

Abstract
Niemann-Pick disease is caused by a genetic deficiency in acid sphingomyelinase (ASM) leading to the intracellular accumulation of sphingomyelin and cholesterol in lysosomes. In the present study, we evaluated the effects of direct intracerebral transplantation of neural progenitor cells (NPCs) on the brain storage pathology in the ASM knock-out (ASMKO) mouse model of Type A Niemann-Pick disease. NPCs derived from adult mouse brain were genetically modified to express human ASM (hASM) and were transplanted into multiple regions of the ASMKO mouse brain. Transplanted NPCs survived, migrated, and showed region-specific differentiation in the host brain up to 10 weeks after transplantation (the longest time point examined). In vitro, gene-modified NPCs expressed up to 10 times more and released five times more ASM activity into the culture media compared with nontransduced NPCs. In vivo, transplanted cells expressed hASM at levels that were barely detectable by immunostaining but were sufficient for uptake and cross-correction of host cells, leading to reversal of distended lysosomal pathology and regional clearance of sphingomyelin and cholesterol storage. Within the host brain, the area of correction closely overlapped with the distribution of the hASM-modified NPCs. No correction of pathology occurred in brain regions that received transplants of nontransduced NPCs. These results indicate that the presence of transduced NPCs releasing low levels of hASM within the ASMKO mouse brain is necessary and sufficient to reverse lysosomal storage pathology. Potentially, NPCs may serve as a useful gene transfer vehicle for the treatment of CNS pathology in other lysosomal storage diseases and neurodegenerative disorders.
Keywords: neural progenitor cell, transplantation, metabolic, neurodegenerative, storage, repair
Introduction
Type A Niemann-Pick disease (NPD-A) is an inherited lysosomal storage disease caused by a deficiency in acid sphingomyelinase (ASM). Absence of ASM enzyme activity results in progressive lysosomal accumulation of the nondegraded substrate sphingomyelin and cholesterol in most tissues, including the brain. Clinically, NPD-A patients fail to thrive and develop organomegaly and a rapidly deteriorating neurodegenerative course with death by 3 years of age (Schuchman and Desnick, 2001).
Enzyme replacement therapy using recombinant ASM protein delivery is a promising treatment for visceral storage pathology in NPD-A (Taylor and Wolfe, 1997; Torchiana et al., 1998; Miranda et al., 2000; Jin et al., 2002). However, this approach has proven ineffective in treating the CNS, because the blood-brain barrier prevents entry of circulating enzymes into the brain (Sly and Volger, 2002). Intracranial transplantation of cells as enzyme delivery vehicles is one viable approach for bypassing the blood-brain barrier altogether (Taylor and Wolfe, 1997; Jin et al., 2002). After transplantation, enzyme expressing donor-derived cells may supply sufficient ASM for uptake by neighboring host brain tissue and enzymatic “cross-correction” (Sando and Neufeld, 1977). To effectively treat the CNS aspect of NPD-A disease, a cell delivery vehicle is required that can migrate throughout the diseased brain, survive in vivo long term, and release therapeutic enzyme levels for uptake by surrounding neurons and other neural cells. In this regard, neural stem cells (NSCs) or neural progenitor cells (NPCs) have aroused a great deal of interest for their therapeutic potential in neurological disorders (Gage et al., 1995; McKay, 1997). In vitro, both types of cells have the capacity for proliferation and self-renewal and are multipotent, giving rise to differentiated cells of neuronal or glial lineage (Reynolds and Weiss, 1992; Gritti et al., 1996; Palmer et al., 1997). After transplantation into the brain, NSCs migrated throughout the CNS, differentiated exclusively into neural cell types, and survived for several months (Flax et al., 1998; Fricker et al., 1999; Englund et al., 2002). Genetically engineered NSCs or NPCs also survived long term and maintained expression of foreign genes after transplantation (Buchet et al., 2002), including delivery of therapeutic metabolic enzyme levels in an animal model of mucopolysaccharidosis VII (MPS VII) (Sly disease) (Meng et al., 2003).
A murine model of NPD-A, the ASM knock-out (ASMKO) mouse, mimics the human phenotype with prominent lysosomal pathology throughout the brain and other organs (Horinouchi et al., 1995). The aim of this study was to determine whether an allograft of mouse NPCs could deliver therapeutic levels of ASM and reduce storage pathology in ASMKO mice. Our results demonstrate that NPCs survive, migrate, and show regionally restricted differentiation in the diseased brain similar to the normal brain. NPCs retrovirally transduced to express the human ASM protein and implanted into adult ASMKO mouse brain led to a significant reduction in lysosomal storage pathology with reversal of sphingomyelin and cholesterol accumulation. This study highlights the therapeutic potential of neural cell therapy as a vehicle for delivery of enzyme to treat storage pathology in the ASMKO brain and potentially for other neurometabolic and neurodegenerative disorders.
Materials and Methods
Isolation and culture of mouse NPCs. Adult mouse brains (C57BL/6), minus the cerebella, were cut into 1-2 mm pieces. The tissue was mechanically and enzymatically dissociated with papain-protease-DNase solution, and then the digestion was stopped. Dissociated cells were partially purified from contaminating debris using Percoll density gradients (Palmer et al., 1999). The dissociated cells were collected and washed thoroughly. Isolated cells were grown on uncoated plates in DMEM-F12 supplemented with N2 medium containing epidermal growth factor (EGF) (20 ng/ml; Peprotech, Rockhill, NJ), basic fibroblast growth factor 2 (FGF-2) (20 ng/ml; Peprotech), and 5 μg/ml heparin. Mouse progenitor cell proliferation and neurosphere formation were detected 3-5 d after plating, and neurospheres increased in size with time in culture. Cultures were passaged multiple times, and cells could be frozen, thawed, and recultured without significant loss in cell number. For more details on the isolation of the adult mouse neural progenitors, see Ray et al. (1999).
In vitro immunocytochemistry. Cells were plated at a density of 25 × 105 cells/cm2 (high-density culture) onto polyornithine and laminin-coated glass chamber slides (Biocoat, Bedford, MA). The cells were incubated overnight in N2 medium at 37°C. The next day, the cultures were fixed with 4% paraformaldehyde and stained with one of the following antibodies to characterize neural differentiation: the neuronal marker mouse anti-microtubule-associated protein (anti-MAP2a+b) (1:250; Sigma, St. Louis, MO), the astroglial marker rabbit anti-glial fibrillary acidic protein (anti-GFAP) (1:2500; Dako, High Wycombe, UK), mouse anti-galactocerebroside (anti-GalC) (1:500; Chemicon International, Temecula, CA), a monoclonal antibody that stains oligodendrocyte progenitors, and mouse anti-human ASM (anti-hASM) (1:500; Genzyme, Cambridge, MA). The primary antibodies were detected with fluorescent-labeled secondary antibodies. Labeled cells were visualized using a Nikon (Tokyo, Japan) E800 microscope under epifluorescence.
Virus preparation. An ASM retroviral vector was constructed by inserting the full-length human ASM cDNA into the pLXIN and pDON retroviral vectors (Miller and Rosman, 1989; Kim et al., 1998). The ASM gene was inserted as a SacI-EcoRV fragment into the HpaI and XhoI sites of the retroviral vector pLXIN (Clontech, Palo Alto, CA) or in the retroviral vector pDON-AI (Takara Biomedicals, Shiga, Japan) digested with SalI and HpaI. The construction was performed as a three-part ligation to enable the correct orientation of the ASM gene to be cloned. A total of 1.5 × 107 human embryonic kidney 293 cells were triple transfected with plasmids encoding the Moloney murine leukemia virus gag/pol genes, pVPack-gag/pole (Stratagene, La Jolla, CA), vesicular stomatitis virus G-protein (VSV-G) as an envelope protein, pVPack-VSV-G (Stratagene), and a replication-defective vector expressing the human ASM gene and neomycin resistance gene (either pDON-ASM or pLXIN-ASM). Equal amounts of each vector were transfected using calcium phosphate. Ten micrograms of DNA (total) were transfected per 2.5 × 106 cells in a 6 cm plate. Fifteen hours after transfection, media were removed and replaced with 3 ml of media per 6 cm plate. Forty-eight hours after medium change, supernatants were pooled, filtered through a 0.45 μm filter, and spun in an SW28 rotor at 20,000 rpm for 2 hr at 4°C. Supernatants were removed, and virus was resuspended in 800 μl of defined N2 supplemented medium.
