

Abstract
Kainate receptors (KARs) play an important role in synaptic physiology, plasticity, and pathological phenomena such as epilepsy. However, the physiological implications for neuronal networks of the distinct expression patterns of KAR subunits are unknown. Using KAR knock-out mice, we show that subunits glutamate receptor (GluR) 5 and GluR6 play distinct roles in kainate-induced gamma oscillations and epileptiform burst activity. Ablation of GluR5 leads to a higher susceptibility of the network to the oscillogenic and epileptogenic effects of kainate, whereas lack of GluR6 prevents kainate-induced gamma oscillations or epileptiform bursts. Based on experimental and simulated neuronal network data as well as the consequences of GluR5 and GluR6 expression for cellular and synaptic physiology, we propose that the functional interplay of GluR5-containing KARs on axons of interneurons and GluR6-containing KARs in the somatodendritic region of both interneurons and pyramidal cells underlie the oscillogenic and epileptogenic effects of kainate.
Keywords: kainate receptors, gamma oscillations, interneurons, hippocampus, knock-out mice, epilepsy
Introduction
Kainate receptors (KARs) are widely expressed in the hippocampal formation with the various subunits [glutamate receptor (GluR) 5-7, KA1-2] showing distinct expression patterns (Wisden and Seeburg, 1993; Bureau et al., 1999). Contrasting our knowledge of KAR expression, our understanding of their role in synaptic and network function is limited. Functional KARs are expressed at both presynaptic and postsynaptic sites, and their activation has a multitude of effects (Chittajallu et al., 1996; Castillo et al., 1997; Clarke et al., 1997; Rodriguez-Moreno et al., 1997, 2000; Vignes and Collingridge, 1997; Cossart et al., 1998, 2001; Frerking et al., 1998, 1999; Contractor et al., 2000, 2001; Schmitz et al., 2000, 2001a; Semyanov and Kullmann, 2001) (for review, see Lerma et al., 2001; Lerma, 2003).
The distinct distribution pattern of KAR subunits strongly suggests that KARs fulfill different roles in the neuronal network that depend on their localization. Surprisingly, the roles of specific KAR subunits in generating rhythmic activity in neuronal networks have never been investigated. Such oscillations, especially in the gamma frequency band (20-80 Hz), play an important role in learning, memory, and cognition (Gray et al., 1989; Singer, 1993). KAR antagonists prevent induction of mossy fiber long-term potentiation (Bortolotto et al., 1999), which is considered important for learning and memory (Muller et al., 2002). Moreover, kainate injection is used as an animal model for epileptogenesis (Ben-Ari, 1985; Ben-Ari and Cossart, 2000), and in some types of epilepsy (Prince, 1978), learning and memory deficits as well as cognitive decline is observed (Teitelbaum et al., 1990; Viskontas et al., 2000). In the present study, we use genetically engineered homozygous mice lacking various KAR subunits [henceforth GluR5-/- (Mulle et al., 2000), GluR6-/- (Mulle et al., 1998), GluR7-/-, and KA2-/- (Contractor et al., 2003)] to investigate an involvement of KARs in gamma oscillations and their underlying cellular and synaptic mechanisms. We demonstrate that kainate-induced extracellular gamma oscillations arise from synchronized intracellular currents and show that ablation of the GluR5 subunit renders the hippocampal network more susceptible to both the oscillogenic and epileptogenic effects of kainate. In contrast, lack of the GluR6 subunit prevents both kainate-induced gamma oscillations and epileptiform bursts. Our data support distinct roles for KARs containing GluR5 expressed on axons of interneurons and for GluR6-containing KARs expressed in the somatodendritic region of both interneurons and pyramidal cells.
Materials and Methods
Hippocampal slice preparation. Mice were anesthetized by isoflurane volatile inhalation and decapitated according to National Institutes of Health guidelines. Horizontal hippocampal slices were prepared from 20- to 25-d-old (450 μm thickness, extracellular field recordings and concomitant “blind” patch-clamp recordings) or 13- to 16-d-old (250 μm thickness, all other patch-clamp recordings) animals of either sex as described previously (Fisahn et al., 2002). Slices were maintained at room temperature (22°C) in a submerged holding chamber. For extracellular field and concomitant patch-clamp recordings, slices were transferred to an interface chamber and maintained at 35°C at the interface between warm, humidified, carbogen gas (95% O2/5% CO2) and artificial CSF (ACSF) containing the following (in mm): 130 NaCl, 3.5 KCl, 24 NaHCO3, 1.25 NaH2PO4, 1.5 CaCl2, 1.5 MgCl2, and 10 glucose. For patch-clamp recordings, slices were transferred to a submerged recording chamber and superfused with ACSF at room temperature.
Electrophysiology. Extracellular field recordings were made in st. radiatum of the hippocampal area CA3 and patch-clamp recordings from somata of pyramidal cells and identified interneurons of area CA3 as described previously (Fisahn et al., 1998, 2002). The anatomical identity of interneurons was confirmed post hoc and most closely resemble basket cells, a subgroup of interneurons that synapses onto the somatic domain of pyramidal cells as well as other interneurons and play a major role in controlling the frequency of rhythmic network activity (Freund and Buzsaki, 1996).
Briefly, glass microelectrodes (resistance, 3-4 MΩ for both extracellular and patch-clamp recordings) were made from thin-walled borosilicate glass TW150F (World Precision Instruments, Sarasota, FL). They contained the following recording solutions: for extracellular field recordings, electrodes were filled with oxygenated ACSF; for patch-clamp recordings of IPSCs (in mm), 140 cesium methylsulfate, 10 HEPES, 0.6 EGTA, 2 ATPNa2, 0.3 GTPNa2, 2 MgCl2; for patch-clamp recordings of kainate-induced inward currents, depolarizations, and Ih (in mm), 130 K-gluconate, 2 ATPNa2, 0.3 GTPNa2, 10 HEPES, 10 BAPTA, 1 MgCl2. Biocytin (0.5%) was added to the internal solution when recording from interneurons to allow post hoc identification. For patch-clamp recordings of IsAHP, the solution contained the following (in mm): 140 potassium methylsulfate, 10 HEPES, 0.6 EGTA, 2 ATPNa2, 0.3 GTPNa2, 2 MgCl2. Osmolarity was adjusted to 270-280 mOsmol, pH 7.3. Drugs were purchased from Tocris Cookson (Bristol, UK) [kainate, metabotropic GluR antagonist α-methyl-4-carboxyphenylglycine (S-MCPG), DNQX, d-APV,GABAB receptor (GABABR) antagonist (+)-(2S)-5,5-dimethyl-2-morpholineacetic acid (SCH50911), hyperpolarization-activated ion channel blocker 4-(N-ethyl-N-phenylamino)-1,2-dimethyl-6-(methylamino) pyrimidinium chloride (ZD7288)] or Sigma [TTX, AMPA receptor (AMPAR) antagonist 1-(4-aminophenyl)-4-methyl-7,8-methylenedioxy-5H-2,3-benzodiazepine (GYKI53655), picrotoxin, biocytin]. All drugs were diluted directly from frozen stock solutions into the superfusion medium. Patch-clamp recordings were made using an Axopatch-1D amplifier with pClamp acquisition software (Axon Instruments, Foster City, CA). When recording evoked IPSCs (eIPSCs), slices were stimulated at sites close to the stratum pyramidale-stratum radiatum border by patch pipettes filled with oxygenated ACSF (as described above). Current pulses (∼100 μsec, 40-100 μA amplitude) were delivered at a frequency of 0.1 Hz.
Interface chamber versus submerged chamber recordings. Recording extracellular field activity in an interface chamber allows for a better signal-to-noise ratio. However, to elucidate the cellular effects of kainate, patch-clamp recordings of single cells are necessary (dual extracellular-intracellular recordings using blind patch clamp in the interface chamber were only made to obtain data on the temporal relationship of intracellular and extracellular events). Despite the different recording configurations used in interface and submerged chambers (see Hippocampal Slice Preparation), the oscillation-inducing effects and the cellular effects of kainate can be compared in a meaningful manner. Bath temperature only affects the peak frequency of kainate-induced gamma oscillation (24°C, 17.1 ± 0.4 Hz; 29°C, 23.8 ± 0.5 Hz; 34°C, 34.6 ± 0.7 Hz; 39°C, 47.7 ± 1.1 Hz; n = 3) (supplemental material, available at www.jneurosci.org). Moreover, different slice thickness (250 vs 450 μm; n = 3) and animal age (p10 vs p28; n = 3) had no effect on kainate-induced gamma oscillations (supplemental material, available at www.jneurosci.org). This demonstrates that the neuronal network in a P10 mouse is already sufficiently developed and interconnected to generate gamma oscillations and correlates well with the suggested role of gamma oscillations in learning, memory, and cognition (Gray et al., 1989; Singer, 1993; Bragin et al., 1995).
KAR knock-out mice. One potential concern in knock-out studies is compensation by other receptor subunits. However, to date, there is no evidence for functional or molecular compensation in the KAR knockout mice. Specifically, Mulle et al. (1998) have shown that there is no increased expression of KA1 in GluR6-/- animals. In addition, studies in dorsal root ganglion (Mulle et al., 2000) have shown no compensation after targeted elimination of the principal kainate receptor subunit in these neurons. Our studies also show that elimination of the principal subunit in CA3 pyramidal cells leads to loss of the kainate-mediated current, suggesting no functional ionotropic-mediated compensation. We did not specifically look at the expression of the KA1 and KA2 receptor subunit in our study. However, given that these receptor subunits by themselves do not form functional receptors, the overall expression of these subunits should not influence our conclusion, which relied on elimination of kainate receptor-mediated depolarization.
