

Abstract
We examined the mechanism underlying increased quantal content after block of activity at the mouse neuromuscular junction in vivo. We found that, when quantal content was measured in solution containing normal extracellular calcium, block of activity had no effect on either quantal content or the response to repetitive stimulation. However, when quantal content was measured in low extracellular calcium, there was a large increase in quantal content after block of activity. The increase in quantal content was accompanied by increased depression during repetitive stimulation. The explanation for these findings was that there was a shift in the calcium dependence of release after block of activity that manifested as an increase in probability of release in low extracellular calcium. Block of presynaptic P/Q channels eliminated the increase in probability of release. We propose that activity-dependent regulation of presynaptic calcium entry may contribute to homeostatic regulation of quantal content.
Keywords: transmitter release, synaptic plasticity, neuromuscular junction, synaptic activity, quantal content, calcium channel, homeostatic
Introduction
The ways in which activity regulates synaptic strength have been studied extensively but are still only partially understood. It has been proposed recently that there is a series of changes in synaptic function after manipulations of activity that serve to maintain the firing rates of networks within certain boundaries (Davis and Bezprozvanny, 2001; Burrone and Murthy, 2003; Turrigiano and Nelson, 2004). This type of synaptic plasticity is known as homeostatic regulation, and its cardinal feature is that changes in synaptic strength oppose changes in synaptic activity. Homeostatic regulation appears to involve a number of distinct mechanisms, including changes in cell excitability, changes in the number of vesicles released (quantal content), and changes in the response to an individual quantum (quantal size) (Davis and Bezprozvanny, 2001; Burrone and Murthy, 2003; Turrigiano and Nelson, 2004). Most studies of homeostatic regulation of synaptic function have focused on quantal size because accurate measurement of quantal content is difficult, if not impossible, for neurons receiving inputs from many synapses.
The mammalian neuromuscular junction is a classic synaptic preparation ideally suited to studies of synaptic function in vivo, because there is only one presynaptic input and one neurotransmitter. The simplicity of the preparation allows for easy differentiation between changes in synaptic strength that are caused by changes in quantal size versus changes in quantal content. The first report of what would now be termed homeostatic regulation of synaptic strength came from a study at the neuromuscular junction (Snider and Harris, 1979). In that study, an increase in quantal content was found after block of synaptic activity. Here, we set out to develop a more detailed understanding of the mechanism underlying homeostatic regulation of quantal content after block of activity at the neuromuscular junction in vivo.
Materials and Methods
TTX cuff placement on the left sciatic nerve was performed under intraperitoneal anesthesia (140 mg/kg ketamine and 15 mg/kg xylazine). TTX cuffs were fashioned from SILASTIC tubing (Dow Corning, Midland, MI) and were connected to an Alzet osmotic pump model 1002 (Durect, Cupertino, CA) containing 850 μm tetrodotoxin dissolved in 0.9% NaCl containing 200 U/ml penicillin and 200 U/ml streptomycin. Two- to 4-month-old mice were used for experiments. They were killed using CO2 inhalation, and the tibialis anterior muscle was removed. The tibialis anterior muscle was chosen because it had a favorable endplate band configuration for recording. It is innervated primarily by fast motor units that are only active during movement in rodents (Roy et al., 1991). We chose 8-10 d as the time point to study quantal content because, at that time, nerve damage by the TTX cuff with subsequent reinnervation could not confound the results. In pilot experiments, we found that, after sciatic nerve crush at the point at which we normally place the TTX cuff, reinnervation of muscle begins at 10 d. All animal surgery and EMG analysis was performed in accordance with Emory University Institutional Animal Care and Use Committee guidelines.
For most experiments, the recording chamber was continuously perfused with Ringer's solution containing the following (in mmol/l): 118 NaCl, 3.5 KCl, 2 CaCl2, 0.7 MgSO4, 26.2 NaHCO3, 1.7 NaH2PO4, and 5.5 glucose, pH 7.3-7.4 (20-22°C, equilibrated with 95% O2 and 5% CO2). For lowering the probability of release, some recordings were done in low (1 mm) Ca2+ solutions in which the Mg2+ concentration was kept at 0.7 mm. In other experiments, MgSO4 was raised to 8 mm to further lower the probability of release without an additional decrease in the concentration of divalent cations. All of the other components of the recording solution were unchanged. The concentrations of extracellular Ca2+ used to determine the Ca2+ dependence of release were 0.6, 0.9, and 1.2 mm and were used in conjunction with 8 mm Mg2+.
