

Abstract
Here, we investigated the intracellular mechanisms that underlie the relative invulnerability of adult versus developing dorsal root ganglion (DRG) sensory neurons. In culture, adult neurons were resistant to stimuli that caused apoptosis of their neonatal counterparts. In both adult and neonatal neurons, death stimuli induced the apoptotic c-Jun N-terminal protein kinase (JNK) pathway, but JNK activation only caused death of neonatal neurons, indicating that adult neurons have a downstream block to apoptosis. Expression of the dominant-inhibitory p53 family member, ΔNp73, rescued JNK-induced apoptosis of neonatal neurons, suggesting that it might participate in the downstream apoptotic block in adult neurons. To test this possibility, we examined adult DRG neurons cultured from p73+/- mice. Adult p73+/- DRG neurons were more vulnerable to apoptotic stimuli than their p73+/+ counterparts, and invulnerability could be restored to the p73+/- neurons by increased expression of ΔNp73. Moreover, although DRG neuron development was normal in p73+/- animals in vivo, axotomy caused death of adult p73+/- but not p73+/+ DRG neurons. Thus, one way adult neurons become invulnerable is to enhance endogenous survival pathways, and one critical component of these survival pathways is p73.
Keywords: p73, p53 tumor suppressor, sensory neurons, dorsal root ganglion, sciatic nerve injury, nerve growth factor, neuronal apoptosis, JNK, PI3-kinase, DNA damage
Introduction
A fundamental question is how developing neurons, which are programmed to undergo apoptosis unless they establish the appropriate connectivity, become resistant to apoptotic stimuli as they mature, a transition that ensures their survival for the remainder of an animal's lifetime. This transition is perhaps best characterized in the peripheral nervous system. During the developmental cell death period, peripheral neurons are absolutely dependent on trophic factors produced by their targets, and those developing neurons that fail to compete successfully for sufficient target support die by apoptosis. Moreover, any insult that interrupts the supply of target-derived growth factors causes apoptosis of newly connected developing neurons. For example, axotomy or target removal leads to rapid and extensive cell death in both central and peripheral neurons (for review, see Snider et al., 1992), and exogenous replacement of target-derived growth factors rescues these axotomized neurons (Hendry and Campbell, 1976; Yip et al., 1984). This contrasts to the situation in adulthood, where peripheral neurons no longer depend on target-derived growth factors for survival (Angeletti et al., 1971; Bjerre et al., 1975; Goedert et al., 1978; Otten et al., 1979), and axotomy results only in delayed and restricted neuronal loss (Tandrup et al., 2000; Ma et al., 2001). The mechanisms that underlie the relative invulnerability of adult neurons in vivo are not well understood.
Culture studies have confirmed that the enhanced invulnerability of adult peripheral neurons is a cell-intrinsic phenomenon. For example, acutely isolated adult sympathetic or sensory neurons do not require exogenous neurotrophins for survival in culture (Lazarus et al., 1976; Chun and Patterson, 1977; Lindsay, 1988), a finding that might be partially explained by endogenous neurotrophin synthesis in older neurons (Acheson et al., 1995). However, these older neurons are also less sensitive to apoptosis induced by ionizing radiation (Tong et al., 1997), and removal of exogenous neurotrophins activates the same initial apoptotic signaling events in neonatal sympathetic neurons as in neurons that have been “aged” 3 weeks in culture (Easton et al., 1997; Vogelbaum et al., 1998), suggesting the existence of additional cell-intrinsic mechanisms that confer apoptotic resistance on the older neurons.
What might these cell-intrinsic mechanisms be? Potential differences might include an impairment in apoptotic signaling pathways and/or an enhancement in intrinsic neuronal survival pathways that might “block” apoptosis. Potential insights into the specifics of such pathways derive from extensive work on naturally occurring cell death in NGF-dependent sympathetic neurons. During development, the ultimate survival of any given sympathetic neuron is a function of survival signals deriving from the tyrosine receptor kinase A (TrkA)/NGF receptor and apoptotic signals deriving from the p75 panneurotrophin receptor (Bamji et al., 1998; Majdan et al., 2001) (for review, see Miller and Kaplan, 2001). TrkA mediates survival primarily via a phosphatidylinositol 3 (PI3)-kinase-Akt-dependent pathway, whereas activation of a c-Jun N-terminal protein kinase (JNK)-Bax apoptotic pathway occurs after either p75NTR activation or NGF withdrawal. Interestingly, work with sympathetic neurons that have been aged for 3 weeks in culture and that lose their NGF dependence show that after NGF withdrawal, one of the major downstream apoptotic events, translocation of Bax to the mitochondria, no longer occurs (Easton et al., 1997; Putcha et al., 2000). These findings suggest that in the mature, resistant neurons, either an essential link between the cell surface and Bax is no longer present and/or a potent survival signal inhibits the apoptotic pathway at this level.
In this regard, we identified previously a key apoptotic checkpoint in developing sympathetic neurons that involves the p53 tumor suppressor family. Specifically, we have shown that p53 functions as a proapoptotic molecule during developmental sympathetic neuron death (Aloyz et al., 1998) and that a second family member, ΔNp73, is an essential prosurvival protein during the same developmental window (Pozniak et al., 2000). The p73 gene encodes two classes of protein products: full-length isoforms that are structurally homologous to p53 (TAp73α, TAp73β) and N-terminal truncated isoforms that lack the transactivation domain (ΔNp73α, ΔNp73β) (for review, see Irwin and Kaelin, 2001). In developing sympathetic neurons, NGF potently upregulates the prosurvival ΔNp73 isoform, and ΔNp73 maintains sympathetic neuron survival both in vivo and in culture by p53-dependent and -independent mechanisms (Pozniak et al., 2000; Lee et al., 2004).
In the CNS, ΔNp73 variants are also the predominant isoforms, acting as potent prosurvival proteins in cultured cortical neurons exposed to a variety of apoptotic insults (Pozniak et al., 2002). In this regard, we have shown recently that p73-/- animals display a prolonged and steady loss of cortical neurons from the first to the eighth postnatal week in vivo, suggesting that p73 is important for the maintenance of mature cortical neurons (Pozniak et al., 2002). On the basis of these findings, we hypothesized that ΔNp73 may well provide a key prosurvival mechanism for adult neurons. In this study, we tested this hypothesis and show that ΔNp73 is an essential component of a cell-intrinsic pathway that is required for the survival of adult sensory neurons both in culture and in vivo after axonal injury.
Materials and Methods
Animals. Mice deficient in p73 have been described previously (Pozniak et al., 2000, 2002; Yang et al., 2000). These mice were maintained through heterozygote breeding pairs in a C3H background, and genotyping was performed as described previously (Pozniak et al., 2000).
Primary neuronal cultures. Newborn dorsal root ganglia (DRGs) were removed rapidly, placed in F12 media (Invitrogen, San Diego, CA), and digested in 0.125% collagenase in F12 media for 20 min followed by 10 min in 0.25% trypsin, all at 37°C. For adult (6 weeks of age) animals, DRGs were dissected and digested in 0.125% collagenase in F12 media for 1.5 hr followed by 30 min in 0.25% trypsin. Ganglia were washed in F12 media and then gently triturated through a glass-polished pipette, and the cell suspension was centrifuged through a 15% BSA solution (Sigma, St. Louis, MO). The cell pellet was resuspended in Ultraculture media containing 2 mm glutamine, 100 U/ml penicillin, 100 μg/ml streptomycin, 50 ng/ml NGF, 5 μm cytosine arabinoside, and 20 μm fluorodeoxyuridine. Neurons were maintained for 24 hr and then treated as follows. For NGF deprivation, cultures were rinsed with Ultraculture, followed by the addition of Ultraculture containing goat anti-NGF antibodies (Abs) (Sigma). For cell survival experiments, cultures were rinsed with Ultraculture, followed by the addition of Ultraculture containing 50 ng/ml NGF alone or in combination with either 50 μm 2-(4-Morpholinyl)-8-phenyl-1(4H)-benzopyran-4-one (LY294002) or 10 μm camptothecin. For cultures treated with anti-NGF, LY294002, or camptothecin, the number of phase-bright neurons within a defined area was counted on the day of treatment and at 24 hr intervals thereafter. Cells with round, phase-bright bodies and intact neurites were counted as surviving neurons. Survival is expressed as a percentage of the original number of neurons. Approximately 200 neurons were counted for each experiment. Statistical differences between conditions were examined with a Student's t test.
