

Abstract
Interactions between NMDA receptors (NMDARs) and the PDZ [postsynaptic density-95 (PSD-95)/Discs large/zona occludens-1] domains of PSD-95/SAP90 (synapse-associated protein with a molecular weight of 90 kDa) family proteins play important roles in the synaptic targeting and signaling of NMDARs. However, little is known about the mechanisms that regulate these PDZ domain-mediated interactions. Here we show that casein kinase II (CK2) phosphorylates the serine residue (Ser1480) within the C-terminal PDZ ligand (IESDV) of the NR2B subunit of NMDAR in vitro and in vivo. Phosphorylation of Ser1480 disrupts the interaction of NR2B with the PDZ domains of PSD-95 and SAP102 and decreases surface NR2B expression in neurons. Interestingly, activity of the NMDAR and Ca2+/calmodulin-dependent protein kinase II regulates CK2 phosphorylation of Ser1480. Furthermore, CK2 colocalizes with NR1 and PSD-95 at synaptic sites. These results indicate that activity-dependent CK2 phosphorylation of the NR2B PDZ ligand regulates the interaction of NMDAR with PSD-95/SAP90 family proteins as well as surface NMDAR expression and may be a critical mechanism for modulating excitatory synaptic function and plasticity.
Keywords: NR2B, PSD-95, CK2, PDZ ligand, phosphorylation, trafficking
Introduction
The NMDA-type ionotropic glutamate receptors (NMDARs) play critical roles in neuronal development, excitotoxicity, and synaptic plasticity (Bliss and Collingridge, 1993; Seeburg, 1993; Choi, 1994; Hollmann and Heinemann, 1994). Functional NMDARs are heteromultimers mainly consisting of NR1 and NR2 (NR2A-NR2D) subunits (Wenthold et al., 2003). The NR2 subunits determine synaptic localization and function of NMDARs because deletion of the C-terminal tail of NR2 perturbs synaptic NMDAR localization, impairs NMDAR-mediated synaptic activity, and alters synaptic plasticity (Mori et al., 1998; Sprengel et al., 1998). The C-terminal T/SXV motifs (where X represents any amino acid) of NR2A and NR2B can directly interact with the PDZ [postsynaptic density-95 (PSD-95)/Discs large/zona occludens-1] domains of PSD-95, SAP102 (synapse-associated protein with a molecular weight of 102 kDa), and PSD-93, and such interactions induce clustering of NMDARs in transfected heterologous cells (Kornau et al., 1995; Kim et al., 1996; Lau et al., 1996; Muller et al., 1996; Niethammer et al., 1996). PSD-95 inhibits NR2B-mediated internalization and enhances surface NMDAR clustering (Roche et al., 2001), whereas targeted disruption of the PSD-93 gene reduces surface NR2A and NR2B expression and the NMDAR-mediated EPSC (Tao et al., 2003). However, NMDAR clusters are recruited to new synapses without PSD-95 (Washbourne et al., 2002), and disruption of PSD-95 clustering had no effect on synaptic localization of NMDARs (Passafaro et al., 1999). Similarly, PSD-95 mutant mice exhibit defects in synaptic plasticity and spatial learning despite the proper synaptic localization of NMDARs (Migaud et al., 1998). These results suggest that PDZ domain-mediated interactions between NMDARs and PSD-95/SAP90 family proteins are not required for synaptic targeting of NMDARs. Instead, these interactions are important for stabilizing and/or promoting surface NMDAR expression and linking NMDAR to cytoplasmic signaling pathways.
Although our understanding of the functional roles of PDZ domain-mediated interactions between NMDARs and PSD-95/SAP90 family proteins has increased, little is known about the mechanisms that dynamically regulate these interactions. For the AMPA receptor (AMPAR), PKC phosphorylation of the C-terminal PDZ ligand of the AMPA receptor subunit (GluR2) disrupts its interaction with the PDZ domains of glutamate receptor interacting protein (GRIP) but not PICK1 (protein interacting with C kinase-1) (Matsuda et al., 1999; Chung et al., 2000) and regulates AMPAR trafficking (Chung et al., 2000; Perez et al., 2001; Braithwaite et al., 2002) and synaptic plasticity (Daw et al., 2000; Xia et al., 2000; Kim et al., 2001; Chung et al., 2003; Seidenman et al., 2003). Moreover, protein kinase A (PKA) phosphorylation of the C-terminal PDZ ligand (T/SXV motif) of the inward rectifier potassium channel Kir2.3 disrupts its interaction with PSD-95 (Cohen et al., 1996). We hypothesized that phosphorylation of the C-terminal T/SXV motif of NR2B may regulate the interactions between NMDARs and PSD-95/SAP90 family proteins. We found that casein kinase II (CK2) directly phosphorylates the -2 serine residue (Ser1480) within the C-terminal T/SXV motif of NR2B. Such phosphorylation disrupts the interaction of NMDARs with the PDZ domains of PSD-95 and SAP102 and reduces surface NMDAR expression in neurons. Furthermore, NMDAR activity regulates CK2 phosphorylation of Ser1480 partially via Ca2+/calmodulin-dependent protein kinase II (CaMKII). These results indicate that activity-dependent phosphorylation of the PDZ ligand may be a critical regulator of NMDAR function and synaptic plasticity.
Materials and Methods
Generation and characterization of anti-NR2B-pS1480 antibodies. Anti-NR2B-pS1480 antibody was raised against the synthetic peptide KFNGSSNGHVYEKLSSIESDV corresponding to amino acids 1463-1482 of NR2B with phosphoserine included at Ser1480. Anti-NR2B-pS1480 antibodies were affinity purified from sera by sequential chromatography on Affi-Gel (Bio-Rad, Hercules, CA) columns covalently linked to BSA-conjugated unphosphorylated and Ser1480-phosphorylated NR2B peptides. Antibody characterization was performed on the immunoblots of rat hippocampal P2 membranes and human embryonic kidney 293T (HEK293T) cell crude membrane fractions. HEK293T cells were transfected with pRK5, pGW1-NR2B (Kim et al., 1996), or pGW1-NR2BS1480A using calcium phosphate coprecipitation (Chung et al., 2000). Rat hippocampal P2 membranes were prepared as described previously (Luo et al., 1997). Crude membrane fractions of HEK293T cells were isolated as described previously (Chung et al., 2000). Protein samples were loaded onto an SDS-PAGE gel, transferred to polyvinylidene difluoride (PVDF) membranes (Immobilon-P membrane; Millipore, Bedford, MA), and analyzed by immunoblotting with anti-NR2B-pS1480 and anti-NR2B C-terminal antibodies. To test the specificity, anti-NR2B-pS1480 antibodies were preincubated with either unphosphorylated or Ser1480-phosphorylated NR2B peptides (20 μg/ml), or PVDF membrane was treated with λ-phosphatase (1200 U/ml; New England Biolabs, Beverly, MA) for 30 min at 30°C before immunoblotting.
Site-directed mutagenesis. NR2B Ser1480 in pGW1-NR2B was mutated to alanine using QuikChange Site-Directed Mutagenensis (Stratagene, La Jolla, CA) and the following synthetic oligonucleotides: sense 5′ctttctagtattgaggctgatgtctgagtgag3′, antisense 5′ctcactcagacatcagcctcaatactagaaag3′). NR2B Ser1480 in pRK5-GFP-NR2B (provided by Dr. S. Vicini, Georgetown University, Washington, DC) was mutated to a glutamate residue using the following oligonucleotides: sense 5′ctttctagtattgaggaggatgtctgagtgag3′, antisense 5′ctcactcagacatcctcctcaatactagaaag3′).
Purification of crude membranes from cortical neurons. Neurons were homogenized by sonication in 1 ml of resuspension buffer [10 mm sodium phosphate, pH 7.0, 100 mm NaCl, 5 mm EGTA, 5 mm EDTA, 1 μm okadaic acid, 50 mm NaF, 10 mm sodium pyrophosphate, 1 mm sodium orthovanadate, and protease inhibitor mixture (2 μg/ml aprotinin, 1 μg/ml leupeptin, 2 μg/ml antipain, 10 μg/ml benzamide, and 1 mm PMSF)]. The homogenates were centrifuged at 14,000 × g to obtain crude membrane pellets, which were resuspended in SDS sample buffer.
Immunoblot analysis. Qualitative immunoblots were visualized by enhanced chemiluminescence development using Renaissance substrate (PerkinElmer Life Sciences, Newton, MA). Quantitative immunoblots were visualized by enhanced chemifluorescence development (Amersham Biosciences, Piscataway, NJ) and quantified on a Storm Imaging system (Molecular Dynamics, Sunnyvale, CA) with Image Quant software (Amersham Biosciences). To quantify the relative degree of phosphorylation, we calculated the ratio of the intensity of the labeling with anti-NR2B-pS1480 antibody to the intensity of the labeling with anti-NR2B C-terminal antibody. The phosphorylation ratio of control samples was taken as 100%, and the phosphorylation ratio of test samples was normalized to the ratio of control samples to obtain the percentage of phosphorylation.
Activation of NMDARs in neurons. High-density cortical neuronal cultures from 18-d-old embryonic rats were prepared as described previously (Ghosh and Greenberg, 1995). dl-APV (200 μm; Tocris, Ellisville, MO) was added to glia-conditioned culture medium 4 d after plating and maintained for 3 weeks as reported previously (Liao et al., 2001). To activate NMDARs, these neurons were transferred to artificial CSF (ACSF) (in mm: 10 HEPES-free acid, 150 NaCl, 3 KCl, 10 glucose, and 2 CaCl2, pH 7.4) containing 100 μm picrotoxin, 5 μm strychnine, and 100 μm glycine [APV withdrawal (APV wd)] for 15 min at 37°C with or without 1 μm TTX (Tocris). Control neurons were transferred to ACSF containing 1 mm MgCl2 and 200 μm dl-APV to continue to block NMDARs (APV control). In 0 mm Ca2+ experiments, neurons were incubated in the APV withdrawal ACSF without calcium.
