

Abstract
The proinflammatory and lipopolysaccharide (LPS)-inducible cytokine tumor necrosis factor α (TNFα) has been shown to enhance primary sensory nociceptive signaling. However, the precise cellular sites of TNFα and TNF receptor synthesis are still a matter of controversy. Therefore, we differentiated the neuronal and non-neuronal sites of TNFα, TNFR1, and TNFR2 mRNA synthesis in dorsal root ganglion (DRG) of control rats and evaluated how their expression is altered under systemic challenge with LPS. In situ hybridization (ISH), RT-PCR analysis of laser-microdissected cells, and immunocytochemistry revealed absence of TNFα from DRG neurons and LPS-induced expression of TNFα exclusively in a subpopulation of non-neuronal DRG cells. Using RT-PCR and Northern blotting TNFR1 and TNFR2 mRNAs were found to be constitutively expressed and increased after LPS. TNFR1 mRNA was expressed in virtually all neurons and in non-neuronal cells with increased levels after LPS in both. TNFR2 was exclusively expressed and regulated in non-neuronal cells. RT-PCR analysis of microdissected DRG neurons and of the sensory neuronal cell line F11 confirmed the neuronal expression of TNFR1 and excluded that of TNFR2. Double ISH revealed varying levels of TNFR1 mRNA in virtually all DRG neurons including putative nociceptive neurons coding for calcitonin gene-related peptide, substance P, or vanilloid receptor 1. Taken together, we provide evidence that non-neuronally synthesized TNFα may directly act on primary afferent neurons via TNFR1 but not TNFR2. This is likely to be relevant under conditions of inflammatory pain and infections accompanied by widespread TNFα synthesis and release and may drive sickness behavior.
Keywords: cytokine, neuroimmune, primary afferent, neuropeptide, vanilloid receptor, pain
Introduction
Tumor necrosis factor α (TNFα), a proinflammatory cytokine with pleiotropic functions, is involved in nociceptive responses causing hyperalgesia (Cunha et al., 1992; Watkins et al., 1995; Ignatowski et al., 1999; Junger and Sorkin, 2000). In chronic constriction injury (CCI), an animal model of injury-induced painful mononeuropathy, inhibition of TNFα synthesis and release results in reduced pain behavior (Lindenlaub et al., 2000). In rat dorsal root ganglion (DRG), acute topical application of TNFα increases the firing rate of sensory neurons in normal rats and rats subjected to chronic DRG compression (Liu et al., 2002).
During an immune response TNFα production is enhanced in cells of the monocyte-macrophage lineage and in T-cells (Bette et al., 1993). In the CNS microglia and astrocytes are believed to be the primary cellular sources of TNFα expression (Hopkins and Rothwell, 1995). In peripheral nerve injury, Schwann cells have been reported to produce TNFα (Wagner and Myers, 1996). TNFα mRNA has been shown to be increased in axotomized rat DRG but its cellular source has not been determined (Murphy et al., 1995). Based on immunocytochemical findings sensory neurons of the DRG have been suggested to contain and produce TNFα protein (Schafers et al., 2003b). However, whether TNFα protein is indeed synthesized by DRG neurons or taken up from extracellular sources is still controversial.
TNFα acts on two distinct subtypes of receptors, TNF receptor type 1 (TNFR1, p55) and TNF receptor type 2 (TNFR2, p75) (MacEwan, 2002). TNFα-induced hyperalgesia seems to depend on TNFR1. TNFR1 but not TNFR2 neutralizing antibodies as well as antisense RNA against TNFR1 can reduce experimentally induced hyperalgesia (Sommer et al., 1998; Parada et al., 2003). TNFR1 and TNFR2 immunoreactivity (IR) has been reported in DRG neurons (Shubayev and Myers, 2001; Pollock et al., 2002; Schafers et al., 2003a). However, direct evidence for TNFR1 and TNFR2 biosynthesis in DRG neurons by demonstrating their mRNAs is still missing. The bacterial lipopolysaccharide (LPS) that has been reported to cause hyperalgesia (Watkins et al., 1994; Walker et al., 1996; Reeve et al., 2000), is known to enhance not only TNFα synthesis and release (Holst et al., 1996) but also the expression of TNFR2 and, to a lesser extent, of TNFR1 (Nadeau and Rivest, 1999). Therefore, it was our aim to discern neuronal and non-neuronal expression patterns of TNFα, TNFR1, and TNFR2 in rat DRG using in situ hybridization and cell-specific laser capture microdissection techniques combined with RT-PCR analysis and to examine their plasticity under conditions of LPS-induced inflammatory pain. The F11 cell line (Platika et al., 1985), a hybrid cell line from rat DRG neurons and mouse neuroblastoma cells, was used to further differentiate which of the TNF receptors is neuronally expressed. To test for the relationship of TNF receptors with presumed nociceptive DRG neurons, we performed cohybridization studies of TNFR1 with the genes encoding calcitonin gene-related peptide (CGRP), substance P (SP), or the vanilloid receptor type 1 (VR1) also referred to as TRPV1, a member of the transient receptor potential (TRP) channel family.
Materials and Methods
Animals. Animal care and procedures were conducted in accordance with institutional and National Institutes of Health guidelines. Adult (200-225 gm) male Wistar rats (Charles River, Sulzfeld, Germany) were housed in clean plastic cages on a 12 hr light/dark cycle with ad libitum access to food and water. Rats were injected intraperitoneally with ultrapure LPS (0.5 mg/kg body weight; Escherichia coli 0111:B4, InvivoGen, San Diego, CA) and killed after 1, 3, or 6 hr LPS stimulation by exposure to CO2. Dorsal root ganglia were rapidly removed from LPS-stimulated rats and normal rats and frozen either in liquid nitrogen for RNA extraction or by immersion in -50°C cold 2-methybutane on dry ice for histological processing and embedded in Tissue-Tek OCT compound (Sakura, Zoeterwoude, The Netherlands).
Cell culture. The F11 cell line (rat dorsal root ganglion-mouse neuroblastoma hybridoma) was provided by Dr. Mark C. Fishman (Massachusetts General Hospital, Harvard Medical School, Boston, MA). F11 cells were grown in Ham's F-12 medium, supplemented with 15% Hyclone (Logan, UT) defined fetal bovine serum, 1× HAT (sodium hypoxanthine, aminopterin, and thymidine) supplement, and 1× antibiotic-antimycotic at 37°C under 5% CO2 in tissue culture flasks. All cell culture reagents were purchased from Invitrogen (Karlsruhe, Germany). At ∼70-80% confluence cells were harvested for RNA and protein extractions.
Laser capture microdissection. We cut 10-μm-thick frozen sections through rat DRG on a Leica (Nussloch, Germany) cryostat, mounted on glass slides and stored at -70°C. Before microdissection, slides were stained with 0.5% cresyl violet (in 60 mm sodium acetate, 340 mm acetic acid), rinsed in distilled water, and dehydrated in 70, 96, twice in 100% isopropanol, and in xylene for 4 min, and finally air-dried. The perikarya of DRG neurons and non-neuronal cells were microdissected separately with a PixCell II laser capture microdissection (LCM) system (Arcturus, San Diego, CA) using the following settings: laser spot size 7.5 μm, pulse power 70 mW, pulse width 0.6 msec. After capturing the caps were plugged into 0.5 ml plastic tubes containing 100 μl TRIzol (Invitrogen) and stored at -20°C.
RNA isolation. Total RNA from pooled dorsal root ganglia, F11 cells, and microdissected cells was isolated using TRIzol reagent according to the manufacturer's protocol, treated with RNase-free DNase I (Roche Diagnostics, Mannheim, Germany) at 37°C for 30 min and column-purified using RNeasy Mini Kit (Qiagen, Hilden, Germany). To the RNA samples from microdissected cells glycogen (Roche Diagnostics) was added as carrier to a final concentration of 250 μg/ml followed by phenolchloroform purification. After precipitation the RNA pellets were washed in 70% ethanol, dissolved in RNase-free water and stored at -70°C. Poly(A+) RNA was prepared using the poly(A+) tract mRNA Isolation system III (Promega, Mannheim, Germany).
