

Abstract
We investigated whether peroxisome proliferator-activated receptor γ (PPARγ) could be involved in the modulation of the amyloid cascade causing Alzheimer's disease. Inducing expression or activating PPARγ using synthetic agonists of the thiazolinedione family results in a dramatic decrease in the levels of the amyloid-β (Aβ) peptide in the conditioned medium of neuronal and non-neuronal cells. PPARγ does not affect expression or activity of any of the secretases involved in the generation of the Aβ peptide but induces a fast, cell-bound clearing mechanism responsible for the removal of the Aβ peptide from the medium. Although PPARγ expression is generally low in the CNS, induction of PPARγ expression during inflammation could be beneficial for inducing Aβ clearance. We confirm that the Aβ clearance mechanism can indeed be induced by PPARγ activation in primary murine-mixed glia and cortical neuronal cultures. Our results suggest that PPARγ-controlled mechanisms should be explored further as potential drug targets for Alzheimer's disease treatment.
Keywords: PPARγ, TZD, Aβ, APP, NSAIDs, Alzheimer's disease
Introduction
Alzheimer's disease (AD), the most common form of dementia in the elderly, is a neurodegenerative disease characterized by the deposition of extracellular amyloid-β (Aβ) plaques and the formation of intracellular tangles in the CNS. Aβ plaques are mainly composed of Aβ peptides, protein fragments derived by proteolytic cleavage of the amyloid precursor protein (APP). An inflammatory component appears to contribute also to the pathogenesis of AD with activated microglia, the principal immune cells in the brain, extensively associated with the Aβ plaques and secreting proinflammatory cytokines (Perlmutter et al., 1990; McGeer et al., 1993; Tan et al., 1999). An important number of retrospective epidemiological studies have provided evidence that long-term treatment with some nonsteroidal anti-inflammatory drugs (NSAIDs) reduce the risk for AD, delay its onset, and slow the progression of the disease (Rich et al., 1995; McGeer and McGeer, 1996; Stewart et al., 1997; Mackenzie and Munoz, 1998; in t' Veld et al., 2001). Initial in vitro experiments showed that the treatment of microglia and monocytes in culture with NSAIDs attenuated the secretion of proinflammatory cytokines in response to fibrilar Aβ. Furthermore, the conditioned medium of these stimulated microglia was neurotoxic, whereas the medium from microglia stimulated with Aβ but treated with NSAIDs displayed a protective effect in neurons (Klegeris et al., 1999; Combs et al., 2000). Interestingly, the thiazolidinedione (TZD) drugs (Willson et al., 2000), agonists of the peroxisome proliferator-activated receptor γ (PPARγ), showed the same anti-inflammatory neuroprotective effect as NSAIDs (Combs et al., 2000). Because some NSAIDs are agonists of PPARγ (Lehmann et al., 1995), and given the evidence for a direct role of NSAIDs on amyloid pathology (Lim et al., 2000; Weggen et al., 2001; Jantzen et al., 2002; Q. Yan et al., 2003), the possible role of PPARγ in this context deserves additional investigation. PPARγ is a nuclear transcription factor belonging to the PPAR family. These transcription factors play important physiological roles in the regulation of lipid metabolism (Mangelsdorf et al., 1995; Lemberger et al., 1996). PPARγ is involved in adipocyte differentiation (Chawla et al., 1994; Tontonoz et al., 1994; Hu et al., 1995; Rosen et al., 1999), insulin action (Olefsky, 2000; Steppan et al., 2001), and cell proliferation (Wang et al., 2001; Okano et al., 2002). The recent discovery that PPARγ stimulation reduces inflammation in vitro (Lemberger et al., 1996; Colville-Nash et al., 1998; Jiang et al., 1998; Petrova et al., 1999; Ricote et al., 1999; Combs et al., 2000) and in vivo (Heneka et al., 2000; Dehmer et al., 2004), together with the fact that PPARγ agonists (Willson et al., 2000) have been used for years in the treatment of diabetes type II, have raised the hope that PPARγ could become a drug target for the treatment of neurological diseases with an inflammatory component-like AD. Based on the NSAID studies mentioned above, we speculated that activation of PPARγ could have, in addition to its well known immunomodulating role, a more direct effect on the amyloid cascade in AD. We therefore investigated the effects of PPARγ activation, either by expressing PPARγ or by using TZD agonists, on APP processing and Aβ clearance in different cell lines and primary cultures of brain cells. We demonstrate that the activation of PPARγ directly affects the stability of Aβ externally added to the cell, suggesting the activation of a rapid clearance mechanism.
Materials and Methods
Generation of adenoviruses. The Ad5/[green fluorescent protein (GFP), human (h) APP695sw, hAPP-C99, hPPARγ, hPPARα, hPPARδ, human retinoic X receptor (hRXR), mNotch ΔE-myc, and notch intracellular domain (NICD)] recombinant adenoviruses were generated by Galapagos Genomics NV as described previously (Michiels et al., 2002).
Drugs and peptides used in culture. Rosiglitazone (stock solution prepared in ethanol), pioglitazone (solution in DMSO), and the PPARγ antagonist GW9662 (solution in DMSO) were obtained from Alexis Biochemicals. Troglitazone (in DMSO) and retinoic acid (solution in DMSO) were obtained from Biomol Research Laboratories (Plymouth Meeting, PA). Aβ1-40 synthetic peptide was purchased from Sigma (St. Louis, MO).
Antibodies. The antibodies used for Aβ detection were mouse monoclonal WO2 (epitope Aβ4-10; Abeta, Heidelberg, Germany), rabbit polyclonal B7/8 (Aβ peptide), generated as described previously (De Strooper et al., 1995), and mouse monoclonal 4G8 (epitope Aβ17-24; Senetec PLC). For APPsα detection, we used the mouse monoclonal 6E10 antibody (Aβ1-16) obtained from Senetec PLC. For APPsβsw detection, we used the rabbit polyclonal 54 antibody obtained from Mary Savage (Cephalon, West Chester, PA). Full-length APP, β-APP-CTF, and α-APP-CTF were detected using the rabbit polyclonal B63.1 antibody generated against the last 15 amino acids of APP as described (C. Esselens, B. De Strooper, and W. Annaert, unpublished observations). Mouse monoclonal anti-Myc (9E10) antibody was purchased from Sanver Tech (Turnhout, Belgium). Rabbit polyclonal anti-cleaved Notch (val 1744) was obtained from Cell Signal/Westburg. The rabbit polyclonal antibodies anti-hPPARγ (H-100), anti-hPPARδ (H-74), and anti-hRXRα (D-20) were obtained from Santa Cruz Biotechnology (Santa Cruz, CA). Mouse monoclonal anti-β-actin antibody (A5441) was obtained from Sigma, and polyclonal anti-SP1 antibody (5C059) was obtained from Santa Cruz Biotechnology.
Cell culture. Primary murine-mixed glial and cortical neuronal cultures were established from brains of embryonic day 14 or 17 fetal mice as described previously (Cai et al., 2001). Briefly, the dissected brain cortices were suspended in HBSS supplemented with 0.25% trypsin and 0.01% DNaseI and incubated at 37°C for 10 min. The tissues were then transferred to DMEM (Invitrogen, San Diego, CA) supplemented with 10% (v/v) fetal bovine serum (FBS) and dissociated by repeated trituration. The dispersed cells were collected by centrifugation and plated at 75 × 104 cells/well on 6-well cell culture plates (coated with poly-d-lysine) in B27/Neurobasal media (Invitrogen). For neuronal cultures, 24 hr after plating, cells were treated with cytosine arabinoside (3 × 10-7 m) to prevent non-neuronal (glia) cell proliferation and used 48 hr later. Glia cells were allowed to grow for 7 d before starting the experiments. Cells were infected with recombinant adenovirus with doses of virus defined as 1000, 2000, and 3000 multiplicities of infection (MOI) for 24 hr maximum and kept in culture for another 40-42 hr. Neuronal cultures were treated after APPsw infection with TZD drugs for 9 hr.
Human embryonic kidney 293 (HEK293) APPsw (Swedish mutation) cells, kindly provided by Christian Haass (Adolf Butenandt Institute, Ludwig-Maximilians University, Munich, Germany), and HEK293 cells were kept in culture using DMEM with 10% FBS. For viral infection, cells were seeded at 500,000 cells/6-well plate or 112,000 cells/24-well plate and cultured at 37°C in the presence of 5% CO2. After 24 hr of incubation, cultures (60% confluence) were infected with recombinant adenovirus with 1, 3, 10, 30, or 100 MOI. After 48 hr, medium was discarded, and a minimum volume of DMEM supplemented with 1% FBS was added to the cells for different time periods. When TZD drugs were used, cells were treated for 16 hr, and then samples of conditioned medium were analyzed for Aβ detection. IMR-32 cells were grown in culture in Eagle's minimum essential medium (Invitrogen) supplemented with 1 mm sodium pyruvate, 0.1 mm nonessential amino acids, 1.5 gm/l sodium bicarbonate, and 10% FBS. Cells were differentiated toward a more neuronal phenotype by incubating the cells for 7 d in differentiation medium [medium as described above with 1 mm Bt2cAMP and 2.5 μm bromodeoxyuridine (Sigma)] before infection for 48 hr using 1000 MOI recombinant adenovirus.