Inhibition or activation of kinases in neurons. Three-week-old cortical neurons were incubated with DMSO (0.1% v/v; as control) or various kinase inhibitors for 1 hr before APV withdrawal or APV control. Inhibitors were (in μm): 5-10 KN93, 5-10 KN92, 5 chelerythrine, 10 RP-8-bromo-cAMP (RP-8-Br-cAMP), and 10-20 4,5,6,7-tetrabromobenzotriazole (TBB; provided by Dr. D. Shugar, Institute of Biochemistry and Biophysics, Warsaw, Poland). To activate PKC and PKA, respectively, neurons were incubated with 0.4 μm phorbol 12-myristate 13-acetate (TPA) and 20 μm forskolin for 15 min at 37°C. All kinase inhibitors and activators used here were purchased from Calbiochem (San Diego, CA), except RP-8-Br-cAMP (Sigma, St. Louis, MO).
Surface biotinylation. Steady-state surface biotinylation of the neurons was performed with 1 mg/ml sulfo-NHS-LC-biotin (Pierce, Rockford, IL), and the biotinylated surface proteins were purified by incubating with 100 μl of 50% NeutraAvidin agarose (Pierce) as described previously (Chung et al., 2000).
In vitro phosphorylation of NR2B fusion proteins. The vector pGEXNR2B containing the NR2B C-terminal tail (residues 1230-1482) fused in frame with glutathione S-transferase (GST) was constructed. NR2B fusion proteins were purified from Escherichia coli BL21 cells after induction with isopropyl β-d-1-thiogalactopyranoside (IPTG; Sigma) as described in the GST gene fusion system handbook (Amersham Biosciences). All in vitro phosphorylation reactions were performed with 0.2 μg of NR2B fusion proteins and 100 μm ATP or [γ-32P]ATP (500-1000 cpm/pmol) in a 50 μl total volume for 20 min at 30°C and stopped by adding 25 μl of 3× SDS-PAGE sample buffer. Phosphorylation was visualized by autoradiography or by immunoblotting with anti-NR2B-pS1480 antibodies. Phosphorylation of NR2B fusion proteins by CaMKII (50 U; Calbiochem) was performed in CaMKII reaction buffer (10 mm HEPES, pH 7, 10 mm MgCl2, 1 mm CaCl2, 1.2 μm calmodulin; Calbiochem). Phosphorylation reactions by CK2 (20-50 U; New England Biolabs) were performed in CK2 reaction buffer (in mm: 20 Tris-HCl, pH 7.5, 10 MgCl2, 150 NaCl, and 1 DTT). Phosphorylation reactions by G-protein-coupled receptor kinase 2 and 5 (GRK2 and GRK5) were performed in GRK reaction buffer (20 mm Tris-HCl, pH 7.5, 2 mm EDTA, 5 mm MgCl2, and 0.4 μg of GRK2 or GRK5; provided by Dr. J. Benovic, Thomas Jefferson University, Philadelphia, PA) with NR2B fusion proteins or 0.2 μg of purified urea-treated bovine rod outer segments (provided by Dr. J. Benovic). For TBB inhibition, CK2, GRK2, and GRK5 were preincubated with 20 μm TBB before the reaction. For in vitro phosphorylation of NR2B fusion proteins by CaMKII phosphorylated CK2, phosphorylation of CK2 (10 U) by rat brain CaMKII (20 ng) was first performed in a 25 μl total volume of CaMKII reaction buffer with 100 μm ATP for 30 min at 30°C. After the reaction mixture was cooled on ice with 10 μm KN93 (Calbiochem) for 10 min, the mixture was added to 25 μl of CK2 reaction buffer containing 0.2 μg of GST fusion proteins of NR2B and 100 μm ATP. The resulting 50 μl reaction was incubated at 30°C for various time points (0-9 min).
In vitro phosphorylation of CK2 by CaMKII. Phosphorylation of CK2 (2 μg) was performed in a 50 μl total volume of CaMKII reaction buffer containing 100 μm [γ-32P]ATP (500-1000 cpm/pmol), 20 μm TBB, and rat brain CaMKII (20 ng; Calbiochem) and analyzed by autoradiography.
Immunoprecipitation. For immunoprecipitation (IP) of NR2B, neurons were harvested in IP buffer (25 mm Tris-HCl with 100 mm NaCl, 5 mm EDTA, 5 mm EGTA, 1 μm okadaic acid, 50 mm NaF, 1 mm sodium vanadate, and protease inhibitor mixture) containing 1% SDS and then diluted with 5 vol of ice-cold 2% Triton X-100 in IP buffer. NR2B subunits were immunoprecipitated with 4 μg of anti-NR2B C-terminal antibodies for 2-3 hr at 4°C. For coimmunoprecipitation, neurons were harvested in IP buffer containing 2% Triton X-100, and NMDAR complexes were immunoprecipitated with 4 μg of anti-NR1 C-terminal antibodies. Immunoprecipitates were eluted with SDS-PAGE sample buffer and subjected to immunoblot analysis with anti-NR2B C-terminal, anti-NR2B-pS1480, and anti-PSD-95 antibodies. To quantify the coimmunoprecipitation, we calculated the ratio of the intensity of the labeling with anti-PSD-95 antibody to the intensity of the labeling with anti-NR2B C-terminal antibody for coimmunoprecipitation and input. The PSD-95/NR2B ratio of APV control samples was taken as 100%, and the ratio of APV withdrawal samples was normalized to the ratio of APV control samples to obtain the percentage ratio (PSD-95/NR2B).
In vitro binding studies. Full-length SAP102 cDNA was subcloned into the vector pRK5 with an N-terminal myc tag. The HEK293T cells transfected with Myc-SAP102-pRK5 or PSD-95-pGW1 were harvested in IP buffer containing 1% Triton X-100 and then incubated with either unphosphorylated or Ser1480-phosphorylated NR2B peptides conjugated to affinity columns at 4°C for 2 hr. The columns were prepared by coupling 2 mg/ml BSA-conjugated peptides with 2 ml of activated Affi-Gel-10 resin. After extensive washing with IP buffer, the bound proteins were eluted by SDS sample buffer and analyzed by immunoblot with anti-myc and anti-PSD-95 antibodies.
Yeast cotransformation assay for protein interaction. Yeast assays were performed as described previously (Dong et al., 1997) using the PJ69 strain harboring HIS3, ADE2, and β-galactosidase as reporter genes. The C-terminal NR2B (residues 1257-1482) or mutant NR2BS1480E cDNAs were subcloned into the vector pPC97, whereas PDZ domains (1-3) of PSD-95 cDNAs were subcloned into the vector pPC86. The interactions between PSD-95 and NR2B were detected by growth on quadruple minus plates (Leu-, Trp-, His-, Ade-) and assayed for β-galactosidase activity with 5-bromo-4-chloro-3 indolyl-β-d-galactopyranoside as a substrate.
Immunocytochemistry of hippocampal neurons. Low-density hippocampal cultures from 18-d-old embryonic rats were prepared on coverslips as described previously (Goslin, 1991). Permeabilized immunostaining of the hippocampal neurons [21 d in vitro (DIV)] was performed as described previously (Liao et al., 1999) using anti-CK2α (Santa Cruz Biotechnology, Santa Cruz, CA), anti-PSD95 (Upstate, Charlottesville, VA), and anti-NR1 C-terminal antibodies (Liao et al., 1999). For surface immunostaining, hippocampal neurons (7 DIV) were transfected with the vector pRK5-GFP-NR2B wild type or pRK5-GFP-NR2BS1480E using Lipofectamine2000 (Invitrogen, Carlsbad, CA). After APV control or withdrawal, surface immunostaining on the transfected neurons (9 DIV) was performed using anti-green fluorescent protein (GFP) antibody (Chemicon, Temecula, CA) as described previously (Luo et al., 2002). The surface GFP-NR2B subunits were stained with rhodamine Red X (RRX)-conjugated secondary antibodies, whereas the total GFP-NR2B subunits were visualized by GFP fluorescence. Images of the pyramidal neurons were taken with a digital camera of confocal microscope (Zeiss) using identical exposure times to visualize the difference of fluorescence intensity. The ratio of surface GFP intensity over total GFP intensity per unit area was measured using the MetaMorph Imaging System (Universal Imaging, Downingtown, PA).
Data analysis. All data are reported as mean ± SE. Sample size n refers to the number of images processed in immunostaining and the number of dishes analyzed in phosphorylation, biotinylation, and in vitro phosphorylation assay. For phosphorylation, a group-paired t test was used to test the difference between the control and testing groups. For surface immunostaining, Student's t test was used (*p < 0.05; **p < 0.01; ***p < 0.001).
Results
The C-terminal PDZ ligand of NR2B is phosphorylated at Ser1480 in vivo
We generated a phosphorylation site-specific antibody to recognize the in vivo phosphorylated serine residue (Ser1480) within the NR2B PDZ ligand (T/SXV). The resulting antibody, anti-NR2B-pS1480, detected a single protein of 180 kDa, the predicted molecular weight of the NR2B subunit, in the HEK293T cells transfected with the wild-type NR2B but not empty pRK5 vector (mock) (Fig. 1A). Mutation of Ser1480 to alanine (NR2BS1480A) abolished recognition of NR2B by anti-NR2B-pS1480 (Fig. 1A). Incubation of anti-NR2B-pS1480 antibody with Ser1480-phosphorylated NR2B C-terminal peptides (p2B) before immunoblotting blocked recognition of NR2B, whereas incubation with unphosphorylated peptides (2B) did not (Fig. 1B). Anti-NR2B-pS1480 antibody did not recognize NR2B when NR2B was dephosphorylated by λ-phosphatase treatment before immunoblotting (Fig. 1C). These results demonstrate the specificity and phosphorylation dependence of anti-NR2B-pS1480 antibody.
Figure 1.
