

Abstract
Spinal cord injury usually results in permanent paralysis because of lack of regrowth of damaged neurons. Here we demonstrate that adult mice lacking EphA4 (-/-), a molecule essential for correct guidance of spinal cord axons during development, exhibit axonal regeneration and functional recovery after spinal cord hemisection. Anterograde and retrograde tracing showed that axons from multiple pathways, including corticospinal and rubrospinal tracts, crossed the lesion site. EphA4-/- mice recovered stride length, the ability to walk on and climb a grid, and the ability to grasp with the affected hindpaw within 1-3 months of injury. EphA4 expression was upregulated on astrocytes at the lesion site in wild-type mice, whereas astrocytic gliosis and the glial scar were greatly reduced in lesioned EphA4-/- spinal cords. EphA4-/- astrocytes failed to respond to the inflammatory cytokines, interferon-γ or leukemia inhibitory factor, in vitro. Neurons grown on wild-type astrocytes extended shorter neurites than on EphA4-/- astrocytes, but longer neurites when the astrocyte EphA4 was blocked by monomeric EphrinA5-Fc. Thus, EphA4 regulates two important features of spinal cord injury, axonal inhibition, and astrocytic gliosis.
Keywords: spinal cord injury, astrocytic gliosis, anterograde, retrograde, neurite outgrowth, cytokine
Introduction
Injury to the CNS usually results in very limited, if any, regeneration of lesioned axons, with subsequent permanent impairment of function. Although some CNS neurons appear to lose the intrinsic ability to regenerate neurites postnatally (Goldberg et al., 2002), many others, such as corticospinal tract (CST) neurons, appear able to regenerate, but are inhibited from doing so by the environment of the injury site. Major impediments to CNS regeneration are the presence of myelin inhibitors and astrocytic gliosis.
Myelin proteins that inhibit axonal regeneration include Nogo (Caroni and Schwab, 1988; Schnell and Schwab, 1990, 1993; Bandtlow and Schwab, 2000), myelin-associated glycoprotein (MAG) (McKerracher et al., 1994; Mukhopadhyay et al., 1994; DeBellard et al., 1996; Schafer et al., 1996), and oligodendrocyte-myelin glycoprotein (OMgp) (Wang et al., 2002b). Each of these proteins appears to inhibit regeneration by the same mechanism. Nogo, MAG, and OMgp bind to the Nogo receptor (Domeniconi et al., 2002; Liu et al., 2002; Wang et al., 2002a), which interacts with the p75 neurotrophin receptor on the axon (Wang et al., 2002a). This leads to activation of the small GTPase RhoA (Wang et al., 2002a; Yamashita et al., 2002), which inhibits neurite outgrowth. However, although blocking individual myelin proteins or their common receptor in vivo after spinal cord injury results in partial regeneration and improves functional recovery, at best only a small percentage of axons regrow (Bregman et al., 1995; GrandPre et al., 2002; Kim et al., 2003; Simonen et al., 2003; Zheng et al., 2003). Therefore, other impediments to regeneration still need to be overcome before robust regeneration can occur.
The other major barrier to axonal regeneration is glial scarring, the main component of which is astrocytic gliosis (Stichel and Muller, 1998). Normally quiescent astrocytes in the adult show a vigorous response to injury. They become hypertrophic, proliferative, upregulate expression of glial fibrillary acidic protein (GFAP), and form a dense network of glial processes both at and extending from the lesion site. Accompanying these morphological changes are a range of physiological changes, including secretion of a variety of cytokines and production of cell adhesion and extracellular matrix molecules. Some of these products are inhibitory to regeneration, such as chondroitin sulfate proteoglycan (CSPG) (McKeon et al., 1991) and collagen IV (Stichel et al., 1999), and if their deposition is inhibited, axonal regeneration is promoted (Stichel et al., 1999).
Other factors that may be implicated in inhibition of axonal regeneration, but which have received little attention in this regard, are cell surface molecules that are involved in axon guidance during development. One such family of molecules is the Eph receptor tyrosine kinase family, which, together with their ligands, the Ephrins, are implicated in the formation of a variety of neural structures, including the corticospinal tract and anterior commissure (Henkemeyer et al., 1996; Dottori et al., 1998; Coonan et al., 2001; Kullander et al., 2001a; Leighton et al., 2001). Both the Eph receptors and the Ephrins are membrane-bound, and cell-cell contact is required for signaling. In addition, some of the Ephrins are able to transduce signals, so reverse signaling can also occur (for review, see Kullander and Klein, 2002). Because Eph-Ephrin signaling appears to regulate axon guidance through contact repulsion, inducing the collapse of neuronal growth cones (Wahl et al., 2000; Kullander et al., 2001b), and members of this family are upregulated in the adult after neural injury (Moreno-Flores and Wandosell, 1999; Rodger et al., 2001; Willson et al., 2002), the aberrant expression or absence of Eph receptors could prove pivotal in determining the outcome of injury in the adult CNS. This possibility was investigated in this study by comparing neural regeneration after spinal cord hemisection in wild-type and EphA4-/- mice.
Materials and Methods
Mice
Adult EphA4-/- and C57BL/6 mice, 3-12 months old, and maintained as previously described (Coonan et al., 2001), were used in this study. Experiments were approved by the Royal Melbourne Hospital and the University of Melbourne Animal Ethics Committees in accordance with the Australian Code of Practice for the Care and Use of Animals for Scientific Purposes.
Spinal cord lesions
Mice were anesthetized with a mixture of ketamine and xylazine (100 mg/kg and 16 mg/kg, respectively). The spinal cord was exposed via a laminectomy, in which two or three vertebral arches were removed at levels T12-L1, corresponding to the level of the lumbar enlargement. A spinal left hemisection at T12 was performed using a fine corneal blade (cut twice in the same place to ensure complete section), and the overlying muscle and skin were then sutured. A total of 71 wild-type and 83 EphA4-/- mice underwent the spinal hemisection surgery. EphA4-/- mice were more sensitive to the anesthetic than wild-type mice, as a result of which 4% of wild-type mice and 12% of EphA4-/- mice died. Mice of both genotypes that recovered from surgery showed good survival, with no spontaneous deaths recorded, however three wild-type and four EphA4-/- mice were killed because of infection, and two EphA4-/- mice because of full left and right hindlimb paralysis. The success rate of the hemisection model, as assessed by total paralysis of the left hindlimb at 24 hr after surgery, was 96% for wild-type mice and 95% for EphA4-/- mice. Mice that showed only partial paralysis at this time point, indicative of incomplete lesion, were excluded from analysis. Spinal cords of animals that were included in the analysis were also examined histochemically to ensure complete hemisection.
Anterograde tracing
Five weeks after spinal cord lesion, tetramethylrhodamine dextran (“Fluoro-Ruby”; molecular weight, 10,000 kDa) was injected as two 0.2 μl injections into the midlateral aspect of the spinal cord at the level of the cervical enlargement, ipsilateral to the lesion, via a glass pipette attached to a Hamilton syringe. After an additional 7 d survival period, the animals were perfused with 4% paraformaldehyde. Longitudinal serial sections of spinal cord were cut at 50 μm on a freezing microtome, and sections were mounted on gelatinized slides and examined using fluorescence and confocal microscopy. This technique labeled many but not all axons in pathways ipsilateral to the injection site (estimated to be 25%) but none contralateral to the injection site (supplemental material, available at www.jneurosci.org).
The number of labeled axons running rostrally to caudally through a 250-μm-wide box placed in the white matter, at the border of the gray matter, of all intact serial sections (8-10 per spinal cord) was counted at 400× by focusing up and down through the sections at 2.5 mm and 50-100 μm proximal to the lesion site and 50-100 μm, 1 and 5 mm distal to the hemisection. Axons at the lateral edge of the white matter and near the pial surface were therefore excluded from these counts, as were sections in which the entire length of the labeled spinal cord was not available. The lumbar site of the lesion precluded analysis of regrowth longer than 5 mm because of termination of the fibers and commencement of the Cauda Equina. Significance of results was analyzed using the Student's t test.
