

Abstract
Increased food intake is a major factor in the development of obesity, and the control of meal size is a valid approach to reduce food intake in humans. Meal termination, or satiety, is thought to be organized within the caudal brainstem where direct signals from the food handling alimentary canal and long-term signals from the forebrain converge in the solitary nucleus. Cholecystokinin (CCK) released from the gut after ingestion of food has been strongly implicated in nucleus tractus solitarius (NTS)-mediated satiation, but the exact cellular and intracellular signaling events are not understood. Using Western blotting and immunohistochemistry with phosphospecific antibodies, we demonstrate here that peripheral administration of CCK in rats leads to rapid activation of the extracellular signal-regulated kinase (ERK) signaling cascade in NTS neurons and that blockade of ERK signaling with microinfusion of a selective mitogen-activated ERK kinase inhibitor into the fourth ventricle attenuates the capacity of CCK to suppress food intake. In addition, we show that CCK-induced activation of ERK results in phosphorylation of the voltage-dependent potassium channel Kv4.2 and the nuclear transcription factor CREB (cAMP response element-binding protein). The results demonstrate that ERK signaling is necessary for exogenous CCK to suppress food intake in deprived rats and suggest that this pathway may also be involved in natural satiation and the period of satiety between meals through coupling of ERK activation to both cytosolic and nuclear effector mechanisms that have the potential to confer acute and long-term changes in neuronal functioning.
Keywords: satiety, brainstem, U0126, MAPK, CREB, Kv4.2
Introduction
Obesity with its secondary comorbidities such as type 2 diabetes and cardiovascular disease is now recognized as a serious and global health problem. Because food intake is a major determinant of energy balance, a considerable research effort is directed toward developing effective anorectic drugs. The process of satiation that leads to termination of a meal is thought to depend mainly on various signals generated by the interaction of ingested food with the gastrointestinal tract, such as gastric distension and cholecystokinin (CCK) (Gibbs et al., 1973; Smith et al., 1985; Schwartz and Moran, 1996; Smith, 1996; Moran, 2000). Many of these satiety signals reach the nucleus of the solitary tract in the caudal brainstem via vagal sensory nerve fibers (Smith et al., 1985; Walls et al., 1995; Schwartz et al., 1999), where they are integrated with information from the hypothalamus and other forebrain sites to stop additional ingestion (Berthoud, 2004).
Little is known about the mechanisms within the nucleus tractus solitarius (NTS) and caudal brainstem that lead to termination of a meal and a period of satiety. Ingestion of food or stimuli associated with the ingestion of food invariably leads to neuronal activation in the NTS as shown with in vivo electrophysiological recording (Schwartz and Moran, 2002) or with c-Fos immunohistochemistry (Rinaman et al., 1993, 1998; Berthoud et al., 2001; Emond et al., 2001). Because glutamate is the primary neurotransmitter released from vagal afferents (Allchin et al., 1994), and NTS neurons activated by gastrointestinal stimuli have been shown to express both NMDA and AMPA glutamate receptors (Berthoud et al., 2001), excitatory glutamatergic input seems to be responsible for this neuronal activation and c-Fos expression.
Because satiation is experienced in rats with midcollicular decerebration, it must be organized neurologically within the brainstem (Grill and Norgren, 1978; Grill and Smith, 1988), but it is not clear how activation of NTS neurons eventually leads to a change in configuration of oromotor circuitry associated with meal termination. It could involve direct and rapid effects of glutamate receptor activation on neuronal firing rate and, in addition, more sustained changes in membrane excitability and gene expression mediated by intracellular signaling cascades. The extracellular signal-regulated kinases (ERKs) comprise a well characterized signal transduction pathway that is activated by increased intracellular calcium coupled to glutamate receptor stimulation (Xia et al., 1996; Chandler et al., 2001). ERK signaling is initiated through p21Ras, Raf-1, and mitogen-activated ERK kinase (MEK), leading to ERK1/2 phosphorylation. In turn, ERK activates various cytosolic effector proteins, and the nucleartranscription factor cAMP response element-binding protein (CREB), leading to the induction of immediate-early genes like c-fos (Rosen et al., 1994; Sweatt, 2001).
Given the strong induction of c-Fos in NTS neurons by satiety signals, we hypothesized that gastrointestinal signals might engage the ERK signaling pathway and its downstream effector mechanisms to produce satiation and satiety. To test this hypothesis, we assessed activation of the ERK cascade in the rat NTS after peripheral administration of CCK. In addition, we examined the capacity of CCK to suppress food intake after blockade of the ERK signaling cascade.
Materials and Methods
Animals and housing. Adult male Sprague Dawley rats (Harlan, Indianapolis, IN) weighing 300-350 gm were housed individually in hanging wire-mesh cages under standard laboratory conditions (12 hr light cycle, lights on at 7:00 A.M.; 22 ± 2°C). Purina 5001 laboratory chow and water were available ad libitum, except where noted. All animal procedures were approved by the Institutional Animal Care and Use Committee and conformed to the guidelines of the National Institutes of Health.
Peptides and antibodies. CCK (sulfated octapeptide; CCK 26-33; catalog #7183) was purchased from Bachem-Peninsula (San Carlos, CA). MEK inhibitor U0126 (catalog #V1121) and anti-ACTIVE calcium/calmodulin-dependent protein kinase II (CaMKII) (pT286) antibody (catalog #V1111) were obtained from Promega (Madison, WI). Cell Signaling Technology (Beverly, MA) supplied primary antibodies for p44/42 MAP kinase (MAPK) (Thr202/Tyr204; pERK1/2; catalog #9101), MEK 1/2 (Ser217/221; catalog #9121), phosphorylated CREB (Ser133; catalog #9191), p44/42 MAPK (ERK1/2), MEK1/2 (catalog #9122), and CREB (catalog #9192). The phosphorylated pKv4.2 (Thr602-R; catalog #sc-16983-R) and total Kv4.2 (N-15; catalog #sc-11680) primary antibodies were obtained from Santa Cruz Biotechnology (Santa Cruz, CA). With immunoblot analysis, all antibodies gave clear signals at the predicted molecular sizes of the investigated proteins.
