

Abstract
The central nucleus of amygdala (CeA) is important in regulating alcohol consumption and plays a major role in the anxiogenic response to ethanol withdrawal. We showed previously that acute ethanol augments GABAA receptor-mediated IPSPs and IPSCs, possibly by a presynaptic mechanism. Here, we have examined the interaction of acute ethanol with the GABAergic system in chronic ethanol-treated (CET) rats using an in vitro CeA slice preparation and in vivo brain microdialysis. We found that in CeA slices from CET rats, the baseline evoked IPSP and IPSC amplitudes were increased, and paired-pulse facilitation ratios were lower than in naive rats, suggesting an increased GABAergic transmission after chronic ethanol treatment. Interestingly, acute ethanol (5-66 mm) significantly enhanced IPSPs and IPSCs equally in CET and naive rats, indicating a lack of tolerance for this effect of acute ethanol. Analysis of miniature IPSC frequency suggests that the increased GABAergic transmission by both acute and chronic ethanol arises from a presynaptic mechanism involving enhanced vesicular release of GABA. These data are supported by microdialysis studies showing that CET rats presented a fourfold increase in baseline GABA dialysate content compared with naive rats. In vivo administration of ethanol (0.1, 0.3, and 1.0 m) produced a dose-dependent increase in GABA release in the CeA dialysate in both CET and naive rats. These combined findings suggest that acute and chronic ethanol increases GABA release in CeA and support previous reports that the behavioral actions of ethanol are mediated, in part, by increased GABAergic transmission in the CeA.
Keywords: alcohol, acute ethanol, chronic ethanol treatment, inhibition, microdialysis, electrophysiology
Introduction
The central nucleus of amygdala (CeA) is a major component of the extended amygdala and has been identified as a specific site of action for both the acute positive reinforcement of drugs of abuse and for the negative reinforcement associated with drug abstinence (Koob and Le Moal, 2001). Behavioral studies have implicated GABAergic transmission in the CeA in regulating alcohol intake (Hyytia and Koob, 1995; McBride et al., 1998). However, little is known about the neurochemical and cellular mechanisms through which GABAergic transmission in the CeA modulates the behavioral effects of ethanol. We reported recently that the CeA is the first brain region to reveal, without conditional treatments such as GABAB receptor antagonists (Roberto et al., 2003), a consistent ethanol-induced enhancement of GABAergic transmission via actions at both presynaptic and postsynaptic sites. We demonstrated that in CeA slices, acute ethanol augmented GABAA receptor-mediated IPSPs and IPSCs in most CeA neurons, in part through an ethanol-induced increase in presynaptic GABA release (Roberto et al., 2003; Nie et al., 2004). This effect is mediated by the neuropeptide corticotrophin releasing factor (CRF) (Nie et al., 2004). Coupled with the observation that GABAA receptor antagonism in the CeA reduces ethanol self-administration in rats (Hyytia and Koob, 1995), this finding suggests that ethanol reinforcement may occur via increased GABAergic transmission in the CeA. Activation of GABAA receptors in the CeA alters ethanol self-administration only in ethanol-dependent rats (Roberts et al., 1996), suggesting that the behavioral role of GABA neurotransmission in this brain region is altered by chronic ethanol treatment. Because the chronic ethanol treatment paradigm provides information relevant to CNS changes that develop with the long-term alcohol abuse that occurs in humans, in the present investigation, we have assessed: (1) whether synaptic changes occur in GABAergic transmission in CeA after chronic ethanol exposure and (2) whether local administration of ethanol in vivo alters GABA release in the CeA of naive and chronic ethanol-treated (CET) rats.
Materials and Methods
Slice preparation. We prepared amygdala slices from male Sprague Dawley rats (250-300 gm) that were anesthetized with halothane (3%), decapitated, and the brains rapidly removed and placed into ice-cold artificial CSF (ACSF) gassed with 95% O2 and 5% CO2 (Roberto et al., 2003). Coronal slices (400 μm thick) were cut on a Vibroslicer (Campden Instruments, Lafayette, IN) or a Vibratome Series 3000 (Technical Products International, St. Louis, MO), incubated in an interface configuration for ∼30 min, and then completely submerged and continuously superfused with warm (31°C) gassed ACSF. The ACSF had the following composition (in mm): 130 NaCl, 3.5 KCl, 1.25 NaH2PO4, 1.5 MgSO4·7 H2O, 2.0 CaCl2, 24 NaHCO3, and 10 glucose. The inner chamber had a total volume of 0.8 ml; at the superfusion rates used (flow rate, 2-4 ml/min), 90% replacement of the chamber solution could be obtained within 1 min. Drugs were added to the ACSF from stock solutions at known concentrations.
Intracellular recording. We recorded from CeA neurons (medial division of CeA) with sharp micropipettes (3 m KCl) using discontinuous voltage-clamp or current-clamp mode. We used a switching frequency of 3-5 kHz and continuously monitored electrode settling time and capacitance neutralization at the headstage. Data were acquired with an Axoclamp-2A preamplifier (Axon Instruments, Foster City, CA) and stored for later analysis using pClamp software (Axon Instruments). We evoked pharmacologically isolated GABAA-IPSPs and -IPSCs by stimulating locally within the CeA through a bipolar stimulating electrode while superfusing the glutamate receptor blockers 6-cyano-7-nitroquinoxaline-2,3-dione (CNQX) (10 μm) and dl-2-amino-5-phosphonovalerate (APV) (30 μm) together with 1 μm CGP 55845A (a GABAB receptor antagonist). To determine the response parameters for each cell, an input-output (I/O) protocol was performed. We stimulated at a range of currents (typically between 50 and 200 μA) starting at the threshold current required to elicit a GABAA-IPSP and -IPSC, and the stimulus strength was increased in three steps of 30 μA (rate of 1 pulse per 8 sec) until the voltage required to elicit the maximum amplitude was reached. We normalized the four stimulus intensities as 1-4×. Most neurons were held near their resting membrane potential (RMP), and we applied hyperpolarizing and depolarizing current steps (200 pA increments; 750 msec duration) to generate voltage-current (V/I) curves. The evoked IPSP and IPSC amplitudes and V/I responses were quantified by Clampfit software (Axon Instruments).
Paired-pulse facilitation. We examined pair-pulse facilitation (PPF), a phenomenon whereby a secondary evoked synaptic response is increased by a preceding primary stimulation of equal intensity (Andreasen and Hablitz, 1994; Bonci and Williams, 1997), in each CeA neuron using two stimuli at 50 msec interpulse intervals (Roberto et al., 2004). The stimulus strength was adjusted such that the amplitude of the first IPSP or IPSC of the pair was 50% of the maximal amplitude determined from the I/O relationship. We calculated the PPF ratio as the second IPSP or IPSC amplitude over that of the first IPSP or IPSC. We took measures before acute ethanol superfusion (control), during ethanol (5-10 min), and after ethanol washout (20-30 min). All values were expressed as mean ± SEM, and data were subjected to a between-subjects or within-subject ANOVA with repeated measures and to the Newman-Keuls post hoc test, with p < 0.05 considered statistically significant. When appropriate, we used Student's paired or unpaired t test.
