

Abstract
The juxtamembrane domain (JMD) of N-cadherin cytoplasmic tail is an important regulatory region of the clustering and adhesion activities of the protein. In addition, the JMD binds a diversity of proteins capable of modifying intracellular processes including cytoskeletal rearrangement mediated by Rho GTPases. These GTPases also function as regulators of voltage-activated calcium channels, which in turn modulate neuronal excitability. The present study was designed to determine whether there is a direct functional link, via Rho GTPase, between the N-cadherin JMD and these voltage-activated channels. It was found that the infusion of the soluble JMD into chick ciliary neurons causes a substantial decrease in the amplitude of the high-threshold voltage-activated (HVA) calcium current. The activation time is increased while the inactivation process is reduced, suggesting that the decreased current amplitude reflects a reduction in the number of channels available to open. This effect was reversed by inhibition of RhoA or its downstream effector, Rho-associated kinase (ROCK). Because ROCK determines the active state of myosin, these results suggest that the modulation of HVA by the JMD could be mediated by changes in the status of the actin-myosin cytoskeleton.
Keywords: N-cadherin, voltage-activated calcium channel, RhoA GTPase, ciliary ganglion neuron, cell adhesion, ROCK
Introduction
N-cadherin is a cell adhesion molecule (Takeichi, 1990) abundantly localized to the synaptic complex, where it forms adherens-like junctions adjacent to or fused to both the active zone and postsynaptic density (Fannon and Colman, 1996; Uchida et al., 1996; Brusés, 2000). These junctions participate in neuronal physiology in that activity-dependent potentiation of synaptic transmission is correlated with the strengthening of cadherin-mediated adhesion (Bozdagi et al., 2000; Tanaka et al., 2000).
The N-cadherin cytoplasmic tail contains two binding sites for catenins (Yap et al., 1997). The C-terminal site interacts with β-catenin and in recent studies has been identified as an important organizer of the synaptic complex either by linking N-cadherin to the actin cytoskeleton via α-catenin (Togashi et al., 2002) or by direct interaction of the β-catenin postsynaptic density-95/Discs large/zona occludens-1-binding region with synaptic vesicles (Bamji et al., 2003). The juxtamembrane domain (JMD) is a modulator of cadherin activity (Yap et al., 1998; Gumbiner, 2000) and serves as a binding site for a variety of proteins, including the p120-catenin family members (Anastasiadis and Reynolds, 2000), hakai (Fujita et al., 2002), and presenilin 1 (Baki et al., 2001). Of particular importance for our studies is the fact that interactions between the JMD and its binding partners can influence other cellular processes including the activity of small GTPases and cytoskeletal dynamics. For example, p120-catenin promotes changes in the cytoskeleton by inhibiting RhoA GTPases, and these effects can be reverted by a JMD-p120-catenin interaction (Anastasiadis et al., 2000; Noren et al., 2000).
The relationship between small GTPase activity and the JMD has attracted our attention because small GTPase and the cytoskeleton have also been implicated in the modulation of voltage-gated Ca2+ channels (Wilk-Blaszczak et al., 1997; Rueckschloss and Isenberg, 2001; Ward et al., 2004). This type of channel is a major regulator of cytosolic calcium (Dolphin, 1998; Catterall, 2000) and therefore has important implications for neuronal physiology (West et al., 2002; Spafford and Zamponi, 2003). The present study was designed to look for a functional link between these two GTPase-associated mechanisms using the chick ciliary neuron, which abundantly expresses both N-cadherin and high-threshold voltage-activated (HVA) calcium channels of the N type (White et al., 1997).
Materials and Methods
Synthesis and isolation of recombinant proteins. Polypeptides were N-terminally fused with a 6× or 10× histidine tag. The chicken N-cadherin cDNA (Hatta et al., 1988) (kindly provided by Dr. M. Takeichi, Kyoto University, Kyoto, Japan) was used as a template to generate fragments by PCR using Pfu Turbo DNA polymerase (Stratagene, La Jolla, CA). The soluble JMD (sJMD) (from Lys 753 to Gly 839) was amplified with a 5′ end primer (bp 2257-2280) and a 3′ end primer (bp 2497-2517) and cloned into pQE-60 vector (Qiagen, Chatsworth, CA). The enhanced green fluorescence protein (EGFP) coding sequence was removed from pEGFP-N1 vector (Clontech, Palo Alto, CA) by sequentially digesting NotI, blunt ended, and BamHI, and the fragment subcloned into pQE31 (Qiagen) opened with BamHI and SmaI. cDNAs were checked by sequencing analysis.
