

Abstract
Ion channels in the degenerin-epithelial sodium channel (DEG-ENaC) family perform diverse functions, including mechanosensation. Here we explored the role of the vertebrate DEG-ENaC protein, acid-sensing ion channel 2 (ASIC2), in auditory transduction. Contributions of ASIC2 to hearing were examined by comparing hearing threshold and noise sensitivity of wild-type and ASIC2 null mice. ASIC2 null mice showed no significant hearing loss, indicating that the ASIC2 was not directly involved in the mechanotransduction of the mammalian cochlea. However, we found that (1) ASIC2 was present in the spiral ganglion (SG) neurons in the adult cochlea and that externally applied protons induced amiloride-sensitive sodium currents and action potentials in SG neurons in vitro, (2) proton-induced responses were greatly reduced in SG neurons obtained from ASIC2 null mice, indicating that activations of ASIC2 contributed a major portion of the proton-induced excitatory response in SG neurons, and (3) ASIC2 null mice were considerably more resistant to noise-induced temporary, but not permanent, threshold shifts. Together, these data suggest that ASIC2 contributes to suprathreshold functions of the cochlea. The presence of ASIC2 in SG neurons could provide sensors to directly convert local acidosis to excitatory responses, therefore offering a cellular mechanism linking hearing losses caused by many enigmatic causes (e.g., ischemia or inflammation of the inner ear) to excitotoxicity.
Keywords: cochlea, spiral ganglion neurons, noise, hearing, ASIC, acid
Introduction
The degenerin-epithelial sodium channel (DEG-ENaC) family includes at least 17 proteins that all share similar sequences and predicated membrane protein topology. Many ion channels in this family have been implicated in playing a role in mechanical transduction. For example, the pickpocket channel (a Drosophila DEG-ENaC channel) is localized in the sensory dendrites of a subset of peripheral neurons thought to play key roles in proprioception and touch sensation (Adams et al., 1998). The β and γ subunits of the epithelial ENaCs have been localized in specialized mechanosensory endings (e.g., Meissner corpuscles and Merkel cell-neurite complexes) in the skin of rats (Drummond et al., 2000). These localization patterns make them good candidates to participate in mechanosensory transduction. Functional involvement of many DEG-ENaC channels has been confirmed by genetic mutation studies in Caenorhabditis elegans and in mice. It is proposed that multiple genes belonging to the DEG-ENaC family [MEC-4 and MEC-10 (mechanotransducing channel subunits 4 and 10)] are needed to form the mechanoreceptor complex responsible for mechanosensation in C. elegans (Driscoll and Chalfie, 1991; Huang and Chalfie, 1994). Disruptions of UNC-8 and UNC-105 genes impair proprioception (Tavernarakis et al., 1997) and cause difficulties in detecting muscle stretch, respectively. Liu et al. (1996) suggested that UNC-105 is a stretch-activated channel interacting with type IV collagen.
Extracellular protons are the most commonly used stimuli to test ion channels in one branch of the DEG-ENaC family in mice, the acid-sensing ion channels (ASICs). Despite the name implication, ASICs are known to be involved in a wide range of neuronal functions, including mechanosensation (Price et al., 2000, 2001), memory and learning (Wemmie et al., 2002), synaptic plasticity related to fear conditioning (Wemmie et al., 2003), development of mechanical hyperalgesia in muscle afferents (Sluka et al., 2003), and modulation of retinal functions (Ettaiche et al., 2004). Among the four ASICs cloned so far, ASIC2 and ASIC3 have been considered candidate channels constituting part of the mechanoreceptor because deletion of these ASIC genes in mice affects sensitivity of specific components of mechanotransduction (Price et al., 2000, 2001). These results prompted a number of groups to comprehensively investigate direct contributions of ASIC2 and ASIC3 in mammalian mechanosensation, mechanonociception, and hearing (Drew et al., 2004; Hildebrand et al., 2004; Roza et al., 2004). Data published recently by these groups showed that the two types of ASIC channels are not required for normal mechanosensory transductions, at least at the threshold level. In this paper, we show that, although ASIC2s were not required for hearing sensitivity at the threshold level, these channels contributed to suprathreshold hearing functions by specifically affecting noise-induced temporary threshold shifts (TTSs). Immunolabeling showed ASIC2 expression in spiral ganglion (SG) neurons in the adult cochlea. Comparisons of proton-evoked excitatory responses recorded from wild-type and ASIC2-/- mice identified ASIC2 to be the major contributor to the proton-activated inward currents in the SG neurons. Therefore, the ASIC2 in the cell membrane of SG neurons could provide cellular sensors needed to directly convert extracellular acidosis to excitatory responses.
Materials and Methods
Recording membrane currents and voltages of SG neurons by whole-cell voltage and current clamps. Cell culture procedures and cell selection criteria of SG neurons isolated from mice cochlea were the same as those described previously for culturing gerbil SG neurons (Lin, 1997). All of the chemicals used in the experiments were purchased from Sigma (St. Louis, MO) unless otherwise specified. Patch-clamp recordings were made from SG neurons obtained from either acutely dissociated adult SG neurons or cultured SG neurons [for mice younger than postnatal day 10 (P10)]. The normal external solution contained the following (in mm): 150 NaCl, 3 KCl, 4 CaCl2, and 5 HEPES, pH 7.4. In ion substitution experiments, NaCl was replaced by an equal concentration of choline chloride. For recording proton-gated currents, tetrodotoxin (0.5 μm), tetraethylammonium (10 mm), and nimodipine (5 μm) were added to block voltage-gated Na+, K+, and Ca2+ currents, respectively. External solutions buffered at different pH levels were delivered by a programmable focal superfusion system (ALA Scientific Instruments, Westbury, NY).
Membrane currents or voltages were amplified by an Axon 200B amplifier (Axon Instruments, Union City, CA), filtered at 2 kHz (80 dB/decade), and digitized at a rate of at least 10 kHz. pClamp software (version 7; Axon Instruments) was used for data acquisitions and analyses. Borosilicate glass capillary pipettes (World Precision Instruments, Sarasota, FL) with a resistance of ∼1.5 MΩ were made on a horizontal microelectrode puller (P-97; Sutter Instruments, Novato, CA). The access resistance was compensated by at least 80%. Voltage-clamp and junction potential errors were corrected by the standard method. All results described were repeated from at least 12 SG neurons unless indicated otherwise.
