

Abstract
The specification and organization of glutamatergic synaptic transmission require the coordinated interaction among glutamate receptors and their synaptic adaptor proteins closely assembled in the postsynaptic density (PSD). Here we investigated the interaction between NMDA receptors and metabotropic glutamate receptor 5 (mGluR5) in the integral regulation of extracellular signal-regulated protein kinase (ERK) and gene expression in cultured rat striatal neurons. We found that coapplication of NMDA and the mGluR5 agonist (S)-3,5-dihydroxyphenylglycine synergistically increased ERK phosphorylation. Interestingly, the synergistic increase in ERK phosphorylation was dependent on the cross talk between NMDA receptor-associated synaptic adaptor protein PSD-95 and the mGluR5-linked adaptor protein Homer1b/c but not on the conventional Ca2+ signaling derived from NMDA receptors (Ca2+ influx) and mGluR5 (intracellular Ca2+ release). This was demonstrated by the findings that the synergistic phosphorylation of ERK induced by coactivation of NMDA receptors and mGluR5 was blocked by either a Tat peptide that disrupts NMDA receptor/PSD-95 binding or small interfering RNAs that selectively reduce cellular levels of Homer1b/c. Furthermore, ERK activated through this PSD-95/Homer1b/c-dependent and Ca2+-independent pathway was able to phosphorylate the two key transcription factors Elk-1 and cAMP response element-binding protein, which further leads to facilitation of c-Fos expression. Together, we have identified a novel Ca2+-independent signaling pathway to ERK by the synergistic interaction of NMDA receptors and mGluR5 via their adaptor proteins in the PSD of neurons, which underlies a synapse-to-nucleus communication important for the transcriptional regulation.
Keywords: glutamate, Elk-1, CREB, Fos, PSD-95, Homer
Introduction
The NMDA receptor (NMDAR) controls intracellular Ca2+ signaling (McBain and Mayer, 1994; Dingledine et al., 1999). The group I metabotropic glutamate receptors (mGluRs), on the other hand, increase the hydrolysis of membrane phosphoinositide (PI), via activating G-protein-dependent phospholipase Cβ1 (PLCβ1), to yield diacylglycerol (DCG), which activates protein kinase C (PKC), and inositol-1,4,5-triphosphate (IP3), which releases intracellular Ca2+ ([Ca2+]i) (Nakanishi, 1994; Conn and Pin, 1997). In addition to the conventional Ca2+ signaling, NMDARs are organized into multiprotein complexes with synaptic proteins within the postsynaptic density (PSD) (Ziff, 1997; Sheng and Kim, 2002). A prominent organizing protein in complexes is PSD-95 (Kornau et al., 1995), which couples the C terminus of the modulatory NMDAR subunit NR2B to various cytoplasmic proteins and enzymes (Sheng, 2001; Sheng and Kim, 2002). Similarly, group I mGluRs are organized with the synaptic proteins named Homer (Brakeman et al., 1997). Through long C-terminal intracellular tails, group I mGluRs bind all members of Homer families, including inducible immediate-early gene Homer1a and constitutively expressed Homer1b/c, Homer2, and Homer3 (Brakeman et al., 1997; Xiao et al., 1998, 2000). Emerging evidence indicates that the coordinated protein-protein interaction among receptors and submembraneous scaffolding/signaling proteins in the PSD enables the PSD apparatus to transmit variable extracellular signals to precise postsynaptic activity and plasticity (Sheng and Kim, 2002). In particular, Homer1b/c, which can promote multimerization through its coiled-coil domain and leucine zipper motifs (Xiao et al., 1998), is enriched along the interface of the PSD and the subjacent cytoplasm (Xiao et al., 1998). This physical arrangement may allow Homer1b/c to couple NMDARs inside of the PSD to group I mGluRs at its periphery (Sheng and Kim, 2000) and may thus promote a potential synergistic effect after concomitant stimulation of the two receptors.