Whole-cell patch-clamp recording of miniature IPSCs. We recorded from another set of CeA neurons using the “blind” method of whole-cell patch clamp in the presence of 10 μm CNQX, 30 μm APV, 1 μm CGP 55845A, and 1 μm tetrodotoxin (TTX). To isolate spontaneous miniature GABAA-IPSCs (mIPSCs), recordings were made with electrodes filled with an internal solution containing (in mm): 135 KCl, 10 HEPES, 2 MgCl2, 0.5 EGTA, 5 ATP, and 1 GTP, pH 7.2-7.4, osmolarity 275-290 mOsm (Roberto et al., 2003). We pulled patch pipettes on a Flaming/Brown puller from borosilicate glass (input resistance, 2-3 MΩ). The mIPSC data were acquired with an Axoclamp-2A preamplifier (Axon Instruments) and analyzed using Mini 5.1 software (Synaptosoft, Leona, NJ). We evaluated ethanol effects on the frequency and amplitude of mIPSCs within individual neurons using cumulative probability analysis, with statistical significance determined by the Kolmogorov-Smirnov nonparametric two-sample test (p < 0.05 is considered significant).
Chronic ethanol treatment. We used the standard ethanol vapor inhalation method of The Scripps Research Institute Alcohol Research Center (La Jolla, CA) for developing ethanol-dependent rats (Rogers et al., 1979; Roberto et al., 2004). Male Sprague Dawley rats were housed two to four per cage with a 6 A.M. to 6 P.M. light cycle and ad libitum access to food and water. The animals were randomly divided into two groups and placed either into ethanol vapor chambers or into air-only chambers (sham controls). In the ethanol-dependent group, we exposed rats to continuous ethanol vapors for at least 2 weeks before they were used for slice or microdialysis experiments. We determined blood alcohol levels (BALs) of the dependent animals from tail blood sampled three times per week. When necessary, we adjusted the ethanol vapor concentration after BAL measurement to achieve a constant target BAL of 150-200 mg/dl. The mean BAL of all ethanol-dependent animals was 178.4 ± 16 mg/dl (n = 38). The dependent animals did not show signs of deteriorating health, impaired locomotor activity, or abnormality in body posture and gait. The mean body weight of CET animals was 250 gm (n = 16), compared with a mean body weight of 270 gm (n = 10) for sham or naive animals. Naive rats were treated exactly like CET rats except for exposure to air rather than ethanol vapor.
To assess ethanol dependence, at the end of the ethanol exposure period, we measured the performance of a separate sample of CET rats (n = 6) on several behavioral tests for ethanol withdrawal severity. We measured the withdrawal signs (ventromedial distal limb flexion response, tail stiffness, and abnormal body posture and gait) (Macey et al., 1996; Roberto et al., 2004) at 2, 4, 6, and 8 hr after ethanol withdrawal in the ethanol-dependent group and compared them with similar measures from the naive group. Moderate behavioral signs of ethanol withdrawal were evident in all six rats tested at 2-8 hr after the termination of the ethanol treatment, a time frame corresponding to the electrophysiological recordings and microdialysis samples.
On electrophysiology experiment days, animals were maintained in the vapor chambers until preparation of the amygdala slices in ethanol-free ACSF. We made recordings from slices of CET or naive rats 2-8 hr after cutting the slices, corresponding to the time period for the hyper-excitable behavioral response to withdrawal characteristic of ethanol dependence. The experimenter was “blind” to the chronic ethanol treatment protocol.
Microdialysis. Sprague Dawley rats (250-370 gm; Charles River, Hollister, CA) were anesthetized with isofluorane (1-2%) and stereotaxically implanted with a stainless steel microdialysis guide cannula (21 gauge; Plastics One, Roanoke, VA) that terminated at the dorsal surface of the medial CeA (anteroposterior, -2.3 mm; mediolateral, +4.0 mm; ventral, -6.4 mm from dura) (Paxinos and Watson, 1986). After a 7 d recovery period, the animals were lightly anesthetized (1-2% isofluorane), and microdialysis probes (1 mm active length; 0.6 μl/min ACSF perfusate flow rate) (Frantz et al., 2002) were inserted and secured to the guide cannulas. Rats regained consciousness within 5 min of probe insertion, and a 3 hr postimplantation period was given before dialysate collection. We subsequently collected microdialysate samples at 10 min intervals during a 60 min baseline period, followed by a 30 min period during which ACSF containing 0.1, 0.3, and 1.0 m ethanol was perfused through the probe (Roberto et al., 2004). We allowed a 40 min recovery period after each ethanol pulse, during which ethanol-free ACSF was the perfusate. To avoid technical artifacts arising from switching the perfusate solutions, the first sample collected after each perfusate change was not included in the final analyses. Based on previous work characterizing the delivery of ethanol by reverse dialysis (Robinson et al., 2000), the efficiency of ethanol delivery in the present study was estimated to be ∼10%.
For microdialysis, in the CET group, as for slice experiments, rats were continuously exposed to ethanol vapors for at least 2 weeks before microdialysis was initiated. The mean BAL of these CET animals was 165.8 mg/dl (n = 7). Sham, naive rats were treated similarly but without ethanol vapors. We implanted the stainless steel microdialysis guide cannula 7 d before the beginning of the chronic ethanol treatment. On experiment days, animals were maintained in the vapor chambers until the microdialysis probes were inserted and secured to the guide cannulas. We began collection of the dialysate samples 3 hr after implantation, as above, 2-8 hr after removal of the rats from the vapor chambers. Dialysate samples were frozen at -70°C until they were derivatized with a fluorescent probe (naphthalene-2,3-dicarboxaldehyde) and analyzed for amino acid content using capillary electrophoresis with laser-induced fluorescence detection. We conducted all procedures in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals. Postmortem verification of probe placement revealed that in all cases the active area of the microdialysis probe was located in the medial portion of the CeA.