Immunohistochemical labeling of ASIC2 in the inner ear. CD-1 mice from embryonic day 11.5 (E11.5) to adult were killed by CO2 inhalation. Inner ears were dissected out surgically and fixed in 4% paraformaldehyde for 2-3 hr. Mice older than P8 were fixed first by cardiac perfusion of 4% paraformaldehyde. Cochlear samples from mice older than P4 were decalcified for at least 2 weeks in a 120 mm EDTA solution at 4°C. Frozen sections of the cochlea (8 μm in thickness) were prepared according to the standard procedure and were labeled with antibodies against ASIC2 (catalog number AB5460; Chemicon, Temecula, CA). The antigenic peptide corresponds to amino acid residues 2-18 of rat ASIC2, which is specific for ASIC2a. Therefore, this antibody should specifically recognize ASIC2a. Details of the immunolabeling procedure are given in our previous publication (Lin, 1997). Briefly, cochlear sections were fixed for 1 hr with 4% paraformaldehyde prepared in 0.1 m PBS, pH 7.3. They were incubated with the antibody against ASIC2 for 24 hr at 4°C. The antibody was diluted (1:200) in a solution containing the following: 20% fetal calf serum, 80% PBS, 0.02% Triton X-100, and 0.05% thimerosal. After staining with the primary antibody, cochlear sections were rinsed with PBS three times and labeled with the secondary antibody (antimouse IgG conjugated with FITC; 1:200 dilution) for 2 hr at room temperature or 24 hr at 4°C. Results were observed and documented with a Zeiss (Thornwood, NY) inverted microscope (Axiovert 135TV) equipped with epifluorescence and a cooled CCD camera (AxioCam; Zeiss). Controls were obtained by preincubating antibodies with antigenic peptide (ratio of antibody/peptide was 1:1) for 1 hr before the start of the immunolabeling procedures. Results were photographed using a cooled CCD camera (AxioCam; Zeiss). Relative ASIC2 labeling intensity (see Fig. 8) was quantified by calculating the ratio of pixel intensities of ASIC2-labeled areas to those of the background in the same cochlear sections.
Figure 8.

Examples of click-evoked ABR waveforms recorded using a series of sound intensities ranging from 80 dB (top data traces) to 25 dB (bottom data traces) changing in a 5 dB step. Measurements were done before (A) and after noise exposures for wild-type (B) and ASIC2 null (C) mice. Hearing thresholds were determined by following amplitude and latency changes of the wave III or IV in the ABRs until a minimal sound level was determined, at which point just a detectable ABR was observed.
Testing of noise susceptibility of ASIC2 knock-out mice. Heterozygote breeding pairs of ASIC2 knock-out (KO) mice were provided by Dr. Margaret Price (University of Iowa, Iowa City, IA). Transcripts of ASIC2a and its splice variant ASIC2b were absent in ASIC2 null mice (Price et al., 2000). Genotypes of ASIC2 null mice were confirmed by methods published previously (Price et al., 2000, 2001). Wild-type and ASIC2 knock-out mice (20 animals in each group) were exposed to white-band noise of 110 dB sound pressure level (SPL) or 125 dB SPL for 1 hr to create temporary and permanent threshold shifts (PTSs), respectively. The long-term effect of noise exposures on hearing sensitivity was monitored by measuring thresholds of auditory brainstem responses (ABRs) for at least 3 weeks. To prevent subjective bias in measurements, the genotype of the mice was kept confidential to the ABR tester. Sound stimuli, consisting of tone bursts of various frequencies (10 msec in duration and a rise-fall time of 0.5 msec) or clicks were delivered through a high-frequency sound transducer (Intelligent Hearing Systems, Miami, FL). ABRs were recorded and analyzed by conventional methods using Tucker-Davis System II hardware and software (Tucker-Davis Technologies, Alachua, FL). The hearing threshold was determined by following the amplitude and latency changes of the wave III or IV in the ABRs until a minimal sound level was determined, at which point just a detectable ABR waveform is observed.
Results
Protons opened amiloride-sensitive cation channels and evoked excitatory responses in SG neurons; ASIC2 contributed the major portion of the proton-evoked currents in SG neurons
One commonly used method of testing the presence of ASIC channels is to directly test membrane responses to a lowering of extracellular pH. Figure 1 shows typical proton-elicited responses obtained from isolated SG neurons in culture. Virtually all cultured SG neurons we tested (124 of 131) showed acid-evoked inward currents. Extracellular solutions that had different pH values (range, pH 7.0-4.5) were applied sequentially to each SG neuron with washout perfusions applied between proton applications (Fig. 1A). SG neurons were voltage clamped at -70 mV and bathed normally in an extracellular solution buffered at pH 7.4. A small inward current was elicited at pH 7.0 (Fig. 1A). When solutions with more acidic pH were delivered, subsequent responses were larger in amplitude and apparently contained both transient and sustained components. The dose-response to protons is given in Figure 1B, which shows that the half-maximal current elicited by protons was obtained at approximately pH 6.5. The half-maximal average peak current density was 0.44 ± 0.07 pA/μF (n = 15). Because current density at ∼0.08 pA/μF is able to induce action potentials in cultured SG neurons (Lin, 1997), these voltage-clamp results suggest that small pH decreases should be able to elicit excitatory responses in SG neurons. This was confirmed by results obtained in current-clamp experiments. Figure 1C shows that a lowering of pH from 7.4 to 6.8 elicited the firing of a train of action potentials (n = 10), demonstrating the direct and strong excitatory effects to SG neurons by a moderate increase of proton concentration. In contrast, the same pH changes (from 7.4 to 6.8) failed to generate the firing of action potentials in SG neurons obtained from ASIC2 knock-out mice (see Fig. 3D). Heterologously expressed DEG-ENaC channels are cation channels commonly inhibited by the diuretic amiloride. Figure 2A shows the effect of substituting Na+ in the external solution with a much larger molecule, choline+. The acid-evoked responses (a lowering of pH from 7.4 to 5.5) were abolished in a reversible manner (Fig. 2A), suggesting that anions are not the ionic carriers for the currents. The proton-elicited currents in SG neurons were also blocked by amiloride. One example of the block and inhibitory dose responses to pH 4.5 are given in Figure 2B. The blocking by amiloride was totally reversible (Fig. 2B, inset). The inhibitory dose responses (Fig. 2B) were obtained from data gathered from nine SG neurons and fit with the following equation: %response = (1 - [amiloride]n/(IC50n + [amiloride]n)) × 100, using half-maximal inhibition concentration (IC50) of 13.6 μm and a coefficient factor (n) of 1.1. Results of the curve fit are given by the smooth curve in Figure 2B.
Figure 1.
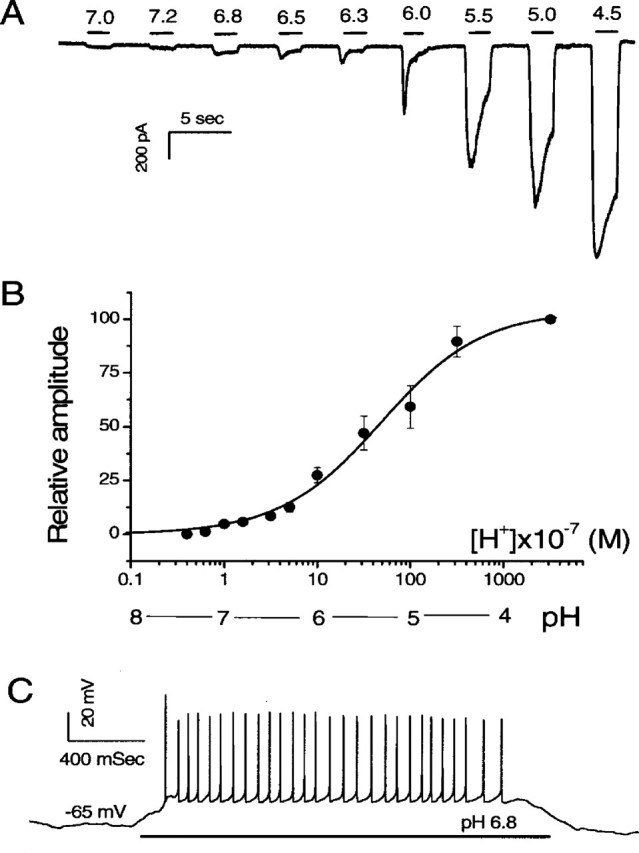
Proton-elicited responses in cultured SG neurons. A, Inward currents elicited by increasing concentrations of protons. The pH values and approximate duration of the application of external solutions to the cell are indicated by the numbers and horizontal bars above the data trace, respectively. Normal external solution was perfused between the proton applications. The currents were recorded from an SG neuron voltage clamped at -70 mV. B, Dose-response curve of the normalized peak current. The data were fitted with a smooth curve using the following equation: %response = 100 × [pH]n/([pH]50n + [pH]n), in which [pH]50 is the pH value at which half-maximal currents was obtained, and n is the coefficient factor. Black dots and error bars give the average data and SEMs, respectively. C, Action potentials elicited by increasing concentration of protons in an SG neuron. The approximate duration of the application of the pH 6.8 solution is indicated by a horizontal bar underneath the data trace. The neuron was current clamped at its resting membrane potential of -65mV; therefore, no DC was injected.
