

Abstract
Although Ca2+ release from internal stores has been proposed to be important for the induction of long-term synaptic plasticity, the importance of Ca2+ stores localized in presynaptic terminals remains unclear. Here, we have selectively applied pharmacological antagonists to either the presynaptic or postsynaptic cell in paired whole-cell recordings from hippocampal CA3 pyramidal neurons in slice culture. We demonstrate directly the necessary role of presynaptic, but not postsynaptic, ryanodine-sensitive Ca2+ stores in the induction of NMDA receptor (NMDAR)-dependent long-term depression (LTD). Using two-photon laser scanning microscopy, we further find that release from the ryanodine-sensitive stores during prolonged synaptic stimulation generates a slowly rising Ca2+ signal in the presynaptic terminal that is required for the induction of LTD. Moreover, this form of LTD has a significant presynaptic component of expression because it causes a marked decrease in the rate of release from CA3 neuron presynaptic terminals of FM 1-43, a fluorescent probe of synaptic vesicle cycling. Thus, Ca2+ release from presynaptic ryanodine-sensitive stores is critical in the induction of a presynaptic component of NMDAR-dependent LTD.
Keywords: long-term depression, CA3, internal calcium stores, ryanodine-sensitive stores, hippocampus, synaptic plasticity
Introduction
Ca2+ release from internal stores, mediated by either the IP3 receptor (IP3R) or ryanodine receptor (RyR) Ca2+ release channel, amplifies and dynamically regulates cytosolic Ca2+ levels (Berridge et al., 2003). In neurons, Ca2+ release from internal stores has been implicated in postsynaptic signaling, presynaptic control of transmitter release, and long-term synaptic plasticity (Berridge, 1998; Fitzjohn and Collingridge, 2002), although its role in these events remains controversial. This is partly because of technical difficulties that limit direct measurement of store-dependent Ca2+ signals in small dendritic processes and presynaptic terminals in brain slices. Here, we take advantage of recent advances in imaging techniques to investigate the role of Ca2+ release in long-term depression (LTD) at the CA3-CA3 pyramidal neuron synapse in hippocampal slice culture (Montgomery et al., 2001).
A role for Ca2+ release from internal stores has been previously suggested for LTD at several hippocampal synapses (Reyes and Stanton, 1996; Wang et al., 1997; Caillard et al., 2000; Nishiyama et al., 2000). At least two forms of hippocampal LTD have been identified: an NMDA receptor (NMDAR)-dependent form (Stanton and Sejnowski, 1989; Dudek and Bear, 1992; Mulkey and Malenka, 1992) and a metabotropic glutamate receptor (mGluR)-dependent form (Bolshakov and Siegelbaum, 1994; Oliet et al., 1997). In both cases, a rise in postsynaptic Ca2+ is required for the induction of plasticity (Bolshakov and Siegelbaum, 1994; Oliet et al., 1997). Moreover, at CA3-CA1 synapses in acute hippocampal slices, IP3R-mediated Ca2+ release in the postsynaptic CA1 pyramidal neuron (Reyes and Stanton, 1996; Wang et al., 1997; Caillard et al., 2000; Nishiyama et al., 2000) and RyR-mediated Ca2+ release in either the postsynaptic CA1 neuron (Reyes and Stanton, 1996; Wang et al., 1997; Caillard et al., 2000; Nishiyama et al., 2000) or presynaptic CA3 neuron (Reyes and Stanton, 1996) have been implicated in the induction of NMDAR-dependent LTD. The importance of Ca2+ stores has also been shown genetically because deletion of the RyR3 ryanodine receptor subunit abolishes LTD at CA3-CA1 synapses (Futatsugi et al., 1999). However, a specific Ca2+ signal attributable to RyR- or IP3R-mediated Ca2+ release during the induction of LTD has not yet been identified, nor has the presynaptic contribution of Ca2+ stores been directly investigated.
We have focused on the importance of Ca2+ stores in the induction of LTD at the excitatory collateral synapse formed between CA3 pyramidal neurons in organotypic slice cultures of the rat hippocampus (Stoppini et al., 1991; Debanne et al., 1998; Montgomery and Madison, 2002). Dual whole-cell patch-clamp recordings from pairs of synaptically connected CA3 neurons allowed us to deliver pharmacological agents selectively into either the presynaptic or postsynaptic cell. Two-photon laser scanning microscopy (TPLSM) permitted us to image Ca2+ in individual presynaptic terminals of CA3 neurons during the induction of LTD.
Here, we confirm that robust, NMDAR-dependent LTD exists at CA3-CA3 synapses (Montgomery and Madison, 2002) and show that the induction of this LTD requires Ca2+ release from presynaptic ryanodine-sensitive stores. Moreover, we provide direct evidence that NMDAR-dependent LTD produces a change in presynaptic function, based on two-photon imaging of the release of the fluorescent styryl dye FM 1-43 (Betz and Bewick, 1992) from presynaptic terminals. We suggest that the RyR-dependent Ca2+ signal may gate the response of the presynaptic terminal to a retrograde message, permitting only those terminals that have been activated to undergo LTD. Given the autoassociative nature of the CA3-CA3 connection, this form of LTD may be required to prevent the saturation of transmission at this synapse because of facilitatory processes such as LTP.
Materials and Methods
Slice culture preparation. Slice cultures were made using the general membrane insert technique (Stoppini et al., 1991). Postnatal day 8 Sprague Dawley rats were decapitated, and their brains were quickly removed and placed in chilled minimal essential medium (catalog #12370-037; Invitrogen, San Diego, CA) supplemented with 1% penicillin-streptomycin solution (catalog #15140-122; Invitrogen). The hippocampus was carefully dissected and cut into 400-μm-thick slices using a tissue chopper. Three slices were placed on one Millicell CM membrane insert (catalog #PICM03050; Millipore, Bedford, MA), the underside of which was bathed in 1 ml of warmed 5% CO2 equilibrated culture medium [50% minimal essential medium (catalog #12370-037; Invitrogen), 25% HBSS (catalog #24020-117; Invitrogen), 25% horse serum (catalog #A-3311-L; HyClone, Logan, UT), 0.5% l-glutamine solution (catalog #25030-081; Invitrogen), and 1% penicillin-streptomycin solution (catalog #15140-122; Invitrogen)]. The slice cultures were first placed in a 37°C incubator for 2 d and then moved to a 34°C incubator. Slice cultures were fed by exchanging the culture medium (1 ml; formulation described above) every 2-3 d. Experiments were performed on slice cultures after 1-6 weeks in vitro.
Electrophysiology. Paired whole-cell recordings were obtained with an Axoclamp-2A amplifier (presynaptic cell; Axon Instruments, Foster City, CA) and an EPC-9 amplifier (postsynaptic cell; HEKA Elektronik, Lambrecht/Pfalz, Germany). Patch pipettes were filled with the following internal solution (in mm): 115 KMeSO4, 20 KCl, 10 HEPES, 4 MgCl2, 4 Na2ATP, 10 NaPhosphocreatine, 0.4 Na2GTP, and 0.1 EGTA, titrated to pH 7.2-7.3 with KOH [all reagents were purchased from Sigma (St. Louis, MO), except KMeSO4 from ICN Biomedicals (Cleveland, OH)]. In some experiments, the internal solution was supplemented with one of the following: 2-5 μm thapsigargin (0.1-0.3% DMSO), 30 μm cyclopiazonic acid (CPA; 0.3% DMSO), 300 μm ryanodine (0.3% DMSO), 0.3% DMSO, or 400 μm ruthenium red. Recordings were held ∼1 hr before application of LTD induction protocols to allow sufficient time for drug diffusion throughout the cell. Based on imaging experiments, in which we tracked the time course of diffusion of the calcium dye Oregon Green BAPTA-1 to presynaptic terminals and distal postsynaptic dendrites, a 1 hr period results in a concentration of dye at distal dendrites and presynaptic terminals equal to ∼50-60% of the concentration in the patch pipette. Because most of the drugs we used were similar in size or smaller than this calcium dye (molecular weight, 1114 Da), a 1 hr time period should be adequate for a sufficient concentration of drug to reach its target.
Patch pipettes were pulled to resistances between 3 and 6 MΩ, and access resistances of recordings were between 8 and 30 MΩ. Experiments in which postsynaptic access resistance was not stable were not included in the analysis. Monosynaptic connections were identified by their constant short latency [<3 msec between action potential (AP) peak and EPSC rise]. Individual slice cultures were cut out of the membrane using a scalpel and placed in a submerged perfusion chamber in artificial CSF (ACSF) solution (in mm: 119 NaCl, 2.5 KCl, 1 MgCl2, 2 CaCl2, 1.25 Na2HPO4, 26.2 NaHCO3, and 10 glucose) equilibrated with 95% O2/5% CO2. Slices were perfused at a rate of 2 ml/min. In some experiments, one of the following drugs was added to the ACSF: 50 μm d-APV, 200 μm 1-aminoindan-1,5-dicarboxylic acid (AIDA), 5 μm nitrendipine, 2-3 μm CPA, 50 μm nickel chloride, or 10 μm ryanodine. In experiments using bath application of CPA and ryanodine, slice cultures were perfused with these agents for 20-30 min before the LTD induction protocol to ensure adequate delivery. All experiments using nitrendipine were conducted in a darkened room with the nitrendipine-containing ACSF shielded from light. Thapsigargin, ryanodine, and ruthenium red were purchased from Calbiochem (La Jolla, CA). CPA and nickel chloride were purchased from Sigma. d-APV, AIDA, and nitrendipine were purchased from Tocris (Bristol, UK).