Polypeptides were generated and purified according to the manufacturer's (Qiagen) recommendations. Briefly, plasmids were transfected into M15 cells, and protein expression was induced by isopropylthio-β-d-galactoside (1 mm). Thereafter, the cells were grown for 4 hr at 37°C and lysed by sonication, and the recombinant protein was purified on a nickel column. Samples were examined by SDS-PAGE and dialyzed against intracellular solution. Protein concentration was determined by BCA assay (Pierce, Rockford, IL).
Cell dissociation. Ciliary ganglia were dissected from St 40 (Hamburger, 1951) White Leghorn chicken embryos in extracellular solution (in mm): 130 NaCl, 5 KCl, 1 MgCl2, 5 CaCl2, 10 HEPES, and 11 glucose, pH 7.4. The ganglia were incubated (30 min at 37°C) with type IV collagenase (0.4 mg/ml) and hyaluronidase (10,000 U/ml) (Worthington Biochemical, Freehold, NJ) and trypsin inhibitor (125 μl/ml; Sigma-Aldrich, St. Louis, MO), rinsed in extracellular solution containing 15% fetal bovine serum (Invitrogen, San Diego, CA), mechanically dissociated with a fire-polished Pasteur pipette, and transferred to a concanavalin-treated bottom glass perfusion chamber mounted on an inverted Axiovert 100M microscope (Zeiss Oberkochen, Germany). The perfusion flow was 1-2 ml/min, and experiments were run at 24°C.
Electrophysiology. Ca2+ currents were recorded at a -80 mV holding potential in whole-cell configuration of the patch-clamp technique, using a Multiclamp 700A amplifier (Axon Instruments, Foster City, CA) connected to a Digidata 1322A (Axon Instruments) analog-to-digital converter. Patch pipettes were made of borosilicate glass capillaries (Garner Glass 8250) using a two-stage vertical puller (David Kopf 730), heat polished (5-8 MΩ) and filled with intracellular solution (in mm): 140 CsCl, 10 NaOH, 1 MgCl2, 0.5 BAPTA, 20 HEPES, 5 ATP-Mg, 0.2 GTP-Na, and 4.7 mannitol, pH 7.4. Series resistance was compensated by 80-90% before each recording. The isolating Ca2+ current solution contained the following (in mm): 140 tetraethylammonium chloride, 5 CaCl2, 1 MgCl2, 10 HEPES, 0.0003 tetrodotoxin, and 11 glucose, pH 7.4. Currents were elicited and acquired using pClamp9 software (Axon Instruments). A standard P/4 protocol was used for linear leakage and capacitance subtraction. Currents were low-pass filtered at a cutoff frequency of 2 kHz and were acquired at 20 kHz.
Data analysis and statistics. Raw data were analyzed and plotted using Clampfit 9.0, SigmaStat 3.0.1, and SigmaPlot 8.0.2 (SPSS, Chicago, IL). Calcium current amplitudes were normalized to membrane capacitance (pA/pF). Statistical significance was assessed by Student's t test and was assumed at p < 0.01. Current activation and deactivation data were fitted, respectively, to a Boltzmann and a single-exponential function.
Toxins and drugs. Clostridium botulinum C3 exoenzyme (Biomol International, Plymouth Meeting, PA) and Y-27632 (Calbiochem, La Jolla, CA) were dissolved in water and used at 0.5 and 10 μm, respectively.
Results
The sJMD inhibits high-voltage-activated Ca2+ current
Inward Ca2+ current (ICa) was recorded from the somata of freshly dissociated ciliary neurons. Figure 1A (top) depicts a family of ICa traces elicited by a series of 10 mV voltage steps (bottom) of 100 msec duration over a range of membrane holding potentials from -80 to +50 mV. The whole-cell calcium current was recorded under conditions that prevent contamination by other currents (see Materials and Methods) and therefore is attributable entirely to Ca2+ influx. The ICa is characterized by an initial peak, the activation time of which decreases as the depolarizing voltage step increases. This initial peak is followed by a partially inactivating component that extends throughout the duration of the voltage pulse.