Figure 3.
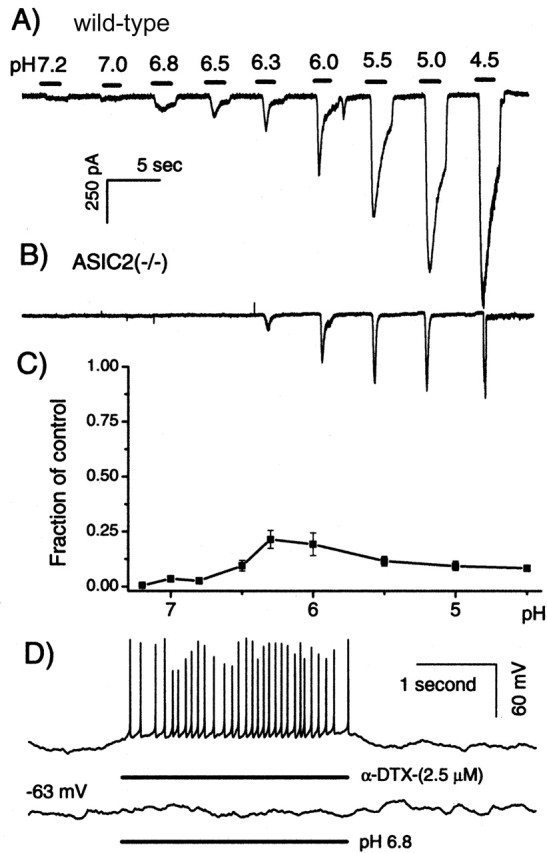
Comparison of proton-elicited responses obtained from wild-type and ASIC2 knock-out mice. Inward currents elicited by increasing concentrations of protons in the wild-type (A) and ASIC2 KO (B) mice. pH values and the approximate duration of the external solution applied are indicated by numbers and black bars above the data traces, respectively. Time and amplitude scales are the same for both A and B. C, Ionic fluxes elicited by protons obtained in ASIC2 knock-out mice normalized to that of the wild-type mice. Total ionic fluxes were obtained by integrating inward currents induced by protons. The x-axis gives the pH value of external solutions applied to the SG neurons. Error bars indicate SEM. D, α-DTX (top data trace) and proton-elicited (bottom data trace) voltage responses obtained from the same SG neurons under current clamp. The average resting membrane potential of the SG neuron was approximately -63 mV. The approximate duration of the applications of α-DTX and proton are indicated by horizontal bars under the data traces.
Figure 2.
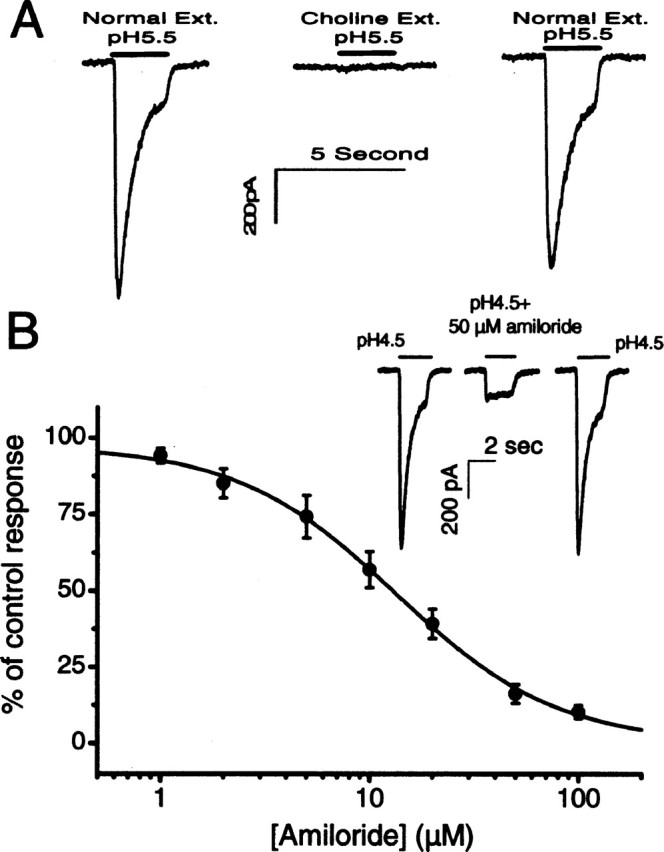
Ionic specificity and pharmacological block of the proton-elicited responses in SG neurons. A, Comparison of inward currents recorded when Na+ ions were substituted by choline+ in the external (Ext.) solution. pH values and approximate duration of perfusions are given by the numbers and horizontal bars above the data traces. B, Inhibitory dose-response of amiloride. Black dots and error bars give the average data and SEMs, respectively. The smooth curve was obtained by fitting the data with the following equation: %response = (1 - [amiloride]n/(IC50n + [amiloride]n)) × 100, in which IC50 is the half-maximal inhibition concentration of amiloride, and n is the coefficient factor. Inset, One example of the pH 4.5-elicited currents blocked by 50 μm amiloride.
Availability of ASIC2 knock-out mice allowed us to directly test the contribution of ASIC2 in proton-elicited responses in SG neurons. Voltage-clamp recordings similar to those described in Figure 1 were repeated using SG neurons isolated from the cochlea of ASIC2 null mice. In sharp contrast to results obtained from wild-type animals (Fig. 3A, data trace), solutions with pH values >6.3 failed to evoke any detectable inward current (Fig. 3, compare A, B) (n = 7). More acidic stimuli evoked more transient currents that quickly inactivated completely to the baseline (Fig. 3B). Figure 3C shows the proton-elicited relative current fluxes (defined as current amplitude multiplied by the duration of the current) obtained in ASIC2 null mice normalized to that of the wild-type mice. The absence of ASIC2 greatly reduced proton-induced current flows in SG neurons (Fig. 3C) (n = 7). At moderate pH drops (pH >6.5), the inward currents induced by protons were absent in SG neurons obtained from the cochlea of ASIC2 null mice. In sharp contrast to data obtained from wild-type animals (Fig. 1C), SG neurons isolated from ASIC2 null mice failed to show firings of action potential to a pH change from 7.4 to 6.8 (Fig. 3D, bottom data trace). As a positive control, firings of action potentials were induced by applications of α-dendrotoxin (α-DTX) to these neurons (n = 6). These results suggest that ASIC2 contributed the major component of proton-evoked currents in SG neurons, especially at moderate pH changes down to 6.5. Alternatively, ASIC2 could be a required component in a heteromultimerically assembled complex responding to extracellular protons. Its absence greatly altered the kinetics of the proton-elicited responses.