Retrograde tracing
The lumbar spinal cord below the lesion was exposed via a lower lumbar laminectomy. Fast Blue [2% (w/v), 0.3 μl per injection; EMS-POLYLOY; GmBH, Gross-Umstadt, Germany], which labels the neuronal soma of axons damaged by the injection, was injected into two sites of the spinal cord ipsilateral to the lesion site with a glass micropipette attached to a Hamilton syringe. After a 5 d survival period, the animals were perfused with 4% paraformaldehyde in PBS. The brain and spinal cord were removed and postfixed for 24 hr in 20% sucrose in fixative before being serially sectioned at 50 μm on a freezing microtome in the coronal-transverse plane. Injections were considered successful by confirmation of a unilateral injection site in the operated spinal cord longitudinal sections. Qualitative and quantitative comparisons of labeled neurons were made by mapping the locations of labeled cells in every fourth section of a series using a computer-linked digitizing system (MD3 microscope digitizer and MD-plot software; Minnesota Datametrics Corporation).
Behavioral analysis
The hopping gait of the EphA4-/- mice precluded use of commonly used behavioral assessments, such as the Basso, Beattie, Bresnahan scale. The behavioral assessments chosen, and especially the grip strength test and climbing on an angled grid, allow direct comparison of EphA4-/- mice and wild-type mice, independent of hopping or reciprocal gait. Both the grid walking and grip test rely on sensory as well as motor input.
Stride length. Before and after hemisection, mice were footprinted by painting their hindpaws with nontoxic ink and placing them in a tunnel on blotting paper (wild type, n = 7; EphA4-/-, n = 9 mice). Stride length was determined by measurement of multiple successive steps and results were expressed as a percentage of each animal's own baseline stride length.
Grid walking. The ability of wild-type (n = 5) and EphA4-/- (n = 7) mice to walk on a horizontal or angled (75° from horizontal) wire grid (1.2 × 1.2 cm grid spaces; 35 × 45 cm total area) was determined to assess their locomotion (Ma et al., 2001). The mice were tested 1, 2, and 3 months after the spinal cord hemisection and compared with nonlesioned mice from each group. On the horizontal grid each mouse was allowed to walk freely around the grid for 5 min, during which a minimum 2 min of walking time was required. On the angled grid, each mouse was measured over 10 climbs. If the left hindpaw protruded entirely through the grid, with all toes and heel extended below the wire surface, it was counted as a misstep. The total number of steps taken with the left hindlimb was also counted. The results were expressed as the percentage of accurate footsteps and significance was analyzed using the Student's t test.
Sensory and motor ability-grasp test
The ability of hemisected and nonlesioned wild-type (n = 5) and EphA4-/- (n = 7) mice to grasp a 2 mm diameter rod was tested on the left hindlimb. The hindlimbs of the mice were lifted 2 cm from the table top while allowing the forelimbs to remain in contact with the table. Grasp ability was tested by lightly touching the left footpad with the rod and assessing the response based on a scale from 0 to 4: 0, no movement of paw and toes; 1, partial movement of the paw, no movement of the toes; 2, partial grasp, slight movement of toes and paw; 3, weak full grasp, not maintained with gentle rod movement; 4, strong grasp, maintained with gentle rod movement. Mice were graded at least three times in parallel with the grid tests described. Results were expressed as the mean ± SEM of the score of each group, and significance was analyzed using the Student's t test.
Immunohistochemistry and astrocyte counts
Standard immunohistochemical procedures, using rabbit anti-GFAP (1: 500; Dako, Carpenteria, CA), mouse anti-CSPG (1:200; Sigma, St. Louis, MO), and rabbit anti-EphA4 were followed. The rabbit anti-EphA4 antibody, obtained from Dr. J. Coonan (Walter and Eliza Hall Institute, Melbourne, Australia), was prepared against a peptide corresponding to amino acids 938-953 of the intracellular SAM domain of EphA4 (Gen-Bank accession number NM007936) using standard procedures (Cooper and Paterson, 2000). The number of hypertrophic astrocytes, as well as the total number of GFAP-expressing astrocytes, were counted in a 0.25 mm2 grid at and 2.5 mm proximal to the lesion site, in every third serial longitudinal 8 μm section. Hypertrophic astrocytes were defined as intensely stained GFAP-positive cells with a large cell body and multiple thick long processes. Nonhypertrophic astrocytes stained less intensely for GFAP and had a small cell body with thin, less complex processes. Hypertrophic astrocytes were more than twice the size of nonhypertrophic astrocytes.
Astrocyte and neuronal cultures and neurite length measurement
Purified astrocyte and neuronal cultures were prepared as previously described (Turnley et al., 2002). For analysis of neurite length, E16 cortical neurons were plated at 5000 per well in chamber slides (Falcon; Becton Dickinson, Franklin Lakes, NJ) containing wild-type or EphA4-/- astrocyte monolayers or which were poly-dl-ornithine—laminin coated. In some experiments, astrocytes were pretreated for 1 hr with monomeric EphrinA5-Fc (0.15, 1.5, 10 μg/ml) or complexed EphrinA5-Fc (1.5 μg/ml complexed with 0.15 μg/ml anti-human IgG (Vector Laboratories, Burlingame, CA) for 30 min at room temperature before addition). After 22 hr, cells were fixed and immunostained for the neuronal marker βIII-tubulin (1:2000; Promega, Madison, WI). Neurite length was measured using image analysis as previously described (Turnley and Bartlett, 1998). Significance of differences in the mean neurite lengths was analyzed using the Student's t test.
For biochemical analysis of astrocytes, factors as indicated were added to 80% confluent monolayers in 10 cm plates (Falcon) for the times indicated. EphrinA5-Fc (a gift from Prof. Andrew Boyd, Queensland Institute of Medical Research, Brisbane, Australia) was precomplexed as above.
Immunoprecipitation and Western analysis
Cells were lysed in lysis buffer (50 mm HEPES, pH 7.5, 150 mm NaCl, 10% glycerol, 1% Triton X-100, 1 mm EDTA, 1 mm EGTA, pH 8.0, and 1.5 mm MgCl) containing protease inhibitors [Complete protease inhibitor cocktail tablet (Roche), 200 μm sodium vanadate, and 2 mm PMSF] and a sample kept aside for analysis of total protein levels. The remainder of the lysate was used for immunoprecipitation in EphA4 or Rho activation assays. For Western analysis, gel sample reducing buffer was added to cell lysates and boiled. For other experiments, an aliquot of lysates was taken for Western analysis and the remainder of the lysate was used for immunoprecipitation with mouse anti-phosphotyrosine antibody (1:100; Cell Signaling Technology, Beverly, MA). Antibodies were first coupled to anti-mouse IgG agarose (Pierce, Rockford, IL) by 1 hr preincubation then proteins in the lysate supernatant were immunoprecipitated with aliquots of the antibody-agarose complex for 2 hr at 4°C. The immunoprecipitates were washed three times with HNTG buffer (20 mm HEPES, pH 7.5, 0.15 mm NaCl, 0.1% Triton X-100, and 10% glycerol), resolved by SDS-PAGE gel electrophoresis through 12 or 8-16% gels (Gradipore, Sydney, Australia) and electrophoretically transferred to a nitrocellulose membrane (Bio-Rad, Hercules, CA). Membranes were blocked for 2 hr in TBS containing 0.05% Tween 20 and 6% skim milk powder then incubated with rabbit anti-EphA4 antibody (kindly provided by Dr. D. Wilkinson, National Institute for Medical Research, London, UK). Total EphA4 and β-actin expression levels were determined in nonlesioned and 7 d postlesioned spinal cords by Western analysis using rabbit anti-EphA4 antibody as above and mouse anti-β-actin antibody (Sigma). Densitometry was performed on the autoradiographs using NIH Image software to determine relative levels of the EphA4 bands and normalized to β-actin levels.
Rho activation assay
Cells were exposed to indicated stimuli, and after treatment, cells were lysed in lysis buffer as above. Rho activation assays were performed using the Rhotekin RBD assay, according to the manufacturer's instructions (Upstate Biotechnology, Lake Placid, NY). Endogenous Rho-GTP was precipitated from cell lysates at 4°C for 1 hr, using a GST-tagged fusion protein, corresponding to residues 7-89 of mouse Rhotekin Rho-binding domain. Beads were collected by centrifugation, washed with lysis buffer and resuspended in sample buffer. The eluted protein samples were resolved on 12% SDS-PAGE and electrophoretically transferred to nitrocellulose membrane. Rho was detected using mouse anti-Rho antibody (1:500; Pierce) overnight, followed by anti-mouse secondary antibody linked to horseradish peroxidase (1:40,000; Cell Signaling). Immunoreactive bands were detected with ECL as above. Densitometry was performed on the autoradiographs using NIH Image software to determine relative levels of activated compared with total Rho.