Amino acid analysis. We determined microdialysate amino acid content using capillary electrophoresis with laser-induced fluorescence detection (Roberto et al., 2004). We derivatized the amino acids by mixing 5 μl of microdialysate with 9 μl of 40 mm borate buffer, pH 10.5, containing 3.8 mm KCN and 1 μl of 5 mm naphthalene-2,3-dicarboxaldehyde in methanol. This mixture was allowed to react at room temperature in the dark for 30 min before placing the samples in the refrigerated (10°C) sample tray of the capillary electrophoresis instrument (Agilent Technologies, Wilmington, DE). We subsequently loaded derivatized dialysate onto a 90 cm fused silica capillary (30 μm inner diameter; sample loading by 50 millibar pressure for 10 sec) and separated the amino acids using +15 kV and a background electrolyte solution consisting of 100 mm borate buffer, pH 9.2, containing 30 mm SDS and 2 mm hydroxypropyl-β-cyclodextrin. We detected the amino acids using a laser-induced fluorescence detector (Zetalif; Picometrics, Ramon Ville, France) with a 442 nm HeCd laser (30 mW; Melles Griot, Carlsbad, CA). External calibration standards were run in duplicate and were interspersed throughout the sample run. The limits of quantitation were ∼1nm for each of the analytes. We obtained all reagents and amino acid standards from Sigma-Aldrich (St. Louis, MO).
Results
Electrophysiological studies
All experiments were performed 2-8 hr after preparation (in ethanol-free ACSF) of the CeA slices from either CET or naive control rats. We recorded from 80 CeA neurons with a mean RMP of -75 ± 2 mV and a mean input resistance of 105 ± 4MΩ. Chronic ethanol treatment did not significantly affect these properties.
We evoked pharmacologically isolated GABAA-IPSPs and -IPSCs by stimulating locally within the CeA. Acute ethanol (5-66 mm) dose-dependently increased the amplitude of evoked GABAA-IPSPs (Fig. 1) in CET rats. The highest ethanol concentration tested, 66 mm, actually enhanced GABAA-IPSPs to a lesser extent than did 44 mm, reminiscent of the inverted U-shaped dose-response curves obtained from CeA neurons (Roberto et al., 2003) and accumbens neurons (Nie et al., 2000) of naive rats. We found that the apparent EC50 value for the ethanol effect in CET rats was 18 mm (Fig. 1), equivalent to the 20 mm EC50 value in naive rats (Roberto et al., 2003).
Figure 1.

Concentration-response relationship for ethanol enhancement of mean GABAA-IPSP and -IPSC amplitudes in CeA neurons taken from CET rats, expressed as percentage of control (ethanol superfused for 7-10 min; number of cells in parentheses). The logistic curve, plotted by Origin Software (Microcal Software, Northampton, MA), using y = (A1 - A2)/(1 + (x/xo)*p + A2), gives an apparent EC50 value of 18 mm ethanol for IPSP and IPSC enhancement. Parameters of the logistic curve were set at the following: upper asymptote fixed at 151 and lower at 101%. Rate was fixed at 3.7, with “center” unfixed. Error bars represent SEM. Because of the inverted U-shaped curve (data not shown) of the total concentration-response curve, we eliminated the 66 mm point to obtain a valid logistic curve up to 44 mm.
In addition, superfusion of 44 mm ethanol had no significant (p > 0.1) effect on basic membrane properties of CeA neurons from either naive or CET rats (data not shown). A 44 mm concentration of ethanol significantly enhanced (to 149%; mean across four different stimulus intensities; n = 8) evoked GABAA-IPSP and -IPSC amplitudes (Fig. 2A) in naive rats, as we reported previously (Roberto et al., 2003). In neurons from CET rats, acute 44 mm ethanol also significantly (p < 0.05) enhanced (to 150%; mean across four stimulus intensities; n = 16) GABAA-IPSPs and -IPSCs (Fig. 2B) to an equivalent extent as in slices from naive rats (Fig. 2A), suggesting lack of tolerance at this time point. Interestingly, baseline GABAA-IPSP and -IPSC input-output curves generated by equivalent stimulus intensities were significantly (p < 0.05) higher in slices from CET rats compared with those from naive rats (Fig. 3A,B), suggesting an increased efficiency of GABAergic transmission.
Figure 2.

Acute ethanol increases GABAergic transmission in CeA slices. A, Top, Representative recordings of GABAA-IPSCs in a CeA slice from a naive rat recorded before, during, and after superfusion of 44 mm ethanol (EtOH). Bottom, Pooled data showing that superfusion of ethanol significantly (p < 0.05; n = 8) increased the mean GABAA-IPSP and -IPSC amplitudes in naive CeA neurons. B, Top, GABAA-IPSC recordings in a CeA slice from a CET rat. Bottom, Acute ethanol significantly (p < 0.05; n = 16) increased the mean GABAA-IPSP and -IPSC amplitudes, with recovery on ethanol washout (Wash) (20-30 min). Error bars represent SEM.
Figure 3.
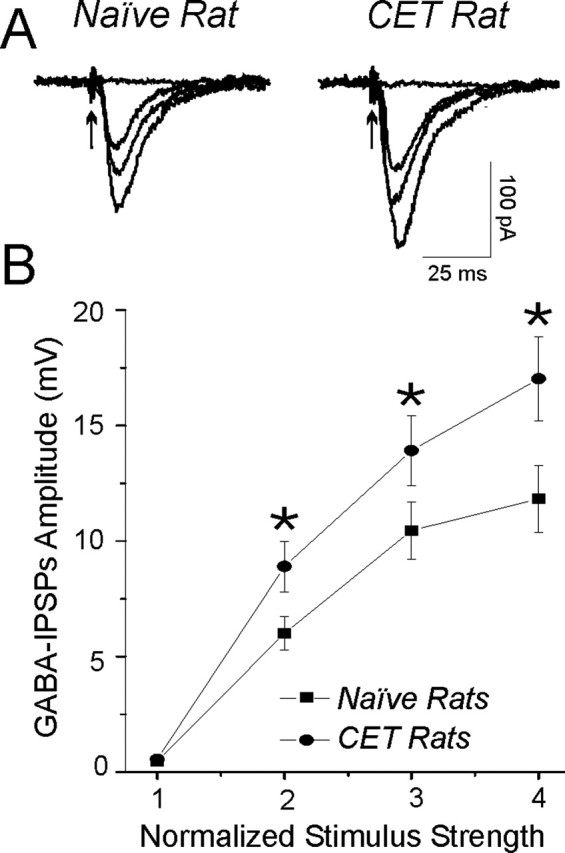
Basal GABAergic transmission is enhanced in CET rats. A, Superimposed traces of four representative GABAA-IPSCs evoked (arrow) by four different stimulus intensities in slices from naive (left) and CET (right) rats. Note that the GABAA-IPSCs are larger in dependent rats using equivalent stimulus intensities. B, Input-output curves of mean GABAA-IPSP and -IPSC amplitudes. The mean baseline GABAergic transmission is significantly (*p < 0.05) increased in slices from CET rats (n = 16) compared with naive rats (n = 9). Error bars represent SEM.