ASIC2 expressed early and its spatial expression patterns changed dramatically during cochlear development
The results presented above showed that ASIC2 was the major contributor to proton-induced currents in SG neurons. We therefore concentrated our efforts on detecting ASIC2 expression patterns in the cochlea. The specificity of the antibody we used for labeling was supported by a nearly total absence of immunoreactivity in cochlear sections preabsorbed with immunogenic peptide (Figs. 4, 5, ctrl panels). Results showed that immunoreactivities to ASIC2 were strong in specific regions of the cochlea, and the spatial expression pattern of ASIC2 changed dramatically during cochlear development (Figs. 4, 5). ASIC2 expression was detected as early as E11.5 in the otocyst (data not shown). Before birth, ASIC2 immunoreactivity was confined primarily to all epithelial cells (except for the hair cells) located on the lumen side of the endolymphatic space. An example obtained from a P18.5 cochlear section is shown in Figure 4, and the enlargement given on the top right shows labeling of cells in the apical turn of the cochlea. ASIC2 expression in SG neurons was relatively weak before birth (Fig. 4, arrows 1). Postnatally, the intensity of the ASIC2 labeling on the lumen side of the membranous labyrinth began to decrease (Figs. 4, 5). Cellular regions showed that immunoreactivity shifted gradually toward the top of supporting cells and the spiral limbus (Figs. 4, 5). Although ASIC2 immunoreactivity at SG neurons was nearly absent in the early developing cochlea, it started to increase after E18.5, and the average immunolabeling density at SG neurons more than doubled after birth. In Figures 4 and 5, locations of SG neurons in the cochlea are indicated by arrows 1. Antibody to ASIC2 also labeled the lumen side of all cells forming the vestibular cavity at early embryonic stages (Fig. 4, left) and at the top of the macula of saccule and utricle (Fig. 4, arrowhead). Labeling at the vestibular ganglion (Scarpa's) neurons was also observed (Fig. 5, labeled neurons indicated by arrows 2).
Figure 4.

ASIC2 immunolabeling results of a prenatal cochlear sections (E18.5). The negative control (E18.5 ctrl) was obtained by preincubating the primary antibody with antigenic peptide. The enlarged view given in the top right panel shows detailed labeling patterns in areas of the cochlea outlined by a frame. Arrow 1 points to the location of SG neurons, and the arrowhead indicates developing macula of utricle. Scale bar, ∼800 μm. SV, Scala vestibule; SM, scala media; ST, scala tympani.
Figure 5.
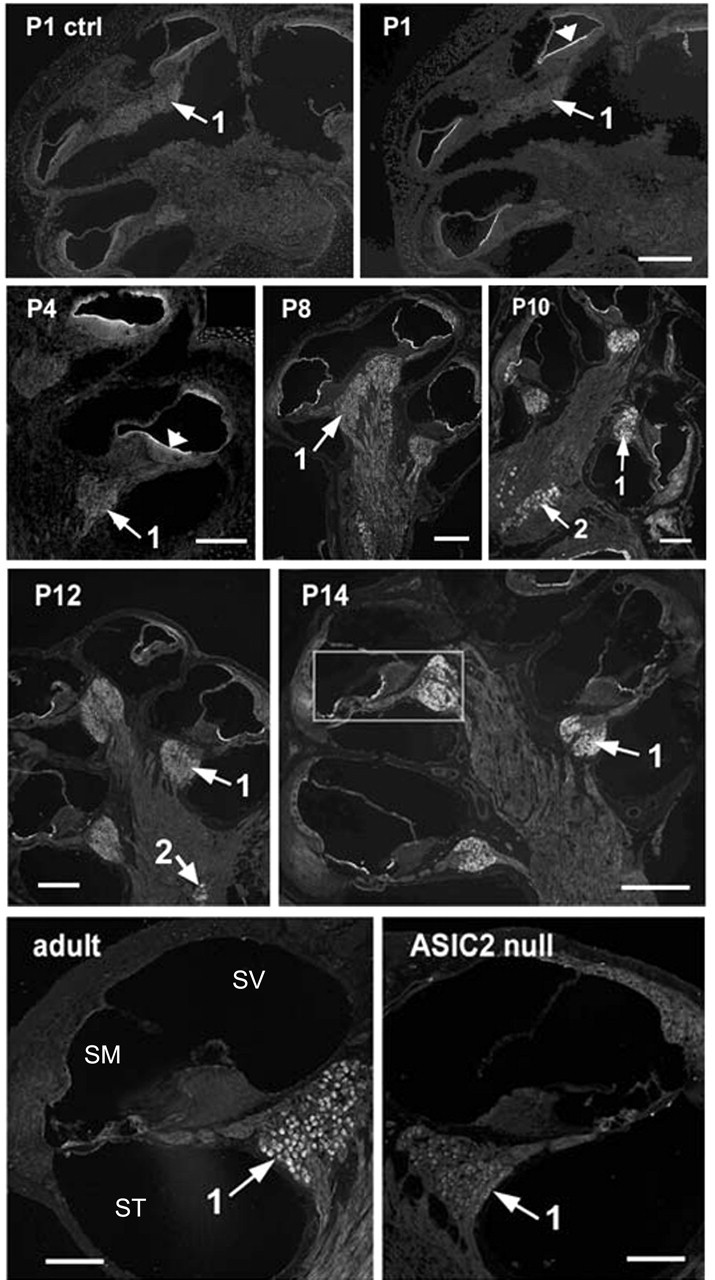
Immunoreactivities of postnatal cochlear sections to an antibody against ASIC2. Immunolabeling of cochlear sections obtained at P1, P4, P8, P10, P12, P14, and adult. P1 control (ctrl) shows negative control results obtained by preincubating first antibody with antigenic peptide. ASIC2 null shows labeling results using cochlear sections obtained from adult ASIC2 null mouse (bottom right). The organ of Corti, outlined by a box in P14, is enlarged and shown in Figure 6 for details. Arrows 1 point to the location of spiral ganglion neurons, and arrows 2 point to the location of vestibular (Scarpa's) ganglion neurons. SV, Scala vestibule; SM, scala media; ST, scala tympani.
After P14, ASIC2 was strongly expressed in SG neurons in the cochlea. In addition to strong labeling in the soma of SG neurons, punctuated immunolabeling spots along the peripheral processes (Fig. 6A,B, arrows) and neurites under the base of inner hair cells (Fig. 6B, arrows near the base of the inner hair cell) were also observed. Our results also demonstrated that cells expressing ASIC2 concentrated on the top of supporting cells and interdental cells after birth, and its expression was restricted to the SG neurons in the mature cochlea (Fig. 7). In cochlear sections obtained from ASIC2 null mice, immunoreactivity to ASIC2 antibodies was reduced to near background level in all regions of the cochlea (Fig. 5, ASIC2 null). These results showed that ASIC2 was prominently expressed in the cochlea, and its spatial and temporal expressions changed dynamically during development, concentrating almost exclusively on both the soma and processes of SG neurons after the maturation of hearing functions.
Figure 6.
