

Abstract
Genetically encoded Ca2+ indicators (GECIs) based on fluorescent proteins (XFPs) and Ca2+-binding proteins [like calmodulin (CaM)] have great potential for the study of subcellular Ca2+ signaling and for monitoring activity in populations of neurons. However, interpreting GECI fluorescence in terms of neural activity and cytoplasmic-free Ca2+ concentration ([Ca2+]) is complicated by the nonlinear interactions between Ca2+ binding and GECI fluorescence. We have characterized GECIs in pyramidal neurons in cultured hippocampal brain slices, focusing on indicators based on circularly permuted XFPs [GCaMP (Nakai et al., 2001), Camgaroo2 (Griesbeck et al., 2001), and Inverse Pericam (Nagai et al., 2001)]. Measurements of fluorescence changes evoked by trains of action potentials revealed that GECIs have little sensitivity at low action potential frequencies compared with synthetic [Ca2+] indicators with similar affinities for Ca2+. The sensitivity of GECIs improved for high-frequency trains of action potentials, indicating that GECIs are supralinear indicators of neural activity. Simultaneous measurement of GECI fluorescence and [Ca2+] revealed supralinear relationships. We compared GECI fluorescence saturation with CaM Ca2+-dependent structural transitions. Our data suggest that GCaMP and Camgaroo2 report CaM structural transitions in the presence and absence of CaM-binding peptide, respectively.
Keywords: two-photon, X-Rhod-5F, Fluo4-FF, GCaMP, Camgaroo2, Inverse Pericam
Introduction
Ca2+ is a ubiquitous second messenger in the brain, and its regulation at the level of influx, binding to effector proteins, and extrusion dictates the diverse physiological responses it controls (Berridge, 1998). Imaging methods using synthetic Ca2+ indicators can probe [Ca2+] dynamics with sub-micrometer spatial and sub-millisecond temporal resolution (Yasuda et al., 2004). [Ca2+] imaging can be used to monitor the activity of neurons (Callaway and Ross, 1995; Helmchen et al., 1996; Svoboda et al., 1997; Maravall et al., 2000) and synapses (Muller and Connor, 1991; Yuste and Denk, 1995; Oertner et al., 2002; Nimchinsky et al., 2004) and to report the dynamics of populations of neurons in brain slices (Yuste et al., 1992) and in vivo (O'Malley et al., 1996; Stosiek et al., 2003).
Synthetic Ca2+ indicators have traditionally been introduced into neurons by bulk loading of the membrane permeable AM-ester (Tsien et al., 1982; Yuste et al., 1992) or dextran (O'Donovan et al., 1993; O'Malley et al., 1996) derivative, or by loading individual cells through a pipette with a cell-impermeant indicator salt (Tank et al., 1988; Muller and Connor, 1991; Yuste and Denk, 1995; Oertner et al., 2002; Nimchinsky et al., 2004).
Despite their success, synthetic indicators have significant limitations. First, they are difficult to load into populations of neurons in the intact brain (but see Stosiek et al., 2003). Second, they cannot be targeted specifically to particular cell types or subcellular compartments. Third, once loaded, synthetic indicators are cleared from the cytoplasm, making long-term imaging experiments difficult.
A major breakthrough was the development of genetically encoded Ca2+ indicators (GECIs) based on fluorescent proteins (XFPs) (Miyawaki et al., 1997, 1999; Baird et al., 1999; Griesbeck et al., 2001; Nagai et al., 2001). GECIs can be delivered efficiently to specific subsets of neurons and neuronal compartments using transfection, viral infection, transgenic, and knock-in techniques (J. W. Wang et al., 2003; Yu et al., 2003; Hasan et al., 2004; Y. Wang et al., 2004).
GECIs have already been used to measure [Ca2+] transients in worms (Kerr et al., 2000; Suzuki et al., 2003), flies (Fiala et al., 2002; Reiff et al., 2002; J. W. Wang et al., 2003; Yu et al., 2003; Y. Wang et al., 2004), zebrafish (Higashijima et al., 2003), and mice (Hasan et al., 2004; Ji et al., 2004). However, GECI dynamics are more complicated than those of synthetic Ca2+ indicators. Interpreting functional imaging experiments using GECIs requires a quantitative understanding of the relationship between [Ca2+], neural activity, and GECI fluorescence.
Here, we analyze the properties of three GECIs based on circularly permuted XFPs [Inverse Pericam (Nagai et al., 2001), GCaMP (Nakai et al., 2001), and Camgaroo2 (Griesbeck et al., 2001)] in brain slice neurons (see Fig. 1A). First, we relate GECI fluorescence to neural activity [i.e., action potential (AP) number and frequency] and compare the fluorescence responses with those measured with synthetic Ca2+ indicators. Our data show that GECIs are nonlinear indicators of neural activity with poor sensitivity at low firing rates. Second, we relate GECI fluorescence to [Ca2+]. Our data show that GECI fluorescence has complex relationships with [Ca2+], with supralinear and sublinear regimes. Third, we compare our in situ biochemical characterization with data from solution biochemistry. We find that some GECIs (GCaMP and Camgaroo2) report Ca2+-dependent calmodulin (CaM) structural transitions.
Figure 1.
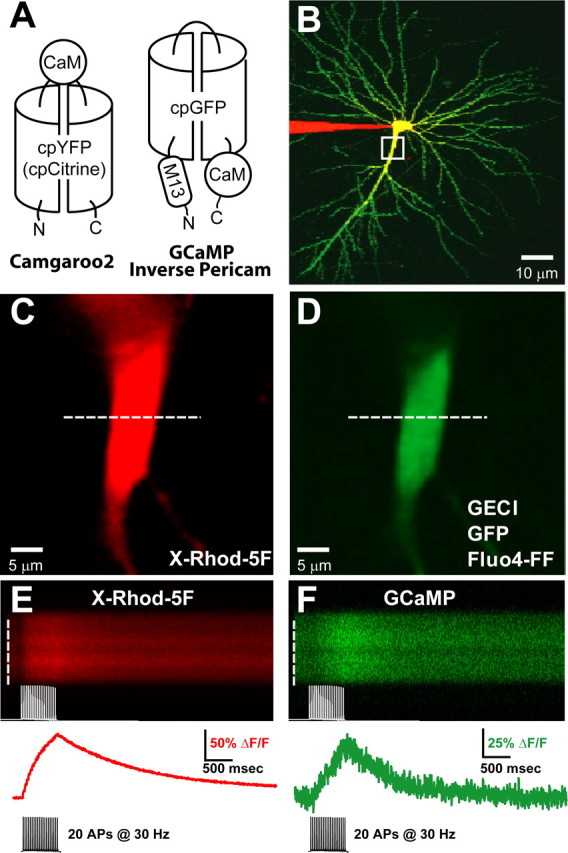
Dual-color imaging of AP-evoked fluorescence responses in CA1 pyramidal cells transfected with GECIs. A, Molecular topology of GECIs used in this study. Binding Ca2+ causes changes in GECI fluorescence. Camgaroo2 (left) is based on the cpYFP (yellow fluorescent protein) molecule cpCitrine and has CaM inserted between the two halves of the cpCitrine barrel. GCaMP and Inverse Pericam (right) are based on cpGFP and contain CaM inserted C terminal to the cpGFP barrel and the CaM-binding peptide M13 inserted N terminal. B, CA1 pyramidal neuron expressing GCaMP (green; 9 d after transfection) and patched with a pipette filled with 500 μm X-Rhod-5F (red). The yellow region indicates overlapping green and red fluorescence. C, D, Magnified image of primary apical dendrite (boxed region in B; location of line scan, dashed white line), showing red X-Rhod-5F (C) and green GECI (D) fluorescence. E, F, Red and green fluorescence transients evoked by a train of 20 APs at 30 Hz. Fluorescence was averaged across the spatial extent of the dendrite (white dashed lines; top) to produce the fluorescence response (red and green traces; bottom) used for subsequent analysis.
