

Abstract
Neuronal activity is accompanied by transmembranous ion fluxes that cause cell volume changes. In whole mounts of the guinea pig retina, application of glutamate resulted in fast swelling of neuronal cell bodies in the ganglion cell layer (GCL) and the inner nuclear layer (INL) (by ∼40%) and a concomitant decrease of the thickness of glial cell processes in the inner plexiform layer (IPL) (by ∼40%) that was accompanied by an elongation of the glial cells, by a thickening of the whole retinal tissue, and by a shrinkage of the extracellular space (by ∼18%). The half-maximal effect of glutamate was observed at ∼250 μm, after ∼4 min. The swelling was caused predominantly by AMPA-kainate receptor-mediated influx of Na+ into retinal neurons. Similar but transient morphological alterations were induced by high K+ and dopamine, which caused release of endogenous glutamate and subsequent activation of AMPA-kainate receptors. Apparently, retinal glutamatergic transmission is accompanied by neuronal cell swelling that causes compensatory morphological alterations of glial cells. The effect of dopamine was elicitable only during light adaptation but not in the dark, and glutamate and high K+ induced strong ereffects in the dark than in the light. This suggests that not only the endogenous release of dopamine but also the responsiveness of glutamatergic neurons to dopamine is regulated by light-dark adaptation. Similar morphological alterations (neuronal swelling and decreased glial process thickness) were observed in whole mounts isolated immediately after experimental retinal ischemia, suggesting an involvement of AMPA-kainate receptor activation in putative neurotoxic cell swelling in the postischemic retina.
Keywords: glutamate, dopamine, ATP, neuroglia, circadian phase, ischemia, retina
Introduction
Glial cells play crucial roles in supporting neuronal survival and information processing (Newman and Reichenbach, 1996): they regulate the extracellular homeostasis of relevant ions including K+, control the water content of the extracellular space, and mediate the rapid termination of neuronal activity by transmitter uptake from the synaptic cleft (Matsui et al., 1999). In the retina, neuronally released K+, water, and glutamate are cleared from the extracellular space mainly by the radial glial (Müller) cells. Retinal glial cells display a complex pattern of K+ channel expression in their plasma membranes that directs the flow of neuronally released K+ into the glial cell bodies and farther into the vitreous and the blood vessels (Kofuji et al., 2002). Parallel water fluxes were suggested to be coupled to the transglial K+ fluxes (Nagelhus et al., 1999).
The K+ and water fluxes, generated by neuronal activity and mediated by glial cells, are accompanied by swelling of cells and shrinkage of the extracellular space caused by the decreased extracellular osmolality (Dietzel et al., 1980; Jendelová and Syková, 1991; Syková, 1991). Cell volume alterations are important, particularly under pathological conditions such as epilepsy and ischemia in which overexcited neurons release large amounts of glutamate and K+ (Adachi et al., 1972; Glass and Dragunow, 1995; Nicholson and Syková, 1998). Increased extracellular glutamate may evoke slow swelling of retinal glial cells resulting from the influx of Na+, which drives the glial glutamate transporters (Izumi et al., 1996), and high K+ may cause swelling of astrocytes mediated by the Na-K-2Cl cotransport (Walz and Hinks, 1985; MacVicar et al., 2002). Spreading depression, a profound transient and progressing depolarization of neurons and glia (Somjen, 2001), is accompanied by cell swelling and can be blocked by antagonists of NMDA receptors, whereas blockers of AMPA-kainate receptors are essentially ineffective (Anderson and Andrew, 2002).
With the exceptions of the relatively slow glutamate-evoked swelling of glial cells (Izumi et al., 1996) and the cell volume changes during spreading depression, little is known about fast neurotransmitter-induced morphological alterations of glial and neuronal cells in the normal vertebrate retina. Here we show that retinal glial cells quickly and strongly change their shape when glutamate is applied to retinal whole mounts and that this effect is induced by glutamate-evoked neuronal cell swelling. At least one other neurotransmitter, dopamine, exerts a similar effect on retinal cell morphology. Because dopamine plays a crucial role in retinal light adaptation and in signaling nightday transitions of retinal activity (Morgan and Boelen, 1996), we studied the glutamate-, high K+-, and dopamine-induced morphological alterations during light versus dark adaptation and found significant differences between these two functional states. Furthermore, because glutamate excitotoxicity has been implicated in the pathomechanism of neuronal degeneration in the postischemic retina (Osborne et al., 2004), we investigated the morphological alterations of neuronal and glial retinal cells immediately after pressure-induced retinal ischemia.
Materials and Methods
Materials. LY341495, l-anti-endo-3,4-methanopyrrolidinedicarboxylate (MPDC), and d,l-threo-β-benzyloxyaspartate (TBOA) were from Tocris (Bristol, UK). ATP and GABA were purchased from Serva Electrophoresis (Heidelberg, Germany). All other substances were from Sigma-Aldrich (Taufkirchen, Germany), unless stated otherwise.
Whole-mount preparation and dye loading. The experiments were performed on acutely isolated retinal whole mounts of adult guinea pigs. The animals were kept in a 12 hr light/dark cycle; food and water were available ad libitum. All experiments were performed at room temperature. The animals were anesthetized deeply with urethane (2 gm/kg, i.p.) before decapitation. Pieces of retinal whole mounts (5 × 5 mm) were explanted and mechanically fixed in a perfusion chamber, with their vitreal surface up. The whole mounts were loaded with Mitotracker Orange (chloromethyltetramethylrosamine, 10 μm; Molecular Probes, Eugene, OR) resolved in extracellular solution; stem solution was made in dimethylsulfoxide (DMSO). For some records, whole mounts were loaded simultaneously with Mitotracker Deep Red and FM 1-43 (Molecular Probes), a vital dye that stains cell membranes. After an incubation time of 1-3 min, the tissues were examined using a confocal laser scanning microscope (LSM 510 META; Zeiss, Oberkochen, Germany) by viewing from the vitreal side. Mitotracker Orange was excited at 543 nm; emission was recorded with a 560 nm long-pass filter. Mitotracker Deep Red and FM 1-43 were excited at 633 and 488 nm, respectively; emissions were recorded by using 650 and 560 nm long-pass filters. Reflection images of the retina were acquired using a 650 nm HeNe laser and a 340 nm long-pass filter. Images were taken from a 230 × 230 μm area. Most experiments were performed in normal room light; experiments in the dark were made after 24 hr adaptation of the animals. Isolation of the retina and mechanical fixing of the whole mounts were performed under red light. To compare responses during light and dark conditions, reflection images of the unstained retina were recorded between 1 and 3 P.M. (light) and 6 and 8 P.M. (dark), respectively.
Solutions. The recording chamber was perfused continuously with extracellular solution; test substances were added by fast changing (10 sec) of the perfusate. The extracellular solution consisted of (in mm): 110 NaCl, 3 KCl, 2 CaCl2, 1 MgCl2, 1 Na2HPO4, 10 HEPES, 11 glucose, and 25 NaHCO3, adjusted to pH 7.4 with Tris, and bubbled with carbogen (95% O2, 5%CO2). The nominally Ca2+-free extracellular solution contained 0.1 mm CaCl2 and 1 mm EGTA. The tissue was preincubated for at least 10 min with calcium-free solution before test substances were added. Na+-free solution was achieved by replacing NaCl by choline chloride; elevation of the K+ concentration was made by equimolar reduction of Na+. For preparation of the low-Cl- solution, sodium gluconate or sodium methylsulfate was substituted for NaCl. Low- and high-osmolarity solutions were made by reducing and increasing, respectively, the NaCl content of the bath solution.