Retroviral transduction of NPCs. NPCs were transduced with one of the replication-defective retroviral vectors expressing the hASM gene and neomycin resistance gene from long-terminal repeat, prepared using standard methods (Pear et al., 1993). A cell suspension of NPCs (2 × 106 cells/ml) in N2 medium was mixed with concentrated retroviral vector (1 × 106/ml); polybrene was added to a final concentration of 8 μg/ml and incubated for 3-4 hr. The cell suspension was then centrifuged, resuspended, and plated in N2 medium containing EGF, FGF, and heparin. The transduced cultures were expanded and grown under selection in the presence of 100 μg/ml G418 (Invitrogen, San Diego, CA), starting at 5 d after transduction. Samples of culture media taken from transduced and nontransduced cells as well as cell pellets were assayed for ASM activity and hASM protein, as described below.
ASM enzyme assay. Enzymatic activity was measured using a modification of the method described by Gal et al. (1975), based on the hydrolysis of HAD-PC (2-(N-hexadecanoylamido)-4-nitrophenylphosphorylcholine). The product, HAD-NP (2-(N-hexadecanoylamido)-4-nitrophenolate), has an absorbance maximum at 415 nm that was measured with a spectrophotometer. The specific activity of the enzyme within each sample was calculated using the change in the absorbance reading between sample and blank. An assay was performed on cell pellets and 24 hr conditioned medium and cell pellets. The cells were scraped off plates in assay buffer and put through a freeze-thaw three times. Cell debris was centrifuged, and the assay was performed on the supernatant from the pellet. Results were expressed as units of activity per liter for conditioned medium or per milligram of protein for cell pellets.
hASM ELISA. Costar (Corning, Corning, NY) 9018 plates were coated (100 μl/well) with a monoclonal anti-human ASM antibody (2 μg/ml) diluted in 50 mm sodium carbonate buffer, pH 9.6, and incubated overnight at 2-8°C. Excess coating antibody was removed, and blocking diluent (KPL, Gaithersburg, MD) was added for 1 hr at 37°C. Plates were washed with a microplate washer (Molecular Devices, Sunnyvale, CA) for two cycles. Standards, controls, and samples diluted in standard dilution buffer (PBS, 0.05% Tween 20, 1% highly purified BSA) were pipetted in duplicate and allowed to incubate for 1 hr at 37°C. Plates were washed as described above. One hundred microliters of biotinylated hASM antibody (diluted 1:20,000 in standard dilution buffer) were added to each well, allowed to incubate for 1 hr at 37°C, and then removed with a microplate washer. Streptavidin-HRP (Pierce Biotechnology, Rockford, IL), diluted 1:10,000, was then added (100 μl/well) and allowed to incubate for 30 min at room temperature. Plates were washed as described above and then incubated with SureBlue TMB (KPL) for 15 min at 36-38°C. The reaction was stopped with stop solution (KPL), and absorbance values were then read at 450 nm with a Spectra Max 340 plate reader (Molecular Devices). Data analysis was completed using Softmax Pro 4.3 software (Molecular Devices). Assay sensitivity was 1.25 ng/ml. Results are expressed as nanograms of protein per milliliter of sample.
Protein assay. The protein concentration for each sample was determined using a BCA protein assay kit (Pierce Biotechnology) with bovine serum albumin as standard.
Labeling methods and preparation of cells for transplantation. To enable the detection of cells in vivo, expanded cultures were incubated with 0.5 μm bromodeoxyuridine (BrdU), which was added to the culture medium 72 hr before the preparation of cells for transplantation. Cultures were trypsinized for 1 min with 0.25% trypsin-EDTA, and detached cells were washed with 0.1 m PBS, collected by centrifugation at 2500 rpm for 3 min, and resuspended in 1 ml of PBS. Cell viability was checked by Trypan blue exclusion method. The cell suspension was centrifuged a second time and resuspended in a smaller volume of 0.1 m PBS to give the equivalent of 100,000 viable cells/μl.
Animals. The ASMKO mouse model was created by gene targeting, as described previously (Horinouchi et al., 1995), and back-crossed onto the C57BL/6 strain. Affected (homozygous) mice were distinguished from normal, wild-type animals and heterozygotes using a PCR-based assay (Gal et al., 1975). To generate ASMKO animals for neonatal studies, pairs of homozygous mice were used for breeding. Animals were maintained on a 12 hr light/dark cycle and given water and food ad libitum. All procedures were performed under an approved Institutional Animal Care and Use Committee protocol and in accordance with the use of animals in laboratory research.
Transplantation and tissue processing. Single-cell suspensions of BrdU-labeled NPCs were prepared (100,000 cell/μl) and stereotaxically injected unilaterally into multiple regions of the brains of ASMKO mice (1 μl/injection site). Host animals were implanted at three different ages: neonatal (4-5 d postnatal), juvenile (5 weeks), and adult (9-13 or 26 weeks). Neonatal mice received injections into the hippocampus and thalamus; juvenile and adult mice were injected in the hippocampus, thalamus, and striatum. No immunosuppression was used.
Neonatal mice were anesthetized by placing them on ice for 6 min and then maintaining hypothermia during unilateral injection into one hemisphere using a stereotaxic instrument. Coordinates were initially determined from cresyl violet-stained cryostat sections of age-matched animals. Cell transplants were placed 0.6 mm lateral to midline [mediolateral (ML)], 0.6 mm posterior to bregma [anteroposterior (AP)], and 1.2 and 2.2 mm [dorsoventral (DV)] below the dura for hippocampal and thalamic transplants, respectively. Adult and juvenile mice were anesthetized by inhalation of 3% isofluorane with 30% oxygen, prepared for surgery, and placed in a stereotaxic instrument.
Transplantation sites were 1.2 mm ML, 1.7 mm AP, and 1.0 and 3.5 mm DV for hippocampal and thalamic transplants, respectively. For the striatum, the coordinates used were 1.5 mm ML, 1.1 mm AP, and 2.5 mm DV.
After survival times of 1, 2, 4, 6, or 10 weeks after transplantation (Table 1), injected animals were anesthetized and killed by cardiac perfusion with buffered formaldehyde. Brains were sectioned (50 μm) in the coronal plane with a vibrating blade microtome (Leica, Nussloch, Germany). For immunofluorescence staining, sections were pretreated for BrdU detection (Gage et al., 1995) and stained with rat anti-BrdU (1:100; Accurate, Westbury, NY) and with one of the following antibodies: neuronal marker mouse anti-neuronal-specific nuclear protein (NeuN) (1: 100; Chemicon), glial progenitor marker rabbit anti-NG2 (1:500; Chemicon), or astroglial marker rabbit anti-GFAP (1:2500, Dako, Glostrup, Germany). Expression of hASM by the transplanted cells was evaluated using immunostaining with a biotinylated mouse anti-human ASM antibody (1:200; Genzyme). Secondary antibodies used for single or double labeling were donkey anti-species specific conjugated with FITC and/or Texas Red. Sections were visualized under a fluorescent microscope and also imaged using a confocal microscope to aid in detection of double-labeled cells.