Baseline recordings were made by firing a single AP in the presynaptic cell at 0.1 Hz by injecting 10-15 msec current steps (1 nA), while maintaining the postsynaptic cell in voltage-clamp mode at -70 mV. LTD was induced by switching the postsynaptic cell into current-clamp mode and firing the presynaptic cell at 5 Hz for 3 min (total, 900 APs). The recordings were then returned to the baseline configuration for the duration of the recording. Traces were acquired using Pulse (HEKA), digitized at 5 kHz and filtered at 2 kHz. Because of the variability in the size of the unitary EPSC in paired recording experiments, we used a sliding boxcar window to average 20 successive EPSCs (representing a 200 sec time period) and have displayed the mean EPSC amplitude versus time by shifting the boxcar over each successive EPSC. LTD was measured by averaging 20 data points centered on the time point 15 min after the end of the induction protocol. This analysis was done for each pair, and the average reported is the mean value from the different paired recording experiments. All mean values from paired recording experiments reported either in bar graphs or in the text are calculated from the 15 min time point, as described above. In some experiments, presynaptic neurons were stimulated using an ACSF-filled glass extracellular electrode placed in the CA3 cell body layer. In extracellular stimulation experiments, LTD was analyzed as the value of the data point 15 min after the end of the induction protocol. Errors are reported as SEM. Statistical significance was assessed using Student's t test, except as noted in the text and figure legends. All experiments were performed at room temperature.
Imaging. Calcium imaging was performed by patch clamping individual CA3 pyramidal neurons in whole-cell mode using the same internal solution described above, with the following change: EGTA was replaced with 500 μm Oregon Green 488 BAPTA-1 (Molecular Probes, Eugene, OR). Based on fluorescence intensity, we estimate that the dye concentration at the presynaptic terminal was ∼300 μm. In some experiments, we used the lower-affinity calcium dye Fluo-5F at a concentration of 300 μm in the patch pipette to explore the effects of buffer affinity and concentration on the measured calcium signals. In experiments in which we examined the importance of calcium stores, the internal solution was supplemented with one of the following: 30 μm CPA (0.3% DMSO), 300 μm ryanodine (0.3% DMSO), or 0.3% DMSO.
After ∼1 hr loading, a visual search of the basal dendritic region of the cell revealed portions of the axonal arbor. Following the course of an axon branch would often lead to presynaptic varicosities along its length. TPLSM was performed using a BX50WI upright microscope with a 60×, 0.9 numerical aperture objective (Olympus, Melville, NY) and a 1024-MP system (Bio-Rad, Hercules, CA) with a Tsunami Ti:sapphire laser (Spectra-Physics, Mountain view, CA) tuned to 812 nm. Images were acquired using LaserSharp software (Bio-Rad).
Each frame encompassing a presynaptic terminal was 128 × 128 pixels and was acquired once every 10 sec. These sequences of images of fluorescence intensity were exported and analyzed using custom-written software in Interactive Data Language (Research Systems, Boulder, CO) to measure baseline fluorescence and fluorescence changes. One region of interest (ROI) was centered on the terminal and a second ROI off the cell to record the background. This background was subtracted from the terminal signal to obtain the fluorescence value. Fast and slow phases of the calcium rise were measured as the difference between the fluorescence values at time points specified (see Fig. 4 E). For each trace, fluorescence values for three consecutive time points (10 sec intervals) were averaged during the baseline (indicated by “1”) (see Fig. 4 E), at the peak of the fast phase (“2”), and at the end of the slow phase (“3”) during the 5 Hz train and after decay of the Ca2+ signal back to baseline after the train (“4”). The amplitude of the fast phase was measured as the difference between the resultant values at times 2 and 1 (fast phase, 2-1). The amplitude of the slow phase was measured as the difference between the resultant average values at times 3 and 2 (slow phase, 3-2). All averages reported are the mean value of the different experiments, and the error is SEM.
Figure 4.

Presynaptic terminal Ca2+ rises in two distinct phases during the LTD induction protocol. A, Image of a CA3 pyramidal neuron filled with Oregon Green 488 BAPTA-1. The white box delineates the approximate region shown at higher magnification in B. Scale bar (in C), 50 μm. B, Basal dendritic region showing thin, smooth axons and thicker, spiny dendrites. The arrows point to an example of one axon crossing from top right to bottom left of field. Scale bar (in C), 10 μm. C, High-magnification view of presynaptic terminal (arrow). Scale bar, 1 μm. D, Single images taken from the terminal shown in C, before (1), during the beginning (2), during the end (3), and after (4) the LTD induction protocol. Fluorescence changes coded with a thermal look-up table, small changes coded by “cooler” colors, and larger changes coded by “warmer” colors are shown. Scale bar (in C), 1 μm. E, Ca2+ rise measured as ΔF/F (change in fluorescence intensity divided by initial baseline fluorescence intensity) in the terminal shown in C during the LTD induction protocol. The striped bar marks delivery of LTD induction protocol (5 Hz stimulation for 3 min). The numbers along the trace show times at which images in D were obtained. F, Inset, Presynaptic Ca2+ signal during induction of LTD detected in a cell loaded with 300 μm Fluo-5F, a relatively low-affinity Ca2+ dye. A similar fast and slow component to the Ca2+ signal is detected. Data were averaged from seven cells.
Preparation, acquisition, and analysis of FM 1-43 imaging data were performed using a protocol similar to one published previously (Zakharenko et al., 2001). Slice cultures were loaded by bath application of FM 1-43 (25 μm) for 2 min with no stimulation, followed by a 2 min loading stimulation (10 Hz). After a total of 4 min of exposure to FM 1-43, slices were washed with ACSF containing the sulfonated β-cyclodextrin ADVASEP-7 (100 μm; Cydex, Overland Park, KS) for 20 min to remove any extracellular dye (Kay et al., 1999). Dye release was then determined using 1.5 Hz stimulation in the continued presence of ADVASEP-7, following the same protocol published previously (Zakharenko et al., 2001).
Results
NMDAR-dependent LTD is present at CA3-CA3 synapses
Although LTD has been documented and studied at a variety of central synapses, relatively little is known about LTD at recurrent CA3-CA3 synapses in the hippocampus. We recorded from pairs of synaptically connected CA3-CA3 pyramidal neurons in organotypic slice cultures of the rat hippocampus using a protocol similar to that developed by Madison and colleagues (Pavlidis and Madison, 1999; Pavlidis et al., 2000; Montgomery et al., 2001). Stimulation of the presynaptic cell that evoked a single AP elicited an EPSC in the postsynaptic cell. The amplitude of this EPSC was quite variable (range, ∼10-300 pA), even in a single synaptic pair, most likely because of the probabilistic nature of release from a relatively small number of presynaptic terminals (Fig. 1A, left).
Figure 1.

CA3-CA3 synapses exhibit NMDAR-dependent LTD. A, Left, Peak EPSCs recorded from a synaptically connected pair of CA3-CA3 pyramidal neurons before and after induction of LTD (induction protocol shown by striped bar). Letters a and b mark time periods during the recording from which sample traces are shown (on the right). Right, Presynaptic and postsynaptic responses before (a) and after (b) induction of LTD. The bottom traces are current-clamp records showing an overlay of 10 consecutive APs elicited in the presynaptic cell by current injection. The top traces are voltage-clamp records (Vm = -70 mV) from the postsynaptic cell showing an overlay of 10 consecutive EPSCs. Calibration: vertical, 40 mV, 100 pA; horizontal, 20 msec. B, Averaged (normalized) peak EPSC during experiment before and after LTD induction (n = 11 pairs). C, Sliding boxcar average (window size, 20 points) of data in the presence (top trace) and absence (bottom trace) of 50 μm d-APV. D, LTD at CA3-CA3 synapses does not require activation of mGluR5 receptors. Averaged (normalized) peak EPSCs before and after induction of LTD (induction stimulation shown by bar) in the presence (open squares; n = 6) and absence (closed circles; n = 6) of 10 μm MPEP. In this experiment, extracellular field stimulation was used to elicit EPSCs and induce LTD. No sliding boxcar was used.
When the presynaptic neuron was stimulated at a low frequency of 0.1 Hz, the mean amplitude of the postsynaptic response was stationary over time (Fig. 1A, left). However, when we stimulated the presynaptic neuron at a frequency of 5 Hz for a period of 3 min (900 APs), a protocol used to evoke LTD at CA3-CA1 synapses in acute hippocampal slices (Bolshakov and Siegelbaum, 1994), we observed a robust and long-lasting decrease in the size of the evoked EPSC that lasted at least 30 min (Fig. 1A). On average, 15 min after the induction of LTD, the magnitude of the evoked EPSC was significantly reduced to 32 ± 7% (n = 11 pairs; p < 0.001) of its baseline value (Fig. 1B,C; see Fig. 3C). Because of the variability in the size of the unitary EPSC, we used a boxcar average to display the time course of the EPSC amplitude during the experiments (Fig. 1C). LTD was only observed when the postsynaptic cell was allowed to depolarize under current-clamp conditions during the induction protocol; when the postsynaptic cell was voltage clamped to -70 mV during the induction protocol, no LTD was observed (data not shown). This result indicates that the induction of LTD at CA3-CA3 synapses requires postsynaptic activity.
Figure 3.