Endplate recordings were performed as follows. After dissection, the tibialis anterior muscle was partially bisected and folded apart to flatten the muscle. After pinning, muscle strips were stained with 10 μm 4-Di-2ASP [4-(4-diethylaminostyryl)-N-methylpyridinium iodide] (Magrassi et al., 1987) and imaged with an upright epifluorescence microscope (DMR; Leica, Bannockburn, IL). At this concentration, 4-Di-2ASP staining enabled visualization of surface nerve terminals as well as individual surface muscle fibers. All of the endplates were imaged and impaled within 100 μm as described previously (Rich et al., 2002). We used two-electrode voltage clamp to measure endplate current (EPC) and miniature EPC (MEPC) amplitude as described previously (Rich et al., 2002). In experiments in normal external calcium, muscle fibers were crushed away from the endplate band and voltage clamped to -45 mV to avoid movement after nerve stimulation (Barstad and Lilleheil, 1968; Glavinovic, 1979; Argentieri et al., 1992). However, in experiments performed in low calcium, muscle contraction was not an issue, so muscle fibers were not crushed, and fibers were clamped at -70 mV.
Determination of the role of P/Q Ca2+ channels in evoked release was performed by measuring quantal content in individual muscles before and after application of ω-agatoxin GIVA (Peptide Institute, Osaka, Japan). After recording the quantal content from at least five endplates in a tibialis anterior muscle in the absence of ω-agatoxin, each muscle was placed in 2 ml of Ringer's solution containing 1 μm ω-agatoxin GIVA for 1 hr. The solution was continuously bubbled with 95% O2 and 5% CO2. After 1 hr, the muscle was put into a recording chamber that was slowly perfused with Ringer's solution containing no ω-agatoxin GIVA. Quantal content was then measured from at least five endplates. All of the endplate recordings after the application of ω-agatoxin GIVA were made within 30 min of placing the muscle in the recording chamber. During this 30 min period, quantal content did not increase because of washout of ω-agatoxin GIVA. Determination of quantal content and response to repetitive stimulation were performed as outlined previously (Rich et al., 2002). In most experiments, the quantal content was calculated directly by dividing EPC amplitude by the average MEPC amplitude. However, when the quantal content was <0.5 (after blockade of P/Q channels), the method of failures was used to calculate quantal content (Del Castillo and Katz, 1954). Between quantal contents of 0.3 and 0.5, there was good agreement between the direct measure of quantal content and the method of failures. When the method of failures was used, 100-200 stimulations were used to ensure an adequate sample size.
Data were tested for group differences (i.e., TTX cuff vs control) using nested ANOVA (Neter et al., 1990). The n given for all data refers to the number of muscles studied. At least five muscle fibers were sampled from each muscle studied. Bonferroni correction was applied in instances in which multiple comparisons were made between treatment groups. Mean values ± 1 SEM are shown.
Results
Alterations in synaptic function after block of synaptic activity depend on external Ca2+ concentration
We measured endplate currents and calculated quantal contents at endplates in which evoked transmitter release was blocked by TTX for 8-10 d and at control endplates that were untreated. Our use of the crushed muscle fiber preparation permitted measurement of endplate currents at normal Ca2+ concentrations (2 mm). Under these conditions, we observed that neither endplate currents nor quantal contents were different between TTX and control endplates (Fig. 1A). This observation was surprising, given previous reports of increases in quantal contents after block of nerve activity with TTX (Snider and Harris, 1979; Tsujimoto and Kuno, 1988; Tsujimoto et al., 1990). However, previous studies were performed with endplates bathed in solution containing low external Ca2+-raised Mg2+, and, when we reproduced those conditions, we found a significant increase in quantal content (Fig. 1B) (p < 0.01). To rule out the possibility that cuff placement, rather than block of activity, caused an increase in quantal content, we placed cuffs containing normal saline in three mice. When quantal content was recorded 8-10 d later in solution containing low external Ca2+-raised Mg2+, there was no effect of saline cuff placement (2.3 ± 0.6 vs 2.2 ± 0.8; saline cuff vs control; n = 3 muscles for each treatment; p = 0.87). These findings confirm that block of activity alters synaptic transmission and demonstrate that expression of this effect depends on external Ca2+.
Figure 1.