Time course and dose-response effects of CCK. Twenty-four rats were handled for several days and adapted to the intraperitoneal injection procedure with mock injections. At the end of the adaptation procedure, the rats were assigned to eight groups of three rats each, matched for mean body weight. On test days, between 9:00 and 10:00 A.M. (3-4 hr after lights on), one group of overnight food-deprived rats received injections of saline and the other seven groups received injections of 10 μg/kg CCK dissolved in saline (1 ml/kg, i.p.). Groups of rats were killed by guillotine in a separate room 0, 2, 4, 8, 15, 40, or 100 min after injection, and brains were extracted rapidly, blocked, and snap frozen in isopentane cooled in dry ice. The tissue was then held at -80°C until ready for protein extraction.
For dose-response studies, 16 rats were adapted as above. Four rats matched for mean body weight were assigned to each treatment group. On test days between 9:00 and 10:00 A.M., overnight food-deprived rats received injections of saline alone or 2, 10, or 100 μg/kg CCK dissolved in saline (1 ml/kg, i.p.). Rats were killed for Western blot analysis by guillotine 15 min after the injection, and brains were quickly harvested and frozen as above.
Immunohistochemical detection of phosphorylated ERK1/2. Two groups of rats, four rats per group, were housed singly, adapted to handling, and fasted overnight. On the test day, rats received intraperitoneal injections of saline or CCK (10 μg/kg in saline) at 1 ml/kg. Twelve minutes after the injection, rats were anesthetized with pentobarbital sodium (70 mg/kg, i.p.) and 3 min later perfused transcardially with heparinized (20 U/ml) saline, followed by ice-cold 4% phosphate-buffered paraformaldehyde, pH 7.4. The brains were extracted, blocked, and postfixed for a minimum of 2 hr. Tissues were then cryoprotected in 15% sucrose in 0.1 m PBS overnight at 4°C. Twenty-five micrometer coronal sections of medulla were cut on a cryostat, separated into five series, and held in PBS for processing immediately or stored in a cryoprotectant solution (50% PBS, 30% ethylene glycol, and 20% glycerol) at -20°C. One series (∼15 sections) from each rat, covering the rostrocaudal extent of the NTS, was processed for phosphorylated ERK (pERK) immunohistochemistry. With appropriate washings between incubations, free-floating sections were rinsed with fresh 0.5% sodium borohydride in PBS, followed by pretreatment in 3% hydrogen peroxide-methanol (1:4) before blocking in a solution of 5% normal goat serum and 1% bovine serum albumin in PBS with 0.5% Triton X-100 (PBSTx) to minimize background staining. Anti-rabbit phosphorylated p44/42 MAPK (pERK1/2) in primary diluent (0.1% gelatin and 0.05% sodium azide in PBSTx) was applied at a 1:200 concentration overnight at room temperature. Biotinylated goat anti-rabbit secondary antibody (1:500; Jackson ImmunoResearch, West Grove, PA) followed thorough washing in PBS, after which sections were incubated in an avidin-biotin complex (1:500; Vectastain ABC Elite kit; Vector Laboratories, Burlingame, CA). Cytoplasmic pERK was visualized by the dark brown/black reaction product of the cobalt/nickel-enhanced diaminobenzidine substrate kit (Pierce, Rockford, IL). Nonspecific staining was absent when the primary antibody was omitted. ERK1/2-immunopositive neurons were counted manually using conventional light microscopy with a 20× objective. The number of labeled neurons was counted bilaterally on 30-μm-thick frontal sections at three rostrocaudal levels of the NTS, immediately rostral to, at the mid level, and immediately caudal to the area postrema. The average count of labeled neurons for the three sections was used as a score for each animal. Only neurons with nuclei that were in the plane of the section and stained above background were included in the data analysis.
Fourth ventricular cannulations. Animals were anesthetized with ketamine/xylazine/acepromazine (80/5/1.6 mg/kg, s.c.) and given atropine (1 mg/kg, i.p.). A 24 gauge stainless steel guide cannula was aimed at the fourth ventricle (2.5 mm anterior to the posterior occipital suture, on the midline, 5.0 mm below the dura). A 30 gauge beveled injector was designed to protrude for 1.0 mm from the guide cannula. Rats were given 10 d to recover, after which cannula placement and patency were verified using the 5-thio-glucose test (Ritter et al., 1981), consisting of injecting 5-thio-glucose (210 μg in 3 μl of sterile saline) and measuring plasma glucose concentration after 30 min. Only animals responding with an increase of plasma glucose concentration of at least 80 mg/ml were used for experiments.
Effects of MEK inhibitor on CCK-induced suppression of food intake and stimulation of pERK. Sixteen cannulated rats were used in a crossover, counterbalanced design to study the effect of the MEK inhibitor U0126 on CCK-induced reduction of food intake. At 8:00 A.M. on test days, overnight fasted rats were given U0126 (2 μg in 50% DMSO/sterile saline) or vehicle, by infusing 3 μl into the fourth ventricle over a 2 min period. This dose of U0126 was based on previous studies (Schafe et al., 2000; Coogan and Piggins, 2003). After infusion, the rats were returned to their home cages and 1 hr later given intraperitoneal injections of saline or CCK (2 μg/kg). Allowed ad libitum access to chow, food intake was measured at 60 min by weighing the food cups and taking spillage into consideration.
At the end of behavioral testing, rats were subjected again to one of the four treatments (n = 4 per treatment) and killed 15 min after the intraperitoneal CCK or saline injection to harvest NTS tissue. In addition, 12 naive rats (n = 3 per treatment) without previous behavioral testing were used for Western blotting. Because there was no difference in the treatment effect on pERK, data from the two separate groups were pooled (n = 7 per treatment).
Tissue dissection for protein purification. Frozen brains were mounted on chucks and slowly warmed to -20°C in a cryostat. Three hundred-micrometer-thick sections were cut, and the NTS was dissected on a cold plate by means of cylindrical stainless steel punch tools (Palkovits, 1973). Tissue from each rat was then solubilized in 2% SDS, and total protein was determined by the bicinchoninic acid assay. The solubilized protein was aliquotted and stored at -80°C. Punching an area of the NTS bordered by the solitary tracts, the area postrema, the dorsal motor nucleus, and the nucleus cuneatus and gracilis on six slices, spanning a rostrocaudal level from -13.0 to -14.8 mm from bregma (Paxinos and Watson, 1986), yielded an average of 200 μg of total protein per rat.