Materials and Methods
DNA constructs, transfection, and slice culture. Camgaroo2 was from R. Tsien (University of California San Diego, San Diego, CA) (Griesbeck et al., 2001), GCaMP was from J. Nakai (Department of Information Physiology, National Institute for Physiological Sciences, Myodaiji, Okazaki, Japan) (Nakai et al., 2001), Inverse Pericam was from A. Miyawaki (RIKEN) (Nagai et al., 2001), and enhanced green fluorescent protein (EGFP) was from Clontech (Cambridge, UK) (pEGFP-N1). The CaM sequences in GCaMP and Camgaroo2 are derived from rat and Xenopus, respectively, but have 100% protein sequence similarity. Inverse Pericam contains a CaM with a mutation in the third calcium-binding loop (E104Q) (Evenas et al., 1998). The M13 domain used in GCaMP and Inverse Pericam is derived from smooth muscle myosin light chain kinase CaM-binding peptide (Peersen et al., 1997). All experimental protocols were conducted according to National Institutes of Health guidelines for animal research and were approved by the Institutional Animal Care and Use Committee at Cold Spring Harbor Laboratory. Cultured hippocampal slices were prepared from postnatal day 6 (P6) or P7 rats (Stoppini et al., 1991) and transfected using particle-mediated biolistic gene transfer (McAllister and Stevens, 2000) at 5-9 d in vitro. Expression was for 7-14 d.
Electrophysiology. Transfected or neighboring untransfected cells (20-50 μm deep in the slice) were selected for whole-cell recording (see Fig. 1 B). Recordings were performed in artificial CSF (ACSF) consisting of (in mm) 127 NaCl, 25 NaHCO3, 25 d-glucose, 2.5 KCl, 4 MgCl2, 4 CaCl2, and 1.25 NaH2PO4, aerated continuously with 95% O2/5%CO2. Intracellular solution consisted of 130 mm KMeSO3, 10 mm HEPES, 4 mm MgCl2, 4 mm Na2ATP, 0.4 mm NaGTP, 10 mm Na-phosphocreatine, 3 mm ascorbate, and 500 μm X-Rhod-5F and/or Fluo4-FF; pH was adjusted to 7.3 with KOH. Recordings were at room temperature in ACSF. To evoke APs, cells were held in the current-clamp configuration, and 3-5 nA of current was injected for 2 msec through the recording electrode. The resting membrane potentials and input resistances of transfected cells (GECIs and GFP) and untransfected cells were not different (data not shown).
Imaging. We used a custom-built two-photon laser scanning microscope (2PLSM) with an Olympus objective (60×, 0.9 numerical aperture) and a Ti:sapphire laser (Mira; Coherent, Santa Clara, CA) tuned to λ = 910 nm. Fluorescence was detected in epi- and trans-fluorescence modes (Mainen et al., 1999) with photomultiplier tubes (R3896; Hamamatsu, Hamamatsu City, Japan). Dichroic mirrors (565 nm) were used to separate green and red fluorescence. BG22 colored glass filters and 607/45 barrier filters were placed, respectively, in the “green” and “red” pathways to eliminate transmitted or reflected excitation light (all filters and dichroics were from Chroma, Brattleboro, VT). Laser power was modulated with microsecond temporal resolution using an electro-optical modulator (EOM; model 350-80 LA; Conoptics, Danbury, CT). Image acquisition was controlled by ScanImage (Pologruto et al., 2003).
For most experiments, neurons were loaded with a “red” synthetic Ca2+ indicator (X-Rhod-5F; 500 μm; KD = 1.9 μm at room temperature). In some cases, we also used a “green” synthetic indicator (Fluo4-FF; 500 μm; KD = 10.4 μm at room temperature; synthetic indicators were purchased from Molecular Probes, Eugene, OR) (Yasuda et al., 2004). During loading, the amplitudes of [Ca2+] transients produced by single APs were monitored. Data collection started only after equilibration was achieved (∼20 min after break-in) (supplemental material, available at www.jneurosci.org) (Maravall et al., 2000). After initiation of whole-cell recording, the baseline green fluorescence reporting the concentration of cytoplasmic green proteins (GFP and GECIs) decreased slowly over time (∼40% over 50 min), consistent with washout.
For most experiments, fluorescence (green and red) was collected while scanning at 500 Hz in a line that intersected the apical dendrite (see Fig. 1C,D). Fluorescence signals were averaged over the dendrite to obtain the fluorescence time course, F (see Fig. 1 E, F). Photo multiplier tube dark noise (shutter closed) was collected for 50 msec at the beginning of each image (see Fig. 1 E, F), and the mean dark noise was subtracted from F. Baseline fluorescence (F0) was the average fluorescence over a 200 msec window before the stimulus. ΔF/F was calculated as ΔF/F = (F-F0)/F0.
Free-calcium concentration changes, Δ[Ca2+], and fluorescence saturation changes, Δϕ, were calculated from ΔF/F using a method that relies on estimating the fluorescence increase that would arise from a saturating Δ[Ca2+][(ΔF/F)max], using Equations 1a and 1b, respectively (Maravall et al., 2000):
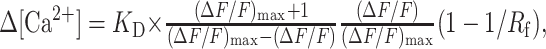 |
1a |
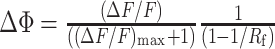 |
1b |
F is the fluorescence from the indicator. ϕ is the degree to which the fluorescence of the indicator is saturated, expressed as a fraction (range, 0-1). KD is the effective dissociation constant of the indicator for Ca2+, defined as the [Ca2+] in which fluorescence is half-saturated (i.e., ϕ = 0.5 when [Ca2+] = KD). Rf is the dynamic range of the indicator, and Rf = Fmax/Fmin. KD and Rf were previously determined for synthetic Ca2+ indicators (Yasuda et al., 2004).
Because of the weak dependence of Δ[Ca2+] on Rf, this method can be used to quantify Δ[Ca2+] with good accuracy for indicators with large Rf, such as X-Rhod-5F and Fluo4-FF. The Rf value of X-Rhod-5F differs substantially from the maximal change in fluorescence [(ΔF/F)max] obtained in cells when compared with Fluo4-FF (Table 1), because of differential dye interactions in the cell (Maravall et al., 2000). Estimates of resting calcium [Ca2+]0 and resting fluorescence saturation Φ0 can also be obtained using this method (Maravall et al., 2000).
Table 1.
GECI and synthetic indicator properties
|
Indicator |
(ΔF/F)max |
Reported KD (μm) |
Measured KD (μm) |
Reported n |
Measured n |
Stimulus for SNR ~2 |
|---|---|---|---|---|---|---|
| X-Rhod-5F (1) | 2.5 | 1.9 | 1.3 ± 0.5 | 1 | 1.2 ± 0.21 | 1-2 APs |
| Fluo4-FF (1) | 22 | 10.4 | n/a | 1 | n/a | 1 AP |
| GCaMP (2) | 1.8 | 0.24 | 1.7 ± 1 | 3.3 | 3.3 ± 1.2 | 5 APs at 20 Hz |
| Camgaroo2 (3) | ~2 | 5.3 ± 0.3 | 8 ± 1.6 | 1.24 | 1.4 ± 0.42 | 33 APs at 50 Hz |
| Inverse Pericam (4) |
−0.25 |
0.2 |
0.9 ± 1.4 |
1 |
3.8 ± 2.1 |
>20 APs at 30 Hz |
Data are from the present study and (1) Yasuda et al., 2004; (2) Nakai et al., 2001; (3) Griesbeck et al., 2001; and (4) Nagai et al., 2001. The reported values for the Hill coefficient (n) and dissociation constant (KD) are from the literature (references next to indicator name). All other values are from the present study.