Calcium imaging. Whole mounts of isolated retinal pieces were placed, with their vitreal surface up, in the perfusion chamber and incubated for 1 hr at room temperature in extracellular solution containing two different calcium-sensitive fluorescence dyes, Fluo-4 AM (11 μm) and fura-Red AM (17 μm; Molecular Probes). Stock solutions of the dyes were made in DMSO plus pluronic F-127 (2%). After loading with fluorescence dyes, retinal whole mounts were perfused with extracellular solution for at least 10 min before application of test substances. The fluorescence dyes were excited at 488 nm; the emission of Fluo-4 was recorded with a bandpass filter between 505 and 550 nm, and the emission of fura-Red was recorded with a 650 nm long-pass filter. Images were taken every 5 sec.
Imaging of single dye-filled glial cells and of the intrinsic optical signal. To determine the glutamate-evoked elongation of Müller cells and the thickening of the retinal tissue, single Müller cells in acutely isolated retinal slices (thickness, 1 mm) were iontophoretically filled with a saturated solution of Lucifer yellow. The pipettes (5 MΩ) were located near the vitreal surface of the slices, and the dye was injected by applying negative 10 V pulses of 300 msec duration at 2.5 Hz. Lucifer yellow was excited at 430 nm, and emission >470 nm was recorded every second by using a conventional imaging system (Till Photonics, Gräfelfing, Germany). Simultaneously, the intrinsic optical signals of the slices were recorded as transmission of infrared light at 890 nm.
Measurement of the extracellular space volume. Microelectrodes selective for tetramethylammonium (TMA) were used to measure TMA+ concentration in the extracellular space reflecting the volume of this space. TMA+-selective microelectrodes were prepared as described for K+-selective electrodes (Syková, 1992); the ion exchanger was Corning 477317 (Fluka, Milwaukee, WI), but the ion-sensing barrel was backfilled with 100 mm TMA chloride instead of 150 mm KCl. The microelectrode was connected to the reference electrode, which was filled with NaCl (150 mm), and both electrodes were placed in the inner plexiform layer by using infrared microscopy. TMA+ (100 μm) was added to the extracellular solution. The electrodes were calibrated before and after each experiment in a sequence of solutions of 150 mm NaCl plus 3 mm KCl with the addition of different concentrations of TMA chloride. Because the thinness of the guinea pig retina did not allow us to use the real-time iontophoretic method to determine the absolute values of the extracellular space volume (Nicholson and Syková, 1998), only relative changes of the extracellular space volume were calculated from the changes of the TMA+ baseline concentrations.
Ischemia model. Retinal ischemia was induced in one eye of adult guinea pigs, and the other eye remained untreated and served as control. Ketamine/xylazine anesthesia was induced, and the anterior chamber of the treated eye was cannulated from the pars plana with a 27 gauge infusion needle connected to a bag containing normal saline. The intraocular pressure was increased to 160 mmHg for 60 min by elevating the saline bag; immediately after the end of ischemia, retinal whole mounts and slices were acutely isolated.
Data analysis. The diameters of Müller cell trunks in the IPL were measured by analyzing the Mitotracker Orange fluorescence or the reflection data by using a computer program based on an erosion algorithm programmed with Delphi 6 personal free trial version (Borland, Scotts Valley, CA). In each whole mount investigated, the glial cell profiles present in three areas of 57.5 μm2 were measured, and the mean areas of ∼80 trunks in the IPL were calculated, assuming a circular cross section. The diameters of nerve cell bodies were measured manually using the LSM software, and the cross-sectional areas were calculated. To evaluate the calcium responses, the fluorescence ratio FFluo-4/Ffura-Red was calculated. Subsequently, the values were normalized to the prestimulus baseline value by calculation of the ratio F/F0, where F0 represents the mean baseline fluorescence ratio that was calculated using the mean of the last 10 values before agonist application. Statistical analysis (unrelated or paired t test) was made using the Prism program (Graph Pad Software, San Diego, CA); n represents the number of whole mounts or slices investigated. Statistical significance was accepted at p < 0.05.
Results
Glutamate-evoked morphological alterations
To detect activity-dependent cellular reshaping within the retina of the guinea pig, confocal recordings were made in acutely isolated retinal whole mounts within the GCL, IPL, and INL, respectively (Fig. 1A). The vital dye, Mitotracker Orange, selectively stained glial (Müller) cells in the whole mounts (Uckermann et al., 2004). The view of the GCL (Fig. 1B, left) displays dark round somata of neuronal cells and green fluorescent cross sections through glial cell end feet surrounding the neuronal cell bodies. The optical section through the IPL (Fig. 1C, left) show cross sections through vitreal trunks of retinal glial cells (large green profiles) and a pattern of small red structures, representing synaptic elements that reflect light. The view of the INL reveals unstained neuronal cell bodies and green fluorescent glial cell bodies (Fig. 1D, left).
Figure 1.

Glutamate evokes morphological alterations in whole-mount guinea pig retinas. A, In a retinal slice, Mitotracker Orange (10 μm) selectively stained glial (Müller) cells (green). NFL, Nerve fiber layer; OPL, outer plexiform layer; ONL, outer nuclear layer. B, Bath application of l-glutamate (1 mm) resulted in a reversible swelling of the neuronal cell bodies within the GCL-NFL (left). The view of the GCL-NFL shows unstained somata that are surrounded by glial cell end feet that are green labeled by Mitotracker Orange. The images were recorded before (control), 10 min after the onset, and 40 min after the end of agonist application. Middle, Scatter plot of the cross-sectional areas of neuronal cell bodies (in square micrometers) within the GCL-NFL that were measured before and during glutamate exposure (n = 208 somata from 4 whole mounts), clearly showing agonist-evoked cell body swelling. Right, Mean (±SD) cross-sectional areas of neuronal cell bodies within the GCL-NFL before (control), 10 min after the onset, and 40 min after the end of glutamate application. (Data were obtained from 88 cells from 3 whole mounts.) C, Recordings were made with the focal plane at the IPL displaying profiles through Müller cell stem processes (green stained by Mitotracker) and synaptic structures between them (red). Application of l-glutamate (1 mm) for 10 min decreased reversibly the thickness of the glial cell profiles. The recovery was recorded after 30 min washout. Middle, Histogram showing cross-sectional areas of glial cell profiles (in square micrometers) in the IPL (n = 105 cells from 3 whole mounts) that were measured before and during agonist application. Right, Mean (±SD) cross-sectional areas of glial cell profiles within the IPL before (control) and 10 min after application of l-glutamate (1 mm) or d-glutamate (1 mm). The data were obtained in five independent experiments. D, l-Glutamate-evoked morphological alterations in the INL. After a 10 min agonist exposure, the neuronal cell bodies (dark round structures) were swollen, whereas the green-stained glial cell bodies displayed a decreased thickness. Scale bars, 20 μm. Significant differences versus control: •p < 0.05; •••p < 0.001. Significant effect of washout: ○○p < 0.01.