Figure 1.

N-cadherin sJMD inhibits HVA Ca2+ current. A, Control Ca2+ currents elicited by stepping the membrane holding potential from -80 to +50 mV at a 10 mV interval. B, Calcium currents of a neuron infused with intracellular solution containing 1 μm sJMD. Note the inhibitory effect on the current amplitude. C, Current traces for control (•) and experimental (○) conditions are superimposed. Note the decrease in current amplitude attributable to the sJMD. D, Top, Ciliary ganglion neuron infused with 1 μm EGFP. Bottom, EGFP does not change the macroscopic calcium current. E, Average current density-voltage plots for calcium currents recorded in control (n = 10) and in the presence of the sJMD (n = 9). Note that the sJMD shifts the calcium current amplitude toward lower values. Values are expressed as mean ± SE. Scale bar, 10 μm.
To assess their effects on calcium current, in vitro-synthesized polypeptides were infused into the cell through the patch pipette. EGFP was used to evaluate the efficiency of this delivery. This treatment resulted in spread of the fluorescent protein throughout the cell without changing the ICa (Fig. 1D). When the sJMD was used, however, there was a substantial decrease in the amplitude of ICa (Fig. 1A,B). This is clearly shown in Figure 1C, where the ICa elicited under control and experimental conditions is superimposed.
Figure 1E illustrates the average current density-voltage plots for control and for sJMD-infused neurons, showing a decrease in current amplitude at each voltage tested. The control plot shows the calcium current activating near -30 mV and peaking at 0 mV, confirming that the ciliary neuron possesses HVA N-type calcium channels (White et al., 1997). The current reverses near +60 mV, as expected for calcium current recorded in the presence of intracellular cesium. The sJMD shifts the plot toward lower current density values, with the greatest current inhibition occurring at 0 mV. In contrast, the sJMD does not change the threshold, peak, or reversing potential of ICa.
Effects of the sJMD on the voltage gating and kinetics of HVA Ca2+ current
Figure 2A shows a normalization and superimposition of the peak Ca2+ currents illustrated in Figure 1C. It is evident that the ICa inhibition caused by the sJMD occurs concomitantly with a slowing in activation and a reduction in inactivation of the calcium current. Moreover, the polypeptide changes the current deactivation, as indicated by an increase in the decay time of the calcium tail current.
Figure 2.

Effects of sJMD on the voltage gating and kinetics of HVA Ca2+ current. A, Superimposed control and normalized sJMD calcium current traces. Note that current suppression occurs with a delay in activation time, a slowing of inactivation, and an increase in tail deactivation time. B, Raw data fitted to a Boltzmann function. Note that the sJMD does not change the voltage dependence of the ion channel steady-state inactivation. C, Ten to 90% activation time as a function of voltage step. Note that the sJMD delays voltage-dependent current activation. D, Iend/Ipeak inactivation index as a function of voltage step. Note that the sJMD decreases Ca2+-dependent inactivation at intermediate voltage while promoting a closed-state ion channel at both lower- and upper-end voltage values. E, Tau tail deactivation parameter as a function of voltage step. Note that the sJMD prolongs Ca2+channel closure as indicated by an increase in deactivation time (control, n = 10; sJMD, n = 9). Values are expressed as mean ± SE.
To gain insight into the possible mechanisms by which the sJMD suppresses the HVA Ca2+ current amplitude, the voltage dependence of the current gating was investigated, and its kinetic parameters were quantified. Figure 2B shows Boltzmann fits to the average normalized current values derived from ICa triggered by 10 mV incremental voltage steps starting from -70 to 0 mV. The fitted function parameters for the control data were Vmid (membrane potential at which the current is half-maximal; -8.9 ± 0.4 mV) and Vc (voltage required to change ICa± e-fold; -3.1 ± 0.4 mV). In the presence of the sJMD, the Vmid is -9.1 ± 0.3 mV, and the Vc is -3.2 ± 0.3 mV. The two sample groups are not statistically significant, indicating that the sJMD is able to decrease ICa amplitude without affecting the voltage dependence of the calcium channel steady-state inactivation.