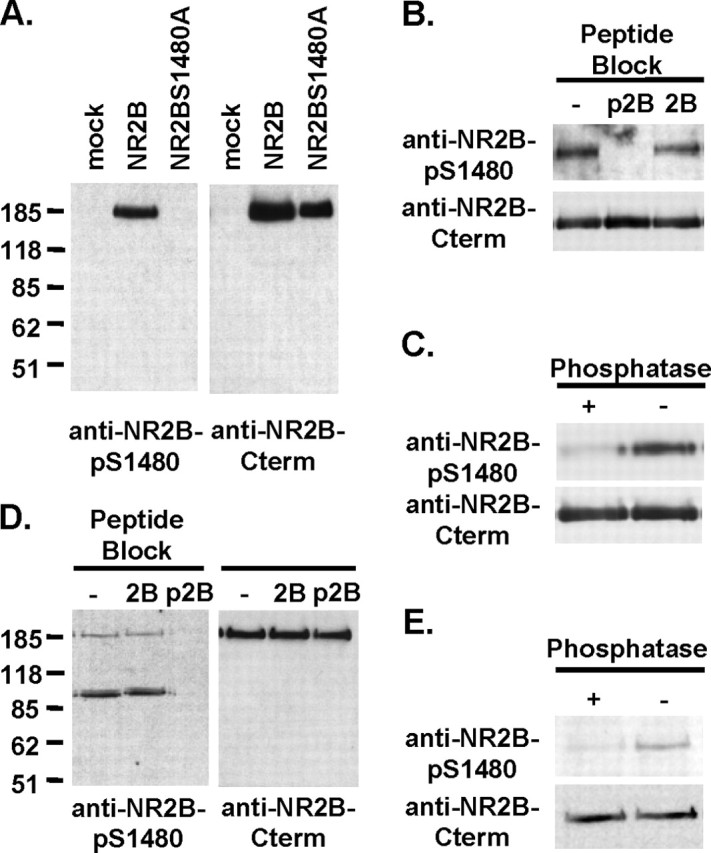
The C-terminal PDZ ligand of NR2B is phosphorylated at Ser1480 in vivo. A-C, Characterization of the phosphorylation site-specific anti-NR2B-pS1480 antibody using immunoblots of HEK293T cell crude membranes expressing NR2B. After immunoblotting with anti-NR2B-pS1480 antibody, the blot was stripped and reblotted with phosphorylation-independent anti-NR2B C-terminal antibody. A, Anti-NR2B-pS1480 antibody recognized NR2B in cells transfected with wild-type NR2B (NR2B) but not empty vector (mock) or the mutant NR2B (NR2BS1480), in which Ser1480 was mutated to alanine. B, Anti-NR2B-pS1480 antibody did not recognize NR2B when anti-NR2B-pS1480 antibody was preincubated with Ser1480-phosphorylated NR2B C-terminal peptide (p2B) but not unphosphorylated peptide (2B). C, Anti-NR2B-pS1480 antibody no longer recognized NR2B when the blot was pretreated with λ-phosphatase before immunoblotting. D, E, Western blot for NR2B-pS1480 in rat brain membrane homogenates. D, Anti-NR2B-pS1480 antibody recognized NR2B in rat brain. Preincubation of anti-NR2B-pS1480 antibody with Ser1480-phosphorylated NR2B C-terminal peptide (p2B) but not unphosphorylated peptide (2B) blocked NR2B recognition by anti-NR2B-pS1480 antibody. E, Anti-NR2B-pS1480 antibody no longer recognized NR2B when the blot was pretreated with λ-phosphatase before immunoblotting.
In rat hippocampal membrane homogenates, anti-NR2B-pS1480 antibody detected a protein of 185 kDa, which comi-grated with the NR2B subunit as detected by anti-NR2B C-terminal antibody, and two additional proteins of 90 kDa (Fig. 1D). Incubation of anti-NR2B-pS1480 antibody with p2B peptides but not 2B peptides before immunoblotting abolished recognition of NR2B (Fig. 1 D). Anti-NR2B-pS1480 antibody did not recognize NR2B when the homogenates were dephosphorylated by λ-phosphatase treatment before immunoblotting (Fig. 1 E). These results indicate that the PDZ ligand of NR2B is phosphorylated at Ser1480 in vivo.
Activity of NMDARs regulates phosphorylation of NR2B Ser1480
To examine whether neuronal activity regulates phosphorylation of NR2B Ser1480, the NMDAR antagonist (APV) was added to cultured cortical neurons at day 4 after plating, and the neurons were maintained in APV for 3 weeks as described previously (Liao et al., 2001). This chronic blockade of NMDAR activity has been shown to increase NMDAR clusters in these neurons (Liao et al., 2001). Quantitative immunoblot analysis with anti-NR2B-pS1480 and anti-NR2B C-terminal antibodies revealed that chronic APV treatment significantly decreased phosphorylation of NR2B Ser1480 compared with the APV-untreated control neurons (51 ± 11% of control; n = 4; p < 0.05) (Fig. 2A), without affecting the level of NR2B expression (97 ± 9% of control; n = 4; p > 0.05).
Figure 2.

NMDAR activity regulates phosphorylation of the NR2B PDZ ligand. Quantitative immunoblot analysis with anti-NR2B-pS1480 and anti-NR2B C-terminal antibodies was performed on the crude membranes of cultured cortical neurons. A, Chronic APV treatment of cultured cortical neurons decreased phosphorylation of NR2B Ser1480 (APV; n = 4; t test; *p < 0.05) compared with the control neurons maintained in APV-free medium (Ctl), without affecting the level of NR2B expression. B-D, Synaptic NMDARs were activated by removing APV for 15 min from the chronic APV-treated neurons (APV wd). The APV control neurons were maintained in APV (APV ctl). B, IP of NR2B subunits using anti-NR2B C-terminal antibody. Synaptic activation of NMDARs by APV withdrawal increased phosphorylation of NR2B Ser1480. C, APV withdrawal significantly increased phosphorylation of NR2B Ser1480 compared with the APV control (n = 6; t test; ***p < 0.001). This increase was partially blocked by 1 μm TTX (APV wd+TTX; n = 4; t test; **p < 0.01) and was abolished by APV withdrawal without calcium (APV wd+0 mm Ca2+; n = 4; t test; p > 0.05). D, APV withdrawal significantly increased Ser1480 phosphorylation of surface biotinylated NR2B subunits compared with APV control (APV ctl; n = 4; t test; *p < 0.05).
To examine rapid activity-dependent changes in the phosphorylation of NR2B Ser1480, chronic APV-treated cortical neurons were incubated with ACSF containing picrotoxin (GABAA receptor blocker), strychnine (glycine receptor antagonist), and glycine (NMDAR co-agonist) for 15 min as described previously (Liao et al., 2001). This APV withdrawal treatment allows rapid activation of synaptic NMDARs by the spontaneous release of glutamate. Control neurons were maintained in ACSF containing APV (APV control). NR2B subunits were immunoprecipitated with anti-NR2B C-terminal antibodies from these cultures and immunoblotted with anti-NR2B-pS1480 and anti-NR2B C-terminal antibodies. Rapid activation of NMDARs by APV withdrawal increased phosphorylation of NR2B Ser1480 compared with APV control (271 ± 43% APV control; n = 4; p < 0.05) (Fig. 2B,D). Similar levels of increased phosphorylation were detected for surface NR2B subunits (340 ± 53% APV control; n = 4; p < 0.05) (Fig. 2D). Interestingly, activation of both synaptic and extrasynaptic NMDARs by bath application of glutamate (100 μm) or NMDA (50 μm) in APV-untreated neurons caused dephosphorylation of Ser1480 [NMDA, 31 ± 2% of control (n = 3), p < 0.05; glutamate, 30 ± 5% of control (n = 3), p < 0.05; data are shown in supplemental material, available at www.jneurosci.org].
High-density cortical neuronal cultures have high levels of spontaneous activity (Murphy et al., 1992). To test whether phosphorylation of NR2B Ser1480 is attributable to NMDAR activation by spontaneous synaptic responses, APV withdrawal was performed in the presence of TTX, which blocks sodium channel-mediated action potentials. APV withdrawal increased the phosphorylation of NR2B Ser1480 (489 ± 64% of APV control; n = 6; p < 0.001). In contrast, APV withdrawal in the presence of TTX caused a much less dramatic increase in the phosphorylation of Ser1480 (262 ± 39% of APV control; n = 4; p < 0.01) (Fig. 2C). In addition, no increase in the phosphorylation of Ser1480 was observed when we performed APV withdrawal in Ca2+-free ACSF to block the release of presynaptic vesicles (92 ± 16% of APV control; n = 4; p > 0.05) (Fig. 2C). These results demonstrate that synaptic NMDAR activation dynamically modulates phosphorylation of the NR2B PDZ ligand.
CaMKII regulates phosphorylation of NR2B Ser1480
Synaptic NMDAR activation has been shown to stimulate CaMKII activity (Fukunaga et al., 1992). To determine whether CaMKII can regulate phosphorylation of NR2B Ser1480, chronic APV-treated cortical neurons were preincubated with DMSO (0.1% v/v), KN93 (CaMKII inhibitor; 5-10 μm), or KN92 (inactive analog of KN93; 5-10 μm) for 1 hr before APV withdrawal. APV withdrawal in DMSO-treated neurons increased phosphorylation of NR2B Ser1480 (330 ± 49% of APV DMSO control; n = 5; p < 0.001) and activated CaMKII as detected by increased autophosphorylation of CaMKII at Thr286 (Fig. 3A). KN93 treatment completely inhibited the activation of CaMKII and partially blocked the APV withdrawal-induced phosphorylation of Ser1480 (206 ± 26% of APV DMSO control; n = 4; p < 0.05), whereas KN92 treatment did not (320 ± 50% of APV DMSO control; n = 4; p < 0.05) (Fig. 3A). These results indicate that CaMKII is partially required for activity-induced phosphorylation of NR2B Ser1480.
Figure 3.