Bath application of glutamate (1 mm) for 10 min resulted in a reversible swelling of nerve cell bodies in the GCL (Fig. 1B, left) and INL (Fig. 1D), accompanied by a reversible decrease of the thickness of the glial cell profiles in the IPL (Fig. 1C, left) and INL (Fig. 1D). Without agonist, the cross-sectional areas of the profiles in the IPL remained constant for at least 30 min (100.6 ± 3.4%; n = 10; not significant). Histograms showing the cross-sectional areas of nerve cell bodies in the GCL (Fig. 1B, middle) and glial cell profiles in the IPL (Fig. 1C, middle) under control conditions versus glutamate exposure reveal this opposite modulation of the morphological parameters of glial and neuronal elements. The cross-sectional area of the neuronal cell bodies in the GCL increased by ∼40% compared with control in the presence of glutamate (1 mm) (n = 3; p < 0.05) (Fig. 1B, right), whereas the thickness of the glial cell profiles in the IPL decreased by ∼40% (n = 3; p < 0.01) (Fig. 1C, right).
The glutamate-evoked decrease of the cross-sectional area of glial cell bodies and trunks was accompanied by an elongation of the cells, suggesting that the decrease of glial cell process thickness may be compensated to maintain the volume of the cells. Recordings of the intrinsic optical signals of retinal slices revealed that the inner retinal layers, especially, became thicker during exposure to glutamate (Fig. 2Aa). Recordings of the fluorescence signals of single dye-filled glial cells showed that the distance between the cell soma (which lies within the INL) and the cell end foot (which abutts the vitreous body, in situ) became longer (Fig. 2Ab,Ac). The inner retinal part of the glial cells elongated by 16.9 ± 4.2% (n = 4; p < 0.05) in the presence of glutamate, whereas the outer retinal part remained essentially unaltered (Fig. 2B). In additional experiments, the cross-sectional area of the glial cell profiles in the IPL was recorded to monitor the morphological alterations in the retina.
Figure 2.

Glutamate (1 mm) evokes thickening of retinal slices and elongation of single dye-filled glial (Müller) cells. A, Example of one slice. Aa, Intrinsic optical signals in dependence on the recording time. Agonist application is indicated by the thick bar. There is an artifact at the time of removal of the agonist. Ab, Time dependence of the fluorescence signal of one Lucifer yellow-filled glial (Müller) cell. Ac, Examples of fluorescence records of the dye-filled cell at three different time points indicated by the arrows in Ab. The distance between soma and end foot of the cell is indicated. Scale bars, 20 μm. B, Lengths of the outer and inner retinal parts of fluorescence dye-filled glial (Müller) cells, measured after 10 min of glutamate exposure. The data were obtained in four independent experiments and are expressed as percentage of control (100%). •p < 0.05.
The glutamate effect on glial cell shape was dose and time dependent and stereo specific: l-glutamate decreased the area of the glial cell profiles, whereas application of d-glutamate (1 mm for 10 min) resulted in a slight reversible increase of their thickness (Fig. 1C, right). Glutamate concentrations >100 μm decreased the profile area; the half-maximal effect was observed at ∼250 μm, and concentrations >500 μm displayed maximal effects (Fig. 3A). The glutamate effect became visible 1-2 min after the onset of agonist application, was maximal after 10 min of agonist exposure, and remained constant as long as glutamate was present in the bath (for at least 30 min) (Fig. 3B). The recovery after washout of glutamate was slow; after a 30 min washout period, the profile area was restored to 89.5 ± 2.2% of control (n = 3; p < 0.01) (Fig. 3C).
Figure 3.

The glutamate-induced fast morphological alterations of retinal glial cells are evoked by activation of AMPA-kainate receptors. The cross-sectional areas of the glial cell profiles in the IPL are given as percentage of control (100%). A, Concentration dependence of the glutamate effect on the thickness of the profiles. Half-maximal effect was observed at ∼250 μm. B, Time dependence of the glutamate (1 mm) effect on the thickness of the profiles. Half-maximal effect was observed after 4 min. C, Time dependence of the recovery of the profile thickness during washout of glutamate. After a 10 min incubation of glutamate, the washout was started (washout time 0). Half-maximal washout effect was observed after 17 min. D, The effect of glutamate (1 mm) on the thickness of the profiles was mimicked by kainate (100 μm) but not by AMPA (50 μm) or NMDA (100 μm). E, Time dependence of the effect of kainate (100 μm) on the thickness of the glial cell profiles. Half-maximal effect was observed after 2 min. F, The effect of glutamate (1 mm) on the thickness of the profiles was blocked by CNQX (50 μm; a competitive inhibitor of non-NMDA glutamate receptors) but not by LY341495 (100 μm; a blocker of metabotropic glutamate receptors), MK-801 (10 μm; a blocker of NMDA receptors), MPDC (100 μm; an inhibitor of Na+-dependent high-affinity glutamate transporters), or TBOA (100 μm; a blocker of excitatory amino acid transporters). In A and D-F, the agonists with or without antagonists were simultaneously applied for 10 min. The data were obtained in 3-12 independent experiments. Significant differences versus control: •p < 0.05; ••p < 0.01; ○○○p < 0.001. Significant effect of the blocker: ○○○p < 0.001.
Receptor subtype involved in glutamate-evoked morphological alteration
To determine the subtypes of glutamate receptors involved in the morphological alterations of retinal glial cells, several different receptor agonists were tested. Among the glutamate receptor agonists tested (kainate, AMPA, NMDA), only kainate was able to mimic the effect of glutamate (Fig. 3D), and among the glutamate receptor and uptake blockers tested (CNQX, LY341495, MK-801, MPDC, TBOA), only the AMPA-kainate receptor-selective blocker CNQX inhibited the effect of glutamate (Fig. 3F). The data suggest that activation of AMPA-kainate receptors mediated the glutamate effect on glial cell morphology. Kainate exerted its effect faster than glutamate: the maximal effect was observed after 4 min (compared with 10 min during glutamate exposure) (Fig. 3E).
Glial (Müller) cells of the guinea pig retina do not express functional AMPA-kainate receptors. This conclusion is based on the following observations. (1) In whole-cell patch-clamp experiments, both in situ and on acutely isolated cells, extracellular application of glutamate (1 mm) or kainate (1 mm) did not modify the whole-cell conductance (T. Pannicke, personal communication). (2) In calcium imaging experiments on acutely isolated Müller cells, extracellular application of glutamate (1 mm) did not induce any alteration of the intracellular free calcium concentration, whereas extracellular ATP (100 μm) evoked a transient calcium response (data not shown). Comparable results were obtained in fluorimetric calcium imaging experiments on retinal whole mounts (Fig. 4). (3) Bath application of glutamate (1 mm) evoked intracellular calcium rises in neuronal cell bodies within the GCL (Fig. 4, right top) and in the synaptic structures in the IPL (Fig. 4, right bottom), whereas glial cell structures (both end feet in the GCL and profiles in the IPL) did not show any significant response; application of ATP (200 μm) did evoke transient calcium rises in glial cells but not in neuronal cells (Fig. 4, left). It is concluded that the glutamate effect on glial cell morphology is mediated indirectly by AMPA-kainate receptors on retinal neurons.
Figure 4.

Glutamate evokes intracellular calcium rises in retinal neurons but not in glial cells. Example of records in one whole mount of the guinea pig retina. Middle, Confocal views of the GCL-NFL (top) and IPL (bottom), respectively. The views were recorded before (control) and at the peak calcium responses after application of ATP (200 μm) and glutamate (1 mm), respectively. In the case of ATP application, glial cell end feet and profiles responded with transient calcium rises (left, gray traces), whereas neuronal cell bodies and synaptic structures were unresponsive (left, black traces). On application of glutamate, neuronal cell bodies and synaptic structures showed calcium responses (right, black traces), whereas glial cell end feet and profiles were unresponsive (right, gray traces).