Western blotting. An aliquot of the frozen sample was diluted with an equal volume of 2× electrophoresis sample buffer (final concentration; 50 mm Tris HCl, pH 6.7, 4% w/v glycerol, 4% SDS, 1% 2-mercaptoethanol, and 0.02 mg/ml bromphenol blue) and boiled for 10 min. Twenty micrograms were separated by size on a 10% SDS-polyacrylamide gel using the Laemmli buffer system and transferred to Immobilon-P polyvinylidene difluoride (PVDF) membranes in Towbin-transfer buffer (25 mm Tris, 192 mm glycine, 20% methanol, and 0.01% SDS). After transfer, blots were washed with PBS containing 0.05%Tween 20 (PBST). The membranes were blocked in PBST containing 5% nonfat dry milk and 1% bovine serum albumin for 1 hr at room temperature with agitation. Immunoblotting with phosphospecific antibodies was performed after transfer of proteins to PVDF membranes. Membranes were incubated for 1 hr at room temperature with a specific phosphorylated rabbit polyclonal primary antibody (pERK1/2, pMEK1/2, and pCREB were used at a 1:1000 dilution, whereas pCaMKII and pKv4.2 were used at a concentration of 1:500), followed by incubation with a horseradish peroxidase-conjugated secondary antibody (diluted to 1:1000; Southern Biotechnology, Birmingham, AL) for 1 hr at room temperature. After this incubation, the membranes were washed in PBST, theantigen-antibody-peroxidase complex detected by enhanced chemiluminescence according to the manufacturer's instructions, and visualized by exposure to Hyperfilm ECL (Amersham Biosciences, Piscataway, NJ). Membranes were then stripped by incubation in stripping buffer (100 mm 2-mercaptoethanol, 2% SDS, and 62.5 mm Tris-HCl, pH 6.7) for 30 min at 50°C with gentle agitation. Membranes were blocked and reprobed with total rabbit polyclonal antibodies to ERK1/2, MEK1/2, CREB, or to goat anti-Kv4.2 (all at 1:1000 concentrations, except for Kv4.2 at 1:5000) as described above. Film autoradiograms were analyzed and quantified by computer-assisted densitometry. For the ERK1/2 antibodies, only the ERK2 bands were quantified for densitometry.
Data analysis. Western blots were quantified using densitometry [HP Scanjet 5200 (Hewlett Packard) and Quantity One software 4.4.1 (Bio-Rad, Hercules, CA)]. Densitometry scores were analyzed by two-way ANOVA, followed by Bonferroni-adjusted least-squared difference post hoc comparisons (SYSTAT-10). Counts of pERK-positive neurons in the NTS were analyzed by Student's t test. Results are presented as means ± SEM.
Results
Exogenous CCK administration activates ERK signaling cascade in the NTS
Our time course study showed that intraperitoneal administration of 10 μg of CCK increased pERK1 and pERK2 but not total ERK1 and ERK2 (Fig. 1A). As shown in Figure 1B, quantitative analysis revealed a significant effect of treatment (F(7,16) = 35.273; p < 0.01), with pERK2 significantly increased at 8, 15, and 40 min. A maximal, approximate threefold increase (p < 0.01) of pERK2 was detected 15 min after the injection of CCK, and after 100 min levels had returned to baseline values.
Figure 1.

Time course of CCK-induced pERK1/2 in rat NTS. A, Representative immunoblots of pERK1/2 (top) and total ERK 1/2 (bottom) in NTS tissue isolated from individual rats after intraperitoneal administration of 10 μg/kg CCK at the times indicated. B, Mean ± SEM (n = 3) of optical density of pERK2. Bars that do not share the same letter are significantly different from each other (p < 0.01; ANOVA, followed by Bonferroni-adjusted least-squares difference post hoc tests).
A dose-response study using the optimal interval of 15 min between injection and assay showed a dose-dependent effect of CCK on phosphorylation of ERK1 and ERK2 but not on total ERK1 and ERK2 (Fig. 2A). Quantitative analysis on pERK2 (Fig. 2B) revealed a significant dose effect (F(3,13) = 75.8; p < 0.01), with even the smallest dose of 2 μg/kg producing a highly significant twofold increase (p < 0.01).
Figure 2.

Dose-response of CCK-induced activation of pERK1/2, measured 15 min after CCK administration. A, Representative immunoblots of pERK1/2 (top) and total ERK1/2 (bottom) in NTS tissue isolated from individual rats after intraperitoneal administration of saline or different doses of intraperitoneal-administered CCK. B, Mean ± SEM (n = 4) of optical density of ERK2. Bars that do not share the same letter are significantly different from each other (p < 0.01; ANOVA, followed by Bonferroni-adjusted least-squares difference post hoc tests).
To better characterize activation of the ERK signaling pathway in NTS neurons, we determined the level of phosphorylation and thus activation of other known members of the ERK signal transduction cascade. CaMKII has been demonstrated to activate ERK1/2 signaling through phosphorylation and inhibition of SynGAP (Komiyama et al., 2002). We determined CaMKII activation by immunoblotting with an antibody that recognizes phosphorylated CaMKII at Thr286. No increase in CamKII phosphorylation was observed after any dose of CCK (Fig. 3A). CaMKII did not follow a separate temporal pattern of activation because we could not detect any increase in CaMKII phosphorylation in time course experiments (data not shown).
Figure 3.

Dose-response of CCK-induced activation of pCaMKII (A), pMEK1/2 (B), and pCREB (C), measured 15 min after CCK administration. The top of each panel shows representative immunoblots, and the bottom shows quantitative analysis (n = 4). Bars that do not share the same letter are significantly different from each other (p < 0.01; ANOVA, followed by Bonferroni-adjusted least-squares difference post hoc tests).
In neurons, ERK activation is coupled to Raf kinase-dependent activation of MEK, and this kinase is the upstream activator of ERK1/2. As shown in Figure 3B, CCK dose-dependently increased phosphorylation of MEK in the NTS (F(3,12) = 38.67; p < 0.01).
ERK1/2 activation is known to initiate genomic responses in the nucleus of neurons, and a well known nuclear effector of the ERK pathway is CREB. Using an antibody that selectively recognizes CREB phosphorylation at Ser133, we show that all three doses of CCK significantly increased pCREB (F(3,13) = 12.987; p < 0.01) to a similar degree (Fig. 3C).
To identify specific NTS neurons that exhibit CCK-induced phosphorylation of ERK1/2, we used immunohistochemistry on frozen medulla sections. As shown in Figure 4A-C, after saline injection only a few ERK-immunopositive neurons were present in the NTS and the rest of the brainstem. In contrast, rats given injections of 10 μg/kg CCK showed many more pERK1/2-immunopositive cells in the NTS and in the ventrolateral medulla (data not shown). Proximal dendrites of some darkly stained, spindle-shaped neurons could clearly be seen. Quantitative analysis revealed a CCK-induced, significant threefold increase (p < 0.01) of the number of pERK-positive cells compared with saline controls (Fig. 4D).