RT-PCR. cDNA was synthesized using SUPERSCRIPT II RNase H- reverse transcriptase (Invitrogen). For PCR amplification of rat TNFR1, TNFR2, TNFα, and GAPDH, the following gene-specific primers were used: rat TNFR1 (M63122; nt. 983-1382), forward primer: gggattcagctcctgtcaaa, reverse primer: atgaactccttccagcgtgt; (M63122, nt. 704-1902) forward primer: tcccctgtaaggagaaacagaa, reverse primer: gctttttctccacaatcacctc. Rat TNFR2 (AF498039, nt. 579-1034), forward primer: gttctctgacaccacatcatcc, reverse primer: gtcaataggtgctgctgttcaa; (AF498039, nt. 541-1242) forward primer: aatggaaacgtgatatgcagtg, reverse primer: gctacagacgttcacgatgc. Rat TNFα (NM 012675, nt. 15-708) forward primer: catgatccgagatgtggaact, reverse primer: tcacagagcaatgactccaaag. rat GAPDH (AF106860, nt. 119-345), forward primer: cgaccccttcattgacctcaactacatg, reverse primer: ccccggccttctccatggtggtgaagac. Hot start PCR was performed using 2 μl cDNA in a total volume of 50 μl containing 0.2 μm primer mix, 1× PCR buffer, 1.5 mm MgCl2, 200 μm dNTPs mixture, and 1 U AmpliTaq Gold (Roche Diagnostics) and the following program: 5 min at 95°C, 25-40 cycles at (30 sec at 94°C, 30 sec at 57°C, and 30-90 sec at 72°C) and 10 min extension at 72°C.
Probes. The PCR products were subcloned into pGEM-T (Promega), their sequence identify was confirmed by double-strand DNA sequencing. Plasmid constructs were linearized by NotI and AatII for TNFα; NotI and NcoI for TNFR1; NdeI and AatII for TNFR2; SalI and ApaI for GAPDH. Riboprobes in antisense and sense orientation were generated from linearized plasmid constructs by in vitro transcription using the appropriate RNA polymerases and 35S-UTP, 32P-UTP, or digoxigenin-UTP as label. After transcription, the probes were subjected to mild alkaline hydrolysis as described (Schafer et al., 1993).
In situ hybridization histochemistry. In situ hybridization was performed as described previously (Schafer et al., 1993). Briefly, radioactive probes were diluted to 5 × 104 dpm/μl in hybridization solution (3× SSC, 50 mm NaPO4 pH 7.4, 10 mm dithiothreitol, 1× Denhardt's solution, 0.25 gm/l yeast tRNA, 10% dextran sulfate, and 50% formamide). Formaldehyde-fixed 14-μm-thick frozen sections were hybridized with 25 μl hybridization solution containing radioactive RNA probes at 60°C in formamide humid chamber box for 16 hr. After a series of washes, the sections were dehydrated in 50% and 70% isopropanol. For autoradiographic detection slides were coated with Kodak (Eastman Kodak, Rochester, NY) NTB-2 nuclear emulsion and exposed at 4°C for 7 d to 14 d in the dark. Slides were developed in Kodak D19 solution and counter-stained with cresyl violet. Photographic documentation was performed under dark- and bright-field illumination using an Olympus (Hamburg, Germany) AX70 microscope.
Double in situ hybridization histochemistry. For colocalization of two mRNAs, digoxigenin-labeled riboprobes for αCGRP, SP, and VR1 were added to the radioactive hybridization solutions at a final concentration of 1 ng/μl (Mika et al., 2003). Hybridization and washing procedures were performed as described above except dehydration in 50% and 70% ethanol. Detection of nonradioactive hybrids was performed according to the manufacturer's instruction (nonradioactive in situ hybridization application manual; Roche Molecular Biochemical, Mannheim, Germany) using alkaline phosphatase-conjugated anti-digoxigenin Fab fragments diluted to 1 U/ml and a chromogene solution containing 0.2 mm 5-bromo-4-chloro-3-indolyl phosphate and 0.2 mm nitroblue tetrazolium salt (Roche Diagnostics). Color development was monitored under microscopic control. For detection of 35S-labeled probes, the slides were coated with K5 Emulsion (ILFORD Imaging, Mobberley Chershire, UK) diluted 1:1 in water. After 7 d exposure at 4°C in the dark slides were developed as described above.
Grain counting analysis. Computer-aided grain counting was performed on autoradiograms counterstained with cresyl violet under bright-field conditions at 670× magnification using a digital Camera (Spot RT Slider, Diagnostics Instruments Inc., Sterling Heights, MI) and the microcomputer imaging device Image Analysis software (Imaging Research Inc, St. Catharines, Ontario, Canada). All image analysis was performed by the same observer who was not aware of the treatment group. From three sections per group, neuronal (n = 391, controls; n = 335, LPS) and non-neuronal (n = 129, controls; n = 133, LPS) cell populations were analyzed. The cross-sectional area of each perikaryon with a visible nucleus was outlined, and the number of grains above each outlined cell was determined using a threshold algorithm. To distinguish cell size-specific changes, DRG neurons were divided into small (<600 μm2), medium (600-1200 μm2), and large (>1200 μm2) subpopulations. Grain number was corrected using predetermined background grain density (0.12 ± 0.02 grains/μm2) on adjacent sections hybridized with the sense probe. Measurements were expressed as estimated number of silver grains per cell. Differences between groups were analyzed by one-way ANOVA followed by the Mann-Whitney U test. p values <0.05 were considered statistically significant.
Northern blot analysis. Aliquots of 10 μg total RNA or 2 μg poly(A+) RNA were size-fractionated on a formaldehyde-denatured 1.5% agarose gel, transferred to a nylon membrane, and cross-linked. Membranes were prehybridized with NorthernMax solution (Ambion, Cambridgeshire, UK) at 68°C for 30 min and hybridized with 1-4 × 106 cpm/ml probe at 68°C overnight. After washing twice for 5 min with preheated 2× SSC containing 0.1% SDS and twice for 15 min with preheated 0.1 × SSC containing 0.1% SDS at 68°C, the membranes were exposed to Hyperfilm-MP (Amersham Pharmacia Biotech, Freiburg, Germany) for 20 min to 16 hr. The membranes were rehybridized with 1-4 × 106cpm/ml GAPDH RNA probe to normalize for RNA loading differences. The density of the specific bands was measured by using Scion (Frederick, MD) Image Beta 4.02.
Western blot analysis. Protein was extracted from the TRIzol-treated F11 cells after RNA isolation according to the manufacturer's protocol. Protein samples (30 μg/lane) were separated by 12% SDS-PAGE and transferred to Hybond C extra membrane (Amersham Biosciences, Freiburg, Germany). After blocking nonspecific binding sites with 3% bovine serum albumin (SERVA, Heidelberg, Germany) in TBST (Tris-buffered saline containing 0.1% Tween 20) and endogenous avidin and biotin with a blocking kit (Vector Laboratories, Burlingame, CA), membranes were incubated with a goat anti-rat TNFRp55 antibody (Santa Cruz Biotechnology, Heidelberg, Germany) diluted 1:5000 in TBST for 1 hr at room temperature. After five washes for 5 min each, the membranes were incubated for 1 hr with an anti-goat IgG secondary antibody (Dianova, Hamburg, Germany) diluted 1:500,000 at room temperature. After several washes in TBST, detection was performed using the ABC complex (Vector Laboratories) for 1 hr at room temperature and enhanced chemiluminescence reagents (Amersham Biosciences).