To assess the effect of the sJMD on Ca2+ current activation, we measured the time elapsed between 10 and 90% of the transient peak amplitude. Figure 2C illustrates the average 10-90% rising time values at each voltage step for both control and experimental conditions. The sJMD increased the activation time-voltage relationship, with a significant maximal effect occurring at 0 mV (average 10-90% rising time: control, 3.5 ± 0.4 msec; sJMD, 5.6 ± 0.4 msec).
To determine the extent to which the sJMD impacts the ICa inactivation, the Iend/Ipeak ratio was calculated indicating the amount of peak current that decays by the end of the voltage pulse. Figure 2D shows the average voltage Iend/Ipeak plots obtained from control and experimental recordings. For control cells, the “inactivation ratio” follows an inverse pattern of the current amplitude. That is, the ratio decreases with the current proceeding toward its peak amplitude and progressively increases as the current approaches its reversal potential. This current behavior indicates that the ICa in ciliary neurons is susceptible to calcium-dependent inactivation. For neurons infused with the sJMD, the average Iend/Ipeak is increased for maximum current amplitude between -10 and 30 mV (control, 0.63 ± 0.02; sJMD, 0.81 ± 0.02). This change is consistent with the suppression of calcium influx because of the inhibition of HVA calcium channels. Surprisingly, the inactivation ratio for relatively small amplitudes of ICa (e.g., -20, +40, and +50 mV values) is accompanied by a more robust inactivation than expected. These findings suggest that the decrease in ICa amplitude caused by the sJMD may be attributable to a reduced number of channels that are available to open.
To quantify the effect of the sJMD on the process of current deactivation, a single-exponential function was fitted over a 10 msec decay time of the peak tail current. Figure 2E shows the average tau calculated from tail currents recorded in controls and in the presence of the sJMD at membrane voltages (0-50 mV) in which current inactivation is decreasing. It is evident from these results that the sJMD first increases tau at each voltage step tested and then prevents the control plateau from occurring at the upper end of the voltage stimuli. In summary, the decrease in ICa caused by the sJMD is accompanied by an increase in activation time, a decrease in inactivation, and an increase in current deactivation. These events occur without affecting the voltage dependence of the ion channel steady-state inactivation.
RhoA and Rho-associated kinase inhibition antagonize the effect of the sJMD on Ca2+ current
As described in Introduction, it is plausible that the effect of the sJMD on the calcium current reflects the activation of RhoA. This hypothesis was tested using the C. botulinum C3 exoenzyme, which irreversibly inactivates RhoA (Aktories et al., 1987). The sJMD was coinfused with C3 into ciliary neurons for 2 min, followed by a voltage step (from -80 to 0 mV) known to elicit maximal Ca2+ current (Fig. 3A, top trace). On average, the amplitude was reduced compared with controls (data not shown). A close inspection of the current trace reveals that ICa tends to rise throughout the voltage stimulus, indicating that C3 causes an increase in the current amplitude, despite the presence of the sJMD. The effect of the toxin became more evident (Fig. 3A, bottom trace) after 5 min of infusion, in that ICa increased substantially in amplitude and its kinetics tended to be similar to those of control currents (Fig. 3B, inset). Comparison of the average current density-voltage plots (Fig. 3B) constructed from currents recorded in the presence or in the absence of C3 indicates a significant reversal by the RhoA inhibitor of the suppression of ICa elicited by the sJMD. The involvement of RhoA in the control of the HVA calcium conductance is further supported by the finding that C3 reverts the inhibition of ICa caused by the sJMD without changing both the Vmid and Vc (Fig. 3C), indicating that the voltage dependence of the channel steady-state inactivation is not affected. In contrast, the components of the current, which were affected by the sJMD (decrease in the current amplitude, slowness in ICa activation, upward shift in inactivation, and delay of the deactivation process), are all partially reverted by blocking RhoA activity with the C3 exoenzyme (Fig. 3D-F).
Figure 3.