Table 1.
Cell transplant recipients by age group at time of transplantation, whether they received a unilateral or bilateral transplant, survival time, genotype, type of cell transplanted, and number of mice that were processed histologically
|
Age |
Transplant region |
Survival time (in weeks) |
Genotype |
Cells transplanted |
Number of mice |
|---|---|---|---|---|---|
| Adult | |||||
| 8 weeks | Unilateral | 4 | KO | NPC plus ASM | 2 |
| 4 | KO | NPC | 2 | ||
| 9-13 weeks | 1 | KO | NPC plus ASM | 2 | |
| 2 | KO | NPC plus ASM | 4 | ||
| 4 | KO | NPC plus ASM | 2 | ||
| 6 | WT | NPC | 7 | ||
| 6 | WT | NPC plus ASM | 6 | ||
| 6 | KO | NPC | 2 | ||
| 6 | KO | NPC plus ASM | 3 | ||
| 26-28 weeks | 2 | KO | NPC | 2 | |
| 2 | KO | NPC plus ASM | 2 | ||
| 8 weeks | Bilateral | 4 | KO | NPC-NPC plus ASM | 4 |
| 26-28 weeks | 2 | KO | NPC-NPC plus ASM | 3 | |
| Juvenile | 6 | KO | NPC | 5 | |
| 5 weeks | 6 | WT | NPC | 1 | |
| Neonate | 7 | KO | NPC | 8 | |
| 4-5 d |
|
10 |
WT |
NPC plus ASM |
6 |
KO, Knock-out; WT, wild type.
Lysosomal pathology. Sections (50 μm) were thoroughly washed in phosphate buffer, dehydrated in graded alcohol, and flat embedded in 2-hydroxyethyl methacrylate (JB4-Plus embedding kit; Electron Microscopy Sciences, Fort Washington, PA). Polymerization was performed at 4°C in a cold room overnight. Sections (3 μm thick) were cut with a rotary microtome (MT 990; Boeckeler Instruments, Tucson, AZ) and then mounted on glass slides. Slides were stained with Richardson's staining for 1 min, rinsed, air dried, and mounted with Permount mounting medium (Electron Microscopy Sciences).
Filipin staining and quantitation. Every 12th 50 μm section was incubated with 10 mg/ml filipin complex (Sigma) in PBS for 3 hr, and then the sections were rinsed three times with PBS, mounted on slides, cover-slipped, and examined using a fluorescent microscope. Sections from each animal that had transplanted cells within the hippocampus and thalamus (n = 4) were evaluated for the percentage of reduction in fluorescent deposits using the Metamporph Image Analysis System (Universal Imaging, Downingtown, PA) (Table 2). Regional measurements were made of the area of filipin reduction at low magnification (4×) of the cortex, hippocampus, and thalamus in a section close to the injection site. The percentage of filipin reduction was determined by comparing the intensity of filipin staining on the injected side with the threshold of staining on the contralateral control side in the hippocampus and thalamus.
Table 2.
Frequency of correction as shown by filipin clearance by age group at the time of transplantation and survival time after transplantation.
|
Age (in weeks) |
Total number of mice |
Survival time (in weeks) |
Number of mice with reduced filipin |
Number of positive transplants |
|---|---|---|---|---|
| 8 | 6 | 4 | 6 | 6 |
| 9-10 | 5 | 6 | 4 | 4 |
| 12 | 8 | 1, 2, or 4 | 8 | 8 |
| 26-28 |
5 |
2 |
4 |
4 |
Transplants containing BrdU-immunoreactive cells were classified as positive.
Lysenin immunostaining. Sections (50 μm) were washed thoroughly in phosphate buffer containing 0.02% saponin. The sections were then incubated with 10 μg/ml lysenin, a spingomyelin-specific binding protein (Yamaji et al., 1998), in 0.5% BSA-PBS containing 0.02% saponin overnight at 4°C, washed with PBS plus 0.02% saponin, and then incubated with rabbit anti-lysenin antiserum (1:250) for 1 hr. The sections were washed and incubated with biotinylated secondary antibody (1:250; donkey anti-rabbit IgG) for 2 hr followed by strepavidin-fluorescein for 2 hr. The sections were then analyzed using a fluorescent microscope. The percentage of lysenin staining reduction was determined by comparing the intensity of lysenin staining on the treated side with the threshold of staining on the contralateral control side using the MetaMorph Image Analysis System.
Reverse transcriptase-PCR. Brains were perfused with saline, and tissue punches (diameter, 1 mm) were taken from the transplant site and frozen at -80°C. Total RNA was isolated from tissue samples using RNeasy (Qiagen, Valencia, CA). cDNA was synthesized using 4 μg of total RNA in a 33 μl reaction. The Superscript first-strand synthesis system for reverse transcriptase (RT)-PCR (Invitrogen) was used. The RT-PCR used 5 μl of cDNA, two sets of primers within exon 2 of the hASM gene (571 bp product), and PCR SuperMix (Invitrogen), for amplification. The PCR was run for 35 cycles; a 7 min incubation at 72°C was added at the end to ensure complete extension.
Results
In vitro characterization of adult mouse progenitor cells
After isolation from the adult forebrain, proliferating NPCs grew as free-floating spheres on uncoated plates. Some of the larger neurospheres attached to the plate, and small phase bright cells with multiple small processes migrated out to form adherent monolayers (Fig. 1A). Cells could be maintained and passaged as neurospheres or grown as monolayers. A small percentage of cells in the cultures spontaneously differentiated into cells with antigenic properties, as well as morphology, of neuronal, astroglial, and oligodendroglial lineages (Fig. 1B,C). Single spheres could be dissociated and replated under identical conditions to yield more than one neurosphere. Both primary and secondary neurospheres contained cells that differentiated to give rise to all three neural lineages (data not shown).
Figure 1.

Neural progenitor cells isolated from adult mouse brain express neural lineage-specific markers in vitro. A, Phase-contrast image of proliferating cultures. B, Fluorescent micrograph showing expression of neuronal (MAP2a+b; green) or astroglial (GFAP; red) markers. C, Fluorescent micrograph showing expression of an oligodendroglial (GalC) marker; cultures were counterstained for 4′,6′-diamidino-2-phenylindole (B; blue). Scale bars: A, 100 μm; (in B) B, C, 10 μm.
Survival and differentiation of adult mouse progenitor cells after transplantation into the ASMKO mouse
Studies were conducted to evaluate the survival, dispersion, and differentiation of adult mouse neural progenitors grafted into the ASMKO mouse brain. Proliferating BrdU-labeled cells were transplanted into the striatum, hippocampus, or thalamus of the ASMKO mouse brain (100,000 cells/site) of juvenile and adult mice and into the hippocampus and thalamus of neonatal mice. After a 6-10 week survival period, brain sections were processed by immunohistochemistry for BrdU and cell phenotypic markers. Transplanted cells were found in all animals. Cells transplanted in different recipient regions survived at least 6 weeks in juvenile and adult animals and 10 weeks in neonates (longest time points examined). There was a broad dispersion of transplanted cells away from the injection sites, in both the rostrocaudal and mediolateral axis in the hippocampus (Fig. 2A-D), thalamus (Fig. 2E-H), and striatum (data not shown). The distance and distribution pattern were consistent with migration of cells away from the implant site. BrdU-labeled cells were detected along a total distance of 1.32 ± 0.66 mm (striatum), 1.6 ± 0.85 mm (hippocampus), and 1.7 ± 0.9 mm (thalamus) in the rostrocaudal axis in both adult and juvenile ASMKO mice. There were no apparent differences in the engraftment and dispersion of transplanted cells in ASMKO mice versus wild-type mice or among regions. Cell distribution in neonates was slightly greater than in older animals, averaging 1.95 ± 1.12 mm in the above three regions. Interestingly, in the hippocampus of neonatal transplanted animals, the majority of the transplanted cells homed to the granular cell layer (Fig. 3A), a region undergoing neurogenesis at the time of transplantation (Altman and Bayer, 1990), and coexpressed the neuronal marker NeuN (Figs. 2I,3B).