Kainate receptor knock-out mice originated from a mixed 129/Sv and C57/Bl6 background that were backcrossed for at least 10 generations to 129Sv/Ev mice to provide an isogenic 129Sv/Ev strain. After six generations of mating to 129Sv/Ev, >99% of the genetic background is 129Sv/Ev. To minimize the numbers of animals used, knock-out mice from homozygote crossings were compared with control recordings taken from 129Sv/Ev wild-type (WT) crossings.
Data analysis. Commercially available and in-house algorithms using Axograph (Axon Instruments) and KaleidaGraph software (Synergy Software, Reading, PA) were used for analyses. Power spectra were obtained over 60-sec-long recording periods using a Fast Fourier Transform algorithm. Data are represented as mean ± SEM. The Student's t test was used for statistical analyses. The paired-pulse ratio (PPR) was calculated as the meanP2/meanP1 (Kim and Alger, 2001), where P1 and P2 were the amplitude of the first and second evoked event, respectively. P2 amplitude was measured after subtraction of the remaining P1 “tail” current. Spikelets were detected as unitary events with a fixed amplitude and a rise time <3 msec.
Computer modeling. The network model used was adapted from the one described by Traub et al. (2003). Briefly, it contains 3072 pyramidal neurons and 384 interneurons (basket, axo-axonic, and dendrite contacting). Each neuron is multicompartmental and has branching dendrites and a segment of axon. Details of intrinsic cell properties and connectivity (except for between-interneuron gap junctions) are given in the study by Traub et al. (2000). The network contains the following synaptic interactions: AMPA receptor-mediated to pyramidal cells and interneurons, and GABAA receptor (GABAAR)-mediated to pyramidal cells and interneurons (with the exception that axo-axonic cells do not contact interneurons). The network also contains gap junctions between the axons of pyramidal cells (Schmitz et al., 2001b) and between the dendrites of interneurons [with properties as described by Traub et al. (2001) and effects as described by Traub et al. (2003)]. Importantly, the model was altered to allow a two component synaptic excitation of interneurons: a fast AMPA component (tau, 1 msec) and a slower and smaller kainate component (tau, 5 msec). The time course (in milliseconds) of interneuron EPSC conductance (in nanosiemens) in the computer model was: c = f (t) = 1.25t exp(-t) + 0.015t exp(-t/5). For additional details on parameters and/or a copy of the program, please contact R.D.T. (roger.traub@downstate.edu).
Results
Kainate-induced gamma oscillations in WT and knock-out mouse hippocampus
Extracellular field recordings were initially used to characterize rhythmic network activity induced by ionotropic glutamate receptor agonists in the CA3 area of hippocampal slice preparations from WT mice. Bath application of the KAR agonists kainate (100-600 nm; n = 20) (Fig. 1A,D) (Hormuzdi et al., 2001) and domoate (100 nm; n = 16; data not shown) induced gamma oscillations in the CA3 hippocampal area, the power of which increased with agonist concentration, whereas its peak frequency remained unaffected (4.3 ± 2.2 10-9V2 at 600 nm kainate; 38.4 ± 5.3 Hz; n = 14) (Fig. 1D). In contrast, superfusion of the AMPAR agonist AMPA (1-10 μm; n = 6) failed to alter baseline network activity beyond a small increase in noise across the whole frequency spectrum (data not shown).
Figure 1.
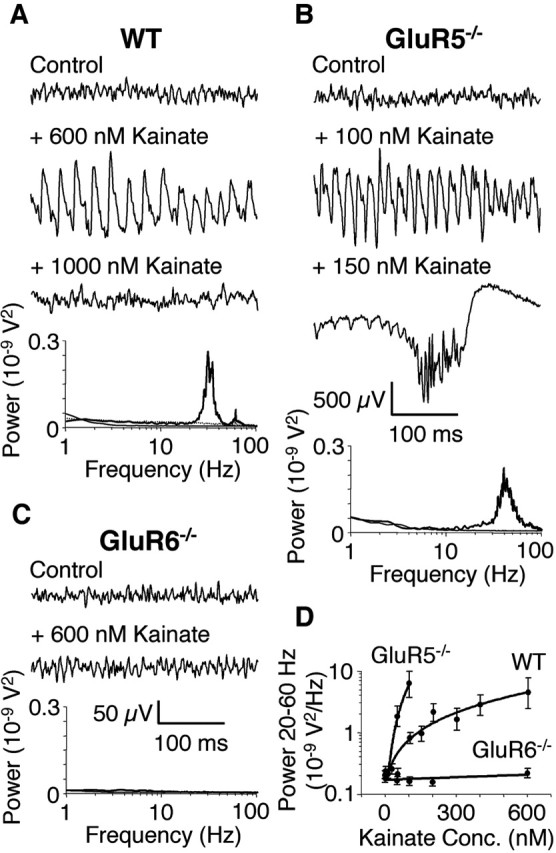
Kainate-induced gamma oscillations are disrupted in GluR6-/- but not GluR5-/- hippocampal slices. A, Example traces of extracellular field recordings in the CA3 area. In WT slices, no rhythmic network activity is seen in control conditions (no drug; thin line). Bath application of kainate induces gamma oscillations (kainate concentration that resulted in the maximal amplitude oscillations is shown; thick line). Increasing the kainate concentration leads to a breakdown of gamma oscillations (dotted line). Power spectra of the recorded traces are shown below. B, In GluR5-/- slices, maximal amplitude gamma oscillations are induced by much lower concentrations of kainate compared with WT (thick line). Increasing the kainate concentration leads to the occurrence of epileptiform burst activity (dotted line; 500 μV scale bar applies only to 150 nm kainate trace). Power spectra of the recorded traces are shown below (the power spectrum of the trace showing an epileptiform burst is too large to be displayed). C, In GluR6-/- slices, kainate fails to induce either gamma oscillations or epileptiform bursts (thick line). Power spectra of the recorded traces are shown below. D, Summary diagram showing the dependence of gamma oscillation power (integrated between 20 and 60 Hz) on kainate concentration. WT and GluR5-/- data points are shown only for kainate concentrations that resulted in gamma oscillations (n = 6 for WT, GluR5-/-, and GluR6-/-).
Kainate-induced gamma oscillations persisted in the presence of the NMDAR antagonist d-APV (50 μm; n = 6) and the broad-spectrum mGluR antagonist (S)-MCPG (200 μm; n = 6) (Fig. 2A). Importantly, kainate-induced gamma oscillations persisted in the presence of the selective AMPAR antagonist GYKI53655 (50 μm; n = 6) but were abolished by the AMPAR/KAR antagonist DNQX (20 μm; n = 6) (Fig. 2A). Also, the ability of kainate to induce gamma oscillations remained unaffected by previous application of GYKI53655 (50 μm), besides a decrease in peak frequency to 19.7 ± 4.1 Hz (n = 3), which was confirmed in a computer model of kainate-induced gamma oscillations adapted from Traub et al. (2003) (see Materials and Methods). Kainate-induced gamma oscillations persisted both in the presence of the cholinergic antagonist atropine (20 μm; n = 6) (Fig. 2B), a concentration sufficient to block muscarine-induced gamma oscillations (Fisahn et al., 1998), as well as in M1-/- mice in which muscarine-induced gamma oscillations were abolished (Fisahn et al., 2002), confirming that kainate-induced gamma oscillations do not rely on cholinergic-muscarinic mechanisms also known to induce this activity in hippocampus (Fisahn et al., 1998). Finally, kainate-induced gamma oscillations were abolished by the GABAAR antagonist bicuculline (3 μm; n = 6) (Fig. 2B), revealing a dependence on intact inhibitory neurotransmission via GABAARs. Elimination of synaptic inhibition via GABAARs in the computer model leads to cessation of gamma oscillations and confirms the experimental data.
Figure 2.
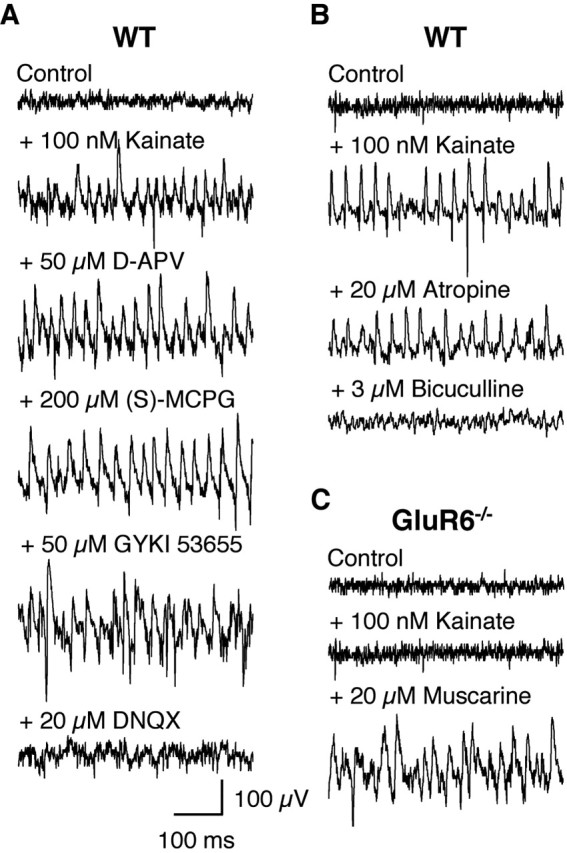
Pharmacology of kainate-induced gamma oscillations in hippocampal slices. A, Example traces of extracellular field recordings in the CA3 area. Kainate-induced gamma oscillations do not depend on NMDAR, mGluR, or AMPAR activation but on KARs (n = 6). B, Kainate-induced gamma oscillations do not depend on cholinergic receptors but do require intact inhibitory GABAergic neurotransmission (n = 6). C, In GluR6-/- slices, kainate-induced gamma oscillations are absent but can still be induced by muscarine (n = 4).