Cultures of sympathetic neurons were prepared from newborn p73+/+ and p73+/- mice. Superior cervical ganglia from individual mice were dissected and triturated as described previously (Majdan et al., 2001). Neurons were then plated on collagen-coated 96-well culture dishes (Falcon Plastics, Franklin Lakes, NJ) in Ultraculture media containing 2 mm glutamine, 100 U/ml penicillin, 100 gm/ml streptomycin, 3% FBS (Invitrogen), and 50 ng/ml mouse 2.5 S NGF prepared from mouse salivary gland (Cedarlane, Hornby, Ontario, Canada). One day after plating, neurons were fed with the same media containing 0.5% cytosine arabinoside (Sigma). For cell survival experiments, cultures were rinsed with Ultraculture, followed by the addition of Ultraculture containing 50 ng/ml NGF alone or in combination with either 50 μm LY294002 or 10 μm camptothecin. Two days later, cultures were rinsed and fixed in 4% paraformaldehyde and stained with Hoechst. Apoptosis was assessed by counting the number of neurons that displayed evidence of condensed apoptotic nuclei. Statistical differences between values were examined with a Student's t test.
Adenovirus infections. For JNK activation experiments, semiconfluent human embryonic kidney (HEK) 293A cells were rinsed with DMEM and infected with 20 multiplicities of infection (MOI) of adenovirus expressing wild-type mixed-lineage kinase 3 (MLK3)/green fluorescent protein (GFP) (Roux et al., 2002) or GFP alone. Alternatively, DRG cultures were rinsed with Ultraculture 24 hr after plating, followed by the addition of Ultraculture containing 50 ng/ml NGF and 100 MOI of adenovirus expressing wild-type MLK3/GFP or GFP alone. After 24 hr, cell lysates were prepared, and the level of JNK phosphorylation was assessed by immunoblotting (see below). For survival experiments with DRG neurons, cultures were rinsed with Ultraculture 24 hr after plating, followed by the addition of Ultraculture containing 50 ng/ml NGF and 100 MOI of adenovirus expressing wild-type MLK3/GFP or GFP alone. Two days later, cultures were switched to media containing 50 ng/ml NGF alone for an additional 3 d. Cultures were then rinsed and fixed in 4% paraformaldehyde and stained with Hoechst. Apoptosis was assessed by counting 200 infected neurons and determining the number of infected neurons displaying evidence of condensed apoptotic nuclei. Statistical differences between values were examined with a Student's t test. For the ΔNp73 rescue experiments, cultures of adult p73+/- neurons were rinsed with Ultraculture 24 hr after plating, followed by the addition of Ultraculture containing 50 ng/ml NGF and 100 MOI of adenovirus expressing ΔNp73β/GFP (Pozniak et al., 2000) or GFP alone. Two days later, cultures were switched to media either lacking NGF or containing NGF with LY294002 or camptothecin. Apoptosis was assessed as for the MLK3 experiments. For double-infection experiments with sympathetic neurons, cultures were rinsed with Ultraculture 4 d after plating, followed by the addition of Ultraculture containing 10 ng/ml NGF, 50 mm KCl, and 100 MOI of adenovirus expressing wild-type MLK3/GFP either alone or in combination with 100 MOI of ΔNp73β/GFP or 100 MOI of GFP. One day later, cultures were washed free of virus and replaced with Ultraculture media containing 10 ng/ml NGF. Two days later, cultures were then rinsed and fixed in 4% paraformaldehyde and stained with Hoechst, and apoptosis was assessed as described above.
Reverse transcription-PCR. Whole DRGs were dissected from neonatal and adult (6 weeks of age) wild-type mice and quickly frozen. Total RNA was isolated from tissues using the Trizol reagent and was reverse transcribed using M-MuLV reverse transcriptase (Fermentas, Burlington, Ontario, Canada). PCR was performed using site-specific primers. Primer sequences were as follows: p73 core, 5′-TGCCCCGGCTAGCCACCTCATCC-3′ and 5′-CCTCGTCCCCGTGGCGTCTCTTCT-3′; TAp73, 5′-GAGCACCTGTGGAGTTCTCTAGAG-3′ and 5′-GGTATTGGAAGGGATGACAGGCG-3′; ΔNp73, 5′-GTCGGTGACCCCATGAGACAC-3′ and 5′-GGGGCAGCGCTTAACAATGT-3′; p53, 5′-AACCGCCGACCTATCCTTACCATC-3′ and 5′-AGGCCCCACTTTCTTGACCATTGT-3′; glyceraldehyde-3-phosphate dehydrogenase (GAPDH), 5′-ACGGCAAGTTCAATGGCACAGTCA-3′ and 5′-GCTTTCCAGAGGGGCCATCCACAG-3′. Expected PCR product sizes were as follows: p73 core, 434 bp; TAp73, 297 bp; ΔNp73, 429 bp; p53, 432 bp; GAPDH, 425 bp.
Surgical procedures. Sciatic nerve resections were performed on adult (6 weeks of age) p73 heterozygous mice and their wild-type littermates. Adult animals were anesthetized with an intraperitoneal injection of a ketamine/xylazine mixture (100 mg/kg per 10 mg/kg). The right sciatic nerve was exposed at midthigh level, and a 5 mm segment of the nerve was resected. The wound was sutured, and the animals were allowed to recover for 1 month.
Immunohistochemistry. Mice were terminally anesthetized with sodium pentobarbital and then perfused transcardially with a solution of 4% paraformaldehyde in 0.1 m phosphate buffer, pH 7.4. The L4 DRGs were removed, postfixed for 2 hr in the same fixative, and then immersed for 3 d in 30% phosphate-buffered sucrose. Ganglia were embedded in OCT compound, frozen in isopentane, and cryosectioned at 10 μm onto chrom-alum gelatin-coated slides. Sections were stained with cresyl violet or incubated for 1 hr in 0.1 m PBS containing 10% normal goat serum and 0.25% Triton X-100, followed by incubation with primary antibody overnight at 4°C. Primary antibodies were mouse anti-neurofilament 200 (Sigma) and rabbit anti-calcitonin gene-related peptide (CGRP) (Chemicon, Temecula, CA). After rinsing in PBS, sections were incubated with either Alexa 594-conjugated anti-mouse or Alexa 350-conjugated anti-rabbit secondary antibodies (Molecular Probes, Eugene, OR) for 2 hr at room temperature. Sections were washed in PBS, mounted, and viewed under a Zeiss (Thornwood, NY) fluorescence microscope with 20× magnification. For isolectin B4 (IB4) staining, after the blocking step, sections were incubated for 2 hr with FITC-labeled IB4 lectin (Sigma) in 0.1 m PBS containing 0.25% Triton X-100. Sections were then rinsed and coverslipped with geltol (Immunon, Shandon, Pittsburgh, PA) and analyzed with a Zeiss Axioplan microscope using 20× objective and the Northern Eclipse computer-based image analysis software (Empix Imaging, Ontario, Canada).
Electron microscopy. The L4 dorsal (sensory) spinal roots from adult p73 heterozygous mice and their wild-type littermates were examined for the number of nerve fibers in plastic-embedded material. Dorsal root specimens were fixed in 4% paraformaldehyde and 1% gluteraldehyde in 0.1 m phosphate buffer. They were then postfixed in aqueous 1% OsO4 and dehydrated through a graded series of acetone followed by infiltration and embedding in the Epon-Araldite epoxy resin (Canemco, Quebec, Canada). Semithin sections were cut and stained with Toluidene Blue for light microscopy examination. Specimen blocks with appropriate cross sections were selected for ultrathin sectioning. Sections ∼80 nm thick were cut with a diamond knife and stained with uranyl acetate and lead citrate for transmission electron microscopy (TEM) imaging.
The sections were examined in a JEM-1230 TEM (JEOL, Peabody, MA) operating at 80 kV. Digital electron micrographs were recorded directly using CCD camera with a computer system (AMT Advantage CCD ORCA camera; AMT, Deben UK, Suffolk, UK). Images with offset overlapping edges covering the entire area of the dorsal root were systematically recorded. The total number of nerve fibers (both myelinated and unmyelinated) in the L4 dorsal roots of heterozygous and wild-type mice was determined from electron micrographs at 5000×. Statistical differences between wild-type and heterozygous values were examined with a Student's t test.