Mechanism of glutamate-evoked morphological alterations of glial cells
The glutamate effects on glial cell morphology may be caused by different mechanisms such as transmembrane osmolyte fluxes and accompanying water shifts (which may alter the volume of the cells) or alterations of the cytoskeleton (which may change the cell shape). Neuronal activity is accompanied by increases of the extracellular K+ concentration, and glial cells mediate retinal K+ fluxes from the tissue into the vitreous via K+ channels (Newman and Reichenbach, 1996). To detect a possible involvement of K+ channels, the K+ channel blockers TEA and Ba2+ were tested. TEA (1 mm) has been shown to block distinct types of K+ channels in retinal glial cells (Bringmann et al., 1997), whereas Ba2+ (1 mm) fully blocks the main membrane conductance mediated by inwardly rectifying K+ channels (Reichelt and Pannicke, 1993). The glutamate-evoked shrinkage of the glial cell profiles in the IPL was not blocked by either TEA or Ba2+ (Fig. 5A), suggesting that the effect was not mediated predominantly by ion fluxes through K+ channels. Furthermore, a blocker of the Na-K-2Cl cotransport, furosemide, was without effect (Fig. 5A). The glutamate effect was blocked in Na+-free or low-Cl- extracellular solutions (Fig. 5A), however, which suggests that an influx of Na+ into retinal neurons, possibly via open AMPA-kainate receptor channels, and a simultaneous influx of Cl- ions is a step in the signaling cascade mediating the morphological changes of retinal glial cells. Two different Cl- channel blockers [flufenamic acid and 5-nitro-2-(3-phenylpropylamino)benzoic acid (NPPB)], as well as picrotoxin, did not inhibit the glutamate-evoked morphological alterations (Fig. 5A). A Ca2+ influx into retinal cells seems not to be required, because neither Ca2+-free extracellular solution nor a solution that was supplemented with tetrodotoxin (1 μm) plus Cd2+ (100 μm) modified the glutamate action on glial cells (Fig. 5B). An increase of the osmolarity of the bath solution (by increasing its Na+ content) caused a thickening of the glial cell trunks, whereas a decrease of the osmolarity was without significant effect (Fig. 5C). Similarly, a hypotonic solution that was prepared by diluting the extra-cellular solution to 60% ionic strength was without effect (Fig. 5D). A similar lack of effect of hypotonic solution on the cell volume has been described recently in glial cells of the rat retina (Pannicke et al., 2004). Addition of taurine to the extracellular solution did not change the glutamate-evoked morphological alterations (Fig. 5E).
Figure 5.

Glutamate-evoked morphological alterations of retinal glial cells are mediated by Na+ and Cl- fluxes. The cross-sectional areas of the glial cell profiles in the IPL are given as percentage of control (100%). A,Na+-free or low-Cl- extracellular solutions reversed the morphological effect of glutamate. The K+ channel blockers TEA (10 mm) and Ba2+ (1 mm), the Na-K-2Cl cotransport blocker furosemide (5 mm), and the Cl- channel blockers flufenamic acid (500 μm), NPPB (100 μm), and picrotoxin (200 μm) did not inhibit the glutamate effect. The low-Cl- extracellular solutions were made by exchanging NaCl with either sodium gluconate or sodium methylsulfate. B, Ca2+-free extracellular solution or a solution that was supplemented with TTX (1 μm) plus Cd2+ (100 μm) did not alter the glutamate-evoked effects on glial cell morphology. C, Effect of solutions with different osmolarities on the cross-sectional area of glial cell trunks. The osmolarity was changed by alterations of the NaCl concentration in the extracellular solution. D, Effect of an extracellular solution that was diluted to 60% ionic strength. E, Addition of taurine (20 mm) to the extracellular solution did not change the glutamate-evoked morphological alterations. F, Nocodazol (10 μg/ml; preincubation for 2 hr at room temperature), a microtubuli destabilizing drug, did not alter the glutamate effect on the glial cell morphology. Similarly, cytochalasin D (1 μg/ml; preincubation for 15 min), which disrupts the actin cytoskeleton, and ML7 (50 μm; preincubation for 1.5 hr), an inhibitor of the myosin light-chain kinase, did not inhibit the action of glutamate. The effects of glutamate (1 mm) and ions were measured after a 10 min exposure. Recovery was measured after 15 min washout. Means ± SEM of three to six experiments. Significant differences versus control: •p < 0.05; ••p < 0.01; •••p < 0.001. Significant effects of the blocker and of washout: ○p < 0.05.
To test whether the morphological changes of glial cells depend on their cytoskeleton, different inhibitors were tested. Nocodazol, a microtubules-destabilizing drug, did not change the effect of glutamate on glial cells (Fig. 5F). Similarly, depolymerization of the actin cytoskeleton by cytochalasin D was without effect on the action of glutamate (Fig. 5F). To test the requirement of myosin-actin interactions for the morphological changes, the myosin light-chain kinase inhibitor ML7 was preincubated, but this also failed to inhibit the action of glutamate (Fig. 5F). Apparently the observed responses do not require alterations of the microtubule organization or other alterations of the cytoskeleton.
Another possible explanation of how retinal glial cell processes in the IPL might become thinner during glutamate application is “simple” mechanical compression by the swollen cell somata and processes that should be reflected in a shrinkage of the extracellular space. To investigate volume alterations of the extracellular space, the concentration of TMA+ in the IPL was measured by using TMA+-containing bath solution and TMA+-sensitive microelectrodes. Bath application of l-glutamate resulted in a reversible increase of the extracellular TMA+ concentration by ∼22% after 10 min agonist exposure (Fig. 6B), which suggests a decrease of the extracellular space volume by ∼18%. The time dependence of the TMA+ concentration changes was found to be similar to the glutamate-evoked morphological changes of glial cell processes, during both agonist exposure and washout (Fig. 6A).
Figure 6.

Glutamate application causes a decline of the volume of the extracellular space in the IPL of the guinea pig retina. The volume was measured by using TMA+-containing bath solution and TMA+-sensitive microelectrodes. A, The trace shows that bath application of glutamate (1 mm) results in a reversible increase of the electrode potential, which reflects an increase of the extracellular TMA+ concentration in the IPL. The gray bars show the mean time-dependent changes of the cross-sectional area of glial cell profiles. The black bar indicates agonist exposure. B, Mean (±SEM) TMA+ concentration in the IPL before (control) and 10 min after the onset of glutamate (1 mm) exposure (n = 5). ••p < 0.01.
K+-evoked morphological alterations
Elevation of the K+ concentration in the bath solution from 3 to 50 mm resulted in a decrease of the area of glial cell profiles in the IPL (Fig. 7A). Additionally, dark spherical structures appeared in the IPL between the glial cell profiles, likely representing swollen synaptic elements. In contrast to the effect of glutamate, the effect of the high-K+ solution was fast and transient: the maximal effect (a decrease by ∼50%) was observed after 3 min (Fig. 7B). When the high-K+ solution was applied for >3 min, the thickness of the glial trunks returned slowly to the control value, and a full recovery was observed after 120 min (Fig. 7B). The K+-evoked effect on glial cell shape was inhibited in the presence of the AMPA-kainate receptor blocker, CNQX, or of riluzole, an inhibitor of endogenous glutamate release (Fig. 7C). The data suggest that the high-K+ effect on glial cells was caused by stimulation of endogenous glutamate release and AMPA-kainate receptor activation. The effect of high K+ was partially inhibited (by ∼50%) by simultaneous application of the K+ channel blocker Ba2+ or TEA (Fig. 7D). Simultaneous application of both blockers caused no enhancement of the blocking effect (data not shown). Similarly, in the presence of a Ca2+-free extracellular solution, the effect of high K+ was partially blocked (Fig. 7D), and TEA did not show any additional inhibiting effect (Fig. 7D). This suggests that open K+ channels mediate a high-K+-evoked cell depolarization that then results in extracellular Ca2+-dependent endogenous glutamate release.