Cell proliferation assay
The [3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl]tetrazolium bromide (MTT) assay, which determines mitochondrial activity in living cells, is commonly used as a proliferation assay (Mosmann, 1983). Living cells transform the tetrazolium ring into dark blue formazan crystals, which can be quantified by reading the optical density (OD); an increase in OD correlates with an increase in cell number over time. Wild-type and EphA4-/- astrocytes were plated on 96 well plates (Falcon) at 3 × 103 cells/well in DMEM supplemented with 10% calf serum in the presence or absence of either leukemia inhibitory factor (LIF; 1000 U/ml; Chemicon, Temecula, CA) or interferon-γ (IFNγ; 100 U/ml; Becton Dickinson). The MTT assay was performed at 2, 24, 48, and 72 hr after plating. MTT (0.25 mg/ml; Sigma) was incubated with the cells at each time point for 2 hr at 37°C, the cells were then lysed with an equal volume of acidic isopropanol (0.04 m HCl in absolute isopropanol), and the OD of the formazan product was measured at 550-650 nm.
Astrocyte scratch wound assay
Wild-type and EphA4-/- astrocytes were plated in DMEM (Invitrogen, San Diego, CA) supplemented with 10% FCS on poly-dl-ornithine-coated 8 well chamber slides (Falcon) and grown to confluence. The medium was then removed, and the monolayer was scratched with a sterile 20-200 μl plastic pipette tip. The cells were washed twice with sterile PBS, and medium was replaced, with or without LIF or IFNγ. After 3 d of incubation, cells were fixed and immunostained for β-actin, GFAP, and DAPI, and the number of astrocytes entering the wound area was determined by counting the number of nuclei per square millimeter.
Results
Tracing of lesioned axons indicates extensive regeneration by 6 weeks
An anterograde tracing technique was used whereby the tracer, tetramethylrhodamine dextran, was injected into the cervical spinal cord, well above the lumbar lesion site. This allowed us to assess general regeneration of individual axons from multiple different tracts without making any assumptions about the effect of EphA4 on particular pathways. Use of this technique in unlesioned wild-type and EphA4-/- mice showed equivalent labeling of descending axonal pathways ipsilateral to the injection site but none contralateral to the injection site (supplemental material, available at www.jneurosci.org).
At 6 d after lesion, in both wild-type and EphA4-/- mice, anterograde labeling revealed no labeled fibers within the lesion site, although there were axons with growth cones near the lesion site in the EphA4-/- mice (Fig. 1a,b). By 6 weeks after spinal hemisection, however, many anterogradely labeled axons crossed the EphA4-/- lesion site (Fig. 2a) (supplemental material, available at www.jneurosci.org), unlike in the wild-type lesion (Fig. 2b). Only sections in which the entire length of spinal cord was intact were included in the results and axons that were close to the pial surface were excluded from the counts. Of the anterogradely labeled axons counted that reached the lesion site (sum total 133 ± 16.8 in EphA4-/- mice compared with 58 ± 5.3 in wild-type, in a 250-μm-wide box over the white matter bordering the gray matter; p < 0.05), 70% of EphA4-/- axons crossed it (as measured 100 μm distally) and, of these, 75% were maintained at 1 mm and 15% at 5 mm distal to the injury (Fig. 2a,c). In contrast, in wild-type mice ∼4% of the fibers crossed the lesion site and virtually none of these was detected at 1 mm and 5 mm distal. Furthermore, in lesioned mice 2.5 mm upstream of the lesion site, there was no significant difference in the number of labeled axons between wild-type and EphA4-/- animals (25.4 ± 2.7 and 31.7 ± 2.2 axons per section, respectively; p = 0.13); it was only close to the lesion site that the difference between wild-type and EphA4-/- numbers became significant. Because considerably more axons in the EphA4-/- mice reached the lesion site, the magnitude of the difference is even more pronounced and demonstrates that there is considerable inhibition of regrowth of wild-type axons upstream of the lesion. Although the regenerating axons that crossed the lesion site at 6 weeks appeared “wavy” (Fig. 2av), as is typical during regeneration, the vast majority could be traced as running in an unhindered rostral to caudal line (Fig. 2a). Although some fibers showed branching or deviation, particularly after the lesion site (Fig. 2aiii, aiv), a montage of confocal micrographs covering the entire lesion area and across the midline (Fig. 2a) (supplemental material, available at www.jneurosci.org) revealed that no fibers crossed from the unlesioned side and contributed to the labeled fiber bundle running through or distal to the lesion. Thus, the large number of labeled axons passing through and beyond the lesion site in EphA4-/- mice can only be attributed to genuine regrowth of severed axons.
Figure 1.

At 6 d after injury, EphA4-/- axons approach but do not cross the lesion site. Anterograde tracing and confocal analysis of lesioned EphA4-/- spinal cords 6 d after hemisection (a) show large numbers of labeled axons 2.5 mm proximal to the lesion (ia) and a small number of axons with growth cones (aiii, arrows) approaching the lesion site, which is indicated by the dotted line (i) and shown more clearly in a hematoxylin and eosin (H&E)-stained section (ii). b, Wild-type spinal cord also shows very few axons approaching the lesion site. bia shows labeling 2.5 mm upstream of the lesion site. biii, An enlargement of bi shows few axons upstream of the lesion site. In both panels, rostral is to the right and caudal to the left, and the lesion site is indicated by dotted lines. Enlarged areas are indicated by boxed areas and arrows. Scale bars: i, 250 μm; ii, 200 μm; iii, 50 μm.
Figure 2.

Extensive axonal regeneration in EphA4-/- mice at 6 weeks after injury. Anterograde tracing and confocal analysis of lesioned EphA4-/- spinal cords 6 weeks after hemisection showed that a large percentage of EphA4-/- axons crossed the lesion site (a, c) and extended caudally (*p < 0.001), unlike wild-type (EphA4+/+) axons that did not cross the lesion site (b, c). A montage of confocal images of EphA4-/- spinal cord (ai) showed that the regenerating axons passed through the lesion site (indicated by dotted line and by H&E-stained section in aii and extended caudally in a straight line with some “waviness” seen immediately after lesion in aiii, iv, and v). In both panels, rostral is to the right and caudal to the left, and the lesion site is indicated by dotted lines. Enlarged areas are indicated by boxed areas and arrows. ii in both cases shows an adjacent H&E-stained section demonstrating the lesion site. Scale bars: i, 250 μm; ii, 200 μm; iii—v, 50 μm. Asterisk in ai indicates the midline.
Because the anterograde tracing used in this study labeled many axons in multiple descending spinal pathways, retrograde tracing was used to identify which specific axonal tracts had regenerated. This revealed that in the EphA4-/- but not the wild-type mice multiple axonal tracts showed regeneration. Labeled neurons were present in motor cortex (corticospinal tract) and the red nucleus (rubrospinal tract), as well as in the hypothalamus, the vestibular and reticular nuclei, and the periaqueductal gray matter (Fig. 3a,b), the same regions that were labeled in the nonlesioned control EphA4-/- and wild-type mice (Fig. 3c). In the wild-type mice, only a small number of bilaterally projecting reticulospinal neurons were labeled after lesion. Interestingly, unlike wild-type mice, unlesioned EphA4-/- mice showed a bilateral projection of the rubrospinal pathway (Fig. 3c), indicating that deletion of the EphA4 gene results in developmental guidance defects of the rubrospinal pathway, as has previously been shown for the corticospinal tract (Dottori et al., 1998; Coonan et al., 2001) and the anterior commissure (Dottori et al., 1998).
Figure 3.