Figure 2.

Survival and differentiation of NPCs in the ASMKO mouse brain 6 weeks after transplantation. Coronal sections show the distribution of BrdU-positive (red) NPCs after transplantation into the adult mouse hippocampus (A-D) and thalamus (E-H). Photomicrographs were taken at 300 μm intervals. There was clustering of cells at the implantation site (C, G), but many of the transplanted cells migrated throughout the hippocampus and thalamus, respectively. Confocal images show BrdU-immunoreactive cells (red) colocalized with NeuN immunoreactivity (green) within the granular cell layer of the hippocampus (I). Yellow indicates transplanted cells double labeled for NeuN and BrdU. Other BrdU-immunoreactive cells colocalized with GFAP-astrocyte immunoreactivity (J) or with NG2-oligodendrocyte immunoreactivity (K) in the molecular layer of the hippocampus. Scale bars: (in D) A-D, 100 μm; (in H) E-H, 100 μm; I, 5 μm; J, K, 10 μm.
Figure 3.

Distribution and differentiation of NPCs after transplantation into the neonatal mouse hippocampus. A, B, Coronal section of the dentate gyrus of the hippocampus showing the remarkable localization of transplanted BrdU-labeled cells (A) within the granular cell layer (B). BrdU-immunoreactive cells (red) colocalized with NeuN immunoreactivity (green) mostly along the inner margins of the granular cell layer of the dentate gyrus. Inset, A confocal image showing transplanted cells within the granular cell layer double labeled for NeuN (green) and BrdU (red). Scale bars: (in B) A, B, 100 μm; inset, 5 μm.
In the hippocampus of adult animals, transplanted cells that migrated into neuronal layers of the dentate gyrus expressed the neuronal marker NeuN (Fig. 2I). Within the granular cell layer, ∼50% of the BrdU-labeled cells expressed NeuN. Few, if any, of the transplanted BrdU-labeled nuclei expressed NeuN outside the granular cell layer of the hippocampus in any age groups. Several glial-associated markers were chosen to classify the fate of BrdU-labeled cells. The presence of oligodendrocyte progenitors was determined by colocalization with the proteoglycan marker NG2 (Stallcup, 2002). Staining for GFAP-positive cells identified astrocytes. In all regions of the hippocampus outside the granule cell layer, as well as in the thalamus and striatum, ∼5% of BrdU cells differentiated into astrocytes (Fig. 2J), and 30% differentiated into oligodendrocyte progenitors expressing NG2 (Fig. 2K).
Expression of ASM in vitro and in vivo
Based on the good migratory capacity of the transplanted NPCs, we next investigated the therapeutic potential of NPCs as a vehicle to deliver missing enzyme to the ASMKO brain. Preliminary experiments found that NPCs expressed low levels of ASM protein and secreted little, if any, enzyme. Therefore, NPCs were transduced with a retroviral vector intended to overexpress hASM. The transduced cells were expanded by growing them under selection for the expression of the neomycin gene. After 1 week, the majority of cells expressed the human ASM protein, as detected by staining for the human-specific ASM antibody (Fig. 4A). The punctate staining within the cytoplasm of the transduced cells, shown at high magnification (Fig. 4B), was consistent with targeting of hASM to the lysosomes. ASM activity and hASM protein were measured in the cell pellet from transduced and nontransduced NPCs and in conditioned medium collected over a 24 hr period. The level of hASM protein expression increased more than sevenfold in transduced cells versus nontransduced cells (from 20.13 to 151.99 ng/ml in conditioned medium and from 68.23 to 460.57 ng/mg protein in cell pellets). ASM enzyme activity within the cell pellet increased >10-fold over normal values after transduction (from 0.012 to 0.162 U/mg protein) and released at least fivefold more ASM activity into the culture media (from 0.24 to 1.4 U/l medium); background levels of ASM activity in media alone were 0.24 U/l.
Figure 4.

Retroviral transduction of mouse NPCs with the human acid sphingomyelinase gene. A, B, Immunostaining of transduced NPCs for human acid sphingomyelinase (red) 1 week after selection. High magnification shows lysosomal localization of hASM (B). Nuclei are counterstained by 4′, 6′-diamidino-2-phenylindole (blue). C, Expression of hASM at the implant site. Coronal section of the thalamus of ASMKO recipients at 2 weeks after transplantation showing the presence of ASM-immunopositive cells in the area close to the injection site (arrowhead points to single ASM-labeled cells shown at higher magnification in inset). D, RT-PCR expression of hASM mRNA in tissue isolated from the transplant site of seven animals compared with two (untreated) control animals. Four of the seven transplanted animals had distinct bands of hASM mRNA (571 bp band) expression compared with control tissue samples in which no hASM band was detected. Scale bars: A, B, 10 μm; C, 100 μm; inset, 25 μm.
To evaluate ASM enzyme expression in tissue samples from transplanted animals, a group of 15 adult animals (transplanted at 10-12 weeks of age) were killed at various time points (1, 2, and 4 weeks) and processed for histological and RT-PCR analyses. Anti-human ASM antibodies were used to detect the expression of enzyme in brain sections. Although some NPCs were positive for hASM protein, particularly at the site of implantation (Fig. 4C), fewer cells expressed ASM than were labeled with BrdU (data not shown). This discrepancy likely reflects the low level of ASM expressed by the cells, the relative insensitivity of the immunofluorescent method to detect protein, and the possible downregulation of gene expression. However, it was possible to demonstrate hASM mRNA expression by reverse transcriptase-nested PCR in four of seven transplanted animals. The absence of expression in some of the transplanted animals may be attributable to the location and sample size of the punch tissue relative to the implant site. No hASM band was detected in tissue samples from nonimplanted, control contralateral brain regions (Fig. 4D).
Reversal of storage pathology and cholesterol accumulation
We examined brains for changes in lysosomal storage deposits in the areas around the transplant. Animals were transplanted at 12 or 26 weeks of age and survived for 4 or 2 weeks after transplantation, respectively. These ages represent animals with fairly advanced disease, because the ASMKO mouse by 10 weeks of age has already developed significant lysosomal accumulation in neurons throughout the brain. Storage pathology and secondary cholesterol accumulation were prominent in the granule and pyramidal cell layers of the hippocampus and thalamus of nontransplanted, control animals or in these areas contralateral to the transplant. Examination of these regions on the implanted side showed a marked reduction in lysosomal distention associated with the presence of surviving NPCs within the hippocampus at 4 weeks after transplantation into 12-week-old animals (Fig. 5B) compared with the contralateral, untreated hippocampus (Fig. 5A). Similarly, marked reduction in lysosomal pathology was seen in the thalamus at 2 weeks after transplantation into 26-week-old animals (Fig. 5D) compared with the contralateral thalamus that received non-ASM-transduced NPCs (Fig. 5C).
Figure 5.

Reduced amount of lysosomal pathology in the hippocampus and thalamus of an ASMKO mouse brain that received ASM-transduced NPC transplants. A, B, Photomicrographs of the granular cell layer of the hippocampus from nontransplanted (A) and transplanted (B) ASMKO mice at 4 weeks after transplantation. C, D, Photomicrographs of the thalamus transplanted with nontransduced cells (C) and ASM-transduced cells (D) of ASMKO mice at 4 weeks after transplantation. Note the decrease in cytoplasmic distension in neural cells in the ASMKO mouse transplanted with ASM-expressing NPCs (B, D), whereas there remains profound vacuolation in cells on the nontransplanted side (A) or the side transplanted with nontransduced cells (C) as a result of the accumulation of distended lysosomes (arrows). Insets show higher magnifications of cells indicated by arrows. Scale bars: (in B) A-D, 20 μm; insets, 5 μm.