Pharmacological analysis of effects of calcium channel antagonists, glutamate receptor antagonists, and disruption of calcium stores on induction of LTD. A, B, Results from boxcar averaged paired recordings showing effects of selective voltage-gated calcium channel antagonist. A, Bath applied nitrendipine (5 μm) does not prevent the induction of LTD (n = 3 pairs). B, Bath applied Ni2+ (50 μm) prevents the induction of LTD (n = 6 pairs). C, Summary of pharmacology experiments. Each bar plots mean reduction in EPSC after induction of LTD. Error bars indicate SEM. Control, Normal extent of LTD in the absence of pharmacological agents; RR, LTD with postsynaptic ruthenium red (400 μm; n = 3); RyPost, LTD with postsynaptic ryanodine (300 μm; n = 4); AIDA, LTD with bath-applied AIDA (200 μm; n = 3); Nitren, LTD with bath-applied nitrendipine (5 μm; n = 3); Ni, LTD with bath-applied Ni2+ (50 μm; n = 6); APV, LTD with bath-applied d-APV (50 μm; n = 5); RyPre, LTD with presynaptic ryanodine (300 μm; n = 4); CPA, LTD with presynaptic CPA (30 μm; n = 4); Thap, LTD with presynaptic thapsigargin (2-5 μm; n = 5). The asterisk indicates that an agent caused a significant inhibition in the amount of LTD (ANOVA, p < 0.002; with post hoc Bonferroni test, p < 0.05).
To distinguish whether this form of LTD requires NMDAR or mGluR activation, we applied specific pharmacological antagonists. When the 5 Hz stimulation protocol was applied in the presence of the NMDAR antagonist d-APV (50 μm), the induction of LTD was blocked. The size of the evoked EPSC was not significantly different from its baseline value before the 5 Hz stimulation protocol (EPSC amplitude was 94 ± 23% of baseline; n = 5 pairs; p > 0.33) (Fig. 1C; see Fig. 3C). In contrast, inhibition of group I mGluRs using either the mGluR5 antagonist 2-methyl-6-(phenylethynyl)-pyridine (MPEP; 10 μm) (Fig. 1D) (Gasparini et al. 1999) or the mGluR1 antagonist AIDA (200 μm) (see Fig. 3C) (Moroni et al., 1997) had no effect on the magnitude of LTD measured 15 min after its induction. Thus, in the presence of either MPEP or AIDA, the 5 Hz stimulation reduced the EPSP to 54 ± 9% (Fig. 1D) (p < 0.05; n = 6) or 48 ± 18% of baseline (see Fig. 3C)(p < 0.05, n = 3), respectively. This is in agreement with the recent findings of Montgomery and Madison (2002) that CA3-CA3 LTD induced by a prolonged 15 min period of 1 Hz stimulation is NMDAR dependent but mGluR independent.
LTD depends on ryanodine-sensitive Ca2+ stores
Although the presence of Ca2+ stores and their role in synaptic plasticity is well documented at parallel fiber-Purkinje cell synapses (Finch and Augustine, 1998; Inoue et al., 1998; Takechi et al., 1998; Daniel et al., 1999) and in the generation of large-amplitude miniature IPSCs (Llano et al., 2000) in the cerebellum, their importance at pyramidal neuron synapses in the hippocampus is more controversial (Harvey and Collingridge, 1992; Emptage et al., 1999, 2001; Nakamura et al., 1999; Svoboda and Mainen, 1999; Rose and Konnerth, 2001). We have therefore tested for the presence and role of Ca2+ stores in the induction of LTD at the CA3-CA3 synapse. First, we used a pharmacological approach, involving bath application of Ca2+ store antagonists and extracellular field stimulation, previously used to demonstrate a role for Ca2+ release in the induction of LTD at CA3-CA1 synapses (Reyes and Stanton, 1996).
In these pharmacological experiments, we used extracellular field stimulation of CA3 inputs at 5 Hz for 3 min, which produced a robust LTD of very similar magnitude to that obtained using paired recordings as described above. On average, the evoked EPSC was reduced to 44 ± 6% of its baseline value 15 min after induction of LTD (n = 8 cells; p < 0.0001) (Fig. 2A). To test for a role of Ca2+ stores in LTD, we applied the LTD induction protocol in the presence of CPA (2-3 μm), an inhibitor of the smooth endoplasmic reticulum Ca2+-ATPase (SERCA) pump that depletes both ryanodine-sensitive and IP3-sensitive stores (Kurebayashi and Ogawa, 1991). Under these conditions, the induction of LTD was blocked. The size of the evoked EPSC measured 15 min after the induction protocol was not significantly different from the baseline EPSC amplitude (117 ± 11% of baseline; n = 10 cells; p > 0.15) (Fig. 2A).
Figure 2.
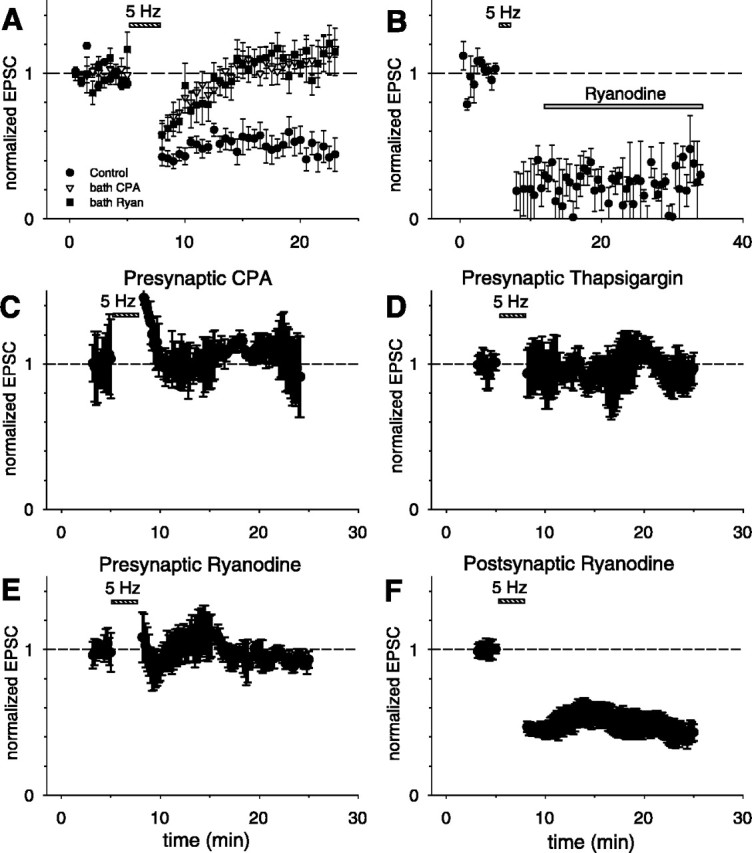
LTD requires presynaptic, but not postsynaptic, ryanodine-sensitive Ca2+ stores. A, Average (normalized) peak EPSC elicited by extracellular stimulation before and after LTD induction in normal external solution (filled circles; n = 8 cells), in presence of external CPA (2-3 μm; open inverted triangles; n = 10 cells), or in presence of external ryanodine (10 μm; filled squares; n = 8 cells). B, Bath application of ryanodine (10 μm; n = 4 cells) 5 min after the end of the LTD induction protocol does not inhibit LTD. C-F, Results from boxcar averaged paired recordings showing effects of selective presynaptic or postsynaptic application of antagonists through recording electrodes. C, Presynaptic application of CPA (30 μm; n = 4 pairs). D, Presynaptic application of thapsigargin (2-5 μm; n = 5 pairs). E, Presynaptic application of ryanodine (300 μm; n = 4 pairs). F, Postsynaptic application of ryanodine (300 μm; n = 4 pairs).
We next investigated whether ryanodine-sensitive Ca2+ stores are involved in the induction of LTD, as has been previously shown for CA3-CA1 synapses (Reyes and Stanton, 1996; Wang et al., 1997; Caillard et al., 2000; Nishiyama et al., 2000). When the 5 Hz stimulation protocol was applied in the presence of 10 μm ryanodine, a concentration that depresses Ca2+ release selectively from ryanodine-sensitive stores (Zucchi and Ronca-Testoni, 1997), LTD was inhibited. The size of the evoked EPSC measured 15 min after the induction protocol was not significantly different from the baseline value (115 ± 16% of baseline; n = 8 cells; p > 0.25) (Fig. 2A). To distinguish whether ryanodine-sensitive stores are necessary for the induction or expression of LTD, we applied ryanodine to the slice 5 min after the end of the induction protocol. This treatment did not reverse LTD. Thus, the magnitude of the EPSC remained depressed at 33 ± 9% of its baseline value 15 min after the induction protocol (n = 4; p < 0.001) (Fig. 2B). This finding is consistent with a previous study at CA3-CA1 synapses that demonstrated a requirement for ryanodine-sensitive stores during the induction, but not the expression, of LTD (Reyes and Stanton, 1996). Taken together, these experiments strongly suggest that ryanodine-sensitive Ca2+ stores are required for LTD induction at CA3-CA3 synapses.
Presynaptic, but not postsynaptic, ryanodine-sensitive stores are required for LTD
Studies using bath application of Ca2+ store antagonists cannot determine whether the stores important for LTD are located presynaptically, postsynaptically, or in glia, all of which contain internal Ca2+ stores that are thought to affect synaptic transmission (Reyes and Stanton, 1996; Wang et al., 1997; Emptage et al., 1999, 2001; Llano et al., 2000; Liang et al., 2002). Here, we have tested directly the potential presynaptic role of ryanodine-sensitive Ca2+ stores required for the induction of LTD by applying drugs to the presynaptic cell through the whole-cell recording electrode.