Confocal laser scanning microscopy. Deparaffinized serial sections of adult DRG were collected from controls (n = 5) and from animals 3 hr after LPS treatment (n = 5) and immersion-fixed in Bouin-Hollande solution or in 4% phosphate-buffered formaldehyde, pH 7.4, for 24 hr. Immunofluorescence was performed by overnight incubation at room temperature with a polyclonal goat anti-murine TNFα antibody (PAK-mTNFα-g50; 1:100 dilution; Strathmann Biotec AG, Hamburg, Germany) together with a monoclonal IgG-purified antibody recognizing rat ED1, recognizing a macrophage—monocyte-specific antigen (1:5 dilution; Serotec, Oxford, UK). TNFα immunoreactivity was visualized with biotinylated anti-goat antibody (1:200 dilution; Dianova, Hamburg, Germany) at 37°C for 45 min, respectively, followed by an incubation with streptavidin-conjugated Alexa Fluor 488 diluted 1:200 (MoBiTec, Göttingen, Germany) at 37°C for 2 hr. Rat ED1 immunoreactivity was visualized with an anti-mouse antibody conjugated to Alexa Fluor 647 diluted 1:200 (MoBiTec) at 37°C for 2 hr. Specificity controls for TNFα immunostaining were performed by omission of the primary antisera, by incubation with TNFα preimmune serum, and by staining tissue sections from TNFα knock-out mice (kindly provided by B. Dietzschold, Thomas Jefferson University, Philadelphia, PA). Antibodies from other sources against murine or rat TNFα (Santa Cruz Biotechnology, Roche, Strathmann) were also tested, but all of them exhibited nonspecific immunostaining in rat DRG. Immunostained sections were analyzed with an Olympus Fluoview confocal laser-scanning microscope and documented as false-color confocal images.
Results
Cellular distribution of TNFα mRNA in rat dorsal root ganglion after LPS treatment
To determine the cellular sites of TNFα synthesis in the DRG and its regulation by LPS, we first investigated the expression pattern of the TNFα gene at the mRNA level using in situ hybridization. In sections through normal rat DRG, specific hybridization signals for TNFα could not be detected (Fig. 1A). However, 1 hr after systemic LPS treatment, signals for TNFα were observed in many small cells scattered throughout the DRG (Fig. 1B). Microscopic analysis of cresyl violet-counterstained sections revealed that the silver grains were located over non-neuronal cells identified by their small darkly stained nuclei (Fig. 1C). When sections were hybridized with TNFα riboprobes in sense orientation, only background labeling was observed (data not shown). To confirm the non-neuronal expression and to exclude a low basal expression of TNFα in primary sensory neurons, we performed RT-PCR on RNA extracts of microdissected DRG neurons (Fig. 1D-F) and non-neuronal cells (Fig. 1G-I). As shown in Figure 1J, no specific PCR product for TNFα could be detected in RNA extracts of microdissected DRG neurons from control or LPS-treated rats. Specific TNFα amplicons were also undetectable in RNA extracts of microdissected non-neuronal cells from normal rats. Only after LPS treatment a PCR product for TNFα could be observed in non-neuronal RNA extracts (Fig. 1J), which was confirmed by double-strand DNA sequencing.
Figure 1.
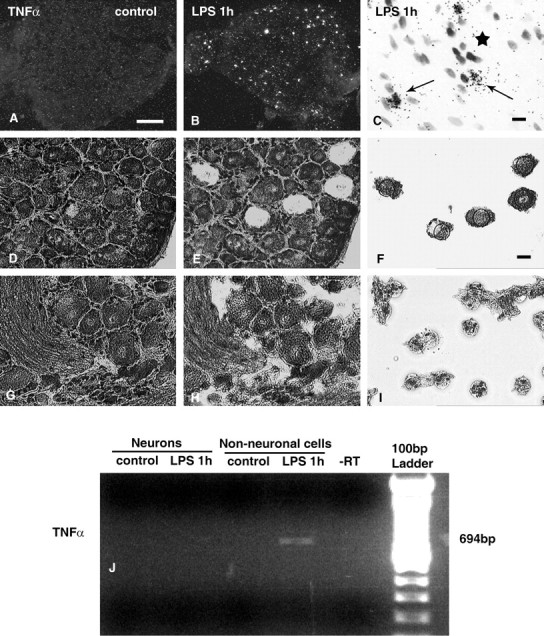
Detection of TNFα mRNA in rat DRG by in situ hybridization and in microdissected cells by RT-PCR. Dark-field autoradiograms of cryosections through rat lumbar (L4) DRG after hybridization with a 35S-labeled riboprobe specific for rat TNFα illustrate the lack of positive signals for TNFα mRNA in lumbar DRG of a normal rat (A) but presence in a DRG of a rat 1 hr after systemic LPS (B). High-resolution bright-field micrograph depicts the accumulation of silver grains representing TNFα mRNAover non-neuronal cells 1 hr after LPS treatment (C, arrows). Note the absence of specific hybridization signals over neuronal perikarya (C, asterisk). Two hundred laser-microdissected neurons (D, E) and non-neuronal cells (G-I) from rat DRG were subjected to RT-PCR for TNFα (J). Specific bands representing TNFα transcripts are detectable only in the extracts of microdissected non-neuronal cells 1 hr after systemic LPS. Scale bars: A, 200 μm; C, F, 10 μm.
Detection of TNFα immunoreactivity in rat dorsal root ganglion after LPS treatment
In Bouin-Hollande fixed DRG sections from rats 3 hr after LPS, specific TNFα IR was detected in a subpopulation of non-neuronal cells surrounding primary afferent perikarya (Fig. 2A). Double immunofluorescence staining revealed that the TNFα-IR cells were also positive for ED1 antigen (Fig. 2B,C), an established marker for cells of the macrophage-monocyte lineage (Damoiseaux et al., 1994). Specific TNFα IR was absent from neuronal perikarya that exhibited faint nonspecific immunostaining (Fig. 2A). In DRGs from control rats we failed to detect any specific TNFα IR (data not shown). In formaldehyde-fixed DRG sections of both control and LPS-treated rats, we observed weak immunostaining of primary afferent perikarya and fibers when using the specific TNFα antisera. However, incubation with the preimmune serum produced the identical neuronal staining pattern, strongly suggesting that the neuronal immunostaining was nonspecific (data not shown).
Figure 2.
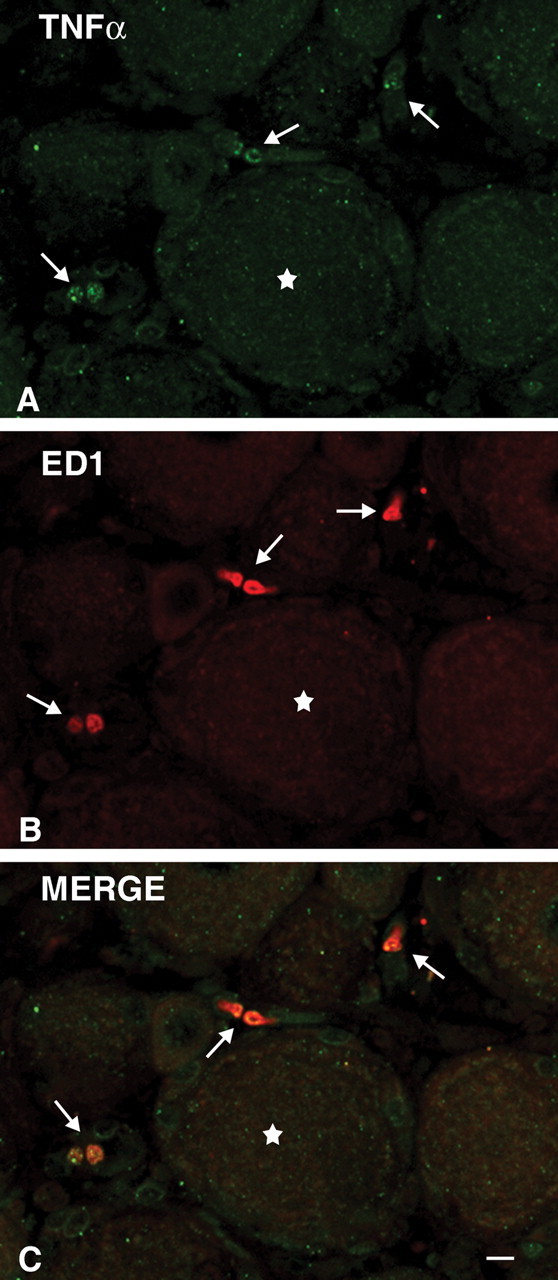
Detection of TNFα immunoreactivity in rat dorsal root ganglion ED1-positive cells after LPS treatment. TNFα immunoreactivity (green) was detected in rat DRG non-neuronal cells (arrows) but not in DRG neurons (asterisk) 3 hr after LPS treatment (A). Dual immunofluorescence with an ED1 antibody (red) (B) identified the TNFαIR cells as members of the macrophage-monocyte lineage (C, arrows). Scale bar, 10 μm.