The use of two Ca2+ indicators allows the measurement of [Ca2+] with one indicator and the fluorescence saturation, ϕ, of the other. This is useful when the biochemical properties of one indicator are known, while the properties of the other are unknown (see supplemental material, available at www.jneurosci.org). We measure [Ca2+] using the synthetic indicator with known properties and binding stochiometry (using Eq. 1a), whereas ϕ is measured for the unknown indicator. We validated this approach using two synthetic indicators with known properties, treating one as unknown (see Fig. 5 and supplemental material, available at www.jneurosci.org) (Yasuda et al., 2004).
Figure 5.

Comparison of genetically encoded and synthetic Ca2+ indicator fluorescence saturation. Fluorescence saturation curves (Φ vs [Ca2+]; mean ± SEM) were fit (solid lines) to a general Hill model (Eq. 1). A, X-Rhod-5F fluorescence saturation measured simultaneously with [Ca2+] (using Fluo4-FF). The data show excellent agreement with the expected values for a synthetic Ca2+ indicator (Table 1). B-D, GECI fluorescence saturation measured simultaneously with [Ca2+] using X-Rhod-5F (Table 1).
Fluorescence saturation curves (ϕ vs [Ca2+]) (see Fig. 5) were obtained using this dual-indicator method and were fit to a generalized Hill model:
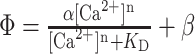 |
2 |
KD is always defined as [Ca2+] when ϕ = 0.5, regardless of n. When n = 1, the Hill model reduces to the familiar hyperbolic binding curve. When n > 1, the binding is non-hyperbolic (sigmoidal). α and β are scaling and nonspecific binding factors, respectively. They can be used to normalize the fluorescence saturation curves to determine the biochemical parameters (KD and n) correctly.
After fitting the fluorescence saturation curves with Equation 2 to determine α, β, KD, and n, the normalized ϕ (ϕ′) is calculated as:
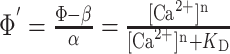 |
3 |
Once ϕ′ is determined, another fit is performed (ϕ′ vs [Ca2+]) to determine the reported biochemical parameters, KD and n (α, β = 0) (Table 1).
Fluorescence transients in response to periodic AP stimuli were analyzed to determine the sensitivity for detecting individual AP fluorescence responses during trains of APs. Fluorescence curves were fit by a fifth-degree polynomial. This best-fit polynomial was subtracted from the original data to remove low-frequency trends. Power spectra were computed from the de-trended fluorescence time series (see Fig. 4).
Figure 4.

Frequency response of genetically encoded and synthetic Ca2+ indicator fluorescence. Power spectra of fluorescence time series (from Fig. 3A-E) were calculated, and the spectra of X-Rhod-5F (A) and GCaMP (B) from a 20 Hz AP train are shown. A, X-Rhod-5F fluorescence power spectrum showing pronounced peaks at the AP train frequency (fundamental; 20 Hz) and its harmonics. B, GCaMP fluorescence power spectrum does not reveal a definite peak, even at the AP train frequency (20 Hz).
Fluorescence recovery after photobleaching. Fluorescence recovery after photobleaching (FRAP) experiments were performed to measure the mobility of GECIs in the cell cytoplasm. Dendritic spines were imaged using 2PLSM at high magnification (field of view, 1.5 × 1.5 μm2) in a line scan across the spine parallel to the parent dendrite (see Fig. 6 A, B). A 338 msec baseline was acquired, followed by a bleach period that lasted 50 msec and a recovery period that lasted for 4 sec. During the bleach period, the laser power was increased 5- to 10-fold using the EOM (the degree of bleaching did not affect the recovery time constant significantly; data not shown). Fluorescence recovery curves were fit with a single exponential with time constant τ (see Fig. 6 D, E). All analysis was done in MATLAB (MathWorks, Natick, MA).
Figure 6.

FRAP reveals that GECIs have mobilities similar to GFP. A, A region of the apical dendrite from a cell transfected with GFP used for FRAP measurement. B, A magnified image of the FRAPed spine (boxed region in A), with the region used for line scan FRAP indicated by a black line. C, Fluorescence across marked region in B showing bleaching (50 msec bleach time) and recovery in raw fluorescence traces. D, Normalized fluorescence across marked region in B before and after photobleaching. The recovery time constant is measured by fitting the fluorescence recovery to a single exponential. E, Cumulative probability distribution of recovery lifetimes (circles) for individual spines from cells expressing GFP, GCaMP, Camgaroo2, and Inverse Pericam. A Kolmogorov-Smirnov pairwise comparison revealed the distributions to be identical (p > 0.3). F, Mean recovery times were identical (ANOVA; p > 0.3) for all GECIs and were similar to GFP. Box plots (black lines) show mean value (central line) as well as 95% confidence intervals (black trapezoids above and below mean line).
Results
GECI responses to APs
To characterize the response of GECIs to various neural stimuli, we imaged proximal apical dendrites (5-9 μm diameter; 13-38 μm from soma) of pyramidal cells in cultured hippocampal slices (Fig. 1). APs were generated in the soma and propagated reliably into proximal apical dendrites (Callaway and Ross, 1995), triggering Ca2+ influx through voltage-sensitive Ca2+ channels (Jaffe et al., 1992; Helmchen et al., 1996; Johnston et al., 1996; Sabatini and Svoboda, 2000; Sabatini et al., 2002; Yasuda et al., 2003).
We compared AP-evoked fluorescence changes produced by GECIs and X-Rhod-5F (Fig. 2A). X-Rhod-5F has a lower affinity for Ca2+ than GCaMP (Nakai et al., 2001) and Inverse Pericam (Nagai et al., 2001) and similar affinity to Camgaroo2 (Griesbeck et al., 2001) (Table 1). Under our experimental conditions, the buffering of Ca2+ by X-Rhod-5F did not significantly perturb GECI fluorescence (supplemental material, available at www.jneurosci.org). In response to a single AP, the synthetic indicator (X-Rhod-5F) produced robust, rapid onset fluorescence changes (Fig. 2A,B) (ΔF/F amplitude, 0.2 ± 0.04; mean ± SD) that decayed with a characteristic time constant (500 ± 85 msec). In contrast, GCaMP and Inverse Pericam produced only very small fluorescence responses (ΔF/F amplitude, 0.05 ± 0.03 and 0.15 ± 0.08, respectively); these were detected above the noise only when averaging over many (8-16) trials (Fig. 2A-C, E). Fluorescence changes produced by Camgaroo2 in response to single APs were not detectable (Fig. 2D). Thus, despite its relatively low affinity for Ca2+, X-Rhod-5F is more sensitive compared with GECIs for detection of single APs.
Figure 2.
Fluorescence responses to APs. A, Single AP-evoked fluorescence transients averaged over 13 trials. The scale bar applies to all traces. Right, Expanded view (boxed region from left) of X-Rhod-5F and GCaMP responses to one AP (line above trace indicates stimulus duration). B-E, Responses to trains of APs for X-Rhod-5F (B), GCaMP (C), Camgaroo2 (D), and Inverse Pericam (E). A scaled copy of the X-Rhod-5F single AP fluorescence response has been overlayed on the single AP response in C-E (gray trace). Each trace is the average of four to eight trials from the same cell.
Next, we measured fluorescence responses to trains of APs. GCaMP began to show clear fluorescence responses to trains >5 APs at 20 Hz (Fig. 2C), and fluorescence changes increased with stimulus strength (i.e., the number of APs and frequency of the train). Camgaroo2 required intense trains (>10 APs at 20 Hz) (Fig. 2D) to produce robust signals. Although Camgaroo2 had the largest dynamic range [(ΔF/F)max] among GECIs (Table 1 and supplemental material, available at www.jneurosci.org), only a fraction (<50%) of this range was accessed by even the most intense trains of APs (Fig. 2D). Inverse Pericam showed some response to low-frequency trains, but response amplitudes changed little with stimulus strength (Fig. 2E), demonstrating that this indicator has a limited dynamic range (Table 1 and supplemental material, available at www.jneurosci.org).