Western blot analysis. HEK293A cells or DRG neurons were rinsed briefly in cold TBS and then lysed in TBS lysis buffer (137 mm NaCl, 20 mm Tris, pH 8.0, 1% v/v NP-40, and 10% v/v glycerol) supplemented with Mini Complete protease inhibitor mixture (Roche Diagnostics, Mannheim, Germany) and 1.5 mm sodium vanadate. Lysates were scraped into Eppendorf tubes (Eppendorf Scientific, Westbury, NY) and rocked for 10 min at 4°C. Samples were then cleared by centrifugation. Protein concentration was determined by the BCA assay (Pierce, Rockford, IL) using BSA as a standard. Samples were diluted in sample buffer and placed in a 95°C heat block for 5 min. Equal amounts of protein were separated on 10% SDS-PAGE gels and transferred onto nitrocellulose membranes. For all antibodies, blots were blocked in 3% skim milk in TBS overnight at 4°C. Primary antibodies used included anti-JNK1 (Santa Cruz Biotechnology, Santa Cruz, CA), anti-phospho-JNK (Promega, Madison, WI), anti-MLK3 (Santa Cruz Biotechnology), anti-GFP (Clontech, Cambridge, UK). For Western blots, secondary antibodies used were HRP-conjugated anti-mouse (1:10,000) and anti-rabbit (1: 10,000) polyclonal Abs (Roche Diagnostics). All incubations were performed in 3% skim milk in TBS plus 0.1% Tween 20 (Sigma). For detection, blots were washed with TBS, and antibody localization was visualized using the ECL chemiluminescence kit (Amersham, Bucking-hamshire, UK).
Results
Adult sensory neurons are resistant to stimuli that induce apoptosis of neonatal sensory neurons
To study the intracellular mechanisms that promote the survival of adult neurons, in vitro studies have relied primarily on embryonic neurons that have been aged in culture (Easton et al., 1997; Vogelbaum et al., 1998; Putcha et al., 2000), despite the possibility that neurons may develop differently in culture than in vivo. To overcome this limitation, we examined instead the survival requirements of sensory neurons acutely isolated from adult (6 weeks of age) rat DRGs and directly compared them to cultured neurons isolated from neonatal (postnatal day 0) DRGs. In all of these experiments, neurons were isolated from lumbar DRGs, and low-density cultures were established and maintained in serum-free culture conditions containing few non-neuronal cells. Under these conditions, the adult cultures were comprised of phase-bright neurons of varying sizes, consistent with neuronal heterogeneity in the DRG.
Initially, we confirmed the previously reported finding (Lindsay, 1988; Vogelbaum et al., 1998) that adult DRG neurons no longer require neurotrophins for their survival. Adult or neonatal DRG neurons were cultured in the presence of NGF for 1 d and then switched into media lacking NGF but with the addition of function-blocking anti-NGF antibodies. Quantitation of surviving, phase-bright neurons 2 d later revealed that, whereas only 40 ± 9% of neonatal neurons were still alive, the large majority of adult neurons (77 ± 11%) survived the NGF withdrawal (Fig. 1A). We then asked whether this enhanced survival of adult DRG neurons in the absence of NGF reflected a more general resistance of adult neurons to apoptotic stimuli. We first asked whether adult DRG neurons were more resistant to apoptosis caused by inhibition of the PI3-kinase survival pathway; established cultures of neonatal or adult neurons were switched into media containing NGF plus 50 μm of the pharmacological PI3-kinase inhibitor LY294002. Western blot analysis demonstrated the specificity of this concentration of LY294002 to inhibit the phosphorylation of the PI3-kinase target Akt in these neurons (data not shown), as we published previously for sympathetic neurons (Vaillant et al., 1999). We then quantitated survival; after 2 d of PI3-kinase inhibition, only 25 ± 14% of neonatal DRG neurons remained alive, whereas the majority (62 ± 6%) of adult neurons survived (Fig. 1B). As a second, more robust, apoptotic insult, we treated neurons with the DNA damaging agent camptothecin, which causes p53-dependent neuronal apoptosis (Xiang et al., 1998). Established cultures of adult or neonatal DRG neurons were treated with 10 μm camptothecin, and survival was quantitated 2 d later. This analysis demonstrated that, surprisingly, adult DRG neurons were relatively resistant to this apoptotic stimulus; after 2 d, 72 ± 8% of adult neurons survived versus 26 ± 7% of neonatal neurons (Fig. 1C).
Figure 1.

Adult sensory neurons are more resistant to apoptotic insults than their neonatal counterparts. A-C, Adult (black squares) or neonatal (open squares) sensory neurons of the L4 DRG were cultured for 24 hr in NGF and then cultured for an additional 2 d after NGF withdrawal and in the presence of anti-NGF antibodies (A), in the presence of NGF but with the addition of 50 μm LY294002, an inhibitor of PI3-kinase (B), or in the presence of NGF but with the addition of 10 μm camptothecin (C), a DNA-damaging agent. As controls, sister cultures were maintained in NGF over the same time period (open circles). Phase-bright neurons with intact neurites in multiple identified fields were counted at 0, 1, and 2 d for a total of at least 200 neurons per experiment, and the amount of survival was plotted relative to the initial number of neurons in the same fields at the time the treatment was initiated (percentage survival). Results are the cumulative results of at least three independent experiments, and the error bars represent SEM (t test; p < 0.005; comparison at 2 d time point for all experiments).
Activation of the JNK pathway is sufficient to cause apoptosis of neonatal, but not adult, neurons, and JNK-mediated neonatal neuron apoptosis can be rescued by ΔNp73
A generalized apoptotic resistance in adult neurons could result from an impairment in apoptotic signaling pathways and/or enhanced adult survival pathways. To distinguish these two possibilities, we examined one common component of the apoptotic pathways induced by a variety of stimuli, activation of the N-terminal jun-kinase. Specifically, adult or neonatal DRG neurons were established and then either withdrawn from NGF or treated with 50 μm LY294002 or 10 μm camptothecin for 24 hr. Western blot analysis revealed that JNK phosphorylation was increased in neonatal DRG neurons in all of these conditions (Fig. 2A). An increase inJNK phosphorylation was also observed in adult DRG neurons treated in the same way (Fig. 2B), although the magnitude of the increase was smaller. Thus, apoptotic pathways upstream of JNK were intact in both populations, but adult neurons apparently had a downstream block to apoptosis.
Figure 2.

JNK activation occurs in both adult and neonatal sensory neurons, but exogenous JNK activation preferentially causes apoptosis of neonatal neurons. A, B, Western blot analysis with an antibody specific for the phosphorylated, activated form of JNK (p-JNK) in lysates of neonatal (A) or adult (B) neurons that were established in NGF for 1 d and then either withdrawn from NGF (anti-NGF) or treated with 50 μm LY294002 or 10 μm camptothecin for 24 hr. Equal amounts of protein were present in all lanes, as determined by reprobing the same blots for total JNK protein. The numbers to the left of the blots refer to the molecular weights of the two bands, which correspond to p46 and p54 isoforms of JNK. C, Western blot analysis for the activated, phosphorylated form of JNK (pJNK) in equal amounts of protein from lysates of HEK293 cells (left panel) or neonatal DRG cultures (right panel) that were infected with replication-deficient recombinant adenoviruses expressing either GFP alone (Ad-GFP) or both GFP and the upstream kinase MLK3 (Ad-MLK3). Uninfected cells were also examined as an additional control. Blots were reprobed with anti-JNK to demonstrate that equal amounts of protein were present as well as for MLK3 and GFP. Note that overexpression of MLK3 causes activation of both p46 and p54 isoforms of JNK. D, Quantitation of the percentage of GFP-positive infected neurons with apoptotic nuclei, as determined by Hoechst staining 3 d after infection. Neonatal or adult sensory neurons were established, infected with adenoviruses expressing either GFP alone or GFP plus MLK3, and apoptosis was quantitated 3 d later. Note that the majority of neonatal neurons become apoptotic in response to MLK3 overexpression (t test; **p < 0.05). Error bars represent SD. E, MLK3-induced neonatal neuron death is rescued by ectopic expression of ΔNp73. Quantitation of the percentage of GFP-positive infected neonatal sympathetic neurons with apoptotic nuclei, as determined by Hoechst staining 3 d after infection. Neonatal sympathetic neurons were established, infected with adenoviruses expressing GFP, MLK3/GFP, and/or ΔNp73/GFP, and then apoptosis was quantitated 3 d later. Note that ΔNp73 rescued MLK3-mediated death (ANOVA; *p < 0.001). Error bars represent SD.