To determine whether the facilitatory effect of ethanol on GABAergic transmission involved presynaptic changes in GABA release, we first measured PPF (50 msec interpulse intervals) of GABAA-IPSPs and -IPSCs in CeA neurons before, during, and after ethanol application. Changes in PPF are inversely related to transmitter release, such that manipulations that increase probability of release are associated with a reduction of PPF (Andreasen and Hablitz, 1994; Bonci and Williams, 1997). We found that the baseline PPF ratio of GABAA-IPSPs and -IPSCs was significantly (p < 0.05) decreased in neurons from dependent rats compared with naive rats, suggesting that GABA release was augmented after chronic ethanol (Fig. 4). Furthermore, acute ethanol (44 mm) significantly (p < 0.05) decreased the PPF of GABAA-IPSPs and -IPSCs to the same extent in both naive and CET rats, predicting both increased GABA release and a lack of tolerance for this acute ethanol effect (Fig. 4).
Figure 4.
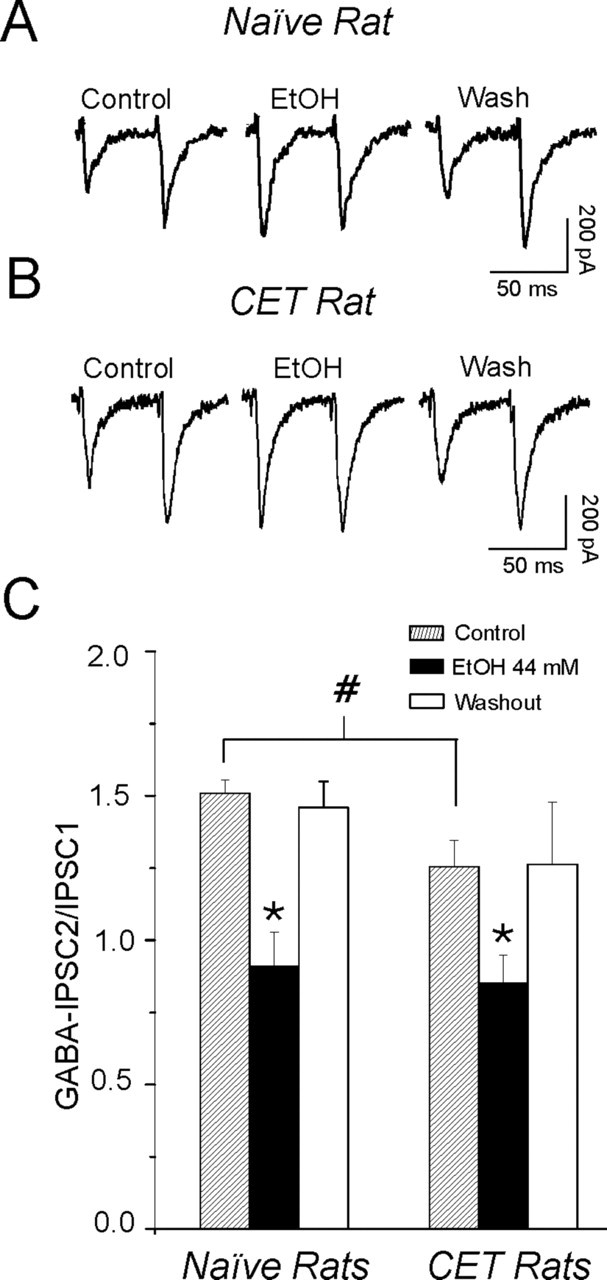
Baseline PPF of IPSPs and IPSCs is decreased after chronic ethanol treatment, and acute ethanol decreases PPF ratios. A, Current traces of GABAA-IPSCs in response to two stimuli 50 msec apart in a CeA neuron from a naive rat. Wash, Washout. Acute ethanol (EtOH) reduced PPF ratio (at 50 msec interpulse intervals; expressed as the second IPSP or IPSC amplitude over the first IPSP or IPSC amplitude) of GABAA-IPSPs and -IPSCs. B, Current traces of PPF of GABAA-IPSCs in a CeA neuron from a CET rat. Acute ethanol reduced the PPF ratio. C, Pooled data of mean PPF ratios in neurons of naive and CET rats. Baseline PPF of GABAA-IPSPs and -IPSCs was significantly (#p < 0.05) reduced in CET rats compared with naive rats. Furthermore, acute ethanol significantly (*p < 0.001) decreased the PPF ratio in neurons from both naive (by 40%; n = 9) and CET (by 32%; n = 9) rats. Error bars represent SEM.
To further characterize the increased GABAergic release in CET rats and the presynaptic effect of acute ethanol, we recorded pharmacologically isolated spontaneous GABAA-mIPSCs using whole-cell patch clamp in the presence of 1 μm TTX to eliminate the action potential-dependent release of transmitter. An increase in the frequency of mIPSCs denotes an increased probability of vesicular transmitter release, and an increase in the amplitude of mIPSCs generally reflects enhancement in the sensitivity of postsynaptic receptors (De Koninck and Mody, 1994; Otis et al., 1994). We first compared the baseline frequency and amplitude of mIPSCs in both naive and CET rats (Figs. 5, 6A,B). Notably, the mean frequency of mIPSCs was significantly (p < 0.001) higher in CeA neurons of CET rats (0.72 ± 0.11 Hz; n = 15) than in those of naive rats (0.22 ± 0.04 Hz; n = 9) (Fig. 6A), as indicated by the shift to the left of the cumulative frequency curves of Figure 5D compared with Figure 5C, suggesting an increased GABA release after chronic ethanol treatment. In CET rats, the mean amplitude of mIPSCs (25 ± 4 pA; n = 9) was also significantly increased compared with naive (15 ± 2 pA; n = 15) (Figs. 5E,F, 6B) rats.
Figure 5.
