protein PSD-Zip45 (Homer 1c) and its differential regulation by NMDA receptors and calcium channels. J Neurosci 21: 9561-9571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park E, Na M, Choi J, Kim S, Lee JR, Yoon J, Park D, Sheng M, Kim E (2003) The Shank family of postsynaptic density proteins interacts with and promotes synaptic accumulation of the beta PIX guanine nucleotide exchange factor for Rac1 and Cdc42. J Biol Chem 278: 19220-19229. [DOI] [PubMed] [Google Scholar]
- Prange O, Murphy TH (2001) Modular transport of postsynaptic density-95 clusters and association with stable spine precursors during early development of cortical neurons. J Neurosci 21: 9325-9333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qualmann B, Boeckers TM, Jeromin M, Gundelfinger ED, Kessels MM (2004) Linkage of the actin cytoskeleton to the postsynaptic density via direct interactions of Abp1 with the ProSAP/Shank family. J Neurosci 24: 2481-2495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rao A, Kim E, Sheng M, Craig AM (1998) Heterogeneity in the molecular composition of excitatory postsynaptic sites during development of hippocampal neurons in culture. J Neurosci 18: 1217-1229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sala C, Piech V, Wilson NR, Passafaro M, Liu G, Sheng M (2001) Regulation of dendritic spine morphology and synaptic function by Shank and Homer. Neuron 31: 115-130. [DOI] [PubMed] [Google Scholar]
- Sala C, Futai K, Yamamoto K, Worley PF, Hayashi Y, Sheng M (2003) Inhibition of dendritic spine morphogenesis and synaptic transmission by activity-inducible protein Homer1a. J Neurosci 23: 6327-6337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Satoh K, Yanai H, Senda T, Kohu K, Nakamura T, Okumura N, Matsumine A, Kobayashi S, Toyoshima K, Akiyama T (1997) DAP-1, a novel protein that interacts with the guanylate kinase-like domains of hDLG and PSD-95. Genes Cells 2: 415-424. [DOI] [PubMed] [Google Scholar]
- Scannevin RH, Huganir RL (2000) Postsynaptic organization and regulation of excitatory synapses. Nat Rev Neurosci 1: 133-141. [DOI] [PubMed] [Google Scholar]
- Sheng M, Kim E (2000) The Shank family of scaffold proteins. J Cell Sci 113: 1851-1856. [DOI] [PubMed] [Google Scholar]
- Sheng M, Pak DTS (2000) Ligand-gated ion channel interactions with cytoskeletal and signaling proteins. Annu Rev Physiol 62: 755-778. [DOI] [PubMed] [Google Scholar]
- Shin H, Hsueh YP, Yang FC, Kim E, Sheng M (2000) An intramolecular interaction between Src homology 3 domain and guanylate kinase-like domain required for channel clustering by postsynaptic density-95/SAP90. J Neurosci 20: 3580-3587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soltau M, Richter D, Kreienkamp HJ (2002) The insulin receptor substrate IRSp53 links postsynaptic shank1 to the small G-protein cdc42. Mol Cell Neurosci 21: 575-583. [DOI] [PubMed] [Google Scholar]
- Takeuchi M, Hata Y, Hirao K, Toyoda A, Irie M, Takai Y (1997) SAPAPs. A family of PSD-95/SAP90-associated proteins localized at postsynaptic density. J Biol Chem 272: 11943-11951. [DOI] [PubMed] [Google Scholar]
- Tu JC, Xiao B, Yuan JP, A. LA, Leoffert K, Li M, Linden DJ, Worley PF (1998) Homer binds a novel proline-rich motif and links group 1 metabotropic glutamate receptors with IP3 receptors. Neuron 21: 717-726. [DOI] [PubMed] [Google Scholar]
- Tu JC, Xiao B, Naisbitt S, Yuan JP, Petralia RS, Brakeman P, Doan A, Aakalu VK, Lanahan AA, Sheng M, Worley PF (1999) Coupling of mGluR/Homer and PSD-95 complexes by the Shank family of postsynaptic density proteins. Neuron 23: 583-592. [DOI] [PubMed] [Google Scholar]
- Usui S, Konno D, Hori K, Maruoka H, Okabe S, Fujikado T, Tano Y, Sobue K (2003) Synaptic targeting of PSD-Zip45 (homer 1c) and its involvement in the synaptic accumulation of F-actin. J Biol Chem 10: 10. [DOI] [PubMed] [Google Scholar]
- Wilson HL, Wong AC, Shaw SR, Tse WY, Stapleton GA, Phelan MC, Hu S, Marshall J, McDermid HE (2003) Molecular characterisation of the 22q13 deletion syndrome supports the role of haploinsufficiency of SHANK3/PROSAP2 in the major neurological symptoms. J Med Genet 40: 575-584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao B, Tu JC, Petralia RS, Yuan JP, Doan A, Breder CD, Ruggiero A, Lanahan AA, Wenthold RJ, Worley PF (1998) Homer regulates the association of group 1 metabotropic glutamate receptors with multivalent complexes of homer-related, synaptic proteins. Neuron 21: 707-716. [DOI] [PubMed] [Google Scholar]
- Yao I, Iida J, Nishimura W, Hata Y (2003) Synaptic localization of SAPAP1, a synaptic membrane-associated protein. Genes Cells 8: 121-129. [DOI] [PubMed] [Google Scholar]
- Zitzer H, Honck HH, Bachner D, Richter D, Kreienkamp HJ (1999) Somatostatin receptor interacting protein defines a novel family of multidomain proteins present in human and rodent brain. J Biol Chem 274: 32997-33001. [DOI] [PubMed] [Google Scholar]