To directly test this hypothesis, we exogenously activated JNK and asked whether this was, on its own, sufficient to cause apoptosis of neonatal versus adult neurons. To perform these experiments, we used a bicistronic recombinant adenovirus that coexpressed GFP and wild-type MLK3, an upstream kinase that efficiently activates JNK when overexpressed in peripheral neurons (Maroney et al., 1999). Confirmation of the efficacy of this adenovirus was obtained by transducing HEK293A cells or neonatal DRG cultures and assaying for expression of MLK3 and activation of JNK by Western blot analysis (Fig. 2C). We then infected established neonatal or adult neurons with adenoviruses expressing MLK3/GFP or GFP alone and scored GFP-positive neurons for apoptotic nuclei as indicated by Hoechst staining. This analysis revealed that GFP alone had no effect on neuronal survival. However, overexpression of MLK3 was sufficient to cause the apoptosis of the majority of neonatal, but not adult, neurons (Fig. 2D), suggesting that adult neurons have a relative impairment in the apoptotic pathways downstream of JNK.
One protein that acts downstream of JNK in neonatal sympathetic neurons, and that plays an essential prosurvival role, is the dominant-inhibitory p53 family member ΔNp73 (Pozniak et al., 2000; Lee et al., 2004). To determine whether enhanced ΔNp73 levels or function could explain the downstream block to JNK-mediated apoptosis in adult neurons, we overexpressed ΔNp73 in neonatal neurons and asked whether this could rescue MLK3-mediated apoptosis. To perform this experiment, we turned to neonatal sympathetic neurons, which apoptose in response to JNK pathway activation (Aloyz et al., 1998; Eilers et al., 1998) and can be reliably double infected by recombinant adenovirus. Neonatal neurons were infected with recombinant adenoviruses expressing GFP, MLK3, and/or ΔNp73, and neuronal survival was quantitated by assessing nuclear morphology with Hoechst 3 d after infection. These experiments demonstrated that, as seen with neonatal sensory neurons (Fig. 2D), overexpression of MLK3 in neonatal sympathetic neurons was sufficient to induce apoptosis in the presence of NGF (Fig. 2E). The magnitude of this apoptotic effect was unaffected by coinfection with a control, GFP-expressing adenovirus. In contrast, when neurons were coinfected with adenoviruses expressing MLK3 and ΔNp73, infected sympathetic neurons were rescued from apoptosis (Fig. 2E). Thus, enhanced levels of ΔNp73 were sufficient to block neonatal neuron apoptosis in response to JNK pathway activation, suggesting that upregulation of ΔNp73 function would be sufficient to uncouple extracellular apoptotic stimuli from an apoptotic response in adult neurons.
p73 is required for the relative invulnerability of cultured adult DRG neurons
These studies indicated that enhanced ΔNp73 levels and/or function would be sufficient to confer relative invulnerability to apoptotic stimuli on adult neurons. We therefore asked whether ΔNp73 was necessary for the invulnerability of adult sensory neurons. To do this, we initially characterized the expression of full-length (TAp73) versus N-terminal truncated (ΔNp73) isoforms in dorsal root ganglia isolated from adult mice. Reverse transcription (RT)-PCR analysis with specific primers demonstrated that mRNAs encoding both TAp73 and ΔNp73 isoforms were expressed in adult DRGs as they are in the developing brain (Fig. 3A,B) (Pozniak et al., 2002). A similar analysis demonstrated that these isoforms are also both expressed in the neonatal DRG (Fig. 3A) (data not shown). The specificity of this analysis was demonstrated by showing that these RT-PCR products were not present in the p73-/- brain (Fig. 3B).
Figure 3.

Expression of p73 in L4 DRG. RT-PCR analysis for p73 mRNA in total RNA isolated from the neonatal (P1 DRG) and adult (6-week-old DRG) mouse dorsal root ganglia. Total RNA from the E16 cortex or from the total brain (P1) of p73+/+ versus p73-/- animals were all used as controls. Primers were specific to the core region that is shared among all p73 isoforms (p73 core) (A), to the full-length TA-containing isoforms (TAp73) (A), to the N-terminal truncated isoforms (ΔNp73) (B), or as a positive control to p53. RT-PCR for GAPDH was used to demonstrate that RNA was present in all samples. Note that all p73 isoforms are expressed in the developing brain, as we reported previously (Pozniak et al., 2000, 2002), and are not present in the p73-/- brain RNA samples.
We next asked whether p73 was required for the relative resistance of adult sensory neurons to death-inducing stimuli. To perform these experiments, we chose to directly compare adult DRG neurons from p73+/+ and p73+/- mice. To ensure that p73 heterozygosity did not developmentally influence sensory neuron repertoire, we characterized 6-week-old p73+/+ versus p73+/- L4 DRGs [most p73-/- animals die at postnatal day 10 (P10)]. Sectioning and Nissl-staining of ganglia from 6-week-old p73+/+ and p73+/- mice revealed that they were similar in size and morphology (Fig. 4A). Immunostaining of these sections for neurofilament 200 kD (NF200), which marks predominantly large diameter myelinated neurons, CGRP, which marks predominantly small diameter peptidergic nociceptors, and IB4, which marks small diameter nonpeptidergic nociceptors, demonstrated that the sensory neuron repertoire was also similar (Fig. 4B). Confirmation that the actual number of neurons was similar in p73+/+ versus p73+/- L4 DRGs was obtained by quantitating axons in dorsal root transverse sections using electron microscopy, a method that provides a reliable index of neuronal number in the L4 DRG (Coggeshall et al., 1997). This analysis revealed that p73+/+ and p73+/- dorsal roots contain 3411 ± 170 (n = 3) and 3301 ± 179 (n = 3) unmyelinated and 2376 ± 104 (n = 3) and 2335 ± 131 (n = 3) myelinated axons, respectively, numbers that were statistically similar between the genotypes (Fig. 4C,D).
Figure 4.

Deletion of one p73 allele does not affect the development of sensory neurons of the L4 DRG. A, Cresyl violet-stained sections of the L4 DRG from p73+/+ versus p73+/- adult littermates. Scale bar, 100 μm. B, Immunocytochemical analysis of sections through the p73+/+ and p73+/- L4 DRG with antibodies specific to NF200 (top panels), which marks predominantly large-diameter myelinated neurons, CGRP (middle panels), which marks predominantly small-diameter peptidergic nociceptors, and IB4 (bottom panels), which marks small-diameter nonpeptidergic nociceptors. Scale bars, 50 μm. C, Electron micrographs of transverse sections through the L4 dorsal roots of adult p73+/+ versus p73+/- animals. Scale bars, 2 μm. D, Quantitation of sections similar to those shown in C for the number of myelinated and unmyelinated axons in the L4 dorsal roots of p73+/+ and p73+/- animals.
Having demonstrated that p73 heterozygosity had no impact on the repertoire or number of DRG neurons in vivo, we then asked whether the decreased levels of p73 in p73+/- neurons made them more vulnerable to apoptotic insults in culture. Adult DRG neurons were isolated from p73+/+ and p73+/- mice, established in the presence of NGF for 24 hr, treated with the DNA damaging agent camptothecin (10 μm), and the number of phase-bright neurons counted over the ensuing 2 d. As seen with adult rat DRG neurons (Fig. 1B), the survival of adult p73+/+ mouse DRG neurons was only slightly diminished (80 ± 7% surviving neurons) by camptothecin at 48 hr (Fig. 5A). In contrast, p73+/- neurons showed a significantly enhanced sensitivity to camptothecin treatment relative to the p73+/+ neurons (Fig. 5A), with only 54 ± 6% surviving at 48 hr. A similar difference was seen when we inhibited the PI3-kinase survival pathway in these two populations of neurons using 50 μm LY294002. At 48 hr, survival was 72 ± 7 versus 40 ± 6% for p73+/+ versus p73+/- neurons, respectively (Fig. 5B). Thus, adult DRG neurons from p73+/- heterozygous mice are more vulnerable to apoptotic stimuli than their wild-type counterparts.
Figure 5.