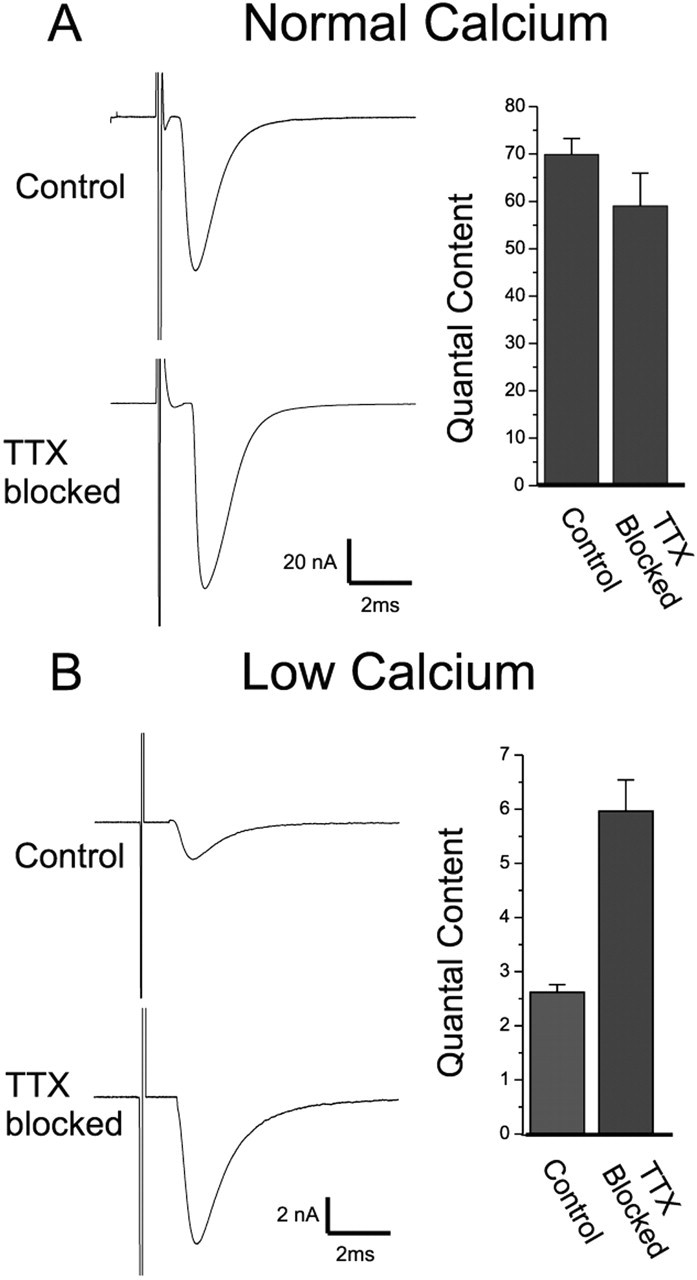
Blocking activity increases quantal content in low, but not normal, extracellular Ca2+. A, Average EPCs and quantal content measured in normal calcium after block of activity. Shown are representative EPCs evoked in normal Ca2+ (2 mm) and normal Mg2+ (0.7 mm) that were obtained from averaging 20-30 nerve stimulations. There was little difference in either EPC amplitude or quantal content between the control and TTX-blocked endplates (p = 0.22 for EPC amplitude; p = 0.15 for quantal content; n = 20 control muscles, 12 TTX-blocked muscles). B, Average EPCs and quantal content measured in low Ca2+ (1 mm) and high Mg2+ (8 mm) after block of activity. Both the EPC amplitude and the quantal content are much larger after block of synaptic activity (p < 0.01 for both measures; n = 14 control muscles, 10 TTX blocked muscles). Note the difference in calibration for EPC traces in A and B. Control, endplates in which synaptic activity is normal; TTX blocked, endplates in which synaptic activity was blocked for 8-10 d with a TTX cuff around the nerve.
The finding that quantal content was increased in low external Ca2+-raised Mg2+ but unchanged in normal external Ca2+ suggested that probability of release might be increased. To infer the probability of release, we examined short-term synaptic plasticity during repetitive stimulation. Synapses with a high probability of release undergo depression, whereas those with a low probability of release undergo facilitation (Zucker and Regehr, 2002). In normal Ca2+, the amount of depression by the 10th pulse of a 50 Hz train was similar in both control and TTX-blocked endplates (Fig. 2) (p = 0.56). However, in solution containing low extracellular Ca2+-normal Mg2+, facilitation present on the 10th pulse of a 50 Hz train was reduced in TTX-blocked endplates relative to control (p < 0.01) (Fig. 2). Thus, both initial quantal content and short-term plasticity were only altered after block of synaptic activity, when measured in solution containing low extracellular calcium. Both changes were consistent with an increase in probability of release when extracellular Ca2+ was low.
Figure 2.