Radioactive labeling. IMR-32 differentiated cells and primary murine cortical cultures were labeled with 100 μCi/ml 35S-methionine for 5-7 hr. After labeling, Aβ was immunoprecipitated from the media with 4G8 or B7/8 antibodies, and APP was immunoprecipitated from cell extracts prepared in double-immune precipitation buffer with B63.1 antibody. Samples were resolved in SDS-PAGE gel and analyzed by phosphor-imaging.
Gel electrophoresis and immunoblotting. For Notch ΔEmyc, NICD, APPsw full-length, β-APP-CTF, and α-APP-CTF detection, cells were lysed in 1% Triton, and postnuclear fractions were isolated by centrifugation at 10,000 × g at 4°C for 15 min. Protein concentrations were determined by the Bradford assay (1976). Proteins were resolved in 10% Bis-Tris SDS-PAGE gels (Invitrogen) and transferred to polyvinylidene difluoride (PVDF) membranes for Western blot detection with c-Myc, anti-NICD (val 1744), or B63 antibodies. For Aβ intracellular detection, cells were lysed in 200 μl of ice-cold radioimmunoprecipitation buffer (0.1% SDS, 0.5% natrium deoxycholate, 1% NP-40, and 5 mm EDTA in TBS, pH 8.0), and postnuclear fractions were isolated by centrifugation at 10,000 × g at 4°C for 10 min. All material was used for Aβ immunoprecipitation using B7/8 antibody. Immunoprecipitated samples were loaded in 12% Bis-Tris SDS-PAGE gels (Invitrogen) and transferred to PVDF membranes for immunoblotting using WO2 antibody. Nuclear cell extracts were prepared as described previously (Andrews and Faller, 1991), and protein concentrations were determined by the Bradford assay (1976). Samples were loaded on 7% Tris-acetate gels (Invitrogen) and transferred to PVDF membranes for immunoblot detection using specific antibodies. Samples of conditioned medium were resolved in 12% Bis-Tris for secreted Aβ detection, or in 4-12% Bis-Tris for APPsα and APPsβ detection, and transferred to PDVF membranes. Immunoblot analysis was performed using WO2, 6E10, or 54 antibodies, respectively. Detection of signal was performed using the chemiluminescence kit (PerkinElmer Life Sciences, Emeryville, CA).
γ-Secretase cell-free assay. HEK293 APPsw cells were transduced for 48 hr. Sixteen hours after infection, cells were harvested. Membranes were prepared as described previously (Nyabi et al., 2003). Briefly, cells were homogenized with a ball-bearing cell cracker (10 μm), and postnuclear fractions were isolated by centrifugation (800 × g for 10 min). Microsomal fractions were isolated by ultracentrifugation (100,000 × g for 1 hr) and washed twice with 0.02% saponin. The pellet was solubilized in 0.5% 3-[(3-cholamidopropyl)dimethylammonio]-1-propanesulfonate choline acetyltransferase buffer, pH 7, for 1 hr at 4°C, and extracts were cleared (100,000 × g for 1 hr). Solubilized membranes (5 μg) were incubated without or with substrate (APP C100-flag) for 16 hr at 37°C.
Aβ stability assay. HEK293 cells were infected for 48 hr, followed by 16 hr in DMEM and 1% FBS before adding the Aβ1-40 synthetic peptide for different time points. Conditioned medium was collected and subjected to immunoblotting analysis for Aβ stability.
Statistical analysis. Values were expressed as means ± SEM.
Results
hPPARγ expression reduces steady-state levels of Aβ peptide in the culture medium of HEK293 cells
HEK293 cells stably transfected with APPsw (Citron et al., 1992; Lannfelt et al., 1993; Rossor et al., 1993) were transduced in duplicate with recombinant adenovirus driving GFP (control), hAPPsw, or hPPARγ expression. Aβ levels in the medium were assessed by Western blot after 16 hr (Fig. 1A, lanes 1-6). High levels of hPPARγ expression resulted in a remarkable reduction of Aβ in the culture medium. In contrast, additional expression of hAPPsw caused, as expected, an increase in Aβ production (Fig. 1A).
Figure 1.
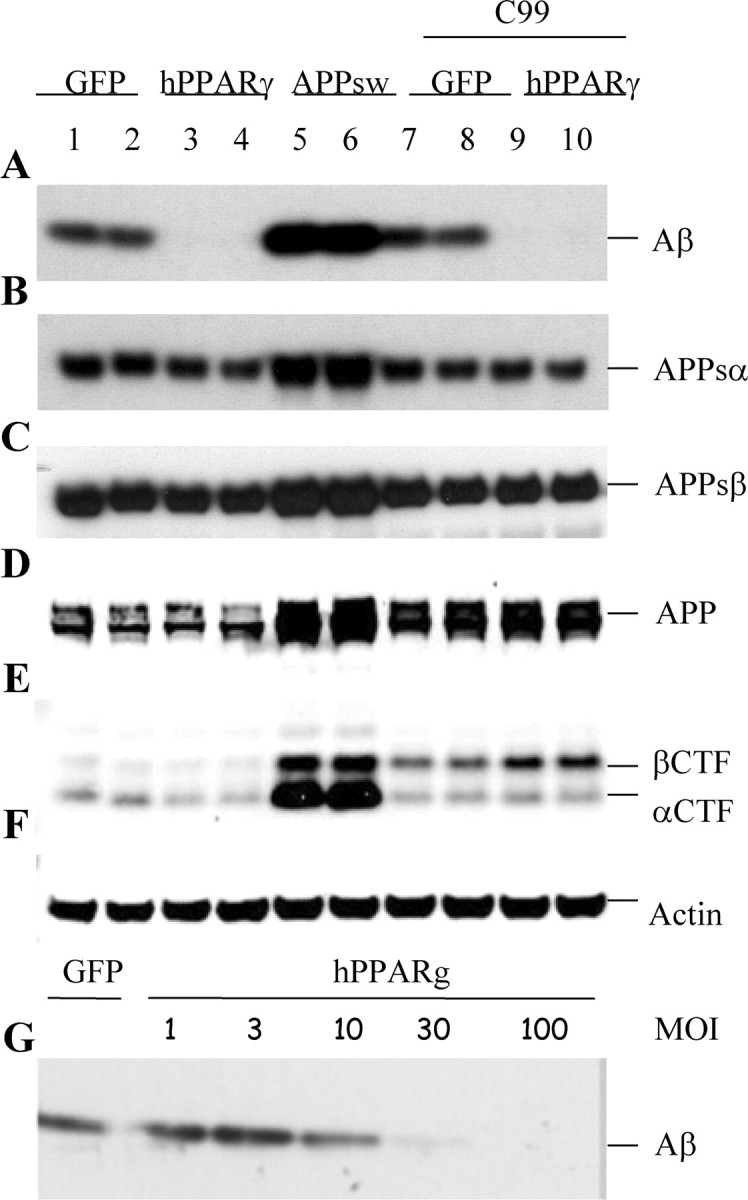
Analysis of the processing of APP in hPPARγ-overexpressing cells. HEK293 APPsw cells were transduced with recombinant adenovirus (MOI 100) driving expression of GFP (lanes 1, 2), hPPARγ (lanes 3, 4), hAPP695sw (lanes 5, 6), or APP-C99 together with GFP (lanes 7, 8) or hPPARγ (lanes 9, 10). Cells and conditioned medium were collected 16 hr after infection. A, Western blot detection of secreted Aβ with WO2 antibody. B, Western blot detection of APPsα with 6E10 antibody. C, Western blot detection of APPsβsw with 54 antibody. D, E, Full-length APP, β-APP-CTF, and α-APP-CTF were detected by Western blot using B63 antibody. F, Levels of endogenous β-actin are shown as a loading control for the cell extracts. G, Cells were transduced with adenovirus driving expression of either GFP or hPPARγ with different MOI (1, 3, 10, 30, and 100). Aβ levels were detected with WO2 antibody.
Both Aβ40 and Aβ42 levels were decreased to a similar extent by hPPARγ expression as assessed by a sandwich ELISA (data not shown). Furthermore, we could demonstrate an inverse correlation between the levels of Aβ present in the medium and the levels of hPPARγ expression in the cell (Fig. 1G).
hPPARγ expression does not modulate expression nor activity of the α- and β- secretases
APP is cleaved either by the α- or the β-secretase, resulting in the release of the APP ectodomain (APPsα or APPsβ, respectively). The respective APP C-terminal fragments remaining in the cell membrane (α-APP-CTF and β-APP-CTF) become substrates for the γ-secretase complex (see below) (De Strooper et al., 1998). It is possible that PPARγ expression modulates the activity of these secretases because increased α-secretase or decreased β-secretase could indeed result in decreased Aβ generation. Using specific antibodies, we found comparable levels of APPsα and APPsβ in the medium (Fig. 1B,C, lanes 1-6). In addition, the membrane-bound α-APP-CTF and β-APP-CTF were not significantly affected by PPARγ expression (Fig. 1D, lanes 1-6). To further confirm that the effect of PPARγ on Aβ levels was not mediated by an effect on the direct precursor of the peptide, β-APP-CTF, we expressed a synthetic APP-C99 construct together with hPPARγ or GFP and tested again the levels of Aβ in culture (Fig. 1A-F, lanes 7-10). High levels of PPARγ again caused a down-regulation of Aβ despite the high expression of APP-C99 (Fig. 1E, lanes 7-10). We further confirmed by Western blotting that levels of expression of the aspartic protease BACE1 or β-secretase (Hussain et al., 1999; Sinha et al., 1999; Vassar et al., 1999; R. Yan et al., 1999), and of the candidate α-secretases ADAMs 9, 10, and 17 (Pan and Rubin, 1997; Koike et al., 1999; Lammich et al., 1999) were not affected by hPPARγ expression (data not shown). We conclude, therefore, that neither the expression nor the activity of α- or β-secretase nor the stability of the APP-C99 fragment were affected in hPPARγ-expressing cells.