Our experiments provide several lines of evidence that indicate that presynaptic ryanodine-sensitive Ca2+ stores are necessary for the induction of LTD. First, when we applied to the presynaptic cell CPA (at a concentration of 30 μm in the presynaptic electrode), the induction of LTD was blocked; the average evoked EPSC measured 15 min after the induction protocol was 101 ± 24% of baseline (n = 4 pairs; p > 0.70) (Figs. 2C, 3C). In contrast, inclusion of the DMSO vehicle (see Materials and Methods) by itself in the presynaptic electrode had no effect on LTD; the average EPSC measured 15 min after the induction protocol was reduced to 40 ± 14% of baseline (n = 4 pairs; p < 0.001). Second, presynaptic application of another SERCA pump inhibitor, thapsigargin (2-5 μm), also prevented the induction of LTD, with an average EPSC response equal to 93 ± 28% of baseline (n = 5 pairs; p > 0.53) (Figs. 2D, 3C). Finally, application of ryanodine (300 μm in the pipette) to the presynaptic cell also blocked the induction of LTD, with an average response that was 92 ± 15% of baseline (n = 5 pairs; p > 0.55) (Figs. 2E, 3C). (We used 300 μm ryanodine in the pipette because 10 μm, the concentration used in bath applications, was ineffective, presumably because of loss of the membrane-permeable compound as it diffused from the whole-cell pipette in the soma down the axon to the presynaptic terminals).
In contrast to the inhibitory effect of presynaptic ryanodine, application of ryanodine (300 μm in the postsynaptic electrode) to the postsynaptic cell did not alter the induction of LTD. The average EPSC 15 min after the induction protocol was reduced to 43 ± 12% of baseline (n = 4 pairs; p < 0.01) (Figs. 2F, 3C). The lack of an effect of postsynaptic ryanodine is unlikely to reflect its failure to diffuse into the dendrites because we observed rapid diffusion of a variety of dyes (see below) into distal dendrites and presynaptic ryanodine was effective in blocking LTD. In addition, we found that postsynaptic application of ruthenium red (400 μm in the pipette), a hydrophilic membrane-impermeant inhibitor of Ca2+ release from ryanodine-sensitive stores that prevents LTD at other hippocampal synapses (Caillard et al., 2000), failed to block LTD (EPSC reduced to 45 ± 19% of baseline; n = 3, p < 0.01) (Fig. 3C). These experiments strongly suggest that the requirement for functional ryanodine-sensitive Ca2+ stores during the induction of CA3-CA3 LTD is limited to the presynaptic cell. A lack of involvement of postsynaptic ryanodine-sensitive Ca2+ stores in LTD was previously reported at CA3-CA1 synapses (Reyes and Stanton, 1996; cf. Nishiyama et al., 2000). However, our results do not exclude the possibility that ryanodine-insensitive Ca2+ stores in the postsynaptic cell are necessary for LTD.
LTD depends on T- or R-type, but not L-type, voltage-gated Ca2+ channels
RyRs can participate in a form of Ca2+ store mobilization known as Ca2+-induced Ca2+ release (CICR). In this process, Ca2+ influx through a plasma membrane channel triggers the subsequent release of Ca2+ from internal stores through RyRs. In cardiac muscle, Ca2+ influx through the L-type voltage-gated Ca2+ channel (VGCC) has been shown to be the dominant mechanism for providing RyRs with this trigger source of Ca2+ (Ogawa et al., 1999). However, additional sources, including influx through low voltage-activated T-type Ca2+ channels, have been implicated under certain situations (Ogawa et al., 1999). To test what mechanisms may be at work in CA3-CA3 synapses during the induction of LTD, we repeated our paired whole-cell recording induction protocol with the L-type VGCC antagonist nitrendipine present in the bath solution. Application of nitrendipine (5 μm) did not prevent the induction of LTD, because the average EPSC was reduced to 22 ± 9% of baseline (n = 3 pairs; p < 0.01) (Fig. 3A,C). In contrast, bath application of 50 μm Ni2+, a concentration that is thought to be a relatively selective T- or R-type VGCC antagonist (Fox et al., 1987; Turner et al., 1995; Huguenard, 1996), prevented the induction of LTD. Fifteen minutes after the LTD induction protocol was delivered in the presence of Ni2+, the EPSC remained at 96 ± 11% of its initial value (n = 6 pairs; p > 0.57) (Fig. 3B, C). This is in accordance with evidence that T-type VGCCs are required for the induction of LTD at several other synapses in the hippocampus (Christie et al., 1997; Oliet et al., 1997; Wang et al., 1997).
Although N- and P/Q-type VGCCs are believed to mediate fast synaptic transmission between most central neurons, less is known about the role of T- and R-type VGCCs (Turner et al., 1995; Meir et al., 1999). In light of our data demonstrating that Ni2+ can prevent LTD induction, we examined its effect on basal synaptic transmission at CA3-CA3 synapses. We discovered that 50 μm Ni2+ inhibited the EPSC to 68 ± 7% (n = 4 cells; p < 0.01) of its initial level (data not shown), suggesting that T- or R-type VGCCs might contribute to transmitter release at this synapse. This result complicates the interpretation of our finding that Ni2+ blocked the induction of LTD. Thus, blockade of the T- or R-type channels could act to block induction of LTD either by inhibiting Ca2+ release from internal presynaptic stores or by reducing the extent of postsynaptic activity required to trigger postsynaptic signal transduction cascades (because of the reduction in the EPSC). Results presented below using Ca2+ imaging, however, do suggest a contribution of T- or R-type channels to Ca2+ release from presynaptic stores.
Presynaptic terminal Ca2+ rises in two phases during the LTD protocol
To test directly whether or not the LTD induction protocol mobilizes Ca2+ from internal presynaptic stores, we used TPLSM to image Ca2+ transients in individual presynaptic terminals from CA3 pyramidal neurons. We loaded presynaptic CA3 neurons with the Ca2+ indicator Oregon Green 488 BAPTA-1 (500 μm) in the pipette solution under whole-cell recording conditions. After ∼1 hr of whole-cell recording, portions of the axonal arbor could be observed interspersed among spiny basal dendrites (Fig. 4A). Based on their small diameter, smooth appearance, and presence of varicosities, axons were clearly distinguishable from dendrites (Fig. 4B,C). A previous study on the same preparation has shown that the vast majority of these varicosities are presynaptic terminals of en passant synapses (Emptage et al., 2001).
Imaging of presynaptic terminals of a CA3 neuron during the LTD induction protocol (APs at 5 Hz for 3 min) revealed a large increase in terminal Ca2+ with two distinct phases (Figs. 4D,E, 5A). A fast rising phase is complete within 10 sec of the start of the 3 min period of 5 Hz stimulation (the time resolution of these experiments). During this fast phase, the fluorescence signal significantly increased by 248 ± 25% (n = 11; p < 0.001) over its initial resting level. This fast phase is then followed by a slow continuous rise in Ca2+ levels over the remaining 3 min period of 5 Hz stimulation. During the slow phase, the fluorescence signal significantly increased by an additional 121 ± 48% (n = 11; p < 0.05) over its resting level. To examine whether the slow phase was an artifact of altered Ca2+ buffering attributable to the high concentration in the patch pipette of Oregon Green BAPTA-1, which has a high Ca2+ affinity of 170 nm, we repeated the measurements with a lower concentration of the low-affinity Ca2+ dye Fluo-5F (Kd = 2.3 μm). We observed a rapid and slow phase of the Ca2+ transient in the presynaptic terminals during the 3 min period of 5 Hz stimulation using 300 μm Fluo-5F in the pipette solution (the lowest concentration that gave a reasonable signal in the presynaptic terminal), essentially identical to the results with Oregon Green BAPTA-1 (Fig. 4F). Thus, we conclude that the slow rise in Ca2+ represents a genuine signal that is not an artifact of our measuring conditions.
Figure 5.

Slow phase of Ca2+ signal depends on release from presynaptic ryanodine-sensitive stores, and the fast phase depends partially on influx via T-type or R-type VGCCs. A, Average presynaptic terminal Ca2+ rise during LTD induction protocol (n = 11). Arrows point to fast and slow phases. Selective presynaptic application of CPA (30 μm; n = 15; B) or ryanodine (300 μm; n = 5; C) selectively inhibits the slow phase. D, Bath application of d-APV (50 μm) does not affect either the fast or slow phase (n = 3). E, Bath application of Ni2+ (50 μm) prevents the slow phase and partially inhibits the fast phase (n = 4). F, Summary data from all terminal imaging experiments. The asterisk indicates condition that is significantly different from control (post hoc Fisher LSD test; p < 0.05).