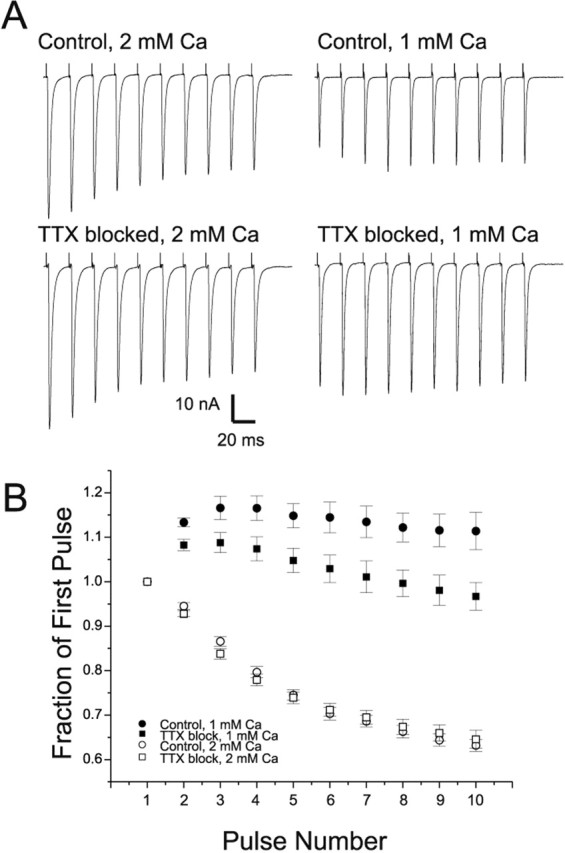
Repetitive stimulation suggests that probability of release is increased in low, but not normal, calcium after block of synaptic activity. A, The average EPCs from 6-10 trains of 50 Hz pulses are shown for control and TTX-blocked endplates in normal (2 mm Ca2+-0.7 mm Mg2+) Ca2+ and low (1 mm Ca2+-0.7 mm Mg2+) Ca2+. In normal Ca2+, there was a similar degree of depression in control and TTX-blocked endplates. In low Ca2+, although facilitation occurred in both control and TTX-blocked endplates, the facilitation was not as great in the TTX-blocked endplate and was offset by depression later in the train, such that the 10th pulse had the same amplitude as the first pulse. Calibration shown is the same for normal and low Ca2+. Note that the low Ca2+ solution used in this study had normal Mg2+, as opposed to the solution used in Figure 1.B, Normalized average EPCs during trains of 50 Hz pulses are shown for control (n = 8 muscles) and TTX-blocked (n = 6 muscles) endplates in low and normal extracellular Ca2+. In low Ca2+, both control and TTX-blocked endplates showed facilitation during repetitive stimulation. However, the degree of facilitation decreased in the TTX-blocked endplates (p < 0.01 for 2nd pulse; p < 0.05 for last pulse). In normal Ca2+, there was a similar degree of depression in control and TTX-blocked endplates (p = 0.18 for 2nd pulse; p = 0.57 for 10th pulse).
We next examined how the Ca2+ dependence of release had been altered to selectively increase probability of release in low extracellular Ca2+. Possibilities were that the slope of the Ca2+-release relationship had been altered or that the relationship had been shifted in a parallel manner. Previous studies of the Ca2+ dependence of quantal content at the neuromuscular junction have found a slope of between 3 and 4 on a double-logarithmic plot (Dodge and Rahamimoff, 1967; Cull-Candy et al., 1980; Smith, 1988). We measured quantal content at three concentrations of low external Ca2+ in the presence of raised extracellular Mg2+. At each Ca2+ concentration, quantal content was close to three times greater at the TTX-blocked endplates. The slope of the line fitting the data was close to 4 for both control and TTX-blocked endplates. However, data from the TTX-blocked end-plates were shifted to the left (Fig. 3).
Figure 3.
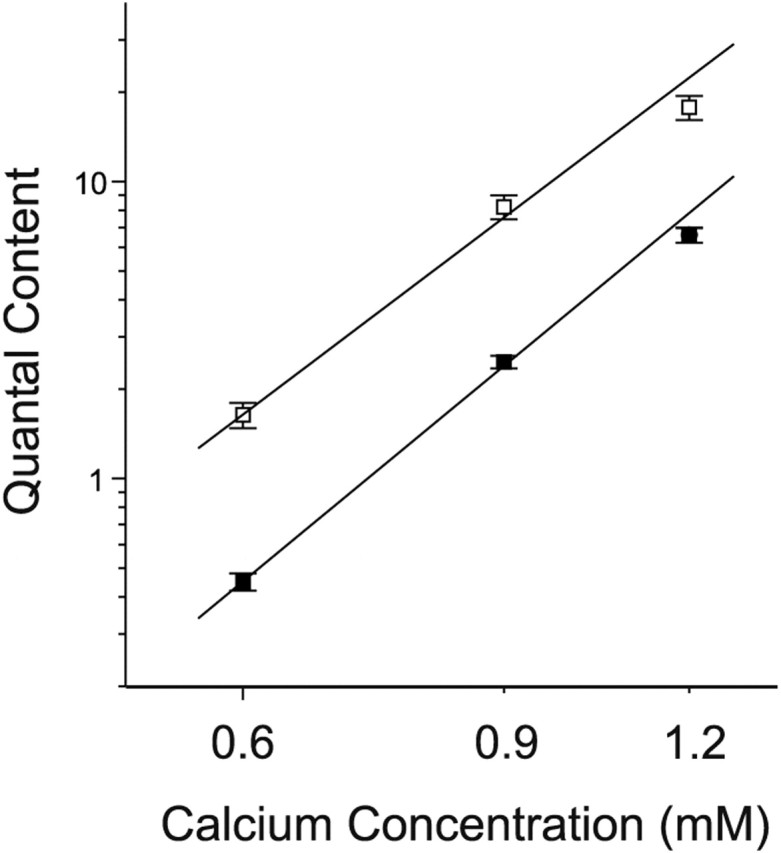
There is a leftward shift in the Ca2+ dependence of quantal content after block of synaptic activity. The quantal content of control and TTX-blocked endplates is plotted versus calcium concentration on a double-logarithmic plot. Magnesium concentration was 8 mm in all of the solutions used. The slope was 3.9 in control (r = 0.998; p = 0.04) and 3.5 in TTX-blocked (r = 0.995; p = 0.07) endplates. Open squares, TTX-blocked; filled squares, control.