Sphingomyelin, the major accumulating storage substrate in NPD, has a very strong affinity for cholesterol, leading to sequestration of large amounts of cholesterol in the lysosomes of ASMKO mice and human patients (Viana et al., 1990; Slotte, 1999; Leventhal et al., 2001). Filipin is an autofluorescent molecule isolated from Streptomyces filipinensis that binds to cholesterol complexes (Leventhal et al., 2001; Sarna et al., 2001). Uninjected ASMKO brains had very high levels of filipin staining-cholesterol accumulation within cells, whereas normal mouse brains were unstained with filipin (data not shown). In animals receiving transplants at 26 weeks of age, the distribution of BrdU-labeled transplanted cells (Fig. 6A) was colocalized with an area of clearance of filipin-cholesterol staining (Fig. 6B,D) compared with the untreated, contralateral side (Fig. 6C). Cholesterol accumulation was corrected in all sites in which ASM-expressing transplanted cells were present, including the granular cell layer of the hippocampus (Fig. 7B), the pyramidal cells of the subiculum and CA1 (Fig. 7D), and the thalamus (Fig. 7F) at 6 weeks after transplantation, whereas the contralateral control side demonstrated high levels of filipin staining attributable to abundant cholesterol accumulation in lysosomes (Fig. 7A,C,E). In this study, we also evaluated differentiation of ASM-transduced NPCs grafted into the ASMKO mouse brain. The ASM-transduced NPCs showed region-specific differentiation into the three neural cell types after transplantation into the ASMKO similar to what was described with nontransduced NPCs (Fig. 2). There were no apparent differences in the engraftment and differentiation of ASM-transduced versus nontransduced transplanted NPCs cells in ASMKO mice.
Figure 6.

Regional clearance of cholesterol deposits at 2 weeks after transplantation visualized by filipin staining in the ASMKO mouse brain that received NPCs. A, Coronal view of the adult mouse hippocampus showing the broad dispersion of transplanted BrdU-immunoreactive cells. B, Adjacent sections stained with filipin showing that the regional clearance of fluorescent deposits matches closely with the distribution of the transplanted cells indicated by arrowheads. D, Higher magnification showing the substantial decrease of filip in deposits within the granular cell layer (gcl), the molecular layer, and the CA1 region of the hippocampus from the transplanted region (indicated by arrowheads in B) compared with the nontransplanted side (C). Scale bars: (in A) A, B, 200 μm; (in C) C, D, 100 μm.
Figure 7.
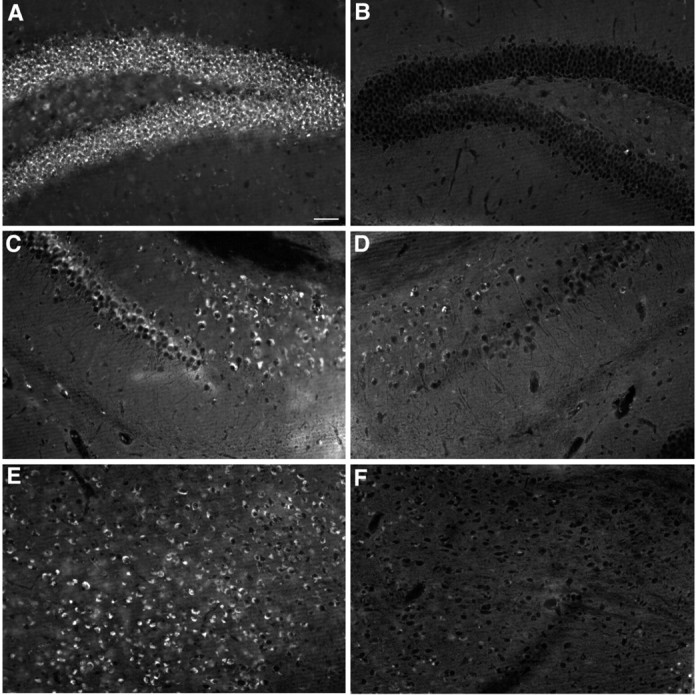
Clearance of cholesterol accumulation visualized by filipin staining in different regions of ASMKO mouse brains that received ASM-expressing NPCs 4 weeks after transplantation. Coronal views of the dentate gyrus (A, B), subiculum (C, D), and thalamus (E, F) show the presence of fluorescent deposits, representing accumulated cholesterol within cells contralateral to the transplanted NPCs (A, C, E) and clearance of the fluorescent deposits ipsilateral to the transplanted ASM-expressing cells (B, D, F). Scale bar: (in A) A-F, 50 μm.
By examining sections at various distances from the graft, we found that the area of pathological correction overlapped closely with the distribution of the transplanted NPCs. All animals that received transplants of NPCs expressing ASM and had established transplants showed significant clearance of filipin staining around the area of the graft. The area of clearance extended 2.1 ± 0.18 mm in the rostrocaudal axis. In a section close to the injection site, the area of clearance was 4.65 ± 0.66 mm2 including the cortex, hippocampus, and thalamus on the injected side. The percentage of reduction in fluorescence visualized by filipin was 90.02 ± 3.75 and 91.69 ± 3.49% in the hippocampus and thalamus, respectively, compared with the contralateral control side. Clearance of cholesterol storage deposits was seen as early as 1 week after transplantation in animals that received transplants at 12 weeks of age and 2 weeks after transplantation in animals that received transplants at 26 weeks of age, an extremely advanced age for this animal model. Lysosomal pathology and filipin-cholesterol remained reduced within the transplanted regions up to 6 weeks after transplantation (the latest time point examined).
To evaluate directly whether transduction of ASM gene into NPCs was necessary, animals (n = 8) received transplants into the hippocampus and thalamus of ASM-expressing NPCs on one side and nontransduced NPCs on the contralateral side. The survival and distribution of ASM-transduced and nontransduced NPCs were similar. BrdU-labeled transplanted cells were distributed throughout the hippocampus from a single implant site (Fig. 8A,B). However, storage pathology was only reduced on the side receiving transplants of ASM-expressing NPCs, whereas nontransduced NPCs had no obvious impact on the degree of storage pathology. Filipin staining demonstrated a marked decrease in fluorescent deposits in the CA1 pyramidal cell layer (Fig. 8D), granular cell layer, and CA4 pyramidal cell layer (Fig. 8F) on the side that received ASM-expressing cells, compared with more extensive fluorescent staining on the side that received nontransduced NPCs (Fig. 8C,E). There was a substantial reduction (>90%) of filipin staining in the pyramidal CA1 and granular neurons on the transplant side expressing ASM (Fig. 8D,F, insets) compared with the contralateral side (Fig. 8C,E, insets).
Figure 8.