Given the wide expression of KAR subunits in the hippocampus, we next investigated which were necessary for kainate-induced gamma oscillations. In slices from GluR7-/- (3.9 ± 2.5 10-9V2 at 600 nm kainate; 39.7 ± 6.1 Hz; n = 10; data not shown) and KA2-/- (4.6 ± 2.7 10-9V2 at 600 nm kainate; 40.1 ± 5.8 Hz; n = 10; data not shown) mice, kainate-induced gamma oscillations were indistinguishable from WT, ruling out a role for either of these subunits. Interestingly, in GluR5-/- hippocampal slices, the concentration of kainate sufficient to elicit gamma oscillations of maximum and comparable power was significantly lower (6.0 ± 2.6 10-9V2 at 100 nm kainate; 39.7 ± 5.9 Hz; n = 6) than observed in WT (Fig. 1A,D), whereas the pharmacological characteristics of the activity in GluR5-/- remained comparable (data not shown). In WT, when the kainate concentration was increased further, gamma oscillations broke down and ceased (1000 nm; n = 6) (Fig. 1A), whereas a similar manipulation in GluR5-/- resulted in epileptiform activity as evidenced by interictal bursting (150 nm; n = 6) (Fig. 1B). Epileptiform bursts were not observed in WT hippocampal slices at concentrations of kainate up to 1000 nm. In contrast, kainate failed to induce either gamma oscillations or epileptiform bursts in GluR6-/- hippocampal slices (100-600 nm; n = 6) (Fig. 1C,D). Superfusion with 20 μm muscarine reliably induced gamma oscillations in GluR6-/- hippocampal slices (n = 4) (Fig. 2C), confirming that the inability of the hippocampal network to generate rhythmic activity did not arise from altered neuronal circuitry in the GluR6-/- hippocampus.
Extracellular oscillations depend on inhibitory synaptic events
Rhythmic extracellular activity depends on and reflects ensemble synaptic activity, particularly inhibitory events but also excitatory events and action potentials. Concomitant recordings of extracellular gamma oscillations and whole-cell voltage clamp from single CA3 pyramidal cells demonstrate that IPSCs occur randomly under control conditions with the extracellular field showing only background noise. After superfusion with 100 nm kainate, IPSCs occur rhythmically in the gamma frequency range in parallel with the developing extracellular gamma activity (n = 4) (Fig. 3A). When GABAA receptor activation is blocked with 5 μm bicuculline, IPSCs are markedly reduced and consequently, the extracellular gamma oscillation disappears (n = 4) (Fig. 3A).
Figure 3.
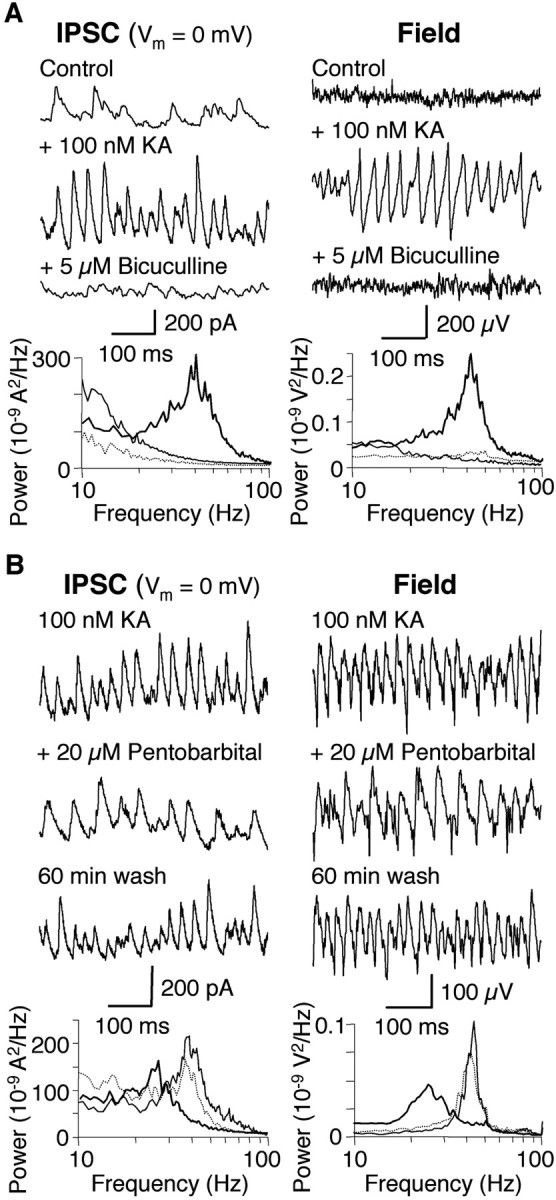
Kainate-induced gamma oscillations depend on inhibitory synaptic currents. Concomitantly recorded intracellular IPSCs (whole-cell patch-clamp mode) and extracellular field activity in WT hippocampal slices show rhythmicity in the gamma frequency range after superfusion with 100 nm kainate (thick line). A, Blocking GABAA receptors with 5 μm bicuculline terminates extracellular and intracellular gamma oscillations (n = 4; dotted line). Power spectra are shown below the example traces. B, Lengthening the time course of IPSCs by application of the benzodiazepine pentobarbital (20 μm) reversibly decreases the frequency of the intracellular and extracellular gamma oscillation (n = 3; thick line; wash, dotted line). Power spectra are shown below the example traces.
Additional confirmation of the dependence of extracellular gamma oscillations on inhibitory events was obtained by increasing the time course of IPSCs using barbiturates. Concomitant recordings of extracellular and intracellular activity show that 20 μm pentobarbital reversibly decreases the frequency of IPSCs occurring in the presence of 100 nm kainate. Consequently, this results in a reversible decrease in the frequency of extracellular gamma oscillations (n = 3) (Fig. 3B), similar to what has been described in muscarine-induced gamma oscillations (Fisahn et al., 1998) and demonstrates the dependence of extracellular gamma oscillations on inhibitory synaptic currents.
Events measured intracellularly during ongoing kainate-induced gamma oscillations occur at precise time points within an extracellularly recorded oscillation cycle. Concomitant recordings of extracellular and intracellular activity and their temporal correlation show that IPSCs occur in antiphase and EPSCs in phase with the extracellular gamma oscillation (n = 4 for each) (Fig. 4A). Investigations in the whole animal have shown that specific interneuron populations with different synaptic target profiles fire action potentials phase locked to specific time points in the extracellular oscillation cycle and are likely to have precise roles in oscillatory activity (Klausberger et al., 2003, 2004).
Figure 4.

Kainate-induced inward current and depolarization are absent in GluR6-/- pyramidal cells and interneurons. A, Concomitant recording of IPSCs and EPSCs (whole-cell patch clamp) and extracellular gamma oscillations. The corresponding cross-correlation analyses show IPSCs are in antiphase and EPSCs are in phase with the extracellular gamma oscillation (n = 4). B, In the absence of AMPAR antagonists, kainate (arrows) induces inward currents in CA3 pyramidal cells of WT but fails to induce any inward current at even the highest kainate concentration tested in pyramidal cells of GluR6-/- hippocampal slices (voltage clamp; Vh = -60 mV). In current clamp (pyramidal cells held below firing threshold), bath application of 100 nm kainate (arrows) leads to a depolarization and increase in firing frequency in WT (Vm = -62.2 ± 2.0 mV) but not GluR6-/- pyramidal cells (Vm = -58.8 ± 1.7 mV). Action potentials are truncated for figure clarity. C, In voltage clamp (Vh = -60 mV), bath application of 100 nm kainate (arrows) induces an inward current in WT (-50.8 ± 9.5 pA) but not GluR6-/- interneurons (-1.9 ± 1.3 pA). In current clamp (interneurons held below firing threshold), bath application of 100 nm kainate (arrows) leads to a depolarization and increase in firing frequency in WT (Vm = -61.7 ± 0.9 mV) but not GluR6-/- interneurons (Vm = -58.0 ± 2.0 mV). Action potentials are truncated for figure clarity. D, Summary histograms of the experiments in C. The kainate-induced inward current is absent in GluR6-/- interneurons (n = 5; GluR5-/-, n = 5; WT, n = 8; p = 0.002). Kainate-induced depolarization and increase in firing frequency are significantly bigger in GluR5-/- (Vm = -63.3 ± 2.0 mV) than in WT interneurons. Partly compromising inhibitory neurotransmission by application of 10 μm picrotoxin results in increased depolarization and firing frequency in WT interneurons (n = 11 for WT; n = 9 for GluR5-/-; n = 5 for GluR6-/-; n = 4 for WT plus picrotoxin; *p < 0.02 for depolarization; p < 0.0001 for frequency).