γ-Secretase activity is not affected by PPARγ activation
The α-APP-CTF and β-APP-CTF, products from the α- and the β-secretase activities, are further cleaved by presenilin/γ-secretase to generate the secreted p3 and Aβ peptides, respectively, and the cytoplasmic APP intracellular domain (AICD). The fact that APP-CTFs did not accumulate in the hPPARγ-transduced cells already suggests normal activity of the γ-secretase complex (Fig. 1E). However, given the evidence in the literature that NSAIDs can modulate γ-secretase (Weggen et al., 2001; Eriksen et al., 2003) and knowing that they are also agonists of PPARγ (Lehmann et al., 1995), we checked the possibility of PPARγ regulating γ-secretase activity in more detail.
We first evaluated whether the generation of the AICD was affected by PPARγ expression. We used a reporter assay based on a C99-Gal4 BD-VP16 AD construct (Karlstrom et al., 2002) that contains the transmembrane domain of APP fused via its AICD domain to the Gal4-binding domain and to the VP16 activation domain. After cleavage, this domain is released from the cell membrane and translocates to the nucleus, where it binds to the Gal4 upstream activator sequence, activating the expression of a luciferase reporter gene. No differences were observed in cells transduced with either control GFP vector or with the hPPARγ vector (data not shown), indicating that AICD production was not altered by hPPARγ expression at least within the limits of detection of this assay. Next we used a cell-free assay to measure directly the generation of Aβ (Nyabi et al., 2003) (Fig. 2A). Membranes purified from GFP control (Fig. 2A, lanes 1, 2) and hPPARγ-transduced cells (Fig. 2A, lanes 3, 4) were incubated either without (Fig. 2A, lanes 1, 3) or with a flag-tagged substrate for γ-secretase, representing the C-terminal 100 amino acids from APP (APP C100-flag) (Fig. 2A, lanes 2, 4) for 16 hr at 37°C. Aβ production was not altered by hPPARγ (Fig. 2A, lanes 2, 4). The substrate is also incubated alone (Fig. 2A, lane 5), showing no degradation. This result implies that downregulation of the Aβ levels observed in hPPARγ-expressing cells is not attributable to a total or partial inactivation of the γ-secretase complex.
Figure 2.

Analysis ofγ-secretase cleavage of APP and of Notch. A, Membranes from HEK293 APPsw cells expressing GFP (lanes 1, 2) or hPPARγ (lanes 3, 4) were purified and incubated without (lanes 1, 3) or with (lanes 2, 4) APP C100-flag substrate at 37°C for 16 hr. Aβ was detected with WO2 antibody. hPPARγ does not affect Aβ generation in this assay. B-D, HEK293 APPsw cells were coinfected with mNotchΔE-myc and either GFP (lanes 2, 3) or hPPARγ adenovirus (lanes 4, 5). Cells coinfected with NICD and GFP (lane 6) are shown as a positive control for the detection of NICD. Cells only infected with GFP (lane 1) are used as a negative control. Cells were treated for 4 hr with lactacystin (10 μm) to avoid proteasome degradation of NICD. B, Detection of Notch ΔE-myc and NICD using an anti-myc antibody. Levels of the substrate NotchΔE and NICD were comparable in GFP- and hPPARγ-transduced cells, showing a normal processing by γ-secretase. C, Detection of NICD with antibody (val 1744) specific for the cleaved form of Notch. D, Conditioned medium was collected, and Aβ levels were detected with WO2 antibody to confirm the activity of PPARγ in this experiment.
Notch processing is not affected by hPPARγ expression
An important substrate of the γ-secretase complex is the Notch receptor. After Notch cleavage, NICD is released and translocates to the nucleus, where it activates transcription of genes involved in cell-fate decisions (Jarriault et al., 1995). To investigate in a more direct way the effect of PPARγ on γ-secretase activity, we transduced HEK293 APPsw cells with mNotchΔE-myc recombinant adenovirus in combination with either hPPARγ (Fig. 2B-D, lanes 4, 5) or GFP (Fig. 2B-D, lanes 2, 3). mNotchΔE-myc is a truncated Notch protein lacking most of the ectodomain that undergoes proteolytic cleavage by the γ-secretase complex, releasing NICD (Kopan et al., 1996; De Strooper et al., 1999). HEK293 APPsw cells infected with GFP alone (Fig. 2B-D, lane 1) or together with NICD (Fig. 2B-D, lane 6) were used as controls. Similar levels of mNotchΔE-myc (the substrate) and NICD (the cleavage product) were detected with the anti-myc antibody (Fig. 2B). Using an antibody against cleaved Notch (Val 1744) that recognizes a neo-epitope in NICD after γ-secretase cleavage (Fig. 2C), we confirmed that similar levels of NICD were generated in GFP- or hPPARγ-expressing cells. We noticed the presence of two NICD-like bands (Fig. 2B,C). Because the two bands were detected by both the anti-NICD antibody specific for the γ-secretase cleavage site at the N terminus and by anti-myc, recognizing the epitope at the C terminus of NICD, both bands must represent full-length NICD. Therefore, we suggest that they correspond to different posttranslational modifications of NICD in HEK293 cells.
To prove that hPPARγ was active in the transduced cells, we also checked the levels of secreted Aβ in the conditioned medium. Confirming the results displayed in Figure 1, cells overexpressing hPPARγ showed strongly reduced levels of the Aβ peptide (Fig. 2D). Thus, γ-secretase cleavage of mNotchΔE-myc is not affected by hPPARγ overexpression.
We finally assessed by Western blotting the expression levels of all the γ-secretase components described in the literature thus far (i.e., presenilin, nicastrin, Pen 2, and Aph 1a) (De Strooper et al., 1998; Yu et al., 2000; Francis et al., 2002; Goutte et al., 2002; De Strooper, 2003). No changes were observed in hPPARγ-transduced compared with GFP-transduced cells (data not shown).
We conclude that neither γ-secretase activity on APP or Notch nor the expression of the known components constituting γ-secretase complex are affected by hPPARγ expression.
hPPARγ does not cause accumulation of intracellular Aβ-peptide
We next investigated whether the decreased levels of secreted Aβ could be explained by a deficit in Aβ secretion. We transduced HEK293 APPsw cells with recombinant GFP or hPPARγ adenovirus and analyzed postnuclear extracts measuring intracellular Aβ levels at four different postinfection time points by Western blot (Fig. 3B). Aβ in GFP-transduced cells tends to slightly increase over time, whereas Aβ is downregulated in hPPARγ-overexpressing cells at 3 hr after infection, and levels remain low and even tend to slightly decrease over time. Levels of secreted Aβ were also measured in this experiment at the same four postinfection time points (Fig. 3A), confirming our previous results. Thus, analogous with the secreted Aβ, the levels of intracellular Aβ are also decreased after hPPARγ overexpression, ruling out the possibility of intracellular Aβ accumulation.
Figure 3.

Aβ intracellular accumulation is decreased after overexpression of hPPARγ. HEK293 APPsw cells were transduced with GFP or hPPARγ recombinant adenovirus. At four different postinfection time points, conditioned medium was collected and cells were lysed in 1% Triton buffer. A, Detection of Aβ and APPsα was performed using WO2 antibody. B, Total cell extract was subjected to immunoprecipitation with B7/8 antibody and immunoblotted with WO2 for intracellular Aβ detection. Both secreted and intracellular Aβ were downregulated in hPPARγ cells.
Downregulation of Aβ is not mediated by a secreted Aβ-degrading enzyme
The steady-state levels of Aβ are the result of a dynamic equilibrium between production and degradation of the peptide. Because production of Aβ does not seem to be affected, we further investigated whether the turnover of Aβ was increased in hPPARγ-expressing cells. We first tested whether a secreted Aβ-degrading enzyme could be involved in Aβ turnover. In a cell-free in vitro assay, conditioned medium from GFP-transduced HEK293 APPsw cells that contain high levels of Aβ was incubated in a 50:50 ratio with fresh medium (Fig. 4A, lane 1) or with medium from hPPARγ-transduced HEK293 APPsw cells (Fig. 4A, lane 2) at 37°C for 2 hr. The Aβ peptide was not degraded faster after incubation with medium from hPPARγ-overexpressing cells. Longer incubation up to 16 hr also did not result in any measurable effect on Aβ (data not shown). There was no Aβ detectable in medium from hPPARγ-transduced cells (Fig. 4A, lane 3) or in medium alone (Fig. 4A, lane 4), as expected. These results rule out basically that a secreted protease is responsible for the observed Aβ degradation. However, it could be argued, as a limitation of the in vitro assay, that a potential soluble protease may need cell-bound factors to be active. To test this possibility, we added conditioned medium from hPPARγ-transduced cells to GFP-transduced HEK293 control cells coinfected with APPsw adenovirus (Fig. 4B, lanes 1-4). As controls, we added medium from GFP-transduced HEK293 cells (Fig. 4B, lanes 5-8) or unconditioned fresh medium (Fig. 4B, lanes 9-12). The level of Aβ in the conditioned medium was not different at any of the four time points investigated (Fig. 4B). Together, these results show that the effect of hPPARγ on Aβ levels is not mediated by an Aβ-degrading enzyme secreted to the conditioned medium.