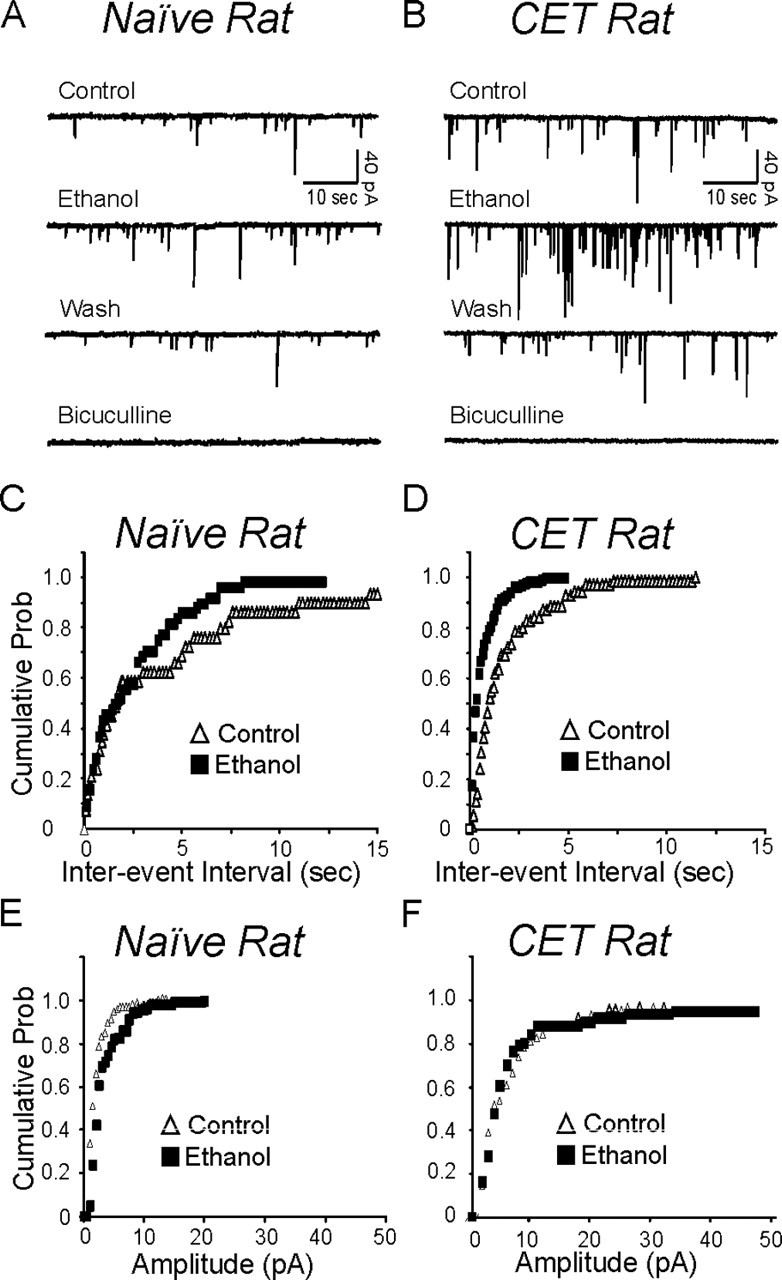
Whole-cell voltage-clamp recordings of mIPSCs from representative CeA neurons in the presence of the following (in μm): 1 TTX, 30 APV, 10 CNQX, and 1 CGP 55845A. A, Current traces from a naive rat. Acute superfusion of 44 mm ethanol onto this cell increased the frequency and amplitude of the mIPSCs, with recovery on washout (Wash). Subsequent superfusion of 30 μm bicuculline totally blocked these mIPSCs. B, Traces of mIPSCs from a CeA neuron of a CET rat. A 44 mm concentration of ethanol increased the frequency and amplitude of the mIPSCs, with recovery on washout. C, Cumulative frequency histogram for the naive neuron of A showing a shift to the left, indicating a shorter interevent interval (higher frequencies) during the application of 44 mm ethanol. D, Cumulative frequency histogram from the neuron of B showing a significant increase in mIPSC frequency. Data shown are means from a 2 min recording. E, Cumulative amplitude histogram from the neuron of A showing an increase in the distribution of mIPSC amplitudes. Note the shift to the right (especially in the first part of the curve, 1-10 pA) and elongation (beyond 15 pA) of the ethanol curve, compared with the control curve.F, Cumulative amplitude histogram from the neuron of B showing an increase in the distribution of mIPSC amplitude (note elongation of the ethanol curve beyond 30-50 pA). Prob, Probability.
Figure 6.
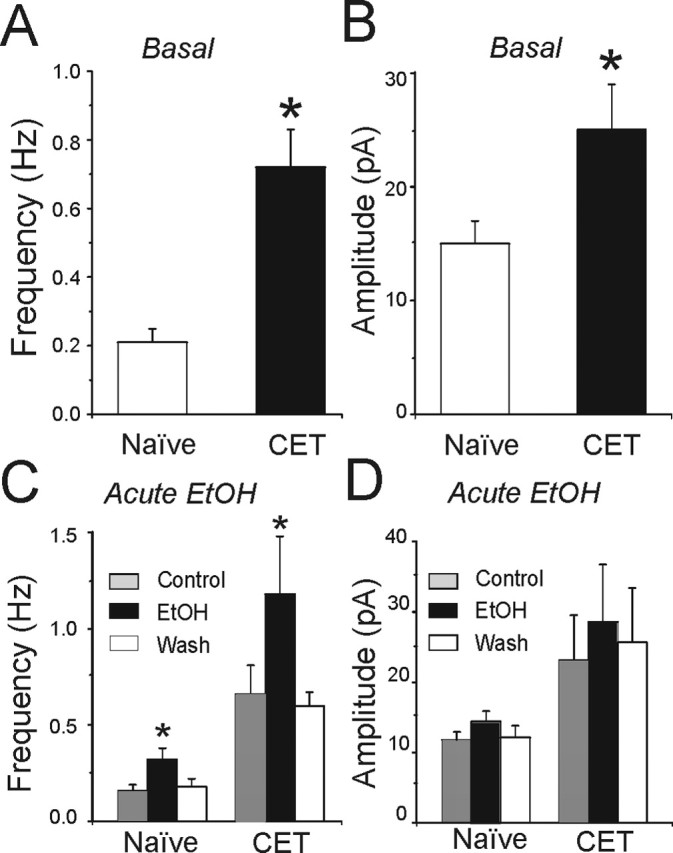
Pooled data. Chronic ethanol treatment increased the mean frequency and amplitude of spontaneous mIPSCs, and acute ethanol further increased the mean frequency of mIPSCs. A, Average (mean ± SEM) frequency of mIPSCs (in TTX) for CeA neurons from naive rats (n = 9) and CET rats (n = 15) (*p < 0.001). B, The same group of neurons showed an increase in mean amplitude of mIPSCs from CET rats (*p < 0.05). C, Acute superfusion of 44 mm ethanol (EtOH) significantly increased the mean frequency of mIPSCs in neurons from both naive (n = 7) and CET (n = 9) rats, with recovery on washout (Wash). D, Mean amplitudes of mIPSCs from the same group of neurons as in C. Acute ethanol did not significantly increase the mean mIPSC amplitude (p > 0.05) in either group. The asterisk denotes statistical significance by the Kolmogorov-Smirnov nonparametric two-sample test (p < 0.05).
We also evaluated the effect of acute ethanol on mIPSCs. Superfusion for 6-10 min of 44 mm ethanol increased the mean frequency of mIPSCs to 200 ± 12% of control and significantly shifted the cumulative frequency distribution to shorter interevent intervals in seven CeA neurons of naive rats (means: control, 0.16 ± 0.3 Hz; ethanol, 0.31 ± 0.07 Hz; p < 0.05) (Figs. 5A,C, 6C), supporting the PPF data indicating an increased presynaptic release of GABA by acute ethanol. These mIPSCs were totally blocked by superfusion of bicuculline (Fig. 5A). In CeA neurons of CET rats (n = 9), acute ethanol still produced a significant (p < 0.05) increase in the frequency of mIPSCs to 179 ± 25% of control (means: control, 0.67 ± 0.2 Hz; ethanol, 1.2 ± 0.3 Hz) (Figs. 5B,D, 6C), again suggesting a lack of tolerance.