EphA4 (-/-) mice show multiple tract regeneration and improved function. Identification of regenerating neuronal populations was determined by retrograde tracing using Fast Blue (a-c), and each neuron was plotted using an MD3 microscope digitizer and MD-plot software. Unlike lesioned wild-type (WT) mice (b), multiple axonal tracts regenerated in the lesioned EphA4-/- (KO) mice (a, b), with a pattern similar to that of unlesioned controls (c). Regenerated neurons included corticospinal neurons in layer 5 of the cortex (ai, b), rubrospinal neurons in the red nucleus (RN) (aii, b), as well as neurons in the hypothalamus (Hyp), the vestibular (VN) and reticular nuclei, and the periaqueductal gray (PAG) matter. Scale bars: (in a) 200 μm. Functional analysis of lesioned mice showed that EphA4-/- mice recovered substantial function within 1 month (*p < 0.005; n = 7 WT and 9 KO mice). One day after lesion, stride length (d), hindpaw grasping (e), and the ability to walk on a horizontal or angled (75°) grid (f) were minimal. Stride length was regained in KO mice within 3 weeks, whereas wild-type mice reached a plateau at 70% recovery. Grasping and grid-walking were significantly (*p < 0.001; n = 5 WT and 7 KO mice) improved in KO compared with WT by 1 month, continuing to improve up to 3 months.
Functional recovery of EphA4-/- mice
The axonal regeneration observed in EphA4-/- mice also had a functional correlate. Mice were behaviorally assessed, first by measuring their stride length (Bregman et al., 1995) before and from 24 hr to 4 weeks after spinal hemisection. At 24 hr both EphA4-/- and wild-type mice showed minimal function. EphA4-/- mice regained 100% of their baseline stride length within 3 weeks, whereas wild-type mice showed only 70% recovery (Fig. 3d) and did not improve thereafter. At up to 3 weeks, mice of both genotypes were unable to move their ankles or toes and displayed toe dragging. By 1 month EphA4-/- but not wild-type mice showed ankle and toe movement and began to bear weight on the plantar surface of the affected hindpaw.
Given that wild-type mice also showed improvement in stride length, two other functional measures were used that did not improve significantly in wild-type mice and which required more sophisticated sensory and motor skills; ability to walk or climb on a grid and hindpaw grip strength. One month after hemisection, the ipsilateral hindpaw grip strength (Fig. 3e), and ability to walk or climb on a grid (Fig. 3f) were dramatically improved in EphA4-/- mice compared with wild-type. These functions continued to improve up to 3 months after lesion. Nonlesioned EphA4-/- and wild-type mice both achieved maximal scores in these tests.
Lack of astrocytic gliosis in EphA4-/- mice
A striking feature of the hemisected EphA4-/- spinal cord was the virtual absence of astrocytic gliosis, as assessed by GFAP expression, compared with the wild-type (Fig. 4a,b,d,e). At day 7, the vast majority (90.4%) of the GFAP-positive astrocytes at the wild-type lesion site were hypertrophic and stained very strongly for GFAP, whereas only 7.4% of EphA4-/- astrocytes were hypertrophic (Fig. 4g). Overall, the total number of GFAP-positive cells was fewer at the EphA4-/- hemisection over the first 7 d after lesion, and this was strikingly the case proximal to the lesion site (Fig. 4h). In nonlesioned cases there was no difference in astrocyte numbers between EphA4-/- and wild-type mice (wild-type 836.8 ± 108.3/mm2 compared with EphA4-/- 825.6 ± 98.1/mm2). The lack of glial response resulted in a marked reduction in the size of the glial scar of EphA4-/- mice at 6 weeks after lesion as assessed by immunostaining for a component of the glial scar, CSPG (Fig. 4c,f). Analysis of macrophage-microglia activation at the lesion site 1 week and 6 weeks after lesion, by use of an antibody to the microglial marker CD11b, showed no apparent differences in the response of these cells to the injury in mice of either genotype (supplemental material, available at www.jneurosci.org).
Figure 4.

Astrocytic gliosis and the glial scar are greatly diminished in EphA4-/- mice after injury. Immunostaining for GFAP expression at the lesion site 4 d after spinal cord lesion showed a florid astrocytic gliosis in wild-type mice (a) that was virtually absent in EphA4-/- mice (d). Under higher magnification, the vast majority of astrocytes in wild-type mice were revealed to be hypertrophic (white arrows) (b, g), unlike EphA4-/- astrocytes (black arrows) (e, g). The total number of astrocytes increased with time after lesion, with greater numbers in EphA4+/+ spinal cords (h). Immunostaining for chondroitin sulfate proteoglycan, a component of the glial scar, 6 weeks after lesion, revealed that the scar was diminished in the EphA4-/- mice (f) compared with the wild-type animals (c). Scale bars: a, d, 200 μm; b, e, 50 μm; c, f, 200 μm. *p < 0.0001 in g and h.
Because EphA4 expression appeared to regulate both the level of regeneration and gliosis after lesioning, we next examined whether EphA4 expression was upregulated after spinal hemisection. Immunostaining and Western analysis (Fig. 5a,d) revealed that EphA4 expression occurred at very low levels, undetectable by immunostaining in nonlesioned animals except on some motor neurons (supplemental material, available at www.jneurosci.org). However, expression and phosphorylation were upregulated after spinal lesion (Fig. 5d), and almost exclusively on GFAP-expressing astrocytes at the lesion site (Fig. 5a-c). Low levels of EphA4 were found on anterogradely labeled axons proximal to the lesion site (supplemental material, available at www.jneurosci.org). A ligand for EphA4, EphrinB3, was also expressed on regenerating axons, as well as on some astrocytes (supplemental material, available at www.jneurosci.org).
Figure 5.

Expression of EphA4 on astrocytes inhibits neurite outgrowth. After spinal hemisection, EphA4 (a) and GFAP (b) are coexpressed as assessed by immunofluorescence on reactive astrocytes at the lesion site (c; a merged image of a and b). EphA4 was also expressed on some neurons (a-c, arrow). Western blot analysis (d) showed upregulation and phosphorylation of EphA4 (p-EphA4) at the lesion site (les) in comparison with unlesioned control (con) mice; * shows a nonspecific band present in all lanes. β-Actin was used as a loading control and EphA4-/- spinal cord as an EphA4 expression control. The EphA4 expression on astrocytes was inhibitory to cortical neuronal neurite outgrowth, because βIII-tubulin-positive cortical neurons on EphA4-/- astrocytes (e, g) had significantly longer neurites than on EphA4-expressing (EphA4+/+) astrocytes (f, g) after 22 hr (*p < 0.0001). EphA4-/- neurite outgrowth was also enhanced on EphA4-/- and EphA4+/+ astrocytes, compared with that of wild-type neurons (g; **p < 0.0001). h, The inhibition of neurite outgrowth by EphA4 on astrocytes could be blocked in a dose-dependent manner by addition of monomeric (mono) EphrinA5-Fc, but this had no effect on neurites grown on laminin. Multimerized (multi) EphrinA5-Fc inhibited neurite outgrowth both on astrocytes and on laminin (*p < 0.0001). Scale bars, 50 μm.
EphA4 expression on astrocytes inhibits neurite outgrowth
We then investigated whether EphA4 expression on astrocytes inhibits neurite outgrowth of cortical neurons in vitro. E16 cortical neurons were plated onto monolayers of either wild-type or EphA4-/- astrocytes, and the length of the longest neurite was measured 22 hr later. This revealed a twofold to threefold increase in outgrowth on EphA4-/- astrocytes compared with wild-type astrocytes (Fig. 5e-g). This effect appeared to be directly attributable to expression of EphA4 on the astrocytes, because similar results were obtained when neurons were grown on 293T cells transfected with EphA4 (neurite length on nontransfected 293T cells was 80.4 ± 3.3 μm compared with 30.2 ± 1.9 μm on EphA4-transfected cells). The increased neurite outgrowth of EphA4-/- neurons compared with wild-type neurons, on both wild-type and EphA4-/-astrocytes (Fig. 5g), suggests that EphA4 expressed on the neurons may also inhibit neurite outgrowth, as has been previously suggested (Wahl et al., 2000; Kullander et al., 2001b), and may contribute to the regeneration observed in EphA4-/- mice. Inhibition of neurite outgrowth on astrocytes was potently blocked in a dose-dependent manner by the addition of monomeric EphrinA5-Fc, which strongly binds to EphA4 in the astrocyte monolayer; however, it had no effect on neurons grown on laminin-coated glass slides (Fig. 5h). Conversely, addition of complexed EphrinA5-Fc inhibited neurite outgrowth on glass slides, as previously described (Wahl et al., 2000; Kullander et al., 2001b) and further inhibited outgrowth on astrocytes (Fig. 5h). This indicates that blocking of EphA4 on astrocytes, but not on neurons, enhances neurite outgrowth, whereas activation of EphA4 on both neurons and astrocytes inhibits neurite outgrowth. Both results point directly to the activation of EphA4 by a ligand as being the mechanism for neurite inhibition. In vivo, a possible activator of the neurite responses to EphA4 expression on the astrocytes was EphrinB3, which has been shown to transduce signals (Palmer et al., 2002) and which was expressed by regenerating axons in the spinal cord (supplemental material, available at www.jneurosci.org).