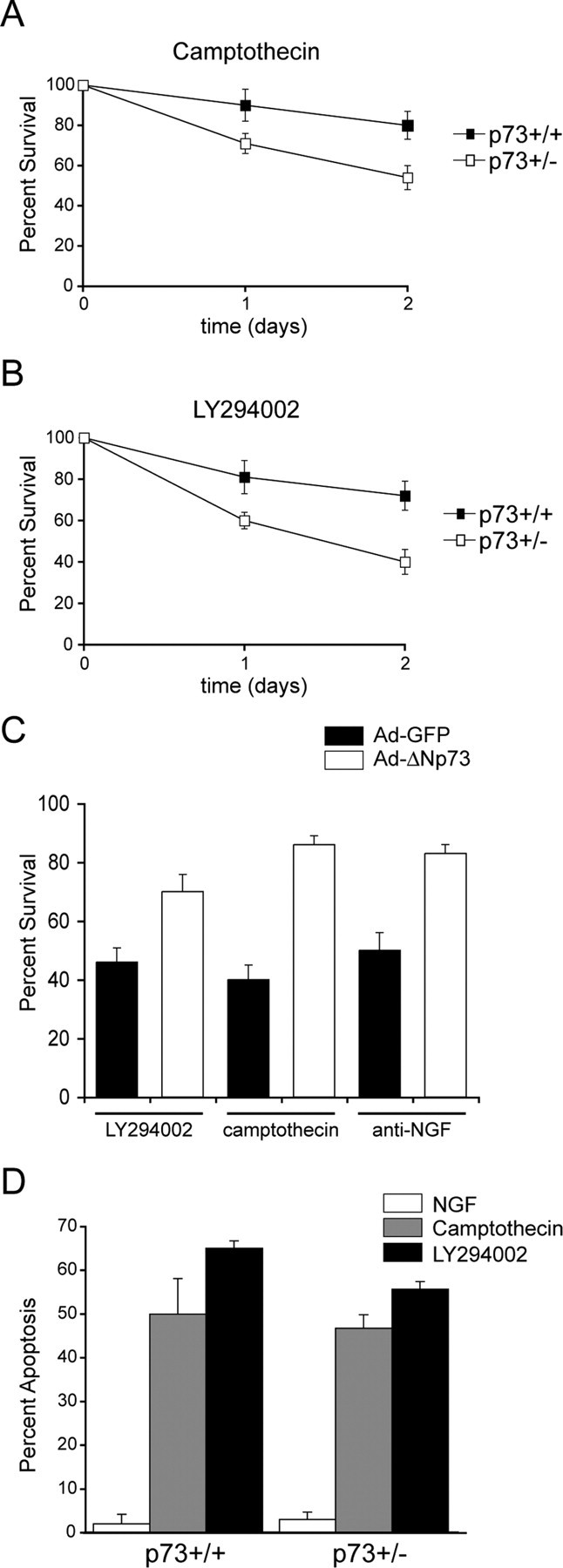
A, B, p73+/- adult sensory neurons are more vulnerable to apoptotic stimuli. Adult sensory neurons cultured from p73+/+ versus p73+/- animals were established in NGF for 1 d and then were maintained in NGF but with the addition of 10 μm camptothecin, a DNA-damaging agent (A) or 50 μm LY294002, an inhibitor of PI3-kinase (B). Phase-bright neurons with intact neurites in multiple identified fields were counted at 0, 1, and 2 d, and the amount of survival plotted relative to the initial number of neurons in the same fields at the time the treatment was initiated (percentage survival). Results are the cumulative results of at least three independent experiments, and the error bars represent SEM (t test; p < 0.005; comparison between groups at 2 d time point). C, The enhanced vulnerability of p73+/- adult sensory neurons can be rescued by expression of ΔNp73. Adult p73+/- neurons were established in NGF, infected with recombinant adenoviruses expressing either GFP or ΔNp73β and GFP, and then either withdrawn from NGF (anti-NGF) or maintained in the presence of NGF with the addition of 50 μm LY294002 or 10 μm camptothecin. Two days later, the survival of GFP-positive neurons was quantitated and expressed as a percentage of the total number of GFP-positive neurons. Error bars represent SEM. D, p73+/- and p73+/+ neonatal sympathetic neurons are equally sensitive to apoptotic stimuli. Neonatal sympathetic neurons cultured from p73+/+ versus p73+/- animals were established in NGF for 5 d and then maintained in either 10 ng/ml NGF or 10 ng/ml NGF with the addition of either 10 μm camptothecin or 50 μm LY294002. The number of apoptotic nuclei was quantified as determined by Hoechst staining 48 hr after treatment. Error bars represent SEM.
Although these data argued that p73 was essential for the relative invulnerability of adult neurons, and that even a small decrease in levels was sufficient to make them more sensitive to apoptotic stimuli, it was still formally possible that the p73+/- neurons differed from their wild-type counterparts in some other parameter. To address this possibility, we performed a rescue experiment. If the increased sensitivity of p73+/- adult neurons to apoptotic stimuli was attributable to a decrease in levels of ΔNp73, then we should be able to rescue this phenotype by exogenously increasing expression of ΔNp73. To perform this rescue experiment, established cultures of adult p73+/- DRGs were infected with recombinant adenoviruses expressing ΔNp73/GFP or GFP alone, and 1 d later, neurons were either withdrawn from NGF or treated with 10 μm camptothecin or 50 μm LY294002. Quantitation of the number of GFP-positive, phase-bright neurons 48 hr later revealed that the overexpression of ΔNp73, but not GFP, rescued the enhanced susceptibility of p73+/- neurons (Fig. 5C), arguing that ΔNp73 was essential for making adult DRG neurons resistant to apoptotic stimuli, at least in culture.
One other population of neurons that requires ΔNp73 for their survival is neonatal sympathetic neurons (Pozniak et al., 2000; Lee et al., 2004). Moreover, exogenously increased expression of ΔNp73 is sufficient to rescue sympathetic neurons from apoptosis induced by NGF withdrawal (Pozniak et al., 2000; Lee et al., 2004). We therefore asked whether p73+/- neonatal sympathetic neurons were also more sensitive to apoptotic insults than their wild-type counterparts. Neurons were cultured from neonatal p73+/- versus p73+/+ littermates, these neurons were then exposed to either 10 μm camptothecin or 50 μm LY294002, and apoptosis was quantified 48 hr later by assessing nuclear morphology. As shown previously (Crowder and Freeman, 1998; Park et al., 1998; Vaillant et al., 1999), both of these apoptotic stimuli were sufficient to cause sympathetic neuron apoptosis in the presence of NGF (Fig. 5D). However, unlike adult DRG neurons, apoptosis of p73+/- sympathetic neurons was not enhanced relative to p73+/+ sympathetic neurons (Fig. 5D).
p73 is necessary for the survival of adult DRG neurons after peripheral nerve injury in vivo
The anatomical analysis of the L4 DRG presented above demonstrated that the development of sensory neurons is normal in the p73+/- animals, but our culture data implied that adult neurons might be impaired in their survival after injury. To directly test this idea, we resected the sciatic nerve of 6-week-old p73+/+ versus p73+/- animals and, 1 month later, asked whether sensory neuron survival was affected by counting myelinated and unmyelinated axons in electron micrographs of the L4 DRG dorsal roots. This analysis revealed that, as previously reported in rats (Coggeshall et al., 1997; Tandrup et al., 2000), the amount of axonal loss in wild-type mice (p73+/+ littermates) was minimal 1 month after this type of axonal injury (Fig. 6A,B). The number of small, unmyelinated axons was reduced from 3411 ± 170 to 3061 ± 309 (n = 3) (a decrease of ∼10%), whereas the number of large, myelinated axons was unaffected (2376 ± 105, n = 3, vs 2314 ± 132, n = 3). In contrast, although the number of myelinated axons was unaffected after axotomy of p73+/- mice (2335 ± 130, n = 3, vs 2292 ± 154, n = 3), there was a substantive and significant 30% loss of unmyelinated axons in p73+/- animals (2144 ± 209, n = 3, vs 3061 ± 309, n = 4, p73+/- vs p73+/+, respectively). Thus, a decrease in the levels of p73 is sufficient to cause enhanced neuronal vulnerability both in vivo and in vitro, arguing that this molecule plays a key role in rendering adult neurons relatively invulnerable to injury.
Figure 6.