Extracellular signal-regulated protein kinases 1 and 2 (ERK1/2 or p44/p42) in postmitotic neurons are involved in the regulation of multiple cellular activities, including inducible gene expression (Volmat and Pouyssegur, 2001). Hyperphosphorylation of ERK1/2 on their Thr202 and Tyr204 sites has been demonstrated in many cell lines in response to a wide range of extracellular stimuli, including glutamate stimulation (Peyssonnaux and Eychene, 2001; Wang et al., 2004). Agonist binding of NMDARs activates ERK1/2 (Sgambato et al., 1998; Mao et al., 2004), probably via a signaling mechanism involving several Ca2+-sensitive kinases (Perkinton et al., 1999, 2002). Stimulation of group I mGluRs also activate ERK1/2 via a less-characterized pathway (Vanhoutte et al., 1999; Thandi et al., 2002). The multiple positive signaling pathways to ERK1/2 through different glutamate receptors imply the potential integral regulation of ERK1/2 by concurrent activation of these receptors.
In this study, we examined the interaction between NMDARs and group I mGluRs and have identified a novel synergistic effect of coactivation of the two receptors on ERK1/2 in neurons. Interestingly, the synaptic proteins in the postsynaptic density, PSD-95 and Homer1b/c, couple the converging signals from NMDARs and mGluR5 to ERK1/2 in a Ca2+-independent manner. Furthermore, we demonstrated that the activated ERK1/2 is involved in an efficient cascade for synapse-to-nucleus communication regulating gene expression.
Materials and Methods
Primary striatal neuronal cultures. Standardized procedures preparing primary striatal neuronal cultures from 18 d rat embryos or neonatal 1 d rat pups (Charles River, New York, NY) were used in this study (Mao and Wang, 2002, 2003a). Predominant GABAergic neurons were obtained using this procedure, as evidenced by the fact that >90% of total cells were immunoreactive to glutamic acid decarboxylase-65/67, GABA, and the specific marker for neurons [microtubule-associated protein-2a + 2b (MAP2)] but not for glia (glial fibrillary acidic protein). Cells were usually cultured for 15-18 d before use.
Western blot analysis. Cell lysates from cultures were sonicated, protein concentrations were determined, and the equal amount of protein (20 μg in 20 μl/lane) was loaded on NuPAGE Novex 4-12% gels (Invitrogen, Carlsbad, CA) for separation of proteins. Proteins were transferred to polyvinylidene fluoride membrane (Immobilon-P; 0.45 mm; Millipore, Bedford, MA) and blocked in blocking buffer (5% nonfat dry milk in PBS and 0.1% Tween 20) for 1 hr. The blots were incubated in primary rabbit polyclonal antibodies against pERK1/2(Thr202/Tyr204) (1:1000; Cell Signaling, Beverly, MA), ERK1/2 (1:1000; Cell Signaling), phosphorylated cAMP response element-binding protein (pCREB) (1:500; Santa Cruz Biotechnology, Santa Cruz, CA), CREB (1:500; Santa Cruz Biotechnology), phosphorylated Elk-1 (pElk-1) (1:100; Santa Cruz Biotechnology), Elk-1 (1:500; Santa Cruz Biotechnology), c-Fos (1:1000; Oncogene Research Products, San Diego, CA), or Homer2a/b (1:500; Santa Cruz Biotechnology), or in rat antibodies against Homer1b/c (1:1000; Chemicon, Temecula, CA) overnight at 4°C. This was followed by a 1 hr incubation in goat anti-rabbit or anti-rat horseradish peroxidase-linked secondary antibodies (Jackson ImmunoResearch, West Grove, PA) at 1:5000. Immunoblots were developed with the enhanced chemiluminescence (ECL) reagents (Amersham Biosciences, Piscataway, NJ) and captured into Kodak (Rochester, NY) Image Station 2000R. Kaleidoscopeprestained standards (Bio-Rad, Hercules, CA) were used for protein size determination. The density of immunoblots was measured using the Kodak 1D Image Analysis software, and all bands were normalized as percentage of control values.