Figure 4.

Levels of secreted Aβ from GFP-transduced cells are not changed after incubation with conditioned medium from hPPARγ cells. A, HEK293 APP695sw cells were infected with GFP or hPPARγ adenovirus. Sixteen hours after infection, medium was collected. Medium from GFP-transduced cells was incubated with fresh medium (lane 1) or with hPPARγ medium (lane 2) at 37°C for 2 hr. Aβ stability was detected by immunoprecipitation (B7/8 antibody) and immunoblot analysis (WO2 antibody). As additional controls, we also included 1% FBS medium (lane 4) and medium from hPPARγ-transduced cells (lane 3) with no Aβ detectable, as expected. B, HEK293 cells were cotransduced with hAPP695sw and GFP adenovirus. After 48 hr of infection, cells were incubated with medium taken 16 hr after infection from hPPARγ-transduced HEK293 cells (lanes 1-4), medium taken 16 hr after infection from GFP-transduced HEK293 cells (lanes 5-8), or with 1% FBS DMEM fresh medium (lanes 9-12) for the times indicated. Samples from media were subjected to immunoblot analysis with WO2 antibody to detect APPsα and Aβ. HEK293 nontransduced with hAPP695sw did not have detectable levels of Aβ in the medium (lanes 1, 5). There were no changes in Aβ stability after incubation of GFP cells with the medium from hPPARγ cells.
Next we investigated the effect on secreted and intracellular Aβ levels in PPARγ-transduced HEK293 APPsw cells by the addition of a proteasome inhibitor (10 μm lactacystin), a proton pump ATPase inhibitor (0.1 μm bafilomycin), or an alkalinization agent (30 mm NH4Cl) (data not shown). None of the mentioned treatments rescued the levels of secreted or intracellular Aβ to control levels (GFP-transduced HEK293 APPsw cells). Therefore, we conclude that PPARγ effect on Aβ levels is not mediated by proteasome degradation or by a pH-sensitive protease (such as lysosomal degradation).
Clearance of the Aβ40 peptide is increased after hPPARγ expression
We next investigated the possibility that hPPARγ expression results in an increased cell-dependent Aβ clearance (and rapid degradation). For that purpose, we added exogenously synthetic Aβ40 peptide to the conditioned medium of HEK293 cells and incubated the cells over a 9 hr period (Fig. 5A). In the hPPARγ-expressing cells, a decline in Aβ40 synthetic peptide is already observed at 7 and 20 min, with very little remaining Aβ at 3 hr. In GFP-transduced cells, the levels of Aβ40 peptide remain stable. No increase in Aβ was detected in extracts from hPPARγ-expressing cells, implying that the peptide was effectively and rapidly degraded once it was taken up by the cells (data not shown). These results show that the stability of the Aβ peptide in the medium is specifically affected in cells with increased levels of expression of the hPPARγ.
Figure 5.

Aβ40 peptide is rapidly cleared from medium of cells expressing hPPARγ. HEK293 cells were infected with either GFP or hPPARγ adenovirus. Aβ40 synthetic peptide was added to the medium 16 hr after infection. Conditioned medium was collected at 7 min, 20 min, 1 hr, 3 hr, or 9 hr, and Aβ was detected by Western blot with WO2 antibody. Endogenous levels of secreted Aβ are not detectable in this assay (Fig. 4 B, lanes 1, 5). The levels of the Aβ in the medium decrease gradually with time in hPPARγ-overexpressing cells. B, Logarithmic representation of two experiments. The medium value of Aβ from all GFP-transduced cells is set as 100% and considered as an initial amount. All values refer to the initial amount.
Aβ peptide downregulation is specific for the PPARγ isoform of the PPAR family
We next checked whether other members of the PPAR family had similar effects on Aβ levels (Willson et al., 2000). In addition, we also questioned whether increased expression of the hRXR, known to form the active heterodimer transcription factors in the PPAR family (Miyata et al., 1994; Blumberg and Evans, 1998), could also affect the levels of Aβ present in culture. For this purpose, HEK293 APPsw cells were transduced with GFP control, hPPARγ-, hPPARδ-, hPPARα-, or hRXR-expressing recombinant adenoviruses. We controlled the expression of the different constructs by immunoblotting the nuclear fractions of the transduced cells with specific antibodies (Fig. 6B-D). No antibody is available to detect expression of the hPPARα gene in Western blotting. Therefore, we had to assume that hPPARα was expressed to a similar extent as its homologs in this experiment. Regardless, as shown in Figure 6A, levels of Aβ secreted into the medium were not significantly altered by any of the other hPPAR or hRXR transcription factors. Thus, we conclude that the effect of hPPARγ on Aβ degradation is specific.
Figure 6.
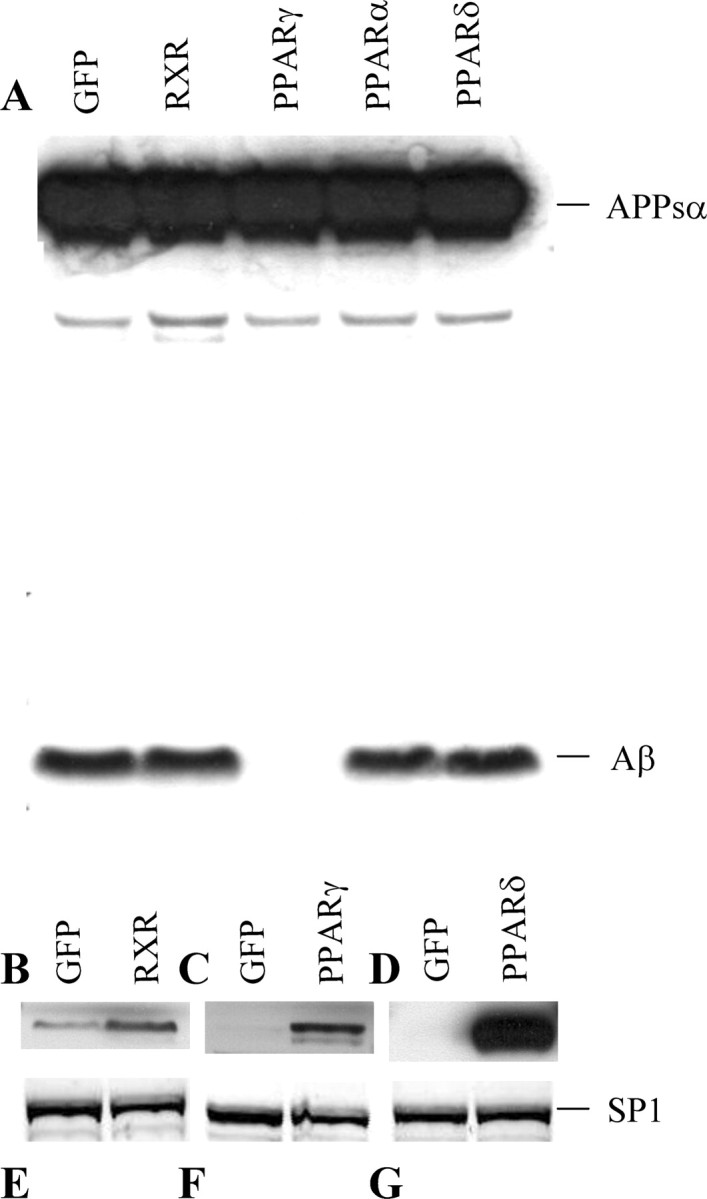
Downregulation of Aβ levels is a specific effect of the hPPARγ isoform of the PPAR family. HEK293 APPsw cells were infected either with GFP, hPPARγ, hPPARα, hPPARδ, or hRXR recombinant adenoviruses. A, Samples from medium were analyzed by Western blot, and APPsα and Aβ were detected with WO2 antibody. B-D, Levels of expression of hRXR, hPPARγ, and hPPARδ were detected by Western blot from nuclear extracts using specific antibodies. E-G, Levels of expression of the SP1 transcription factor are detected as a loading control. The results show that Aβ is downregulated specifically after hPPARγ overexpression.