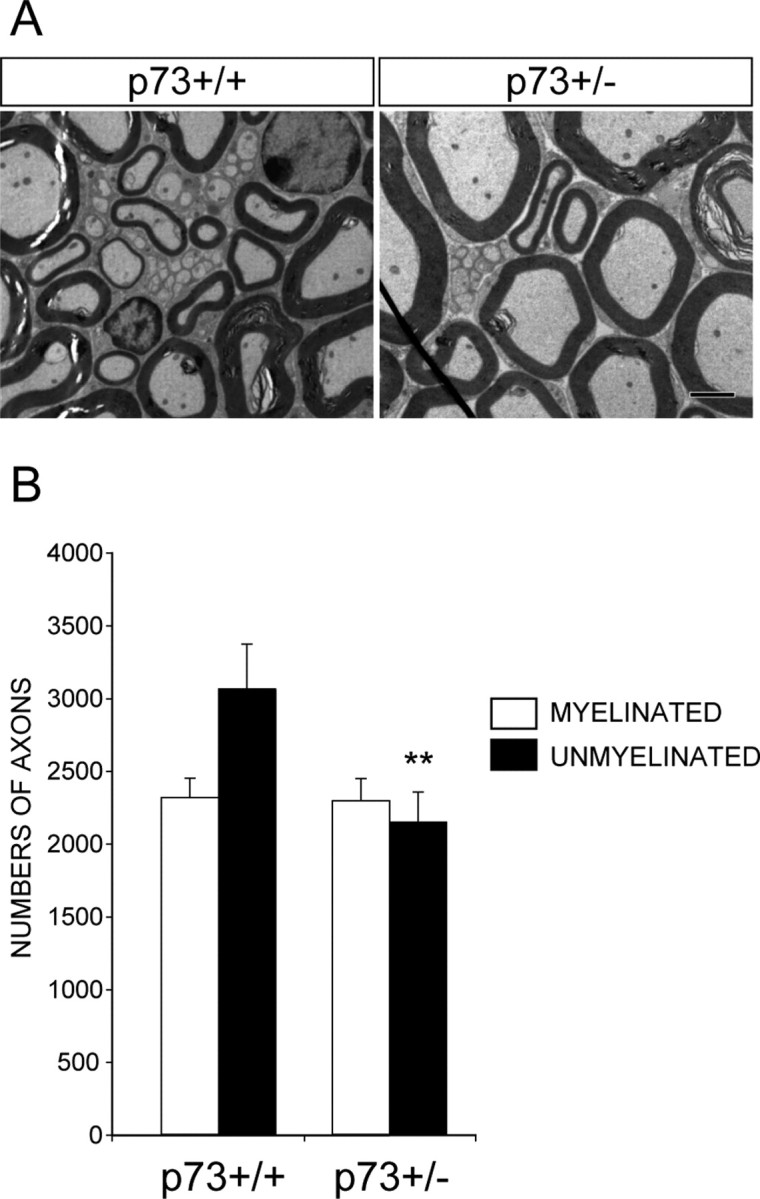
The absence of one p73 allele leads to enhanced death of small diameter, unmyelinated adult sensory neurons after axonal injury. A, Electron micrographs of transverse sections through the L4 dorsal roots of p73+/- versus p73+/+ animals 1 month after sciatic nerve resection. Scale bar, 2 μm. B, Quantitation of the number of myelinated and unmyelinated axons present in cross sections similar to those shown in A (t test; **p < 0.005). Error bars represent SD.
Discussion
In this study, we present data indicating that the relative invulnerability of adult neurons is attributable to a cell-intrinsic enhancement of neuronal survival pathways, and that a key component of these survival pathways is ΔNp73. More specifically, our data support three major conclusions. First, we provide evidence that adult sensory neurons are much more resistant to a variety of apoptotic stimuli than are their neonatal counterparts, and that this resistance is mediated downstream of JNK. Second, we show that increased expression of ΔNp73 is sufficient to rescue JNK-mediated apoptosis of neonatal neurons, and that the loss of one allele of p73 renders adult sensory neurons more vulnerable to apoptotic stimuli in culture. This enhanced vulnerability is attributable to decreased ΔNp73 because the invulnerability of adult sensory neurons can be restored by exogenous expression of ΔNp73 in p73+/- neurons. Third, we demonstrate that these findings are relevant in vivo; the loss of even one p73 allele, while having no overt effects on developing sensory neurons, enhanced the death of small unmyelinated DRG neurons after axonal injury in adults. Thus, p73 represents one key mechanism used by neurons to ensure that they survive the lifetime of the animal.
We demonstrated previously that the ΔNp73 isoforms are highly potent survival proteins, inhibiting the death of cultured sympathetic and cortical neurons as mediated by a variety of stimuli, including NGF withdrawal and DNA damage (Pozniak et al., 2000, 2002). Moreover, we previously demonstrated enhanced death of p73-/- postnatal sympathetic and cortical neurons in vivo, indicating that this gene was essential for the survival of at least some populations of developing neurons (Pozniak et al., 2000, 2002; Lee et al., 2004). However, the first indication that p73 might be important for survival of mature neurons was our finding of ongoing loss of cortical neurons between 3 and 8 weeks postnatally in p73-/- animals. Although there are many potential explanations for such a finding, this observation suggested that p73 might play a previously unsuspected role in determining the maintenance of adult neurons. In that regard, the data documented here demonstrate such a cell-intrinsic role for p73 in peripheral neurons, and our previous in vivo work on the p73-/- cortex suggests that this role may well generalize to CNS neurons.
How does p73 determine the long-term survival of adult neurons? We propose that it is the ΔNp73 isoforms that are important for this function because (1) ΔNp73 isoforms are the only isoforms detectable in the brain at the protein level, (2) as discussed above, ΔNp73 is a very potent survival protein when overexpressed, whereas overexpression of TAp73 causes neuronal apoptosis (Lo et al., 2003), and (3) as shown here, ΔNp73 rescues the enhanced vulnerability of p73+/- adult sensory neurons. We also propose that one of the ways that ΔNp73 determines survival of mature neurons is by functionally antagonizing p53. p53 has been shown to play a key role in mediating the apoptosis of mature neurons in vivo in response to a wide variety of stimuli, including excitotoxicity, DNA damage (for review, see Miller et al., 2000; Morrison and Kinoshita, 2000), and, with regard to the current study, axonal injury (Martin and Liu, 2002). Support for this model derives from the fact that ΔNp73 rescues sympathetic neurons from apoptosis induced by p53 overexpression (Pozniak et al., 2000), and from our recent finding that deletion of even one p53 allele is sufficient to partially rescue the enhanced sympathetic neuron apoptosis seen in developing p73-/- animals (Lee et al., 2004). Together, we believe these findings make a compelling argument that p53 is one of the targets of ΔNp73. However, it is also clear that ΔNp73 has additional targets (Lee et al., 2004), and we propose that these additional targets include other full-length, proapoptotic p53 family members such as p63, the third p53 family member (Yang et al., 1998, 1999), which is predominantly expressed in the nervous system as a full-length TAp63 isoform (Jacobs et al., 2004). In that regard, we propose that it is ultimately the balance of full-length, proapoptotic p53/TAp63 family members relative to the prosurvival N-terminal truncated ΔNp73/ΔNp63 isoforms that determines the life versus death of any given neuron.
How does such a p53 family-dependent checkpoint integrate with our current knowledge of adult neuron survival and apoptosis? Although most of what we know about neuronal survival and death pathways derives from studies of developing neurons, studies on peripheral neurons aged in culture have provided some insight. In particular, aged, NGF-independent sympathetic neurons still activate the JNK pathway after NGF withdrawal, but they do not translocate Bax in the mitochondrion, and Bcl2+/- neurons are just as resistant to NGF withdrawal as Bcl2+/+ neurons, implying that a major block to apoptosis occurs between JNK and Bax (Greenlund et al., 1995; Putcha et al., 2000). Is there any evidence that the p53 family might act at such an upstream point? Although p53 is thought to mediate at least some of its proapoptotic actions at the transcriptional level, for example by enhanced transcription of Bax (Miyashita and Reed, 1995), there is also evidence that it can promote apoptosis directly at the level of the mitochondrion in a cell-free system (Schuler et al., 2000; Mihara et al., 2003). Moreover, we demonstrate here that ΔNp73 can rescue apoptosis induced by JNK pathway activation, and our recent studies in sympathetic neurons demonstrate that ΔNp73 can block the mitochondrial apoptotic transition after NGF withdrawal, potentially by inhibiting full-length p53 family members and/or binding directly to JNK itself (Lee et al., 2004). Thus, all of these data are consistent with the hypothesis that adult neurons have a major block to apoptosis downstream of JNK but upstream of the mitochondrion, and that this major checkpoint involves ΔNp73. It is likely that additional downstream checkpoints also exist, potentially involving the Bcl2 and/or inhibitor of apoptosis protein families (Wiese et al., 1999; Yuan and Yankner, 2000). In that regard, if one way that adult neurons become resistant to apoptosis is by enhancing survival pathways, it makes sense to have multiple checkpoints at different levels to ensure that mature neurons are only eliminated when they have received multiple and continuous apoptotic insults.