Detection of TNF receptor transcripts in rat dorsal root ganglion extracts: effects of LPS
To investigate the expression of TNF receptors in rat DRG under basal conditions and after systemic injection of LPS, RT-PCR analysis was performed on total RNA extracts using gene-specific primers. Results are illustrated in Figure 3. TNFR1 transcripts could be detected in DRG extracts from control rats and appeared to be increased 6 hr after LPS treatment. In contrast, only a faint band representing TNFR2 mRNA could be detected in extracts of control rats. TNFR2 mRNA levels peaked at 3 hr after LPS injection and decreased to control levels at 6 hr after LPS. To determine the changes of TNFR mRNA levels indicated by the RT-PCR data, we performed Northern blot analysis at time points when TNFR mRNA levels were maximal as shown in Figure 3. For TNFR1 a single transcript of 2.3 kb was observed, whereas three different TNFR2 mRNA species of 5.4, 4.4, and 3.1 kb were detected. Densitometry revealed a 2.5-fold increase of TNFR1 mRNA in DRG 6 hr after LPS (Fig. 4A), whereas TNFR2 mRNA levels, measuring all three transcripts, were increased ∼2.7-fold 3 hr after LPS treatment (Fig. 4B).
Figure 3.

RT-PCR analysis of TNF receptor transcripts in extracts of dorsal root ganglion from control and LPS-treated rats. Ethidium bromide-stained agarose gel loaded with PCRs from DRG extracts. Specific amplicons for TNFR1 (top panel) are present in all lanes with a subtle increase at 6 hr after LPS. TNFR2 mRNA (middle panel) is expressed at very low levels in control rats, but increases to peak levels at 3 hr after LPS treatment and decreases to control levels at 6 hr after LPS. GAPDH was used as a housekeeping gene (bottom panel). -RT, Omission of reverse transcriptase as negative control. M, DNA size marker.
Figure 4.

Northern blot analysis of TNFR1 and TNFR2 in rat DRG. A, For the detection of TNFR1 mRNA, 10 μg of total RNA isolated from DRGs of control- or LPS-injected rats were loaded per lane. Hybridization with a TNFR1-specific 32P-labeled riboprobe yielded a single transcript of 2.3 kb. Densitometry of autoradiograms revealed a 2.5-fold increase of TNFR1 mRNA 6 hr after LPS as compared with control. GAPDH was used as a housekeeping gene to normalize the sample loading. Exposure time: 16 hr for TNFR1, 2 hr for GAPDH. B, Hybridization of blots loaded with 2 μg of poly(A+) RNA of rat DRG isolated from control rats or rats 3 hr after LPS with a TNFR2-specific 32P-labeled riboprobe yielded three transcripts of 5.4, 4.4, and 3.1 kb. Densitometry of the total signal per lane revealed a 2.7-fold increase of TNFR2 mRNA 3 hr after LPS as compared with control. GAPDH was used as housekeeping gene for normalization of the samples differences. Exposure time: 16 hr for TNFR2, 20 min for GAPDH.
Detection of TNF receptor transcripts in microdissected DRG neurons
To determine which TNF receptor types are expressed in neurons, we performed LCM in combination with RT-PCR (Fig. 5). Perikarya of DRG neurons were captured from rat DRG sections, and total RNA was isolated. RT-PCR analysis revealed that TNFR1 mRNA was constitutively expressed in microdissected DRG neurons, whereas TNFR2 mRNA was not (Fig. 5D).
Figure 5.

RT-PCR analysis of TNFR1 and TNFR2 transcripts in RNA extracts of microdissected DRG neurons. Laser capture microdissection was performed on 10-μm-thick cresyl violet-stained cryosections of rat DRGs from normal rats. Pictures were taken from before (A) and after (B) microdissection and from microdissected material (C). RT-PCR of RNA extracts from 150 microdissected neuronal perikarya revealed specific amplicons for TNFR1 but not for TNFR2 (D). GAPDH was used as a housekeeping gene detected as positive control. RT-PCR without reverse transcriptase (-RT) was performed to exclude genomic DNA contamination. A 100 bp DNA ladder was used as size marker (M).
Analysis of TNF receptors in the F11 cell line
To lend further support to the observation that TNFR1 rather than TNFR2 is expressed in primary sensory neurons, we examined the cell line F11 for TNF receptor expression. As shown in Figure 6A, RT-PCR analysis revealed that rat TNFR1 mRNA but not rat TNFR2 mRNA was constitutively expressed in F11 cells. In addition, using Western blot analysis a specific band of 55 kDa representing TNFR1 protein could be demonstrated in F11 protein extracts as shown in Figure 6B.
Figure 6.

RT-PCR and Western blot analysis of TNF receptors in F11 cell line. A, In RNA extracts of F11 cells, constitutive expression of TNFR1 but not of TNFR2 mRNA is detected by RT-PCR. (B) Western blotting shows a single band of 55 kDa representing TNFR1 protein (lane a). Incubation with the secondary antibody revealed no immunostaining (lane b).
Cellular distribution and LPS-induced changes of TNF receptor mRNAs in rat DRG
To identify the DRG cells expressing TNF receptors, we performed ISH analysis on serial sections of rat lumbar dorsal root ganglion (Fig. 7). Hybridization experiments with TNFR1 or TNFR2 riboprobes in sense orientation as negative controls did not yield any specific labeling (data not shown). TNFR1 mRNA was predominantly expressed in neuronal cells (Fig. 7A,B,E,F). High-resolution microscopic analysis revealed that most DRG neurons expressed TNFR1 mRNA (Fig. 7E). TNFR1 mRNA levels appeared to differ between DRG neurons without exhibiting preference to small- or large-diameter neurons. In addition, silver grains representing TNFR1-specific hybrids were also observed over non-neuronal cells (Fig. 7E,F). Systemic LPS treatment appeared to increase TNFR1 mRNA levels in non-neuronal cells and to a smaller extent in DRG neurons as well (Fig. 7F). Quantitative image analysis of cellular TNFR1 mRNA levels using grain counting confirmed this observation (Fig. 8). In DRG neurons, a significant increase in TNFR1 mRNA levels was measured 6 hr after LPS treatment (Fig. 8A). This change was independent of their size category, small [169 ± 6 grains per cell (LPS; n = 77) vs 133 ± 8 grains per cell (control, n = 51; p < 0.05)], medium [281 ± 6 grains per cell (LPS; n = 152) vs 240 ± 7 grains per cell (control; n = 157; p < 0.05)], and large [478 ± 12 grains per cell (LPS; n = 106) vs 412 ± 12 grains per cell (control; n = 183; p < 0.05)]. In non-neuronal cells (Fig. 8B), the LPS-induced increase of TNFR1 mRNA cells was relatively higher [41 ± 0.7 grains per cell (LPS; n = 133) vs 27 ± 0.8 grains per cell (control; n = 129; p < 0.05)]. TNFR2 mRNA was detected at very low levels in DRGs from control rats (Fig. 7C,G). Strong scattered signals for TNFR2 were detected 3 hr after LPS treatment (Fig. 7D,H). Under high-resolution bright-field illumination, the silver grains for TNFR2 transcripts were exclusively observed over non-neuronal cells (Fig. 7G,H).