Similar levels of Aβ downregulation are obtained through activation of endogenous hPPARγ
Obviously, both from a cell biological and a pharmacological point of view, it is important to demonstrate that a similar effect on Aβ could be obtained by activating endogenously expressed PPARγ. We therefore treated cells in culture with several synthetic PPARγ ligands from the TZD drug family (Berger et al., 1996; Willson et al., 2000) (rosiglitazone, troglitazone, and pioglitazone) or with 9-cis-retinoic acid (9-cis-RA), an agonist of the RXR (Mangelsdorf et al., 1992). TZD drugs are direct agonists of the PPARγ transcription factor, whereas 9-cis-RA activates the heterodimeric complex formed by PPAR-RXR (Mukherjee et al., 1997). We incubated HEK293 APPsw cells with increasing concentrations of troglitazone or pioglitazone (1, 3, 10, and 30 μm), rosiglitazone (30 μm), or 9-cis-RA (30 μm). As a control for solvent interactions, we treated the cells also with DMSO or ethanol alone. Levels of Aβ and APPsα were assessed by Western blot (Fig. 7A-D). As shown in Figure 7A-D, Aβ is decreased after treatment with troglitazone (1, 3, 10, and 30 μm), pioglitazone (1, 3, 10, and 30 μm), 9-cis-RA (30 μm), and rosiglitazone (30 μm) in a dose-dependent manner. The levels of APPsα in the conditioned medium are not significantly affected, demonstrating the absence of toxicity of the treatments and equal loading (Fig. 7A-D). Thus, activation of the endogenous levels of PPARγ present in HEK293 APPsw cells, through its specific agonists or through 9-cis-RA, activating the PPARγ-RXR heterodimer, resulted in a notable reduction of the Aβ levels, comparable with that seen with high levels of hPPARγ expression in the cell. To confirm that the TZD effect on Aβ is mediated by PPARγ transcriptional activity, we incubated the TZD PPARγ-activated cells with the PPARγ antagonist GW9662 to reverse the effects (Huang et al., 1999; Willson et al., 2000). As shown in Figure 8, treatment with this antagonist restored Aβ levels to control ones (DMSO treatment), confirming the role of PPARγ activation in Aβ downregulation.
Figure 7.

Downregulation of Aβ is reached through TZD activation of endogenous PPARγ. A-D, HEK293 APPsw cells were transduced with GFP adenovirus and incubated for 16 hr with several TZD drugs or only transduced with hPPARγ adenovirus. Levels of APPsα and Aβ were detected by Western blot (WO2 antibody) from samples of conditioned medium. A, Cells were treated with 1, 3, 10, and 30 μm troglitazone with DMSO (solvent) or transfected with hPPARγ without treatment. B, Cells were treated with 1, 3, 10, and 30 μm pioglitazone with DMSO or transfected with hPPARγ. C, Cells were treated with 30 μm 9-cis-RA with DMSO or transfected with hPPARγ. D, Cells were treated with 30 μm rosiglitazone, ethanol (solvent), or transfected with hPPARγ. A-D, Aβ quantification from two to four independent experiments. Values of Aβ were normalized for APPsα (Aβ/APPsα) and referred to the GFP control value set as 100%.
Figure 8.

The PPARγ antagonist GW9662 effectively blocks the TZD effect on Aβ levels. HEK293 APPsw cells are incubated for 16 hr either with troglitazone (1, 3, and 10 μm) alone or together with GW9662 (1 μm). DMSO (solvent) treatment shows the control levels of Aβ. Levels of APPsα and Aβ were detected by Western blot (WO2 antibody) from samples of conditioned medium.
Aβ is downregulated in a differentiated human neuroblastoma cell line, in primary cultures of murine glia cells, and in murine cortical neurons through an hPPARγ-activated pathway
We next tested whether the effect of PPARγ on Aβ levels could be confirmed in cells and cell lines that are more relevant for AD. First, we used the human neuron-related cell line IMR-32 (Donnelly-Roberts et al., 1998). The cells were differentiated and then transduced with either GFP or hPPARγ adenovirus and metabolically labeled with 35S-methionine for 5 hr. No secreted Aβ was detected in hPPARγ-expressing cells, whereas control cells secreted detectable levels of Aβ (Fig. 9A). It should also be noted that in these experiments APP was expressed at endogenous levels. We next tested whether Aβ could be downregulated in primary cultures of murine-mixed glia cells cotransduced with APPsw together with either GFP or hPPARγ. Levels of Aβ present in the culture medium are clearly downregulated after hPPARγ expression in glia cells (Fig. 9B). Finally, we found in primary cortical neuronal cultures cotransduced with APPsw and hPPARγ and treated with increasing concentrations of troglitazone (3, 10, and 30 μm) a decrease of Aβ levels. Probably reflecting the relative low efficiency of transduction of primary neurons, we obtained an ∼30% reduction of Aβ burden when treated with 30 μm troglitazone (Fig. 9C). These data confirm in three additional and relevant cell lines that an Aβ clearance pathway can be activated by hPPARγ.
Figure 9.
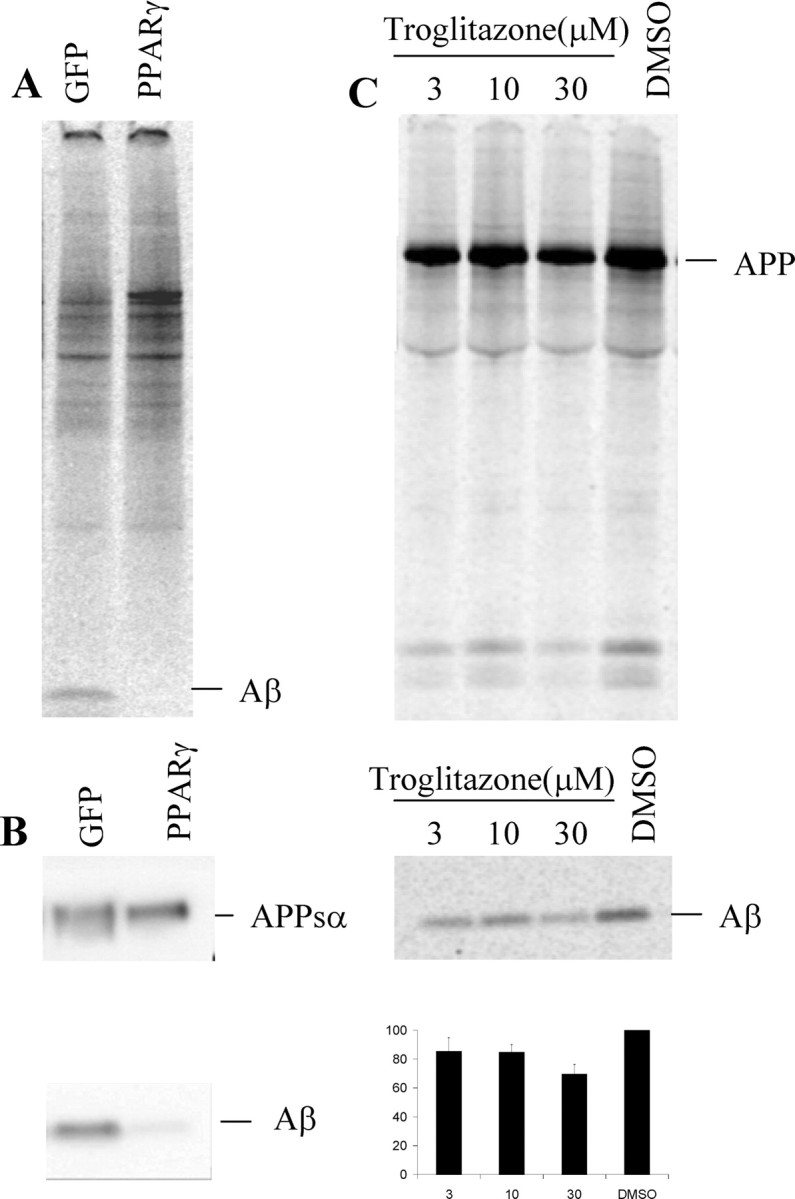
Aβ is downregulated in differentiated human neuroblastoma IMR-32 cells in primary murine-mixed glial cultures and in primary cortical cultures after hPPARγ induction. A, Differentiated IMR-32 cells were infected with either GFP or hPPARγ recombinant adenovirus and metabolically labeled with 35S-methionine for 5 hr. Aβ was immunoprecipitated from medium with 4G8 antibody and analyzed by phosphorimaging. B, Primary murine-mixed glial cultures are cotransduced with APPsw and either GFP or hPPARγ recombinant adenovirus. Secreted Aβ was detected with WO2 antibody. C, Primary cortical cultures are transduced with APPsw together with hPPARγ and treated with increasing concentrations of troglitazone (3, 10, and 30 μm).
Discussion
The work presented here shows a PPARγ-specific effect on the regulation of the Aβ steady-state levels in several cell lines and, more importantly, in primary neuronal and mixed glial cell cultures. The effect is conserved in human and in rodent cells. We convincingly showed that high levels of expression of PPARγ strongly reduce the levels of secreted and intracellular Aβ in a dose-dependent manner. This effect is (1) specific for the PPARγ receptor, because overexpression of any of the other isoforms of the PPAR family did not significantly alter the levels of Aβ; (2) physiologically relevant, because similar downregulation of Aβ is reached viaendogenous PPARγ activation by TZD agonists; (3) mediated by the transcriptional activity of PPARγ, as demonstrated by the reversion of the effect on Aβ using the GW9662 PPARγ antagonist; and (4) independent on levels of expression of APP or on the presence of the Swedish mutation used in some of our assays, as shown by our experiments with differentiated IMR-32 cells expressing endogenous wild-type APP.