Ethanol superfusion also increased the mIPSC amplitudes in some cells of both naive and CET rats (Figs. 5A,B,E,F, 6D), suggesting a possible postsynaptic as well as a presynaptic ethanol effect in these cells. However, averaged over all seven neurons from naive rats, 44 mm ethanol increased the mean amplitude of mIPSCs to 130 ± 12% of control (n = 7), which was not statistically significant (p < 0.5) (Fig. 6D). In CET rats, acute ethanol induced a similar but insignificant increase in the amplitude of mIPSCs to 124 ± 8% of control (n = 9) (Fig. 6D)
Microdialysis study
We measured the neurochemical effects of chronic ethanol treatment and locally administered ethanol on the release of the inhibitory amino acids GABA, glycine, and taurine within the CeA of freely moving rats withdrawn from the vapor chambers for 2-8 hr. The mean baseline GABA dialysate concentration in naive rats (n = 11) was 15.8 ± 3.9 nm (Fig. 7). Interestingly, there was an approximately fourfold increase in baseline dialysate GABA concentrations in CeA of CET rats (57.4 ± 15 nm; n = 7) compared with naive rats (p < 0.001) (Fig. 7). In naive rats, subsequent local ethanol administration (by reverse dialysis) produced a significant (p < 0.05) dose-related increase in dialysate GABA levels (net increase of 3.2 ± 0.8, 9 ± 2.6, and 16.8 ± 3 nm induced by 0.1, 0.3, and 1.0 m ethanol perfusates, respectively; n = 11) (Fig. 7). In each case, dialysate GABA levels returned to pre-ethanol baseline levels when the perfusion medium was replaced with ethanol-free ACSF. Furthermore, in CET rats local administration of 0.1, 0.3, and 1.0 m ethanol also significantly (p < 0.05) increased dialysate GABA levels (net increase of 8.8 ± 6.8, 25.8 ± 17.8, and 37.3 ± 15.6 nm, respectively) (Fig. 7). Although the net increase in dialysate GABA for each perfusate ethanol concentration was greater in CET animals than in naive rats, it should be noted that these increases are superimposed on higher baseline GABA levels in the CET group. To evaluate the effects of locally administered ethanol on GABA efflux in the context of the different baseline GABA levels between groups, we also compared the data as the percentage change from baseline levels in each treatment history group. In this context, the 0.1, 0.3, and 1.0 m ethanol perfusates increased dialysate GABA levels to 127 ± 8, 175 ± 26, and 237 ± 26% of baseline in ethanol-naive animals and to 114 ± 9, 139 ± 21, and 170 ± 26% of baseline in CET animals. Two-way ANOVA, with ethanol history (naive vs CET) as the between-subjects factor, on dialysate GABA expressed as the percentage change from baseline, revealed a significant effect of perfusate ethanol on dialysate GABA levels (p < 0.0001) but no significant interaction between ethanol history and the effects of locally administered ethanol on dialysate GABA levels. The same statistical conclusion was reached when a twoway ANOVA was performed on the data expressed as the absolute change in dialysate GABA levels. Thus, these findings are consistent with our electrophysiological data suggesting a lack of tolerance to acute ethanol and an increase in ethanol-induced GABA release in CeA of CET rats.
Figure 7.

Acute and chronic ethanol increase dialysate levels of GABA in CeA in vivo. In both naive and CET rats, ethanol administration into the CeA significantly (*p < 0.05) and dose-dependently increased mean local dialysate GABA levels. Furthermore, the mean baseline dialysate GABA level was significantly (#p < 0.001) increased in CET compared with naive rats.
Baseline dialysate levels of taurine and glycine did not significantly differ between CET (3.0 ± 0.5 and 2.4 ± 0.3 μm, respectively; n = 7) and naive rats (3.3 ± 0.7 and 2.5 ± 0.4 μm, respectively; n = 11) (data not shown). Local ethanol administration increased dialysate taurine in both CET and naive rats, although this effect was significant (p < 0.001) only at the highest (1 m) ethanol dose (data not shown). There was no significant effect of ethanol administration on dialysate glycine levels in either CET or naive rats (data not shown).
Discussion
Considerable evidence from biochemical, electrophysiological, and behavioral studies points to a GABAergic component in the behavioral effects of ethanol (for review, see Harris et al., 1992; Grobin et al., 1998; Chester and Cunningham, 2002). However, whereas many studies have evaluated the effects of GABA agonists and antagonists on ethanol-related phenomena, few have investigated the influence of ethanol on GABA release itself. Using electrophysiological and microdialysis techniques, we report here that chronic ethanol administration enhances basal GABAergic transmission and GABA tone at CeA synapses via vesicular release. Interestingly, despite increased basal GABA release, there is a lack of tolerance to acute ethanol-induced GABA release in CET rats under the conditions of our study.
Acute and chronic ethanol consumption differentially affects GABAergic function in the brain (Allan and Harris, 1987). Although there is still controversy over the mechanisms involved (Siggins et al., 1999), considerable evidence suggests that acute ethanol facilitates GABAergic neurotransmission (Mihic, 1999). Conversely, some data suggest that chronic ethanol consumption may reduce GABAergic function, consistent with the development of tolerance to the acute effects of ethanol (Allan and Harris, 1987; Davis and Wu, 2001). Indeed, adaptations within the GABA system have been suggested to participate in both the development of behavioral tolerance and dependence as well as many aspects of acute and protracted ethanol withdrawal (Grobin et al., 1998; Papadeas et al., 2001; Davies, 2003). Melis et al. (2002) reported that a single in vivo injection of ethanol (2 gm/kg, i.p.) induced a long lasting facilitation of GABAergic transmission in ventral tegmental area (VTA) slices. We reported recently that acute ethanol consistently enhances GABAergic transmission in the CeA of naive rats, probably in large part by increasing the release of GABA (Roberto et al., 2003) through the activation of CRF1 receptors (Nie et al., 2004).
Here, we have extended our electrophysiological study of acute ethanol to the CeA of ethanol-dependent rats. We recorded from CeA slices that were bathed in ethanol-free ACSF for 2-8 hr and saw no observable postsynaptic hyperexcitability. Interestingly, in these slices from CET rats, the basal evoked GABAA-IPSP and -IPSC amplitudes and the baseline mIPSC frequencies were significantly greater and baseline PPF ratios lower than in naive rats, suggesting an increased GABAergic transmission after chronic ethanol treatment. We hypothesize that the overall increase in basal GABA release may be related to changes in presynaptic function elicited by chronic ethanol treatment, representing a compensatory mechanism counteracting an increased excitability after chronic ethanol to maintain system allostasis (Koob and Le Moal, 2001). One possible explanation for this increased GABA release after chronic ethanol treatment could be because of a reduced sensitivity and/or downregulation of GABAB receptors that are known to reduce GABA release and ethanol effects in other brain regions (Wan et al., 1996; Peris et al., 1997; Siggins et al., 1999). In future studies, it will be important to determine the role of GABAB receptors in acute and chronic ethanol effects.