Kainate-induced effects in pyramidal cells and interneurons
To confirm that exogenous kainate selectively activated KARs and not AMPARs, we next performed whole-cell patch-clamp recordings in voltage-clamped CA3 pyramidal cells (Vh = -60 mV). In the absence of AMPAR antagonists, 100 nm and 3 μm kainate induced inward currents of -43.2 ± 6.5 pA (n = 5) and -223.9 ± 38.6 pA (n = 4) in WT pyramidal cells, respectively. Under the same conditions, 100 nm kainate induced an inward current of -35.5 ± 7.8 pA) in GluR5-/- pyramidal cells. In contrast, 3 μm kainate failed to induce an inward current in GluR6-/- pyramidal cells (-0.8 ± 0.8 pA; p < 0.005; n = 3; 100 μm picrotoxin, 50 μm d-APV, 1 μm TTX present) (Fig. 4B). In the presence of the AMPAR antagonist GYKI53655 (50 μm), the inward current induced by 100 nm kainate persisted in both WT (-46.8 ± 8.1 pA) and GluR5-/- (-28.6 ± 7.0 pA), whereas it remained negligible in GluR6-/- pyramidal cells (-0.5 ± 0.3 pA; n = 6 each; 100 μm picrotoxin, 50 μm d-APV, 1 μm TTX present; data not shown). The kainate-induced inward current in WT and GluR5-/- pyramidal cells was abolished by the AMPAR/KAR antagonist DNQX (20 μm; n = 5 each; data not shown). Therefore, kainate, in the concentrations used in this study, does not activate AMPARs on CA3 pyramidal cells. Consistent with the observed inward current, under current-clamp conditions, kainate (100 nm) depolarized (11.7 ± 1.2 mV) and increased action potential firing (0.8 ± 0.1 Hz) in WT pyramidal cells initially held just below firing threshold (Vm = Vrmp = -62.2 ± 2.0 mV; n = 6). In contrast, kainate failed to depolarize (0.5 ± 0.5 mV; p < 0.0002) or increase action potential firing (0.02 ± 0.02 Hz) in GluR6-/- pyramidal cells (Vm = Vrmp = -58.8 ± 1.7 mV; p < 0.0002; n = 5; no other drugs present) (Fig. 4B). Together, these data demonstrate that a GluR6-dependent membrane depolarization is likely an essential component for generation of gamma oscillatory activity.
Similarly, in visually identified hippocampal interneurons at the CA3 stratum pyramidale-radiatum border, kainate (100 nm) under voltage-clamp conditions (Vh = -60 mV) induced pronounced inward currents in WT (-50.8 ± 9.5 pA; n = 8) and GluR5-/- preparations (-71.4 ± 11.6 pA; n = 5). In contrast, kainate had only a minimal effect in GluR6-/- interneurons (Fig. 4C,D) (-1.9 ± 1.3 pA; n = 5; 100 μm picrotoxin, 50 μm GYKI53655, 50 μm d-APV, 1 μm TTX present). Under current-clamp conditions (interneurons initially held just below firing threshold), kainate (100 nm) caused a membrane depolarization and an increase in action potential firing in WT (9.5 ± 2.0 mV; 3.2 ± 0.6 Hz; Vm = Vrmp = -61.7 ± 0.9 mV; n = 11) and GluR5-/- interneurons (16.8 ± 1.7 mV; 11.9 ± 1.4 Hz; Vm = Vrmp = -63.3 ± 2.0 mV; n = 9; no other drugs present) (Fig. 4C,D). Conversely, kainate failed to depolarize GluR6-/- interneurons (0.4 ± 0.3 mV; 0.0 ± 0.0 Hz; Vm = Vrmp = -58.0 ± 2.0 mV; n = 5; no other drugs present) (Fig. 4C). Surprisingly, the kainate-induced depolarization and firing increase in GluR5-/- interneurons was significantly larger than observed in WT (Fig. 4D). Because the isolated inward currents were not significantly different in WT and GluR5-/- (Fig. 4D), this discrepancy may result from an additional effect arising via the excitatory network. It is possible that GluR5-containing KARs play a role in promoting spontaneous GABA release from interneurons as described previously (Semyanov and Kullmann, 2001). Consequently, a lack of GluR5 would result in reduced GABA release and a concomitant decrease in hippocampal inhibitory tone permitting a greater depolarization by excitatory afferents. Consistent with this hypothesis, partial blockade of GABAARs with picrotoxin (10 μm) resulted in a significantly larger kainate-induced depolarization and increased firing frequency in WT interneurons and approached the values observed in GluR5-/- interneurons (13.6 ± 1.4 mV; 11.2 ± 2.1 Hz; n = 4) (Fig. 4D). Together, these data suggest that KARs containing GluR6 (but not GluR5) are essential for the depolarization and increased excitability of pyramidal cells and interneurons and highlight a potential novel role for GluR5 receptors in regulating the inhibitory tone of local circuit interneurons.
Kainate-induced increases in sIPSC amplitude and frequency are absent in GluR6-/-
Results from extracellular field recordings (Fig. 2A) demonstrated that AMPA receptor activation is not a critical component for kainate-induced gamma oscillations, ruling out a major role for AMPAreceptor-mediated synaptic activity. In contrast, functional GABAA receptors are critical for kainate-induced gamma oscillations (Figs. 2B, 3A). To determine the impact of the kainate-induced increase in interneuron excitability on the local circuitry, we next recorded sIPSCs and mIPSCs in CA3 pyramidal cells. Under voltage clamp (Vh = 0 mV), kainate increased the mean sIPSC amplitude in WT (from 20.8 ± 1.1 to 46.0 ± 4.1 pA; n = 6) and in GluR5-/- (from 8.6 ± 0.9 to 17.6 ± 2.4 pA; n = 6). In GluR6-/-, sIPSC amplitude remained unchanged by kainate (Fig. 5 A, B) (18.6 ± 3.1 to 19.0 ± 3.5 pA; n = 5; 50 μm GYKI53655, 50 μm d-APV, and 20 μm GABABR antagonist SCH50911 present). Similarly, kainate significantly increased sIPSC frequency in WT (14.9 ± 1.0 to 18.3 ± 0.1 Hz; n = 6) and GluR5-/- pyramidal cells (8.0 ± 1.0 to 17.0 ± 0.3 Hz; n = 6), whereas sIPSC frequency was unchanged in GluR6-/- (10.7 ± 2.4 to 12.2 ± 2.3 Hz; n = 5) (Fig. 5A,C). It is interesting to note that the basal level of IPSC amplitude and frequency is significantly lower in GluR5-/- compared with WT. This could indicate a lowered excitability in the inhibitory axonal network and a resulting lowered efficacy of GABAergic synapses as recently described by Jiang et al. (2001).
Figure 5.

Kainate-induced changes in sIPSC are absent in GluR6-/- but not WT and GluR5-/- hippocampal pyramidal cells. A, In voltage clamp (Vh = 0 mV), bath application of kainate induces an increase in sIPSC amplitude and frequency in both WT and GluR5-/- but not GluR6-/- pyramidal cells. sIPSCs are observed as upward deflections at 0 mV. B, Summary histogram for sIPSC amplitude analysis and representative cumulative probability plots of the experiment in A (control, black bars and thin lines; kainate, white bars and thick lines). Kainate increases sIPSC amplitude in WT. In GluR5-/-, sIPSC control amplitude is less than half of WT control amplitude. Kainate increases sIPSC amplitude in GluR5-/- to WT control levels. In GluR6-/-, sIPSC amplitude remains unchanged by kainate at WT control levels (n = 6 for WT and GluR5-/-; n = 5 for GluR6-/-; *p = 0.02 for WT; p < 0.09 for GluR5-/-). C, Summary histogram for sIPSC frequency analysis and representative cumulative probability plots of the experiment in A (control, black bars and thin lines; kainate, white bars and thick lines). Kainate increases sIPSC frequency in WT as well as GluR5-/-. However, sIPSC control frequency in GluR5-/- is approximately half of WT sIPSC control frequency. In GluR6-/-, sIPSC frequency remains unchanged by kainate near WT control levels (n = 6 for WT and GluR5-/-; n = 5 for GluR6-/-; *p < 0.1 for WT; p < 0.02 for GluR5-/-).
In contrast, 100 nm kainate was without effect on mIPSC amplitude or frequency in WT (10.0 ± 0.8 to 10.2 ± 0.7 pA; 4.4 ± 0.4 to 4.3 ± 0.3 Hz) and GluR5-/- pyramidal cells (8.0 ± 0.2 to 7.3 ± 0.3 pA; 6.8 ± 0.4 to 6.8 ± 0.5 Hz; n = 4 each; data not shown; 50 μm GYKI53655, 50 μm d-APV, 20 μm SCH50911, 1 μm TTX present; no significant differences between WT and GluR5-/-). The failure of kainate to alter mIPSC frequency in WT pyramidal cells is consistent with previous findings (Cossart et al., 1998; Frerking et al., 1998, 1999; Semyanov and Kullmann, 2001) (but see Rodriguez-Moreno et al., 1997) and argues against a direct action of kainate on possible terminal KARs to regulate spontaneous transmitter release at interneuron-to-pyramidal cell synapses.
A kainate-induced decrease in eIPSC amplitude is absent in GluR6-/-
Previous studies have indicated that kainate increases sIPSC frequency but decreases eIPSC amplitude (Rodriguez-Moreno et al., 1997; Cossart et al., 1998; Frerking et al., 1998, 1999). These seemingly contradictory results have been explained either by direct actions of kainate on GABA release via presynaptic KARs (Rodriguez-Moreno et al., 1997; Rodriguez-Moreno and Lerma, 1998) or by a kainate-induced increase in GABA release, which acts on both presynaptic and postsynaptic GABARs (Frerking et al., 1998, 1999). In the latter scenario, both presynaptic GABABRs and postsynaptic GABAARs are activated, which subsequently depresses further GABA release and decreases the postsynaptic input resistance (increase postsynaptic shunting), respectively (Frerking et al., 1999).