Figure 7.

High extracellular K+ evokes transient morphological alterations in the guinea pig retina that are caused by stimulation of endogenous glutamate release. A, Elevation of the K+ concentration in the bath solution from 3 to 50 mm caused a decrease of the cross-sectional areas of glial cell profiles in the IPL (recorded before and after 5 min of high-K+ exposure). Cross sections through Müller cell processes were red-stained by Mitotracker Deep Red, and cell membranes were stained green by FM 1-43. Scale bar, 5 μm. B, Time dependence of the high-K+ (50 mm) effect. C, The effect of high K+ was inhibited by coapplication of the AMPA-kainate receptor blocker CNQX (50 μm) and of the glutamate release blocker riluzole (500 μm), respectively. D, The effect of high K+ was partially inhibited by coapplication of Ba2+ (1 mm) or TEA (10 mm) and in the presence of a Ca2+-free extracellular solution (with or without 10 mm TEA). E, The presence of glutamine in the extracellular solution partially restored the effect of high K+ on the glial morphology during a second application. The whole mounts were superfused for 10 min with a high-K+ (50 mm) solution that decreased the thickness of glial cell profiles by ∼40% (left). Subsequently, the high-K+ solution was washed out for 45 min in the absence or presence of glutamine (250 μm) (recovery; middle), and then the high-K+ solution was again applied for 10 min (right). As control, glutamate (1 mm) was tested. The cross-sectional areas of the glial cell profiles in the IPL are given as percentage of control (100%). In C and D, the effects were measured after 10 min exposures. Data were obtained in 3-17 independent experiments. Significant effect versus control: •p < 0.05; ••p < 0.01; •••p < 0.001. Significant effects of the blocker: ○p < 0.05; ○○○p < 0.001.
The bath application of the high-K+ solution caused a nearly complete release of endogenous glutamate stores because a second application of this solution, after a 45 min washout period, caused only a slight, nonsignificant effect on glial cell process diameter (by 5%) (Fig. 7E). When the slices were superfused with a solution containing glutamine (the precursor of neuronal glutamate synthesis), however, the second application of the high-K+ solution induced a significantly stronger effect on glial cells (by 22%; p < 0.05). As control, glutamate (1 mm) was applied and decreased the thickness of the glial cell processes by ∼40%.
Dopamine-evoked morphological alterations
Another way that neuronal AMPA-kainate receptor activation may induce effects on glial cells might be the stimulation of receptors on glial cells by neuronally released bioactive substances. Retinal glial cells express purinergic P2 receptors (Newman, 2001) that may be activated by neuron- or glia-derived ATP (Santos et al., 1999; Newman, 2001); however, bath application of ATP (200 μm) had no effect on Müller cell morphology and did not block the effect of glutamate (Fig. 8A). A segregation of ATP- and glutamate-evoked signaling is also indicated by the results of the calcium imaging experiments shown above (Fig. 4). Similarly, GABA (1 mm) application had no significant effects on glial cell morphology and failed to modify the action of glutamate (Fig. 8A). In contrast, application of dopamine (100 μm) resulted in a significant decrease of the area of glial cell profiles in the IPL (by ∼10%) and amplified the effect of glutamate (Fig. 8A). The dopamine effect is mediated by stimulation of endogenous glutamate release and subsequent AMPA-kainate receptor activation, as shown by its inhibition in the presence of CNQX (Fig. 8B). When dopamine was applied for longer time periods, it became evident that its effect on glial cells was transient, with a maximal effect after ∼10 min and no remaining effect after ∼25 min (Fig. 8C).
Figure 8.

Dopamine evokes morphological alterations via stimulation of endogenous glutamate release. The areas of the glial cell profiles in the IPL are given as percentage of control (100%). A, Effects of ATP (200 μm), GABA (1 mm), or dopamine (100 μm) at control conditions and during co-exposure with glutamate (1 mm). B, The AMPA-kainate receptor blocker CNQX (50 μm) inhibits the dopamine effect on glial cell morphology. C, Time dependence of the dopamine (100 μm) effect. In A and B, the effects were measured after a 10 min exposure. Means ± SEM of 3-10 experiments. Significant differences versus control: •p < 0.05; •••p < 0.001. Significant effect of dopamine during glutamate exposure: ○p < 0.05.
Dopamine plays a crucial role in retinal light adaptation and in the signaling of night-day transitions of retinal activity, with increased dopamine release in the light (Morgan and Boelen, 1996). Therefore, we investigated the effects of agonists and high K+ on glial cell morphology both during normal room light and under dark conditions. Under control conditions, the area of glial cell profiles in the IPL was slightly but not significantly smaller in the dark than in the light (Fig. 9A), probably a reflection of a slightly elevated endogenous glutamate release in the dark. Application of glutamate in the dark resulted in faster and stronger changes of the glial cell morphology than in the light; the difference was significant (p < 0.05) after a 10 min exposure (Fig. 9B). Furthermore, the high-K+ response after 5 min was stronger in the dark. It is noteworthy that although dopamine evoked an effect on the glial cell morphology in the light after 10 min, it was ineffective in the dark (p < 0.05). This suggests that the dopaminergic- and K+-induced stimulations of endogenous glutamate release are dependent on dark versus light conditions.
Figure 9.

The changes of the glial cell morphology differ in their amplitudes during dark and light. A, Mean (±SEM) areas of the glial cell profiles in the IPL during light and dark. B, Effects of glutamate (1 mm), of a solution containing 50 mm K+, and of dopamine (100 μm) on glial cell morphology. The effects were measured after 5 min (50 mm K+) and 10 min exposures, respectively. The cross-sectional areas of the glial cell stem processes in the IPL are given as percentage of control (100%). The data were obtained in four to nine independent experiments. Significant differences versus control: ••p < 0.01; ○○○p < 0.001. Significant differences between light and dark: ○p < 0.05.
Ischemia-induced morphological alterations
The excitotoxicity of excessively released glutamate is suggested to be one main cause of neuronal cell degeneration in the retina during ischemia-reperfusion (Osborne et al., 2004). To reveal whether transient ischemia causes morphological alterations in the guinea pig retina, retinal whole mounts and slices were acutely isolated immediately after the end of 60 min of pressure-induced ischemia in vivo. As shown above for acute application of glutamate, the cross-sectional areas of the glial cell profiles in the IPL were significantly smaller in the postischemic retina compared with control (by 10%) (Fig. 10A), whereas the cross-sectional areas of neuronal cell bodies in the GCL were significantly thicker (by 12%) (Fig. 10B). These morphological alterations of retinal cells were accompanied by a thickening of the whole retinal tissue compared with control (Fig. 10C, left), with a thickening of the IPL by 13% (Fig. 10C, right). Together, the morphological alterations caused by ischemia were similar to that observed during acute application of glutamate.
Figure 10.