Rho activation and proliferation is decreased in EphA4-/- astrocytes
Given that previous studies have demonstrated that gliosis is mediated by inflammatory cytokines, including IFNγ and LIF (Yong et al., 1991; Balasingam et al., 1994; Sugiura et al., 2000), we then investigated whether these cytokines play a role in the upregulation of EphA4 on astrocytes. IFNγ and LIF upregulated EphA4 expression by 56 and 69%, respectively, whereas interleukin-1 (IL-1) and tumor necrosis factor-α (TNFα) had no effect (Fig. 6a). To directly address the question of whether EphA4 expression is accompanied by downstream activation that could lead to astrocytic responses, we examined whether EphA4 is phosphorylated. Both IFNγ and LIF upregulated EphA4 phosphorylation twofold, in a similar manner to the addition of a soluble multimeric EphA4 ligand, EphrinA5-Fc (Fig. 6a). In addition, this led to a marked increase in activation of the small GTPase, Rho, a major regulator of cytoskeletal changes (Hall, 1998), downstream of Eph receptor signaling (Wahl et al., 2000; Shamah et al., 2001). Increased Rho activation occurred both in wild-type spinal cord tissue removed from the lesion site (Fig. 6b) and in cultured astrocytes (Fig. 6c); no such response was observed using cells or tissue removed from lesioned EphA4-/- animals. Activation of Rho in astrocytes, as well as in neurons and oligodendrocytes, has also recently been reported in spinal cord after injury (Dubreuil et al., 2003).
Figure 6.
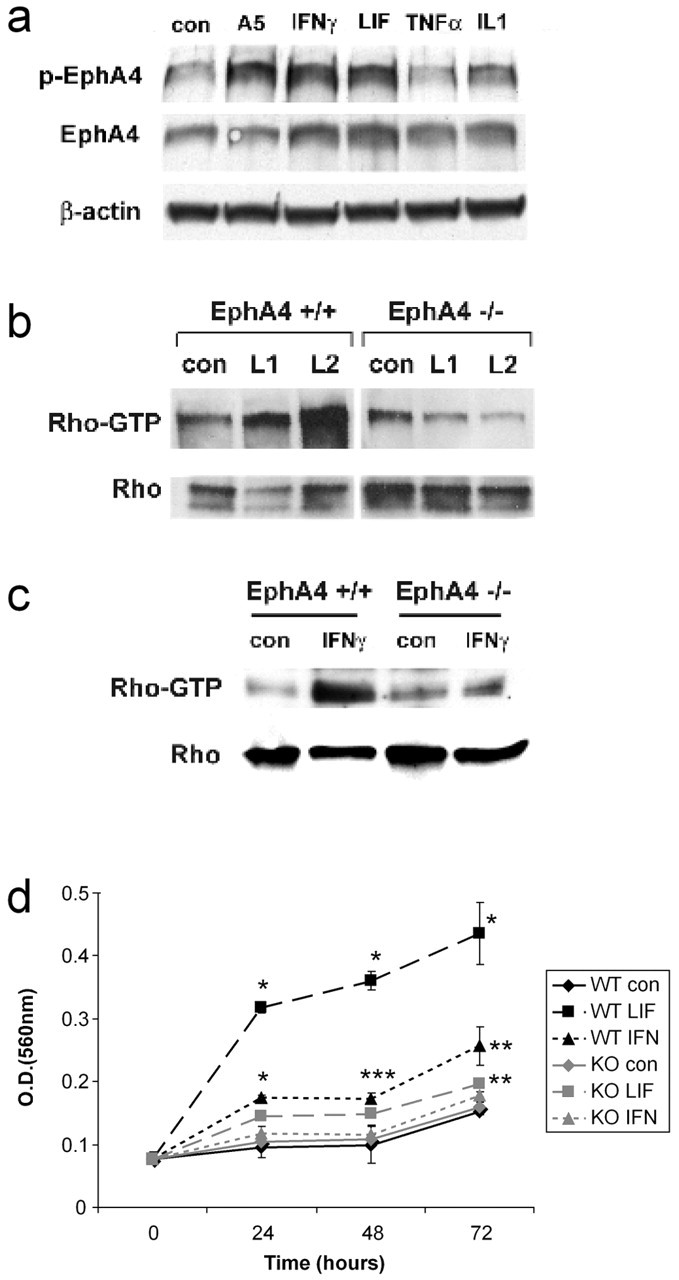
Inflammatory cytokines upregulate EphA4 and Rho activation and astrocyte proliferation. a, Expression of EphA4 was upregulated in cultured astrocytes after 72 hr by IFNγ and LIF but not TNFα or Il-1, compared with untreated controls (con). These cytokines also induced EphA4 phosphorylation (p-EphA4), similar to EphrinA5-Fc (A5). b, EphA4 phosphorylation leads to activation of Rho (RhoGTP), a cytoskeletal regulator. Rho was activated at the lesion site in wild-type but not EphA4-/- lesioned spinal cords (L1, L2), whereas in culture (c), IFNγ, which is an inducer of astrocytic gliosis, activated Rho in wild-type but not EphA4-/- astrocytes. d, An in vitro astrocyte proliferation assay showed that under basal conditions (con), both wild-type (WT) and EphA4-/- (KO) astrocytes proliferated similarly over 72 hr. WT astrocytes showed increased proliferation in response to LIF and IFNγ, whereas the EphA4-/- astrocyte response to these factors was markedly decreased and only significant for LIF at 72 hr. Results are representative of n = 3 separate experiments; *p < 0.001; **p < 0.005; ***p < 0.05.
EphA4-/- astrocyte proliferation and scratch wound repair are decreased in response to cytokines
The failure of cytokine-induced Rho activation in EphA4-/- astrocytes correlated with a failure of cytokine-induced astrocyte proliferation (Fig. 6d). Although basal levels of astrocyte proliferation were not significantly different in wild-type or EphA4-/- astrocytes, wild-type but not EphA4-/- astrocytes proliferated in response to LIF and IFNγ (Fig. 6d), indicating that EphA4 expression is a required cofactor for LIF- and IFNγ-induced astrocyte proliferation. Furthermore, unlike wild-type astrocytes, Epha4-/- astrocytes failed to repair a scratch wound by 72 hr, even in the presence of LIF or IFNγ (Fig. 7A-G). Because LIF and IFNγ are cytokines involved in inducing astrocytic gliosis in vivo, this provides a mechanism for the failure of overt gliosis after spinal cord injury in the EphA4-/- mice.
Figure 7.

EphA4-/- astrocytes fail to repair a scratch wound in vitro. Scratch wounds were performed on wild-type (A, C, E) and EphA4-/- (B, D, F) astrocyte monolayers. After 72 hr under basal conditions (A, B) or in the presence of IFNγ (C, D) or LIF (E, F), cells were immunostained for β-actin and DAPI. The number of cells migrating into the wound area was determined by counting the number of DAPI-stained nuclei (G) (*p < 0.001 compared with basal conditions, representative of n = 3 separate experiments). Scale bar: (in F) 250 μm.
Discussion
These studies indicate that EphA4 has the potential to inhibit axonal regeneration by three different mechanisms. The first of these, as demonstrated by in vitro assays, is the direct inhibition of neurite outgrowth mediated by EphA4 on the astrocytes binding to a receptor-ligand on the axon. Such an action of EphA4 may provide a mechanism for the inhibition of neurite outgrowth on astrocytes observed in the presence of IFNγ (Fok-Seang et al., 1998), which we have shown upregulates EphA4 expression. These results suggest that EphA4 is yet another directly inhibitory molecule produced during astrocytic gliosis, in addition to other inhibitory components, such as extracellular matrix and myelin-derived molecules. Although Eph molecules are generally described as receptors and Ephrins as ligands, both are membrane-bound and signaling can occur in either direction (for review, see Kullander and Klein, 2002). A likely candidate for transducing the inhibitory signal is Ephrin-B3, which was expressed on the axons. Ephrins have previously been shown to mediate reverse signaling in cortical neurons in response to Eph stimulation (Palmer et al., 2002).