CaMKII, but not PKC or PKA, regulates Ser1480 phosphorylation of NR2B during synaptic NMDAR activation. Quantitative immunoblot analysis with anti-NR2B-pS1480 and anti-NR2B C-terminal antibodies was performed on the crude membranes of cultured cortical neurons. A, The chronic APV-treated cortical neurons were incubated with DMSO (0.1% v/v), KN93 (5-10 μm; CaMKII inhibitor), or KN92 (5-10 μm; inactive analog of KN93) for 1 hr before APV withdrawal. Anti-CaMKII-pT286 antibody (CaMKII-pT286) detected autophosphorylation of CaMKII at Thr286. KN93 completely blocked CaMKII activation and partially blocked phosphorylation of NR2B Ser1480 induced by APV withdrawal (APV wd plus DMSO and APV wd plus KN93; n = 4; t test; *p < 0.05). B, The chronic APV-treated cortical neurons were incubated with DMSO (0.1% v/v), chelerythrine (5 μm; PKC inhibitor), or RP-8-Br-cAMP (10 μm; PKA inhibitor) for 1 hr before APV withdrawal. Chelerythrine and RP-8-Br-cAMP had no effect on APV withdrawal-induced phosphorylation of NR2B Ser1480 (APV wd plus DMSO and APV wd plus chelerythrine or RP-8-Br-cAMP; n = 3; t test; p > 0.05). C, In vitro phosphorylation reactions of GST fusion proteins of the NR2B C-terminal tail and GluR1 C-terminal tail were performed with CaMKII (50 U). Phosphorylation of NR2B Ser1480 and GluR1 Ser831 (a previously characterized CaMKII site) was detected by immunoblotting with anti-NR2B-pS1480 and anti-GluR1-pS831 antibodies, respectively. CaMKII directly phosphorylated GluR1 Ser831 but not NR2B Ser1480. The asterisk indicates the C-terminally truncated GluR1 fusion protein, which is recognized by anti-GluR1-pS831 antibody but not by anti-GluR1 C-terminal antibody.
Synaptic NMDAR activation has also been shown to stimulate PKA (Roberson and Sweatt, 1996) and PKC (Klann et al., 1993) activity. However, treatment of neurons with chelerythrine (PKC inhibitor; 5 μm) or RP-8-Br-cAMP (PKA inhibitor; 10 μm) did not inhibit APV withdrawal-induced phosphorylation of NR2B Ser1480 [APV wd plus chelerythrine, 253 ± 25% of APV DMSO control (n = 3); APV wd plus RP-8-Br-cAMP, 290 ± 36% of APV DMSO control (n = 3); p < 0.05] compared with DMSO treatment (APV wd plus DMSO, 243 ± 24% of APV DMSO control; n = 3; p < 0.05) (Fig. 3B). Furthermore, treatment of neurons with forskolin (20 μm) or TPA (0.4 μm) to stimulate PKA and PKC, respectively, had no effect on the phosphorylation of Ser1480 (data not shown).
To test whether CaMKII directly phosphorylates NR2B Ser1480, we performed in vitro phosphorylation reactions using GST fusion proteins of the NR2B C-terminal tail (residues 1230-1482) with the purified rat brain CaMKII. CaMKII did not phosphorylate Ser1480 but robustly phosphorylated Ser831 of GluR1, a previously characterized CaMKII site (Mammen et al., 1997) (Fig. 3C). These results together suggest that it is not likely that CaMKII, PKA, or PKC phosphorylate NR2B at Ser1480 in vivo and that a kinase(s) downstream of CaMKII phosphorylates this site after synaptic NMDAR activation.
Synaptic NMDAR activation increases CK2 phosphorylation of NR2B PDZ ligand
The amino acid sequence surrounding NR2B Ser1480 (IESDV) is a strong consensus site for CK2 (Meggio et al., 1994). To determine whether CK2 is the relevant in vivo kinase for NR2B Ser1480, chronic APV-treated cortical neurons were incubated with DMSO (0.1% v/v) or TBB (10-20 μm) for 1 hr before APV withdrawal. TBB is a cell-permeable, highly specific CK2 inhibitor that inhibits 85% of CK2 activity at this concentration in vitro (Sarno et al., 2001). APV withdrawal of DMSO-treated neurons increased CaMKII activity and phosphorylation of NR2B Ser1480 (348 ± 50% of APV DMSO control; n = 4; p < 0.05) (Fig. 4A). In contrast, TBB treatment abolished APV withdrawal-induced phosphorylation of Ser1480 (124 ± 16% of APV DMSO control; n = 4; p > 0.05) but not autophosphorylation of CaMKII Thr286 (Fig. 4A). Moreover, CK2 directly phosphorylated GST fusion proteins of the NR2B C-terminal tail at Ser1480 in vitro (Fig. 4B). These results suggest that CK2 acts downstream of CaMKII and phosphorylates NR2B Ser1480 after synaptic NMDAR activation.
Figure 4.

CK2 functions downstream of CaMKII during synaptic NMDAR activation and directly phosphorylates Ser1480. A, The chronic APV-treated cortical neurons were incubated with DMSO (0.1% v/v) or TBB (10-20 μm; CK2 inhibitor) for 1 hr before APV withdrawal. Quantitative immunoblot analysis with anti-NR2B-pS1480 and anti-NR2B C-terminal antibodies revealed that TBB completely blocked APV withdrawal-induced phosphorylation of NR2B Ser1480 (APV wd plus DMSO and APV wd plus TBB; n = 4; t test; *p < 0.05) but not autophosphorylation of CaMKII at Thr286. B, C, Immunoblot analysis of in vitro phosphorylation reactions of GST fusion proteins of the NR2B C-terminal tail by CK2 (20 U; B) and CK2, GRK2, and GRK5 (0.4 μg each; C). B, CK2 directly phosphorylated NR2B Ser1480. C, GRK2 and GRK5 phosphorylated NR2B Ser1480 very weakly compared with the same amount of CK2. D, Autoradiograph and Coomassie blue staining of an SDS-PAGE gel containing in vitro phosphorylation reactions of the GST fusion proteins of the NR2B C-terminal tail by CK2 (0.4 μg) with or without TBB (20 μm). TBB inhibited CK2 activity. E, Autoradiograph and Coomassie blue staining of an SDS-PAGE gel containing in vitro phosphorylation reactions of rhodopsin by GRK2 (0.4 μg) with or without TBB (20 μm). TBB did not inhibit GRK2 activity. F, Immunostaining of 3-week-old low-density cultured hippocampal neurons with anti-CK2α (CK2α) and anti-NR1 C-terminal antibodies (NR1). CK2α subunits colocalized with NR1 in the dendritic shafts and some spines. G, Immunostaining of 3-week-old low-density cultured hippocampal neurons with anti-CK2α (CK2α) and anti-PSD-95 antibody (PSD-95). CK2α subunits also colocalized with PSD-95 in some dendritic spines.
Because GRK2 and GRK5 also preferentially phosphorylate serine or threonine residues surrounded by acidic amino acids (Palczewski, 1997), we also performed an in vitro phosphorylation reactions with purified active GRK2 and GRK5. GRK2 and GRK5 phosphorylated NR2B Ser1480 very weakly compared with the same amount of CK2 (Fig. 4C). To test whether the CK2 inhibitor TBB can block GRK2 and GRK5, we performed in vitro phosphorylation reaction with GRK2 and GRK5 in the presence of TBB (20 μm) using rhodopsin as a substrate. TBB dramatically decreased CK2 phosphorylation of NR2B Ser1480 (Fig. 4D) but had no effect on the phosphorylation of rhodopsin by GRK2 (Fig. 4E) and GRK5 (data not shown). These results suggest that GRK2 and GRK5 do not regulate phosphorylation of NR2B Ser1480 after synaptic NMDAR activation.
To examine the subcellular localization of CK2 in relation to the distribution of NMDARs, we performed immunostaining on 3-week-old cultured hippocampal neurons using anti-CK2α antibody and anti-NR1 C-terminal antibody. CK2 catalytic α subunits were localized in soma and throughout dendrites as small clusters (Fig. 4F). CK2 colocalized with NR1 in dendritic shafts and some spines (Fig. 4F). To test whether excitatory synapses contain CK2, we performed immunostaining with anti-PSD-95 antibody. CK2α subunits also colocalized with PSD-95 in some dendritic spines (Fig. 4G). These results suggest that CK2 exists in close proximity to NMDARs to phosphorylate NR2B Ser1480.
CK2 is cyclic nucleotide independent and cannot be directly activated by calcium (Litchfield, 2003). Previous studies show that phosphorylation of its catalytic α and regulatory β subunits by CK2 itself or other kinases can regulate CK2 activity (Blanquet, 1998; Litchfield, 2003). Because CaMKII was partially required for APV withdrawal-induced phosphorylation of NR2B Ser1480 by CK2 (Figs. 3, 4), we investigated whether CaMKII can stimulate CK2 activity by directly phosphorylating its catalytic α or regulatory β subunits. We performed in vitro phosphorylation reactions of CK2 holoenzyme with CaMKII in the presence of TBB (20 μm) to minimize the autophosphorylation of CK2 itself. CaMKII directly phosphorylated the β but not the α subunit of CK2 (Fig. 5A). To test whether CaMKII phosphorylation of CK2β can increase the catalytic activity of CK2, we first in vitro-phosphorylated CK2 with CaMKII and then added KN93 to inhibit CaMKII activity. This reaction mixture was incubated with GST fusion proteins of the NR2B C-terminal tail and CK2 reaction buffer at 30°C for 0-9 min. Preincubation of CaMKII with CK2 overall increased CK2 phosphorylation of NR2B at Ser1480 compared with CK2 alone (Fig. 5B). However this increase was statistically significant at the 6 min time point (146 ± 11% of CK2 alone; n = 4; p < 0.05) but not at the 3 and 9 min time points (n = 4; p > 0.05) (Fig. 5B). These results indicate that direct phosphorylation of CK2β by CaMKII has no effect on the catalytic activity of CK2 in vitro.
Figure 5.

Preincubation of CaMKII with CK2 increases phosphorylation of NR2B Ser1480 by CK2 in vitro. A, Autoradiograph and Coomassie blue staining of an SDS-PAGE gel containing in vitro phosphorylation reactions of purified CK2 holoenzyme (2 μg) with TBB (20 μm) by purified rat brain CaMKII (50 U). CaMKII directly phosphorylated CK2 β-subunits. B, Immunoblot analysis of in vitro phosphorylation reactions of the GST fusion proteins of the NR2BC-terminal tail by CK2 (20U), which was first phosphorylated by CaMKII (50U) in vitro. After CaMKII phosphorylation of CK2, KN93 was added to block CaMKII activity. Preincubation of CaMKII with CK2 overall does not significantly increase CK2 phosphorylation of NR2B Ser1480 compared with CK2 alone. However, the increase was statistically significant at the 6 min time point (n = 4; t test; *p < 0.05).