Figure 7.
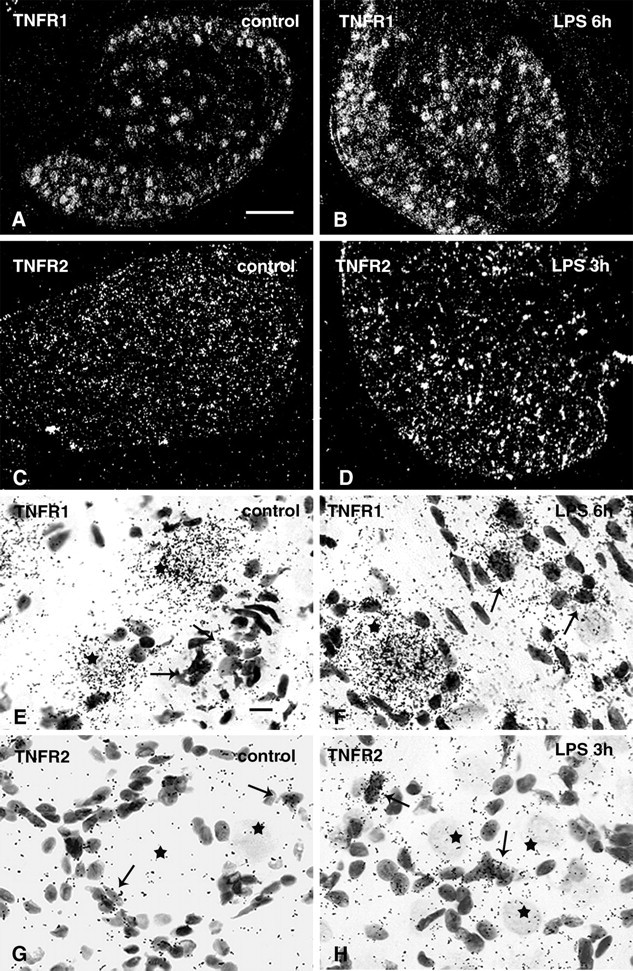
Cellular distribution of TNFR1 and TNFR2 mRNAs in rat DRG. Low-power dark-field autoradiograms of emulsion-coated sections illustrate constitutive and wide spread expression of TNFR1 mRNA in a cryosection of a control rat (A). TNFR1 hybridization signals appear to be increased 6 hr after systemic LPS treatment (B). In controls, hybridization signals for TNFR2 are very weak (C), but have increased strongly 3 hr after systemic LPS (D). High-resolution bright-field images of an emulsion-coated section hybridized with the TNFR1-specific probe shows strong labeling over most large neuronal perikarya (E, F, asterisk) and weaker labeling of the small non-neuronal cell bodies (E, arrows). Note the increase of labeling intensity for TNFR1 6 hr after systemic LPS treatment, in particular over the small non-neuronal cellbodies (F, arrows). Hybridization signals for TNFR2 are present only over non-neuronal cells (G, H, arrows), but not over neuronal perikarya (G, H, asterisk). Systemic LPS treatment enhances TNFR2 mRNA expression only in non-neuronal cells (H, arrows). Exposure times: 7 d (A, B, E, F), 10 d (C, D, G, H). Scale bars: A, 200 μm; E, 10 μm.
Figure 8.

Grain-counting analysis of cellular TNFR1 mRNA levels in rat DRG cells. The average number of silver grains per cell representing TNFR1 mRNA was determined for neurons (A) and for non-neuronal cells (B) in rat DRG. Measurements in serial sections from controls (n = 3) and from rats 6 hr after LPS treatment (n = 3) were pooled. TNFR1 mRNA is significantly increased (*p < 0.05) in neurons independent of their size category, small (<600 μm2), medium (600-1200 μm2), and large (>1200 μm2), as well as in non-neuronal cells (B).
TNFR1 expression in putative nociceptive neurons expressing CGRP, SP, or VR1
To test whether and at which level TNFR1 mRNA is expressed in distinct classes of DRG nociceptive neurons, we performed double labeling in situ hybridization for TNFR1 and the nociceptive receptor VR1 as well as the neuropeptides SP and CGRP known to be involved in primary afferent transmission of pain (Caterina et al., 1997; Nohr et al., 1999). TNFR1 mRNA was found in all presumed nociceptive neurons expressing CGRP (Fig. 9A), SP (Fig. 9B), or VR1 (Fig. 9C). However, TNFR1 mRNA levels exhibited substantial differences within each of these phenotypic categories of DRG neurons with no obvious level preference to any of them. In addition, TNFR1 was expressed in the majority of the remaining DRG neurons (Fig. 9A-C), where TNFR1 mRNA levels also varied. Thus, there was no obvious relationship of TNFR1 mRNA levels with any category of nociceptive and non-nociceptive classes of DRG neurons.
Figure 9.
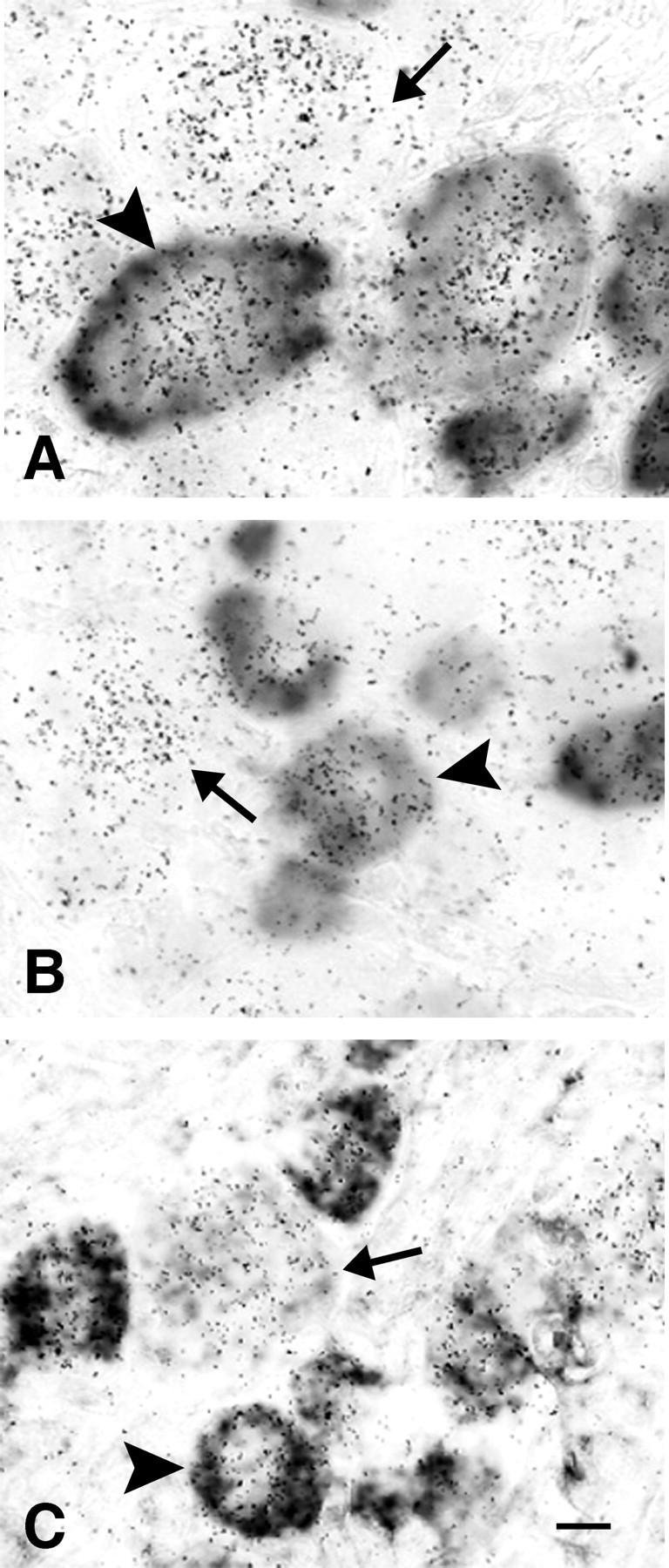
Coexpression of TNFR1 mRNA with αCGRP mRNA, SP mRNA, or VR1 mRNA. Double-labeling in situ hybridization with 35S-labeled probes for TNFR1 and digoxigenin-labeled riboprobes of αCGRP mRNA (A), SP mRNA (B), or VR1 mRNA (C) reveals the coexpression of TNFR1 with nociceptive markers in presumed pain-transmitting neurons (arrowheads), but also TNFR1 expression in other sensory neurons (arrows). Exposure time of autoradiograms is 7 d. Scale bar: C, 10 μm.