The second mechanism may be by activation of EphA4 on the regenerating axons, similar to on E16 cortical neurons. However, we found EphA4 to be highly expressed only on astrocytes and motor neurons, and to be present at low levels on descending axons in lesioned adult spinal cord, suggesting that this mechanism may not play as great a role as the first in axon regeneration.
The third, and totally unexpected, mechanism by which EphA4 exerts an inhibitory effect involves its vital role in activating astrocytes, leading to gliosis and the formation of a glial scar. Such activation appears to be dependent on responsiveness to cytokine stimulation and may be dependent on Rho activation. This cytokine-induced response may be attributable to the upregulation of EphA4 receptor expression on the astrocytes, allowing enhanced ligand binding and receptor activation. It is also possible that the cytokine-induced astrocyte proliferation and hypertrophy may be caused by transactivation of EphA4, as has been shown for FGF2- and PDGF-induced phosphorylation of EphrinB molecules (Bruckner et al., 1997; Chong et al., 2000), leading to Rho activation and cytoskeletal rearrangement. The difference in glial activation seems to be astrocyte specific as there was no apparent difference in macrophage-microglial activation. Ephs and Ephrins have been reported to play a role in interactions between astrocytes and meningeal fibroblasts, excluding fibroblasts from the glial scar (Bundesen et al., 2003). However, whether EphA4 plays any role in regulation of fibroblast reaction at the lesion site remains to be determined.
The overall functional improvement in the EphA4-/- mice is likely to be caused by a lack of astrocytic gliosis and regeneration of ipsilateral axons. The recovery of grip strength and the ability to successfully walk on a grid reflect functional reconnection of both descending motor tracts and ascending sensory tracts. However, although the recovery in stride length is a useful assay, given that wild-type animals also showed significant improvement in the absence of regeneration of ipsilateral descending axons, as previously described (Bregman et al., 1995), it is possible that only the enhanced recovery displayed by the EphA4-/- mice can be attributed to regeneration and that the basal improvement is attributable to other causes. The significant improvement in mice of both genotypes probably reflects the hemisection model used, with the intact right side of the spinal cord allowing movement of the right side of the body, which in turn contributed to movement of the left side, particularly through the hip joint. The stride length was primarily a measure of hip movement, and there was no ankle or toe movement at the early time points in mice of either genotype. Ankle and toe movement did, however, improve in the EphA4-/- mice by 1 month, by which time they were beginning to bear weight on their plantar surface, a phenomenon that was not observed in the wild-type mice. Another potential cause of stride length recovery is activation of spinal reflexes, such as the central pattern generator (Duysens and Van de Crommert, 1998). However, more sensitive methods, such as grid walking and grasp strength, that did not produce marked improvement in wild-type mice and that did not rely on spinal cord reflexes were also assessed. Although grasp strength showed some modest recovery in the EphA4-/- mice at 1 month after injury, with partial grasp and slight movement of toes and paw, substantial regeneration would be required for their ability to maintain a strong grasp on a gently moving rod. The EphA4-/- mice did not approach this score until 2 months after injury and further improved at 3 months, at a time when regenerating axons had passed the lesion site. The improvement in grid walking also showed a similar time course. Neither of these measures significantly improved in wild-type mice, suggesting that a functional level of reconnection of both descending and ascending tracts had occurred in the EphA4-/- mice.
Although the numbers of labeled axons that were counted crossing the lesion site were relatively low, this was an underestimate of the actual number of axons crossing the lesion site. The labeling index of the axons was estimated to be ∼25% or less, so that less than one in four axons crossing the lesion site was labeled with tracer. Axons were also counted in a 250 μm wide box over the white matter, bordering the gray matter, which covered an average of 75% of the width of the white matter. Furthermore, axon counts were performed on only ∼40% of spinal cord sections; only sections that were intact for the entire length, from the site of tracer injection to at least 5 mm below the lesion site were used. Therefore, the actual number of regenerating axons crossing the lesion site was likely to be in the thousands; a number sufficient for functional improvement.
It is unlikely that unlesioned ipsilateral projections contributed to the functional recovery within the observed time frame. The double cutting procedure of the surgery ensured that in most mice all ipsilateral projections were severed. In a small group (∼10%) of both wild-type and EphA4-/- mice, a small number of the most lateral projections were spared. These were easily distinguished from the regenerating fibers as they followed an uninterrupted, unbranched course, without any apparent collateral sprouting and they were consequently excluded from counts of regenerating fibers. The presence of these small numbers of fibers did not contribute to improved function in wild-type mice.
However, given that a small number of CST axons cross the midline in EphA4-/- mice (Dottori et al., 1998; Coonan et al., 2001), and our retrograde tracing experiments showed bilateral projections of both the CST and the rubrospinal tract in unlesioned EphA4-/- mice, it is possible that during development a small number of contralateral projections crossed the midline at a level below the lesion site. After lesion, if only projections from the contralateral side were labeled, we would have seen an asymmetric distribution of labeled neurons in the brain, but as we observed bilateral labeling it indicates that the ipsilateral projections also regenerated. Whether a small number of contralateral projections could contribute to functional recovery is unclear, but as total hemiparalysis was demonstrated for at least the first few days after the lesion, it is unlikely that these fibers played a large role in functional recovery. However, collateral sprouting does appear to play a role in functional recovery in other spinal cord injury models (Galea and Darian-Smith, 1997; Fouad et al., 2001; Bareyre et al., 2004). We did not see any evidence of sprouting from the contralateral side above the site of the lesion. The injection site for the anterograde tracer was at the level of C2-C3, and collaterals from the intact side would have had to have commenced at or above this level to be labeled and tracked down past the lesion. Axons that cross the midline in EphA4-/- animals do not appear to continue down the spinal cord. Rather, they terminate at the level at which they cross (Coonan et al., 2001). We also did not observe any anterogradely labeled axons cross the midline to the contralateral side in lesioned or unlesioned animals.
The observed axonal regeneration and functional recovery also highlight the potential value of modulating the astrocyte response to injury. Whereas total ablation of astrocytes enhances tissue damage after neural injury (Bush et al., 1999; Faulkner et al., 2004), in this study astrocytic gliosis was diminished but not totally abolished. This appeared to still allow the positive aspects of gliosis in aiding wound repair, although decreasing the negative effects, such as the physical barrier of hypertrophic astrocytes and the formation of the glial scar. Similar enhancement of regeneration has recently been described in an entorhinal cortex lesion model in GFAP and vimentin double null mice, which also produced a very mild gliosis (Wilhelmsson et al., 2004). The decreased CSPG expression at the lesion site in the EphA4-/- mice is also likely to have allowed enhanced regeneration, as was observed when CSPG glycosaminoglycan side chains were removed from the lesion site by enzymatic treatment (Grimpe and Silver, 2004).
The effects of EphA4 on neuronal regeneration observed in this study may explain the relative failure of blocking or deleting other putative inhibitory molecules, such as Nogo (Kim et al., 2003; Simonen et al., 2003; Zheng et al., 2003), and demonstrates the presence of another major inhibitory influence in addition to myelin and other inhibitory components. The finding that EphA4 mediates glial activation and is in turn regulated by inflammatory cytokines such as IFNγ and LIF, provides an exciting possibility for overcoming the inhibitory effects of EphA4 and promoting axonal regrowth. Not only is it possible to block Eph activation using monomeric forms of cognate ligands (Davis et al., 1994), such as EphrinA5 (Fig. 5h), but it may also be possible to promote neuronal regeneration by blocking the expression of EphA4 on astrocytes at the injury site by blocking the action of specific cytokines. Given that preliminary results indicate that EphA4 expression is induced on astrocytes in primates after spinal cord damage (Y. Goldshmit and M. P. Galea, unpublished observations), there is also a real possibility of effectively promoting significant axonal regeneration after spinal cord injury in humans by developing pharmaceuticals that effectively block expression or activation of specific Eph receptors.