Figure 4.

Immunohistochemical detection of CCK-induced pERK1/2 in rat NTS. A, B, Low- and high-magnification images showing many pERK-immunopositive neurons (black) in NTS induced by intraperitoneal administration of 10 μg/kg CCK, 15 min before fixation. The inset shows the enlarged pERK-immunopositive neuron with staining of proximal dendrites. C, Few pERK-ir neurons are present after saline control injections. D, Quantitative analysis of the number of pERK-immunopositive neurons in NTS after saline (n = 4) or CCK (n = 4) administration. **p < 0.01; t test. ap, Area postrema; c, central canal; com, commissural subnucleus; ts, solitary tract; x, dorsal motor nucleus.
CCK stimulates phosphorylation of Kv4.2 potassium channel in the NTS
Besides acting through CREB in the nucleus, the ERK signal transduction cascade is known to phosphorylate and activate a number of cytosolic effector proteins. One of these effector proteins is the A-type voltage-dependent potassium channel Kv4.2, which is specifically phosphorylated at Thr602 by ERK, resulting in decreased opening of the channel and a relative depolarization of the neuron (Watanabe et al., 2002; Yuan et al., 2002). We show here that, using an antibody specific for this site, CCK (2 μg/kg, i.p.) significantly (p < 0.01) stimulates phosphorylation of Kv4.2 in the NTS and that previous fourth ventricular treatment with U0126 significantly (p < 0.01) attenuates this effect (Fig. 5). The differences in pKv4.2 levels between the saline/vehicle controls and the CCK/U0126 groups (p = 0.08) and the saline/U0126 and CCK/U0126 groups (p = 0.07) were marginally significant using the Bonferroni adjustment. Total Kv4.2 protein was not changed by CCK. These results clearly show that a low dose of CCK administered intraperitoneally increases phosphorylation of the Kv4.2 potassium channel in an ERK-dependent manner in NTS neurons.
Figure 5.
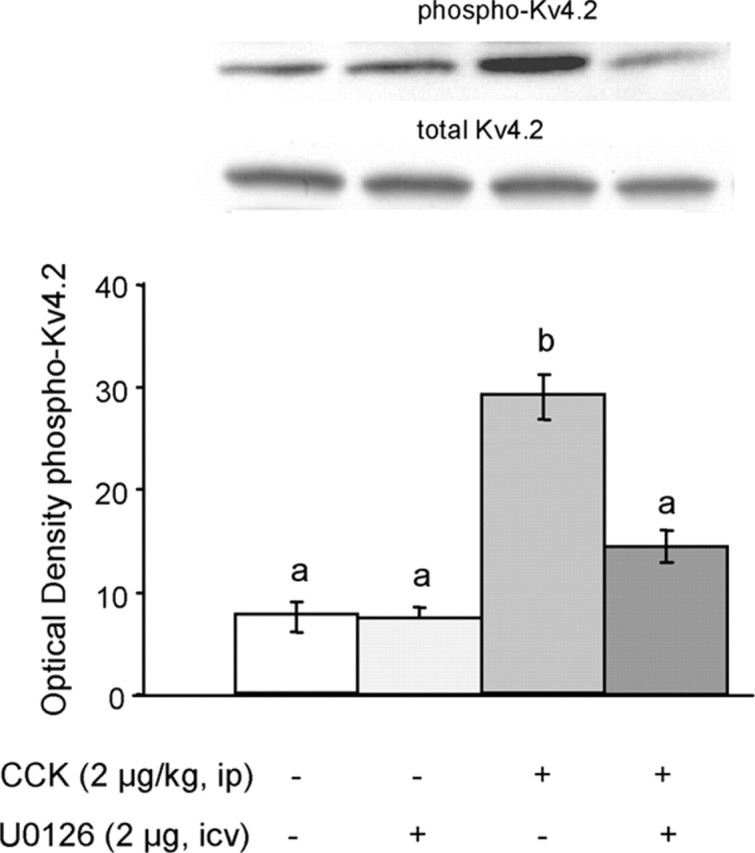
CCK-induced, ERK-dependent activation of pKv4.2, a voltage-sensitive potassium channel. Top, Representative immunoblots of NTS tissue from individual rats administered with intraperitoneal saline or 2 μg/kg CCK, in the presence or absence of the MEK/ERK inhibitor U0126 injected into the fourth ventricle. Bottom, Means + SEM (n = 5) of optical density of pKv4.2. Bars that do not share the same letter are significantly different from each other (p < 0.01; ANOVA, followed by Bonferroni-adjusted least-squares difference post hoc tests). Note that the differences in pKv4.2 levels between the saline/vehicle controls and the CCK/U0126 groups (p = 0.08) and the saline/U0126 and CCK/U0126 groups (p = 0.07) were marginally significant.
Inhibition of the MEK→ ERK signaling cascade decreases the potency of CCK to reduce food intake
If activation of ERK is a necessary step for the expression of CCK-induced satiety, then inhibition of this protein kinase cascade should abolish or attenuate the effects of CCK. We used the MEK1/2 inhibitor U0126 (2 μg) infused into the fourth ventricle to pharmacologically inhibit the ERK cascade in the NTS. As shown in Figure 6, CCK (2 μg/kg, i.p.) produced the expected significant reduction of food intake in overnight food-deprived rats, compared with saline. Previous infusion of U0126 significantly decreased the potency of exogenous CCK to induce satiety by ∼50% (p < 0.01) but did not completely abolish it when 1 hr food intake was examined (Fig. 6). Importantly, when U0126 was administered in the absence of CCK, it did not affect food intake, suggesting that at the dose used it had no aversive effects. Furthermore, fourth ventricular injection of the DMSO vehicle alone did not produce malaise, because the amount of food ingested in 1 hr compares well with values from previous studies without fourth ventricular injections, and no abnormal behavior was detected.
Figure 6.

Blockade of ERK signaling attenuates food intake suppression by intraperitoneal CCK. In 16 hr food-deprived rats, 2 μg/kg CCK decreased 1 hr food intake compared with saline. Previous in fusion of U0126 (2 μg) in to the fourth ventricle significantly at tenuated the ability of CCK to suppress food intake by ∼50%. Infusion of U0126 alone did not affect food intake. Means ± SEM (n = 16) of 1 hr chow intake are shown. Bars that do not share the same letter are significantly different from each other (p < 0.01; ANOVA, followed by Bonferroni-adjusted least-squares difference post hoc tests).