RhoA inhibition antagonizes both the suppression of Ca2+ current amplitude and its kinetics changes triggered by the sJMD. A, Peak calcium current traces recorded at 2 min (top trace) and at 5 min (bottom trace) while perfusing the neuron with sJMD (1 μm) and C3 exoenzyme (500 nm). Note that C3 reverts the calcium current inhibition caused by the sJMD. B, Average current density-voltage plots for control (n = 10), sJMD (n = 9), and sJMD plus C3 exoenzyme (n = 7) recordings. Note that inhibition of RhoA recovers the calcium current density. Inset, Superimposed peak currents for control, sJMD, and sJMDp plus C3 exoenzyme. C, Raw data fitted to a Boltzmann function. Note that the rescue of current amplitude by C3 does not involve a change in the voltage dependence of the ion channel steady-state inactivation (control: Vmid = -8.9 ± 0.4 mV, Vc = -3.1 ± 0.4 mV; sJMD: Vmid = -9.1 ± 0.3 mV, Vc = -3.2 ± 0.3 mV; sJMD plus C3: Vmid = -9.0 ± 0.3 mV, Vc = -3.0 ± 0.3 mV). D, Ten to 90% activation time. E, Iend/Ipeak inactivation index. F, Tau tail deactivation parameter, as a function of voltage step. Note that inhibition of RhoA opposes all current changes attributable to the sJMD. Values are expressed as mean ± SE.
Rho-associated kinase (ROCK) is involved in a variety of cellular functions downstream of RhoA (Amano et al., 1996). To test whether ROCK could be a downstream mediator of the ICa inhibition caused by the sJMD, ciliary neurons were infused with an intracellular solution containing both the sJMD and the ROCK inhibitor Y-27632 (Hirose et al., 1998). Figure 4A shows a peak calcium current trace recorded after 1 min of infusing with sJMD plus Y-27632. An increase in ICa amplitude is observed throughout the duration of the voltage step, similar to the increase observed for the current recorded in the presence of C3 plus sJMD (Fig. 3A). After 2 min, the current has grown in amplitude and has accelerated both its activation and inactivation processes (Fig. 4A). Figure 4B compares the ICa recorded in control, sJMD, and sJMD plus Y-27632 conditions, showing that Y-27632 counteracts the ICa inhibition mediated by the sJMD. Accordingly, Figure 4C shows that neurons infused with sJMD plus Y-27632 exhibit average peak calcium current densities that are significantly greater than those recorded in the presence of the sJMD alone.
Figure 4.
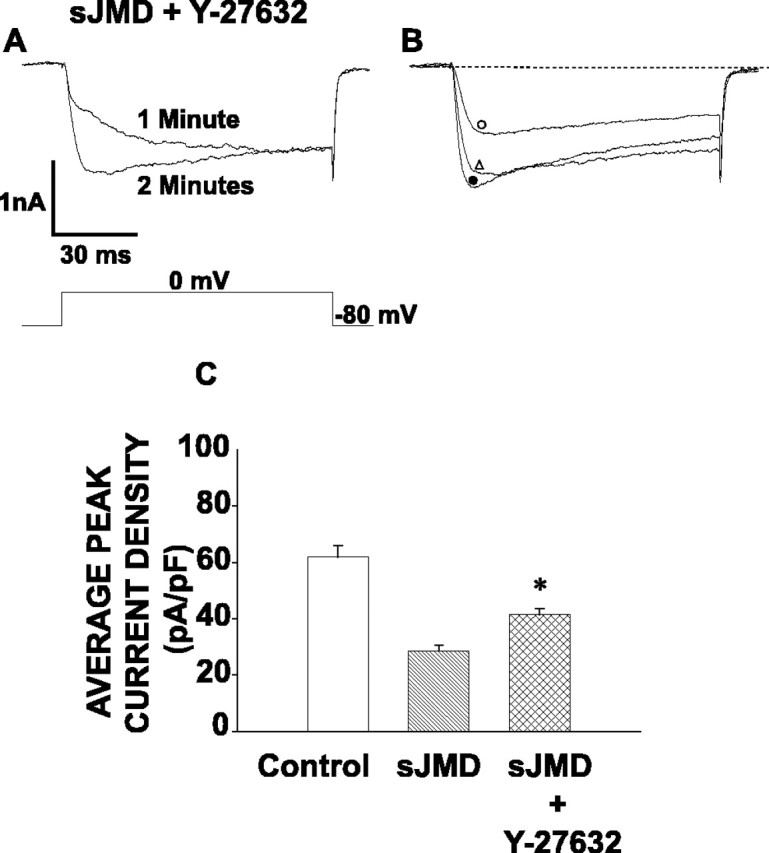
ROCK inhibition counteracts Ca2+ current suppression attributable to sJMD. A, Peak calcium current traces recorded at 1 min (top trace) and at 2 min (bottom trace) while perfusing with sJMD (1 μm) and Y-27632 (10 μm). Note that the ROCK inhibitor reverts current inhibition caused by the sJMD. B, Examples of superimposed calcium currents recorded in controls (•) and in the presence of sJMD (○) or sJMD plus Y-27632 (▵). Note that the inhibition of ROCK in the presence of the sJMD leads to a substantial recovery in ICa amplitude. C, Average peak current density for control (62.3 ± 4.1 pA/pF; n = 10), sJMD (27.9 ± 3.0 pA/pF; n = 9), and sJMD plus Y-27632 (41.4 ± 2.2 pA/pF; n = 7). Note that Y-27632 counteracts sJMD inhibition of average peak calcium current density. Values are expressed as mean ± SE.