Phosphorylation of NR2B PDZ ligand disrupts its interaction with SAP102 and PSD-95
To investigate whether phosphorylation of Ser1480 in the NR2B C-terminal PDZ ligand can regulate the interaction of NR2B with PSD-95/SAP90 family proteins, we tested the in vitro binding of PSD-95 or SAP102 to Ser1480-phosphorylated (p2B) or unphosphorylated NR2B C-terminal (2B) peptides. HEK293T cell lysates expressing PSD-95 and SAP102 were incubated with NR2B peptide-conjugated affinity columns, and bound proteins were eluted and analyzed by immunoblotting. PSD-95 and SAP102 interacted with the 2B peptide but not the p2B peptide (Fig. 6A). The interaction of PSD-95 and SAP102 with the p2B peptide was recovered when the p2B peptide was dephosphorylated by λ-phosphatase before the binding experiment (Fig. 6A). These results indicate that phosphorylation of Ser1480 disrupts the interaction of NR2B with the PDZ domains of PSD-95 and SAP102.
Figure 6.

Phosphorylation of the NR2B PDZ ligand disrupts its interaction with SAP102 and PSD-95. A, In vitro binding studies of PSD-95 (top) or SAP102 (bottom) with NR2B peptides immobilized on Affi-Gel resin. Extracts of HEK293T cells expressing PSD-95 or SAP102 were incubated with unphosphorylated (2B), Ser1480-phosphorylated 2B (p2B), or the λ-phosphatase-treated p2B peptides. The bound PSD-95 or SAP102 proteins were detected by immunoblot with anti-PSD-95 antibody or anti-SAP102 antibody, respectively. Both PSD-95 and SAP102 failed to interact with Ser1480-phosphorylated NR2B. B, IP of NMDAR complexes from the chronic APV-treated cortical neurons after APV control (APV ctl) or APV wd using anti-NR1 C-terminal antibodies. Coimmunoprecipitation of NR2B and PSD-95 was analyzed by immunoblot with anti-PSD-95 (PSD-95), NR2B, and anti-NR2B-pS1480 antibodies (NR2B-pS1480). Synaptic activation of NMDAR increased phosphorylation of NR2B Ser1480 and decreased NMDAR interaction with PSD-95 (n = 5; t test; *p < 0.05).
Because APV withdrawal significantly increased phosphorylation of NR2B Ser1480 (Fig. 2), synaptic NMDAR activation may regulate the interaction of NMDARs with the PSD-95/SAP90 family proteins. To test this, NMDARs were immunoprecipitated with anti-NR1 C-terminal antibodies from cortical neurons treated with APV control or withdrawal. Compared with APV control, synaptic NMDAR activation increased phosphorylation of NR2B Ser1480 but decreased the amount of PSD-95 that coimmunoprecipitated with NR2B subunits (Fig. 6B). This decrease was statistically significant as shown by the ratio of the intensity of PSD-95 over NR2B [IP (APV wd), 45 ± 14% of APV control (n = 5); p < 0.05] (Fig. 6B). In contrast, APV withdrawal did not change the overall amount of PSD-95 and NR2B in total lysate [input (APV wd), 84 ± 12% of APV control (n = 5); p > 0.05] (Fig. 6B). These results demonstrate that activity-induced phosphorylation of NR2B Ser1480 attenuates the interaction of NMDAR with PSD-95 in vivo.
Phosphorylation of NR2B PDZ ligand decreases surface expression of NR2B
Disruption of the interactions between NR2B and PSD-95/SAP102 by phosphorylation of the NR2B PDZ ligand may regulate surface membrane trafficking of NMDARs. To test this, we first mutated Ser1480 to glutamate, which mimics the negative charge of phosphorylated Ser1480.In the yeast two-hybrid system, this phosphorylation mimic mutant, NR2B S1480E C terminus, failed to interact with PDZ domains (1-3) of PSD-95, whereas the wild-type NR2B C terminus interacted well (Fig. 7B). Next, we transfected primary hippocampal neurons with wild-type subunits or the phosphorylation mimic mutant NR2B subunits, which were tagged extracellularly with GFP (GFP-NR2B and GFP-NR2B S1480E). Surface GFP-NR2B subunits were labeled live with anti-GFP antibody and visualized by RRX-conjugated secondary antibody, whereas total GFP-NR2B was visualized by GFP fluorescence. Surface immunostaining showed surface expression of both wild-type GFP-NR2B and mutant GFP-NR2B S1480E on the plasma membrane of dendrites and soma as distinct clusters in hippocampal pyramidal neurons (Fig. 7A). However, surface clusters of mutant GFP-NR2B S1480E were less intense and smaller compared with wild-type NR2B (Fig. 7A). The ratio of surface GFP intensity over total GFP intensity per unit area of mutant NR2B S1480E was significantly lower than that of wild-type NR2B in the dendrites [wild type, 0.135 ± 0.024 per unit area (n = 19); S1480E, 0.034 ± 0.005 per unit area (n = 26); p < 0.001] (Fig. 7C). These data demonstrate that phosphorylation of NR2B Ser1480 decreases the surface expression of NMDARs.
Figure 7.

Mutation of Ser1480 to glutamate decreases surface expression of NR2B in neurons. A, Surface immunostaining of GFP-NR2B wild-type and mutant subunit (NR2B S1480E) in which Ser1480 was replaced with glutamate to mimic phosphorylated Ser1480 (left; scale bar, 10 μm). The right panels show higher magnification of the insets in the left panel. B, In the yeast two-hybrid system, PDZ domains of PSD-95 failed to interact with phosphorylation mimic mutant NR2B S1480E and deletion mutant NR2B (NR2B Δ5aa), in which the PDZ ligand was deleted. C, Quantification of surface GFP staining of wild-type NR2B (n = 19) and mutant NR2B S1480E (n = 26) as the ratio of surface GFP intensity over total GFP intensity per unit area (t test; ***p < 0.001).
Discussion
The present study demonstrates that CK2 phosphorylation of Ser1480 within the NR2B C-terminal PDZ ligand regulates PDZ domain-mediated interaction and surface expression of NMDAR. Interestingly, activity of NMDAR and CaMKII regulates CK2 phosphorylation of Ser1480. Thus, activity-dependent phosphorylation of the PDZ ligand provides a powerful way to control NMDAR trafficking and excitatory synaptic function.
Phosphorylation of the NR2B PDZ ligand by CK2
Various kinases and phosphatases modulate ion channel properties (Greengard et al., 1991; Durand et al., 1993; Lieberman and Mody, 1994; Wang and Salter, 1994; Lieberman and Mody, 1999) and forward trafficking of NMDARs (Lan et al., 2001; Scott et al., 2001), although only a few phosphorylation sites of NMDAR subunits have been identified so far (Hall and Soderling, 1997; Leonard and Hell, 1997; Tingley et al., 1997). NR2B Ser1480 is the first CK2 phosphorylation site identified among the NMDAR subunits. CK2 is highly enriched in brain and phosphorylates various synaptic and nuclear substrates implicated in neuronal development and survival, neurite extension, synaptic transmission, and synaptic plasticity (Blanquet, 2000). CK2 can regulate NMDAR channel gating (Lieberman and Mody, 1999), although the role of CK2 phosphorylation of NR2B Ser1480 in NMDAR channel properties is not known. We also show that CK2 colocalizes with NMDAR in dendrites and at some excitatory synapses, consistent with studies demonstrating enrichment of CK2 activity in synaptosomes (Girault et al., 1990).
Interestingly, CK2 phosphorylation of NR2B Ser1480 is regulated by activation of NMDAR itself. Suppression of NMDAR activity by chronic APV treatment decreases Ser1480 phosphorylation, whereas rapid activation of synaptic NMDARs by APV withdrawal increases Ser1480 phosphorylation. Similarly, induction of NMDAR-dependent long-term potentiation (LTP) in rat hippocampal slices leads to a calcium-dependent increase in CK2 activity (Charriaut-Marlangue et al., 1991). Chronic APV treatment of cultured cortical neurons increases the proportion of “silent synapses” containing only NMDAR (Liao et al., 1999), delaying synapse development. Chronic APV treatment may also affect the morphology of dendritic spines, because NMDAR activity regulates spine morphology (Engert and Bonhoeffer, 1999; Maletic-Savatic et al., 1999). However, APV withdrawal from the chronic APV-treated neurons allows activation of only synaptic NMDARs by synaptically released glutamate and stimulation of CK2 activity for which there is no known activator so far. Because APV withdrawal has been shown to convert silent synapses to “conducting synapses” containing both NMDAR and AMPAR (Liao et al., 1999; Liao et al., 2001), this treatment may allow us to study whether CK2 phosphorylation of NR2B Ser1480 plays a role in synapse maturation. In contrast to APV withdrawal, bath application of glutamate or NMDA activates both synaptic and extrasynaptic NMDARs and induces rapid internalization of AMPARs through activation of phosphatases (Beattie et al., 2000; Ehlers, 2000; Lee et al., 2002). Not surprisingly, glutamate and NMDA treatment causes dephosphorylation of NR2B Ser1480.
It is unclear how synaptic NMDAR activation leads to calcium-dependent CK2 phosphorylation of NR2B Ser1480 because calcium does not directly activate CK2 (Hathaway and Traugh, 1982). Because the CaMK inhibitor KN93 partially blocks APV withdrawal-induced phosphorylation of NR2B Ser1480, calcium/calmodulin-dependent protein kinases may regulate CK2 activity. However, we found that direct phosphorylation of CK2β by CaMKII does not increase CK2 activity in vitro, although the role of CaMKI and CaMKIV in CK2 activity remains to be tested. Alternatively, calmodulin may mediate synaptic targeting of CK2 by interacting with CK2β (Grein et al., 1999) and NR2B (Wyszynski et al., 1997). Recently, induction of NMDAR-dependent LTP in rat hippocampal slices has shown to increase both CaMKII activity and CaMKII association with NR2A and NR2B and decrease PSD-95 association with NR2A and NR2B (Gardoni et al., 2001). Thus, an NMDAR-mediated rise in calcium may stimulate the binding of CaMKII to NR2B (Leonard et al., 1999; Bayer et al., 2001) and/or CaMKII phosphorylation of NR2B Ser1303 (Omkumar et al., 1996; Bayer et al., 2001). These events may induce a conformational change in the NR2B tail, which attenuates PSD-95 binding and exposes the PDZ ligand for CK2 phosphorylation. Once Ser1480 is phosphorylated, NMDAR may remain unbound by PSD-95.