Single and double immunofluorescent labeling. The immunofluorescent labeling on eight-chamber glass slides was performed as described previously (Mao and Wang, 2002, 2003a). Briefly, cultures were fixed in cold 4% paraformaldehyde (10 min), followed by incubation in 4% normal donkey serum and 1% bovine serum albumin (20 min), to block non-specific staining. The cells were treated with primary rabbit antibodies against pERK1/2 or ERK1/2 (Cell Signaling) overnight at 4°C. Sections were then incubated for 1 hr with donkey anti-rabbit secondary antibodies (1:200) conjugated to FITC (Jackson ImmunoResearch). For double labeling, the cells were treated with a mixture of primary antibodies containing rabbit anti-mGluR5 antibodies (1:200; Upstate, Charlottesville, VA) and mouse anti-MAP2 antibodies (1:250; Chemicon) or mouse anti-Tau1 antibodies (1:200; Chemicon) overnight at 4°C. Sections were then incubated for 1 hr with donkey anti-rabbit secondary antibodies conjugated to FITC and donkey anti-mouse secondary antibodies conjugated to tetramethylrhodamine isothiocyanate (Jackson ImmunoResearch) at 1:200. The immunofluorescent images were analyzed using confocal microscopy using a Nikon C1 laser scanning confocal microscope.
Coimmunoprecipitation. Rat striatal cell proteins were prepared under weakly denaturing conditions known to permit the NMDAR-PSD-95 interaction (Takagi et al., 2000). Briefly, striatal cultures were scraped into a microtube containing ice-cold sample buffer (in mm): 10 Tris-HCl, pH 7.4, 5 NaF, 1 Na3VO4, 1 EDTA, and 1 EGTA. The sample was homogenized by sonication. The homogenate was centrifuged at 800 × g for 10 min at 4°C. The supernatant was again centrifuged at 11,000 × g at 4°C for 30 min to obtain the P2 pellet. The P2 pellet, a fraction enriched with synaptic structures, was resuspended in sample buffer and solubilized in sodium deoxycholate (a final concentration of 1%). Samples were held at 37°C for 30 min, and Triton X-100 was added to a final concentration of 0.1%. Insoluble proteins were sedimented by centrifugation at 100,000 × g at 4°C for 20 min. The supernatants were used for coimmunoprecipitation. NR2 subunits were precipitated using rabbit polyclonal antibodies against NR2A or NR2B (Upstate) and 50% protein A agarose/Sepharose bead slurry (Amersham Biosciences). Proteins were separated on NuPAGE Novex 4-12% gels (Invitrogen) and probed with anti-NR2B, anti-NR2A, or anti-PSD-95 (clone K28/43; Upstate) antibodies. HRP-conjugated secondary antibodies and ECL were used to detect proteins. Negative controls with antigen preabsorption were performed for antibodies used in immunoprecipitation.
PI hydrolysis. PI hydrolysis was analyzed by measuring the accumulation of tritiated inositol monophosphate. Cultures were incubated for 24 hr with 2 μCi/ml myo-[3H]-inositol. Cells were washed three times with 0.5 ml of HEPES-buffered balanced salt solution (HBS) (in mm: 154 NaCl, 5.6 KCl, 2 CaCl2, 2 MgSO4, 5.5 glucose, and 20 HEPES-KOH or HEPES-NaOH, pH 7.4) and incubated in 0.75 ml of this solution. LiCl (10 mm) was added for 30 min before agonist addition. After drug treatment, the solutions were aspirated, and 0.75 ml of ice-cold methanol was added to terminate the reaction. Cells were scraped and transferred to tubes containing 0.75 ml of chloroform. Samples were briefly sonicated and vortex mixed before the aqueous and organic phases were separated by centrifugation at 4000 rpm for 10 min. The upper aqueous phase (in 0.75 ml aliquot) was added to anion exchange columns containing Dowex-1 (format form) for the separation of [3H]-inositol-containing compounds (inositol monophosphates, bisphosphates, and triphosphates). [3H]-inositol monophosphate was eluted into scintillation vials and measured by a liquid scintillation counter.