How sensitive are different types of indicators to physiological activity patterns? One would like to know which minimal neural signals could be detected above the noise with a particular indicator. In general, this needs to be determined separately for any experimental situation, because the signal/noise ratio (SNR) depends on the concentration of the indicator, Ca2+ handling in the cell or compartment of interest, the excitation levels, the volume of cytoplasm sampled, limits on illumination intensity imposed by phototoxicity, the noise sources, and so on (Yasuda et al., 2004). However, under the relatively ideal imaging conditions in the cultured brain slice, we can compare the performance of different indicators side by side.
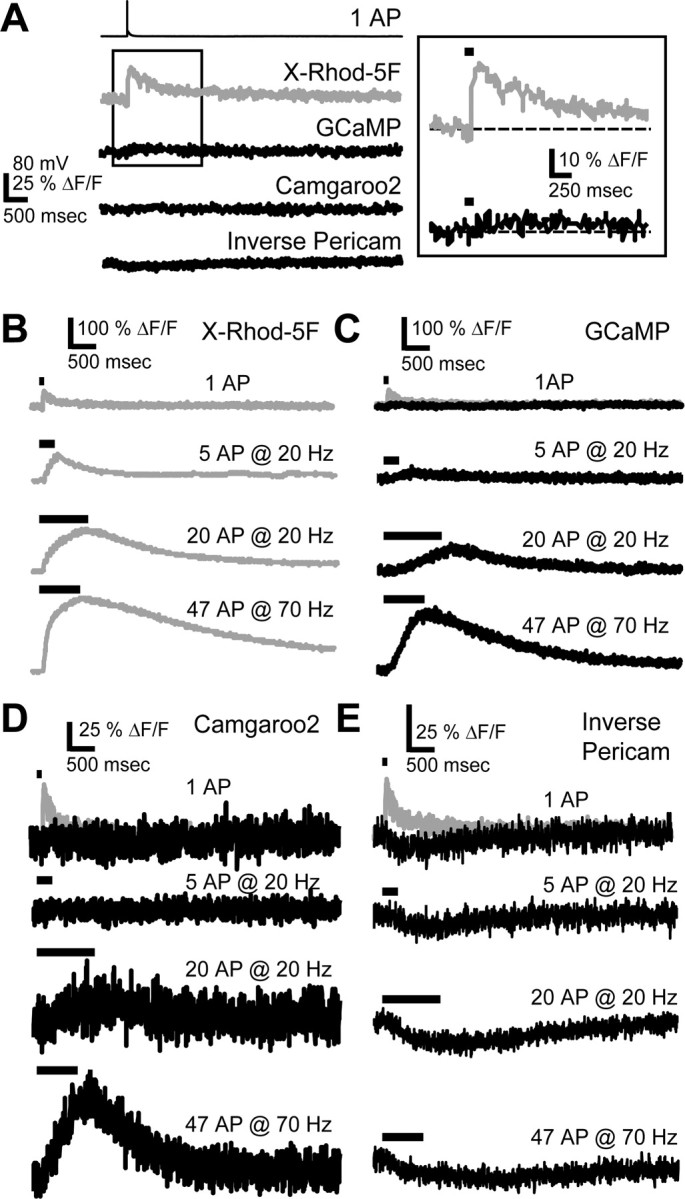
To characterize the sensitivity of the different indicators, we determined which bursts of APs could be detected with a particular SNR (Fig. 3). Fluorescence changes that correspond to SNR = 1, 2, and 3 (Fig. 3A-E, horizontal dashed lines) are translated into AP number and frequency (Fig. 3F-J). These measurements confirm that Fluo4-FF and X-Rhod-5F can detect a single AP with SNR ∼2 (Fig. 3F,G). Much more vigorous stimuli need to be provided to achieve the same SNR with GECIs, e.g., 5 APs at 20 Hz and 33 APs at 50 Hz for GCaMP and Camgaroo2, respectively (Fig. 3H,I). Inverse Pericam had a small SNR even during intense trains (Fig. 3J). In general, GECIs tend to underreport low-frequency activity, including single APs.
Figure 3.
SNRs of fluorescence responses to APs. Data with additional AP trains (frequencies: 20, 30, 50, and 70 Hz) are in supplemental material (available at www.jneurosci.org). A-E, Fluorescence responses to trains of APs delivered at 20 and 70 Hz [black trace is the mean response, colored traces indicate the SEM, and dashed horizontal lines depict different SNRs; 1, blue; 2, green; 3, red]. The SNR is the ratio of ΔF/F to the SD, σ, of ΔF/F from the background fluorescence. The red (A; X-Rhod-5F; N = 8 cells) and green (B; Fluo4-FF; N = 8 cells) synthetic Ca2+ indicators respond to all stimuli, including single APs. GCaMP (C; N = 13 cells), Camgaroo2 (D; N = 6 cells), and Inverse Pericam (E; N = 9 cells) respond poorly (sublinear) to low-frequency AP trains and are supralinear for stronger stimuli. F-J, Left, Activity (AP number and frequency) necessary to elicit a given SNR (closed circles; same colors for SNRs as in A-E). If the indicator never reached a certain SNR, the value was set to the maximum number of APs delivered (open circles). The maximum number of APs delivered was as follows: 20 APs for 20 and 30 Hz, 33 APs for 50 Hz, and 47 APs for 70 Hz. Single APs can yield SNR ∼2 for synthetic Ca2+ indicators (F, G). GCaMP (H), Camgaroo2 (I), and Inverse Pericam (J) require stronger stimuli to obtain the same SNR as synthetic indicators. F-J, Right, Individual trials from single cells (colored lines; mean response in black; smoothed with a 50 msec averaging filter) for a stimulus with SNR ∼2.
Synthetic indicators respond rapidly (<1 msec) to elevations in [Ca2+] (Sabatini and Regehr, 1998; Sabatini and Svoboda, 2000), producing stepwise changes in indicator fluorescence with each AP (Fig. 3A,B). Therefore, fluorescence changes can be used to detect APs. Under some conditions, this strategy may allow optical detection of temporal patterns of spiking activity in neural networks (Smetters et al., 1999). To determine whether GECIs can detect rapid fluorescence changes, we performed frequency domain analysis (i.e., calculated power spectra) of fluorescence responses to periodic trains of APs (Fig. 4) (see Materials and Methods). The power spectrum measures the power of the signal contained at a particular frequency. If the indicator can follow the periodic stimulus, the spectrum should show a large peak at the stimulus frequency. For both X-Rhod-5F (Fig. 4A) and Fluo4-FF (data not shown), the dominant peak of the power spectrum corresponded to the stimulus or fundamental frequency (Fig. 4A, arrow) for even the largest stimulus frequency used (70 Hz; data not shown). Thus, both X-Rhod-5F and Fluo4-FF respond to Ca2+ elevations sufficiently quickly to follow the stimulus patterns reliably. In contrast, GECI power spectra did not reveal a clear peak above the noise at the stimulus frequency, even under the most favorable conditions (20 Hz) (Fig. 4B, GCaMP). Thus, unlike synthetic indicators, GECIs respond too slowly to follow individual APs within a burst.
GECI responses to [Ca2+]
To determine whether GECIs can be used as quantitative indicators of [Ca2+], we measured the relationship between GECI fluorescence and [Ca2+]. To monitor [Ca2+], we calibrated X-Rhod-5F fluorescence (see Materials and Methods and supplemental material, available at www.jneurosci.org). We could then simultaneously measure [Ca2+] (red) and GECI fluorescence saturation, ϕ (green).