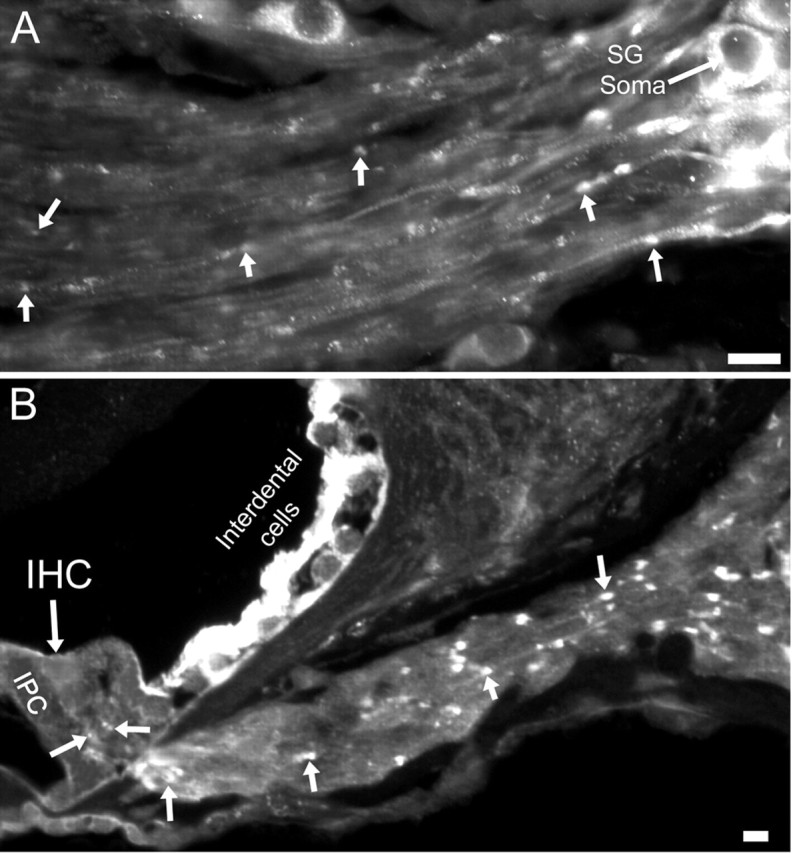
ASIC2 immunoreactivities in the peripheral processes near the soma of SG neurons (A) and in the suspected nerve terminals under and near the bottom of the inner hair cell (B). Arrows point to examples of punctuated ASIC2 labeling along the peripheral processes of SG neurons. Scale bars, ∼20 μm. IHC, Inner hair cell; IPC, inner pillar cell.
Figure 7.
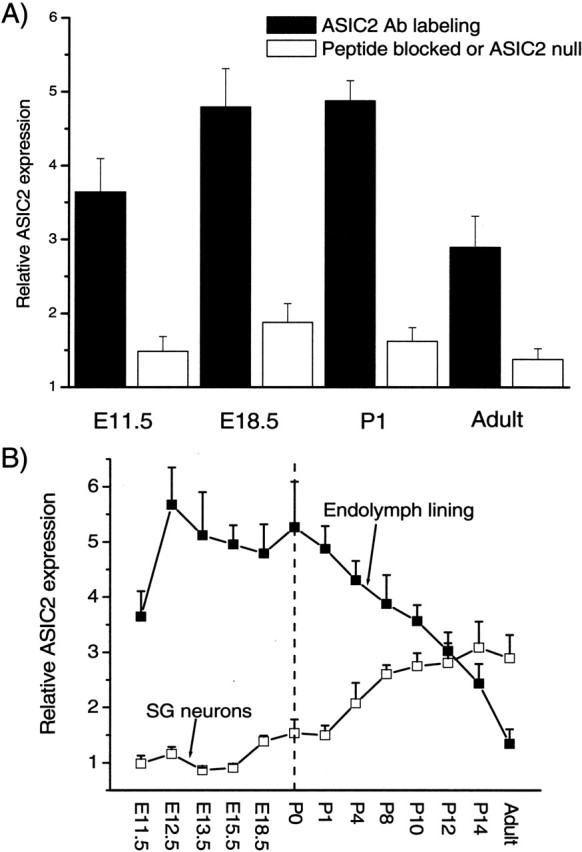
Quantification of ASIC2 labeling intensity in cells lining the endolymphatic space and in SG neurons. A, Relative labeling intensity of ASIC2 (y-axis) at various developmental stages (x-axis) was obtained by comparing labeled regions with background regions in the same cochlear sections. B, The specificity of the labels by the ASIC2 antibody is shown by a large reduction in the relative labeling intensity caused by either a peptide block (unfilled bars) or the absence of labeling in cochlear sections obtained from ASIC2 null mice (last unfilled bar on the right). Error bars indicate SEM.
ASIC2 null mice showed normal hearing sensitivities, but their susceptibilities to noise-induced temporary threshold shifts were decreased
The presence of ASIC2s in the inner ear and our observation that moderate increases of proton concentration gave strong excitatory responses (Figs. 1, 3A) in wild-type animals suggested that ASIC2s could play a role in cochlear functions. We therefore compared hearing threshold and noise sensitivity of wild-type and ASIC2 null mice. Hearing sensitivity of mice was measured by ABRs. Frequency-specific hearing thresholds were measured from tone burst-elicited ABRs, and overall hearing threshold was measured by click-evoked ABRs. Deleting the ASIC2 gene showed no significant effects on the threshold of either click- or tone burst-evoked ABRs (see Fig. 9A). Thresholds for click-evoked ABRs were 29.0 ± 1.2 (n = 20) and 31.5 ± 2.1 dB SPL (n = 20) for ASIC2 wild-type and KO mice, respectively. Changes in the threshold of frequency-specific ABR thresholds from 4 to 24 kHz were also found to be statistically not significant (see Fig. 9A). Although the absence of ASIC2 did not affect hearing threshold, testing of suprathreshold auditory functions revealed that ASIC2 KO mice were significantly more resistant to noise-induced TTSs. Examples of three series of ABR waveforms measured before (Fig. 8A) and after exposures to 110 dB white-band noise obtained in wild-type (Fig. 8B) and ASIC2 null mice (Fig. 8C) are given in Figure 8. Measured 1 hr after exposing animals to noise that was intense enough to cause TTS only (110 dB SPL white-band noise for 1 hr), hearing threshold shifts measured by click-evoked ABR in wild-type and ASIC2 KO mice were 14.3 ± 1.8 (n = 16) and 5.0 ± 1.1 (n = 16) dB, respectively. The hearing thresholds measured at the three most sensitive frequencies (8, 12, and 16 kHz) showed statistically significant differences (Fig. 9B). We followed the recovery of ABR thresholds after noise exposures and found that hearing thresholds of this group of animals returned to preexposure levels in both wild-type and KO mice after ∼3 weeks. Interestingly, we found that, after animals were exposed to 125 dB white-band noise for 1 hr, which caused permanent hearing threshold shift, click-evoked ABR threshold shifts were 36.0 ± 3.1 (n = 10) and 40.5 ± 2.4 (n = 10) dB SPL for the wild-type and ASIC KO mice, respectively. Both click- and tone burst-measured threshold shifts showed no statistically significant differences (Fig. 9C). These results suggested that ASIC2s were specifically involved in generating temporary threshold shifts after noise exposures.