The fast Ca2+ signal depends partially on T- or R-type VGCCs, and the slow phase depends on ryanodine-sensitive stores
To explore a role for release from internal stores in the terminal Ca2+ signal, we imaged the rise in presynaptic Ca2+ during the LTD induction protocol with CPA added to the pipette solution at the same concentration (30 μm) that blocked LTD in our paired recording experiments (Fig. 5B,F). Application of CPA did not affect the initial fast phase of the Ca2+ signal. In the presence of CPA, the Ca2+ signal rose initially by 252 ± 35% (n = 15; p < 0.001) above its resting level, similar to the value of 248% measured without CPA (see above) or to the value of 279 ± 39% (n = 5; p < 0.001) obtained with a pipette solution containing DMSO, the solvent used to dissolve CPA. However, CPA completely blocked the late slow increase in Ca2+, converting it to a slow decline. In the presence of CPA, the fluorescence decreased by 46 ± 20% from its initial peak value during this slow decline (n = 15). This is in contrast to, and significantly different from, the slow increase of 121% determined with our normal pipette solution in the absence of CPA (see above; p < 0.01) or to the slow increase of 178 ± 99% (n = 5; p < 0.01) observed using the DMSO solution. Presynaptic application of ryanodine (300 μm in the pipette), at a concentration that blocked LTD, also significantly blocked the slow phase of the Ca2+ increase, converting it to a slowly declining phase (decreasing by 88 ± 24%; n = 5; p < 0.05) while leaving the fast phase unaffected compared with control (247 ± 39% increase; n = 5, p > 0.98) (Fig. 5C,F). In parallel experiments using the low-affinity calcium indicator Fluo-5F described above, bath application of ryanodine (10 μm) also blocked the slow phase but not the fast phase of the Ca2+ signal (data not shown).
What might provide the Ca2+ trigger for the RyR-mediated slow rise in terminal Ca2+? In light of our pharmacological experiments on LTD, we tested the involvement of NMDARs or T-type/R-type VGCCs using bath application of APV or Ni2+, respectively. Because LTD is blocked by d-APV, a finding normally attributable to postsynaptic NMDAR activation, we wondered whether presynaptic NMDARs might also be involved, as has been recently suggested for cerebellar LTD (Casado et al., 2002). However, we found that d-APV (50 μm), applied in the bath at the same concentration that blocked LTD, had no effect on either the fast phase (286 ± 35% increase; n = 3; p > 0.47) or slow phase (72 ± 48% increase; n = 3; p > 0.62) of the Ca2+ signal compared with control (Fig. 5D,F). In contrast, bath application of the T- and R-type VGCC antagonist Ni2+ at the same concentration that blocked LTD (50 μm) significantly reduced the initial fast phase of the Ca2+ signal by ∼50% (121 ± 27% increase; n = 4; p < 0.05) and completely blocked the slow rise in Ca2+, once again converting it to a slow decline (28 ± 14% decline; n = 4; p < 0.05) (Fig. 5E,F).
Presynaptic Ca2+ stores do not contribute to maintained transmitter release during induction of LTD
What role might Ca2+ release from presynaptic stores play in the induction of LTD? It is generally agreed that release from Ca2+ stores does not contribute to transmitter release evoked by single APs with low-frequency (∼0.1 Hz) stimulation (Reyes and Stanton, 1996; Emptage et al., 2001; Carter et al., 2002). However, it has been suggested that Ca2+ release from internal stores may be involved in short-term synaptic plasticity observed at higher stimulus frequencies (Emptage et al., 2001) (but see Carter et al., 2002). Therefore, Ca2+ release from presynaptic stores may be necessary for maintaining appropriate levels of transmitter release required to produce sufficient postsynaptic activation to induce LTD during the 5 Hz stimulation protocol.
To test this idea, we measured EPSP size during the 5 Hz protocol, either in the presence or absence of antagonists of Ca2+ release (Fig. 6A1-A3). Even in the absence of antagonists, there is a progressive decline in the EPSP amplitude to a steady-state level during the 5 Hz stimulation, presumably resulting from short-term depression because of synaptic vesicle depletion. We characterized several properties of the postsynaptic response during 5 Hz stimulation, including the initial EPSP size, final EPSP size, and time constant of decay of the EPSP. Neither ryanodine nor CPA had any effect on synaptic transmission as measured by these three criteria (Fig. 6B-D). These data suggest that ryanodine-sensitive Ca2+ stores are not required for maintenance of transmitter release during the induction of LTD; rather, these stores must act at a step downstream from the depolarization and activation of the postsynaptic cell.
Figure 6.
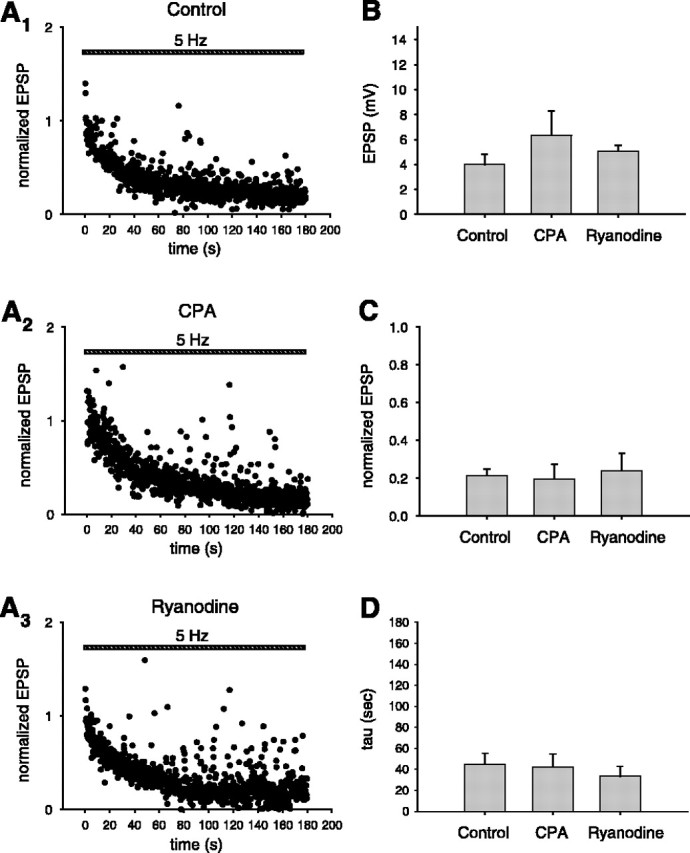
Inhibition of ryanodine-sensitive Ca2+ stores does not reduce postsynaptic depolarization during LTD induction. A1-A3, Average (normalized) peak EPSP during LTD induction protocol (striped bar): A1, control conditions; A2, in the presence of external CPA (2-3 μm); A3, in the presence of external ryanodine (10 μm). B, Size of initial EPSP (average of first 4 responses) during LTD protocol under the three conditions shown in A (ANOVA; p > 0.40). C, Normalized size of steady-state EPSP size at the end of the train (average of last 50 responses) during LTD protocol (ANOVA; p > 0.90). D, Time constant of decay (tau) of EPSP size during LTD induction protocol (ANOVA; p > 0.83).
LTD expression involves a decrease in presynaptic release that is dependent on ryanodine-sensitive stores
The results from our paired recording and imaging experiments argue that presynaptic ryanodine-sensitive stores are required for a slowly rising phase of Ca2+ in the presynaptic terminal that is necessary for the induction of LTD. Given the localization of this slow Ca2+ signal, we next asked whether the expression of CA3-CA3 LTD might have a presynaptic component that requires ryanodine-sensitive stores for its induction. Here, we used the styryl dye FM 1-43, a marker of synaptic vesicle cycling dynamics (Betz and Bewick, 1992; Ryan et al., 1993), to track changes in presynaptic function that may be associated with NMDAR-dependent LTD.
We followed a loading protocol similar to one used previously to investigate presynaptic changes at CA3-CA1 synapses in acute hippocampal slices during LTP (Zakharenko et al., 2001) and during a form of LTD dependent on mGluRs (Zakharenko et al., 2002). FM 1-43 was first loaded into presynaptic terminals by applying the dye (10 μm) in the bath solution during synaptic stimulation (10 Hz for 2 min), which triggers cycles of exocytosis and endocytosis that results in dye uptake into synaptic vesicles. The dye-loading stimulation was performed in the presence of d-APV (50 μm) to block induction of synaptic plasticity at this time. After bath application of the cyclodextrin ADVASEP-7 to remove external dye that is nonspecifically bound to tissue (Kay et al., 1999), brightly fluorescent puncta were visible that corresponded to presynaptic terminals loaded with FM 1-43 (Fig. 7). Presynaptic terminals were then stimulated at 1.5 Hz in the absence of FM 1-43 (but in the presence of d-APV and ADVASEP-7) to unload dye through triggered exocytosis (Fig. 7). Previous studies showed that the rate of dye unloading provides a measure of the efficacy of transmitter release under these conditions (Zakharenko et al., 2001, 2002).
Figure 7.
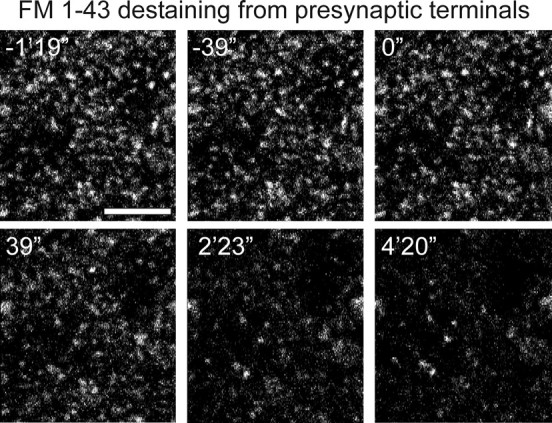
Activity-dependent destaining of FM 1-43 from presynaptic CA3 terminals. Images show FM 1-43 staining for one field of fluorescent puncta that had been previously loaded with dye. Time stamps are marked with reference to the beginning of the 1.5 Hz destaining stimulation, beginning at time 0″ and continuing for 4′ 20″. Scale bar, 10 μm.