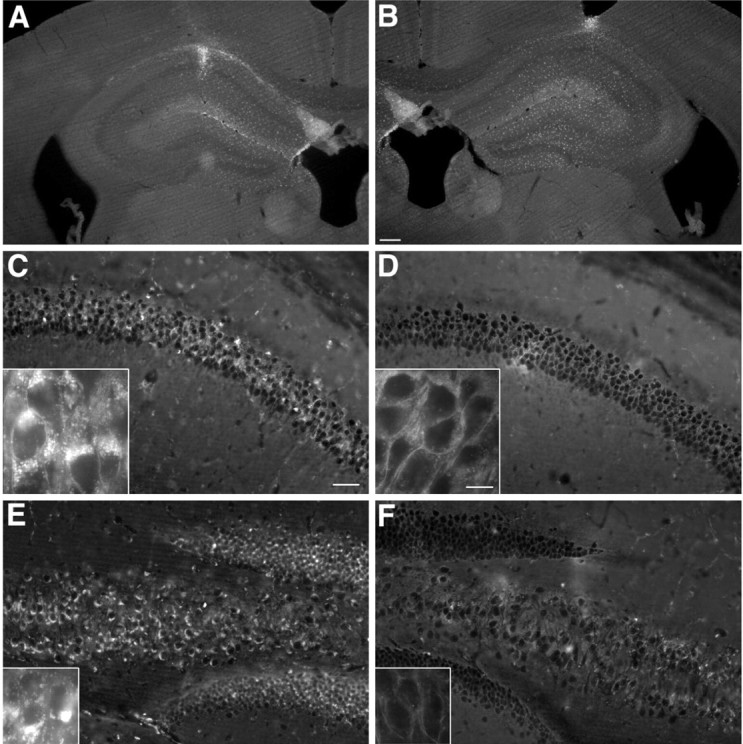
Regional clearance of cholesterol deposits visualized by filipin staining in the hippocampus 4 weeks after bilateral transplantation of ASM-transduced or ASM-nontransduced NPCs. A, B, Coronal view of the adult mouse hippocampus shows the comparable dispersion of transplanted BrdU-positive NPCs that were nontransduced (A) or ASM transduced (B). C-F, Coronal section of the pyramidal CA1 layer and dentate gyrus (C, D) and CA4 pyramidal cell layer (E, F) showing the marked reduction of fluorescent deposits visualized by filipin staining on the side that received ASM-transduced cells (D, F) versus ASM-nontransduced cells (C, E). Insets are higher-magnification views showing more clearly the cytoplasmic clearance of fluorescent deposits from the CA1 pyramidal neurons (D) and granular neurons (F) on the side of the ASM-transduced cells versus the side that received ASM-nontransduced cells (C, E). Scale bars: (in B) A, B, 200 μm; (in C) C-F, 100 μm; insets, 10 μm.
We also used lysenin, a sphingomyelin-specific binding protein, to assess sphingomyelin storage in sections from treated animals. As shown in Figure 9, the amount of lysenin staining was only reduced on the side receiving transplants of ASM-expressing NPCs compared with untreated ASMKO animals (data not shown) or animals treated with nontransduced NPCs. Sections from untreated ASMKO animals and animals treated with nontransduced NPCs had a very strong lysenin signal. There was a marked reduction in lysenin staining (79.22 ± 9.21%) in the striatum (Fig. 9B) and hippocampus (Fig. 9D) on the side that received ASM-expressing cells compared with the side that received nontransduced NPCs (Fig. 9 A,C).
Figure 9.

Depletion of sphingomyelin in the brains of ASMKO mice 4 weeks after bilateral transplantation of ASM-transduced or nontransduced NPCs. The sphingomyelin-specific binding protein lysenin was used to detect this lipid in sections from untreated and treated ASMKO animals. A, B, Coronal views of the striatum (A, B) and the hippocampus (C, D) showing the marked reduction in lysenin staining on the side that received ASM-transduced cells (B, D) versus ASM-nontransduced cells (A, C). Insets are higher-magnification views showing more clearly the marked depletion of sphingomyelin from the cells on the side of the ASM-transduced cells. Scale bars: (in C) A-D, 50 μm; insets, 25 μm.
Discussion
In this study, we used NPCs isolated from the adult mouse brain for allograft transplantation into the brains of normal and diseased ASMKO mice. Our results demonstrate that NPCs engineered to express recombinant human ASM, the missing enzyme in NPD-A disease, can serve as a vehicle for enzymatic cross-correction and reversal of storage pathology of host brain cells in the ASMKO mouse model. NPCs were isolated using a progenitor enrichment protocol based on low buoyancy of multipotent cells on a Percoll gradient, as described previously (Palmer et al., 1999), and were expanded using a combination of FGF-2, EGF, and heparin (Ray and Gage, 1999). The isolated NPCs proliferated to give rise to neurospheres-monolayers that contained cells expressing neuronal, astrocytic, and oligodendrocytic markers in vitro. Previous reports have shown that multipotent neural precursors with the capacity to generate neurons, astroglia, and oligodendroglia are found in the adult brain and possess self-renewal capacity, the defining attribute of stem-like cells (Reynolds and Weiss, 1992; Clarke and Frisen, 2001; Rietze et al., 2001).
The present study shows that NPCs, with these in vitro qualities, have the capacity when transplanted into the brains of ASMKO mice to substantially migrate within the host brain and differentiate into neurons and glia in a region-specific manner, similar to what has been reported previously after transplantation into normal animals (Suhonen et al., 1996; Flax et al., 1998; Shihabuddin et al., 2000). In the neurogenic zone of the hippocampus (granule cell layer), ∼50% of NPCs differentiated into neurons, whereas in non-neurogenic areas of the hippocampus and other implanted regions, ∼40% of NPCs differentiated into glia. In juvenile and adult animals, there seemed to be a nondirected migration of cells away from the transplantation site in all of the regions transplanted, including the hippocampus. Interestingly, in neonatal mice, there was a directed migration and preferential localization of the majority of the transplanted cells in the granular cell layer of the hippocampus, a region undergoing neurogenesis and development at the time of transplantation (Altman and Bayer, 1990). The majority of cells located in the granular cell layer differentiated into neurons. There were no differences in the migration and differentiation patterns of cells transplanted into ASMKO or normal mouse brain. Although NPCs retain migratory capacity independent of host condition (normal or diseased), the host brain does appear to influence the final migration and differentiation pattern as a function of age and site of implant (also independent of host condition).
In vitro, mouse NPCs expressed and released essentially null levels of measurable ASM activity. Retroviral transfection with a vector encoding the human ASM gene was therefore used to increase enzyme levels severalfold. The expression of hASM was maintained with passage and freezing, and thawing of NPCs did not affect the high level of expression of ASM protein. In vivo, however, few cells were positive for anti-hASM immunostaining outside of the immediate implant site. In part, this likely reflects hASM levels below the limit detectable with our available immunostaining methods. RT-PCR, a much more sensitive method, did confirm the presence of hASM mRNA in tissue samples from implanted animals. Reversal of pathology, nevertheless, was seen up to 6 weeks after transplantation (the latest time point examined) only in animals receiving transplants of ASM-expressing cells. These results are consistent with the expression and release of therapeutic levels of ASM by the transplanted cells (see below). Because downregulation of gene expression is often an issue after retroviral transduction (Palmer et al., 1991), we are now exploring the use of lentivirus as a means of generating a more stable set of ASM-expressing NPCs for transplant studies.
Next, we sought to correlate the presence of transplanted ASM-expressing cells with lysosomal pathology and cholesterol-sphingomyelin content in the ASMKO mouse brain. The ASMKO mouse develops severe and widespread lysosomal accumulation in neurons throughout the brain by 10 weeks of age. After transplantation into adults, there was a dramatic reduction in lysosomal pathology within regions containing NPCs. Distended lysosomes were completely or partly cleared from host neurons within regions containing implanted cells. In conjunction with reversal of distended lysosomal pathology, cellular cholesterol accumulation was effectively cleared (>90%) in the ASMKO brain regions that received transduced NPC transplants. Clearance of lysenin-sphingomyelin and filipin-cholesterol deposits occurred rapidly within 1-2 weeks after implant even in older animals with extremely advanced disease. In vitro studies have demonstrated that once sphingomyelin is degraded by ASM, cholesterol is liberated and rapidly effluxes from the lysosomal compartment, entering back into the normal biological pathways (Leventhal et al., 2001; Sarna et al., 2001). The reversal of cholesterol accumulation in the ASMKO brain therefore represents a downstream metabolic correction secondary to clearance of sphingomyelin.