Consistent with previous reports, the effect of kainate on eIPSC amplitude and PPR contradicted its effects on sIPSCs. Under control conditions, eIPSC (Vh = -60 mV) invariably showed paired pulse depression in both WT (PPR, 0.79 ± 0.05; n = 5) and GluR6-/- pyramidal cells (PPR, 0.81 ± 0.04; n = 4). In contrast, eIPSCs invariably showed facilitation in GluR5-/- pyramidal cells (PPR, 1.27 ± 0.12; n = 5), indicative of a lower initial release probability (Fig. 6A,B). Kainate (100 nm) decreased the eIPSC amplitude in both WT (to 52.4 ± 5.7%; n = 5) and GluR5-/- (to 63.8 ± 2.6%; n = 4) but not GluR6-/- pyramidal cells (Fig. 6A,C) (98.3 ± 1.7%; n = 5; 50 μm GYKI53655, 50 μm d-APV, 20 μm SCH50911 present) without altering the PPR in WT (PPR, 0.78 ± 0.04; n = 5), GluR5-/- (PPR, 1.25 ± 0.15; n = 5) and GluR6-/- pyramidal cells (PPR, 0.79 ± 0.06; n = 4) (Fig. 6A,B). Consistent with the hypothesis that the kainate-induced increase in spontaneous GABA release increases postsynaptic shunting, a concomitant reduction in input resistance in both WT and GluR5-/- pyramidal cells (WT, -57.5 ± 5.0%, n = 9; GluR5-/-, -47.2 ± 5.5%, n = 6) and interneurons (WT, -51.3 ± 4.3%, n = 8; GluR5-/-, -44.2 ± 2.2%, n = 7) (Fig. 6D) was observed. In GluR6-/-, the input resistance change is significantly smaller in both pyramidal cells (7.0 ± 12.9%; n = 7) and interneurons (-15.1 ± 7.8%; n = 5) (Fig. 6D). Together, these data argue against a direct action of kainate at the presynaptic terminal in modulating GABA release and suggest a GluR6-dependent mechanism involving postsynaptic shunting via GABAARs (Frerking et al., 1999). A role of GABABRs is excluded by the presence of a GABAB receptor antagonist throughout this series of experiments.
Figure 6.

Kainate-induced changes in eIPSC are absent in GluR6-/- but not WT and GluR5-/- hippocampal pyramidal cells. A, Paired eIPSCs show paired-pulse depression in WT and GluR6-/- but facilitation in GluR5-/- pyramidal cells in control conditions (thin line) as well as after application of 100 nm kainate (thick line). Concomitantly, kainate depresses eIPSC amplitude in WT and GluR5-/- but has no effect in GluR6-/-. B, PPRs remain unchanged after the application of kainate. C, Summary histogram of eIPSC amplitude changes in the experiment in A (n = 5 for WT and GluR6-/-; n = 4 for GluR5-/-; *p < 0.0001; control, black bars; kainate, white bars). D, Kainate (100 nm) decreases the input resistance of WT and GluR5-/- pyramidal cells and interneurons by indirect action via GABAA receptors (postsynaptic shunting; GABAB receptors are absent at this developmental stage). The kainate-induced change in input resistance in GluR6-/- is significantly smaller (pyramidal cells: n = 9 for WT; n = 6 for GluR5-/-; n = 7 for GluR6-/-; interneurons: n = 8 for WT; n = 7 for GluR5-/-; n = 5 for GluR6-/-; *p < 0.0002 for pyramidal cells; p = 0.001 for interneurons).
Kainate-induced effects are PKC independent
Thus far, the vast majority of kainate-induced effects arise via the actions of kainate on ionotropic receptor channels as opposed to metabotropic effects on transmitter release, mediated by second messengers. We next determined whether kainate also acted on other intrinsic conductances and whether they were necessary for induction of gamma oscillations. We confirmed the kainate-induced decrease in the slow afterhyperpolarization current (IsAHP) (Melyan et al., 2002) and its PKC dependency in CA3 pyramidal cells (Vhold = -50 mV). Kainate (100 nm) decreased the charge transfer of IsAHP by 42.0 ± 8.5% (Fig. 7A) (n = 7; depolarizing test pulses to 0 mV, 200 msec duration, 10 times at 0.05 Hz). In WT hippocampal slices, preincubation with the specific PKC inhibitor calphostin C (2 μm; 2 hr; n = 6) blocked the kainate-induced decrease in IsAHP charge transfer (6.4 ± 6.1%; p = 0.005; 50 μm d-APV, 100 μm picrotoxin, 1 μm TTX present) (Fig. 7A). However, kainate-induced inward currents in WT CA3 pyramidal cells of calphostin C-treated slices (-41.8 ± 12.0 pA; n = 4) were similar in magnitude to naive pyramidal cells (-46.8 ± 8.1 pA; n = 6; 100 μm picrotoxin, 50 μm GYKI53655; 50 μm d-APV, 1 μm TTX present) (Fig. 7B). Similarly, kainate-induced gamma oscillations were indistinguishable in naive and calphostin C-treated slices (n = 6) (Fig. 7C). Therefore, kainate-induced gamma oscillations are independent of PKC-dependent mechanisms and modulation of intrinsic currents such as IsAHP, and the ionotropic function of KARs is not compromised by inhibition of PKC.
Figure 7.
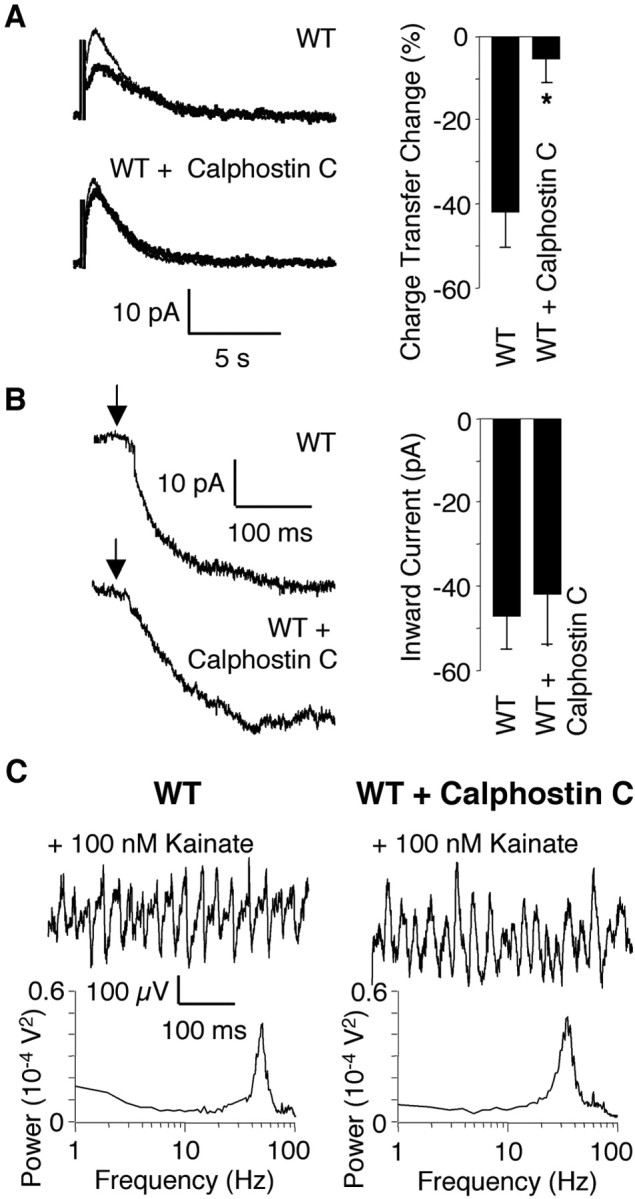
Kainate-induced gamma oscillations are PKC independent. A, In voltage clamp (Vh = -50 mV), IsAHP charge transfer in control conditions (thin line) was decreased by 100 nm kainate (thick line) in WT pyramidal cells (n = 7). Preincubation (2 hr) in the PKC antagonist calphostin C (2 μm) significantly reduced this effect (n = 6; p = 0.005). B, In voltage clamp (Vh = -60 mV) after 2 hr preincubation in 2 μm calphostin C, bath application of 100 nm kainate (arrows) leads to an inward current in WT pyramidal cells that is similar to the inward current induced in naive WT pyramidal cells (n = 6 for WT; n = 4 for WT plus calphostin C). C, Kainate induces extracellular gamma oscillations in both naive and preincubated (2 hr in 2 μm PKC antagonist calphostin C) hippocampal slices. Power spectra are shown below the example traces (n = 6 for WT and WT plus calphostin C).
Finally, we have previously shown that modulation of the hyperpolarization-activated current (Ih) is an essential component for induction of muscarine-induced gamma oscillations (Fisahn et al., 2002). To investigate the possible modulation of Ih by KARs in CA3 pyramidal cells, Ih was activated by hyperpolarizing test pulses to -100 mV (Vhold = -70 mV, 1 sec duration, 5 times at 0.1 Hz; 50 μm d-APV, 100 μm picrotoxin, 1 μm TTX, 1 mm Ba2+ present) as described previously (Fisahn et al., 2002). In both WT (n = 5) and GluR6-/- pyramidal cells (n = 5), kainate (100 nm) failed to modulate the charge transfer of Ih. Likewise, in striking contrast to muscarine-induced gamma oscillations (Fisahn et al., 2002), kainate-induced gamma oscillations were not abolished by superfusion of the Ih antagonist ZD7288 (100 μm; n = 3; data not shown). We conclude that Ih is neither modulated by KARs in CA3 pyramidal cells nor necessary for kainate-induced gamma oscillations.