Regulation of PDZ domain-mediated interaction by phosphorylation of the NR2B PDZ ligand
The inhibition of NR2B interaction with PSD-95 and SAP102 by phosphorylation of the PDZ ligand at Ser1480 can be explained by the structure of type 1 PDZ domain-T/SXV motif interaction (Doyle et al., 1996; Morais Cabral et al., 1996). The addition of a phosphate group to the -2 serine or threonine residue of the T/SXV motif will disrupt the critical hydrogen bond formed between the -2 residue of the T/SXV motif and the histidine within a hydrophobic groove in the PDZ domain (Doyle et al., 1996; Morais Cabral et al., 1996). Similar to our results, PKA phosphorylation of the -2 serine residue of inward rectifier potassium channel Kir2.3 abolishes binding of Kir2.3 to PSD-95 (Cohen et al., 1996). GRK5 phosphorylation of the -2 serine of the β2-adrenergic receptor disrupts binding of the receptor to the PDZ domain of NHERF (Na+/H+ exchanger regulatory factor) protein (Cao et al., 1999). Moreover, the -3 serine of the AMPA receptor subunit GluR2 with its type 2 PDZ ligand (-SVKI) can be phosphorylated by PKC, and such phosphorylation disrupts binding of GluR2 to the PDZ domain of GRIP (Matsuda et al., 1999; Chung et al., 2000). Thus, phosphorylation of the serine or threonine residue within the PDZ ligands may be a common mechanism for regulating PDZ domain-mediated interactions.
Regulation of synaptic signaling by phosphorylation of the NR2B PDZ ligand
PSD-95/SAP90 family proteins interact with many neuronal signaling proteins such as SynGAP (a neuronal RasGTPase activating protein) (Chen et al., 1998; Kim et al., 1998), neuronal nitric oxide synthase (Brenman et al., 1996; Christopherson et al., 1999), and AKAP70/150 (a scaffolding protein for three signaling enzymes: PKA, PKC, and the calcium-dependent phosphatase calcineurin) (Colledge et al., 2000). The targeted mutation of the PSD-95 gene in mice appears to disrupt downstream signaling from NMDAR without affecting synaptic localization of NMDARs (Migaud et al., 1998). In addition, overexpression of PSD-95 enhances synaptic clustering of AMPARs without altering the levels of synaptic NMDARs (El-Husseini et al., 2000). Thus, phosphorylation of the NR2B PDZ ligand may regulate synaptic signaling downstream of NMDAR by modulating interaction of NMDAR with PSD-95/SAP90 family proteins.
Regulation of NMDAR trafficking by phosphorylation of the NR2B PDZ ligand
PDZ domain-mediated interactions facilitate synaptic clustering and surface expression of NMDARs by suppressing the endoplasmic reticulum retention signal in NR1 (Standley et al., 2000) and the internalization signal in the NR2B C terminus, which binds to the adaptor protein-2 (Roche et al., 2001; Lavezzari et al., 2003), and by promoting formation of exocyst-SAP102-NMDAR complexes (Sans et al., 2003). Consistent with these findings, we found that mutation of NR2B Ser1480 to glutamate, which mimics phosphorylated Ser1480, disrupts NR2B interaction with PSD-95/SAP102 and decreases the size and intensity of surface NR2B clusters. Phosphorylation of NR2B Ser1480 may regulate surface expression of NMDAR by multiple mechanisms. Because NMDAR can move laterally between synaptic and extrasynaptic domains of the plasma membrane (Tovar and Westbrook, 2002), phosphorylation of surface NR2B at Ser1480 would disrupt NMDAR interaction with PSD-95/PSD-93 and allow NMDAR to move from synaptic to extrasynaptic regions where they could be internalized by exposing an internalization signal in the NR2B C terminus (Fig. 8A). Alternatively, phosphorylation of intracellular NR2B at Ser1480 would disrupt NMDAR interaction with SAP102 and block surface delivery of NMDAR mediated by SAP102/sec8 - exocyst complex formation (Fig. 8B).
Figure 8.

Model for regulation of NMDAR complex and trafficking by phosphorylation of the NR2B PDZ ligand. Synaptic activation of NMDAR stimulates CaMKII and CK2 activity. A, CK2 phosphorylates the PDZ ligand of surface NR2B at Ser1480 and disrupts the interaction between surface NMDAR and the PDZ domains of PSD-95. This leads to destabilization and internalization of surface NMDAR and decoupling of NMDAR from signaling proteins at excitatory synapses. B, CK2 phosphorylates the PDZ ligand of intracellular NR2B at Ser1480 and disrupts the interaction between intracellular NMDAR with the PDZ domains of SAP102. This leads to disruption of SAP102/Sec8-mediated forward trafficking of NMDAR to the cell surface.
Our studies suggest that NMDAR activity may regulate synaptic targeting of NMDAR by modulating phosphorylation of NR2B Ser1480. We demonstrated that phosphorylation of Ser1480 decreases with chronic APV treatment, whereas it increases after removal of APV. Interestingly, chronic APV treatment increases synaptic accumulation of NMDAR clusters with no preference between NR2A- and NR2B-containing receptors (Rao and Craig, 1997; Liao et al., 1999; Liao et al., 2001; Tovar and Westbrook, 2002), whereas removal of APV to induce spontaneous activity decreases synaptic NMDAR clusters (Rao and Craig, 1997). The increase in NR2A and NR2B and their association with PSD-95 has been suggested to be the potential mechanism of the APV-induced synaptic targeting of NMDAR (Rao and Craig, 1997), although we found no change in the level of NR2B by chronic APV blockade. Thus, the APV-induced synaptic targeting of NMDARs in our cortical cultures (Liao et al., 2001) may be caused by the decrease in Ser1480 phosphorylation and subsequent increase in the interaction between NR2B and PSD-95/SAP102. In contrast to the above studies, APV blockade of NMDARs has been shown to induce trafficking of NR2B away from synapses and trafficking of NR2A subunits to synapses (Aoki et al., 2003; Fujisawa and Aoki, 2003). The differences in activity-dependent synaptic targeting of NR2B/A subunits may be explained by the differences in the duration of APV treatment and in vitro neuronal cultures versus in vivo mature cortical synapses in intact adult brain. Because synaptic targeting of NR2B- and NR2A-containing receptors is regulated differently (Barria and Malinow, 2002; Aoki et al., 2003; Fujisawa and Aoki, 2003) and determines the polarity of synaptic plasticity (Liu et al., 2004), it will be interesting to study whether activity-dependent CK2 phosphorylation of NR2B Ser1480 may affect the synaptic presence of NR2A-containing NMDARs.
Footnotes
This work was supported by the Howard Hughes Medical Institute and the National Institutes of Health (R.L.H.). We thank Dr. D. Shugar for the specific CK2 inhibitor TBB and Dr. J. Benovic for purified GRK2, GRK5, and urea-treated rod outer segments. We also thank Dr. S. Vicini for pRK5-GFP-NR2B cDNA.