Discussion
The essential new findings of this study are: (1) LPS induces expression of TNFα in rat DRG exclusively in non-neuronal cells with no constitutive or LPS-inducible expression of TNFα in DRG neurons; (2) TNFR1 mRNA but not TNFR2 mRNA is constitutively expressed in DRG neurons and in the sensory cell line F11, and TNFR1 mRNA occurs in virtually all DRG neurons, including presumed nociceptive neurons coding for CGRP, SP, or VR1; (3) Both TNFR1 and TNFR2 mRNAs are constitutively expressed in non-neuronal DRG cells, albeit at low levels; (4) After systemic LPS neuronal and non-neuronal expression of TNFR1 mRNA and non-neuronal expression of TNFR2 mRNA in DRG are increased with TNFR2 mRNA remaining to be restricted to non-neuronal cells.
Cellular sources of TNFα in rat dorsal root ganglion
From their distribution pattern and relative abundance, the LPS-induced TNFα mRNA-expressing cells revealed in this study are most likely resident macrophages or dendritic cells but may also include Schwann cells, endoneural fibroblasts, and mast cells. These cell types have been shown to be capable of TNFα synthesis and to occur in the DRG and in peripheral nerves (Gordon and Galli, 1991; Murwani et al., 1996; Wesselingh et al., 1997; Sommer and Schafers, 1998). Our observation of TNFα IR in a subpopulation of non-neuronal cells, which express the ED1 antigen, suggests that cells of the macrophage-monocyte lineage are most likely the main source for TNFα in rat DRG after LPS stimulation.
The absence of TNFα mRNA and protein in DRG neurons revealed in this study is in conflict with previous reports demonstrating, based on immunocytochemistry, that TNFα immunoreactivity is present in a subpopulation of primary afferents and enhanced after chronic constriction injury (CCI) of the sciatic nerve and arguing that this is attributable to biosynthesis of TNFα in DRG neurons (Shubayev and Myers, 2001; Schafers et al., 2002, 2003b). Contrary to this assumption, our present study provides no evidence for but convincing evidence against TNFα gene expression in DRG neurons. Most importantly, ISH failed to demonstrate constitutive or LPS-induced expression of TNFα mRNA in rat DRG neurons but easily revealed LPS-induced TNFα mRNA expression in DRG non-neuronal cells; using the highly sensitive RT-PCR method on RNA extracts of microdissected DRG neurons and non-neuronal cells, we observed TNFα transcripts only in non-neuronal DRG cells and only after LPS treatment while TNFα transcripts were absent from DRG neurons of normal and LPS-treated rats. Our own attempts to localize TNFα protein by immunocytochemistry revealed no specific immunostaining in DRG neurons. In fact, nonimmune sera of rabbit and goat instead of rabbit and goat anti-TNFα antisera resulted in the same nonspecific neuronal immunostaining when processed for indirect immunofluorescence. Only 3 hr after LPS treatment we could detect TNFα immunoreactivity in the ED1-positive non-neuronal subpopulation, whereas neuronal perikarya exhibited only background staining.
Taken together, the total lack of TNFα mRNA from DRG neurons as unequivocally revealed in this study and the absence of specific TNFα IR from neuronal perikarya strongly indicate that TNFα in DRG is produced in non-neuronal cells but not in neurons. Therefore, the view of TNFα biosynthesis in DRG neurons should be dismissed.
Expression of TNFR 1 mRNA but not of TNFR2 mRNA in DRG neurons
Our results obtained from ISH and LCM in combination with RT-PCR provide strong evidence that TNFR1 mRNA but not TNFR2 mRNA is expressed in DRG neurons and that virtually all DRG neurons express TNFR1. The presence of TNFR1 but absence of TNFR2 expression in DRG neurons corresponds well to our observation that TNFR1 but not TNFR2 was expressed in the sensory cell line F11 as revealed by RT-PCR and Western blot analysis. Our finding of virtually pan-neuronal expression of TNFR1 mRNA in rat DRG is in some contrast to the report that TNFR1 immunoreactivity is only found in subpopulations of small- and large-diameter DRG neurons (Schafers et al., 2003a). TNFR1 mRNA expression has been also found in most if not all neurons of mouse trigeminal ganglia (Cunningham et al., 1997) and in all primary sensory neurons of the trigeminal mesencephalic nucleus of the mouse (Bette et al., 2003), suggesting that neuronal expression of TNFR1 may be a widespread phenomenon in sensory pathways.
The absence of TNFR2 mRNA expression in DRG neurons of normal rats and rats challenged by LPS demonstrated in the present study is in contrast to several reports showing positive immunostaining for TNFR2 in small and large DRG neurons of rats and mice with an increase during nerve lesion (Shubayev and Myers, 2001; Pollock et al., 2002; Schafers et al., 2003a). It is puzzling that these reports did not comment on TNFR2 immunostaining in non-neuronal cells of the DRG, where constitutive TNFR2 mRNA was easily detected in the present study. Thus, the concept of biosynthesis of TNFR2 in DRG neurons must be questioned.
Cell-specific plasticity of TNFR1 and TNFR2 expression in the dorsal root ganglion after LPS treatment
The present study demonstrates that systemic application of LPS enhances the expression of TNFR1 mRNA in DRG neurons and the expression of both TNFR1 and TNFR2 mRNA in DRG non-neuronal cells. In contrast, TNFR2 mRNA expression is absent from DRG neurons and remains absent under LPS challenge. Our previous finding demonstrating expression of the LPS receptor toll like receptor 4 (TLR4) in a subset of rat DRG neurons (Li et al., 2002) suggests that increased TNFR1 expression in DRG neurons after LPS may be caused by direct effects of LPS on DRG neurons. The LPS-induced increase of TNFR1 mRNA in DRG neurons may provide a mechanism by which TNFα effects on primary afferents can be amplified.
Possible roles of TNFR1 in DRG neurons and of TNFα, TNFR1, and TNFR2 in DRG non-neuronal cells in pain and other sensory functions
LPS is a potent inducer of proinflammatory cytokines such as TNFα, IL-1, and IL-6 and has been shown to cause hyperalgesia (Wiertelak et al., 1994; Walker et al., 1996; Cahill et al., 1998; Reeve et al., 2000). Thus, LPS-induced expression of TNFα in DRG non-neuronal cells as shown in the present study may be causally related to LPS-induced hyperalgesia. The neuronal expression of TNFR1 but not of TNFR2 in DRG strongly suggests that exogenous and endogenous TNFα from pleiotropic cellular sources is likely to influence primary sensory functions and neurotransmission directly by acting on TNFR1 bearing sensory neurons, at the level of their cell bodies, their intraganglionic collaterals, their collaterals in peripheral nerves, and their peripheral and central terminals (Fig. 10). In fact, in the periphery TNFα is synthesized by a variety of cells during the immune response and under inflammation and in the spinal cord TNFα can be synthesized by and released from glial cells (Watkins et al., 2001). Indeed, neutralizing antibodies against TNFR1 but not against TNFR2 reduce thermal hyperalgesia and mechanical allodynia in the CCI mouse (Sommer et al., 1998), supporting our view that primary sensory neurons are specifically controlled by TNFR1. Intrathecally applied antisense oligodeoxynucleotides to TNFR1 decrease not only TNFR1 protein expression in peripheral nerve terminals of DRG neurons but also reduce inflammatory hyperalgesia in the rat (Parada et al., 2003).