Ectopic action potentials during gamma oscillations
The simulation of kainate-induced gamma oscillations highlights ectopic spikes as an essential component (Fig. 8, bottom trace), similar to what has been shown in other models of gamma oscillations (Traub et al., 2000, 2003). We next wanted to determine whether modulation of ectopic action potentials is a feature of kainate-induced gamma oscillation in the slice preparation (Semyanov and Kullmann, 2001). Indeed, kainate (100 nm) markedly increased the frequency of ectopic action potentials recorded as fixed amplitude spikelets in both WT interneurons (130.4 ± 12.4%; control, 0.26 ± 0.1 Hz; n = 5) and pyramidal cells (230.3 ± 28.6%; control, 0.19 ± 0.1 Hz; n = 6) (Fig. 8). The increase in spikelet frequency seen in WT was significantly reduced in GluR5-/- (43.4 ± 7.8%; control, 0.29 ± 0.1 Hz; n = 6) and GluR6-/- interneurons (10.3 ± 6.7%; control, 0.33 ± 0.2 Hz; n = 5) (Fig. 8). Moreover, it is notable that the kainate-induced increase in spikelet frequency in GluR5-/- interneurons was significantly larger than in GluR6-/- (p < 0.012). Likewise, the increase in spikelet frequency seen in WT pyramidal cells is significantly reduced in GluR6-/- pyramidal cells (-0.1 ± 7.5%; control, 0.22 ± 0.1 Hz; n = 5) (Fig. 8).
Figure 8.
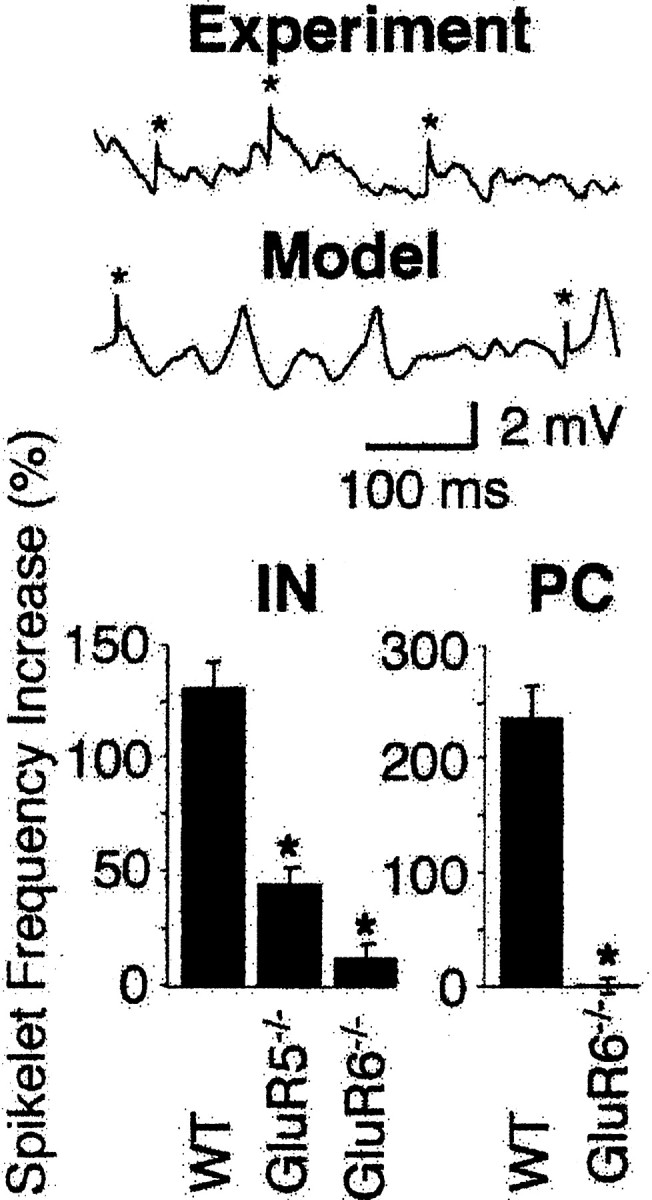
Ectopic action potentials and kainate-induced gamma oscillations. The two example traces show ectopic action potentials recorded as spikelets (*) in pyramidal cells during an ongoing gamma oscillation. The top trace is a physiological recording during kainate-induced gamma oscillations in WT; the bottom trace is computer generated. The summary histograms show the kainate-induced increase in spikelet frequency recorded in interneurons (IN) and pyramidal cells (PC). The increase in spikelet frequency seen in WT interneurons is significantly reduced in GluR5-/- (*p < 0.0002) and GluR6-/- interneurons (p < 0.0001). Note that the increase in spikelet frequency in GluR5-/- is significantly larger than in GluR6-/- (p < 0.012). Likewise, the increase in spikelet frequency seen in WT pyramidal cells is significantly reduced in GluR6-/- (*p < 0.0001) (interneurons: n = 5 for WT, n = 6 for GluR5-/-, n = 5 for GluR6-/-; pyramidal cells: n = 6 for WT, n = 5 for GluR6-/-).
Discussion
In this study, we show that activation of KARs, but not AMPARs, by kainate induces persistent gamma oscillations in the in vitro brain slices of the hippocampus under physiological conditions. Moreover, the lack of the KAR subunits GluR5 or GluR6 produces the following distinct phenotypes of gamma oscillations: heightened susceptibility to epileptiform bursts in response to kainate in the absence of GluR5 and complete failure of kainate to induce gamma oscillations or epileptiform bursts in the absence of GluR6. These distinct phenotypes suggest differential roles played by the GluR5 and GluR6 subunits in kainate-induced gamma oscillations and epileptiform bursts.
GluR5 and GluR6 differentially regulate cellular excitability
KARs have been suggested to exert a strong influence on membrane potential and differ from AMPARs in their ability to encode temporal information (Frerking and Ohliger-Frerking, 2002). Our data support this hypothesis and suggest that GluR5-containing KARs set the inhibitory tone, whereas GluR6-containing KARs provide the necessary bias depolarization of pyramidal cells and interneurons. This arrangement leads to the increase in action potential occurrence in gap junction-connected axonal networks essential for gamma oscillations (Traub et al., 2000). Consistent with this hypothesis, exogenously applied kainate directly depolarizes both WT pyramidal cells and interneurons to increase their action potential firing rate. This increased interneuron excitability translates to an increase in sIPSC amplitude and frequency. All of these effects are absent in GluR6-/-, suggesting that GluR6-containing KARs provide the excitatory drive that sets the polarization level of both CA3 pyramidal cells and interneurons. Our results in WT CA3 pyramidal cells are similar to the kainate-induced increase in sIPSC frequency previously observed in hippocampal area CA1 (Cossart et al., 1998; Frerking et al., 1998). However, in GluR6-/- CA3 pyramidal cells, we failed to observe the kainate-induced sIPSC frequency increase reported by Bureau et al. (1999) for GluR6-/- CA1 pyramidal cells.
In contrast, in GluR5-/-, despite a reduction in the basal level of sIPSC amplitude and frequency, kainate increased both parameters. We suggest that KARs containing GluR5 are intimately involved in setting the level of GABA release and consequent inhibitory tone of the CA3 network, perhaps by depolarizing the axons of inhibitory interneurons. This is consistent with the observations that activation of KARs on mossy fiber (Schmitz et al., 2000) and interneuron axons (Semyanov and Kullmann, 2001) induce an increase in fiber excitability and spontaneous antidromic action potentials. However, because kainate fails to change either the PPR for eIPSCs or the frequency for mIPSCs, GluR5 is most likely not located directly at the presynaptic terminal. Instead, GluR5-containing KARs may be expressed on the axons of interneurons (Semyanov and Kullmann, 2001). The absence of GluR5 would remove a tonic depolarization of interneuron axons resulting in a less excitable inhibitory axonal network and a lowered efficacy of GABAergic synapses (Jiang et al., 2001). Consistent with this hypothesis was the observation of a reduced amplitude and frequency of sIPSCs and a reduced initial release probability of eIPSCs recorded in GluR5-/- pyramidal cells as well as the reduced spikelet frequency increase recorded in GluR5-/- interneurons. The combined effects of the reduction of amplitude and frequency of sIPSCs and initial release probability of eIPSCs would decrease inhibition of both postsynaptic interneurons and pyramidal cells. Although we cannot unequivocally rule out that GluR5-containing KARs are also present on the somato-dendritic axis of pyramidal cells and interneurons, it seems unlikely, given that the kainate-induced inward current was absent in GluR6-/- pyramidal cells and interneurons and that the inward currents in WT and GluR5-/- were virtually identical.
However, the interpretation of the data is made more complex by the fact that KAR subunits GluR6 and GluR5 can form homomeric as well as heteromeric receptors of differing properties, as evidenced by recombinant expression studies (Cui and Mayer, 1999). Although the homomeric receptors desensitize rapidly and completely (Swanson et al., 1997) the heteromeric receptors show reduced desensitization and an enhanced steady-state current in response to kainate (Cui and Mayer, 1999). Although the ratios of homomeric versus heteromeric KAR expression in CA3 pyramidal cells and interneurons is currently unknown, it is possible that altered receptor properties in the GluR5-/- might contribute to some of the effects described in this study, such as the lowered basal level of IPSC amplitude and frequency in GluR5-/-. Future studies are aimed at determining the precise roles for homomeric versus heteromeric kainate receptors on individual cell populations.
Previous reports have observed that kainate either reduces mIPSC frequency (Rodriguez-Moreno et al., 1997) or has no effect (Cossart et al., 1998; Frerking et al., 1998, 1999; Semyanov and Kullmann, 2001). In the present study, we observed no change in mIPSC amplitude and frequency in response to kainate, which supports our hypothesis that kainate does not act directly on the presynaptic terminal to regulate spontaneous transmitter release. Finally, eIPSCs in both WT and GluR6-/- pyramidal cells show paired-pulse depression, whereas GluR5-/- pyramidal cells show paired-pulse facilitation, indicating a lowered initial transmitter release probability in GluR5-/-. Interestingly, although application of kainate reduced the eIPSC amplitude, it failed to affect the PPR in both WT and GluR5-/-. Again, although this argues against kainate acting directly on presynaptic KARs to regulate transmitter release, it suggests that KAR activation depolarizes interneurons to cause an increase in action potential firing and GABA release (Frerking et al., 1999). In the present scenario, somato-dendritically expressed GluR6 in both WT and GluR5-/- are activated by kainate to increase action potential firing and, consequently, GABA release. GABA then activates postsynaptic GABAARs to decrease the input resistance in WT and GluR5-/-, but not GluR6-/-, interneurons and pyramidal cells. Consistent with this hypothesis is the complete lack of modulation of eIPSCs in GluR6-/-. In addition, we cannot rule out that postsynaptic shunting by KAR activation may also contribute to the decreased input resistance of postsynaptic neurons.