Correspondence should be addressed to Dr. Richard L. Huganir, Department of Neuroscience, Howard Hughes Medical Institute, Johns Hopkins University School of Medicine, 904A Preclinical Teaching Building, 725 North Wolfe Street, Baltimore, MD 21205. E-mail: rhuganir@jhmi.edu
Copyright © 2004 Society for Neuroscience 0270-6474/04/2410248-12$15.00/0
References
- Aoki C, Fujisawa S, Mahadomrongkul V, Shah PJ, Nader K, Erisir A (2003) NMDA receptor blockade in intact adult cortex increases trafficking of NR2A subunits into spines, postsynaptic densities, and axon terminals. Brain Res 963: 139-149. [DOI] [PubMed] [Google Scholar]
- Barria A, Malinow R (2002) Subunit-specific NMDA receptor trafficking to synapses. Neuron 35: 345-353. [DOI] [PubMed] [Google Scholar]
- Bayer KU, De Koninck P, Leonard AS, Hell JW, Schulman H (2001) Interaction with the NMDA receptor locks CaMKII in an active conformation. Nature 411: 801-805. [DOI] [PubMed] [Google Scholar]
- Beattie EC, Carroll RC, Yu X, Morishita W, Yasuda H, von Zastrow M, Malenka RC (2000) Regulation of AMPA receptor endocytosis by a signaling mechanism shared with LTD. Nat Neurosci 3: 1291-1300. [DOI] [PubMed] [Google Scholar]
- Blanquet PR (1998) Neurotrophin-induced activation of casein kinase 2 in rat hippocampal slices. Neuroscience 86: 739-749. [DOI] [PubMed] [Google Scholar]
- Blanquet PR (2000) Casein kinase 2 as a potentially important enzyme in the nervous system. Prog Neurobiol 60: 211-246. [DOI] [PubMed] [Google Scholar]
- Bliss TV, Collingridge GL (1993) A synaptic model of memory: long-term potentiation in the hippocampus. Nature 361: 31-39. [DOI] [PubMed] [Google Scholar]
- Braithwaite SP, Xia H, Malenka RC (2002) Differential roles for NSF and GRIP/ABP in AMPA receptor cycling. Proc Natl Acad Sci USA 99: 7096-7101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brenman JE, Chao DS, Gee SH, McGee AW, Craven SE, Santillano DR, Wu Z, Huang F, Xia H, Peters MF, Froehner SC, Bredt DS (1996) Interaction of nitric oxide synthase with the postsynaptic density protein PSD-95 and alpha1-syntrophin mediated by PDZ domains. Cell 84: 757-767. [DOI] [PubMed] [Google Scholar]
- Cao TT, Deacon HW, Reczek D, Bretscher A, von Zastrow M (1999) A kinase-regulated PDZ-domain interaction controls endocytic sorting of the beta2-adrenergic receptor. Nature 401: 286-290. [DOI] [PubMed] [Google Scholar]
- Charriaut-Marlangue C, Otani S, Creuzet C, Ben-Ari Y, Loeb J (1991) Rapid activation of hippocampal casein kinase II during long-term potentiation. Proc Natl Acad Sci USA 88: 10232-10236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen HJ, Rojas-Soto M, Oguni A, Kennedy MB (1998) A synaptic RasGTPase activating protein (p135 SynGAP) inhibited by CaM kinase II. Neuron 20: 895-904. [DOI] [PubMed] [Google Scholar]
- Choi DW (1994) Glutamate receptors and the induction of excitotoxic neuronal death. Prog Brain Res 100: 47-51. [DOI] [PubMed] [Google Scholar]
- Christopherson KS, Hillier BJ, Lim WA, Bredt DS (1999) PSD-95 assembles a ternary complex with the N-methyl-d-aspartic acid receptor and a bivalent neuronal NO synthase PDZ domain. J Biol Chem 274: 27467-27473. [DOI] [PubMed] [Google Scholar]
- Chung HJ, Xia J, Scannevin RH, Zhang X, Huganir RL (2000) Phosphorylation of the AMPA receptor subunit GluR2 differentially regulates its interaction with PDZ domain-containing proteins. J Neurosci 20: 7258-7267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung HJ, Steinberg JP, Huganir RL, Linden DJ (2003) Requirement of AMPA receptor GluR2 phosphorylation for cerebellar long-term depression. Science 300: 1751-1755. [DOI] [PubMed] [Google Scholar]
- Cohen NA, Brenman JE, Snyder SH, Bredt DS (1996) Binding of the inward rectifier K+ channel Kir 2.3 to PSD-95 is regulated by protein kinase A phosphorylation. Neuron 17: 759-767. [DOI] [PubMed] [Google Scholar]
- Colledge M, Dean RA, Scott GK, Langeberg LK, Huganir RL, Scott JD (2000) Targeting of PKA to glutamate receptors through a MAGUK-AKAP complex. Neuron 27: 107-119. [DOI] [PubMed] [Google Scholar]
- Daw MI, Chittajallu R, Bortolotto ZA, Dev KK, Duprat F, Henley JM, Collingridge GL, Isaac JT (2000) PDZ proteins interacting with C-terminal GluR2/3 are involved in a PKC-dependent regulation of AMPA receptors at hippocampal synapses. Neuron 28: 873-886. [DOI] [PubMed] [Google Scholar]
- Dong H, O'Brien RJ, Fung ET, Lanahan AA, Worley PF, Huganir RL (1997) GRIP: a synaptic PDZ domain-containing protein that interacts with AMPA receptors. Nature 386: 279-284. [DOI] [PubMed] [Google Scholar]
- Doyle DA, Lee A, Lewis J, Kim E, Sheng M, MacKinnon R (1996) Crystal structures of a complexed and peptide-free membrane protein-binding domain: molecular basis of peptide recognition by PDZ. Cell 85: 1067-1076. [DOI] [PubMed] [Google Scholar]
- Durand GM, Bennett MV, Zukin RS (1993) Splice variants of the N-methyl-d-aspartate receptor NR1 identify domains involved in regulation by polyamines and protein kinase C. Proc Natl Acad Sci USA 90: 6731-6735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ehlers MD (2000) Reinsertion or degradation of AMPA receptors determined by activity-dependent endocytic sorting. Neuron 28: 511-525. [DOI] [PubMed] [Google Scholar]
- El-Husseini AE, Schnell E, Chetkovich DM, Nicoll RA, Bredt DS (2000) PSD-95 involvement in maturation of excitatory synapses. Science 290: 1364-1368. [PubMed] [Google Scholar]
- Engert F, Bonhoeffer T (1999) Dendritic spine changes associated with hippocampal long-term synaptic plasticity. Nature 399: 66-70. [DOI] [PubMed] [Google Scholar]
- Fujisawa S, Aoki C (2003) In vivo blockade of N-methyl-d-aspartate receptors induces rapid trafficking of NR2B subunits away from synapses and out of spines and terminals in adult cortex. Neuroscience 121: 51-63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukunaga K, Soderling TR, Miyamoto E (1992) Activation of Ca2+/calmodulin-dependent protein kinase II and protein kinase C by glutamate in cultured rat hippocampal neurons. J Biol Chem 267: 22527-22533. [PubMed] [Google Scholar]
- Gardoni F, Schrama LH, Kamal A, Gispen WH, Cattabeni F, Di Luca M (2001) Hippocampal synaptic plasticity involves competition between Ca2+/calmodulin-dependent protein kinase II and postsynaptic density 95 for binding to the NR2A subunit of the NMDA receptor. J Neurosci 21: 1501-1509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghosh A, Greenberg ME (1995) Distinct roles for bFGF and NT-3 in the regulation of cortical neurogenesis. Neuron 15: 89-103. [DOI] [PubMed] [Google Scholar]
- Girault JA, Hemmings Jr HC, Zorn SH, Gustafson EL, Greengard P (1990) Characterization in mammalian brain of a DARPP-32 serine kinase identical to casein kinase II. J Neurochem 55: 1772-1783. [DOI] [PubMed] [Google Scholar]
- Goslin K, Banker G (1991) Culturing nerve cells. London: MIT.
- Greengard P, Jen J, Nairn AC, Stevens CF (1991) Enhancement of the glutamate response by cAMP-dependent protein kinase in hippocampal neurons. Science 253: 1135-1138. [DOI] [PubMed] [Google Scholar]
- Grein S, Raymond K, Cochet C, Pyerin W, Chambaz EM, Filhol O (1999) Searching interaction partners of protein kinase CK2beta subunit by two-hybrid screening. Mol Cell Biochem 191: 105-109. [PubMed] [Google Scholar]
- Hall RA, Soderling TR (1997) Differential surface expression and phosphorylation of the N-methyl-d-aspartate receptor subunits NR1 and NR2 in cultured hippocampal neurons. J Biol Chem 272: 4135-4140. [DOI] [PubMed] [Google Scholar]
- Hathaway GM, Traugh JA (1982) Casein kinases-multipotential protein kinases. Curr Top Cell Regul 21: 101-127. [PubMed] [Google Scholar]
- Hollmann M, Heinemann S (1994) Cloned glutamate receptors. Annu Rev Neurosci 17: 31-108. [DOI] [PubMed] [Google Scholar]
- Kim CH, Chung HJ, Lee HK, Huganir RL (2001) Interaction of the AMPA receptor subunit GluR2/3 with PDZ domains regulates hippocampal long-term depression. Proc Natl Acad Sci USA 98: 11725-11730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim E, Cho KO, Rothschild A, Sheng M (1996) Heteromultimerization and NMDA receptor-clustering activity of Chapsyn-110, a member of the PSD-95 family of proteins. Neuron 17: 103-113. [DOI] [PubMed] [Google Scholar]
- Kim JH, Liao D, Lau LF, Huganir RL (1998) SynGAP: a synaptic RasGAP that associates with the PSD-95/SAP90 protein family. Neuron 20: 683-691. [DOI] [PubMed] [Google Scholar]
- Klann E, Chen SJ, Sweatt JD (1993) Mechanism of protein kinase C activation during the induction and maintenance of long-term potentiation probed using a selective peptide substrate. Proc Natl Acad Sci USA 90: 8337-8341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kornau HC, Schenker LT, Kennedy MB, Seeburg PH (1995) Domain interaction between NMDA receptor subunits and the postsynaptic density protein PSD-95. Science 269: 1737-1740. [DOI] [PubMed] [Google Scholar]
- Lan JY, Skeberdis VA, Jover T, Grooms SY, Lin Y, Araneda RC, Zheng X, Bennett MV, Zukin RS (2001) Protein kinase C modulates NMDA receptor trafficking and gating. Nat Neurosci 4: 382-390. [DOI] [PubMed] [Google Scholar]
- Lau LF, Mammen A, Ehlers MD, Kindler S, Chung WJ, Garner CC, Huganir RL (1996) Interaction of the N-methyl-d-aspartate receptor complex with a novel synapse-associated protein, SAP102. J Biol Chem 271: 21622-21628. [DOI] [PubMed] [Google Scholar]
- Lavezzari G, McCallum J, Lee R, Roche KW (2003) Differential binding of the AP-2 adaptor complex and PSD-95 to the C-terminus of the NMDA receptor subunit NR2B regulates surface expression. Neuropharmacology 45: 729-737. [DOI] [PubMed] [Google Scholar]