[Ca2+]i measurements. [Ca2+]i measurements were performed according to our previous procedures (Mao and Wang, 2002, 2003a). Briefly, the culture was loaded with HBS aerated with 95% O2-5% CO2, pH 7.4, and contained 3 μm fura-2 AM. The fluorescence of fura-2 was sequentially excited at 340 and 380 nm. Emitted lights were collected from the sample through a cooled, intensified CCD video camera (IC-110; Photon Technology International, Lawrenceville, NJ). The fluorescent signal was measured at a single neuronal cell. Baseline was recorded for 3-5 min before bath application of drugs. The [Ca2+]i concentration was calculated from ratios of the intensities of emitted fluorescence at two excitation wavelengths (F340/F380) with Northern Eclipse Image software (Empix Imaging, Mississauga, Ontario, Canada). When needed, fluorescence ratios (340/380) were converted to an absolute [Ca2+]i concentration using the equation [Ca2+]i = Kd(Fmin/Fmax)[(R - Rmin)/(Rmax - R)].
Cell viability assay. Cell viability was measured using a double fluorescein diacetate/propidium iodide staining procedure (Jones and Senft, 1985). Fluorescein diacetate is membrane permeable and freely enters intact cells in which it is hydrolyzed by cytosolic esterase and converted to membrane-impermeable fluorescein with a green fluorescence, exhibited only by live cells. Propidium iodide is nonpermeable to live cells but penetrates the membranes of dying/dead cells, showing red fluorescence. Cells were rinsed twice with 1× PBS and incubated at 37°C for 5 min with 1× PBS (0.5 ml/per well) containing 10 μg/ml fluorescein diacetate (Sigma, St. Louis, MO) and 5 μg/ml propidium iodide (Sigma). Cultures were washed once with PBS and examined under fluorescent light microscopy. The total numbers of viable cells stained by green fluorescein and dead cells stained by red propidium iodide were determined by counting cells in five random fields. Positive control was produced by treating cultures with kainic acid (500-1000 μm; 24 hr).
Small interfering RNA preparation and transient transfections. The small interfering RNA (siRNA) duplexes were chemically synthesized and annealed by Qiagen (Valencia, CA), with 5′ phosphate, 3′ hydroxyl, and two base (dTdT) overhangs on 3′ of each strand. The siRNA duplex sequences targeting the Homer1b/c mRNA (Brakeman et al., 1997) contain sense 5′-UCAGUCUAGACGGCUCAAA-3′ and antisense 5′-UUUGAGCCGUCUAGACUGA-3′. The siRNA sequences targeting the Homer2a/b mRNA (Kato et al., 1998) contain sense 5′-UCGAGACGUCAAGUAAUCA-3′ and antisense 5′-UGAUUACUUGACGUCUCGA-3′. The siRNA corresponding to the enhanced green fluorescent protein (EGFP) reporter gene and control siRNA were purchased from Qiagen (Caplen et al., 2001).
Cotransfections of reporter plasmids and siRNAs were performed on eight-chamber coverglass slides (extra thin for fluorescent detection). Following the manufacturer's instructions, 1 μl of the cationic lipid Lipofectamine 2000 (Invitrogen) was diluted in 50 μl of antibiotic-free and serum-reduced Opti-MEM I medium (Invitrogen) and incubated for 5 min at room temperature. A total of 0.5 μg of pCMS-EGFP vector (Clontech, Palo Alto, CA) and 0.3, 0.6, or 0.9 μg of EGFP siRNAs were mixed in Opti-MEM I (50 μl) and added to the Lipofectamine mixture, and incubation continued for 20 min at room temperature. Cells were quickly washed with Opti-MEM I three times before the addition of the above mixed medium. Cells were transfected for 2-4 hr at 37°C, followed by three rinses in Opti-MEM I. Cells were then incubated in 400 μl of growth medium until analysis. Transfection efficiencies were determined by nuclear DNA staining with 4′,6-diaminino-2-phenylindole (DAPI). Transfections of siRNAs against Homer1b/c were performed on a 24-well plate with Lipofectamine 2000 as described above. Cells were lysed for examining Homer1b/c or Homer2a/b immunoreactivity or used to detect ERK1/2 phosphorylation 24 hr after transfections.