AMPAR activation is not required for kainate-induced gamma oscillations
The observation that blockade of AMPARs fails to abolish kainate-induced gamma oscillations (Fig. 2A) distinguishes this oscillatory activity from other induction paradigms of hippocampal gamma oscillation, such as muscarinic receptor activation (Fisahn et al., 1998, 2002). One condition for the induction of rhythmic activity is increased excitation in the neuronal network and, until now, AMPARs were considered the main providers of that excitation (Traub et al., 2000). However, incorporating our findings into a model of gamma oscillations revealed that in the absence of AMPARs, cellular and synaptic excitation provided by KAR activation alone is sufficient to provide the needed excitatory drive to support gamma oscillations. Moreover, the model also replicates the experimentally observed decrease in peak frequency of kainate-induced gamma oscillations after blocking AMPARs. This emphasizes that simulations of rhythmic network activity need to take into account the existence of two excitatory components, AMPAR-mediated and KAR-mediated, to accurately reflect physiological reality.
Computational models of carbachol-induced gamma oscillations (Traub et al., 2000) as well as kainate-induced gamma oscillations (present study) stress the importance of increased ectopic action potential occurrence for the generation of hippocampal gamma oscillations. By laterally communicating through gap-junction-connected axonal networks, ectopic action potentials are thought to facilitate synchronization in the neuronal network. This is supported by our data on ectopic action potentials recorded as spikelets in interneurons and pyramidal cells and their increased frequency after KAR activation in WT (Fig. 8). The significantly smaller spikelet frequency increase in GluR5-/- and GluR6-/- cells suggests the existence of two populations of spikelets: the first, GluR5 dependent and presumably originating from axon-generated action potentials (Semyanov and Kullmann, 2001), and the second, GluR6 dependent and presumably originating from soma-generated action potentials in adjacent cells. Both axon- and soma-generated action potentials will cross via gap junctions into axons of neighboring cells where they travel antidromically to the soma and are recorded as spikelets or orthodromically to synapses to initiate transmitter release.
Kainate-induced gamma oscillations and the kainate model of epilepsy
A common in vivo animal model used to study temporal lobe epilepsy and epileptiform bursts is generated by systemic or intracerebral administration of the KAR agonist kainate (Nadler, 1981; Ben-Ari, 1985; Ben-Ari and Cossart, 2000). Direct support for the involvement of KARs in the ensuing status epilepticus comes from studies on KAR knock-out mice and human studies. GluR6-deficient mice are less susceptible to seizures after kainate injections than WT mice (Mulle et al., 1998). In addition, GluR5 and GluR6 mRNA levels are decreased in patients suffering from temporal lobe epilepsy (Mathern et al., 1998), and activation of GluR5-containing receptors can reduce the propagation of sei-zures (Khalilov et al., 2002) (but see Smolders et al., 2002). Also, the high level of KAR expression in the hippocampal CA3 area (Wisden and Seeburg, 1993; Bureau et al., 1999) as well as its inherent recurrent connectivity render this region especially sensitive to the epileptogenic and neurotoxic effects of kainate (Westbrook and Lothman, 1983).
The question arises whether there is a connection between the oscillation- and seizure-inducing actions of kainate. The lack of the GluR6 subunit prevented both kainate-induced gamma oscillations and epileptiform bursts in vitro, which corresponds to the finding that GluR6-/- mice are less susceptible to seizures after kainate injections than WT mice (Mulle et al., 1998). In contrast, the lack of the GluR5 subunit rendered hippocampal slices more susceptible to the depolarizing and epileptogenic effects of kainate (Fig. 1D), apparently contradicting the results of Smolders et al. (2002) who showed an anticonvulsant effect of GluR5 antagonists. Interestingly, allelic variants of the human GluR5 subunit, but not of GluR6, confer an increased susceptibility to juvenile absence seizures (Sander et al., 1997).
We conclude that the distinct roles the KAR subunits GluR5 and GluR6 play in rhythmic activity in the healthy brain (i.e., gamma oscillations) also extend to rhythmic activity in the pathological brain (epileptiform bursts), and that small changes in the overall activity of the CA3 area can tilt the balance between excitation and inhibition and cause the neuronal network to switch from gamma oscillations to epileptiform bursts.
Footnotes
This research was supported by a Human Frontier Science Program Long-Term Fellowship (A.F.), the Wellcome Trust (A.C.), the Volkswagen Stiftung, a National Institute of Neurological Disorders and Stroke-National Institutes of Health (NIH) grant (R.D.T.), the Medical Research Council, UK (E.H.B.), NIH Grant NS28709, the McKnight Foundation, the John Adler Foundation (S.F.H.), and a National Institute of Child Health and Human Development intramural award (C.J.M.). This study is dedicated to the memory of Professor Eberhard H. Buhl (1959-2003).
Correspondence should be addressed to Dr. A. Fisahn, Nobel Institute for Neurophysiology, Department of Neuroscience, Retzius väg 8, A3:5, Karolinska Institute, SE-17177 Stockholm, Sweden. E-mail: Andre.Fisahn@neuro.ki.se.
Copyright © 2004 Society for Neuroscience 0270-6474/04/249658-11$15.00/0
References
- Ben-Ari Y (1985) Limbic seizure and brain damage produced by kainic acid: mechanisms and relevance to human temporal lobe epilepsy. Neuroscience 14: 375-403. [DOI] [PubMed] [Google Scholar]
- Ben-Ari Y, Cossart R (2000) Kainate, a double agent that generates seizures: two decades of progress. Trends Neurosci 23: 580-587. [DOI] [PubMed] [Google Scholar]
- Bortolotto ZA, Clarke VR, Delany CM, Parry MC, Smolders I, Vignes M, Ho KH, Miu P, Brinton BT, Fantaske R, Ogden A, Gates M, Ornstein PL, Lodge D, Bleakman D, Collingridge GL (1999) Kainate receptors are involved in synaptic plasticity. Nature 402: 297-301. [DOI] [PubMed] [Google Scholar]
- Bragin A, Jando G, Nadasdy Z, Hetke J, Wise K, Buzsaki G (1995) Gamma (40-100 Hz) oscillations in the hippocampus of the behaving rat. J Neurosci 15: 47-60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bureau I, Bischoff S, Heinemann SF, Mulle C (1999) Kainate receptor-mediated responses in the CA1 field of wild-type and GluR6-deficient mice. J Neurosci 19: 653-663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castillo PE, Malenka RC, Nicoll RA (1997) Kainate receptors mediate a slow postsynaptic current in hippocampal CA3 neurons. Nature 388: 182-186. [DOI] [PubMed] [Google Scholar]
- Chittajallu R, Vignes M, Dev KK, Barnes JM, Collingridge GM, Henley JM (1996) Regulation of glutamate release by presynaptic kainate receptors in the hippocampus. Nature 379: 78-81. [DOI] [PubMed] [Google Scholar]
- Clarke V, Ballyk B, Hoo K, Mandelzys A, Pellizzari A, Bath C, Thomas J, Sharpe E, Davies C, Ornstein P, Schoepp DD, Kamboj RK, Collingridge GL, Lodge D, Bleakman D (1997) A hippocampal GluR5 kainate receptor regulating inhibitory synaptic transmission. Nature 389: 599-603. [DOI] [PubMed] [Google Scholar]
- Contractor A, Swanson GT, Sailer A, O'Gorman S, Heinemann SF (2000) Identification of the kainate receptor subunits underlying modulation of excitatory synaptic transmission in the CA3 region of the hippocampus. J Neurosci 20: 8269-8278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Contractor A, Swanson GT, Heinemann SF (2001) Kainate receptors are involved in short- and long-term plasticity at mossy fiber synapses in the hippocampus. Neuron 29: 209-216. [DOI] [PubMed] [Google Scholar]
- Contractor A, Sailer AW, Darstein M, Maron C, Xu J, Swanson GT, Heinemann SF (2003) Loss of kainate receptor-mediated heterosynaptic facilitation of mossy-fiber synapses in KA2-/- mice. J Neurosci 23: 422-429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cossart R, Esclapez M, Hirsch JC, Bernard C, Ben-Ari Y (1998) GluR5 kainate receptor activation in interneurons increases tonic inhibition of pyramidal cells. Nat Neurosci 1: 470-478. [DOI] [PubMed] [Google Scholar]
- Cossart R, Tyzio R, Dinocourt C, Esclapez M, Hirsch JC, Ben-Ari Y, Bernard C (2001) Presynaptic kainate receptors that enhance the release of GABA on CA1 hippocampal interneurons. Neuron 29: 497-508. [DOI] [PubMed] [Google Scholar]