Figure 10.

Schematic diagram illustrating the cellular and molecular basis of TNFα-mediated signaling in primary sensory neurons and non-neuronal cells in rat DRG. TNFR1 is expressed in all DRG neurons, including peptidergic population and capsaicinsensitized neurons. Both TNFR1 and TNFR2 are expressed in DRG non-neuronal cells. LPS induces expression of TNFα that occurs exclusively in non-neuronal cells. Endogenous or exogenous TNFα is likely to directly act on DRG neurons via TNFR1, both at the level of cell bodies and at the terminals inside the DRG and in the periphery. Similar considerations apply to the spinal cord where TNFα can be synthesized by and released from glial cells. In addition, endogenous and exogenous TNFα can be expected to act on DRG non-neuronal cells via TNFR1 and/or TNFR2. TNFR1-expressing DRG neurons may function as immunosensors to sense TNFα during immune responses and inflammation.
The expression of TNFR1 in small-diameter presumed nociceptive neurons expressing SP, CGRP, or VR1 demonstrated in this study provides evidence how the reported effects of TNFα on nociceptive responses (Watkins et al., 1995; Junger and Sorkin, 2000; Sorkin and Doom, 2000) are exerted directly at the level of distinct classes of primary sensory nociceptive neurons. In fact, TNFα is known to induce the release of SP and CGRP from peripheral terminals (Ding et al., 1995; Hua et al., 1996). Recombinant TNFα excites nociceptors and enhances heat-evoked release of CGRP from peripheral nerve terminals (Opree and Kress, 2000). Given the observation that VR1 neurons express TNFR1 as shown in this study, it is tempting to speculate that TNFα sensitizes VR1 signaling in a similar way as demonstrated for other inflammatory mediators like PGE2 or bradykinin (Sugiura et al., 2002).
Our finding that the expression of the TNFR1 gene is virtually pan-neuronal and not restricted to presumed nociceptive neurons in the rat DRG implies a broader role of TNFα in primary sensory functions than nociception alone as currently assumed. As TNFα contributes to nerve growth factor (NGF)-dependent neuronal cell death during development (Barker et al., 2001), this may also apply to TNFR1 bearing sensory neurons. In fact, neutralizing antibodies against either TNFα or TNFR1 rescued many sensory neurons after NGF deprivation in vitro. Another possibility is that all TNFR1-expressing sensory neurons may function as immunosensors (Weihe et al., 1991) by sensing any TNFα released from a variety of cells in the periphery, either by being in contact with TNFα secreting cells or by being exposed to circulating TNFα. This is likely to be relevant under conditions of inflammatory pain and immune responses during infections accompanied by widespread TNFα synthesis and release and may drive sickness behavior and fatigue syndromes.
Footnotes
This study was supported in part by grants from the Deutsche Forschungsgemeinschaft DFG SFB 297 and the Bundesministerium für Bildung und Forschung 01GS01118. We thank Heike Reichert-Preibsch, Michael Schneider, and Marion Zibuschka for excellent technical assistance.
Correspondence should be addressed to Martin K.-H. Schäfer, Department of Molecular Neuroscience, Institute of Anatomy and Cell Biology, Philipps University Marburg, Robert-Koch-Strasse 8, 35033 Marburg, Germany. E-mail: mkh.schafer@staff.uni-marburg.de.
Copyright © 2004 Society for Neuroscience 0270-6474/04/249623-09$15.00/0
References
- Barker V, Middleton G, Davey F, Davies AM (2001) TNFalpha contributes to the death of NGF-dependent neurons during development. Nat Neurosci 4: 1194-1198. [DOI] [PubMed] [Google Scholar]
- Bette M, Schafer MK, van Rooijen N, Weihe E, Fleischer B (1993) Distribution and kinetics of superantigen-induced cytokine gene expression in mouse spleen. J Exp Med 178: 1531-1539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bette M, Kaut O, Schafer MK, Weihe E (2003) Constitutive expression of p55TNFR mRNA and mitogen-specific up-regulation of TNF alpha and p75TNFR mRNA in mouse brain. J Comp Neurol 465: 417-430. [DOI] [PubMed] [Google Scholar]
- Cahill CM, Dray A, Coderre TJ (1998) Priming enhances endotoxininduced thermal hyperalgesia and mechanical allodynia in rats. Brain Res 808: 13-22. [DOI] [PubMed] [Google Scholar]
- Caterina MJ, Schumacher MA, Tominaga M, Rosen TA, Levine JD, Julius D (1997) The capsaicin receptor: a heat-activated ion channel in the pain pathway. Nature 389: 816-824. [DOI] [PubMed] [Google Scholar]
- Cunha FQ, Poole S, Lorenzetti BB, Ferreira SH (1992) The pivotal role of tumour necrosis factor alpha in the development of inflammatory hyperalgesia. Br J Pharmacol 107: 660-664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cunningham Jr ET, Stalder AK, Sanna PP, Liu SS, Bloom FE, Howes Jr EL, Campbell IL, Margolis TP (1997) Distribution of tumor necrosis factor receptor messenger RNA in normal and herpes simplex virus infected trigeminal ganglia in the mouse. Brain Res 758: 99-106. [DOI] [PubMed] [Google Scholar]
- Damoiseaux JG, Dopp EA, Calame W, Chao D, MacPherson GG, Dijkstra CD (1994) Rat macrophage lysosomal membrane antigen recognized by monoclonal antibody ED1. Immunology 83: 140-147. [PMC free article] [PubMed] [Google Scholar]
- Ding M, Hart RP, Jonakait GM (1995) Tumor necrosis factor-alpha induces substance P in sympathetic ganglia through sequential induction of interleukin-1 and leukemia inhibitory factor. J Neurobiol 28: 445-454. [DOI] [PubMed] [Google Scholar]
- Gordon JR, Galli SJ (1991) Release of both preformed and newly synthesized tumor necrosis factor alpha (TNF-alpha)/cachectin by mouse mast cells stimulated via the Fc epsilon RI. A mechanism for the sustained action of mast cell-derived TNF-alpha during IgE-dependent biological responses. J Exp Med 174: 103-107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holst O, Ulmer AJ, Brade H, Flad HD, Rietschel ET (1996) Biochemistry and cell biology of bacterial endotoxins. FEMS Immunol Med Microbiol 16: 83-104. [DOI] [PubMed] [Google Scholar]
- Hopkins SJ, Rothwell NJ (1995) Cytokines and the nervous system. I: Expression and recognition. Trends Neurosci 18: 83-88. [PubMed] [Google Scholar]
- Hua XY, Chen P, Fox A, Myers RR (1996) Involvement of cytokines in lipopolysaccharide-induced facilitation of CGRP release from capsaicin-sensitive nerves in the trachea: studies with interleukin-1beta and tumor necrosis factor-alpha. J Neurosci 16: 4742-4748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ignatowski TA, Covey WC, Knight PR, Severin CM, Nickola TJ, Spengler RN (1999) Brain-derived TNFalpha mediates neuropathic pain. Brain Res 841: 70-77. [DOI] [PubMed] [Google Scholar]
- Junger H, Sorkin LS (2000) Nociceptive and inflammatory effects of subcutaneous TNFalpha. Pain 85: 145-151. [DOI] [PubMed] [Google Scholar]
- Li Y, Ji A, Schäfer MK (2002) Toll-like receptor 4 is expressed by peptidergic presumed nociceptive neurons in rat dorsal root ganglion. Soc Neurosci Abstr 46: 19. [Google Scholar]