Increased probability of release in low Ca2+ after block of synaptic activity is abolished after block of Ca2+ entry through P/Q channels
The leftward shift in Ca2+ dependence of transmitter release raised the possibility that calcium entry was increased in the presynaptic terminal. Ca2+ entry at the mature neuromuscular junction appears to occur primarily through P/Q Ca2+ channels, because blocking these channels reduces the evoked release by >90% (Uchitel et al., 1992; Hong and Chang, 1995; Giovannini et al., 2002). However, a small portion of transmitter release persists (Hong and Chang, 1995; Giovannini et al., 2002) because of Ca2+ current that is apparently carried by other Ca2+ channel types (Lin and Lin-Shiau, 1997). At developing, reinnervated, or botulinum-treated neuromuscular junctions, significant Ca2+ entry occurs through other Ca2+ channel types (Katz et al., 1996; Rosato Siri and Uchitel, 1999). Thus, an increase in Ca2+ current after the blockade of activity could occur because of either an increase in current through P/Q channels or the upregulation of other Ca2+ channel types. We reasoned that, if TTX blockade acted to increase presynaptic Ca2+ current, then blocking the Ca2+ channels responsible for the increase in Ca2+ entry should eliminate the difference in quantal contents in low Ca2+ between TTX-blocked and control endplates.
To determine whether increased Ca2+ current through P/Q channels mediated increased quantal content in low Ca2+, we measured the quantal content in control and TTX-blocked end-plates from individual mice before and after blocking the P/Q channels with ω-agatoxin GIVA. In these experiments, the control and experimental muscles were taken in pairs from individual mice in which one tibialis anterior muscle was from an untreated leg and the other was from a leg in which nerve activity had been blocked. Both muscles were recorded in the same dish in1mm Ca2+-8 mm Mg2+, before the application of ω-agatoxin. The quantal content was increased by close to 100% in endplates in which nerve activity had been blocked with TTX (p < 0.01) (Fig. 4A1). Both muscles were then placed in a solution containing 1 μm ω-agatoxin. After 1 hr in ω-agatoxin, the quantal content was measured again in each muscle. Blocking P/Q channels reduced quantal content by 96% in the control endplates and 98% in the TTX-blocked ones. After blockade of P/Q channels, quantal content was too low to measure using the direct method (Fig. 4A2), so the method of failures was used. Using the method of failures, quantal content was the same in the control and TTX-blocked endplates (0.11 ± 0.03 vs 0.10 ± 0.04; n = 4 mice; p = 0.8).
Figure 4.

Increased probability of release is eliminated after block of Ca2+ entry through P/Q channels. A1, Average EPCs from representative control (light trace) and TTX-blocked (dark trace) endplates in 1 mm Ca2+-8 mm Mg2+. A2, Representative average EPCs from control (light trace) and TTX-blocked (dark trace) endplates in 1 mm Ca2+-8 mm Mg2+ after block of P/Q channels with 1 μm ω-agatoxin GIVA. The light and dark traces are superimposed and cannot be distinguished. The majority of responses were failures, such that the average EPC is ∼10% of the amplitude of an individual MEPC. A3, Representative average EPCs from control (light trace) and TTX-blocked (dark trace) endplates after block of P/Q channels with 1 μm ω-agatoxin GIVA in 2 mm Ca2+-8 mm Mg2+. B, Quantal content of control and TTX-blocked endplates before and after the P/Q channels were blocked. The concentration of extracellular Ca2+ was doubled from 1 to 2 mm after the addition of ω-agatoxin, to return quantal content to the levels seen before block of P/Q channels. Before P/Q channel blockade, quantal content was significantly higher in TTX-blocked endplates (p < 0.01), but, after P/Q channel blockade, there was no significant difference (n = 5 muscles for both groups).
The similarity of quantal content after block of P/Q channels suggested that an increase in calcium entry through P/Q channels was responsible for the increased probability of release in low Ca2+. However, it was also possible that the severe reduction in quantal content after block of P/Q channels had moved release so far down the calcium-release relationship that we were simply unable to see the increase in quantal content after block of activity. To rule out this possibility, we repeated the experiment in a different set of animals, in which the extracellular Ca2+ was doubled after the P/Q channels were blocked. In this range of extracellular Ca2+, doubling the Ca2+ increases release 16-fold (Dodge and Rahamimoff, 1967; Cull-Candy et al., 1980; Smith, 1988) and would be expected to return the quantal content to the range it was before the P/Q channels were blocked. As expected, doubling the Ca2+ after ω-agatoxin exposure raised the quantal content to levels similar to those recorded before blocking the P/Q channels (Fig. 4A3,B). However, increasing the quantal content did not alter the result: the quantal content was similar in control and TTX-blocked endplates after blockade of the P/Q channels (p = 0.35).