To verify that the MEK inhibitor did, in fact, block CCK-induced activation of the ERK cascade, we performed Western blot analysis of pERK in the NTS. As shown in Figure 7, U0126 significantly decreased the potency of CCK to stimulate pERK by ∼35% (p < 0.02), while not significantly affecting basal levels of pERK in intraperitoneal saline-injected control rats. Thus, the inhibitory effects of CCK on food intake and the stimulatory effects on pERK are attenuated to a similar degree by U0126. These results demonstrate that ERK signaling is necessary and sufficient for exogenous CCK to reduce food intake in food-deprived rats.
Figure 7.
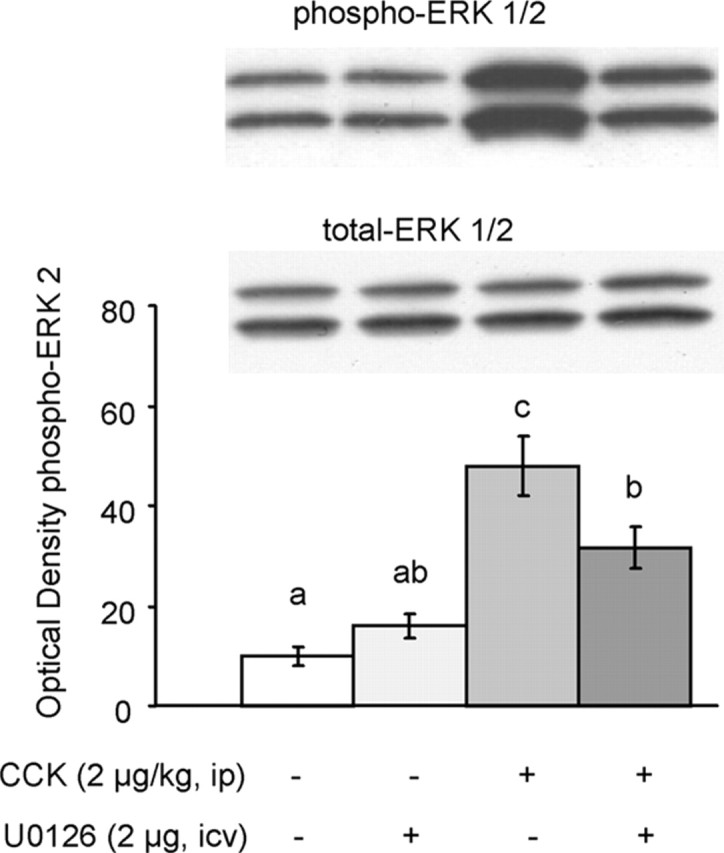
U0126 attenuates CCK-induced increases in ERK1/2 phosphorylation in NTS. A, Representative immunoblots of pERK1/2 and total ERK1/2. B, Quantitative analysis. In 16 hr food-deprived rats, 2 μg/kg CCK significantly increased pERK2 compared with saline. Previous infusion of U0126 (2 μg) into the fourth ventricle significantly attenuated the ability of CCK to increase pERK2 by ∼35%. Infusion of U0126 alone did not affect pERK2. Means ± SEM (n = 7) of optical density of pERK2. Bars that do not share the same letter are significantly different from each other (p < 0.01; ANOVA followed by Bonferroni-adjusted least-squares difference post hoc tests).
Discussion
The neural mechanisms responsible for the termination of a meal by the process of satiation are thought to reside within the brainstem, where gastrointestinal signals generated by ingested food are first processed within the NTS. Here, we show that the archetypical satiety signal CCK activates an intracellular signaling cascade involving MEK, ERK, CREB, and the Kv4.2 potassium channel in rat NTS and that activation of this cascade is necessary for exogenous CCK to suppress food intake in food-deprived rats. These findings suggest, but do not prove, that ERK signaling may also be critically involved in the natural process of satiation and satiety.
Mechanisms of CCK-induced activation of the ERK cascade
There is a large body of evidence demonstrating that CCK excites vagal sensory neurons through the CCK-A receptor (Davison and Clarke, 1988; Schwartz et al., 1993; Patterson et al., 2002) and that this pathway is critical for the control of CCK on meal size and suppression of food intake (Smith et al., 1985; Walls et al., 1995; Schwartz et al., 1999). The primary transmitter released from central terminals of vagal afferents in the NTS is glutamate (Allchin et al., 1994), and most NTS neurons responding to gastric satiety signals express NMDA and AMPA glutamate receptors (Berthoud et al., 2001). Therefore, we can assume that the peripherally administered CCK acts at least partly through this vagal afferent, glutamatergic pathway in our experiments. However, previous experiments using direct brain injections of CCK (Schick et al., 1986) and recent evidence gathered with CCK-A receptor antagonists that do or do not penetrate the blood-brain barrier (Reidelberger et al., 2004) suggest that, in addition to mediation by vagal afferents, exogenous CCK and endogenous CCK from the gut can affect food intake by direct action in the brain.
The immediate action (within milliseconds) of NMDA and AMPA glutamate receptor activation is depolarization and increased neuronal firing rate, and this action alone may be contributing to meal termination. However, both receptors are also coupled to a vast array of intracellular signaling pathways that result in divergent cellular responses with more or less protracted time courses. Increased intracellular calcium is a prominent feature of NMDA receptor activation, and it has also been shown to stimulate the ERK kinase cascade (Perkinton et al., 1999; Chandler et al., 2001). Activation of the ERK pathway can involve the upstream activation of Ras through a complex mechanism thought to involve CaMKII-dependent phosphorylation and inhibition of SynGAP (Komiyama et al., 2002). Although our study did not demonstrate a role for CaMKII in CCK-induced activation of ERK, Ras can be activated through other calcium-dependent as well as calcium-independent mechanisms (Swank and Sweatt, 2001). The specific glutamate receptor input that couples the activation of ERK1/2 in response to CCK is unknown. Interestingly, inhibition of the NMDA subtype of glutamate receptors with a specific antagonist, MK-801 [(+)-5-methyl-10,11-dihydro-5H-dibenzo [a,d] cyclohepten-5,10-imine hydrogen maleate], results in delayed meal termination and increased meal size (Burns and Ritter, 1997; Treece et al., 1998; Zheng et al., 1999; Covasa et al., 2004), suggesting these receptors do play a role in a component of CCK-induced satiety. The role of glutamate receptors in CCK-induced ERK activation needs to be examined in future experiments.