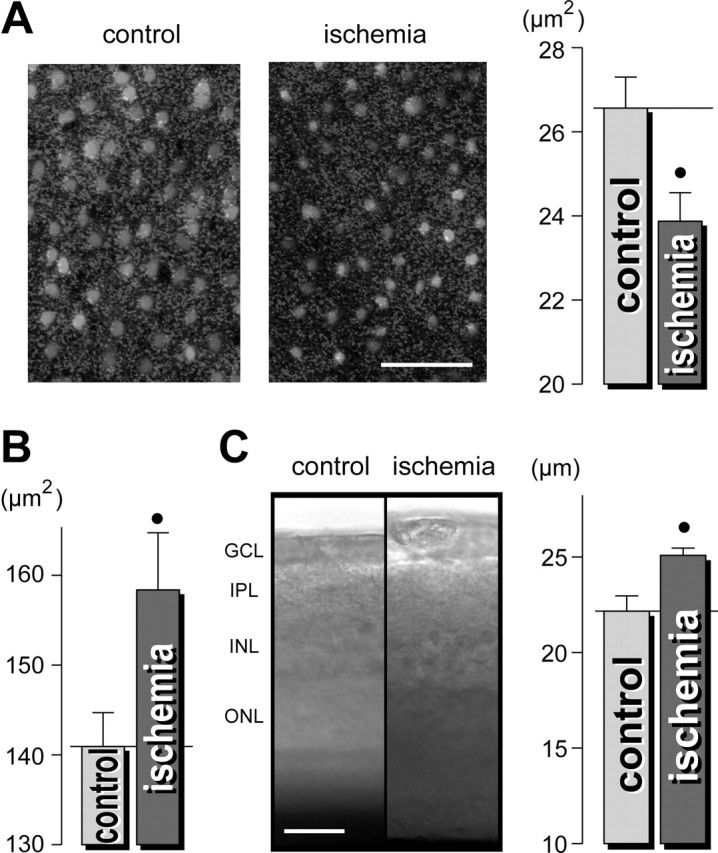
Pressure-induced ischemia of guinea-pig retinas causes morphological alterations that are similar to those observed during acute exposure to glutamate. Immediately after the end of a 1 hr elevation of the intraocular pressure above the systolic blood pressure, the retinas were acutely isolated and the Müller glial cells were labeled with Mitotracker Orange. A, Left, The views of the deep IPL display thinner Müller cell profiles (light round structures) in the ischemic retina compared with the control retina. Right, Mean cross-sectional area of the glial cell profiles in the IPL. B, Mean cross-sectional areas of neuronal cell bodies within the GCL-NFL. C, Acutely isolated ischemic retinas are thicker as control retinas. Left, Examples of retinal slices; the intrinsic optical signals were recorded. Right, Mean thickness of the IPL in retinal slices (in micrometers). The data were obtained in three independent experiments. •p < 0.05. Scale bars, 25 μm.
Discussion
Glutamate-evoked morphological changes
Nonpathological neuronal excitation has been described as inducing a moderate swelling of astrocytes (Amzica and Neckelmann, 1999), likely attributable to uptake of released K+ and glutamate (Kimelberg, 2000; Vargová et al., 2001). Here we show that retinal glial cell processes decrease their thickness when neuronal cell structures swell because of excitation evoked by glutamate, kainate, dopamine, or high K+ that is accompanied by an elongation of the cells and a thickening of the whole retinal tissue. Although the effects of kainate and glutamate continued as long as the agonists were applied, high K+ and dopamine evoked transient responses. The latter effects were mediated by stimulation of endogenous glutamate release and subsequent activation of AMPA-kainate receptors. Most probably, the swelling-inducing effect of glutamate is mediated by an influx of Na+ ions through open AMPA-kainate receptor channels, supported by a Cl- influx and accompanied by water entry into retinal neurons. In addition to the swelling of neuronal cell bodies in the GCL and INL, a swelling of synaptic structures in the IPL may occur. This assumption is supported by the observation of a decreasing volume of the extracellular space during glutamate exposure (Fig. 6), as well as by the high-K+-evoked appearance of spherical structures in the space between the glial cell profiles (Fig. 7A). The glutamate-evoked reshaping of retinal glial cells is probably secondary to the AMPA-kainate receptor activation on neuronal cells because several approaches using electrophysiological and calcium imaging techniques failed to reveal the expression of such receptors on glial cells of the guinea pig retina (Fig. 4). Furthermore, although it has been suggested that retinal glial cells express functional D2 receptors (Biedermann et al., 1995), dopamine is assumed to induce its morphological effect not directly via receptor stimulation on glial cells because the effect was dependent on AMPA-kainate receptor stimulation (Fig. 8C).
Mechanism of glial cell morphological alterations
The mechanism of the glutamate-evoked decrease of the thickness of glial stem processes is unclear. Until now, only a glutamate-evoked swelling of retinal glial cells has been described. It occurred after longer incubation time periods (at least 1 hr) and is thought to be mediated by the Na+ transport that drives the uptake of glutamate (Izumi et al., 1996). Likewise, elevated extracellular K+ has been shown to cause astrocytic cell swelling (Syková and Orkand, 1980; Walz and Hinks, 1985; Anderova et al., 2001; MacVicar et al., 2002). Rapid glutamate- or K+-evoked morphological alterations of retinal glial cells (i.e., thinning and elongation of stem processes) have never been described. Although Na+-free extracellular solution blocked the glutamate effect on the glial cells described here, this effect is certainly not mediated by glutamate transporters, because it was not modified by selective blockers (Fig. 3F) and because an elevated extracellular Na+ concentration resulted in a thickening of glial stem processes (Fig. 5C).
To elucidate how neuronal AMPA-kainate receptor activation may result in glial cell reshaping, different possibilities were investigated. One of the possible mediators is neuronally released K+; however, in contrast to previously described swelling-inducing effects of high extracellular K+ on glial cells (Walz and Hinks, 1985; Syková et al., 1999; MacVicar et al., 2002), we observed a fast and transient decrease of the thickness of glial cell processes (Fig. 7A,B). The high-K+ effects were similar to those of glutamate exposure; however, various ionic blockers of K+ channels failed to inhibit the effect of glutamate (Fig. 5A), whereas blockade of the endogenous glutamate release, or of AMPA-kainate receptors, resulted in an inhibition of the K+ effect (Fig. 7C). The partial block of the K+ effect by Ba2+ or TEA (Fig. 7D) may suggest that K+ channel activity is necessary upstream of glutamate release, i.e., by mediating K+-induced neuronal cell depolarization. In addition to extracellular K+, an involvement of changed osmolarity (Fig. 5C,D) or of alterations of the cytoskeleton (Fig. 5F) in the glial cell reshaping could be ruled out.
Thus, the following chain of events is proposed. (1) Long-term activation of AMPA-kainate receptors by glutamate causes swelling of the synapses in the IPL; this (2) decreases the volume of the extracellular space (Fig. 6) and (3) causes a compression of the glial cell processes in the IPL, which (4) respond with an elongation of their trunks. A similar mechanically mediated effect may be exerted by the swollen neuronal cell bodies in the INL (Fig. 1D). It is noteworthy that the processes of isolated glial cells do not shrink and/or elongate after glutamate or high-K+ exposure (data not shown), supporting the view that the glial responses are induced by the surrounding tissue.