Discussion
Voltage-gated calcium current modulation usually occurs by direct phosphorylation of a channel subunit (Catterall, 2000) or by the binding of either heterotrimeric G-protein subunits (Hille, 1994) or monomeric small GTPases (Wilk-Blaszczak et al., 1997; Ward et al., 2004). The present study reveals a novel mechanism by which the N-cadherin JMD modulates HVA Ca2+ current via RhoA activation and its downstream effector ROCK. Interestingly, the effects of the sJMD on ICa are similar to those observed with cytochalasin D (Rueckschloss and Isenberg, 2001), in that both affect the amplitude and inactivation of the current. Therefore, changes in cytoskeleton dynamics induced by ROCK (Amano et al., 1996) could explain the observed RhoA-mediated suppression of ICa. However, although there is no evidence that ROCK can phosphorylate calcium channels, a direct phosphorylation of channel subunits cannot be ruled out.
Examination of the biophysical parameters of the calcium current suppressed by the sJMD showed substantial inactivation that reflects a decreased number of channels available to open. These observations are strikingly similar to the effect of β-subunit mutations on the Ca2+ channel (Catterall, 2000), suggesting a link between N-cadherin and this channel subunit. The role of the β-subunit on channel function has been assigned to both the stabilization of the pore-forming α-subunit during its trafficking to the surface membrane (Yamaguchi et al., 1998) and to the proper conformation of the α-subunit necessary for a functional ion channel (Neely et al., 1993; Catterall, 1995; Shistik et al., 1995). Although our results do not fit with the time course of trafficking to the membrane, they are consistent with a direct modulation of the channel.
Our focus on the N-cadherin JMD was prompted by the fact that this region of the cadherin cytoplasmic tail affects small Rho GTPases, presumably by interacting with p120-catenin family members known to modulate the activity of these GTPases (Anastasiadis and Reynolds, 2000; Noren et al., 2000). Accordingly, we found that the decrease in ICa amplitude by the sJMD is suppressed by the RhoA inhibitor C3 exoenzyme. The simplest interpretation of our observations and the known regulation of RhoA by p120-catenin would be that JMD-p120-catenin interaction causes the activation of RhoA. However, because the JMD can bind to a variety of proteins, involvement of other components cannot be excluded.
Calcium concentration is tightly controlled in neurons because of its essential role in neurotransmitter release, excitability, and gene transcription (West et al., 2002; Spafford and Zamponi, 2003). Because the voltage-gated calcium channel (Dolphin, 1998) is one of the major regulators of cytosolic calcium (Catterall, 2000), the ability of N-cadherin to modulate Ca2+ channels is likely to be important in the regulation of cell physiology. Thus, it is reasonable to speculate that cell contact-mediated changes involving the N-cadherin complex may be coupled to synaptic physiology through the regulation of HVA Ca2+ channels.
Footnotes
This work was supported by National Institutes of Health Grant NS 40300. We thank C.Curcio and C.Perrin-Tricaud for technical assistance.
Correspondence should be addressed to Dr. Juan L. Brusés, Memorial Sloan-Kettering Cancer Center, 1275 York Avenue, Box 290, New York, NY 10021. E-mail: brusesj@ski.mskcc.org.