It has also been known that ERK activation is coupled to Raf kinase-dependent activation of MEK, and we have clearly demonstrated that CCK stimulates MEK phosphorylation in the same dose-dependent manner because it stimulates ERK phosphorylation. We observed the earliest significant ERK activation with a delay of ∼5-10 min after intraperitoneal administration. Because phosphorylation processes are rapid (seconds to minutes), we assume that it takes a few minutes for CCK to be absorbed and reach a certain titer in the circulation.
CCK-induced phosphorylation of the ERK1/2 cytosolic effector protein Kv4.2
Increases in ERK activation directly translate into downstream phosphorylation of cytosolic effector proteins that govern specific neuronal functions. We show here that CCK significantly increases phosphorylation of the Thr602 site of the voltage-dependent potassium channel Kv4.2 in NTS within 15 min and that this effect is attenuated by pretreatment with the MEK inhibitor U0126. This confirms the ERK-dependent phosphorylation of Kv4.2 at this site reported previously (Yuan et al., 2002). Phosphorylation of Kv4.2 by ERK at this site has been shown to inhibit opening of the channel, resulting in less hyperpolarization and a net increase in neuronal firing rate, as shown by increased excitability of hippocampal neurons (Adams and Sweatt, 2002; Yuan et al., 2002). In the hippocampus, it is thought to play an important role in late-phase long-term potentiation (Watanabe et al., 2002). Thus, this mechanism might render certain NTS neurons more sensitive to glutamatergic inputs toward the end of a meal.
CCK-induced phosphorylation of the ERK nuclear effector protein CREB
We show that exogenous CCK produces a threefold increase in the phosphorylation of the transcription factor CREB at Ser133 with the lowest dose of 2 μg/kg CCK already producing the maximal effect. CREB has been shown to be activated at this phosphorylation site through translocation of activated ERK to the nucleus and an intermediate kinase, ribosomal S6 kinase-2 (p90RSK2) (Xing et al., 1996; Impey et al., 1998). Phosphorylation of CREB at Ser133 by a number of protein kinases is an initiation event for the transcription of immediate-early genes including c-fos (Karin et al., 1997). Induction of c-fos mRNA (Day et al., 1994) and Fos protein (Rinaman et al., 1993; Monnikes et al., 1997; Zittel et al., 1999) by peripheral CCK administration has been shown in numerous reports. It is thought that c-Fos, because of its long kinetics of production and degradation (Herdegen and Leah, 1998), is indicative of a neuron that has initiated some adaptive genomic response leading to a prolonged change in its functional response to future stimuli, commonly referred to as plasticity. Because inhibition of food intake and c-Fos induction typically show clear dose-response relationships, the lack of such a relationship for CREB phosphorylation suggests that upstream effector mechanisms such as Kv4.2 function may be mainly responsible for meal termination, whereas the nuclear effects may be responsible for the next intermeal interval.
In one previous study using immunohistochemistry, peripheral CCK failed to induce increased pCREB in the NTS (Houpt, 1997). However, the high background staining reported in that study most likely prevented detection of increased pCREB immunoreactivity in a subset of neurons.
ERK signaling is necessary for intraperitoneal-administered CCK to suppress food intake
Simply demonstrating CCK-induced activation of ERK signaling in NTS neurons does not implicate this pathway in the suppressive effects of CCK on food intake. CCK may use this pathway to signal satiation-independent effects such as changes in gastrointestinal or other visceral functions (Owyang, 1996). Our demonstration that blockade of the ERK signaling cascade with U0126 attenuates the capacity of exogenous CCK to suppress food intake suggests that this pathway is necessary. The suppression of CCK on food intake was not fully abolished because the dose of U0126 we used did not fully suppress ERK signaling. Given the fact that ERK signaling is necessary for neuronal survival pathways (Campbell et al., 1998), we did not explore higher doses of U0126 to avoid potential nonspecific behavioral responses. Administration of the blocker alone did not change food intake or elicit any abnormal behavior in our experiment, suggesting that this dose of U0126 does not produce malaise or aversion. If endogenous CCK suppresses food intake by activating ERK signaling, one might have expected that U0126 in the absence of exogenous CCK would delay meal termination and increase food intake. However, the large meal after food deprivation may have produced a ceiling effect, making it difficult to see an additional increase. The role of ERK activation in naturally occurring satiation should be tested under non-food deprivation conditions, when meal size is smaller and mainly under control of visceral factors.
ERK cascade inhibition, satiation, and satiety
Using exogenous CCK administration, our study does not directly address the question whether ERK signaling in the NTS is involved in meal termination by endogenous satiety signals. Although this paradigm has been widely used, it is not clear whether there is such a thing as a “physiological” dose of CCK. It is thought that a component of satiation after a large meal or higher doses of exogenous CCK produce nausea or even visceral illness. Our low dose of 2 μg/kg CCK is usually considered not to produce such symptoms and therefore to be physiological (Verbalis et al., 1986). Recent work has suggested that the specific neuronal activation pattern in the NTS may indicate whether a given stimulus is producing satiation or nausea, with moderate recruitment of catecholaminergic neurons associated with normal satiation (Rinaman et al., 1998) and strong recruitment of glucagon-like peptide-1-expressing neurons indicating involvement of nausea and illness (Rinaman, 1999). Furthermore, the suppression of CCK on food intake was significantly attenuated in rats with selective ablation of catecholaminergic neurons in the NTS (Rinaman, 2003). Future studies identifying the neurochemical phenotype of pERK-immunoreactive NTS neurons will be necessary to shed more light on this issue.
In conclusion, our results demonstrate a role for ERK signaling in NTS neurons in food intake suppression by exogenous CCK administration. The coupling of ERK activation to both cytosolic and nuclear effector mechanisms that have the potential to confer acute and longer-term changes in neuronal functioning tempts us to speculate that this pathway may also be involved in natural satiation and the period of satiety between meals.
Footnotes
This work was supported by National Institutes of Health Grant DK47348.
Correspondence should be addressed to Dr. Hans-Rudolf Berthoud, Pennington Biomedical Research Center, 6400 Perkins Road, Baton Rouge, LA 70808. E-mail: berthohr@pbrc.edu.