Dopaminergic stimulation of retinal glutamate release
Among the neurotransmitters tested, only dopamine was found to evoke morphological changes similar to those of glutamate (Fig. 8A); however, its effects were transient (Fig. 8C) and had a significantly lower amplitude. The dopamine effect was mediated by stimulation of the endogenous glutamate release (Fig. 8B) and was found to be dependent on dark-light conditions (Fig. 9B). The relatively small amplitude of the dopamine effect, compared with the effects of high K+ and glutamate, may be explained in different ways. The transient responses during dopamine and high-K+ exposure are likely to be caused by a release of endogenous glutamate stores; once the stores were gradually released, the effects became smaller (and a second application, as in the case of high K+, was ineffective) (Fig. 7E). Although high K+ evoked a full release of endogenous glutamate stores, only a subpopulation of glutamatergic cells in the retina (∼25% of all glutamatergic cells present) may be affected by the stimulation of dopaminergic receptors.
Another explanation for the relatively small effect of dopamine may be the simultaneous stimulation of different subtypes of dopaminergic receptors that may evoke increases as well as decreases of endogenous glutamate release. In retinas of different vertebrate species, dopamine has been shown to enhance or decrease glutamatergic responses, depending on the subtype of dopamine receptor involved. In the rat retina, dopamine inhibits the K+-evoked glutamate release via D2 receptor activation (Kamisaki et al., 1991). In bipolar cells of the tiger salamander retina, dopamine enhances glutamate-gated ionic currents via activation of D1 receptors and subsequent cAMP-dependent protein phosphorylation (Maguire and Werblin, 1994). In bipolar and horizontal cells of the fish, dopamine evokes enhancement of glutamatergic transmission via cAMP-dependent protein phosphorylation and inhibition of the desensitization of AMPA-evoked currents (Heidelberger and Matthews, 1994; Schmidt et al., 1994).
The significant differences in agonist effects during light versus dark conditions were an interesting observation. Although the effects of glutamate and high K+ were stronger in the dark than in the light, the dopamine effect was observed only in the light (Fig. 9B). The reason for this difference is unclear. The endogenous release of dopamine in the retina is inhibited in the dark by glutamate and GABA (Kirsch and Wagner, 1989) and increased in the light by removal of this inhibition and by the excitation received from ON-bipolar cells (Gustincich et al., 1997). The present results suggest that not only is the release of dopamine a subject of light-dark adaptation but also the dopaminergic receptor-expressing glutamatergic cells, responding in light with glutamate release after dopamine exposure, do not respond to dopaminergic stimulation in the dark. Different intracellular levels of cAMP in retinal cells of light- and dark-adapted animals (Vaquero et al., 2001) may play a role in this different responsiveness of glutamatergic cells.
Functional relevance
The dynamic morphological changes of retinal glial cells, with their ability to decrease their thickness and to elongate when neuronal cell structures swell, may limit the volume changes of the extracellular space. Theoretically, a neuronal cell swelling by ∼40% should cause a reduction of the volume of the extracellular space by ∼70%; however, we found a volume decrease of only ∼18% during glutamate exposure (Fig. 6). A stronger extracellular volume decrease, which should severely impair the homeostasis of extracellular ions and neuroactive substances, is apparently prevented by the “compensatory” reshaping of glial cells. This effect should be even more important under pathological conditions, such as epilepsy (Glass and Dragunow, 1995) or ischemia, that cause excessive glutamate release (Adachi et al., 1972; Osborne et al., 2004) and functional down-regulation of glial glutamate uptake (Barnett et al., 2001). Thus, this mechanism may help to decrease the extent of glutamate toxicity. The results of the ischemia-evoked thickening of the retinal tissue (Fig. 10) may suggest that edema formation in the ischemic tissue is caused, at least in part, by glutamate-evoked neuronal cell swelling; however, dynamic morphological alterations of glial cells may also support regular neuronal information processing in the healthy retina. In the rabbit retina, kainate responses were recorded from various types of neuronal cells (Marc, 1999). The light responses of most retinal neurons are mediated primarily by AMPA receptors that desensitize to a maintained plateau (Thoreson and Witkovsky, 1999). The ability of glial cells to change their morphology in dependence on the neuronal activity might be particularly important in the retina, where most of the neurons display sustained potentials.
We suggest that our observation may allow indirect visualization of the sustained functional state of neuronal AMPA receptors, via recording the shape of glial cells, in experiments on retinal whole mounts.
Footnotes
This work was supported in part by grants from the Interdisziplinäres Zentrum für Klinische Forschung Leipzig at the Faculty of Medicine of the University of Leipzig (Project C21); from the Deutsche Forschungsgemeinschaft (Re 849/8-3, Br 1249/2-1); from the Ministry of Education, Sports and Youth of the Czech Republic (J13/98:11130004); and from the Academy of Sciences of the Czech Republic (AVOZ50399906). M.W. was a Marie-Curie Research Fellow in the Department of Neuroscience, Charles University Second Medical School.
Correspondence should be addressed to Dr. Andreas Bringmann, Department of Ophthalmology and Eye Clinic, University of Leipzig, Liebigstrasse 10-14, D-04103 Leipzig, Germany. E-mail: bria@medizin.uni-leipzig.de.
Copyright © 2004 Society for Neuroscience 0270-6474/04/2410149-10$15.00/0
References
- Adachi K, Kashii S, Masai H, Udea M, Morizane C, Kaneda K, Kume T, Akaike A, Honda Y (1972) Mechanism of the pathogenesis of glutamate neurotoxicity in retinal ischemia. Graefes Arch Clin Exp Ophthalmol 236: 766-774. [DOI] [PubMed] [Google Scholar]
- Amzica F, Neckelmann D (1999) Membrane capacitance of cortical neurons and glia during sleep oscillations and spike-wave seizures. J Neurophysiol 82: 2731-2746. [DOI] [PubMed] [Google Scholar]
- Anderova M, Kubinova S, Mazel T, Chvatal A, Eliasson C, Pekny M, Sykova E (2001) Effect of elevated K+, hypotonic stress, and cortical spreading depression on astrocyte swelling in GFAP-deficient mice. Glia 35: 189-203. [DOI] [PubMed] [Google Scholar]
- Anderson TR, Andrew RD (2002) Spreading depression: imaging and blockade in the rat neocortical brain slice. J Neurophysiol 88: 2713-2725. [DOI] [PubMed] [Google Scholar]
- Barnett NL, Pow DV, Bull ND (2001) Differential pertubations of neuronal and glial glutamate transport systems in retinal ischaemia. Neurochem Int 39: 291-299. [DOI] [PubMed] [Google Scholar]
- Biedermann B, Frohlich E, Grosche J, Wagner HJ, Reichenbach A (1995) Mammalian Müller (glial) cells express functional D2 dopamine receptors. NeuroReport 6: 609-612. [DOI] [PubMed] [Google Scholar]