- Cui C, Mayer M (1999) Heteromeric kainate receptors formed by the coassembly of GluR5, GluR6, and GluR7. J Neurosci 19: 8281-8291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fisahn A, Pike F, Buhl EH, Paulsen O (1998) Cholinergic induction of network oscillations at 40 Hz in the hippocampus in vitro. Nature 394: 186-189. [DOI] [PubMed] [Google Scholar]
- Fisahn A, Yamada M, Duttaroy A, Gan JW, Deng CX, McBain CJ, Wess J (2002) Muscarinic induction of hippocampal gamma oscillations requires coupling of the M1 receptor to two mixed cation currents. Neuron 33: 615-624. [DOI] [PubMed] [Google Scholar]
- Frerking M, Ohliger-Frerking P (2002) AMPA receptors and kainate receptors encode different features of afferent activity. J Neurosci 22: 7434-7443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frerking M, Malenka RC, Nicoll RA (1998) Synaptic activation of kainate receptors on hippocampal interneurons. Nat Neurosci 1: 479-486. [DOI] [PubMed] [Google Scholar]
- Frerking M, Petersen CCH, Nicoll RA (1999) Mechanisms underlying kainate receptor-mediated disinhibition in the hippocampus. Proc Natl Acad Sci USA 96: 12917-12922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freund T, Buzsaki G (1996) Hippocampal interneurons. Hippocampus 6: 347-470. [DOI] [PubMed] [Google Scholar]
- Gray CM, König P, Engel AK, Singer W (1989) Oscillatory responses in cat visual cortex exhibit inter-columnar synchronization which reflects global stimulus properties. Nature 338: 334-337. [DOI] [PubMed] [Google Scholar]
- Hormuzdi S, Pais I, LeBeau FEN, Towers SK, Rozov A, Buhl EH, Whittington M, Monyer H (2001) Impaired electrical signaling disrupts gamma frequency oscillations in connexin 36-deficient mice. Neuron 31: 487-495. [DOI] [PubMed] [Google Scholar]
- Jiang L, Xu J, Nedergaard M, Kang J (2001) A kainate receptor increases the efficacy of GABAergic synapses. Neuron 30: 503-513. [DOI] [PubMed] [Google Scholar]
- Khalilov I, Hirsch J, Cossart R, Ben-Ari Y (2002) Paradoxical anti-epileptic effects of a GluR5 agonist of kainate receptors. J Neurophysiol 88: 523-527. [DOI] [PubMed] [Google Scholar]
- Kim J, Alger BE (2001) Random response fluctuations lead to spurious paired-pulse facilitation. J Neurosci 21: 9608-9618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klausberger T, Magill PJ, Marton LF, Roberts JD, Cobden PM, Buzsaki G, Somogyi P (2003) Brain-state- and cell-type-specific firing of hippocampal interneurons in vivo. Nature 421: 844-848. [DOI] [PubMed] [Google Scholar]
- Klausberger T, Marton LF, Baude A, Roberts JD, Magill PJ, Somogyi P (2004) Spike timing of dendrite-targeting bistratified cells during hippocampal network oscillations in vivo. Nat Neurosci 7: 41-47. [DOI] [PubMed] [Google Scholar]
- Lerma J (2003) Roles and rules of kainate receptors in synaptic transmission. Nat Rev Neurosci 4: 481-495. [DOI] [PubMed] [Google Scholar]
- Lerma J, Paternain AV, Rodriguez-Moreno A, Lopez-Garcia JC (2001) Molecular physiology of kainate receptors. Physiol Rev 81: 971-998. [DOI] [PubMed] [Google Scholar]
- Mathern GW, Pretorius JK, Kornblum HI, Mendoza D, Lozada A, Leite JP, Chimelli L, Born DE, Fried I, Sakamoto AC, Assirati JA, Peacock WJ, Ojemann GA, Adelson PD (1998) Altered hippocampal kainate-receptor mRNA levels in temporal lobe epilepsy patients. Neurobiol Dis 5: 151-176. [DOI] [PubMed] [Google Scholar]
- Melyan Z, Wheal HV, Lancaster B (2002) Metabotropic-mediated kainate receptor regulation of IsAHP and excitability in pyramidal cells. Neuron 34: 107-114. [DOI] [PubMed] [Google Scholar]
- Mulle C, Sailer A, Perez-Otano I, Dickinson-Anson H, Castillo PE, Bureau I, Maron C, Gage FH, Mann JR, Bettler B, Heinemann SF (1998) Altered synaptic physiology and reduced susceptibility to kainate-induced seizures in GluR6-deficient mice. Nature 392: 601-605. [DOI] [PubMed] [Google Scholar]
- Mulle C, Sailer A, Swanson GT, Brana C, O'Gorman S, Bettler B, Heinemann SF (2000) Subunit composition of kainate receptors in hippocampal interneurons. Neuron 28: 475-484. [DOI] [PubMed] [Google Scholar]
- Muller D, Nikonenko I, Jourdain P, Alberi S (2002) LTP, memory and structural plasticity. Curr Mol Med 2: 605-611. [DOI] [PubMed] [Google Scholar]
- Nadler JV (1981) Minireview. Kainic acid as a tool for the study of temporal lobe epilepsy. Life Sci 29: 2031-2042. [DOI] [PubMed] [Google Scholar]
- Prince DA (1978) Neurophysiology of epilepsy. Annu Rev Neurosci 1: 395-415. [DOI] [PubMed] [Google Scholar]
- Rodriguez-Moreno A, Lerma J (1998) Kainate receptor modulation of GABA release involves a metabotropic function. Neuron 20: 1211-1218. [DOI] [PubMed] [Google Scholar]
- Rodriguez-Moreno A, Herreras O, Lerma J (1997) Kainate receptors presynaptically downregulate GABAergic inhibition in the rat hippocampus. Neuron 19: 893-901. [DOI] [PubMed] [Google Scholar]
- Rodriguez-Moreno A, López-Garcia JC, Lerma J (2000) Two populations of kainate receptors with separate signaling mechanisms in hippocampal interneurons. Proc Natl Acad Sci USA 97: 1293-1298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sander T, Hildmann T, Kretz R, Furst R, Sailer U, Bauer G, Schmitz B, Beck-Mannagetta G, Wienker TF, Janz D (1997) Allelic association of juvenile absence epilepsy with a GluR5 kainate receptor gene (GRIK1) polymorphism. Am J Med Genet 74: 416-421. [PubMed] [Google Scholar]
- Schmitz D, Frerking M, Nicoll RA (2000) Synaptic activation of presynaptic kainate receptors on hippocampal mossy fiber synapses. Neuron 27: 327-338. [DOI] [PubMed] [Google Scholar]
- Schmitz D, Mellor J, Nicoll RA (2001a) Presynaptic kainate receptor mediation of frequency facilitation at hippocampal mossy fiber synapses. Science 291: 1972-1976. [DOI] [PubMed] [Google Scholar]
- Schmitz D, Schuchmann S, Fisahn A, Draguhn A, Buhl EH, Petrasch-Parwez E, Dermietzel R, Heinemann U, Traub RD (2001b) Axo-axonal coupling. A novel mechanism for ultrafast neuronal communication. Neuron 31: 831-840. [DOI] [PubMed] [Google Scholar]
- Semyanov A, Kullmann DM (2001) Kainate receptor-dependent axonal depolarization and action potential initiation in interneurons. Nat Neurosci 4: 718-723. [DOI] [PubMed] [Google Scholar]
- Singer W (1993) Synchronization of cortical activity and its putative role in information processing and learning. Annu Rev Physiol 55: 349-374. [DOI] [PubMed] [Google Scholar]
- Smolders I, Bortolotto ZA, Clarke VR, Warre R, Khan GM, O'Neill MJ, Ornstein PL, Bleakman D, Ogden A, Weiss B, Stables JP, Ho KH, Ebinger G, Collingridge GL, Lodge D, Michotte Y (2002) Antagonists of GLU(K5)-containing kainate receptors prevent pilocarpine-induced limbic seizures. Nat Neurosci 5: 796-804. [DOI] [PubMed] [Google Scholar]
- Swanson GT, Gereau RW, Green T, Heinemann SF (1997) Identification of amino acid residues that control functional behavior in GluR5 and GluR6 kainate receptors. Neuron 19: 913-926. [DOI] [PubMed] [Google Scholar]
- Teitelbaum JS, Zatorre RJ, Carpenter S, Gendron D, Evans AC, Gjedde A, Cashman NR (1990) Neurologic sequelae of domoic acid intoxication due to the ingestion of contaminated mussels. N Engl J Med 322: 1781-1787. [DOI] [PubMed] [Google Scholar]
- Traub RD, Bibbig A, Fisahn A, LeBeau FEN, Whittington MA, Buhl EH (2000) A model of gamma-frequency network oscillations induced in the rat CA3 region by charbachol in vitro. Eur J Neurosci 12: 4093-4106. [DOI] [PubMed] [Google Scholar]
- Traub RD, Kopell N, Bibbig A, Buhl EH, LeBeau FEN, Whittington MA (2001) Gap junctions between interneuron dendrites can enhance long-range synchrony of gamma oscillations. J Neurosci 21: 9478-9486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Traub RD, Pais I, Bibbig A, LeBeau FEN, Buhl EH, Hormuzdi SG, Monyer H, Whittington MA (2003) Contrasting roles of axonal (pyramidal cell) and dendritic (interneuron) electrical coupling in the generation of gamma oscillations in the hippocampus in vitro. Proc Natl Acad Sci USA 100: 1370-1374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vignes M, Collingridge GL (1997) The synaptic activation of kainate receptors. Nature 388: 179-182. [DOI] [PubMed] [Google Scholar]
- Viskontas IV, McAndrews MP, Moscovitch M (2000) Remote episodic memory deficits in patients with unilateral temporal lobe epilepsy and excisions. J Neurosci 20: 5853-5857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westbrook GL, Lothman EW (1983) Cellular and synaptic basis of kainic acid-induced hippocampal epileptiform activity. Brain Res 273: 97-109. [DOI] [PubMed] [Google Scholar]
- Wisden W, Seeburg PH (1993) A complex mosaic of high-affinity kainate receptors in rat brain. J Neurosci 13: 3582-3598. [DOI] [PMC free article] [PubMed] [Google Scholar]