After induction of LTD with the 5 Hz stimulation protocol (no APV present), the rate of puncta destaining in the CA3 region of slice cultures was significantly decreased (Fig. 8A), consistent with a reduction in the rate of transmitter release. Bath application of d-APV (50 μm) during the LTD stimulation protocol, which blocks the induction of LTD (Fig. 1D), also blocked the decrease in FM 1-43 destaining rate (Fig. 8B). To determine whether the induction of this presynaptic change in FM 1-43 release kinetics required Ca2+ release from presynaptic RyR-dependent stores, we delivered the LTD induction protocol in the presence of ryanodine (10 μm in the bath solution). Under these conditions, both the induction of LTD and the depression of FM 1-43 release were blocked (Fig. 8C). These results demonstrate that NMDAR-dependent LTD at CA3-CA3 synapses involves altered presynaptic function, which requires Ca2+ release from ryanodine-sensitive stores.
Figure 8.

LTD expression involves a decrease in presynaptic FM 1-43 release that depends on ryanodine-sensitive stores. A1, Decay of average fluorescence intensity of puncta during unloading of FM 1-43 with 1.5 Hz stimulation before (filled circles) and after (open circles) the 5 Hz LTD induction protocol (n = 7 slices). The black bar represents duration of 1.5 Hz unloading stimulation. A2, Frequency histogram of unloading halftimes (t1/2) for individual puncta before the LTD protocol. The filled circle on top shows the mean (and SEM) of the distribution (n = 165 puncta). A3, Frequency histogram of halftimes after the LTD induction protocol (significantly different from distribution before 5 Hz stimulation; Kolmogorov-Smirnov 2-sample test; p < 0.001). The open circle shows the mean of the distribution (n = 168 puncta). The filled circle shows the mean of the distribution from A2. B1, Unloading of FM 1-43 when the LTD protocol was given in the presence of d-APV (50 μm; n = 4 slices). B2, Unloading halftime and the mean of the distribution (filled circle) before the LTD induction protocol (n = 280 puncta). B3, Unloading time and the mean of the distribution (open circle) after the LTD induction protocol (Kolmogorov-Smirnovtest; p > 0.05; n = 150 puncta). The filled circle shows the mean of the distribution from B2. Note that the open and filled circles overlap. C1, Unloading of FM 1-43 when the LTD protocol was given in the presence of ryanodine (10 μm; n = 4 slices). C2, Halftime (t1/2) of unloading and the mean of the distribution (filled circle) before the LTD protocol (n = 374 puncta). C3, Halftime and the mean of the distribution (open circle) after the LTD protocol (Kolmogorov-Smirnov test; p > 0.05; n = 297 puncta). The filled circle shows the mean of the distribution from C2.
Discussion
Our study reveals that the induction of LTD at CA3-CA3 synapses in the hippocampus requires presynaptic ryanodine-sensitive Ca2+ stores. Given the role for such stores during the induction of LTD at other hippocampal synapses (Reyes and Stanton, 1996; Wang et al., 1997; Caillard et al., 2000; Nishiyama et al., 2000), our results indicate the conservation of mechanisms of LTD induction at diverse synapses. Dual whole-cell recordings provided direct evidence that the ryanodine-sensitive stores are required only in the presynaptic neuron and not in the postsynaptic neuron during LTD induction. Calcium imaging experiments demonstrated that the LTD induction protocol recruits a slowly rising component of the presynaptic Ca2+ signal that is attributable to calcium release from ryanodine-sensitive stores. Moreover, this signal is necessary for the induction of LTD and the expression of presynaptic changes in vesicle cycling associated with LTD. It is important to point out, however, that our results do not preclude a role for Ca2+ release from ryanodine-insensitive stores in the postsynaptic cell, for example because of IP3Rs, in the induction of LTD at these synapses.
Perhaps the simplest model that is consistent with our data are that LTD requires CICR from presynaptic Ca2+ stores through activation of RyRs themselves. Alternatively, the slow phase of the presynaptic Ca2+ signal may actually result from presynaptic IP3R-dependent Ca2+ release. In this case, the inhibitory effect of ryanodine would be explained if there were a transient opening of the RyRs during the onset of ryanodine application that depleted the IP3R-dependent Ca2+ stores because of the fact that RyRs and IP3Rs can coexist in the same stores (Zacchetti et al., 1991; Wang et al., 1995). Finally, release from internal stores may require a cooperative interaction between RyRs and IP3Rs (Ashby et al., 2003). According to this model, blockade of either type of release channel would abolish the slow phase.
Our attempts to directly assess the role of IP3-sensitive stores in the induction of LTD were inconclusive. The IP3R [and store-operated channel (SOC)] antagonist 2-aminoethoxydiphenyl borate (2-APB) (Maruyama et al., 1997; Ascher-Landsberg et al., 1999) was toxic to neurons in our slice cultures (concentrations from 5 to 250 μm). Although we found that the IP3R antagonist xestospongin C (10 μm) did not block LTD at this synapse (n = 2; data not shown), the lack of adequate positive controls make this result difficult to interpret. However, our finding that group I mGluRs, the type most commonly linked to IP3 production, do not participate in this form of LTD suggests that IP3R-dependent Ca2+ release may not be involved here. This is further consistent with an electron-microscopic study showing a much higher level of expression of RyRs compared with IP3Rs in axons of hippocampal neurons (Sharp et al., 1993). Thus, we believe that the simplest interpretation of our experiments is that induction of LTD requires CICR from presynaptic stores mediated by the RyRs themselves.
The slow time course of the Ca2+ signal associated with release from ryanodine-sensitive stores during the induction of LTD is puzzling because the rise in Ca2+ in response to CICR through RyRs is usually quite rapid (Bers, 2002). The RyRs or ryanodine-sensitive stores in CA3 terminals may exist initially in a nonfunctional state and may slowly activate during the 3 min period of synaptic stimulation during LTD induction. Kuba and colleagues (Narita et al., 1998, 2000) have reported a similar slow ryanodine-sensitive Ca2+ rise in presynaptic terminals of frog motoneurons during a 20-50 Hz train of stimulation, which they attribute to a “priming” effect of the tetanic stimulation. Liang et al. (2002) have imaged Ca2+ in hippocampal mossy fiber presynaptic terminals during stimulation with short trains of APs. They also suggest that regulation of presynaptic ryanodine-sensitive stores might explain the variable presence of a RyR-dependent delayed component to the Ca2+ signal measured in these terminals.
One possibility is that the stores are initially empty and then slowly fill as Ca2+ enters the terminal through VGCCs during the 5 Hz stimulation. Such an effect would be similar to the slow filling of cardiac Ca2+ stores that underlies a slow increase in cardiac contractility (staircase phenomenon) during a tetanus (Brotto and Creazzo, 1996; Huser et al., 1996). Alternatively, there may be a slow modulation of the RyRs themselves. Two particularly intriguing candidate modulators are nitric oxide and cyclic ADP-ribose, both of which are known to alter RyR function (Zucchi and Ronca-Testoni, 1997). Moreover, pharmacological blockade of either pathway inhibits the induction of LTD at CA3-CA1 synapses (Reyes-Harde et al., 1999). Differences in the basal state of RyR function could explain the conflicting results from different studies arguing either for (Emptage et al., 2001) or against (Carter et al., 2002) a role for store-mediated presynaptic Ca2+ release at pyramidal neuron synapses.
It is also possible that the slow Ca2+ signal is not directly attributable to release from RyR-sensitive stores. Rather, pharmacological interference with RyR-dependent store function might lead to secondary changes in Ca2+ signaling. For example, depletion of Ca2+ from RyR-sensitive internal stores during the 5 Hz stimulation may trigger the opening of plasma membrane SOCs, which could give rise to a slow Ca2+ signal (Bouron, 2000; Putney et al., 2001). We have tried to test this hypothesis with the SOC inhibitor 2-APB (Maruyama et al., 1997; Ascher-Landsberg et al., 1999), but, as reported above, this compound was toxic to our slice cultures at relevant concentrations. A second possibility is that the slow Ca2+ signal is attributable to release from mitochondrial stores (Babcock and Hille, 1998; Kaczmarek, 2000), the filling of which is indirectly affected by inhibition of RyR-sensitive stores (perhaps because of changes in resting Ca2+ levels).
What is the function of the slow ryanodine-sensitive rise in Ca2+? One potential clue is that the time course of this slow signal parallels the time course of LTD induction during the 3 min period of 5 Hz stimulation. Shorter periods of stimulation (e.g., 30 sec), which recruit only a small RyR-dependent Ca2+ signal, induce LTP (Winder et al., 1999), not LTD. Thus, the slow Ca2+ rise could serve as an integrator of synaptic activity, informing the presynaptic terminal as to the history of its activity over a time scale of tens of seconds to minutes. The importance of presynaptic signaling is underscored by our finding that LTD is associated with a diminished rate of FM 1-43 release from presynaptic terminals. Given the requirement of postsynaptic NMDAR activation for LTD induction, these results indicate the need for a retrograde messenger that conveys a postsynaptic signal to the presynaptic terminal. In this context, the RyR-dependent slow phase of [Ca2+]i could act as a gate that restricts the action of the retrograde message to those terminals that are coactivated for prolonged periods with postsynaptic NMDAR stimulation. This may explain how a single retrograde messenger, such as nitric oxide, could mediate both LTP (Arancio et al., 1996) and LTD (Reyes-Harde et al., 1999): only LTD-inducing patterns of stimulation would produce a sufficiently prolonged period of presynaptic activity to elicit a sufficiently large ryanodine-sensitive Ca2+ signal to induce LTD, perhaps by gating the response of the presynaptic terminal to nitric oxide, producing depression rather than potentiation. [This explanation may not apply to forms of spike-timing-dependent LTD induced by shorter periods of stimulation (Nishiyama et al., 2000)]. It is unlikely that the slow presynaptic Ca2+ signal is itself generated by the retrograde messenger because the slow Ca2+ rise was not blocked by inhibition of NMDARs with d-APV (Fig. 5D).