- Bringmann A, Faude F, Reichenbach A (1997) Mammalian retinal glial (Müller) cells express large-conductance Ca2+-activated K+ channels that are modulated by Mg2+ and pH, and activated by protein kinase A. Glia 19: 311-323. [DOI] [PubMed] [Google Scholar]
- Dietzel I, Heinemann U, Hofmeier G, Lux HD (1980) Transient changes in the size of the extracellular space in the sensimotor cortex of rats in relation to stimulus-induced changes in potassium concentration. Exp Brain Res 40: 423-432. [DOI] [PubMed] [Google Scholar]
- Glass M, Dragunow M (1995) Neurochemical and morphological changes associated with human epilepsy. Brain Res Rev 21: 29-41. [DOI] [PubMed] [Google Scholar]
- Gustincich S, Feigenspan A, Wu DK, Koopman LJ, Raviola E (1997) Control of dopamine release in the retina: a transgenic approach to neural networks. Neuron 18: 723-736. [DOI] [PubMed] [Google Scholar]
- Heidelberger R, Matthews G (1994) Dopamine enhances Ca2+ responses in synaptic terminals of retinal bipolar neurons. NeuroReport 5: 729-732. [DOI] [PubMed] [Google Scholar]
- Izumi Y, Kirby-Sharkey CO, Benz AM, Mennerick S, Labruyere J, Price MT, Olney JW, Zorumski CF (1996) Swelling of Müller cells induced by AP3 and glutamate transport substrates in rat retina. Glia 17: 285-293. [DOI] [PubMed] [Google Scholar]
- Jendelová P, Syková E (1991) Role of glia in K+ and pH homeostasis in the neonatal rat spinal cord. Glia 4: 56-63. [DOI] [PubMed] [Google Scholar]
- Kamisaki Y, Hamahashi T, Mita C, Itoh T (1991) D2 dopamine receptors inhibit release of aspartate and glutamate in rat retina. J Pharmacol Exp Ther 256: 634-638. [PubMed] [Google Scholar]
- Kimelberg HK (2000) Cell volume in the CNS: regulation and implications for nervous system function and pathology. Neuroscientist 6: 14-25. [Google Scholar]
- Kirsch M, Wagner HJ (1989) Release pattern of endogenous dopamine in teleost retinae during light adaptation and pharmacological stimulation. Vision Res 29: 147-154. [DOI] [PubMed] [Google Scholar]
- Kofuji P, Biedermann B, Siddharthan V, Raap M, Iandiev I, Milenkovic I, Thomzig A, Veh RW, Bringmann A, Reichenbach A (2002) Kir potassium channel subunit expression in retinal glial cells: implications for spatial potassium buffering dagger. Glia 39: 292-303. [DOI] [PubMed] [Google Scholar]
- MacVicar BA, Feighan D, Brown A, Ransom BR (2002) Intrinsic optical signals in the rat optic nerve: role for K+ uptake via NKCC1 and swelling of astrocytes. Glia 37: 114-123. [DOI] [PubMed] [Google Scholar]
- Maguire G, Werblin F (1994) Dopamine enhances a glutamate-gated ionic current in OFF bipolar cells of the tiger salamander retina. J Neurosci 14: 6094-6101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marc RE (1999) Kainate activation of horizontal, bipolar, amacrine, and ganglion cells in the rabbit retina. J Comp Neurol 407: 65-76. [PubMed] [Google Scholar]
- Matsui K, Hosoi N, Tachibana M (1999) Active role of glutamate uptake in the synaptic transmission from retinal nonspiking neurons. J Neurosci 19: 6755-6766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morgan IG, Boelen MK (1996) A retinal dark-light switch: a review of the evidence. Vis Neurosci 13: 399-409. [DOI] [PubMed] [Google Scholar]
- Nagelhus EA, Horio Y, Inanobe A, Fujita A, Haug FM, Nielsen S, Kurachi Y, Ottersen OP (1999) Immunogold evidence suggests that coupling of K+ siphoning and water transport in rat retinal Müller cells is mediated by a coenrichment of Kir4.1 and AQP4 in specific membrane domains. Glia 26: 47-54. [DOI] [PubMed] [Google Scholar]
- Newman EA (2001) Propagation of intercellular calcium waves in retinal astrocytes and Müller cells. J Neurosci 21: 2215-2223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newman EA, Reichenbach A (1996) The Müller cell: a functional element of the retina. Trends Neurosci 19: 307-317. [DOI] [PubMed] [Google Scholar]
- Nicholson C, Syková E (1998) Extracellular space structure revealed by diffusion analysis. Trends Neurosci 21: 207-215. [DOI] [PubMed] [Google Scholar]
- Osborne NN, Casson RJ, Wood JP, Chidlow G, Graham M, Melena J (2004) Retinal ischemia: mechanisms of damage and potential therapeutic strategies. Prog Retin Eye Res 23: 91-147. [DOI] [PubMed] [Google Scholar]
- Pannicke T, Iandiev I, Uckermann O, Biedermann B, Kutzera F, Wiedemann P, Wolburg H, Reichenbach A, Bringmann A (2004) A potassium channel-linked mechanism of glial cell swelling in the postischemic retina. Mol Cell Neurosci 26: 493-502. [DOI] [PubMed] [Google Scholar]
- Reichelt W, Pannicke T (1993) Voltage-dependent K+ currents in guinea pig Müller (glia) cells show different sensitivities to blockade by Ba2+ Neurosci Lett 155: 15-18. [DOI] [PubMed] [Google Scholar]
- Santos PF, Caramelo OL, Carvalho AP, Duarte CB (1999) Characterization of ATP release from cultures enriched in cholinergic amacrine-like neurons. J Neurobiol 41: 340-348. [PubMed] [Google Scholar]
- Schmidt KF, Kruse M, Hatt H (1994) Dopamine alters glutamate receptor desensitization in retinal horizontal cells of the perch (Perca fluviatilis). Proc Natl Acad Sci USA 91: 8288-8291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Somjen GG (2001) Mechanisms of spreading depression and hypoxic spreading depression-like depolarization. Physiol Rev 81: 1065-1096. [DOI] [PubMed] [Google Scholar]
- Syková E (1991) Ionic and volume changes in the microenvironment of nerve and receptor cells. In: Progress in sensory physiology (Ottoson D, ed), pp 1-167. Heidelberg: Springer.
- Syková E (1992) Ion-selective electrodes. In: Monitoring neuronal cells: a practical approach (Stamford J, ed), pp 261-282. New York: Oxford UP.
- Syková E, Orkand RK (1980) Extracellular potassium accumulation and transmission in frog spinal cord. Neuroscience 5: 1421-1428. [DOI] [PubMed] [Google Scholar]
- Syková E, L, Prokopová S, Simonová Z (1999) Glial swelling and astrogliosis produce diffusion barriers in the rat spinal cord. Glia 25: 56-70. [DOI] [PubMed] [Google Scholar]
- Thoreson WB, Witkovsky P (1999) Glutamate receptors and circuits in the vertebrate retina. Prog Retin Eye Res 18: 765-810. [DOI] [PubMed] [Google Scholar]
- Uckermann O, Iandiev I, Francke M, Franze K, Grosche J, Wolf S, Kohen L, Wiedemann P, Reichenbach A, Bringmann A (2004) Selective staining by vital dyes of Müller glial cells in retinal whole mounts. Glia 45: 59-66. [DOI] [PubMed] [Google Scholar]
- Vaquero CF, Pignatelli A, Partida GJ, Ishida AT (2001) A dopamine- and protein kinase A-dependent mechanism for network adaptation in retinal ganglion cells. J Neurosci 21: 8624-8635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vargová L, Jendelová P, Chvátal A, Syková E (2001) Glutamate, NMDA and AMPA induced changes in extracellular space volume and tortuosity in the rat spinal cord. J Cereb Blood Flow Metab 21: 1077-1089. [DOI] [PubMed] [Google Scholar]
- Walz W, Hinks EC (1985) Carrier-mediated KCl accumulation accompanied by water movements is involved in the control of physiological K+ levels by astrocytes. Brain Res 343: 44-51. [DOI] [PubMed] [Google Scholar]