Figure 9.
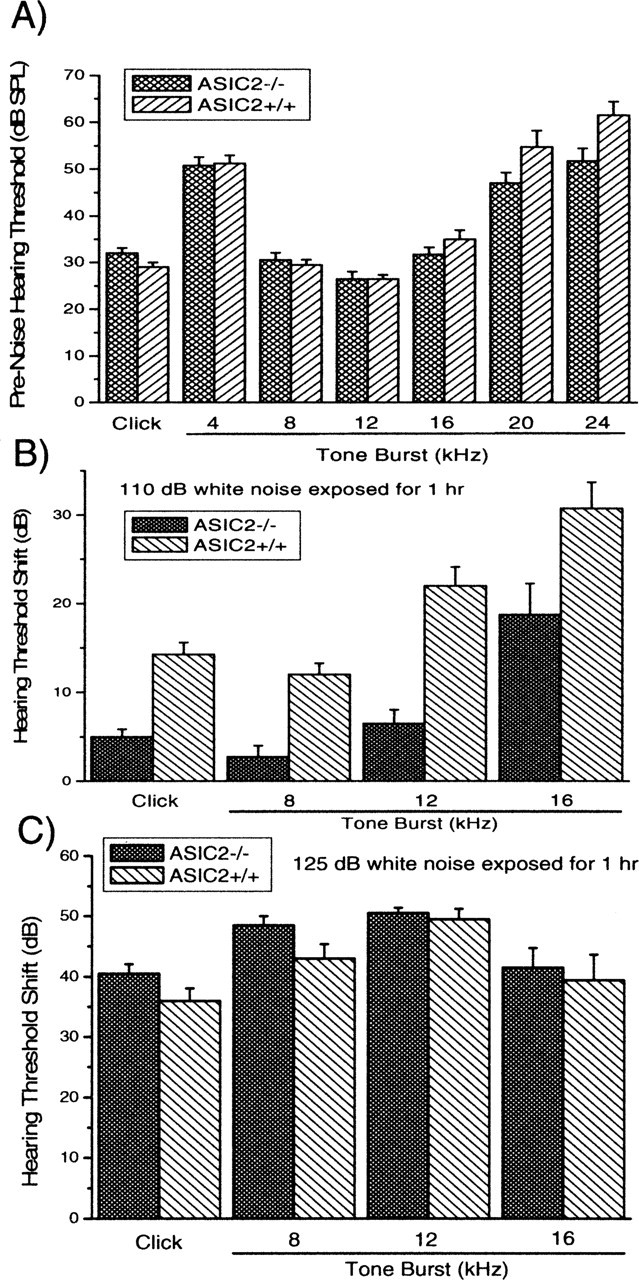
Comparison of hearing thresholds and noise sensitivities of wild-type and ASIC2-/- mice. A, Hearing thresholds of wild-type (ASIC+/+) and ASIC2-/- mice measured by either click (first pair of columns) or tone bursts of various frequencies (indicated by the numbers under the bars). B, Hearing threshold shifts after exposures to 110 dB white-band noise in wild-type and ASIC2-/- mice measured by click (first pair of columns) or tone bursts of various frequencies (indicated by the numbers under the bars). C, Hearing threshold shifts after exposures to 125 dB white-band noise in wild-type and ASIC2-/- mice, measured by click (first pair of columns) or tone bursts of various frequencies (indicated by the numbers under the bars).
Discussion
This study combined several techniques to investigate the contribution of ASIC2 to the excitatory responses of SG neurons and its involvement in hearing functions. We demonstrated direct excitatory effects of extracellularly applied protons to SG neurons in cultures (Figs. 1, 2, 3). We also investigated developmental expression patterns of ASIC2 in the cochlea and contribution of ASIC2 in noise-induced temporary hearing threshold shifts. By comparing responses obtained from both wild-type and ASIC2 null mice (Fig. 3), we identified that ASIC2 was responsible for a major component of proton-induced excitatory responses in SG neurons as indicated by a dramatic reduction in proton-induced inward currents in ASIC2-/- mice. We then focused on characterizing the role of ASIC2 in the cochlea. Our immunolabeling results revealed that ASIC2 was almost exclusively expressed by SG neurons in the mature cochlea, in contrast to a much more widespread expression pattern in the epithelial cells lining the endolymphatic duct in the developing cochlea (Figs. 4, 5). Our finding that ASIC2 null mice had a normal hearing threshold (Fig. 9A) was consistent with a recent report showing that neither ASIC2 (Roza et al., 2004) nor ASIC3 (Hildebrand et al., 2004) was required for normal hearing sensitivity. Therefore, future studies are required to test other candidates forming the mechanotransduction complex, such as vanilloid receptor-related osmotically activated channel (Liedtke et al., 2000), TREK-1 (TWIK-related K+ channel) potassium channel (Maingret et al., 1999), and the NompC channel (Sidi et al., 2003). This is the first report confirming a functional role of ASIC2 in the cochlea. Although we have to unmask a suprathreshold function of ASIC2 with noise exposures, the presence of ASIC2 at the neurites under the base of inner hair cells (Fig. 6B) makes it possible that the ASIC channels may also affect postsynaptic responses at more moderate sound levels if protons are coreleased from the synaptic vesicles. Our observation that ASIC2 null mice had reduced noise-induced TTSs (Fig. 9B) is analogous to previous findings showing that only suprathreshold neuronal functions were markedly altered in ASIC null mice (Wemmie et al., 2002; Ettaiche et al., 2004). For example, disruption of the ASIC1 gene did not affect threshold responses at basal synaptic transmission in the hippocampus. The lack of the gene impaired suprathreshold functions, such as hippocampal long-term potentiation (Wemmie et al., 2002). Recently, it was found that one important function of ASIC2 is to protect against light-induced retinal degeneration, which is another suprathreshold function implicated for ASICs (Ettaiche et al., 2004).
Functional proton-gated channels can be formed by homomultimers as well as heteromultimers. For example, ASIC2 and ASIC3 can form a heteromultimeric channel with properties (kinetics and ionic selectivity) similar to those of native protongated channels found in DRG neurons (Bevan and Yeats, 1991). ASIC1 and ASIC2 subunits also form functional channels in heterologously expressed cell lines to produce acid-evoked currents (Bassilana et al., 1997; Benson et al., 2002). Although the absence of ASIC2 greatly reduced the proton-induced current flows in SG neurons, possibilities remain that ASIC2 may form heteromultimeric channels with other subtypes of ASICs in SG neurons. The greatly altered kinetics of pH-induced currents obtained in ASIC2 null mice may be explained by altered stability of protein complex, loss of interactions of channels with other proteins, or incorrect membrane sorting and targeting as a result of removing ASIC2 subunits from such a heteromultimeric channel complex. By incorporating ASIC2 into heteromultimers, we may also explain our finding that deleting ASIC2s did not completely eliminate the transient inward currents recorded in SG neurons to pH drops more acidic than 6.3. Perhaps the remaining ASIC subunits can still form acid-sensitive channels, albeit responding with a much smaller amplitude and with much faster inactivating kinetic properties in the absence of ASIC2.