Copyright © 2004 Society for Neuroscience 0270-6474/04/2410918-06$15.00/0
References
- Aktories K, Weller U, Chhatwal GS (1987) Clostridium botulinum type C produces a novel ADP-ribosyltransferase distinct from botulinum C2 toxin. FEBS Lett 212: 109-113. [DOI] [PubMed] [Google Scholar]
- Amano M, Ito M, Kimura K, Fukata Y, Chihara K, Nakano T, Matsuura Y, Kaibuchi K (1996) Phosphorylation and activation of myosin by Rho-associated kinase (Rho-kinase). J Biol Chem 271: 20246-20249. [DOI] [PubMed] [Google Scholar]
- Anastasiadis PZ, Reynolds AB (2000) The p120 catenin family: complex roles in adhesion, signaling and cancer. J Cell Sci 113: 1319-1334. [DOI] [PubMed] [Google Scholar]
- Anastasiadis PZ, Moon SY, Thoreson MA, Mariner DJ, Crawford HC, Zheng Y, Reynolds AB (2000) Inhibition of RhoA by p120 catenin. Nat Cell Biol 2: 637-644. [DOI] [PubMed] [Google Scholar]
- Baki L, Marambaud P, Efthimiopoulos S, Georgakopoulos A, Wen P, Cui W, Shioi J, Koo E, Ozawa M, Friedrich Jr VL, Robakis NK (2001) Presenilin-1 binds cytoplasmic epithelial cadherin, inhibits cadherin/p120 association, and regulates stability and function of the cadherin/catenin adhesion complex. Proc Natl Acad Sci USA 98: 2381-2386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bamji SX, Shimazu K, Kimes N, Huelsken J, Birchmeier W, Lu B, Reichardt LF (2003) Role of beta-catenin in synaptic vesicle localization and presynaptic assembly. Neuron 40: 719-731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bozdagi O, Shan W, Tanaka H, Benson DL, Huntley GW (2000) Increasing numbers of synaptic puncta during late-phase LTP: N-cadherin is synthesized, recruited to synaptic sites, and required for potentiation. Neuron 28: 245-259. [DOI] [PubMed] [Google Scholar]
- Brusés JL (2000) Cadherin-mediated adhesion at the interneuronal synapse. Curr Opin Cell Biol 12: 593-597. [DOI] [PubMed] [Google Scholar]
- Catterall WA (1995) Structure and function of voltage-gated ion channels. Annu Rev Biochem 64: 493-531. [DOI] [PubMed] [Google Scholar]
- Catterall WA (2000) Structure and regulation of voltage-gated Ca2+ channels. Annu Rev Cell Dev Biol 16: 521-555. [DOI] [PubMed] [Google Scholar]
- Dolphin AC (1998) Mechanisms of modulation of voltage-dependent calcium channels by G proteins. J Physiol (Lond) 506: 3-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fannon AM, Colman DR (1996) A model for central synaptic junctional complex formation based on the differential adhesive specificities of the cadherins. Neuron 17: 423-434. [DOI] [PubMed] [Google Scholar]
- Fujita Y, Krause G, Scheffner M, Zechner D, Leddy HE, Behrens J, Sommer T, Birchmeier W (2002) Hakai, a c-Cbl-like protein, ubiquitinates and induces endocytosis of the E-cadherin complex. Nat Cell Biol 4: 222-231. [DOI] [PubMed] [Google Scholar]
- Gumbiner BM (2000) Regulation of cadherin adhesive activity. J Cell Biol 148: 399-404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamburger VHH (1951) A series of normal stages in the development of the chick embryo. J Morphol 88: 49-93. [PubMed] [Google Scholar]
- Hatta K, Nose A, Nagafuchi A, Takeichi M (1988) Cloning and expression of cDNA encoding a neural calcium-dependent cell adhesion molecule: its identity in the cadherin gene family. J Cell Biol 106: 873-881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hille B (1994) Modulation of ion-channel function by G-protein-coupled receptors. Trends Neurosci 17: 531-536. [DOI] [PubMed] [Google Scholar]