- Lindenlaub T, Teuteberg P, Hartung T, Sommer C (2000) Effects of neutralizing antibodies to TNF-alpha on pain-related behavior and nerve regeneration in mice with chronic constriction injury. Brain Res 866: 15-22. [DOI] [PubMed] [Google Scholar]
- Liu B, Li H, Brull SJ, Zhang JM (2002) Increased sensitivity of sensory neurons to tumor necrosis factor alpha in rats with chronic compression of the lumbar ganglia. J Neurophysiol 88: 1393-1399. [DOI] [PubMed] [Google Scholar]
- MacEwan DJ (2002) TNF receptor subtype signalling: differences and cellular consequences. Cell Signal 14: 477-492. [DOI] [PubMed] [Google Scholar]
- Mika J, Li Y, Weihe E, Schafer MK (2003) Relationship of pronociceptin/orphanin FQ and the nociceptin receptor ORL1 with substance P and calcitonin gene-related peptide expression in dorsal root ganglion of the rat. Neurosci Lett 348: 190-194. [DOI] [PubMed] [Google Scholar]
- Murphy PG, Grondin J, Altares M, Richardson PM (1995) Induction of interleukin-6 in axotomized sensory neurons. J Neurosci 15: 5130-5138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murwani R, Hodgkinson S, Armati P (1996) Tumor necrosis factor alpha and interleukin-6 mRNA expression in neonatal Lewis rat Schwann cells and a neonatal rat Schwann cell line following interferon gamma stimulation. J Neuroimmunol 71: 65-71. [DOI] [PubMed] [Google Scholar]
- Nadeau S, Rivest S (1999) Effects of circulating tumor necrosis factor on the neuronal activity and expression of the genes encoding the tumor necrosis factor receptors (p55 and p75) in the rat brain: a view from the blood-brain barrier. Neuroscience 93: 1449-1464. [DOI] [PubMed] [Google Scholar]
- Nohr D, Schafer MK, Persson S, Romeo H, Nyberg F, Post C, Ekstrom G, Weihe E (1999) Calcitonin gene-related peptide gene expression in collagen-induced arthritis is differentially regulated in primary afferents and motoneurons: influence of glucocorticoids. Neuroscience 93: 759-773. [DOI] [PubMed] [Google Scholar]
- Opree A, Kress M (2000) Involvement of the proinflammatory cytokines tumor necrosis factor-alpha, IL-1 beta, and IL-6 but not IL-8 in the development of heat hyperalgesia: effects on heat-evoked calcitonin gene-related peptide release from rat skin. J Neurosci 20: 6289-6293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parada CA, Yeh JJ, Joseph EK, Levine JD (2003) Tumor necrosis factor receptor type-1 in sensory neurons contributes to induction of chronic enhancement of inflammatory hyperalgesia in rat. Eur J Neurosci 17: 1847-1852. [DOI] [PubMed] [Google Scholar]
- Platika D, Boulos MH, Baizer L, Fishman MC (1985) Neuronal traits of clonal cell lines derived by fusion of dorsal root ganglia neurons with neuroblastoma cells. Proc Natl Acad Sci USA 82: 3499-3503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pollock J, McFarlane SM, Connell MC, Zehavi U, Vandenabeele P, MacEwan DJ, Scott RH (2002) TNF-alpha receptors simultaneously activate Ca2+ mobilisation and stress kinases in cultured sensory neurones. Neuropharmacology 42: 93-106. [DOI] [PubMed] [Google Scholar]
- Reeve AJ, Patel S, Fox A, Walker K, Urban L (2000) Intrathecally administered endotoxin or cytokines produce allodynia, hyperalgesia and changes in spinal cord neuronal responses to nociceptive stimuli in the rat. Eur J Pain 4: 247-257. [DOI] [PubMed] [Google Scholar]
- Schafer MK-H, Herman JP, Watson SJ (1993) In situ hybridization histochemistry. In: Imaging drug action in the brain (Edythe D. London, ed), pp 337-378. Boca Raton: CRC.
- Schafers M, Geis C, Brors D, Yaksh TL, Sommer C (2002) Anterograde transport of tumor necrosis factor-alpha in the intact and injured rat sciatic nerve. J Neurosci 22: 536-545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schafers M, Sorkin LS, Geis C, Shubayev VI (2003a) Spinal nerve ligation induces transient upregulation of tumor necrosis factor receptors 1 and 2 in injured and adjacent uninjured dorsal root ganglia in the rat. Neurosci Lett 347: 179-182. [DOI] [PubMed] [Google Scholar]
- Schafers M, Geis C, Svensson CI, Luo ZD, Sommer C (2003b) Selective increase of tumour necrosis factor-alpha in injured and spared myelinated primary afferents after chronic constrictive injury of rat sciatic nerve. Eur J Neurosci 17: 791-804. [DOI] [PubMed] [Google Scholar]
- Shubayev VI, Myers RR (2001) Axonal transport of TNF-alpha in painful neuropathy: distribution of ligand tracer and TNF receptors. J Neuroimmunol 114: 48-56. [DOI] [PubMed] [Google Scholar]
- Sommer C, Schafers M (1998) Painful mononeuropathy in C57BL/Wld mice with delayed wallerian degeneration: differential effects of cytokine production and nerve regeneration on thermal and mechanical hypersensitivity. Brain Res 784: 154-162. [DOI] [PubMed] [Google Scholar]
- Sommer C, Schmidt C, George A (1998) Hyperalgesia in experimental neuropathy is dependent on the TNF receptor 1. Exp Neurol 151: 138-142. [DOI] [PubMed] [Google Scholar]
- Sorkin LS, Doom CM (2000) Epineurial application of TNF elicits an acute mechanical hyperalgesia in the awake rat. J Peripher Nerv Syst 5: 96-100. [DOI] [PubMed] [Google Scholar]
- Sugiura T, Tominaga M, Katsuya H, Mizumura K (2002) Bradykinin lowersthe threshold temperature for heat activation of vanilloid receptor 1. J Neurophysiol 88: 544-548. [DOI] [PubMed] [Google Scholar]
- Wagner R, Myers RR (1996) Schwann cells produce tumor necrosis factor alpha: expression in injured and non-injured nerves. Neuroscience 73: 625-629. [DOI] [PubMed] [Google Scholar]
- Walker K, Dray A, Perkins M (1996) Hyperalgesia in rats following intracerebroventricular administration of endotoxin: effect of bradykinin B1 and B2 receptor antagonist treatment. Pain 65: 211-219. [DOI] [PubMed] [Google Scholar]
- Watkins LR, Wiertelak EP, Goehler LE, Smith KP, Martin D, Maier SF (1994) Characterization of cytokine-induced hyperalgesia. Brain Res 654: 15-26. [DOI] [PubMed] [Google Scholar]
- Watkins LR, Goehler LE, Relton J, Brewer MT, Maier SF (1995) Mechanisms of tumor necrosis factor-alpha (TNF-alpha) hyperalgesia. Brain Res 692: 244-250. [DOI] [PubMed] [Google Scholar]
- Watkins LR, Milligan ED, Maier SF (2001) Spinal cord glia: new players in pain. Pain 93: 201-205. [DOI] [PubMed] [Google Scholar]
- Weihe E, Nohr D, Michel S, Muller S, Zentel HJ, Fink T, Krekel J (1991) Molecular anatomy of the neuro-immune connection. Int J Neurosci 59: 1-23. [DOI] [PubMed] [Google Scholar]
- Wesselingh SL, Takahashi K, Glass JD, McArthur JC, Griffin JW, Griffin DE (1997) Cellular localization of tumor necrosis factor mRNA in neurological tissue from HIV-infected patients by combined reverse transcriptase/polymerase chain reaction in situ hybridization and immunohistochemistry. J Neuroimmunol 74: 1-8. [DOI] [PubMed] [Google Scholar]
- Wiertelak EP, Smith KP, Furness L, Mooney-Heiberger K, Mayr T, Maier SF, Watkins LR (1994) Acute and conditioned hyperalgesic responses to illness. Pain 56: 227-234. [DOI] [PubMed] [Google Scholar]