Based on this evidence, we conclude that the effect on Aβ is specific, that the effect on Aβ is not an artifact (e.g., caused by the use of adenoviral transduction), that the mechanism involved is present in many cell types (HEK293, IMR-32, primary mixed glia and neuronal cultures), and, finally, that this mechanism can be activated via drug treatment. Our data contrast with two other studies in which either PPARγ expression was induced in Chinese hamster ovary cells (Sagi et al., 2003) or endogenous PPARγ was activated with TZD in HEK293 cells (Q. Yan et al., 2003). No changes in Aβ levels were observed in these two studies. However, our experiments show that the PPARγ effect on Aβ levels is only observed if sufficient time is given to the cells to activate the clearing mechanism and that relatively high levels of PPARγ expression are required. We hypothesize that differences in the levels of expression of PPARγ or other cellular factors could be possible reasons for the discrepancy between our and previous studies. In any event, we observed in a reproducible and consistent manner that Aβ peptides are reduced in culture media by the action of PPARγ. We want to remark that Aβ was detected with at least three different antibodies directed against different Aβ epitopes, indicating that the loss of Aβ signals in our detection system reflects likely complete degradation of the peptide.
Recent data showed that neuroblastoma cells treated with cytokines produced increased levels of Aβ40 and Aβ42 in culture, which could be reversed by the action of TZD or NSAIDs (Sastre et al., 2003). However, our data indicate that the PPARγ-mediated effect on Aβ is independent of inflammation and additionally provides some evidence of a cellular Aβ clearance mechanism involved. In vivo data from an AD mouse model treated with either pioglitazone, one of the TZD drugs that crosses the blood-brain barrier, or with the NSAID ibuprofen showed a reduction in the number and size of the Aβ plaques after ibuprofen but not pioglitazone treatment (Q. Yan et al., 2003). However, when total Aβ levels from brain homogenates were analyzed, a reduction in the soluble and insoluble Aβ40 and Aβ42 species was observed after pioglitazone treatment as well, agreeing with our observations. In addition, we noticed that the pioglitazone treatment in this mouse study did not have any effect on microglia activation, in discordance with previous in vitro (Combs et al., 2000) and in vivo (Dehmer et al., 2004) data. We suggest that higher doses and other treatment regimens are required before final conclusions on the effect of TZD drugs on amyloid pathology in vivo can be made.
In any event, our data provide evidence that PPARγ can induce an Aβ clearing mechanism in a variety of cells. A previous study proposed PPARγ as a regulator of the transcriptional levels of the β-secretase enzyme BACE1 because the mechanism involved in Aβ downregulation (Sastre et al., 2003). Neuroblastoma cells, when treated with cytokines, showed higher levels of the mRNA transcript, the protein, and the activity of BACE1. The effect of cytokines was again reverted by the action of TZD or NSAIDs (Sastre et al., 2003). Although this mechanism could indeed operate in situations in which significant inflammation occurs, our findings in noninflammatory conditions show that high levels of PPARγ downregulate Aβ without detectable changes in the levels of expression of BACE1 (as measured by Western blot) or in its activity as measured by the generation of the APPsβ and β-APP-CTF. This was further confirmed by the overexpression experiment using an APP-C99 construct, mimicking the cleavage of APP by β-secretase. The results showed clearly that neither the generation nor the stability of APP-C99 is affected by hPPARγ. In addition, normal expression and activity of the γ-secretase complex was also detected, suggesting that changes in the generation of Aβ are not involved in the effects observed in our studies. Moreover, we demonstrated that the cleavage of an artificial Notch substrate or the generation of AICD, a possible important γ-secretase product of APP, is affected by PPARγ. Therefore, we conclude that neither β- or γ-secretase cleavage of APP is changed by PPARγ expression.
We therefore postulate that the mechanism of action of PPARγ is involved with an increase in the rate of Aβ turnover. We confirmed this hypothesis by experiments showing a rapid clearance of Aβ40 synthetic peptide from the culture medium of PPARγ-overexpressing cells. The fact that no increase in the signal of intracellular Aβ was detected in the cell extracts strongly suggests that the Aβ peptide is indeed effectively degraded. Interestingly, the degradation of Aβ seems to depend on the presence of cell-bound factors because medium extracted from PPARγ-overexpressing cells was not able to degrade Aβ peptide on its own. Besides, we could not find evidence for an involvement of the proteasome or lysosomal systems in this Aβ-degradation mechanism, or at least compounds known to block these systems did not affect Aβ clearance in our hands. We hypothesize that PPARγ mediates an activation of a cell-dependent clearance mechanism probably by transcriptional activation of one or several proteins involved in the uptake from the medium and intracellular degradation of the peptide. We want to stress that we believe that this mechanism is physiologically relevant because we could demonstrate its activity not only in HEK293 cells but also in differentiated neuroblastoma IMR-32 cells and in primary murine neuronal and mixed glial cultures (the latter consists mainly of astroglia cells but contains also a small population of microglia and oligodendroglia). Perhaps this clearance mechanism becomes activated in the brain in some pathological situations in which high levels of PPARγ are present. Indeed, high levels of PPARγ have been detected in brains from AD patients (Kitamura et al., 1999). Importantly, we showed that when we simulated such conditions in culture by expressing PPARγ in primary murine cortical neuronal cultures, treatment with TZD compounds indeed resulted in a downregulation of Aβ levels. This not only demonstrates that the PPARγ-regulated Aβ clearance mechanism is present in cells of the central nervous system but also strongly suggests that additional work in vivo is needed to fully explore the potential of TZD compounds in the treatment of AD.
Footnotes
This work was supported by a Pioneer award from the Alzheimer's Association (B.D.S.); the Fund for Scientific Research, Flanders; the Katholieke Universiteit Leuven (Geconcerteerde onderzoeksactie); the European Union (Abnormal proteins in the pathogenesis of neurodegenerative disorders); and the Federal Office for Scientific Affairs, Belgium (IUAP P5/19). We thank Christian Haass for the generous gift of the human embryonic kidney cells stably transduced with amyloid precursor protein (Swedish mutation) (APPsw); Mary Savage (Cephalon) for the 54 antibody specifically recognizingβ-secretase-processed APPsw; Katrien Horre, Kathleen Craessaerts, and Siska Deforce for the preparation of the primary glia and cortical cultures; and Simon Reeve for helpful criticism.
Correspondence should be addressed to Dr. Bart De Strooper, Neuronal Cell Biology and Gene Transfer Laboratory, Center for Human Genetics, Vlaams Interuniversitair Instituut voor Biotechnologie and Katholieke Universiteit Leuven, Herestraat 49, 3000 Leuven, Belgium. E-mail: Bart.destrooper@med.kuleuven.Ac.be.