Copyright © 2004 Society for Neuroscience 0270-6474/04/2410240-08$15.00/0
References
- Adams JP, Sweatt JD (2002) Molecular psychology: roles for the ERK MAP kinase cascade in memory. Annu Rev Pharmacol Toxicol 42: 135-163. [DOI] [PubMed] [Google Scholar]
- Allchin RE, Batten TF, McWilliam PN, Vaughan PF (1994) Electrical stimulation of the vagus increases extracellular glutamate recovered from the nucleus tractus solitarii of the cat by in vivo microdialysis. Exp Physiol 79: 265-268. [DOI] [PubMed] [Google Scholar]
- Berthoud H, Earle T, Zheng H, Patterson LM, Phifer C (2001) Food-related gastrointestinal signals activate caudal brainstem neurons expressing both NMDA and AMPA receptors. Brain Res 915: 143-154. [DOI] [PubMed] [Google Scholar]
- Berthoud H-R (2004) The caudal brainstem and the control of food intake and energy balance. In: Handbook of behavioral neurobiology (Stricker EM, Woods SC, eds), pp 195-240. New York: Plenum.
- Burns GA, Ritter RC (1997) The non-competitive NMDA antagonist MK-801 increases food intake in rats. Pharmacol Biochem Behav 56: 145-149. [DOI] [PubMed] [Google Scholar]
- Campbell SL, Khosravi-Far R, Rossman KL, Clark GJ, Der CJ (1998) Increasing complexity of Ras signaling. Oncogene 17: 1395-1413. [DOI] [PubMed] [Google Scholar]
- Chandler LJ, Sutton G, Dorairaj NR, Norwood D (2001) N-methyl-d-aspartate receptor-mediated bidirectional control of extracellular signal-regulated kinase activity in cortical neuronal cultures. J Biol Chem 276: 2627-2636. [DOI] [PubMed] [Google Scholar]
- Coogan AN, Piggins HD (2003) Circadian and photic regulation of phosphorylation of ERK1/2 and Elk-1 in the suprachiasmatic nuclei of the Syrian hamster. J Neurosci 23: 3085-3093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Covasa M, Ritter RC, Burns GA (2004) NMDA receptor blockade attenuates CCK-induced reduction of real feeding but not sham feeding. Am J Physiol Regul Integr Comp Physiol 286: R826-R831. [DOI] [PubMed] [Google Scholar]
- Davison JS, Clarke GD (1988) Mechanical properties and sensitivity to CCK of vagal gastric slowly adapting mechanoreceptors. Am J Physiol 255: G55-G61. [DOI] [PubMed] [Google Scholar]
- Day HE, McKnight AT, Poat JA, Hughes J (1994) Evidence that cholecystokinin induces immediate early gene expression in the brainstem, hypothalamus and amygdala of the rat by a CCKA receptor mechanism. Neuropharmacology 33: 719-727. [DOI] [PubMed] [Google Scholar]
- Emond M, Schwartz GJ, Moran TH (2001) Meal-related stimuli differentially induce c-Fos activation in the nucleus of the solitary tract. Am J Physiol Regul Integr Comp Physiol 280: R1315-R1321. [DOI] [PubMed] [Google Scholar]
- Gibbs J, Young RC, Smith GP (1973) Cholecystokinin elicits satiety in rats with open gastric fistulas. Nature 245: 323-325. [DOI] [PubMed] [Google Scholar]
- Grill HJ, Norgren R (1978) Chronically decerebrate rats demonstrate satiation but not bait shyness. Science 201: 267-269. [DOI] [PubMed] [Google Scholar]
- Grill HJ, Smith GP (1988) Cholecystokinin decreases sucrose intake in chronic decerebrate rats. Am J Physiol 254: R853-R856. [DOI] [PubMed] [Google Scholar]
- Herdegen T, Leah JD (1998) Inducible and constitutive transcription factors in the mammalian nervous system: control of gene expression by Jun, Fos and Krox, and CREB/ATF proteins. Brain Res Brain Res Rev 28: 370-490. [DOI] [PubMed] [Google Scholar]
- Houpt TA (1997) CREB phosphorylation in the nucleus of the solitary tract and parabrachial nucleus is not altered by peripheral cholecystokinin that induces c-Fos. Brain Res 751: 143-147. [DOI] [PubMed] [Google Scholar]
- Impey S, Obrietan K, Wong ST, Poser S, Yano S, Wayman G, Deloulme JC, Chan G, Storm DR (1998) Cross talk between ERK and PKA is required for Ca2+ stimulation of CREB-dependent transcription and ERK nuclear translocation. Neuron 21: 869-883. [DOI] [PubMed] [Google Scholar]
- Karin M, Liu Z, Zandi E (1997) AP-1 function and regulation. Curr Opin Cell Biol 9: 240-246. [DOI] [PubMed] [Google Scholar]
- Komiyama NH, Watabe AM, Carlisle HJ, Porter K, Charlesworth P, Monti J, Strathdee DJ, O'Carroll CM, Martin SJ, Morris RG, O'Dell TJ, Grant SG (2002) SynGAP regulates ERK/MAPK signaling, synaptic plasticity, and learning in the complex with postsynaptic density 95 and NMDA receptor. J Neurosci 22: 9721-9732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monnikes H, Lauer G, Arnold R (1997) Peripheral administration of cholecystokinin activates c-fos expression in the locus coeruleus/subcoeruleus nucleus, dorsal vagal complex and paraventricular nucleus via capsaicin-sensitive vagal afferents and CCK-A receptors in the rat. Brain Res 770: 277-288. [DOI] [PubMed] [Google Scholar]
- Moran TH (2000) Cholecystokinin and satiety: current perspectives. Nutrition 16: 858-865. [DOI] [PubMed] [Google Scholar]
- Owyang C (1996) Physiological mechanisms of cholecystokinin action on pancreatic secretion. Am J Physiol 271: G1-G7. [DOI] [PubMed] [Google Scholar]
- Palkovits M (1973) Isolated removal of hypothalamic or other brain nuclei of the rat. Brain Res 59: 449-450. [DOI] [PubMed] [Google Scholar]
- Patterson LM, Zheng H, Berthoud HR (2002) Vagal afferents innervating the gastrointestinal tract and CCKA-receptor immunoreactivity. Anat Rec 266: 10-20. [DOI] [PubMed] [Google Scholar]
- Paxinos G, Watson C (1986) The rat brain in stereotaxic coordinates, Ed 2. San Diego: Academic.