The RyR-dependent presynaptic Ca2+ signal may also act to increase the synapse specificity of homosynaptic LTD. A freely diffusible retrograde messenger could impinge on both presynaptic terminals that are activated during the induction of plasticity and presynaptic terminals that are not active during the induction stimulation. If induction of presynaptic LTD required the conjoint action of the retrograde message with the slow Ca2+ signal, LTD would be restricted to only those synapses that were active during its induction. Recently, Nishiyama et al. (2000) proposed a similar role for postsynaptic RyR-sensitive stores because inclusion of ryanodine in the postsynaptic electrode blocked homosynaptic LTD in CA1 neurons induced by spike timing and resulted in the appearance of heterosynaptic LTD. Our results and those of Stanton et al. (2001, 2003), demonstrating a presynaptic component to NMDAR-dependent LTD, raise the interesting question as to its behavioral relevance. Selective deletion of RyRs from presynaptic neurons using region-specific knock-outs would provide a potentially powerful system for exploring the cellular and behavioral consequences of this regulatory pathway.
Footnotes
This work was supported in part by National Institutes of Health Grants MH50733 and NS29832. We thank Arnold Kriegstein, Amy MacDermott, Andrew Marks, Patric Stanton, and Tamily Weissman for helpful comments on this manuscript and Paul Pavlidis for helpful advice.
Correspondence should be addressed to Dr. Steven A. Siegelbaum, Center for Neurobiology and Behavior, Columbia University, 722 West 168th Street, New York, NY 10032. E-mail: sas8@columbia.edu.
Copyright © 2004 Society for Neuroscience 0270-6474/04/249612-11$15.00/0
References
- Arancio O, Kiebler M, Lee CJ, Lev-Ram V, Tsien RY, Kandel ER, Hawkins RD (1996) Nitric oxide acts directly in the presynaptic neuron to produce long-term potentiation in cultured hippocampal neurons. Cell 87: 1025-1035. [DOI] [PubMed] [Google Scholar]
- Ascher-Landsberg J, Saunders T, Elovitz M, Phillippe M (1999) The effects of 2-aminoethoxydiphenyl borate, a novel inositol 1,4,5-trisphosphate receptor modulator on myometrial contractions. Biochem Biophys Res Commun 264: 979-982. [DOI] [PubMed] [Google Scholar]
- Ashby MC, Petersen OH, Tepikin AV (2003) Spatial characterisation of ryanodine-induced calcium release in mouse pancreatic acinar cells. Biochem J 369: 441-445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Babcock DF, Hille B (1998) Mitochondrial oversight of cellular Ca2+ signaling. Curr Opin Neurobiol 8: 398-404. [DOI] [PubMed] [Google Scholar]
- Berridge MJ (1998) Neuronal calcium signaling. Neuron 21: 13-26. [DOI] [PubMed] [Google Scholar]
- Berridge MJ, Bootman MD, Roderick HL (2003) Calcium signalling: dynamics, homeostasis and remodelling. Nat Rev Mol Cell Biol 4: 517-529. [DOI] [PubMed] [Google Scholar]
- Bers DM (2002) Cardiac excitation-contraction coupling. Nature 415: 198-205. [DOI] [PubMed] [Google Scholar]
- Betz WJ, Bewick GS (1992) Optical analysis of synaptic vesicle recycling at the frog neuromuscular junction. Science 255: 200-203. [DOI] [PubMed] [Google Scholar]
- Bolshakov VY, Siegelbaum SA (1994) Postsynaptic induction and presynaptic expression of hippocampal long-term depression. Science 264: 1148-1152. [DOI] [PubMed] [Google Scholar]
- Bouron A (2000) Activation of a capacitative Ca(2+) entry pathway by store depletion in cultured hippocampal neurones. FEBS Lett 470: 269-272. [DOI] [PubMed] [Google Scholar]
- Brotto MA, Creazzo TL (1996) Ca2+ transients in embryonic chick heart: contributions from Ca2+ channels and the sarcoplasmic reticulum. Am J Physiol 270: H518-H525. [DOI] [PubMed] [Google Scholar]
- Caillard O, Ben-Ari Y, Gaiarsa JL (2000) Activation of presynaptic and postsynaptic ryanodine-sensitive calcium stores is required for the induction of long-term depression at GABAergic synapses in the neonatal rat hippocampus amphetamine. J Neurosci 20: RC94(1-5). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carter AG, Vogt KE, Foster KA, Regehr WG (2002) Assessing the role of calcium-induced calcium release in short-term presynaptic plasticity at excitatory central synapses. J Neurosci 22: 21-28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casado M, Isope P, Ascher P (2002) Involvement of presynaptic N-methyl-d-aspartate receptors in cerebellar long-term depression. Neuron 33: 123-130. [DOI] [PubMed] [Google Scholar]
- Christie BR, Schexnayder LK, Johnston D (1997) Contribution of voltagegated Ca2+ channels to homosynaptic long-term depression in the CA1 region in vitro. J Neurophysiol 77: 1651-1655. [DOI] [PubMed] [Google Scholar]
- Daniel H, Levenes C, Fagni L, Conquet F, Bockaert J, Crepel F (1999) Inositol-1,4,5-trisphosphate-mediated rescue of cerebellar long-term depression in subtype 1 metabotropic glutamate receptor mutant mouse. Neuroscience 92: 1-6. [DOI] [PubMed] [Google Scholar]
- Debanne D, Gahwiler BH, Thompson SM (1998) Long-term synaptic plasticity between pairs of individual CA3 pyramidal cells in rat hippocampal slice cultures. J Physiol (Lond) 507: 237-247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dudek SM, Bear MF (1992) Homosynaptic long-term depression in area CA1 of hippocampus and effects of N-methyl-d-aspartate receptor blockade. Proc Natl Acad Sci USA 89: 4363-4367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emptage N, Bliss TV, Fine A (1999) Single synaptic events evoke NMDA receptor-mediated release of calcium from internal stores in hippocampal dendritic spines. Neuron 22: 115-124. [DOI] [PubMed] [Google Scholar]
- Emptage NJ, Reid CA, Fine A (2001) Calcium stores in hippocampal synaptic boutons mediate short-term plasticity, store-operated Ca2+ entry, and spontaneous transmitter release. Neuron 29: 197-208. [DOI] [PubMed] [Google Scholar]
- Finch EA, Augustine GJ (1998) Local calcium signalling by inositol-1,4,5-trisphosphate in Purkinje cell dendrites. Nature 396: 753-756. [DOI] [PubMed] [Google Scholar]
- Fitzjohn SM, Collingridge GL (2002) Calcium stores and synaptic plasticity. Cell Calcium 32: 405-411. [DOI] [PubMed] [Google Scholar]
- Fox AP, Nowycky MC, Tsien RW (1987) Single-channel recordings of three types of calcium channels in chick sensory neurones. J Physiol (Lond) 394: 173-200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Futatsugi A, Kato K, Ogura H, Li ST, Nagata E, Kuwajima G, Tanaka K, Itohara S, Mikoshiba K (1999) Facilitation of NMDAR-independent LTP and spatial learning in mutant mice lacking ryanodine receptor type 3. Neuron 24: 701-713. [DOI] [PubMed] [Google Scholar]
- Gasparini F, Lingenhohl K, Stoehr N, Flor PJ, Heinrich M, Vranesic I, Biollaz M, Allgeier H, Heckendorn R, Urwyler S, Varney MA, Johnson EC, Hess SD, Rao SP, Sacaan AI, Santori EM, Velicelebi G, Kuhn R (1999) 2-Methyl-6-(phenylethynyl)-pyridine (MPEP), a potent, selective and systemically active mGlu5 receptor antagonist. Neuropharmacology 38: 1493-1503. [DOI] [PubMed] [Google Scholar]
- Harvey J, Collingridge GL (1992) Thapsigargin blocks the induction of long-term potentiation in rat hippocampal slices. Neurosci Lett 139: 197-200. [DOI] [PubMed] [Google Scholar]
- Huguenard JR (1996) Low-threshold calcium currents in central nervous system neurons. Annu Rev Physiol 58: 329-348. [DOI] [PubMed] [Google Scholar]
- Huser J, Lipsius SL, Blatter LA (1996) Calcium gradients during excitation-contraction coupling in cat atrial myocytes. J Physiol (Lond) 494: 641-651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inoue T, Kato K, Kohda K, Mikoshiba K (1998) Type 1 inositol 1,4,5-trisphosphate receptor is required for induction of long-term depression in cerebellar Purkinje neurons. J Neurosci 18: 5366-5373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaczmarek LK (2000) Mitochondrial memory banks. Calcium stores keep a record of neuronal stimulation. J Gen Physiol 115: 347-350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kay AR, Alfonso A, Alford S, Cline HT, Holgado AM, Sakmann B, Snitsarev VA, Stricker TP, Takahashi M, Wu LG (1999) Imaging synaptic activity in intact brain and slices with FM1-43 in C. elegans, lamprey, and rat. Neuron 24: 809-817. [DOI] [PubMed] [Google Scholar]