We verified our methods using two synthetic indicators with known properties (Fig. 5A) (supplemental material, available at www.jneurosci.org). The fluorescence saturation curves (Fig. 5, ϕ vs [Ca2+]) can be fit using a Hill equation (Eq. 2; see Materials and Methods) and the Hill coefficient, n, and effective dissociation constant, KD, calculated from the fit (Fig. 5, solid lines) (see Materials and Methods). For X-Rhod-5F, we found KD = 1.3 ± 0.5 μm (Fig. 5A) (r2 = 0.978) and n = 1.2 ± 0.21, in agreement with in vitro calibrations (Yasuda et al., 2004). GCaMP showed a sharp transition (n = 3.3 ± 1.2; r2 = 0.981) with KD = 1.7 ± 1 μm (Fig. 5B). Camgaroo2 had a more gradual curve (n = 1.4 ± 0.42) with KD = 8 ± 1.6 μm (Fig. 5C), with an exaggerated nonresponsive regime for small Δ[Ca2+]. Like GCaMP, Inverse Pericam showed a sharp transition (Fig. 5D) (n = 3.8 ± 2.1; r2 = 0.984), with KD = 0.9 ± 1.4 μm. Thus, GECIs have idiosyncratic and complex fluorescence saturation curves, making their use for quantitative [Ca2+] imaging problematic (see Discussion and Table 1).
GECIs are diffusible in the cytoplasm
Numerous CaM-binding proteins exist in neurons, many of which are not diffusible (Saimi and Kung, 2002). Do GECIs interact with endogenous CaM-binding proteins? Because CaM (and hence GECI) properties are changed by interactions with CaM-binding proteins (Peersen et al., 1997), assessing GECI mobility is important for the interpretation of GECI signals. To address this issue, we measured FRAP in individual dendritic spines (Svoboda et al., 1996; Star et al., 2002) of neurons expressing GECIs or GFP. In all cases, after bleaching, fluorescence recovered to >95% of the baseline fluorescence. In addition, fluorescence recovery time constants for GCaMP (τ = 328 ± 78 msec; N = 3 cells; n = 18 spines), Inverse Pericam (τ = 294 ± 26 msec; N = 5 cells; n = 32 spines), Camgaroo2 (τ = 566 ± 120 msec; N = 6 cells; n = 40 spines), and GFP (τ = 375 ± 79 msec; N = 5 cells; n = 40 spines) were similar. Comparing the distribution of recovery time constants between groups did not reveal any differences (Fig. 6 E, F) (p > 0.3; Kolmogorov-Smirnov and ANOVA pairwise comparisons, respectively). We conclude that GECIs are mostly freely diffusible in the spine cytoplasm (but see Hasan et al., 2004).
Discussion
GECIs based on XFPs hold great promise in cellular and neuroscience systems (Miyawaki et al., 1997, 1999; Baird et al., 1999; Griesbeck et al., 2001; Nagai et al., 2001). We have analyzed the relationship of GCaMP, Camgaroo2, and Inverse Pericam fluorescence to neural activity and cytoplasmic-free Ca2+ concentration, [Ca2+]. We found that all GECIs produce robust signals to high-frequency trains of APs but have poor sensitivity at low firing rates. The relationship of GECI fluorescence with [Ca2+] is complex, with supralinear and sublinear regimes, and depends on the particular indicator. GECIs also do not associate considerably with immobile CaM-binding proteins in the spine cytoplasm.
GECIs as indicators of neural activity
GECIs have great potential as optical sensors of neural activity. Ideal neural activity sensors would be bright and respond linearly to a large range of stimuli (in terms of AP number and frequency). They should also rapidly follow [Ca2+] transients, allowing detection of individual APs. We show that the GECIs studied here fall short of ideal in several ways. First, they have relatively less brightness compared with XFPs. We estimate that GCaMP, Camgaroo2, and Inverse Pericam are ∼25, ∼10, and ∼2 times dimmer than GFP at resting [Ca2+] under comparable conditions of expression in cultured brain slices. Second, GECIs have complex responses to trains of APs, including sublinear and supralinear regimes. At low frequencies, they underreport neural activity compared with higher frequencies (Figs. 2-3). Third, the dynamic ranges and Ca2+ sensitivities are also not optimal for measurements of activity. Saturation of the response is a major problem for Inverse Pericam, whereas Camgaroo2 was far from saturation with even the strongest stimuli used (Fig. 3). The affinity and dynamic range of GCaMP make it the best-suited indicator for detecting spiking activity in pyramidal neurons (Figs. 2-3). Fourth, GECIs fail to report the pattern of APs within bursts, possibly because of their slow kinetics (Fig. 4) (Baird et al., 1999; Griesbeck et al., 2001). Elegant studies in a number of systems have shown that GECIs are already useful for spatial mapping of neural activity (Hasan et al., 2004; Wang et al., 2004), even at the single-cell level (Wang, 2004). However, the supralinearity of GCaMP at low to moderate activity levels could lead to distortions in spatial activity maps, which might contribute to recent controversies (Wang et al., 2003; Wilson et al., 2004).
GECIs as [Ca2+] sensors
GECIs also have great potential to monitor [Ca2+] in targeted compartments. Ideal Ca2+ indicators share most features with ideal reporters of neural activity. In addition, it is necessary to calibrate sensors reliably in terms of [Ca2+] (Grynkiewicz et al., 1985; Kao et al., 1989; Helmchen et al., 1996; Maravall et al., 2000). The nonlinear binding curves will make GECIs challenging to use for quantitative Ca2+ imaging (Fig. 5). This is compounded by their low sensitivity to small, rapid changes in [Ca2+] (Fig. 5). GECIs will find a key niche in allowing selective measurements in particular subcellular compartments (Robert et al., 2001; Demaurex and Frieden, 2003).
The biochemical data acquired here in neurons agrees qualitatively with previous in vitro calibrations (Table 1) (Griesbeck et al., 2001; Nagai et al., 2001; Nakai et al., 2001; Yasuda et al., 2004). One striking discrepancy is between the measured and reported (Nakai et al., 2001) KD of GCaMP for Ca2+ (Table 1). This discrepancy is not likely attributable to an error in our calibration of X-Rhod-5F. The properties of AP-evoked [Ca2+] dynamics in the proximal apical dendrites of CA1 pyramidal neurons are well known (Helmchen et al., 1996; Maravall et al., 2000). We can therefore model [Ca2+] dynamics in dendrites as well as the expected GCaMP fluorescence saturation (supplemental material, available at www.jneurosci.org) independent of X-Rhod-5F fluorescence measurements. If the reported KD of GCaMP applied to our experimental situation, the indicator should saturate in response to a brief train of APs (approximately three APs; 30 Hz) (supplemental material, available at www.jneurosci.org), clearly inconsistent with our data (Fig. 3). Instead, the KD determined here (Table 1) predicts 50% saturation at ∼20 APs at 30 Hz (supplemental material, available at www.jneurosci.org), consistent with our measurements (Fig. 3, Table 1). The discrepancy between the KD determined in vitro and in the cell cytoplasm may arise from differences in the biochemical milieu.
GECIs as CaM activation sensors
Our diffusion measurements suggest that GECIs have mobilities similar to GFP and do not have a significant immobile fraction in spines (Fig. 6). Because CaM-binding sites are likely to be immobile [e.g., fixed to a large protein complex, like an ion channel (Saimi and Kung, 2002)], this implies that the CaM domains on the GECIs do not interact substantially with immobile CaM-binding proteins. Our results differ from those of Hasan et al. (2004). It is possible that they measured FRAP in neuronal regions overlapping with biosynthetic compartments, perhaps explaining the immobile fraction in their data (Hasan et al., 2004).