There are several important aspects of NPC-mediated reversal of pathology that can be noted from these studies. First, the area of clearance tightly overlapped with the regional migration of NPCs, as viewed in adjacent sections. ASM-expressing NPCs were evidently capable of providing therapeutic levels of ASM to surrounding host brain cells. However, NPCs did not secrete sufficient protein to provide a greater sphere of correction to areas outside of the boundary of cell migration, at least within the time limits of the present study. Second, transduction of NPCs to express the hASM protein was necessary and sufficient to have this therapeutic effect. This was clearly demonstrated in animals receiving bilateral transplants of NPCs. The side implanted with cells transduced to express hASM showed reversal of lysosomal storage pathology with clearance of lysenin-sphingomeyllin and filipin-cholesterol. In contrast, there was no obvious change in storage pathology on the side receiving nontransduced NPCs, despite comparable levels of cell survival and migration in the brain. Third, levels of hASM expression that led to correction appeared to be very low (at least below the detection limits of immunostaining), suggesting that only a small amount of enzyme is required for uptake, cross-correction, and efficacy. This finding is consistent with other storage diseases, in which only a fraction of normal levels of enzyme is required to decrease the storage pathology and severity of the disease (Leinekugel et al., 1992). Fourth, implantation of cells into adult animals led to a rapid reduction of pathology within the brain. This demonstrates the ability to reverse preexisting pathology with cell-derived enzyme replacement therapy and the remarkable sensitivity of the ASMKO brain to cell-derived ASM replacement therapy. Other forms of cell therapy, including bone marrow transplantation (Miranda et al., 1998), gene-modified hematopoietic stem cells (Miranda et al., 2000), and mesenchymal stem cells (Jin et al., 2002; Jin and Schuckman, 2003), have also been shown to be efficacious in the ASMKO mouse model, mainly in treating visceral disease and delaying Purkinjie cell loss in the cerebellum.
Our results are consistent with previous studies showing that engraftable neural progenitor cells survive long term in the CNS environment after transplantation and can migrate and differentiate throughout the brain (Svendsen et al., 1997; Englund et al., 2002; Tamaki et al., 2002). Previous studies demonstrated that therapeutic amounts of β-glucuronidase (GUSB) can be delivered to the diseased brain through transplanted cells engineered to super-secrete the normal enzyme for export to surrounding neural tissues (Snyder et al., 1995; Taylor and Wolfe, 1997; Meng et al., 2003). Transplanting GUSB-expressing neural cell lines into the cerebral ventricles of newborn mice, moreover, resulted in widespread correction of lysosomal storage in neurons and glia in diseased mice (Snyder et al., 1995; Meng et al., 2003). In the striatum or cortex of adult MPS VII mice, lysosomal distention was cleared from cells only within the vicinity of grafts of fibroblasts secreting high levels of GUSB (Taylor and Wolfe, 1997).
The present study shows that preexisting lysosomal storage pathology in the adult ASMKO brain can be treated by an ex vivo ASM gene therapy strategy using neural cells engineered to replace the missing enzyme. The extent of correction achieved in the vicinity of the transplant was substantial for the mouse brain, but this will need to be extended significantly to effectively treat a larger brain. Widespread distribution of therapeutic cells in a larger brain will require optimization of the following: (1) a well characterized human-derived source of neural progenitor or neural stem cells, (2) likely use of an ex vivo gene therapy approach to transduce NPCs with human ASM (i.e., to enable stable and increased enzyme production), (3) cell dose per injection site, and (4) strategic selection of transplant site(s) in the brain. It remains to be determined whether reversal of lesions can actually arrest the degeneration in mental development that accompanies most lysosomal storage disorders in human patients. Nevertheless, the encouraging results from the present study hold promise for the use of neural-derived cell therapy in the treatment of NPD-A and other neurometabolic and neurologic disorders.
Footnotes
We thank Drs. Naomi Kleitman, Jasodhara Ray, and Richard Sidman for helpful critique of this manuscript.
Correspondence should be addressed to Lamya Shihabuddin, Genzyme Corporation, One Mountain Road, Framingham, MA 01701. E-mail: Lamya.shihabuddin@genzyme.com.
G. R. Stewart's present address: Medtronic Neurological, 800 53rd Avenue Northeast, Minneapolis, MN 55421.
Copyright © 2004 Society for Neuroscience 0270-6474/04/2410642-10$15.00/0
References
- Altman J, Bayer SA (1990) Migration and distribution of two populations of hippocampal granule cell precursors during the perinatal and postnatal periods. J Comp Neurol 301: 365-381. [DOI] [PubMed] [Google Scholar]
- Buchet D, Serguera C, Zennou V, Charneau P, Mallet J (2002) Long-term expression of beta-glucuronidase by genetically modified human neural progenitor cells grafted into the mouse central nervous system. Mol Cell Neurosci 19: 389-401. [DOI] [PubMed] [Google Scholar]
- Clarke D, Frisen J (2001) Differentiation potential of adult stem cells. Curr Opin Genet Dev 11: 575-580. [DOI] [PubMed] [Google Scholar]
- Englund U, Bjorklund A, Wictorin K (2002) Migration patterns and phenotypic differentiation of long-term expanded human neural progenitor cells after transplantation into the adult rat brain. Brain Res Dev Brain Res 134: 123-141. [DOI] [PubMed] [Google Scholar]
- Flax JD, Aurora S, Yang C, Simonin C, Wills A, Billinghurst LL, Jendoubi M, Sidman RL, Wolfe JH, Kim SU, Snyder EY (1998) Engraftable human neural stem cells respond to developmental cues, replace neurons, and express foreign genes. Nat Biotechnol 16: 1033-1039. [DOI] [PubMed] [Google Scholar]
- Fricker RA, Carpenter MK, Winkler C, Greco C, Gates MA, Bjorklund A (1999) Site-specific migration and neuronal differentiation of human neural progenitor cells after transplantation in the adult rat brain. J Neurosci 19: 5990-6005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gage FH, Ray J, Fisher LJ (1995) Isolation, characterization, and use of stem cells from the CNS. Annu Rev Neurosci 18: 159-192. [DOI] [PubMed] [Google Scholar]
- Gal AE, Brady RO, Hibbert SR, Pentchev PG (1975) A practical chromogenic procedure for the detection of homozygotes and heterozygous carriers of Niemann-Pick disease. N Engl J Med 293: 632-636. [DOI] [PubMed] [Google Scholar]
- Gritti A, Parati EA, Cova L, Frolichsthal P, Galli R, Wanke E, Faravelli L, Morassutti DJ, Roisen F, Nickel DD, Vescovi AL (1996) Multipotential stem cells from the adult mouse brain proliferate and self-renew in response to basic fibroblast growth factor. J Neurosci 16: 1091-1100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horinouchi K, Erlich S, Perl DP, Ferlinz K, Bisgaier CL, Sandhoff K, Desnick RJ, Stewart CL, Schuchman EH (1995) Acid sphingomyelinase deficient mice: a model of types A and B Niemann-Pick disease. Nat Genet 10: 288-293. [DOI] [PubMed] [Google Scholar]