Footnotes
This work was supported by The BHP Community Trust, the National Health and Medical Research Council of Australia, and SpinalCure Australia. Y.G. was supported by an International Postgraduate Research Scholarship from the University of Melbourne. We thank Drs. David Wilkinson and Jason Coonan for anti-EphA4 antibodies and Prof. A. Boyd for Ephrin-Fc fusion proteins. We also thank Rowan Tweedale for assistance with this manuscript and mouse room staff at the Walter and Eliza Hall Institute and the University of Melbourne for excellent care of the mice.
Correspondence should be addressed to either of the following: Ann Turnley, Center for Neuroscience, University of Melbourne, Melbourne, Victoria 3010, Australia, E-mail: turnley@unimelb.edu.au; or Perry Bartlett, The Queensland Brain Institute, The University of Queensland, Brisbane, Queensland 4072, Australia. E-mail: p.bartlett@uq.edu.au.
Copyright © 2004 Society for Neuroscience 0270-6474/04/2410064-10$15.00/0
M.P.G., P.F.B., and A.M.T. contributed equally to this work.
References
- Balasingam V, Tejada-Berges T, Wright E, Bouckova R, Yong VW (1994) Reactive astrogliosis in the neonatal mouse brain and its modulation by cytokines. J Neurosci 14: 846-856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bandtlow CE, Schwab ME (2000) NI-35/250/nogo-a: a neurite growth inhibitor restricting structural plasticity and regeneration of nerve fibers in the adult vertebrate CNS. Glia 29: 175-181. [DOI] [PubMed] [Google Scholar]
- Bareyre FM, Kerschensteiner M, Raineteau O, Mettenleiter TC, Weinmann O, Schwab ME (2004) The injured spinal cord spontaneously forms a new intraspinal circuit in adult rats. Nat Neurosci 7: 269-277. [DOI] [PubMed] [Google Scholar]
- Bregman BS, Kunkel-Bagden E, Schnell L, Dai HN, Gao D, Schwab ME (1995) Recovery from spinal cord injury mediated by antibodies to neurite growth inhibitors. Nature 378: 498-501. [DOI] [PubMed] [Google Scholar]
- Bruckner K, Pasquale EB, Klein R (1997) Tyrosine phosphorylation of transmembrane ligands for Eph receptors. Science 275: 1640-1643. [DOI] [PubMed] [Google Scholar]
- Bundesen LQ, Scheel TA, Bregman BS, Kromer LF (2003) Ephrin-B2 and EphB2 regulation of astrocyte-meningeal fibroblast interactions in response to spinal cord lesions in adult rats. J Neurosci 23: 7789-7800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bush TG, Puvanachandra N, Horner CH, Polito A, Ostenfeld T, Svendsen CN, Mucke L, Johnson MH, Sofroniew MV (1999) Leukocyte infiltration, neuronal degeneration, and neurite outgrowth after ablation of scar-forming, reactive astrocytes in adult transgenic mice. Neuron 23: 297-308. [DOI] [PubMed] [Google Scholar]
- Caroni P, Schwab ME (1988) Two membrane protein fractions from rat central myelin with inhibitory properties for neurite growth and fibroblast spreading. J Cell Biol 106: 1281-1288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chong LD, Park EK, Latimer E, Friesel R, Daar IO (2000) Fibroblast growth factor receptor-mediated rescue of x-ephrin B1-induced cell dissociation in Xenopus embryos. Mol Cell Biol 20: 724-734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coonan JR, Greferath U, Messenger J, Hartley L, Murphy M, Boyd AW, Dottori M, Galea MP, Bartlett PF (2001) Development and reorganization of corticospinal projections in EphA4 deficient mice. J Comp Neurol 436: 248-262. [PubMed] [Google Scholar]
- Cooper H, Paterson Y (2000) Preparation of polyclonal antisera. In: Current protocols in molecular biology (Ausubel F, Brent R, Kingston R, Moore D, Seidman J, Smith J, Struhl K, eds), pp 11.12.11-11.12.19. New York: Wiley.
- Davis S, Gale NW, Aldrich TH, Maisonpierre PC, Lhotak V, Pawson T, Gold-farb M, Yancopoulos GD (1994) Ligands for EPH-related receptor tyrosine kinases that require membrane attachment or clustering for activity. Science 266: 816-819. [DOI] [PubMed] [Google Scholar]
- DeBellard ME, Tang S, Mukhopadhyay G, Shen YJ, Filbin MT (1996) Myelin-associated glycoprotein inhibits axonal regeneration from a variety of neurons via interaction with a sialoglycoprotein. Mol Cell Neurosci 7: 89-101. [DOI] [PubMed] [Google Scholar]
- Domeniconi M, Cao Z, Spencer T, Sivasankaran R, Wang K, Nikulina E, Kimura N, Cai H, Deng K, Gao Y, He Z, Filbin M (2002) Myelin-associated glycoprotein interacts with the Nogo66 receptor to inhibit neurite outgrowth. Neuron 35: 283-290. [DOI] [PubMed] [Google Scholar]
- Dottori M, Hartley L, Galea M, Paxinos G, Polizzotto M, Kilpatrick T, Bartlett PF, Murphy M, Kontgen F, Boyd AW (1998) EphA4 (Sek1) receptor tyrosine kinase is required for the development of the corticospinal tract. Proc Natl Acad Sci USA 95: 13248-13253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dubreuil CI, Winton MJ, McKerracher L (2003) Rho activation patterns after spinal cord injury and the role of activated Rho in apoptosis in the central nervous system. J Cell Biol 162: 233-243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duysens J, Van de Crommert HW (1998) Neural control of locomotion: the central pattern generator from cats to humans. Gait Posture 7: 131-141. [DOI] [PubMed] [Google Scholar]
- Faulkner JR, Herrmann JE, Woo MJ, Tansey KE, Doan NB, Sofroniew MV (2004) Reactive astrocytes protect tissue and preserve function after spinal cord injury. J Neurosci 24: 2143-2155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fok-Seang J, DiProspero NA, Meiners S, Muir E, Fawcett JW (1998) Cytokine-induced changes in the ability of astrocytes to support migration of oligodendrocyte precursors and axon growth. Eur J Neurosci 10: 2400-2415. [DOI] [PubMed] [Google Scholar]
- Fouad K, Pedersen V, Schwab ME, Brosamle C (2001) Cervical sprouting of corticospinal fibers after thoracic spinal cord injury accompanies shifts in evoked motor responses. Curr Biol 11: 1766-1770. [DOI] [PubMed] [Google Scholar]
- Galea MP, Darian-Smith I (1997) Corticospinal projection patterns following unilateral section of the cervical spinal cord in the newborn and juvenile macaque monkey. J Comp Neurol 381: 282-306. [PubMed] [Google Scholar]
- Goldberg JL, Klassen MP, Hua Y, Barres BA (2002) Amacrine-signaled loss of intrinsic axon growth ability by retinal ganglion cells. Science 296: 1860-1864. [DOI] [PubMed] [Google Scholar]
- GrandPre T, Li S, Strittmatter SM (2002) Nogo-66 receptor antagonist peptide promotes axonal regeneration. Nature 417: 547-551. [DOI] [PubMed] [Google Scholar]
- Grimpe B, Silver J (2004) A novel DNA enzyme reduces glycosaminoglycan chains in the glial scar and allows microtransplanted dorsal root ganglia axons to regenerate beyond lesions in the spinal cord. J Neurosci 24: 1393-1397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall A (1998) Rho GTPases and the actin cytoskeleton. Science 279: 509-514. [DOI] [PubMed] [Google Scholar]
- Henkemeyer M, Orioli D, Henderson JT, Saxton TM, Roder J, Pawson T, Klein R (1996) Nuk controls pathfinding of commissural axons in the mammalian central nervous system. Cell 86: 35-46. [DOI] [PubMed] [Google Scholar]
- Kim J-E, Li S, GrandPré T, Qiu D, Strittmatter S (2003) Axon regeneration in young adult mice lacking Nogo-A/B. Neuron 38: 187-199. [DOI] [PubMed] [Google Scholar]