ASICs have been found to play roles in both normal physiologic and pathophysiologic functions. These functions could be related or unrelated to the detection of extracellular pH. Considering the expression patterns of ASIC2 in mature cochlea (Fig. 4, 5), one hypothetical role for ASICs in the cochlea is to modulate postsynaptic responses in SG neurons. It has been suggested that local pH decreases at subcellular structures may be significant enough to activate ASICs, although pH in tissues is tightly regulated at a larger scale by homeostatic mechanisms (Chesler and Kaila, 1992; Wangemann and Schacht, 1996). For example, synaptic vesicles usually contain a high concentration of protons and have a pH of ∼5.7 (Yuste et al., 2000). With intense synaptic activities, local extracellular pH at the synaptic cleft could be transiently lowered after vesicle release (Krishtal et al., 1987; DeVries, 2001; Traynelis and Chesler, 2001). This speculation has been confirmed in cultured hippocampal neurons (Miesenbock et al., 1998) and in hippocampal slices (Krishtal et al., 1987). Therefore, vesicular protons acting on ASICs may modulate synaptic transmission by providing another source for postsynaptic excitation or by suppressing presynaptic Ca2+ currents, as demonstrated in retinal cone receptors (DeVries, 2001). Our observation that an absence of the ASIC2 gene product reduced noise sensitivity in mice, therefore, may be explained by a lack of proton-induced excitotoxicity to SG neurons at sound levels causing TTSs. PTS caused by even higher noise intensities depends on different mechanisms, usually involving irreversible structural damage. The differential effects shown by ASIC2 null mice after exposing them to noises causing TTSs and PTSs (Fig. 9B, C) were consistent with this notion. Presumably, excessive activation of ASIC2s in SG neurons may produce noise-induced excitotoxicities that cause a temporary dysfunction of synaptic transmission. It is known that dendrites of SG neurons may gradually grow back after noise-induced TTS damages (Puel et al., 1998). Conceivably, many cellular stresses (e.g., ischemia and inflammation) in the cochlea may also induce local acidosis. Therefore, the presence of ASIC2 in SG neurons may provide the membrane sensors needed to convert these pathological changes, independent of the mechanical stimuli, to excessive excitatory responses in SG neurons.
The early and dynamic expression of ASIC2 in the cochlea suggested that ASIC2 could play another role in maintaining the microenvironment needed for the maturation of the developing cochlea, especially under stress conditions in which proper homeostatic mechanisms are required. The pH in inner ear fluids is tightly regulated at 7.4 in the cochlea (Wangemann and Schacht, 1996) and 7.0 in the endolymphatic sac (Couloigner et al., 2000). Dysfunction of this acid-base homeostasis in the inner ear fluids is known to affect cochlear functions. Maintenance of the homeostatic mechanism requires concerted actions of many ionic pumpschannels, including the H+-ATPase and anion (Cl--HCO3-) exchanger. Defects in H+-ATPase and Cl--HCO3- have been associated with sensorineural hearing loss in autosomal recessive distal renal tubular acidosis (dRTA) (Karet et al., 1999) and Pendred syndrome (Scott et al., 1999), respectively. Sensorineural hearing losses in dRTA are believed to be caused by mutational defects in a gene encoding the β1 subunit of H+-ATPase (ATP6β1) resulting in the impaired excretion of protons into endolymphatic space (Karet et al., 1999). By acting as sensors for extracellular protons, ASICs could be another membrane protein participating in regulating the acid-base homeostasis of the inner ear. It is interesting to note that ASIC2, ATP6β1, and Cl--HCO3- share overlapping expression patterns in the developing cochlea by lining up epithelial cells constituting the inner border of the endolymphatic space (Karet et al., 1999; Scott et al., 1999).
Although protons are used as a ligand to conveniently test the ASIC channels, cellular functions of ASICs are not necessarily restricted to detecting extracellular protons. For example, ASIC null mice display deficits in spatial learning and synaptic plasticity as tested by fear conditioning and eyeblink conditioning experiments. At the cellular level, impaired hippocampal long-term potentiation is observed (Wemmie et al., 2002). Because ASICs could act as a sensor for noxious stimuli, they have been suggested to play key roles in some pathological conditions as well. Interstitial acidosis associated with seizures and ischemia has been speculated to trigger excessive excitatory activities, thereby worsening the pathological consequences of these conditions (Varming, 1999). Hearing losses could be caused by nonmechanical stimuli (e.g., poor inner ear circulation) because results of inflammation or ischemia are suspected to cause sudden hearing loss. Currently, little is known about the underlying etiology. Expression of ASICs in SG neurons provides candidate cellular sensors to detect homeostatic changes and directly covert pH decreases in extracellular fluid to excitatory responses in SG neurons. Many pathological conditions (e.g., inflammation and ischemia) are accompanied by local acidosis of the extracellular space. Excessive activation of ASICs could potentially significantly impair functions of SG neurons by excitotoxic mechanisms. In these regards, ASICs are promising candidates for further studying many enigmatic hearing losses caused by nonmechanic stimuli.
Footnotes
This work was supported by Grants RO1-DC04709 and R21-DC04492 from the National Institute on Deafness and Other Communication Disorders (X.L.). We thank Dr. Michael J. Welsh for many helpful suggestions and critically reading this manuscript and Kristen Radde-Gallwitz for proofreading the final draft of this manuscript.
Correspondence should be addressed to Xi Lin, Departments of Otolaryngology and Cell Biology, Emory University School of Medicine, 615 Michael Street, Atlanta, GA 30322. E-mail: xlin2@emory.edu.