Unexpectedly, acute ethanol elicited an equivalent increase of both evoked GABAA-IPSP and -IPSC amplitudes and spontaneous mIPSC frequencies in slices from both CET and naive rats, suggesting a lack of tolerance to this effect at this 2-8 hr time point of withdrawal. Of course, the GABA system may not be in a steady state at this time, and our studies do not address the issue of adaptations that might occur with long-term withdrawal. Such studies are now under way in our laboratories. The increased frequency of mIPSCs in CeA neurons from both naive and CET rats suggests an action potential-independent ethanol effect on presynaptic terminals of GABAergic afferents, most likely through increased probability of vesicular GABA release. The acute ethanol-induced effects were concomitant with a decreased PPF, further indicating an increase in GABA release. We speculate that the resistance to tolerance may point to a prominent role for the CeA in the development and maintenance of ethanol dependence. Acute ethanol also had the tendency to increase the mIPSC amplitudes in some cells from both naive and CET rats, suggesting the possibility that ethanol also has a postsynaptic effect (Roberto et al., 2003).
To determine more directly whether acute and chronic ethanol alters GABA release in CeA, we measured dialysate GABA levels in CeA in vivo. One report showed that microdialysate GABA levels in the ventral pallidum or VTA are unaffected by acute peripheral ethanol administration (Cowen et al., 1998), and a similar lack of effect on nucleus accumbens dialysate GABA has been observed with systemic ethanol doses that significantly increase interstitial dopamine and taurine levels (Dahchour et al., 1996). However, ethanol-induced decreases in GABA levels have been observed in the nucleus accumbens of rats selectively bred for high and low sensitivity to ethanol (Piepponen et al., 2002). Although previous microdialysis studies in the CeA have focused on systemic ethanol-induced alterations in dopamine and serotonin (Yoshimoto et al., 2000), ethanol effects on GABA release in CeA were not investigated. In agreement with our in vitro electrophysiological results, we found elevated basal GABA levels in the CeA of CET rats relative to naive controls. Moreover, local administration of acute ethanol increased GABA release in the CeA of both CET and naive rats. Local ethanol administration was chosen rather than systemic, because this approach more closely resembles the ethanol superfusion used in the electrophysiological preparation (Roberto et al., 2003), and for our study of cellular mechanisms, it excludes indirect effects of ethanol at remote brain regions projecting to the CeA. However, because microdialysis data gathered in vivo allows study of the modulatory effects of systemic ethanol on efferents to the CeA, the neurochemical and neurophysiological responses to peripherally administered ethanol are presently under investigation. Ethanol also significantly increased dialysate taurine levels but only at the highest ethanol dose tested, perhaps suggesting a role for taurine in modulating release of other neurotransmitters such as dopamine (Dahchour et al., 1996). Taurine can also mimic GABA or glycine effects in some neurons, although glycine levels were not affected by either acute ethanol or chronic ethanol treatment. In another study, local ethanol administration did not change dialysate glutamate or aspartate content in nondependent rats, but baseline dialysate glutamate was increased in CET compared with naive rats (Roberto et al., 2004).
Our microdialysis findings directly correlate with the present and previous (Roberto et al., 2003) electrophysiological data implicating ethanol-induced GABA release in CeA. These results also support previous behavioral studies (Hyytia and Koob, 1995, Roberts et al., 1996) suggesting that ethanol-GABA interactions in the CeA may play a role in the reinforcing effects of ethanol. Our combined data suggest that acute ethanol increases GABAergic tone, thereby likely producing a net inhibitory effect on CeA neuronal discharge. This activation of the GABA system in CeA may be implicated not only in the sedative, anxiolytic, and anti-convulsant actions of a number of drugs (e.g., benzodiazepines, barbiturates, and ethanol), but also may play an important role in the common neural circuitry for drug reward. Furthermore, the large increase in basal evoked IPSPs and IPSCs, mIPSC frequency, and basal GABA levels after chronic ethanol treatment suggests a novel allostatic mechanism related to ethanol dependence. Thus, the pattern of changes in the release of GABA within the CeA may represent a neuroadaptive mechanism underlying or concurrent with compulsive drug-seeking behavior and the development of dependence.
Footnotes
This work was supported by National Institute on Alcohol Abuse and Alcoholism Grants AA013517 [Integrative Neuroscience Initiative on Alcoholism (INIA)], AA06420, and AA12294 (INIA) and by National Institute on Drug Abuse Grant DA03665. We thank Drs. F. E. Bloom, G. F. Koob, S. J. Henriksen, P. Schweitzer, and P. J. Kenny for critical comments on this manuscript.
Correspondence should be addressed to George R. Siggins, The Scripps Research Institute, CVN-12, 10550 North Torrey Pines Road, La Jolla, CA 92037. E-mail: geobob@scripps.edu.