Copyright © 2004 Society for Neuroscience 0270-6474/04/2410908-10$15.00/0
References
- Andrews NC, Faller DV (1991) A rapid micropreparation technique for extraction of DNA-binding proteins from limiting numbers of mammalian cells. Nucleic Acids Res 19: 2499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berger J, Bailey P, Biswas C, Cullinan CA, Doebber TW, Hayes NS, Saperstein R, Smith RG, Leibowitz MD (1996) Thiazolidinediones produce a conformational change in peroxisomal proliferator-activated receptor-gamma: binding and activation correlate with antidiabetic actions in db/db mice. Endocrinology 137: 4189-4195. [DOI] [PubMed] [Google Scholar]
- Blumberg B, Evans RM (1998) Orphan nuclear receptors-new ligands and new possibilities. Genes Dev 12: 3149-3155. [DOI] [PubMed] [Google Scholar]
- Cai H, Wang Y, McCarthy D, Wen H, Borchelt DR, Price DL, Wong PC (2001) BACE1 is the major beta-secretase for generation of Abeta peptides by neurons. Nat Neurosci 4: 233-234. [DOI] [PubMed] [Google Scholar]
- Chawla A, Schwarz EJ, Dimaculangan DD, Lazar MA (1994) Peroxisome proliferator-activated receptor (PPAR) gamma: adipose-predominant expression and induction early in adipocyte differentiation. Endocrinology 135: 798-800. [DOI] [PubMed] [Google Scholar]
- Citron M, Oltersdorf T, Haass C, McConlogue L, Hung AY, Seubert P, Vigo-Pelfrey C, Lieberburg I, Selkoe DJ (1992) Mutation of the beta-amyloid precursor protein in familial Alzheimer's disease increases beta-protein production. Nature 360: 672-674. [DOI] [PubMed] [Google Scholar]
- Colville-Nash PR, Qureshi SS, Willis D, Willoughby DA (1998) Inhibition of inducible nitric oxide synthase by peroxisome proliferator-activated receptor agonists: correlation with induction of heme oxygenase 1. J Immunol 161: 978-984. [PubMed] [Google Scholar]
- Combs CK, Johnson DE, Karlo JC, Cannady SB, Landreth GE (2000) Inflammatory mechanisms in Alzheimer's disease: inhibition of β-amyloid-stimulated proinflammatory responses and neurotoxicity by PPARγ agonists. J Neurosci 20: 558-567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dehmer T, Heneka MT, Sastre M, Dichgans J, Schulz JB (2004) Protection by pioglitazone in the MPTP model of Parkinson's disease correlates with I kappa B alpha induction and block of NF kappa B and iNOS activation. J Neurochem 88: 494-501. [DOI] [PubMed] [Google Scholar]
- De Strooper B (2003) Aph-1, Pen-2, and Nicastrin with Presenilin generate an active gamma-secretase complex. Neuron 38: 9-12. [DOI] [PubMed] [Google Scholar]
- De Strooper B, Simons M, Multhaup G, Van Leuven F, Beyreuther K, Dotti CG (1995) Production of intracellular amyloid-containing fragments in hippocampal neurons expressing human amyloid precursor protein and protection against amyloidogenesis by subtle amino acid substitutions in the rodent sequence. EMBO J 14: 4932-4938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Strooper B, Saftig P, Craessaerts K, Vanderstichele H, Guhde G, Annaert W, Von Figura K, Van Leuven F (1998) Deficiency of presenilin-1 inhibits the normal cleavage of amyloid precursor protein. Nature 391: 387-390. [DOI] [PubMed] [Google Scholar]
- De Strooper B, Annaert W, Cupers P, Saftig P, Craessaerts K, Mumm JS, Schroeter EH, Schrijvers V, Wolfe MS, Ray WJ, Goate A, Kopan R (1999) A presenilin-1-dependent gamma-secretase-like protease mediates release of Notch intracellular domain. Nature 398: 518-522. [DOI] [PubMed] [Google Scholar]
- Donnelly-Roberts DL, Puttfarcken PS, Kuntzweiler TA, Briggs CA, Anderson DJ, Campbell JE, Piattoni-Kaplan M, McKenna DG, Wasicak JT, Holladay MW, Williams M, Arneric SP (1998) ABT-594 [(R)-5-(2-azetidinylmethoxy)-2-chloropyridine]: a novel, orally effective analgesic acting via neuronal nicotinic acetylcholine receptors: I. In vitro characterization. J Pharmacol Exp Ther 285: 777-786. [PubMed] [Google Scholar]
- Eriksen JL, Sagi SA, Smith TE, Weggen S, Das P, McLendon DC, Ozols VV, Jessing KW, Zavitz KH, Koo EH, Golde TE (2003) NSAIDs and enantiomers of flurbiprofen target gamma-secretase and lower Abeta 42 in vivo. J Clin Invest 112: 440-449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Francis R, McGrath G, Zhang J, Ruddy DA, Sym M, Apfeld J, Nicoll M, Maxwell M, Hai B, Ellis MC, Parks AL, Xu W, Li J, Gurney M, Myers RL, Himes CS, Hiebsch R, Ruble C, Nye JS, Curtis D (2002) aph-1 and pen-2 are required for Notch pathway signaling, gamma-secretase cleavage of betaAPP, and presenilin protein accumulation. Dev Cell 3: 85-97. [DOI] [PubMed] [Google Scholar]
- Goutte C, Tsunozaki M, Hale VA, Priess JR (2002) APH-1 is a multipass membrane protein essential for the Notch signaling pathway in Caenorhabditis elegans embryos. Proc Natl Acad Sci USA 99: 775-779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heneka MT, Klockgether T, Feinstein DL (2000) Peroxisome proliferator-activated receptor-γ ligands reduce neuronal inducible nitric oxide synthase expression and cell death in vivo. J Neurosci 20: 6862-6867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu E, Tontonoz P, Spiegelman BM (1995) Transdifferentiation of myoblasts by the adipogenic transcription factors PPAR gamma and C/EBP alpha. Proc Natl Acad Sci USA 92: 9856-9860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang JT, Welch JS, Ricote M, Binder CJ, Willson TM, Kelly C, Witztum JL, Funk CD, Conrad D, Glass CK (1999) Interleukin-4-dependent production of PPAR-gamma ligands in macrophages by 12/15-lipoxygenase. Nature 400: 378-382. [DOI] [PubMed] [Google Scholar]
- Hussain I, Powell D, Howlett DR, Tew DG, Meek TD, Chapman C, Gloger IS, Murphy KE, Southan CD, Ryan DM, Smith TS, Simmons DL, Walsh FS, Dingwall C, Christie G (1999) Identification of a novel aspartic protease (Asp 2) as beta-secretase. Mol Cell Neurosci 14: 419-427. [DOI] [PubMed] [Google Scholar]
- in t' Veld BA, Ruitenberg A, Hofman A, Launer LJ, van Duijn CM, Stijnen T, Breteler MM, Stricker BH (2001) Nonsteroidal antiinflammatory drugs and the risk of Alzheimer's disease. N Engl J Med 345: 1515-1521. [DOI] [PubMed] [Google Scholar]
- Jantzen PT, Connor KE, DiCarlo G, Wenk GL, Wallace JL, Rojiani AM, Coppola D, Morgan D, Gordon MN (2002) Microglial activation and β-amyloid deposit reduction caused by a nitric oxide-releasing nonsteroidal anti-inflammatory drug in amyloid precursor protein plus presenilin-1 transgenic mice. J Neurosci 22: 2246-2254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jarriault S, Brou C, Logeat F, Schroeter EH, Kopan R, Israel A (1995) Signalling downstream of activated mammalian Notch. Nature 377: 355-358. [DOI] [PubMed] [Google Scholar]
- Jiang C, Ting AT, Seed B (1998) PPAR-gamma agonists inhibit production of monocyte inflammatory cytokines. Nature 391: 82-86. [DOI] [PubMed] [Google Scholar]
- Karlstrom H, Bergman A, Lendahl U, Naslund J, Lundkvist J (2002) A sensitive and quantitative assay for measuring cleavage of presenilin substrates. J Biol Chem 277: 6763-6766. [DOI] [PubMed] [Google Scholar]
- Kitamura Y, Shimohama S, Koike H, Kakimura J, Matsuoka Y, Nomura Y, Gebicke-Haerter PJ, Taniguchi T (1999) Increased expression of cyclooxygenases and peroxisome proliferator-activated receptor-gamma in Alzheimer's disease brains. Biochem Biophys Res Commun 254: 582-586. [DOI] [PubMed] [Google Scholar]
- Klegeris A, Walker DG, McGeer PL (1999) Toxicity of human THP-1 monocytic cells towards neuron-like cells is reduced by non-steroidal anti-inflammatory drugs (NSAIDs). Neuropharmacology 38: 1017-1025. [DOI] [PubMed] [Google Scholar]
- Koike H, Tomioka S, Sorimachi H, Saido TC, Maruyama K, Okuyama A, Fujisawa-Sehara A, Ohno S, Suzuki K, Ishiura S (1999) Membrane-anchored metalloprotease MDC9 has an alpha-secretase activity responsible for processing the amyloid precursor protein. Biochem J 343: 371-375. [PMC free article] [PubMed] [Google Scholar]
- Kopan R, Schroeter EH, Weintraub H, Nye JS (1996) Signal transduction by activated mNotch: importance of proteolytic processing and its regulation by the extracellular domain. Proc Natl Acad Sci USA 93: 1683-1688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lammich S, Kojro E, Postina R, Gilbert S, Pfeiffer R, Jasionowski M, Haass C, Fahrenholz F (1999) Constitutive and regulated alpha-secretase cleavage of Alzheimer's amyloid precursor protein by a disintegrin metalloprotease. Proc Natl Acad Sci USA 96: 3922-3927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lannfelt L, Viitanen M, Johansson K, Axelman K, Lilius L, Almqvist E, Winblad B (1993) Low frequency of the APP 670/671 mutation in familial Alzheimer's disease in Sweden. Neurosci Lett 153: 85-87. [DOI] [PubMed] [Google Scholar]
- Lehmann JM, Moore LB, Smith-Oliver TA, Wilkison WO, Willson TM, Kliewer SA (1995) An antidiabetic thiazolidinedione is a high affinity ligand for peroxisome proliferator-activated receptor gamma (PPAR gamma). J Biol Chem 270: 12953-12956. [DOI] [PubMed] [Google Scholar]