Drug treatments. Cultures were washed with PBS and preincubated at 37°C for 60 min in the humidified atmosphere of 5% CO2 in HBS. For NMDA and glutamate treatments, MgSO4 was omitted from and 10 μm glycine was added to the HBS. For extracellular Ca2+ dependency studies, CaCl2 was omitted from the HBS with substitution of a Ca2+-chelator EGTA (2 mm). An Na+-free HBS was made by iso-osmotically substituting N-methyl-d-glucamine for NaCl and adjusting the pH to 7.2 with HCl. To have parameters comparable, none of the HBSs contained sodium bicarbonate. Cells were treated by adding drug to the HBS. At the end of drug treatment, the cells were quickly washed with ice-cold PBS (pH 7.4; Ca2+ free) and placed on ice immediately. The cell monolayer was rapidly scraped in ice-cold lysis buffer. All drugs were freshly made on the day of the experiment. Drugs were dissolved in 1× PBS with or without an aid of dimethyl sulfoxide (DMSO). Whenever DMSO was used, PBS containing the same concentration of DMSO was used as the control vehicle.
Materials. Chemicals and reagents were obtained from the following sources: NMDA, AMPA, (+)-5-methyl-10,11-dihydro-5H-dibenzo [a,d] cyclohepten-5,10-imine maleate (MK801), dl-2-amino-5-phosphonovaleric acid (AP-5), 6,7-dinitroquinoxaline-2,3-dione (DNQX), 4-(8-methyl-9H-1,3-dioxolo[4,5-hr][2,3]benzodazepin-5-yl)-benzenamine dihydrochloride (GYKI52466), (S)-3,5-dihydroxyphenylglycine (DHPG), (RS)-2-chloro-5-hydroxyphenylglycine (CHPG), 2-methyl-6-(phenyl-ethynyl)pyridine hydrocholoride (MPEP), 7-(hydroxyimino)cy-cloprop-a[b]chromen-1a-carboxylate ethyl ester (CPCCOet), bicuculline, (2S)-3-[[(1S)-1-(3,4-dichlorophenyl)ethyl]amino-2-hydroxypropyl](phenylmethyl)phosphinic acid (CGP 55845), nifedipine, thapsigargin, and 1,4-diamino-2,3-dicyano-1,4-bis[2-aminophenylthio]butadiene (U0126) from Tocris Cookson (Ballwin, MO); 1-[6[[(17β)-3-methoxyestra-1,3,5(10)trien-17-yl]amino]hexyl]-1H-pyrrole-2,5-dione (U73122) and 3-[1-3-(amidinothio)propyl-1H-indol-3-yl]-3-(1-methyl-1H-indol-3-yl) maleimide (Ro-31-8220) from Calbiochem (San Diego, CA); l-glutamate, 2,5-dimethyl-4-[2-(phenylmethyl)ben-zoyl]-1H-pyrrole-3-carboxylic acid methylester (FPL64176), tetrodotoxin (TTX), 2-[1-(3-dimethyl-aminopropyl)-5-methoxyindol-3-yl]-3-(1H-indol-3-yl)maleimide (Gö6983), and phorbol 12-myristate 13-acetate (PMA) from Sigma. Fura-2 AM fluorescent Ca2+ indicator was purchased from Molecular Probes (Eugene, OR). Myo-[3H]-inositol was obtained from American Radiolabeled Chemicals (St. Louis, MO). The synthesis of the membrane-permeable PSD-95-binding (decoy) peptide Tat-NR2Bct (YGRKKRRQRRRKLSSIESDV) and its control peptide with a double point mutation in the C-terminal tSXV motif rendering it incapable of binding PSD-95, Tat-NR2Baa (YGRKKRRQRRRKLSSIEADA), has been described previously (Aarts et al., 2002). Both peptides gain cell permeability by containing an arginine-enriched cell-membrane transduction domain of the human immunodeficiency virus type 1 (HIV-1) Tat protein (YGRKKRRORRR) (Schwarze et al., 1999). A Tat control peptide was synthesized that comprises HIV-1 Tat residues 38-48 (KALGISYGRKK; Tat38-48) outside the transduction domain (Mann and Frankel, 1991).