- Hirose M, Ishizaki T, Watanabe N, Uehata M, Kranenburg O, Moolenaar WH, Matsumura F, Maekawa M, Bito H, Narumiya S (1998) Molecular dissection of the Rho-associated protein kinase (p160ROCK)-regulated neurite remodeling in neuroblastoma N1E-115 cells. J Cell Biol 141: 1625-1636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neely A, Wei X, Olcese R, Birnbaumer L, Stefani E (1993) Potentiation by the beta subunit of the ratio of the ionic current to the charge movement in the cardiac calcium channel. Science 262: 575-578. [DOI] [PubMed] [Google Scholar]
- Noren NK, Liu BP, Burridge K, Kreft B (2000) p120 catenin regulates the actin cytoskeleton via Rho family GTPases. J Cell Biol 150: 567-580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rueckschloss U, Isenberg G (2001) Cytochalasin D reduces Ca2+ currents via cofilin-activated depolymerization of F-actin in guinea-pig cardiomyocytes. J Physiol (Lond) 537: 363-370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shistik E, Ivanina T, Puri T, Hosey M, Dascal N (1995) Ca2+ current enhancement by alpha 2/delta and beta subunits in Xenopus oocytes: contribution of changes in channel gating and alpha 1 protein level. J Physiol (Lond) 489: 55-62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spafford JD, Zamponi GW (2003) Functional interactions between presynaptic calcium channels and the neurotransmitter release machinery. Curr Opin Neurobiol 13: 308-314. [DOI] [PubMed] [Google Scholar]
- Takeichi M (1990) Cadherins: a molecular family important in selective cell-cell adhesion. Annu Rev Biochem 59: 237-252. [DOI] [PubMed] [Google Scholar]
- Tanaka H, Shan W, Phillips GR, Arndt K, Bozdagi O, Shapiro L, Huntley GW, Benson DL, Colman DR (2000) Molecular modification of N-cadherin in response to synaptic activity. Neuron 25: 93-107. [DOI] [PubMed] [Google Scholar]
- Togashi H, Abe K, Mizoguchi A, Takaoka K, Chisaka O, Takeichi M (2002) Cadherin regulates dendritic spine morphogenesis. Neuron 35: 77-89. [DOI] [PubMed] [Google Scholar]
- Uchida N, Honjo Y, Johnson KR, Wheelock MJ, Takeichi M (1996) The catenin/cadherin adhesion system is localized in synaptic junctions bordering transmitter release zones. J Cell Biol 135: 767-779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ward Y, Spinelli B, Quon MJ, Chen H, Ikeda SR, Kelly K (2004) Phosphorylation of critical serine residues in Gem separates cytoskeletal reorganization from down-regulation of calcium channel activity. Mol Cell Biol 24: 651-661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- West AE, Griffith EC, Greenberg ME (2002) Regulation of transcription factors by neuronal activity. Nat Rev Neurosci 3: 921-931. [DOI] [PubMed] [Google Scholar]
- White MG, Crumling MA, Meriney SD (1997) Developmental changes in calcium current pharmacology and somatostatin inhibition in chick para-sympathetic neurons. J Neurosci 17: 6302-6313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilk-Blaszczak MA, Singer WD, Quill T, Miller B, Frost JA, Sternweis PC, Belardetti F (1997) The monomeric G-proteins Rac1 and/or Cdc42 are required for the inhibition of voltage-dependent calcium current by bradykinin. J Neurosci 17: 4094-4100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamaguchi H, Hara M, Strobeck M, Fukasawa K, Schwartz A, Varadi G (1998) Multiple modulation pathways of calcium channel activity by a beta subunit. Direct evidence of beta subunit participation in membrane trafficking of the alpha1C subunit. J Biol Chem 273: 19348-19356. [DOI] [PubMed] [Google Scholar]
- Yap AS, Brieher WM, Gumbiner BM (1997) Molecular and functional analysis of cadherin-based adherens junctions. Annu Rev Cell Dev Biol 13: 119-146. [DOI] [PubMed] [Google Scholar]
- Yap AS, Niessen CM, Gumbiner BM (1998) The juxtamembrane region of the cadherin cytoplasmic tail supports lateral clustering, adhesive strengthening, and interaction with p120ctn. J Cell Biol 141: 779-789. [DOI] [PMC free article] [PubMed] [Google Scholar]