Discussion
More than 25 years ago, it was shown that block of activity at the mammalian neuromuscular junction in vivo increased synaptic strength (Snider and Harris, 1979; Tsujimoto and Kuno, 1988; Tsujimoto et al., 1990). Recent studies have found that a similar increase in synaptic strength occurs after block of activity at synapses between neurons (Manabe et al., 1989; Webb and Cope, 1992; Davis and Bezprozvanny, 2001; Burrone and Murthy, 2003; Turrigiano and Nelson, 2004). Such an increase in synaptic strength is homeostatic, because it opposes the reduction of postsynaptic activity. Both increased quantal size (O'Brien et al., 1998; Turrigiano et al., 1998) and increased quantal content (Snider and Harris, 1979; Tsujimoto and Kuno, 1988; Tsujimoto et al., 1990; Murthy et al., 2001) contribute to homeostatic regulation of synaptic strength. Our study focused on the mechanism underlying homeostatic increases in quantal content.
Our data, as well as previous studies (Tsujimoto et al., 1990; Lnenicka and Hong, 1997; Murthy et al., 2001; Paradis et al., 2001; Moulder et al., 2003), suggest that homeostatic regulation of quantal content occurs because of regulation of probability of release. If homeostatic regulation of synaptic strength is mediated through increases in probability of release rather than increases in the number of synaptic sites, two predictions follow. (1) Although homeostatic regulation affects synapses with high and low probability of release, the functional consequence of homeostatic regulation will be greater at synapses with low probability of release. If synaptic sites have a high probability of release, homeostatic regulation will be less able to further increase the probability of release and will not induce large increases in quantal content. We found this to be the case at the neuromuscular junction in normal extracellular calcium. However, when the neuromuscular junction was made to more closely resemble central synapses by lowering the probability of release, the increase in probability of release after the blocking of activity had a large impact on quantal content. (2) Homeostatic increases in neurotransmitter release should increase depression and decrease facilitation (Zucker and Regehr, 2002). Therefore, not only would resting synaptic strength be altered, but there should also be changes in short-term synaptic plasticity. In low calcium solution, we found altered short-term plasticity after block of activity. The finding that both predictions were borne out supports our hypothesis that homeostatic regulation of quantal content involves modulation of probability of release.
Several mechanisms might underlie an increase in probability of release. These include an increase in the number of vesicles in the readily releasable pool, an increased sensitivity of individual vesicles to calcium, or an increase in calcium entry. There is evidence from an in vitro study of FM1-43 uptake that increased probability of release after block of activity may be caused by an increase in the number of vesicles in the readily releasable pool (Murthy et al., 2001). It is important to note, however, that the size of the readily releasable pool does not always correlate with release probability (Millar et al., 2002). There are no studies to date suggesting that increased calcium sensitivity of synaptic vesicles is involved in homeostatic regulation.
Data from several systems suggest that homeostatic regulation of probability of release involves regulation of calcium entry into the presynaptic terminal. As reported in this study, block of P/Q channels eliminated the difference in probability of release. At synapses between neurons in vitro, there is evidence that chronic depolarization of neurons (perhaps mimicking increased activity) reduces probability of release by reducing presynaptic calcium currents (Moulder et al., 2003). Work at the crayfish neuromuscular junction has shown that increased activity produces weakening of synaptic strength that lasts for days to weeks (Lnenicka and Atwood, 1985, 1989). The weakening appears to be triggered by increased presynaptic activity (Lnenicka and Atwood, 1989) and resultant calcium entry (Hong and Lnenicka, 1993). This triggers a long-term reduction in presynaptic calcium entry through P-type channels (Lnenicka and Hong, 1997). Thus, the mechanism underlying homeostatic regulation of quantal content appears to be conserved between neurons and the neuromuscular junction, as well as between vertebrates and invertebrates.
The molecular signals that underlie activity-dependent regulation of synaptic strength are only beginning to be understood. It has been proposed that CaMKII (calcium/calmodulin-dependent protein kinase II) signaling is involved in the activity-dependent regulation of quantal size and the number of functional synaptic sites (Thiagarajan et al., 2002; Haghighi et al., 2003; Pratt et al., 2003). Our data as well as previous studies (Lnenicka and Hong, 1997; Moulder et al., 2003) suggest that signals that regulate Ca2+ channel isoform expression or function (Atwood and Karunanithi, 2002; Weiss and Burgoyne, 2002; Reid et al., 2003) may be involved in activity-dependent regulation of synaptic function.