Copyright © 2004 Society for Neuroscience 0270-6474/04/2410159-08$15.00/0
References
- Allan AM, Harris RA (1987) Acute and chronic ethanol treatments alter GABA receptor-operated chloride channels. Pharmacol Biochem Behav 27: 665-670. [DOI] [PubMed] [Google Scholar]
- Andreasen M, Hablitz JJ (1994) Paired-pulse facilitation in the dentate gyrus: a patch-clamp study in rat hippocampus in vitro. J Neurophysiol 72: 326-336. [DOI] [PubMed] [Google Scholar]
- Bonci A, Williams JT (1997) Increased probability of GABA release during withdrawal from morphine. J Neurosci 17: 796-803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chester JA, Cunningham CL (2002) GABA(A) receptor modulation of the rewarding and aversive effects of ethanol. Alcohol 26: 131-143. [DOI] [PubMed] [Google Scholar]
- Cowen M, Chen F, Jarrott B, Lawrence AJ (1998) Effects of acute ethanol on GABA release and GABA(A) receptor density in the rat mesolimbic system. Pharmacol Biochem Behav 59: 51-57. [DOI] [PubMed] [Google Scholar]
- Dahchour A, Quertemont E, De Witte P (1996) Taurine increases in the nucleus accumbens microdialysate after acute ethanol administration to naive and chronically alcoholised rats. Brain Res 735: 9-19. [DOI] [PubMed] [Google Scholar]
- Davies M (2003) The role of GABAA receptors in mediating the effects of alcohol in the central nervous system. J Psychiatry Neurosci 28: 263-274. [PMC free article] [PubMed] [Google Scholar]
- Davis KM, Wu JY (2001) Role of glutamatergic and GABAergic systems in alcoholism. J Biomed Sci 8: 7-19. [DOI] [PubMed] [Google Scholar]
- De Koninck Y, Mody I (1994) Noise analysis of miniature IPSCs in adult rat brain slices: properties and modulation of synaptic GABAA receptor channels. J Neurophysiol 71: 1318-1335. [DOI] [PubMed] [Google Scholar]
- Frantz KJ, Hansson KJ, Stouffer DG, Parsons LH (2002) 5-HT(6) receptor antagonism potentiates the behavioral and neurochemical effects of amphetamine but not cocaine. Neuropharmacology 42: 170-180. [DOI] [PubMed] [Google Scholar]
- Grobin AC, Matthews DB, Devaud LL, Morrow AL (1998) The role of GABA(A) receptors in the acute and chronic effects of ethanol. Psychopharmacology (Berl) 139: 2-19. [DOI] [PubMed] [Google Scholar]
- Harris RA, Brodie MS, Dunwiddie TV (1992) Possible substrates of ethanol reinforcement: GABA and dopamine. Ann NY Acad Sci 654: 61-69. [DOI] [PubMed] [Google Scholar]
- Hyytia P, Koob GF (1995) GABAA receptor antagonism in the extended amygdala decreases ethanol self-administration in rats. Eur J Pharmacol 283: 151-159. [DOI] [PubMed] [Google Scholar]
- Koob GF, Le Moal M (2001) Drug addiction, dysregulation of reward, and allostasis. Neuropsychopharmacology 24: 97-129. [DOI] [PubMed] [Google Scholar]
- Macey DJ, Schulteis G, Heinrichs SC, Koob GF (1996) Time-dependent quantifiable withdrawal from ethanol in the rat: effect of method of dependence induction. Alcohol 13: 163-170. [DOI] [PubMed] [Google Scholar]
- McBride WJ, Chernet E, McKinzie DL, Lumeng L, Li TK (1998) Quantitative autoradiography of mu-opioid receptors in the CNS of alcohol-naive alcohol-preferring P and -nonpreferring NP rats. Alcohol 16: 317-323. [DOI] [PubMed] [Google Scholar]
- Melis M, Camarini R, Ungless MA, Bonci A (2002) Long-lasting potentiation of GABAergic synapses in dopamine neurons after a single in vivo ethanol exposure. J Neurosci 22: 2074-2082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mihic SJ (1999) Acute effects of ethanol on GABAA and glycine receptor function. Neurochem Int 35: 115-123. [DOI] [PubMed] [Google Scholar]
- Nie Z, Madamba SG, Siggins GR (2000) Ethanol enhances gamma-aminobutyric acid responses in a subpopulation of nucleus accumbens neurons: role of metabotropic glutamate receptors. J Pharmacol Exp Ther 293: 654-661. [PubMed] [Google Scholar]
- Nie Z, Schweitzer P, Roberts AJ, Madamba SG, Moore SD, Siggins GR (2004) Ethanol augments GABAergic transmission in the central amygdala via CRF1 receptors. Science 303: 1512-1514. [DOI] [PubMed] [Google Scholar]
- Otis TS, De Koninck Y, Mody I (1994) Lasting potentiation of inhibition is associated with an increased number of gamma-aminobutyric acid type A receptors activated during miniature inhibitory postsynaptic currents. Proc Natl Acad Sci USA 91: 7698-7702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papadeas S, Grobin AC, Morrow AL (2001) Chronic ethanol consumption differentially alters GABA(A) receptor alpha1 and alpha4 subunit peptide expression and GABA(A) receptor-mediated 36 Cl(-) uptake in mesocorticolimbic regions of rat brain. Alcohol Clin Exp Res 25: 1270-1275. [PubMed] [Google Scholar]
- Paxinos G, Watson C (1986) The rat brain in sterotaxic coordinates, Ed 2. San Diego: Academic.
- Peris J, Eppler B, Hu M, Walker DW, Hunter BE, Mason K, Anderson KJ (1997) Effects of chronic ethanol exposure on GABA receptors and GABAB receptor modulation of 3H-GABA release in the hippocampus. Alcohol Clin Exp Res 21: 1047-1052. [PubMed] [Google Scholar]
- Piepponen TP, Kiianmaa K, Ahtee L (2002) Effects of ethanol on the accumbal output of dopamine, GABA and glutamate in alcohol-tolerant and alcohol-nontolerant rats. Pharmacol Biochem Behav 74: 21-30. [DOI] [PubMed] [Google Scholar]
- Roberto M, Madamba SG, Moore SD, Tallent MK, Siggins GR (2003) Ethanol increases GABAergic transmission at both pre- and postsynaptic sites in rat central amygdala neurons. Proc Natl Acad Sci USA 100: 2053-2058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberto M, Schweitzer P, Madamba SG, Stouffer DG, Parsons LH, Siggins GR (2004) Acute and chronic ethanol alter glutamatergic transmission in rat central amygdala: an in vitro and in vivo analysis. J Neurosci 24: 1594-1603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts AJ, Cole M, Koob GF (1996) Intra-amygdala muscimol decreases operant ethanol self-administration in dependent rats. Alcohol Clin Exp Res 20: 1289-1298. [DOI] [PubMed] [Google Scholar]
- Robinson DL, Lara JA, Brunner LJ, Gonzales RA (2000) Quantification of ethanol concentrations in the extracellular fluid of the rat brain: in vivo calibration of microdialysis probes. J Neurochem 75: 1685-1693. [DOI] [PubMed] [Google Scholar]
- Rogers J, Wiener SG, Bloom FE (1979) Long-term ethanol administration methods for rats: advantages of inhalation over intubation or liquid diets. Behav Neural Biol 27: 466-486. [DOI] [PubMed] [Google Scholar]
- Siggins GR, Nie Z, Madamba S (1999) A metabotropic hypothesis for ethanol sensitivity of GABAergic and glutamatergic central synapses. In: The “drunken” synapse: studies of alcohol related disorders (Liu Y, Hunt W, eds), pp 135-143. New York: Klewer Academic/Plenum.
- Wan FJ, Berton F, Madamba SG, Francesconi W, Siggins GR (1996) Low ethanol concentrations enhance GABAergic inhibitory postsynaptic potentials in hippocampal pyramidal neurons only after block of GABAB receptors. Proc Natl Acad Sci USA 93: 5049-5054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshimoto K, Ueda S, Kato B, Takeuchi Y, Kawai Y, Noritake K, Yasuhara M (2000) Alcohol enhances characteristic releases of dopamine and serotonin in the central nucleus of the amygdala. Neurochem Int 37: 369-376. [DOI] [PubMed] [Google Scholar]