- Jin HK, Schuchman EH (2003) Ex vivo gene therapy using bone marrow-derived cells: combined effects of intracerebral and intravenous transplantation in a mouse model of Niemann-Pick disease. Mol Ther 8: 876-885. [DOI] [PubMed] [Google Scholar]
- Jin HK, Carter J, Huntley GW, Schuchman EH (2002) Intracerebral transplantation of mesenchymal stem cells into acid sphingomyelinase-deficient mice delays the onset of neurological abnormalities and extends their life span. J Clin Invest 109: 1183-1191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim SH, Yu SS, Park JS, Robbins PD, An CS, Kim S (1998) Construction of retroviral vectors with improved safety, gene expression, and versatility. J Virol 72: 994-1004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leinekugel P, Michel S, Conzelmann E, Sandhoff K (1992) Quantitative correlation between the residual activity of beta-hexosaminidase A and arylsulfatase A and the severity of the resulting lysosomal storage disease. Hum Genet 88: 513-523. [DOI] [PubMed] [Google Scholar]
- Leventhal AR, Chen W, Tall AR, Tabas I (2001) Acid sphingomyelinase-deficient macrophages have defective cholesterol trafficking and efflux. J Biol Chem 276: 44976-44983. [DOI] [PubMed] [Google Scholar]
- McKay R (1997) Stem cells in the central nervous system. Science 276: 66-71. [DOI] [PubMed] [Google Scholar]
- Meng XL, Shen J, Ohashi T, Maeda H, Kim SU, Eto Y (2003) Brain transplantation of genetically engineered human neural stem cells globally corrects brain lesions in the mucopolysaccharidosis type VII mouse. J Neurosci Res 74: 266-277. [DOI] [PubMed] [Google Scholar]
- Miller AD, Rosman GJ (1989) Improved retroviral vectors for gene transfer and expression. Biotechniques 7: 980-990. [PMC free article] [PubMed] [Google Scholar]
- Miranda SR, Erlich S, Friedrich Jr VL, Haskins ME, Gatt S, Schuchman EH (1998) Biochemical, pathological, and clinical response to transplantation of normal bone marrow cells into acid sphingomyelinase-deficient mice. Transplantation 65: 884-892. [DOI] [PubMed] [Google Scholar]
- Miranda SR, He X, Simonaro CM, Gatt S, Dagan A, Desnick RJ, Schuchman EH (2000) Infusion of recombinant human acid sphingomyelinase into Niemann-pick disease mice leads to visceral, but not neurological, correction of the pathophysiology. FASEB J 14: 1988-1995. [DOI] [PubMed] [Google Scholar]
- Palmer TD, Rosman GY, Osborne WR, Miller AD (1991) Genetically modified skin fibroblasts persist long after transplantation but gradually inactivate introduced genes. Proc Natl Acad Sci USA 88: 1330-1334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palmer TD, Takahashi J, Gage FH (1997) The adult rat hippocampus contains primordial neural stem cells. Mol Cell Neurosci 8: 389-404. [DOI] [PubMed] [Google Scholar]
- Palmer TD, Markakis EA, Willhoite AR, Safar F, Gage FH (1999) Fibroblast growth factor-2 activates a latent neurogenic program in neural stem cells from diverse regions of the adult CNS. J Neurosci 19: 8487-8497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pear WS, Nolan GP, Scott ML, Baltimore D (1993) Production of high-titer helper-free retroviruses by transient transfection. Proc Natl Acad Sci USA 90: 8392-8396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ray J, Gage FH (1999) Neural stem cell isolation, characterization and transplantation. In: Modern techniques in neuroscience research (Windhorst U, Johansson H, eds), pp 339-360. New York: Springer.
- Reynolds BA, Weiss S (1992) Generation of neurons and astrocytes from isolated cells of the adult mammalian central nervous system. Science 255: 1707-1710. [DOI] [PubMed] [Google Scholar]
- Rietze RL, Valcanis H, Brooker GF, Thomas T, Voss AK, Bartlett PF (2001) Purification of a pluripotent neural stem cell from the adult mouse brain. Nature 412: 736-739. [DOI] [PubMed] [Google Scholar]
- Sando GN, Neufeld E (1977) Recognition and receptor-mediated uptake of a lysosomal enzyme, alpha-1-iduronidase, by cultured human fibroblasts. Cell 12: 619-627. [DOI] [PubMed] [Google Scholar]
- Sarna J, Miranda SR, Schuchman EH, Hawkes R (2001) Patterned cerebellar Purkinje cell death in a transgenic mouse model of Niemann Pick type A/B disease. Eur J Neurosci 13: 1873-1880. [DOI] [PubMed] [Google Scholar]
- Schuchman EH, Desnick RJ (2001) Niemann-Pick disease type A and B: acid sphingomyelinase deficiencies. In: The metabolic and molecular bases of inherited disease (Scriver CR, Beaudet A, Sly WS, Valle D, eds), pp 3589-3610. New York: McGraw-Hill.
- Shihabuddin LS, Horner PJ, Ray J, Gage FH (2000) Adult spinal cord stem cells generate neurons after transplantation in the adult dentate gyrus. J Neurosci 20: 8727-8735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slotte JP (1999) Sphingomyelin-cholesterol interactions in biological and model membranes. Chem Phys Lipids 102: 13-27. [DOI] [PubMed] [Google Scholar]
- Sly WS, Volger C (2002) Brain-directed gene therapy for lysosomal storage disease: going well beyond the blood-brain barrier. Proc Natl Acad Sci USA 99: 5760-5762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snyder EY, Taylor RM, Wolfe JH (1995) Neural progenitor cell engraftment corrects lysosomal storage throughout the MPS VII mouse brain. Nature 374: 367-370. [DOI] [PubMed] [Google Scholar]
- Stallcup WB (2002) The NG2 proteoglycan: past insights and future prospects. J Neurocytol 31: 423-435. [DOI] [PubMed] [Google Scholar]
- Suhonen JO, Peterson DA, Ray J, Gage FH (1996) Differentiation of adult hippocampus-derived progenitors into olfactory neurons in vivo. Nature 383: 624-627. [DOI] [PubMed] [Google Scholar]
- Svendsen CN, Caldwell MA, Shen J, ter Borg MG, Rosser AE, Tyers P, Karmiol S, Dunnett SB (1997) Long-term survival of human central nervous system progenitor cells transplanted into a rat model of Parkinson's disease. Exp Neurol 148: 135-146. [DOI] [PubMed] [Google Scholar]
- Tamaki S, Eckert K, He D, Sutton R, Doshe M, Jain G, Tushinski R, Reitsma M, Harris B, Tsukamoto A, Gage F, Weissman I, Uchida N (2002) Engraftment of sorted/expanded human central nervous system stem cells from fetal brain. J Neurosci Res 69: 976-986. [DOI] [PubMed] [Google Scholar]
- Taylor RM, Wolfe JH (1997) Decreased lysosomal storage in the adult MPS VII mouse brain in the vicinity of grafts of retroviral vector-corrected fibroblasts secreting high levels of beta-glucuronidase. Nat Med 3: 771-774. [DOI] [PubMed] [Google Scholar]
- Torchiana E, Lulli L, Cattaneo E, Invernizzi F, Orefice R, Bertagnolio B, Di Donato S, Finocchiaro G (1998) Retroviral-mediated transfer of the galactocerebrosidase gene in neural progenitor cells. NeuroReport 9: 3823-3827. [DOI] [PubMed] [Google Scholar]
- Viana MB, Giuguliani R, Leite VH, Barth ML, Lekhwani C, Slade CM, Fensom A (1990) Very low levels of high density lipoprotein cholesterol in four sibs of a family with non-neuropathic Niemann-Pick disease and sea-blue histiocytosis. J Med Genet 27: 499-504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamaji A, Sekizawa Y, Emoto K, Sakuraba H, Inoue K, Kobayashi H, Umeda M (1998) Lysenin, a novel sphingomyelin-specific protein. J Biol Chem 273: 5300-5306. [DOI] [PubMed] [Google Scholar]