Copyright © 2004 Society for Neuroscience 0270-6474/04/2410167-09$15.00/0
References
- Adams CM, Anderson MG, Motto DG, Price MP, Johnson WA, Welsh MJ (1998) Ripped pocket and pickpocket, novel Drosophila DEG/ENaC subunits expressed in early development and in mechanosensory neurons. J Cell Biol 140: 143-152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bassilana F, Champigny G, Waldmann R, de Weille JR, Heurteaux C, Lazdunski M (1997) The acid-sensitive ionic channel subunit ASIC and the mammalian degenerin MDEG form a heteromultimeric H+-gated Na+ channel with novel properties. J Biol Chem 272: 28819-28822. [DOI] [PubMed] [Google Scholar]
- Benson CJ, Xie J, Wemmie JA, Price MP, Henss JM, Welsh MJ, Snyder PM (2002) Heteromultimers of DEG/ENaC subunits form H+-gated channels in mouse sensory neurons. Proc Natl Acad Sci USA 99: 2338-2343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bevan S, Yeats J (1991) Protons activate a cation conductance in a sub-population of rat dorsal root ganglion neurones. J Physiol (Lond) 433: 145-161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chesler M, Kaila K (1992) Modulation of pH by neuronal activity. Trends Neurosci 15: 396-402. [DOI] [PubMed] [Google Scholar]
- Couloigner V, Teixeira M, Hulin P, Sterkers O, Bichara M, Escoubet B, Planelles G, Ferrary E (2000) Effect of locally applied drugs on the pH of luminal fluid in the endolymphatic sac of guinea pig. Am J Physiol Regul Integr Comp Physiol 279: R1695-R1700. [DOI] [PubMed] [Google Scholar]
- DeVries SH (2001) Exocytosed protons feedback to suppress the Ca2+ current in mammalian cone photoreceptors. Neuron 32: 1107-1117. [DOI] [PubMed] [Google Scholar]
- Drew LJ, Rohrer DK, Price MP, Blaver KE, Cockayne DA, Cesare P, Wood JN (2004) Acid-sensing ion channels ASIC2 and ASIC3 do not contribute to mechanically activated currents in mammalian sensory neurones. J Physiol (Lond) 556: 691-710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Driscoll M, Chalfie M (1991) The mec-4 gene is a member of a family of Caenorhabditis elegans genes that can mutate to induce neuronal degeneration. Nature 349: 588-593. [DOI] [PubMed] [Google Scholar]
- Drummond HA, Abboud FM, Welsh MJ (2000) Localization of beta and gamma subunits of ENaC in sensory nerve endings in the rat foot pad. Brain Res 884: 1-12. [DOI] [PubMed] [Google Scholar]
- Ettaiche M, Guy N, Hofman P, Lazdunski M, Waldmann R (2004) Acid-sensing ion channel 2 is important for retinal function and protects against light-induced retinal degeneration. J Neurosci 24: 1005-1012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hildebrand MS, de Silva MG, Klockars T, Rose E, Price M, Smith RJ, McGuirt WT, Christopoulos H, Petit C, Dahl HH (2004) Characterisation of DRASIC in the mouse inner ear. Hear Res 190: 149-160. [DOI] [PubMed] [Google Scholar]
- Huang M, Chalfie M (1994) Gene interactions affecting mechanosensory transduction in Caenorhabditis elegans Nature 367: 467-470. [DOI] [PubMed] [Google Scholar]
- Karet FE, Finberg KE, Nelson RD, Nayir A, Mocan H, Sanjad SA, Rodriguez-Soriano J, Santos F, Cremers CW, Di Pietro A, Hoffbrand BI, Winiarski J, Bakkaloglu A, Ozen S, Dusunsel R, Goodyer P, Hulton SA, Wu DK, Skvorak AB, Morton CC, Cunningham MJ, Jha V, Lifton RP (1999) Mutations in the gene encoding B1 subunit of H+-ATPase cause renal tubular acidosis with sensorineural deafness. Nat Genet 21: 84-90. [DOI] [PubMed] [Google Scholar]
- Krishtal OA, Osipchuk YV, Shelest TN, Smirnoff SV (1987) Rapid extracellular pH transients related to synaptic transmission in rat hippocampal slices. Brain Res 436: 352-356. [DOI] [PubMed] [Google Scholar]
- Liedtke W, Choe Y, Marti-Renom MA, Bell AM, Denis CS, Sali A, Hudspeth AJ, Friedman JM, Heller S (2000) Vanilloid receptor-related osmotically activated channel (VR-OAC), a candidate vertebrate osmoreceptor. Cell 103: 525-535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin X (1997) Action potentials and underlying voltage-dependent currents studied in cultured spiral ganglion neurons of the postnatal gerbil. Hear Res 108: 157-179. [DOI] [PubMed] [Google Scholar]
- Liu J, Schrank B, Waterston RH (1996) Interaction between a putative mechanosensory membrane channel and a collagen. Science 273: 361-364. [DOI] [PubMed] [Google Scholar]
- Maingret F, Fosset M, Lesage F, Lazdunski M, Honore E (1999) TRAAK is a mammalian neuronal mechano-gated K+ channel. J Biol Chem 274: 1381-1387. [DOI] [PubMed] [Google Scholar]
- Miesenbock G, De Angelis DA, Rothman JE (1998) Visualizing secretion and synaptic transmission with pH-sensitive green fluorescent proteins. Nature 394: 192-195. [DOI] [PubMed] [Google Scholar]
- Price MP, Lewin GR, McIlwrath SL, Cheng C, Xie J, Heppenstall PA, Stucky CL, Mannsfeldt AG, Brennan TJ, Drummond HA, Qiao J, Benson CJ, Tarr DE, Hrstka RF, Yang B, Williamson RA, Welsh MJ (2000) The mammalian sodium channel BNC1 is required for normal touch sensation. Nature 407: 1007-1011. [DOI] [PubMed] [Google Scholar]
- Price MP, McIlwrath SL, Xie J, Cheng C, Qiao J, Tarr DE, Sluka KA, Brennan TJ, Lewin GR, Welsh MJ (2001) The DRASIC cation channel contributes to the detection of cutaneous touch and acid stimuli in mice. Neuron 32: 1071-1083. [DOI] [PubMed] [Google Scholar]
- Puel JL, Ruel J, Gervais d'Aldin C, Pujol R (1998) Excitotoxicity and repair of cochlear synapses after noise-trauma induced hearing loss. NeuroReport 9: 2109-2114. [DOI] [PubMed] [Google Scholar]
- Roza C, Puel JL, Kress M, Baron A, Diochot S, Lazdunski M, Waldmann R (2004) Knockout of the ASIC2 channel does not impair cutaneous mechanosensation, visceral mechanonociception and hearing. J Physiol (Lond) 558: 659-669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scott DA, Wang R, Kreman TM, Sheffield VC, Karniski LP (1999) The Pendred syndrome gene encodes a chloride-iodide transport protein. Nat Genet 21: 440-443. [DOI] [PubMed] [Google Scholar]
- Sidi S, Friedrich R, Nicolson T (2003) NompC TRP channel required for vertebrate sensory hair cell mechanotransduction. Science 301: 96-99. [DOI] [PubMed] [Google Scholar]
- Sluka KA, Price MP, Breese NM, Stucky CL, Wemmie JA, Welsh MJ (2003) Chronic hyperalgesia induced by repeated acid injections in muscle is abolished by the loss of ASIC3, but not ASIC1. Pain 106: 229-239. [DOI] [PubMed] [Google Scholar]
- Tavernarakis N, Shreffler W, Wang S, Driscoll M (1997) unc-8, a DEG/ENaC family member, encodes a subunit of a candidate mechanically gated channel that modulates C. elegans locomotion. Neuron 18: 107-119. [DOI] [PubMed] [Google Scholar]
- Traynelis SF, Chesler M (2001) Proton release as a modulator of presynaptic function. Neuron 32: 960-962. [DOI] [PubMed] [Google Scholar]
- Varming T (1999) Proton-gated ion channels in cultured mouse cortical neurons. Neuropharmacology 38: 1875-1881. [DOI] [PubMed] [Google Scholar]
- Wangemann P, Schacht J (1996) Homeostatic mechanism in the cochlea. In: The cochlea (Dallos P, Popper AN, Fay RR, eds), pp 130-184. New York, Berlin: Springer.
- Wemmie JA, Chen J, Askwith CC, Hruska-Hageman AM, Price MP, Nolan BC, Yoder PG, Lamani E, Hoshi T, Freeman Jr JH, Welsh MJ (2002) The acid-activated ion channel ASIC contributes to synaptic plasticity, learning, and memory. Neuron 34: 463-477. [DOI] [PubMed] [Google Scholar]
- Wemmie JA, Askwith CC, Lamani E, Cassell MD, Freeman Jr JH, Welsh MJ (2003) Acid-sensing ion channel 1 is localized in brain regions with high synaptic density and contributes to fear conditioning. J Neurosci 23: 5496-5502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuste R, Miller RB, Holthoff K, Zhang S, Miesenbock G (2000) Synapto-pHluorins: chimeras between pH-sensitive mutants of green fluorescent protein and synaptic vesicle membrane proteins as reporters of neurotransmitter release. Methods Enzymol 327: 522-546. [DOI] [PubMed] [Google Scholar]