- Kullander K, Klein R (2002) Mechanisms and functions of Eph and ephrin signalling. Nat Rev Mol Cell Biol 3: 475-486. [DOI] [PubMed] [Google Scholar]
- Kullander K, Mather NK, Diella F, Dottori M, Boyd AW, Klein R (2001a) Kinase-dependent and kinase-independent functions of EphA4 receptors in major axon tract formation in vivo. Neuron 29: 73-84. [DOI] [PubMed] [Google Scholar]
- Kullander K, Croll SD, Zimmer M, Pan L, McClain J, Hughes V, Zabski S, DeChiara TM, Klein R, Yancopoulos GD, Gale NW (2001b) Ephrin-B3 is the midline barrier that prevents corticospinal tract axons from recrossing, allowing for unilateral motor control. Genes Dev 15: 877-888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leighton PA, Mitchell KJ, Goodrich LV, Lu X, Pinson K, Scherz P, Skarnes WC, Tessier-Lavigne M (2001) Defining brain wiring patterns and mechanisms through gene trapping in mice. Nature 410: 174-179. [DOI] [PubMed] [Google Scholar]
- Liu BP, Fournier A, GrandPre T, Strittmatter SM (2002) Myelin-associated glycoprotein as a functional ligand for the Nogo-66 receptor. Science 297: 1190-1193. [DOI] [PubMed] [Google Scholar]
- Ma M, Basso DM, Walters P, Stokes BT, Jakeman LB (2001) Behavioral and histological outcomes following graded spinal cord contusion injury in the C57BL/6 mouse. Exp Neurol 169: 239-254. [DOI] [PubMed] [Google Scholar]
- McKeon RJ, Schreiber RC, Rudge JS, Silver J (1991) Reduction of neurite outgrowth in a model of glial scarring following CNS injury is correlated with the expression of inhibitory molecules on reactive astrocytes. J Neurosci 11: 3398-3411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKerracher L, David S, Jackson DL, Kottis V, Dunn RJ, Braun PE (1994) Identification of myelin-associated glycoprotein as a major myelin-derived inhibitor of neurite growth. Neuron 13: 805-811. [DOI] [PubMed] [Google Scholar]
- Moreno-Flores MT, Wandosell F (1999) Up-regulation of Eph tyrosine kinase receptors after excitotoxic injury in adult hippocampus. Neuroscience 91: 193-201. [DOI] [PubMed] [Google Scholar]
- Mosmann T (1983) Rapid colorimetric assay for cellular growth and survival: application to proliferation and cytotoxicity assays. J Immunol Methods 65: 55-63. [DOI] [PubMed] [Google Scholar]
- Mukhopadhyay G, Doherty P, Walsh FS, Crocker PR, Filbin MT (1994) A novel role for myelin-associated glycoprotein as an inhibitor of axonal regeneration. Neuron 13: 757-767. [DOI] [PubMed] [Google Scholar]
- Palmer A, Zimmer M, Erdmann KS, Eulenburg V, Porthin A, Heumann R, Deutsch U, Klein R (2002) EphrinB phosphorylation and reverse signaling: regulation by Src kinases and PTP-BL phosphatase. Mol Cell 9: 725-737. [DOI] [PubMed] [Google Scholar]
- Rodger J, Lindsey KA, Leaver SG, King CE, Dunlop SA, Beazley LD (2001) Expression of ephrin-A2 in the superior colliculus and EphA5 in the retina following optic nerve section in adult rat. Eur J Neurosci 14: 1929-1936. [DOI] [PubMed] [Google Scholar]
- Schafer M, Fruttiger M, Montag D, Schachner M, Martini R (1996) Disruption of the gene for the myelin-associated glycoprotein improves axonal regrowth along myelin in C57BL/Wlds mice. Neuron 16: 1107-1113. [DOI] [PubMed] [Google Scholar]
- Schnell L, Schwab ME (1990) Axonal regeneration in the rat spinal cord produced by an antibody against myelin-associated neurite growth inhibitors. Nature 343: 269-272. [DOI] [PubMed] [Google Scholar]
- Schnell L, Schwab ME (1993) Sprouting and regeneration of lesioned corticospinal tract fibres in the adult rat spinal cord. Eur J Neurosci 5: 1156-1171. [DOI] [PubMed] [Google Scholar]
- Shamah SM, Lin MZ, Goldberg JL, Estrach S, Sahin M, Hu L, Bazalakova M, Neve RL, Corfas G, Debant A, Greenberg ME (2001) EphA receptors regulate growth cone dynamics through the novel guanine nucleotide exchange factor ephexin. Cell 105: 233-244. [DOI] [PubMed] [Google Scholar]
- Simonen M, Pedersen V, Weinmann O, Schnell L, Buss A, Ledermann B, Christ F, Sansig G, van der Putten H, Schwab M (2003) Systemic deletion of the myelin-associated outgrowth inhibitor Nogo-A improves regenerative and plastic responses after spinal cord injury. Neuron 38: 201-211. [DOI] [PubMed] [Google Scholar]
- Stichel CC, Muller HW (1998) The CNS lesion scar: new vistas on an old regeneration barrier. Cell Tissue Res 294: 1-9. [DOI] [PubMed] [Google Scholar]
- Stichel CC, Hermanns S, Luhmann HJ, Lausberg F, Niermann H, D'Urso D, Servos G, Hartwig HG, Muller HW (1999) Inhibition of collagen IV deposition promotes regeneration of injured CNS axons. Eur J Neurosci 11: 632-646. [DOI] [PubMed] [Google Scholar]
- Sugiura S, Lahav R, Han J, Kou SY, Banner LR, de Pablo F, Patterson PH (2000) Leukaemia inhibitory factor is required for normal inflammatory responses to injury in the peripheral and central nervous systems in vivo and is chemotactic for macrophages in vitro. Eur J Neurosci 12: 457-466. [DOI] [PubMed] [Google Scholar]
- Turnley AM, Bartlett PF (1998) MAG and MOG enhance neurite outgrowth of embryonic mouse spinal cord neurons. NeuroReport 9: 1987-1990. [DOI] [PubMed] [Google Scholar]
- Turnley AM, Faux CH, Rietze RL, Coonan JR, Bartlett PF (2002) Suppressor of cytokine signaling 2 regulates neuronal differentiation by inhibiting growth hormone signaling. Nat Neurosci 5: 1155-1162. [DOI] [PubMed] [Google Scholar]
- Wahl S, Barth H, Ciossek T, Aktories K, Mueller BK (2000) Ephrin-A5 induces collapse of growth cones by activating Rho and Rho kinase. J Cell Biol 149: 263-270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang KC, Kim JA, Sivasankaran R, Segal R, He Z (2002a) p75 interacts with the Nogo receptor as a co-receptor for Nogo, MAG and OMgp. Nature 420: 74-78. [DOI] [PubMed] [Google Scholar]
- Wang KC, Koprivica V, Kim JA, Sivasankaran R, Guo Y, Neve RL, He Z (2002b) Oligodendrocyte-myelin glycoprotein is a Nogo receptor ligand that inhibits neurite outgrowth. Nature 417: 941-944. [DOI] [PubMed] [Google Scholar]
- Wilhelmsson U, Li L, Pekna M, Berthold CH, Blom S, Eliasson C, Renner O, Bushong E, Ellisman M, Morgan TE, Pekny M (2004) Absence of glial fibrillary acidic protein and vimentin prevents hypertrophy of astrocytic processes and improves post-traumatic regeneration. J Neurosci 24: 5016-5021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willson CA, Irizarry-Ramirez M, Gaskins HE, Cruz-Orengo L, Figueroa JD, Whittemore SR, Miranda JD (2002) Upregulation of EphA receptor expression in the injured adult rat spinal cord. Cell Transplant 11: 229-239. [PubMed] [Google Scholar]
- Yamashita T, Higuchi H, Tohyama M (2002) The p75 receptor transduces the signal from myelin-associated glycoprotein to Rho. J Cell Biol 157: 565-570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yong VW, Moumdjian R, Yong FP, Ruijs TC, Freedman MS, Cashman N, Antel JP (1991) Gamma-interferon promotes proliferation of adult human astrocytes in vitro and reactive gliosis in the adult mouse brain in vivo. Proc Natl Acad Sci USA 88: 7016-7020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng B, Ho C, Li S, Keirstead H, Steward O, Tessier-Lavigne M (2003) Lack of enhanced spinal regeneration in Nogo-deficient mice. Neuron 38: 213-224. [DOI] [PubMed] [Google Scholar]