- Perkinton MS, Sihra TS, Williams RJ (1999) Ca2+-permeable AMPA receptors induce phosphorylation of cAMP response element-binding protein through a phosphatidylinositol 3-kinase-dependent stimulation of the mitogen-activated protein kinase signaling cascade in neurons. J Neurosci 19: 5861-5874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reidelberger RD, Hernandez J, Fritzsch B, Hulce M (2004) Abdominal vagal mediation of the satiety effects of CCK in rats. Am J Physiol Regul Integr Comp Physiol 286: R1005-R1012. [DOI] [PubMed] [Google Scholar]
- Rinaman L (1999) Interoceptive stress activates glucagon-like peptide-1 neurons that project to the hypothalamus. Am J Physiol 277: R582-R590. [DOI] [PubMed] [Google Scholar]
- Rinaman L (2003) Hindbrain noradrenergic lesions attenuate anorexia and alter central cFos expression in rats after gastric viscerosensory stimulation. J Neurosci 23: 10084-10092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rinaman L, Verbalis JG, Stricker EM, Hoffman GE (1993) Distribution and neurochemical phenotypes of caudal medullary neurons activated to express cFos following peripheral administration of cholecystokinin. J Comp Neurol 338: 475-490. [DOI] [PubMed] [Google Scholar]
- Rinaman L, Baker EA, Hoffman GE, Stricker EM, Verbalis JG (1998) Medullary c-Fos activation in rats after ingestion of a satiating meal. Am J Physiol 275: R262-R268. [DOI] [PubMed] [Google Scholar]
- Ritter RC, Slusser PG, Stone S (1981) Glucoreceptors controlling feeding and blood glucose: location in the hindbrain. Science 213: 451-452. [DOI] [PubMed] [Google Scholar]
- Rosen LB, Ginty DD, Weber MJ, Greenberg ME (1994) Membrane depolarization and calcium influx stimulate MEK and MAP kinase via activation of Ras. Neuron 12: 1207-1221. [DOI] [PubMed] [Google Scholar]
- Schafe GE, Atkins CM, Swank MW, Bauer EP, Sweatt JD, LeDoux JE (2000) Activation of ERK/MAP kinase in the amygdala is required for memory consolidation of pavlovian fear conditioning. J Neurosci 20: 8177-8187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schick RR, Yaksh TL, Go VL (1986) Intracerebroventricular injections of cholecystokinin octapeptide suppress feeding in rats—pharmacological characterization of this action. Regul Pept 14: 277-291. [DOI] [PubMed] [Google Scholar]
- Schwartz GJ, Moran TH (1996) Sub-diaphragmatic vagal afferent integration of meal-related gastrointestinal signals. Neurosci Biobehav Rev 20: 47-56. [DOI] [PubMed] [Google Scholar]
- Schwartz GJ, Moran TH (2002) Leptin and neuropeptide y have opposing modulatory effects on nucleus of the solitary tract neurophysiological responses to gastric loads: implications for the control of food intake. Endocrinology 143: 3779-3784. [DOI] [PubMed] [Google Scholar]
- Schwartz GJ, McHugh PR, Moran TH (1993) Gastric loads and cholecystokinin synergistically stimulate rat gastric vagal afferents. Am J Physiol 265: R872-R876. [DOI] [PubMed] [Google Scholar]
- Schwartz GJ, Salorio CF, Skoglund C, Moran TH (1999) Gut vagal afferent lesions increase meal size but do not block gastric preload-induced feeding suppression. Am J Physiol 276: R1623-R1629. [DOI] [PubMed] [Google Scholar]
- Smith GP (1996) The direct and indirect controls of meal size. Neurosci Biobehav Rev 20: 41-46. [DOI] [PubMed] [Google Scholar]
- Smith GP, Jerome C, Norgren R (1985) Afferent axons in abdominal vagus mediate satiety effect of cholecystokinin in rats. Am J Physiol 249: R638-R641. [DOI] [PubMed] [Google Scholar]
- Swank MW, Sweatt JD (2001) Increased histone acetyltransferase and lysine acetyltransferase activity and biphasic activation of the ERK/RSK cascade in insular cortex during novel taste learning. J Neurosci 21: 3383-3391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sweatt JD (2001) The neuronal MAP kinase cascade: a biochemical signal integration system subserving synaptic plasticity and memory. J Neurochem 76: 1-10. [DOI] [PubMed] [Google Scholar]
- Treece BR, Covasa M, Ritter RC, Burns GA (1998) Delay in meal termination follows blockade of N-methyl-d-aspartate receptors in the dorsal hindbrain. Brain Res 810: 34-40. [DOI] [PubMed] [Google Scholar]
- Verbalis JG, McCann MJ, McHale CM, Stricker EM (1986) Oxytocin secretion in response to cholecystokinin and food: differentiation of nausea from satiety. Science 232: 1417-1419. [DOI] [PubMed] [Google Scholar]
- Walls EK, Phillips RJ, Wang FB, Holst MC, Powley TL (1995) Suppression of meal size by intestinal nutrients is eliminated by celiac vagal deafferentation. Am J Physiol 269: R1410-R1419. [DOI] [PubMed] [Google Scholar]
- Watanabe S, Hoffman DA, Migliore M, Johnston D (2002) Dendritic K+ channels contribute to spike-timing dependent long-term potentiation in hippocampal pyramidal neurons. Proc Natl Acad Sci USA 99: 8366-8371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia Z, Dudek H, Miranti CK, Greenberg ME (1996) Calcium influx via the NMDA receptor induces immediate early gene transcription by a MAP kinase/ERK-dependent mechanism. J Neurosci 16: 5425-5436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xing J, Ginty DD, Greenberg ME (1996) Coupling of the RAS-MAPK pathway to gene activation by RSK2, a growth factor-regulated CREB kinase. Science 273: 959-963. [DOI] [PubMed] [Google Scholar]
- Yuan LL, Adams JP, Swank M, Sweatt JD, Johnston D (2002) Protein kinase modulation of dendritic K+ channels in hippocampus involves a mitogen-activated protein kinase pathway. J Neurosci 22: 4860-4868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng H, Kelly L, Patterson LM, Berthoud HR (1999) Effect of brain stem NMDA-receptor blockade by MK-801 on behavioral and fos responses to vagal satiety signals. Am J Physiol 277: R1104-R1111. [DOI] [PubMed] [Google Scholar]
- Zittel TT, Glatzle J, Kreis ME, Starlinger M, Eichner M, Raybould HE, Becker HD, Jehle EC (1999) C-fos protein expression in the nucleus of the solitary tract correlates with cholecystokinin dose injected and food intake in rats. Brain Res 846: 1-11. [DOI] [PubMed] [Google Scholar]