Our ongoing studies on the role of p73 in the nervous system indicate that in addition to the essential role in maintenance of mature neurons that is demonstrated here, ΔNp73 is also important for the survival of a number of populations of developing neurons, including neonatal sympathetic and cortical neurons (Pozniak et al., 2000, 2002; Lee et al., 2004). Moreover, p73-/- mice display reduced numbers of caudal CNS neurons at birth, including facial motor neurons and neurons of the deep cerebellar nuclei (Pozniak et al., 2002), implying that these neuronal populations might also have enhanced apoptosis during their embryonic developmental death period in the absence of p73. However, whereas our findings argue that p73 is important for neuronal survival both in development and in adulthood, data presented here suggest that the requirement for p73 somehow differs in developing versus adult neurons; neonatal sympathetic neurons are not sensitive to the loss of a single p73 allele, whereas adult sensory neurons are sensitive to the loss. One potential explanation for this difference is that developing versus adult neurons differ in how they regulate ΔNp73 levels. For example, when neonatal sympathetic neurons are withdrawn from NGF, ΔNp73 levels fall dramatically (Pozniak et al., 2000), a response that is necessary to ensure the death of neurons that do not compete successfully for target-derived NGF in vivo. Disruption of the essential PI3-kinase survival pathway and/or DNA damage may lead to a similar loss of ΔNp73 expression in neonatal sympathetic neurons, thereby making p73+/- and p73-/- neurons equally vulnerable to apoptotic stimuli. In contrast, if adult neurons maintain higher basal levels of ΔNp73 through a cell-intrinsic mechanism that is essentially impervious to extrinsic cues, these neurons would be relatively invulnerable to apoptotic stimuli. However, a decrease in basal ΔNp73 levels as a function of decreased p73 gene dosage would then be predicted to enhance neuronal vulnerability to these same apoptotic insults.
One of the major implications of this study derives from our finding that the lack of one p73 allele is sufficient to have a major impact on neuronal survival after injury. Although the finding that relatively small potential alterations in protein levels would influence neuronal survival is a prediction of a “rheostat” model as we proposed, it is nonetheless surprising and has broad implications with regard to a genetic propensity for injury-induced damage to the adult nervous system and even with regard to neurodegeneration. In particular, if the levels of ΔNp73 serve as a “buffer,” a small decrease in levels might well be sufficient to enhance the probability that an individual neuron would apoptose in response to an extrinsic cue, something that would ultimately result in an increased rate of neuronal loss over time (Clarke et al., 2000). This is precisely what is seen in the culture studies reported here, where the rate of cell death in response to apoptotic stimuli is increased in p73+/- neurons. Moreover, a similar increase in rate might be responsible for the axotomy results reported here, because the loss of small diameter unmyelinated sensory neurons seen in the p73+/- animals at 1 month is approximately similar in magnitude to the loss that is seen in rats after 8 months (Tandrup et al., 2000). Such a genetically determined increase in the rate of death would be particularly problematic in situations of acute injury, such as during ischemic stroke and potentially even in the aging brain, where the DNA damage response, which is regulated by the p53 family, is now known to be elevated (Lu et al., 2004).
Footnotes
This work was supported by grants from the Canadian Institutes of Health Research (CIHR). D.R.K. is a Canada Research Chair, F.D.M. is a Senior Scientist of CIHR, and G.S.W. was supported by a studentship from the Hospital for Sick Children Research Institute. We thank Yew Meng Heng, Julia Hwang, and Howard Rosenberg of the Electron Microscopy Laboratory, Hospital for Sick Children, for their excellent technical assistance.
Correspondence should be addressed to Dr. Freda D. Miller, Developmental Biology, Hospital for Sick Children, Room Black 3203A, 555 University Avenue, Toronto, Ontario, Canada M5G 1X8. E-mail: fredam@sickkids.ca.
N. Orike's present address: Renovis Inc., 2 Corporate Drive South, San Francisco, CA 94080.
Copyright © 2004 Society for Neuroscience 0270-6474/04/249638-10$15.00/0
G.S.W. and N.O. contributed equally to this work.
References
- Acheson A, Conover JC, Fandl JP, DeChiara TM, Russell M, Thadani A, Squinto SP, Yancopoulos GD, Lindsay RM (1995) A BDNF autocrine loop in adult sensory neurons prevents cell death. Nature 374: 450-453. [DOI] [PubMed] [Google Scholar]
- Aloyz RS, Bamji SX, Pozniak CD, Toma JG, Atwal J, Kaplan DR, Miller FD (1998) p53 is essential for developmental neuron death as regulated by the TrkA and p75 neurotrophin receptors. J Cell Biol 143: 1691-1703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Angeletti PU, Levi-Montalcini R, Caramia F (1971) Analysis of the effects of the antiserum to the nerve growth factor in adult mice. Brain Res 27: 343-355. [DOI] [PubMed] [Google Scholar]
- Bamji SX, Majdan M, Pozniak CD, Belliveau DJ, Aloyz R, Kohn J, Causing CG, Miller FD (1998) The p75 neurotrophin receptor mediates neuronal apoptosis and is essential for naturally occurring sympathetic neuron death. J Cell Biol 140: 911-923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bjerre B, Bjorklund A, Mobley W, Rosengren E (1975) Short- and long-term effects of nerve growth factor on the sympathetic nervous system in the adult mouse. Brain Res 94: 263-277. [DOI] [PubMed] [Google Scholar]
- Chun LL, Patterson PH (1977) Role of nerve growth factor in the development of rat sympathetic neurons in vitro I. Survival, growth, and differentiation of catecholamine production. J Cell Biol 75: 694-704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clarke G, Collins RA, Leavitt BR, Andrews DF, Hayden MR, Lumsden CJ, McInnes RR (2000) A one-hit model of cell death in inherited neuronal degenerations. Nature 406: 195-199. [DOI] [PubMed] [Google Scholar]
- Coggeshall RE, Lekan HA, Doubell TP, Allchorne A, Woolf CJ (1997) Central changes in primary afferent fibers following peripheral nerve lesions. Neuroscience 77: 1115-1122. [DOI] [PubMed] [Google Scholar]
- Crowder RJ, Freeman RS (1998) Phosphatidylinositol 3-kinase and Akt protein kinase are necessary and sufficient for the survival of nerve growth factor-dependent sympathetic neurons. J Neurosci 18: 2933-2943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Easton RM, Deckwerth TL, Parsadanian AS, Johnson Jr EM (1997) Analysis of the mechanism of loss of trophic factor dependence associated with neuronal maturation: a phenotype indistinguishable from Bax deletion. J Neurosci 17: 9656-9666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eilers A, Whitfield J, Babij C, Rubin LL, Ham J (1998) Role of the Jun kinase pathway in the regulation of c-Jun expression and apoptosis in sympathetic neurons. J Neurosci 18: 1713-1724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goedert M, Otten U, Thoenen H (1978) Biochemical effects of antibodies against nerve growth factor on developing and differentiated sympathetic ganglia. Brain Res 148: 264-268. [DOI] [PubMed] [Google Scholar]
- Greenlund LJ, Korsmeyer SJ, Johnson Jr EM (1995) Role of BCL-2 in the survival and function of developing and mature sympathetic neurons. Neuron 15: 649-661. [DOI] [PubMed] [Google Scholar]
- Hendry IA, Campbell J (1976) Morphometric analysis of rat superior cervical ganglion after axotomy and nerve growth factor treatment. J Neurocytol 5: 351-360. [DOI] [PubMed] [Google Scholar]
- Irwin MS, Kaelin WG (2001) p53 family update: p73 and p63 develop their own identities. Cell Growth Differ 12: 337-349. [PubMed] [Google Scholar]
- Jacobs WB, Govoni G, Ho D, Mills AA, Miller FD, Kaplan DR (2004) p63 is an essential proapoptotic molecule during developmental neuronal death. Soc Neurosci Abstr, in press.