- Lee SH, Liu L, Wang YT, Sheng M (2002) Clathrin adaptor AP2 and NSF interact with overlapping sites of GluR2 and play distinct roles in AMPA receptor trafficking and hippocampal LTD. Neuron 36: 661-674. [DOI] [PubMed] [Google Scholar]
- Leonard AS, Hell JW (1997) Cyclic AMP-dependent protein kinase and protein kinase C phosphorylate N-methyl-d-aspartate receptors at different sites. J Biol Chem 272: 12107-12115. [DOI] [PubMed] [Google Scholar]
- Leonard AS, Lim IA, Hemsworth DE, Horne MC, Hell JW (1999) Calcium/calmodulin-dependent protein kinase II is associated with the N-methyl-d-aspartate receptor. Proc Natl Acad Sci USA 96: 3239-3244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao D, Zhang X, O'Brien R, Ehlers MD, Huganir RL (1999) Regulation of morphological postsynaptic silent synapses in developing hippocampal neurons. Nat Neurosci 2: 37-43. [DOI] [PubMed] [Google Scholar]
- Liao D, Scannevin RH, Huganir R (2001) Activation of silent synapses by rapid activity-dependent synaptic recruitment of AMPA receptors. J Neurosci 21: 6008-6017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lieberman DN, Mody I (1994) Regulation of NMDA channel function by endogenous Ca(2+)-dependent phosphatase. Nature 369: 235-239. [DOI] [PubMed] [Google Scholar]
- Lieberman DN, Mody I (1999) Casein kinase-II regulates NMDA channel function in hippocampal neurons. Nat Neurosci 2: 125-132. [DOI] [PubMed] [Google Scholar]
- Litchfield DW (2003) Protein kinase CK2: structure, regulation and role in cellular decisions of life and death. Biochem J 369: 1-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu L, Wong TP, Pozza MF, Lingenhoehl K, Wang Y, Sheng M, Auberson YP, Wang YT (2004) Role of NMDA receptor subtypes in governing the direction of hippocampal synaptic plasticity. Science 304: 1021-1024. [DOI] [PubMed] [Google Scholar]
- Luo J, Wang Y, Yasuda RP, Dunah AW, Wolfe BB (1997) The majority of N-methyl-d-aspartate receptor complexes in adult rat cerebral cortex contain at least three different subunits (NR1/NR2A/NR2B). Mol Pharmacol 51: 79-86. [DOI] [PubMed] [Google Scholar]
- Luo JH, Fu ZY, Losi G, Kim BG, Prybylowski K, Vissel B, Vicini S (2002) Functional expression of distinct NMDA channel subunits tagged with green fluorescent protein in hippocampal neurons in culture. Neuropharmacology 42: 306-318. [DOI] [PubMed] [Google Scholar]
- Maletic-Savatic M, Malinow R, Svoboda K (1999) Rapid dendritic morpho-genesis in CA1 hippocampal dendrites induced by synaptic activity. Science 283: 1923-1927. [DOI] [PubMed] [Google Scholar]
- Mammen AL, Kameyama K, Roche KW, Huganir RL (1997) Phosphorylation of the alpha-amino-3-hydroxy-5-methylisoxazole4-propionic acid receptor GluR1 subunit by calcium/calmodulin-dependent kinase II. J Biol Chem 272: 32528-32533. [DOI] [PubMed] [Google Scholar]
- Matsuda S, Mikawa S, Hirai H (1999) Phosphorylation of serine-880 in GluR2 by protein kinase C prevents its C terminus from binding with glutamate receptor-interacting protein. J Neurochem 73: 1765-1768. [DOI] [PubMed] [Google Scholar]
- Meggio F, Marin O, Pinna LA (1994) Substrate specificity of protein kinase CK2. Cell Mol Biol Res 40: 401-409. [PubMed] [Google Scholar]
- Migaud M, Charlesworth P, Dempster M, Webster LC, Watabe AM, Makhinson M, He Y, Ramsay MF, Morris RG, Morrison JH, O'Dell TJ, Grant SG (1998) Enhanced long-term potentiation and impaired learning in mice with mutant postsynaptic density-95 protein. Nature 396: 433-439. [DOI] [PubMed] [Google Scholar]
- Morais Cabral JH, Petosa C, Sutcliffe MJ, Raza S, Byron O, Poy F, Marfatia SM, Chishti AH, Liddington RC (1996) Crystal structure of a PDZ domain. Nature 382: 649-652. [DOI] [PubMed] [Google Scholar]
- Mori H, Manabe T, Watanabe M, Satoh Y, Suzuki N, Toki S, Nakamura K, Yagi T, Kushiya E, Takahashi T, Inoue Y, Sakimura K, Mishina M (1998) Role of the carboxy-terminal region of the GluR epsilon2 subunit in synaptic localization of the NMDA receptor channel. Neuron 21: 571-580. [DOI] [PubMed] [Google Scholar]
- Muller BM, Kistner U, Kindler S, Chung WJ, Kuhlendahl S, Fenster SD, Lau LF, Veh RW, Huganir RL, Gundelfinger ED, Garner CC (1996) SAP102, a novel postsynaptic protein that interacts with NMDA receptor complexes in vivo. Neuron 17: 255-265. [DOI] [PubMed] [Google Scholar]
- Murphy TH, Blatter LA, Wier WG, Baraban JM (1992) Spontaneous synchronous synaptic calcium transients in cultured cortical neurons. J Neurosci 12: 4834-4845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niethammer M, Kim E, Sheng M (1996) Interaction between the C terminus of NMDA receptor subunits and multiple members of the PSD-95 family of membrane-associated guanylate kinases. J Neurosci 16: 2157-2163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Omkumar RV, Kiely MJ, Rosenstein AJ, Min KT, Kennedy MB (1996) Identification of a phosphorylation site for calcium/calmodulin-dependent protein kinase II in the NR2B subunit of the N-methyl-d-aspartate receptor. J Biol Chem 271: 31670-31678. [DOI] [PubMed] [Google Scholar]
- Palczewski K (1997) GTP-binding-protein-coupled receptor kinases—two mechanistic models. Eur J Biochem 248: 261-269. [DOI] [PubMed] [Google Scholar]
- Passafaro M, Sala C, Niethammer M, Sheng M (1999) Microtubule binding by CRIPT and its potential role in the synaptic clustering of PSD-95. Nat Neurosci 2: 1063-1069. [DOI] [PubMed] [Google Scholar]
- Perez JL, Khatri L, Chang C, Srivastava S, Osten P, Ziff EB (2001) PICK1 targets activated protein kinase Cα to AMPA receptor clusters in spines of hippocampal neurons and reduces surface levels of the AMPA-type glutamate receptor subunit 2. J Neurosci 21: 5417-5428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rao A, Craig AM (1997) Activity regulates the synaptic localization of the NMDA receptor in hippocampal neurons. Neuron 19: 801-812. [DOI] [PubMed] [Google Scholar]
- Roberson ED, Sweatt JD (1996) Transient activation of cyclic AMP-dependent protein kinase during hippocampal long-term potentiation. J Biol Chem 271: 30436-30441. [DOI] [PubMed] [Google Scholar]
- Roche KW, Standley S, McCallum J, Dune Ly C, Ehlers MD, Wenthold RJ (2001) Molecular determinants of NMDA receptor internalization. Nat Neurosci 4: 794-802. [DOI] [PubMed] [Google Scholar]
- Sans N, Prybylowski K, Petralia RS, Chang K, Wang YX, Racca C, Vicini S, Wenthold RJ (2003) NMDA receptor trafficking through an interaction between PDZ proteins and the exocyst complex. Nat Cell Biol 5: 520-530. [DOI] [PubMed] [Google Scholar]
- Sarno S, Reddy H, Meggio F, Ruzzene M, Davies SP, Donella-Deana A, Shugar D, Pinna LA (2001) Selectivity of 4,5,6,7-tetrabromobenzotriazole, an ATP site-directed inhibitor of protein kinase CK2 (casein kinase-2′). FEBS Lett 496: 44-48. [DOI] [PubMed] [Google Scholar]
- Scott DB, Blanpied TA, Swanson GT, Zhang C, Ehlers MD (2001) An NMDA receptor ER retention signal regulated by phosphorylation and alternative splicing. J Neurosci 21: 3063-3072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seeburg PH (1993) The TiPS/TINS lecture: the molecular biology of mammalian glutamate receptor channels. Trends Pharmacol Sci 14: 297-303. [DOI] [PubMed] [Google Scholar]
- Seidenman KJ, Steinberg JP, Huganir R, Malinow R (2003) Glutamate receptor subunit 2 serine 880 phosphorylation modulates synaptic transmission and mediates plasticity in CA1 pyramidal cells. J Neurosci 23: 9220-9228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sprengel R, Suchanek B, Amico C, Brusa R, Burnashev N, Rozov A, Hvalby O, Jensen V, Paulsen O, Andersen P, Kim JJ, Thompson RF, Sun W, Webster LC, Grant SG, Eilers J, Konnerth A, Li J, McNamara JO, Seeburg PH (1998) Importance of the intracellular domain of NR2 subunits for NMDA receptor function in vivo. Cell 92: 279-289. [DOI] [PubMed] [Google Scholar]
- Standley S, Roche KW, McCallum J, Sans N, Wenthold RJ (2000) PDZ domain suppression of an ER retention signal in NMDA receptor NR1 splice variants. Neuron 28: 887-898. [DOI] [PubMed] [Google Scholar]
- Tao YX, Rumbaugh G, Wang GD, Petralia RS, Zhao C, Kauer FW, Tao F, Zhuo M, Wenthold RJ, Raja SN, Huganir RL, Bredt DS, Johns RA (2003) Impaired NMDA receptor-mediated postsynaptic function and blunted NMDA receptor-dependent persistent pain in mice lacking postsynaptic density-93 protein. J Neurosci 23: 6703-6712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tingley WG, Ehlers MD, Kameyama K, Doherty C, Ptak JB, Riley CT, Huganir RL (1997) Characterization of protein kinase A and protein kinase C phosphorylation of the N-methyl-d-aspartate receptor NR1 subunit using phosphorylation site-specific antibodies. J Biol Chem 272: 5157-5166. [DOI] [PubMed] [Google Scholar]
- Tovar KR, Westbrook GL (2002) Mobile NMDA receptors at hippocampal synapses. Neuron 34: 255-264. [DOI] [PubMed] [Google Scholar]
- Wang YT, Salter MW (1994) Regulation of NMDA receptors by tyrosine kinases and phosphatases. Nature 369: 233-235. [DOI] [PubMed] [Google Scholar]
- Washbourne P, Bennett JE, McAllister AK (2002) Rapid recruitment of NMDA receptor transport packets to nascent synapses. Nat Neurosci 5: 751-759. [DOI] [PubMed] [Google Scholar]
- Wenthold RJ, Prybylowski K, Standley S, Sans N, Petralia RS (2003) Trafficking of NMDA receptors. Annu Rev Pharmacol Toxicol 43: 335-358. [DOI] [PubMed] [Google Scholar]
- Wyszynski M, Lin J, Rao A, Nigh E, Beggs AH, Craig AM, Sheng M (1997) Competitive binding of alpha-actinin and calmodulin to the NMDA receptor. Nature 385: 439-442. [DOI] [PubMed] [Google Scholar]
- Xia J, Chung HJ, Wihler C, Huganir RL, Linden DJ (2000) Cerebellar long-term depression requires PKC-regulated interactions between GluR2/3 and PDZ domain-containing proteins. Neuron 28: 499-510. [DOI] [PubMed] [Google Scholar]