Our data suggest that GECI fluorescence changes reflect the intramolecular dynamics of the sensor. What do GECI fluorescence changes report? We compared our measurements with biochemical measurements of the interactions between Ca2+ and CaM. We wondered whether Ca2+ saturation of CaM in the presence and absence of M13 (Falke et al., 1994; Peersen et al., 1997; Mirzoeva et al., 1999) could correspond with the fluorescence saturation curves of GCaMP and Camgaroo2, respectively (Table 1) (free-CaM: KD = 15 μm, n = 1.2; CaM-M13: KD = 1.25 μm, n = 1.7). Although the curves are qualitatively similar, the steepness of the GCaMP fluorescence saturation curve exceeds what would be expected based on the cooperative binding of Ca2+ to CaM in the presence of M13 (Table 1).
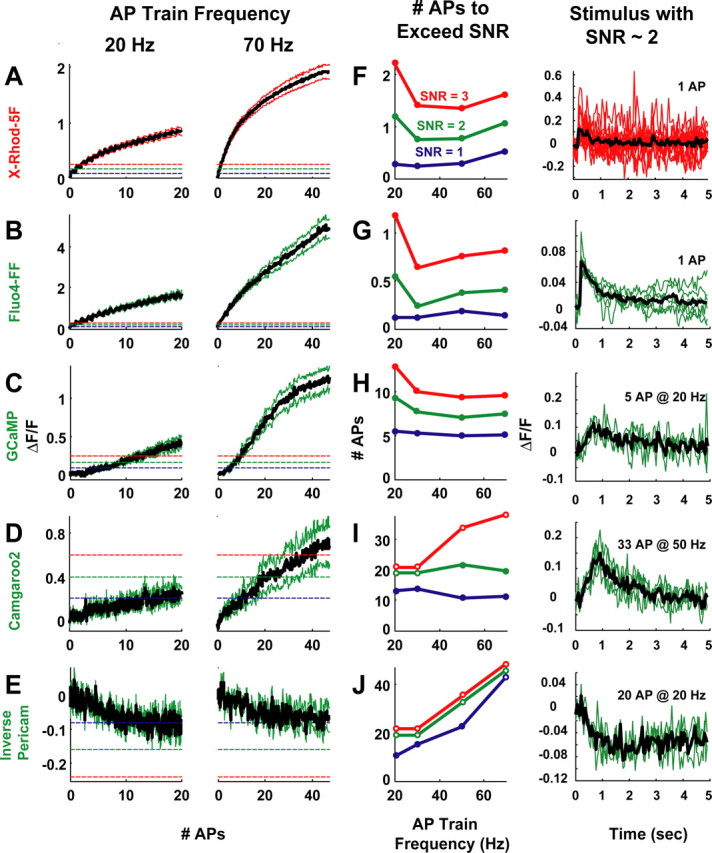
Because the coupling of fluorescence to Ca2+ in GECIs must have a structural basis, we explored whether the fluorescence saturation of GECIs reflects the Ca2+-induced structural transitions of CaM in the presence (GCaMP) and absence (Camgaroo2) of the M13-binding peptide. X-ray scattering can measure the maximum size and radius of gyration of a macromolecular complex (Trewhella, 1997) in solution. X-ray scattering studies of CaM loaded with different amounts of Ca2+ (0, 1, 2, 3, and 4 Ca2+ ions per CaM) in the presence of M13 peptide reveals an abrupt collapse of the size of the CaM-M13 complex as the molecule exceeds 50% (2 Ca2+) saturation (Fig. 7A) (Krueger et al., 1998). This is similar to the steep transition observed for GCaMP. In contrast, free CaM increases in size gradually as Ca2+ ions bind (Fig. 7B) (Trewhella, 1992; Krueger et al., 1998; Komeiji et al., 2002), similar to the behavior of Camgaroo2.
Figure 7.
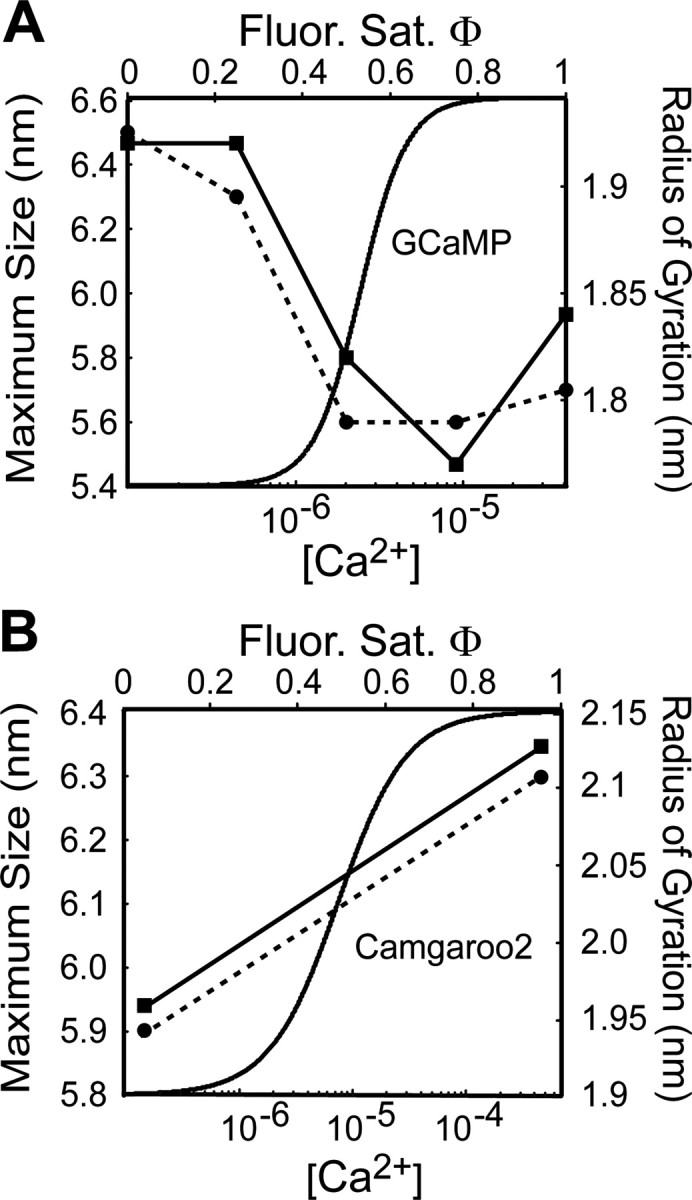
GECI fluorescence changes are coupled to CaM structural transitions. A, Structural transitions of CaM associated with M13 peptide derived from x-ray scattering studies. The abrupt change in fluorescence saturation is coupled to the structural collapse [i.e., decrease in both the maximum size (solid line) and radius of gyration (dashed line); see Discussion for details] of the M13-CaM complex after Ca2+ loading. GCaMP fluorescence is plotted for comparison. Structural data (filled circles and squares) is from the study by Krueger et al. (1998). B, Structural transitions of free-CaM. The occupation of Ca2+-binding sites adds stability to the CaM structure, causing reorientation of the N- and C-terminal domains and increases α-helical content of central helix (Sun et al., 1999). This will cause an elongation of the free-CaM molecule after Ca2+ loading (increase in both the maximum size and radius of gyration; see Discussion for details). Camgaroo2 fluorescence is plotted for comparison. Structural data (filled circles and squares) is from study by Krueger et al. (1998). The solid line (maximum size) and dashed line (radius of gyration) are linear interpolations between the points reflecting the stepwise elongation of free-CaM after Ca2+ loading.