- Lemberger T, Desvergne B, Wahli W (1996) Peroxisome proliferator-activated receptors: a nuclear receptor signaling pathway in lipid physiology. Annu Rev Cell Dev Biol 12: 335-363. [DOI] [PubMed] [Google Scholar]
- Lim GP, Yang F, Chu T, Chen P, Beech W, Teter B, Tran T, Ubeda O, Ashe KH, Frautschy SA, Cole GM (2000) Ibuprofen suppresses plaque pathology and inflammation in a mouse model for Alzheimer's disease. J Neurosci 20: 5709-5714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mackenzie IR, Munoz DG (1998) Nonsteroidal anti-inflammatory drug use and Alzheimer-type pathology in aging. Neurology 50: 986-990. [DOI] [PubMed] [Google Scholar]
- Mangelsdorf DJ, Borgmeyer U, Heyman RA, Zhou JY, Ong ES, Oro AE, Kakizuka A, Evans RM (1992) Characterization of three RXR genes that mediate the action of 9-cis retinoic acid. Genes Dev 6: 329-344. [DOI] [PubMed] [Google Scholar]
- Mangelsdorf DJ, Thummel C, Beato M, Herrlich P, Schutz G, Umesono K, Blumberg B, Kastner P, Mark M, Chambon P (1995) The nuclear receptor superfamily: the second decade. Cell 83: 835-839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGeer PL, McGeer EG (1996) Anti-inflammatory drugs in the fight against Alzheimer's disease. Ann NY Acad Sci 777: 213-220. [DOI] [PubMed] [Google Scholar]
- McGeer PL, Kawamata T, Walker DG, Akiyama H, Tooyama I, McGeer EG (1993) Microglia in degenerative neurological disease. Glia 7: 84-92. [DOI] [PubMed] [Google Scholar]
- Michiels F, van Es H, van Rompaey L, Merchiers P, Francken B, Pittois K, van der Schueren J, Brys R, Vandersmissen J, Beirinckx F, Herman S, Dokic K, Klaassen H, Narinx E, Hagers A, Laenen W, Piest I, Pavliska H, Rombout Y, Langemeijer E (2002) Arrayed adenoviral expression libraries for functional screening. Nat Biotechnol 20: 1154-1157. [DOI] [PubMed] [Google Scholar]
- Miyata KS, McCaw SE, Marcus SL, Rachubinski RA, Capone JP (1994) The peroxisome proliferator-activated receptor interacts with the retinoid X receptor in vivo. Gene 148: 327-330. [DOI] [PubMed] [Google Scholar]
- Mukherjee R, Davies PJ, Crombie DL, Bischoff ED, Cesario RM, Jow L, Hamann LG, Boehm MF, Mondon CE, Nadzan AM, Paterniti Jr JR, Heyman RA (1997) Sensitization of diabetic and obese mice to insulin by retinoid X receptor agonists. Nature 386: 407-410. [DOI] [PubMed] [Google Scholar]
- Nyabi O, Bentahir M, Horre K, Herreman A, Gottardi-Littell N, Van Broeckhoven C, Merchiers P, Spittaels K, Annaert W, De Strooper B (2003) Presenilins mutated at Asp-257 or Asp-385 restore Pen-2 expression and Nicastrin glycosylation but remain catalytically inactive in the absence of wild type Presenilin. J Biol Chem 278: 43430-43436. [DOI] [PubMed] [Google Scholar]
- Okano H, Shiraki K, Inoue H, Yamanaka T, Deguchi M, Sugimoto K, Sakai T, Ohmori S, Fujikawa K, Murata K, Nakano T (2002) Peroxisome proliferator-activated receptor gamma augments tumor necrosis factor family-induced apoptosis in hepatocellular carcinoma. Anticancer Drugs 13: 59-65. [DOI] [PubMed] [Google Scholar]
- Olefsky JM (2000) Treatment of insulin resistance with peroxisome proliferator-activated receptor gamma agonists. J Clin Invest 106: 467-472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan D, Rubin GM (1997) Kuzbanian controls proteolytic processing of Notch and mediates lateral inhibition during Drosophila and vertebrate neurogenesis. Cell 90: 271-280. [DOI] [PubMed] [Google Scholar]
- Perlmutter LS, Barron E, Chui HC (1990) Morphologic association between microglia and senile plaque amyloid in Alzheimer's disease. Neurosci Lett 119: 32-36. [DOI] [PubMed] [Google Scholar]
- Petrova TV, Akama KT, Van Eldik LJ (1999) Cyclopentenone prostaglandins suppress activation of microglia: down-regulation of inducible nitric-oxide synthase by 15-deoxy-delta12,14-prostaglandin J2. Proc Natl Acad Sci USA 96: 4668-4673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rich JB, Rasmusson DX, Folstein MF, Carson KA, Kawas C, Brandt J (1995) Nonsteroidal anti-inflammatory drugs in Alzheimer's disease. Neurology 45: 51-55. [DOI] [PubMed] [Google Scholar]
- Ricote M, Huang JT, Welch JS, Glass CK (1999) The peroxisome proliferator-activated receptor(PPARgamma) as a regulator of monocyte/macrophage function. J Leukoc Biol 66: 733-739. [DOI] [PubMed] [Google Scholar]
- Rosen ED, Sarraf P, Troy AE, Bradwin G, Moore K, Milstone DS, Spiegelman BM, Mortensen RM (1999) PPAR gamma is required for the differentiation of adipose tissue in vivo and in vitro. Mol Cell 4: 611-617. [DOI] [PubMed] [Google Scholar]
- Rossor MN, Newman S, Frackowiak RS, Lantos P, Kennedy AM (1993) Alzheimer's disease families with amyloid precursor protein mutations. Ann NY Acad Sci 695: 198-202. [DOI] [PubMed] [Google Scholar]
- Sagi SA, Weggen S, Eriksen J, Golde TE, Koo EH (2003) The non-cyclooxygenase targets of non-steroidal anti-inflammatory drugs, lipoxygenases, peroxisome proliferator-activated receptor, inhibitor of kappa B kinase, and NF kappa B, do not reduce amyloid beta 42 production. J Biol Chem 278: 31825-31830. [DOI] [PubMed] [Google Scholar]
- Sastre M, Dewachter I, Landreth GE, Willson TM, Klockgether T, van Leuven F, Heneka MT (2003) Nonsteroidal anti-inflammatory drugs and peroxisome proliferator-activated receptor-γ agonists modulate immunostimulated processing of amyloid precursor protein through regulation of β-secretase. J Neurosci 23: 9796-9804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sinha S, Anderson JP, Barbour R, Basi GS, Caccavello R, Davis D, Doan M, Dovey HF, Frigon N, Hong J, Jacobson-Croak K, Jewett N, Keim P, Knops J, Lieberburg I, Power M, Tan H, Tatsuno G, Tung J, Schenk D, et al. (1999) Purification and cloning of amyloid precursor protein beta-secretase from human brain [In Process Citation]. Nature 402: 537-540. [DOI] [PubMed] [Google Scholar]
- Steppan CM, Bailey ST, Bhat S, Brown EJ, Banerjee RR, Wright CM, Patel HR, Ahima RS, Lazar MA (2001) The hormone resistin links obesity to diabetes. Nature 409: 307-312. [DOI] [PubMed] [Google Scholar]
- Stewart WF, Kawas C, Corrada M, Metter EJ (1997) Risk of Alzheimer's disease and duration of NSAID use. Neurology 48: 626-632. [DOI] [PubMed] [Google Scholar]
- Tan J, Town T, Paris D, Mori T, Suo Z, Crawford F, Mattson MP, Flavell RA, Mullan M (1999) Microglial activation resulting from CD40-CD40L interaction after beta-amyloid stimulation. Science 286: 2352-2355. [DOI] [PubMed] [Google Scholar]
- Tontonoz P, Hu E, Spiegelman BM (1994) Stimulation of adipogenesis in fibroblasts by PPAR gamma 2, a lipid-activated transcription factor. Cell 79: 1147-1156. [DOI] [PubMed] [Google Scholar]
- Vassar R, Bennett BD, Babu-Khan S, Kahn S, Mendiaz EA, Denis P, Teplow DB, Ross S, Amarante P, Loeloff R, Luo Y, Fisher S, Fuller J, Edenson S, Lile J, Jarosinski MA, Biere AL, Curran E, Burgess T, Louis JC, et al. (1999) Beta-secretase cleavage of Alzheimer's amyloid precursor protein by the transmembrane aspartic protease BACE. Science 286: 735-741. [DOI] [PubMed] [Google Scholar]
- Wang C, Fu M, D'Amico M, Albanese C, Zhou JN, Brownlee M, Lisanti MP, Chatterjee VK, Lazar MA, Pestell RG (2001) Inhibition of cellular proliferation through IkappaB kinase-independent and peroxisome proliferator-activated receptor gamma-dependent repression of cyclin D1. Mol Cell Biol 21: 3057-3070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weggen S, Eriksen JL, Das P, Sagi SA, Wang R, Pietrzik CU, Findlay KA, Smith TE, Murphy MP, Bulter T, Kang DE, Marquez-Sterling N, Golde TE, Koo EH (2001) A subset of NSAIDs lower amyloidogenic Abeta42 independently of cyclooxygenase activity. Nature 414: 212-216. [DOI] [PubMed] [Google Scholar]
- Willson TM, Brown PJ, Sternbach DD, Henke BR (2000) The PPARs: from orphan receptors to drug discovery. J Med Chem 43: 527-550. [DOI] [PubMed] [Google Scholar]
- Yan Q, Zhang J, Liu H, Babu-Khan S, Vassar R, Biere AL, Citron M, Landreth G (2003) Anti-inflammatory drug therapy alters β-amyloid processing and deposition in an animal model of Alzheimer's disease. J Neurosci 23: 7504-7509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan R, Bienkowski MJ, Shuck ME, Miao H, Tory MC, Pauley AM, Brashier JR, Stratman NC, Mathews WR, Buhl AE, Carter DB, Tomasselli AG, Parodi LA, Heinrikson RL, Gurney ME (1999) Membrane-anchored aspartyl protease with Alzheimer's disease beta-secretase activity. Nature 402: 533-537. [DOI] [PubMed] [Google Scholar]
- Yu G, Nishimura M, Arawaka S, Levitan D, Zhang L, Tandon A, Song YQ, Rogaeva E, Chen F, Kawarai T, Supala A, Levesque L, Yu H, Yang DS, Holmes E, Milman P, Liang Y, Zhang DM, Xu DH, Sato C, et al. (2000) Nicastrin modulates presenilin-mediated notch/glp-1 signal transduction and betaAPP processing. Nature 407: 48-54. [DOI] [PubMed] [Google Scholar]