A key question that remains is what aspect of activity triggers the increase in probability of release. Possibilities include presynaptic activity, postsynaptic activity, and Hebbian regulation in which the timing of presynaptic and postsynaptic spiking is the important aspect of synaptic activity. Additional experiments will be necessary to distinguish among these possibilities.
Footnotes
This work was supported by National Institutes of Health Grant NS-40405 (M.M.R.).
Correspondence should be addressed to Mark Rich, Neurodegenerative Disease Center, Emory University School of Medicine, 615 Michael Street, Atlanta, GA 30322. E-mail: mmrich@emory.edu.
Copyright © 2004 Society for Neuroscience 0270-6474/04/2410687-06$15.00/0
References
- Argentieri TM, Aiken SP, Laxminarayan S, McArdle JJ (1992) Characteristics of synaptic transmission in reinnervating rat skeletal muscle. Pflügers Arch 421: 256-261. [DOI] [PubMed] [Google Scholar]
- Atwood HL, Karunanithi S (2002) Diversification of synaptic strength: presynaptic elements. Nat Rev Neurosci 3: 497-516. [DOI] [PubMed] [Google Scholar]
- Barstad JA, Lilleheil G (1968) Transversely cut diaphragm preparation from rat. An adjuvant tool in the study of the physiology and pharmacology of the myoneural junction. Arch Int Pharmacodyn Ther 175: 373-390. [PubMed] [Google Scholar]
- Burrone J, Murthy VN (2003) Synaptic gain control and homeostasis. Curr Opin Neurobiol 13: 560-567. [DOI] [PubMed] [Google Scholar]
- Cull-Candy SG, Miledi R, Trautmann A, Uchitel OD (1980) On the release of transmitter at normal, myasthenia gravis and myasthenic syndrome affected human end-plates. J Physiol (Lond) 299: 621-638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis GW, Bezprozvanny I (2001) Maintaining the stability of neural function: a homeostatic hypothesis. Annu Rev Physiol 63: 847-869. [DOI] [PubMed] [Google Scholar]
- Del Castillo J, Katz B (1954) Quantal components of the end-plate potential. J Physiol (Lond) 124: 560-573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dodge Jr FA, Rahamimoff R (1967) Co-operative action a calcium ions in transmitter release at the neuromuscular junction. J Physiol (Lond) 193: 419-432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giovannini F, Sher E, Webster R, Boot J, Lang B (2002) Calcium channel subtypes contributing to acetylcholine release from normal, 4-aminopyridine-treated and myasthenic syndrome auto-antibodies-affected neuromuscular junctions. Br J Pharmacol 136: 1135-1145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glavinovic MI (1979) Voltage clamping of unparalysed cut rat diaphragm for study of transmitter release. J Physiol (Lond) 290: 467-480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haghighi AP, McCabe BD, Fetter RD, Palmer JE, Hom S, Goodman CS (2003) Retrograde control of synaptic transmission by postsynaptic CaMKII at the Drosophila neuromuscular junction. Neuron 39: 255-267. [DOI] [PubMed] [Google Scholar]
- Hong SJ, Chang CC (1995) Inhibition of acetylcholine release from mouse motor nerve by a P-type calcium channel blocker, omega-agatoxin IVA. J Physiol (Lond) 482: 283-290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong SJ, Lnenicka GA (1993) Long-term changes in the neuromuscular synapses of a crayfish motoneuron produced by calcium influx. Brain Res 605: 121-127. [DOI] [PubMed] [Google Scholar]
- Katz E, Ferro PA, Weisz G, Uchitel OD (1996) Calcium channels involved in synaptic transmission at the mature and regenerating mouse neuromuscular junction. J Physiol (Lond) 497: 687-697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin MJ, Lin-Shiau SY (1997) Multiple types of Ca2+ channels in mouse motor nerve terminals. Eur J Neurosci 9: 817-823. [DOI] [PubMed] [Google Scholar]
- Lnenicka GA, Atwood HL (1985) Age-dependent long-term adaptation of crayfish phasic motor axon synapses to altered activity. J Neurosci 5: 459-467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lnenicka GA, Atwood HL (1989) Impulse activity of a crayfish motoneuron regulated its neuromuscular synaptic properties. J Neurophysiol 61: 91-96. [DOI] [PubMed] [Google Scholar]
- Lnenicka GA, Hong SJ (1997) Activity-dependent changes in voltage-dependent calcium currents and transmitter release. Mol Neurobiol 14: 37-66. [DOI] [PubMed] [Google Scholar]
- Magrassi L, Purves D, Lichtman JW (1987) Fluorescent probes that stain living nerve terminals. J Neurosci 7: 1207-1214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manabe T, Kaneko S, Kuno M (1989) Disuse-induced enhancement of Ia synaptic transmission in spinal motoneurons of the rat. J Neurosci 9: 2455-2461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Millar AG, Bradacs H, Charlton MP, Atwood HL (2002) Inverse relationship between release probability and readily releasable vesicles in depressing and facilitating synapses. J Neurosci 22: 9661-9667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moulder KL, Cormier RJ, Shute AA, Zorumski CF, Mennerick S (2003) Homeostatic effects of depolarization on Ca2+ influx, synaptic signaling, and survival. J Neurosci 23: 1825-1831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murthy VN, Schikorski T, Stevens CF, Zhu Y (2001) Inactivity produces increases in neurotransmitter release and synapse size. Neuron 32: 673-682. [DOI] [PubMed] [Google Scholar]
- Neter J, Wasserman W, Kutner MH (1990) Applied linear statistical models, Ed 3. Boston: Irwin.