- Kurebayashi N, Ogawa Y (1991) Discrimination of Ca(2+)-ATPase activity of the sarcoplasmic reticulum from actomyosin-type ATPase activity of myofibrils in skinned mammalian skeletal muscle fibres: distinct effects of cyclopiazonic acid on the two ATPase activities. J Muscle Res Cell Motil 12: 355-365. [DOI] [PubMed] [Google Scholar]
- Liang Y, Yuan LL, Johnston D, Gray R (2002) Calcium signaling at single mossy fiber presynaptic terminals in the rat hippocampus. J Neurophysiol 87: 1132-1137. [DOI] [PubMed] [Google Scholar]
- Llano I, Gonzalez J, Caputo C, Lai FA, Blayney LM, Tan YP, Marty A (2000) Presynaptic calcium stores underlie large-amplitude miniature IPSCs and spontaneous calcium transients. Nat Neurosci 3: 1256-1265. [DOI] [PubMed] [Google Scholar]
- Maruyama T, Kanaji T, Nakade S, Kanno T, Mikoshiba K (1997) 2APB, 2-aminoethoxydiphenyl borate, a membrane-penetrable modulator of Ins(1,4,5)P3-induced Ca2+ release. J Biochem (Tokyo) 122: 498-505. [DOI] [PubMed] [Google Scholar]
- Meir A, Ginsburg S, Butkevich A, Kachalsky SG, Kaiserman I, Ahdut R, Demirgoren S, Rahamimoff R (1999) Ion channels in presynaptic nerve terminals and control of transmitter release. Physiol Rev 79: 1019-1088. [DOI] [PubMed] [Google Scholar]
- Montgomery JM, Madison DV (2002) State-dependent heterogeneity in synaptic depression between pyramidal cell pairs. Neuron 33: 765-777. [DOI] [PubMed] [Google Scholar]
- Montgomery JM, Pavlidis P, Madison DV (2001) Pair recordings reveal all-silent synaptic connections and the postsynaptic expression of long-term potentiation. Neuron 29: 691-701. [DOI] [PubMed] [Google Scholar]
- Moroni F, Lombardi G, Thomsen C, Leonardi P, Attucci S, Peruginelli F, Torregrossa SA, Pellegrini-Giampietro DE, Luneia R, Pellicciari R (1997) Pharmacological characterization of 1-aminoindan-1,5-dicarboxylic acid, a potent mGluR1 antagonist. J Pharmacol Exp Ther 281: 721-729. [PubMed] [Google Scholar]
- Mulkey RM, Malenka RC (1992) Mechanisms underlying induction of homosynaptic long-term depression in area CA1 of the hippocampus. Neuron 9: 967-975. [DOI] [PubMed] [Google Scholar]
- Nakamura T, Barbara JG, Nakamura K, Ross WN (1999) Synergistic release of Ca2+ from IP3-sensitive stores evoked by synaptic activation of mGluRs paired with backpropagating action potentials. Neuron 24: 727-737. [DOI] [PubMed] [Google Scholar]
- Narita K, Akita T, Osanai M, Shirasaki T, Kijima H, Kuba K (1998) A Ca2+-induced Ca2+ release mechanism involved in asynchronous exocytosis at frog motor nerve terminals. J Gen Physiol 112: 593-609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narita K, Akita T, Hachisuka J, Huang S, Ochi K, Kuba K (2000) Functional coupling of Ca(2+) channels to ryanodine receptors at presynaptic terminals. Amplification of exocytosis and plasticity. J Gen Physiol 115: 519-532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishiyama M, Hong K, Mikoshiba K, Poo MM, Kato K (2000) Calcium stores regulate the polarity and input specificity of synaptic modification. Nature 408: 584-588. [DOI] [PubMed] [Google Scholar]
- Ogawa Y, Kurebayashi N, Murayama T (1999) Ryanodine receptor isoforms in excitation-contraction coupling. Adv Biophys 36: 27-64. [DOI] [PubMed] [Google Scholar]
- Oliet SH, Malenka RC, Nicoll RA (1997) Two distinct forms of long-term depression coexist in CA1 hippocampal pyramidal cells. Neuron 18: 969-982. [DOI] [PubMed] [Google Scholar]
- Pavlidis P, Madison DV (1999) Synaptic transmission in pair recordings from CA3 pyramidal cells in organotypic culture. J Neurophysiol 81: 2787-2797. [DOI] [PubMed] [Google Scholar]
- Pavlidis P, Montgomery J, Madison DV (2000) Presynaptic protein kinase activity supports long-term potentiation at synapses between individual hippocampal neurons. J Neurosci 20: 4497-4505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Putney Jr JW, Broad LM, Braun FJ, Lievremont JP, Bird GS (2001) Mechanisms of capacitative calcium entry. J Cell Sci 114: 2223-2229. [DOI] [PubMed] [Google Scholar]
- Reyes M, Stanton PK (1996) Induction of hippocampal long-term depression requires release of Ca2+ from separate presynaptic and postsynaptic intracellular stores. J Neurosci 16: 5951-5960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reyes-Harde M, Potter BV, Galione A, Stanton PK (1999) Induction of hippocampal LTD requires nitric-oxide-stimulated PKG activity and Ca2+ release from cyclic ADP-ribose-sensitive stores. J Neurophysiol 82: 1569-1576. [DOI] [PubMed] [Google Scholar]
- Rose CR, Konnerth A (2001) Stores not just for storage. Intracellular calcium release and synaptic plasticity. Neuron 31: 519-522. [DOI] [PubMed] [Google Scholar]
- Ryan TA, Reuter H, Wendland B, Schweizer FE, Tsien RW, Smith SJ (1993) The kinetics of synaptic vesicle recycling measured at single presynaptic boutons. Neuron 11: 713-724. [DOI] [PubMed] [Google Scholar]
- Sharp AH, McPherson PS, Dawson TM, Aoki C, Campbell KP, Snyder SH (1993) Differential immunohistochemical localization of inositol 1,4,5-trisphosphate- and ryanodine-sensitive Ca2+ release channels in rat brain. J Neurosci 13: 3051-3063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stanton PK, Sejnowski TJ (1989) Associative long-term depression in the hippocampus induced by hebbian covariance. Nature 339: 215-218. [DOI] [PubMed] [Google Scholar]
- Stanton PK, Heinemann U, Muller W (2001) FM1-43 imaging reveals cGMP-dependent long-term depression of presynaptic transmitter release. J Neurosci 21: RC167(1-6). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stanton PK, Winterer J, Bailey CP, Kyrozis A, Raginov I, Laube G, Veh RW, Nguyen CQ, Muller W (2003) Long-term depression of presynaptic release from the readily releasable vesicle pool induced by NMDA receptor-dependent retrograde nitric oxide. J Neurosci 23: 5936-5944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stoppini L, Buchs PA, Muller D (1991) A simple method for organotypic cultures of nervous tissue. J Neurosci Methods 37: 173-182. [DOI] [PubMed] [Google Scholar]
- Svoboda K, Mainen ZF (1999) Synaptic [Ca2+]: intracellular stores spill their guts. Neuron 22: 427-430. [DOI] [PubMed] [Google Scholar]
- Takechi H, Eilers J, Konnerth A (1998) A new class of synaptic response involving calcium release in dendritic spines. Nature 396: 757-760. [DOI] [PubMed] [Google Scholar]
- Turner TJ, Lampe RA, Dunlap K (1995) Characterization of presynaptic calcium channels with omega-conotoxin MVIIC and omega-grammotoxin SIA: role for a resistant calcium channel type in neurosecretion. Mol Pharmacol 47: 348-353. [PubMed] [Google Scholar]
- Wang X, Lau F, Li L, Yoshikawa A, van Breemen C (1995) Acetylcholine-sensitive intracellular Ca2+ store in fresh endothelial cells and evidence for ryanodine receptors. Circ Res 77: 37-42. [DOI] [PubMed] [Google Scholar]
- Wang Y, Rowan MJ, Anwyl R (1997) Induction of LTD in the dentate gyrus in vitro is NMDA receptor independent, but dependent on Ca2+ influx via low-voltage-activated Ca2+ channels and release of Ca2+ from intracellular stores. J Neurophysiol 77: 812-825. [DOI] [PubMed] [Google Scholar]
- Winder DG, Martin KC, Muzzio IA, Rohrer D, Chruscinski A, Kobilka B, Kandel ER (1999) ERK plays a regulatory role in induction of LTP by theta frequency stimulation and its modulation by beta-adrenergic receptors. Neuron 24: 715-726. [DOI] [PubMed] [Google Scholar]
- Zacchetti D, Clementi E, Fasolato C, Lorenzon P, Zottini M, Grohovaz F, Fumagalli G, Pozzan T, Meldolesi J (1991) Intracellular Ca2+ pools in PC12 cells. A unique, rapidly exchanging pool is sensitive to both inositol 1,4,5-trisphosphate and caffeine-ryanodine. J Biol Chem 266: 20152-20158. [PubMed] [Google Scholar]
- Zakharenko SS, Zablow L, Siegelbaum SA (2001) Visualization of changes in presynaptic function during long-term synaptic plasticity. Nat Neurosci 4: 711-717. [DOI] [PubMed] [Google Scholar]
- Zakharenko SS, Zablow L, Siegelbaum SA (2002) Altered presynaptic vesicle release and cycling during mGluR-dependent LTD. Neuron 35: 1099-1110. [DOI] [PubMed] [Google Scholar]
- Zucchi R, Ronca-Testoni S (1997) The sarcoplasmic reticulum Ca2+ channel/ryanodine receptor: modulation by endogenous effectors, drugs and disease states. Pharmacol Rev 49: 1-51. [PubMed] [Google Scholar]