These similarities suggest that GCaMP and Camgaroo2 report CaM activation in the presence and absence of M13 peptide, respectively. Many CaM-dependent enzymes, including calcium/calmodulin-dependent protein kinase II (CaMKII), use relief-of-autoinhibition (ROA) by CaM binding to become active. The CaMKII ROA is a structural phenomenon, involving dislocation of the pseudo-substrate region from the substrate-binding site after Ca2+ binding to CaM (Lisman et al., 2002). So an ideal reporter of the activation of CaM-dependent enzymes would read out the structural transitions that underlie ROA in the same compartment as the enzyme. GCaMP behaves like a monitor of CaM acting in ROA. It couples the binding of CaM to a target peptide (M13) with Ca2+ binding, changing its fluorescence as the complex condenses (Fig. 7A). In contrast, Camgaroo2 behaves as a monitor of free-CaM loading with Ca2+ (Fig. 7B). This suggests that GCaMP and Camgaroo2, or variants of these indicators, could be used to explore the physiological signals that are optimized for CaM activation in situ.
Footnotes
This work was supported by the National Institutes of Health and the National Institute of Mental Health. We thank Catherine Zhang and Volker Scheuss for help with experiments; Yuto Komeiji, Madeline Shea, and Hans Vogel for discussions; and Volker Scheuss, Aleksander Sobczyk, Haining Zhong, and Karen Zito for a critical reading of this manuscript.
Correspondence should be addressed to Dr. Karel Svoboda, Howard Hughes Medical Institute/Cold Spring Harbor Laboratory, 1 Bungtown Road, Cold Spring Harbor, NY 11724. E-mail: svoboda@cshl.edu.
Copyright © 2004 Society for Neuroscience 0270-6474/04/249572-08$15.00/0
References
- Baird GS, Zacharias DA, Tsien RY (1999) Circular permutation and receptor insertion within green fluorescent proteins. Proc Natl Acad Sci USA 96: 11241-11246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berridge MJ (1998) Neuronal calcium signaling. Neuron 21: 13-26. [DOI] [PubMed] [Google Scholar]
- Callaway JC, Ross WN (1995) Frequency-dependent propagation of sodium action potentials in dendrites of hippocampal CA1 pyramidal neurons. J Neurophysiol 74: 1395-1403. [DOI] [PubMed] [Google Scholar]
- Demaurex N, Frieden M (2003) Measurements of the free luminal ER Ca(2+) concentration with targeted “cameleon” fluorescent proteins. Cell Calcium 34: 109-119. [DOI] [PubMed] [Google Scholar]
- Evenas J, Malmendal A, Thulin E, Carlstrom G, Forsen S (1998) Ca2+ binding and conformational changes in a calmodulin domain. Biochemistry 37: 13744-13754. [DOI] [PubMed] [Google Scholar]
- Falke JJ, Drake SK, Hazard AL, Peersen OB (1994) Molecular tuning of ion binding to calcium signaling proteins. Q Rev Biophys 27: 219-290. [DOI] [PubMed] [Google Scholar]
- Fiala A, Spall T, Diegelmann S, Eisermann B, Sachse S, Devaud JM, Buchner E, Galizia CG (2002) Genetically expressed cameleon in Drosophila melanogaster is used to visualize olfactory information in projection neurons. Curr Biol 12: 1877-1884. [DOI] [PubMed] [Google Scholar]
- Griesbeck O, Baird GS, Campbell RE, Zacharias DA, Tsien RY (2001) Reducing the environmental sensitivity of yellow fluorescent protein. Mechanism and applications. J Biol Chem 276: 29188-29194. [DOI] [PubMed] [Google Scholar]
- Grynkiewicz G, Poenie M, Tsien RY (1985) A new generation of Ca2+ indicators with greatly improved fluorescence properties. J Biol Chem 260: 3440-3450. [PubMed] [Google Scholar]
- Hasan MT, Friedrich RW, Euler T, Larkum ME, Giese G, Both M, Duebel J, Waters J, Bujard H, Griesbeck O, Tsien RY, Nagai T, Miyawaki A, Denk W (2004) Functional fluorescent Ca2+ indicator proteins in transgenic mice under TET control. PLoS Biol 2: 1-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Helmchen F, Imoto K, Sakmann B (1996) Ca2+ buffering and action potential-evoked Ca2+ signaling in dendrites of pyramidal neurons. Biophys J 70: 1069-1081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Higashijima S, Masino MA, Mandel G, Fetcho JR (2003) Imaging neuronal activity during zebrafish behavior with a genetically encoded calcium indicator. J Neurophysiol 90: 3986-3997. [DOI] [PubMed] [Google Scholar]
- Jaffe DB, Johnston D, Lasser RN, Lisman JE, Miyakawa H, Ross WN (1992) The spread of Na+ spikes determines the pattern of dendritic Ca2+ entry into hippocampal neurons. Nature 357: 244-246. [DOI] [PubMed] [Google Scholar]
- Ji G, Feldman ME, Deng KY, Greene KS, Wilson J, Lee JC, Johnston RC, Rishniw M, Tallini Y, Zhang J, Wier WG, Blaustein MP, Xin HB, Nakai J, Kotlikoff MI (2004) Ca2+-sensing transgenic mice: postsynaptic signaling in smooth muscle. J Biol Chem 279: 21461-21468. [DOI] [PubMed] [Google Scholar]
- Johnston D, Magee JC, Colbert CM, Christie BR (1996) Active properties of neuronal dendrites. Annu Rev Neurosci 19: 165-186. [DOI] [PubMed] [Google Scholar]
- Kao JP, Harootunian AT, Tsien RY (1989) Photochemically generated cytosolic calcium pulses and their detection by fluo-3. J Biol Chem 264: 8179-8184. [PubMed] [Google Scholar]
- Kerr R, Lev-Ram V, Baird G, Vincent P, Tsien RY, Schafer WR (2000) Optical imaging of calcium transients in neurons and pharyngeal muscle of C. elegans Neuron 26: 583-594. [DOI] [PubMed] [Google Scholar]
- Komeiji Y, Ueno Y, Uebayasi M (2002) Molecular dynamics simulations revealed Ca(2+)-dependent conformational change of calmodulin. FEBS Lett 521: 133-139. [DOI] [PubMed] [Google Scholar]
- Krueger JK, Bishop NA, Blumenthal DK, Zhi G, Beckingham K, Stull JT, Trewhella J (1998) Calmodulin binding to myosin light chain kinase begins at substoichiometric Ca2+ concentrations: a small-angle scattering study of binding and conformational transitions. Biochemistry 37: 17810-17817. [DOI] [PubMed] [Google Scholar]
- Lisman J, Schulman H, Cline H (2002) The molecular basis of CaMKII function in synaptic and behavioural memory. Nat Rev Neurosci 3: 175-190. [DOI] [PubMed] [Google Scholar]
- Mainen ZF, Maletic-Savatic M, Shi SH, Hayashi Y, Malinow R, Svoboda K (1999) Two-photon imaging in living brain slices. Methods 18: 231-239. [DOI] [PubMed] [Google Scholar]
- Maravall M, Mainen ZF, Sabatini BL, Svoboda K (2000) Estimating intracellular calcium concentrations and buffering without wavelength ratioing. Biophys J 78: 2655-2667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McAllister AK, Stevens CF (2000) Nonsaturation of AMPA and NMDA receptors at hippocampal synapses. Proc Natl Acad Sci USA 97: 6173-6178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mirzoeva S, Weigand S, Lukas TJ, Shuvalova L, Anderson WF, Watterson DM (1999) Analysis of the functional coupling between calmodulin's calcium binding and peptide recognition properties. Biochemistry 38: 3936-3947. [DOI] [PubMed] [Google Scholar]
- Miyawaki A, Llopis J, Heim R, McCaffery JM, Adams JA, Ikura M, Tsien RY (1997) Fluorescence indicators for Ca2+ based on green fluorescent proteins and calmodulin. Nature 388: 882-887. [DOI] [PubMed] [Google Scholar]