- Lazarus KJ, Bradshaw RA, West NR, Bunge P (1976) Adaptive survival or rat sympathetic neurons cultured without supporting cells or exogenous nerve growth factor. Brain Res 113: 159-164. [DOI] [PubMed] [Google Scholar]
- Lee AF, Ho DK, Zanassi P, Walsh GS, Kaplan DR, Miller FD (2004) Evidence that ΔNp73 promotes neuronal survival by p53-dependent and independent mechanisms. J Neurosci 24: 9174-9184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindsay RM (1988) Nerve growth factors (NGF, BDNF) enhance axonal regeneration but are not required for survival of adult sensory neurons. J Neurosci 8: 2394-2405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lo WD, Akhmametyeva EM, Zhu L, Friesen PD, Chang LS (2003) Induction of apoptosis by the p53-related p73 and partial inhibition by the baculovirus-encoded p35 in neuronal cells. Brain Res Mol Brain Res 113: 1-12. [DOI] [PubMed] [Google Scholar]
- Lu T, Pan Y, Kao SY, Li C, Kohane I, Chan J, Yankner BA (2004) Gene regulation and DNA damage in the ageing human brain. Nature 429: 883-891. [DOI] [PubMed] [Google Scholar]
- Ma J, Novikov LN, Wiberg M, Kellerth JO (2001) Delayed loss of spinal motoneurons after peripheral nerve injury in adult rats: a quantitative morphological study. Exp Brain Res 139: 216-223. [DOI] [PubMed] [Google Scholar]
- Majdan M, Walsh GS, Aloyz R, Miller FD (2001) TrkA mediates developmental sympathetic neuron survival in vivo by silencing an ongoing p75NTR-mediated death signal. J Cell Biol 155: 1275-1285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maroney AC, Finn JP, Bozyczko-Coyne D, O'Kane TM, Neff NT, Tolkovsky AM, Park DS, Yan CY, Troy CM, Greene LA (1999) CEP-1347 (KT7515), an inhibitor of JNK activation, rescues sympathetic neurons and neuronally differentiated PC12 cells from death evoked by three distinct insults. J Neurochem 73: 1901-1912. [PubMed] [Google Scholar]
- Martin LJ, Liu Z (2002) Injury-induced spinal motor neuron apoptosis is preceded by DNA single-strand breaks and is p53- and Bax-dependent. J Neurobiol 50: 181-197. [DOI] [PubMed] [Google Scholar]
- Mihara M, Erster S, Zaika A, Petrenko O, Chittenden T, Pancoska P, Moll UM (2003) p53 has a direct apoptogenic role at the mitochondria. Mol Cell 11: 577-590. [DOI] [PubMed] [Google Scholar]
- Miller FD, Kaplan DR (2001) Neurotrophin signalling pathways regulating neuronal apoptosis. Cell Mol Life Sci 58: 1045-1053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller FD, Pozniak CD, Walsh GS (2000) Neuronal life and death: an essential role for the p53 family. Cell Death Differ 7: 880-888. [DOI] [PubMed] [Google Scholar]
- Miyashita T, Reed JC (1995) Tumor suppressor p53 is a direct transcriptional activator of the human bax gene. Cell 80: 293-299. [DOI] [PubMed] [Google Scholar]
- Morrison RS, Kinoshita Y (2000) The role of p53 in neuronal cell death. Cell Death Differ 7: 868-879. [DOI] [PubMed] [Google Scholar]
- Otten U, Goedert M, Schwab M, Thibault J (1979) Immunization of adult rats against 2.5 S NGF: effects on the peripheral sympathetic nervous system. Brain Res 176: 79-90. [DOI] [PubMed] [Google Scholar]
- Park DS, Morris EJ, Padmanabhan J, Shelanski ML, Geller HM, Greene LA (1998) Cyclin-dependent kinases participate in death of neurons evoked by DNA-damaging agents. J Cell Biol 1998 143: 457-467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pozniak CD, Radinovic S, Yang A, McKeon F, Kaplan DR, Miller FD (2000) An anti-apoptotic role for the p53 family member, p73, during developmental neuron death. Science 289: 304-306. [DOI] [PubMed] [Google Scholar]
- Pozniak CD, Barnabe-Heider F, Rymar VV, Lee AF, Sadikot AF, Miller FD (2002) p73 is required for survival and maintenance of CNS neurons. J Neurosci 22: 9800-9809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Putcha GV, Deshmukh M, Johnson Jr EM (2000) Inhibition of apoptotic signaling cascades causes loss of trophic factor dependence during neuronal maturation. J Cell Biol 149: 1011-1018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roux PP, Dorval G, Boudreau M, Angers-Loustau A, Morris SJ, Makkerh J, Barker PA (2002) K252a and CEP1347 are neuroprotective compounds that inhibit mixed-lineage kinase-3 and induce activation of Akt and ERK. J Biol Chem 277: 49473-49480. [DOI] [PubMed] [Google Scholar]
- Schuler M, Bossy-Wetzel E, Goldstein JC, Fitzgerald P, Green DR (2000) p53 induces apoptosis by caspase activation through mitochondrial cytochrome c release. J Biol Chem 275: 7337-7342. [DOI] [PubMed] [Google Scholar]
- Snider WD, Elliott JL, Yan Q (1992) Axotomy-induced neuronal death during development. J Neurobiol 23: 1231-1246. [DOI] [PubMed] [Google Scholar]
- Tandrup T, Woolf CJ, Coggeshall RE (2000) Delayed loss of small dorsal root ganglion cells after transection of the rat sciatic nerve. J Comp Neurol 422: 172-180. [DOI] [PubMed] [Google Scholar]
- Tong JX, Vogelbaum MA, Drzymala RE, Rich KM (1997) Radiation-induced apoptosis in dorsal root ganglion neurons. J Neurocytol 26: 771-777. [DOI] [PubMed] [Google Scholar]
- Vaillant AR, Mazzoni I, Tudan C, Boudreau M, Kaplan DR, Miller FD (1999) Depolarization and neurotrophins converge on the phosphatidylinositol 3-kinase-Akt pathway to synergistically regulate neuronal survival. J Cell Biol 146: 955-966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogelbaum MA, Tong JX, Rich KM (1998) Developmental regulation of apoptosis in dorsal root ganglion neurons. J Neurosci 18: 8928-8935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiese S, Digby MR, Gunnersen JM, Gotz R, Pei G, Holtmann B, Lowenthal J, Sendtner M (1999) The anti-apoptotic protein ITA is essential for NGF-mediated survival of embryonic chick neurons. Nat Neurosci 2: 978-983. [DOI] [PubMed] [Google Scholar]
- Xiang H, Kinoshita Y, Knudson CM, Korsmeyer SJ, Schwartzkroin PA, Morrison RS (1998) Bax involvement in p53-mediated neuronal cell death. J Neurosci 18: 1363-1373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang A, Kaghad M, Wang Y, Gillett E, Fleming MD, Dotsch V, Andrews NC, Caput D, McKeon F (1998) p63, a p53 homolog at 3q27-29, encodes multiple products with transactivating, death-inducing, and dominant-negative activities. Mol Cell 2: 305-316. [DOI] [PubMed] [Google Scholar]
- Yang A, Schweitzer R, Sun D, Kaghad M, Walker N, Bronson RT, Tabin C, Sharpe A, Caput D, Crum C, McKeon F (1999) p63 is essential for regenerative proliferation in limb, craniofacial and epithelial development. Nature 398: 714-718. [DOI] [PubMed] [Google Scholar]
- Yang A, Walker N, Bronson R, Kaghad M, Oosterwegel M, Bonnin J, Vagner C, Bonnet H, Dikkes P, Sharpe A, McKeon F, Caput D (2000) p73-deficient mice have neurological, pheromonal and inflammatory defects but lack spontaneous tumours. Nature 404: 99-103. [DOI] [PubMed] [Google Scholar]
- Yip HK, Rich KM, Lampe PA, Johnson Jr EM (1984) The effects of nerve growth factor and its antiserum on the postnatal development and survival after injury of sensory neurons in rat dorsal root ganglia. J Neurosci 4: 2986-2992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan J, Yankner BA (2000) Apoptosis in the nervous system. Nature 407: 802-809. [DOI] [PubMed] [Google Scholar]