- O'Brien RJ, Kamboj S, Ehlers MD, Rosen KR, Fischbach GD, Huganir RL (1998) Activity-dependent modulation of synaptic AMPA receptor accumulation. Neuron 21: 1067-1078. [DOI] [PubMed] [Google Scholar]
- Paradis S, Sweeney ST, Davis GW (2001) Homeostatic control of presynaptic release is triggered by postsynaptic membrane depolarization. Neuron 30: 737-749. [DOI] [PubMed] [Google Scholar]
- Pratt KG, Watt AJ, Griffith LC, Nelson SB, Turrigiano GG (2003) Activity-dependent remodeling of presynaptic inputs by postsynaptic expression of activated CaMKII. Neuron 39: 269-281. [DOI] [PubMed] [Google Scholar]
- Reid CA, Bekkers JM, Clements JD (2003) Presynaptic Ca2+ channels: a functional patchwork. Trends Neurosci 26: 683-687. [DOI] [PubMed] [Google Scholar]
- Rich MM, Waldeck RF, Cork LC, Balice-Gordon RJ, Fyffe RE, Wang X, Cope TC, Pinter MJ (2002) Reduced endplate currents underlie motor unit dysfunction in canine motor neuron disease. J Neurophysiol 88: 3293-3304. [DOI] [PubMed] [Google Scholar]
- Rosato Siri MD, Uchitel OD (1999) Calcium channels coupled to neurotransmitter release at neonatal rat neuromuscular junctions. J Physiol (Lond) 514: 533-540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roy RR, Hutchison DL, Pierotti DJ, Hodgson JA, Edgerton VR (1991) EMG patterns of rat ankle extensors and flexors during treadmill locomotion and swimming. J Appl Physiol 70: 2522-2529. [DOI] [PubMed] [Google Scholar]
- Smith DO (1988) Muscle-specific decrease in presynaptic calcium dependence and clearance during neuromuscular transmission in aged rats. J Neurophysiol 59: 1069-1082. [DOI] [PubMed] [Google Scholar]
- Snider WD, Harris GL (1979) A physiological correlate of disuse-induced sprouting at the neuromuscular junction. Nature 281: 69-71. [DOI] [PubMed] [Google Scholar]
- Thiagarajan TC, Piedras-Renteria ES, Tsien RW (2002) alpha- and beta-CaMKII. Inverse regulation by neuronal activity and opposing effects on synaptic strength. Neuron 36: 1103-1114. [DOI] [PubMed] [Google Scholar]
- Tsujimoto T, Kuno M (1988) Calcitonin gene-related peptide prevents disuse-induced sprouting of rat motor nerve terminals. J Neurosci 8: 3951-3957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsujimoto T, Umemiya M, Kuno M (1990) Terminal sprouting is not responsible for enhanced transmitter release at disused neuromuscular junctions of the rat. J Neurosci 10: 2059-2065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turrigiano GG, Nelson SB (2004) Homeostatic plasticity in the developing nervous system. Nat Rev Neurosci 5: 97-107. [DOI] [PubMed] [Google Scholar]
- Turrigiano GG, Leslie KR, Desai NS, Rutherford LC, Nelson SB (1998) Activity-dependent scaling of quantal amplitude in neocortical neurons. Nature 391: 892-896. [DOI] [PubMed] [Google Scholar]
- Uchitel OD, Protti DA, Sanchez V, Cherksey BD, Sugimori M, Llinas R (1992) P-type voltage-dependent calcium channel mediates presynaptic calcium influx and transmitter release in mammalian synapses. Proc Natl Acad Sci USA 89: 3330-3333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Webb CB, Cope TC (1992) Modulation of la EPSP amplitude: the effects of chronic synaptic inactivity. J Neurosci 12: 338-344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiss JL, Burgoyne RD (2002) Sense and sensibility in the regulation of voltage-gated Ca(2+) channels. Trends Neurosci 25: 489-491. [DOI] [PubMed] [Google Scholar]
- Zucker RS, Regehr WG (2002) Short-term synaptic plasticity. Annu Rev Physiol 64: 355-405. [DOI] [PubMed] [Google Scholar]