- Miyawaki A, Griesbeck O, Heim R, Tsien RY (1999) Dynamic and quantitative Ca2+ measurements using improved cameleons. Proc Natl Acad Sci USA 96: 2135-2140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muller W, Connor JA (1991) Dendritic spines as individual neuronal compartments for synaptic Ca2+ responses. Nature 354: 73-76. [DOI] [PubMed] [Google Scholar]
- Nagai T, Sawano A, Park ES, Miyawaki A (2001) Circularly permuted green fluorescent proteins engineered to sense Ca2+ Proc Natl Acad Sci USA 98: 3197-3202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakai J, Ohkura M, Imoto K (2001) A high signal-to-noise Ca(2+) probe composed of a single green fluorescent protein. Nat Biotechnol 19: 137-141. [DOI] [PubMed] [Google Scholar]
- Nimchinsky EA, Yasuda R, Oertner TG, Svoboda K (2004) The number of glutamate receptors opened by synaptic stimulation in single hippocampal spines. J Neurosci 24: 2054-2064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Donovan MJ, Ho S, Sholomenko G, Yee W (1993) Real-time imaging of neurons retrogradely and anterogradely labelled with calcium-sensitive dyes. J Neurosci Methods 46: 91-106. [DOI] [PubMed] [Google Scholar]
- Oertner TG, Sabatini BS, Nimchinsky EA, Svoboda K (2002) Facilitation at single synapses probed with optical quantal analysis. Nat Neurosci 5: 657-664. [DOI] [PubMed] [Google Scholar]
- O'Malley DM, Kao Y-H, Fetcho JR (1996) Imaging the functional organization of zebrafish hindbrain segments during escape behaviors. Neuron 17: 11145-11155. [DOI] [PubMed] [Google Scholar]
- Peersen OB, Madsen TS, Falke JJ (1997) Intermolecular tuning of calmodulin by target peptides and proteins: differential effects on Ca2+ binding and implications for kinase activation. Protein Sci 6: 794-807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pologruto TA, Sabatini BL, Svoboda K (2003) ScanImage: flexible software for operating laser-scanning microscopes. BioMed Eng Online 2: 13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reiff DF, Thiel PR, Schuster CM (2002) Differential regulation of active zone density during long-term strengthening of Drosophila neuromuscular junctions. J Neurosci 22: 9399-9409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robert V, Gurlini P, Tosello V, Nagai T, Miyawaki A, Di Lisa F, Pozzan T (2001) Beat-to-beat oscillations of mitochondrial [Ca2+] in cardiac cells. EMBO J 20: 4998-5007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sabatini BL, Regehr WG (1998) Optical measurement of presynaptic calcium currents. Biophys J 74: 1549-1563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sabatini BL, Svoboda K (2000) Analysis of calcium channels in single spines using optical fluctuation analysis. Nature 408: 589-593. [DOI] [PubMed] [Google Scholar]
- Sabatini BS, Oertner TG, Svoboda K (2002) The life-cycle of Ca2+ ions in spines. Neuron 33: 439-452. [DOI] [PubMed] [Google Scholar]
- Saimi Y, Kung C (2002) Calmodulin as an ion channel subunit. Annu Rev Physiol 64: 289-311. [DOI] [PubMed] [Google Scholar]
- Smetters D, Majewska A, Yuste R (1999) Detecting action potentials in neuronal populations with calcium imaging. Methods 18: 215-221. [DOI] [PubMed] [Google Scholar]
- Star EN, Kwiatkowski DJ, Murthy VN (2002) Rapid turnover of actin in dendritic spines and its regulation by activity. Nat Neurosci 5: 239-246. [DOI] [PubMed] [Google Scholar]
- Stoppini L, Buchs PA, Muller DA (1991) A simple method for organotypic cultures of nervous tissue. J Neurosci Methods 37: 173-182. [DOI] [PubMed] [Google Scholar]
- Stosiek C, Garaschuk O, Holthoff K, Konnerth A (2003) In vivo two-photon calcium imaging of neuronal networks. Proc Natl Acad Sci USA 100: 7319-7324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun H, Yin D, Squier TC (1999) Calcium-dependent structural coupling between opposing globular domains of calmodulin involves the central helix. Biochemistry 38: 12266-12279. [DOI] [PubMed] [Google Scholar]
- Suzuki H, Kerr R, Bianchi L, Frokjaer-Jensen C, Slone D, Xue J, Gerstbrein B, Driscoll M, Schafer WR (2003) In vivo imaging of C. elegans mechanosensory neurons demonstrates a specific role for the MEC-4 channel in the process of gentle touch sensation. Neuron 39: 1005-1017. [DOI] [PubMed] [Google Scholar]
- Svoboda K, Tank DW, Denk W (1996) Direct measurement of coupling between dendritic spines and shafts. Science 272: 716-719. [DOI] [PubMed] [Google Scholar]
- Svoboda K, Denk W, Kleinfeld D, Tank DW (1997) In vivo dendritic calcium dynamics in neocortical pyramidal neurons. Nature 385: 161-165. [DOI] [PubMed] [Google Scholar]
- Tank DW, Sugimori M, Connor JA, Llinas RR (1988) Spatially resolved calcium dynamics of mammalian Purkinje cells in cerebellar slice. Science 242: 773-777. [DOI] [PubMed] [Google Scholar]
- Trewhella J (1992) The solution structures of calmodulin and its complexes with synthetic peptides based on target enzyme binding domains. Cell Calcium 13: 377-390. [DOI] [PubMed] [Google Scholar]
- Trewhella J (1997) Insights into biomolecular function from small-angle scattering. Curr Opin Struct Biol 7: 702-708. [DOI] [PubMed] [Google Scholar]
- Tsien RY, Pozzan T, Rink TJ (1982) Calcium homeostasis in intact lymphocytes: cytoplasmic free calcium monitored with a new, intracellularly trapped fluorescent indicator. J Cell Biol 94: 325-334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang JW, Wong AM, Flores J, Vosshall LB, Axel R (2003) Two-photon calcium imaging reveals an odor-evoked map of activity in the fly brain. Cell 112: 271-282. [DOI] [PubMed] [Google Scholar]
- Wang Y, Guo H, Pologruto TA, Hannan F, Hakker I, Svoboda K, Zhong Y (2004) Stereotyped odor-evoked activity in the mushroom body of Drosophila revealed by GFP-based Ca2+ imaging. J Neurosci 24: 6507-6514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson RI, Turner GC, Laurent G (2004) Transformation of olfactory representations in the Drosophila antennal lobe. Science 303: 366-370. [DOI] [PubMed] [Google Scholar]
- Yasuda R, Sabatini BL, Svoboda K (2003) Plasticity of calcium channels in dendritic spines. Nat Neurosci 6: 948-955. [DOI] [PubMed] [Google Scholar]
- Yasuda R, Nimchinsky EA, Scheuss V, Pologruto TA, Oertner TG, Sabatini BL, Svoboda K (2004) Imaging calcium concentration dynamics in small neuronal compartments. Sci STKE 2004: pl5. [DOI] [PubMed] [Google Scholar]
- Yu D, Baird GS, Tsien RY, Davis RL (2003) Detection of calcium transients in Drosophila mushroom body neurons with camgaroo reporters. J Neurosci 23: 64-72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuste R, Denk W (1995) Dendritic spines as basic functional units of neuronal integration. Nature 375: 682-684. [DOI] [PubMed] [Google Scholar]
- Yuste R, Peinado A, Katz LC (1992) Neuronal domains in developing neocortex. Science 257: 665-669. [DOI] [PubMed] [Google Scholar]