Statistics. The results are presented as mean ± SEM. The data were evaluated using a one- or two-way ANOVA, as appropriate, followed by a Bonferroni (Dunn) comparison of groups using least squares-adjusted means. Probability levels of <0.05 were considered statistically significant.
Results
ERK1/2 phosphorylation by coactivation of NMDARs and group I mGluRs
Activation of NMDARs with NMDA or group I mGluRs with the selective agonist DHPG consistently caused concentration-dependent phosphorylation of ERK1/2 (Fig. 1A). NMDA and DHPG when applied alone at 1 and 3 μm did not change basal levels of pERK1/2. Interestingly, when NMDA and DHPG were coincubated, robust phosphorylation of ERK1/2 occurred at this low concentration range (Fig. 1A). No significant changes were seen in cellular levels of ERK after NMDA and DHPG application alone or in combination at all concentrations surveyed. These data showed a synergistic upregulation of ERK1/2 phosphorylation after costimulation of NMDARs and group I mGluRs. To evaluate the time course of the pERK1/2 induction, NMDA and DHPG at 1-3 μm were added to the cultures for different durations. As shown in Figure 1B, a rapid and transient ERK1/2 phosphorylation occurred without changes in ERK1/2 levels. A different time course evaluation was also performed in which the two drugs (3 μm) were incubated for 1 min and cultures were then lysed 1, 4, 10, or 30 min after the termination of drug treatment. The data from this study (data not shown) were similar to those obtained by the NMDA/DHPG incubation at different durations. To define the subcellular distribution of the induced pERK1/2, we performed immunofluorescent labeling on culture slides. Because the maximal phosphorylation of ERK1/2 was obtained after NMDA/DHPG incubation at 3 μm for 5 min (see above), immunofluorescent images were analyzed on cultures treated with the two drugs at 3 μm for 5 min. Strong pERK1/2 immunostaining was revealed in the nuclear envelop, whereas weak to moderate staining was revealed in the cytoplasm and neural processes after NMDA/DHPG stimulation (Fig. 1C).
Figure 1.

ERK1/2 phosphorylation by coactivation of NMDARs and group I mGluRs with the specific agonists at their low dose range in cultured rat striatal neurons. A, Coadministration of NMDA and DHPG at 1 and 3 μm (5 min) induced a robust increase in pERK1/2 but not ERK1/2 levels, whereas the two agonist applications alone at the same concentrations for 5 min did not change pERK1/2 and ERK1/2 levels. Representative immunoblots are shown left of the quantified data of pERK2 analyzed from separate experiments (mean ± SEM; n = 8-10). +p < 0.05 versus NMDA and DHPG alone at the corresponding concentration. B, NMDA/DHPG caused a rapid and transient increase in pERK1/2 levels without changing ERK1/2 levels. The two drugs at 1-3 μm were added to cultures and incubated for different durations. Representative immunoblots are shown left of the quantified data (mean ± SEM; n = 5). *p < 0.05 versus basal levels. C, Confocal immunofluorescent images illustrating subcellular distributions of pERK1/2 induced by NMDA/DHPG (3 μm; 5 min). Strong pERK1/2 immunostaining is present in the nucleus, and weak to moderate staining is present in the cytoplasm and neural processes.
AMPA receptors cooperate with NMDARs in many normal and inducible cellular activities. To examine whether subsequent activation of AMPA receptors occurred to contribute to NMDA/DHPG-stimulated ERK1/2 phosphorylation, we evaluated the effect of the AMPA receptor-selective antagonist GYKI52466 on NMDA/DHPG action. Pretreatment with GYKI52466 at a dose (100 μm) sufficient to block the AMPA-induced ERK1/2 phosphorylation did not change the ERK1/2 phosphorylation induced by NMDA/DHPG (Fig. 2A). Neither did another AMPA receptor antagonist, DNQX, at 100 μm, a dose sufficient to block the AMPA phosphorylation of ERK1/2 (data not shown). Thus, subsequent activation of AMPA receptors, if there is any, is not involved in the NMDA/DHPG effect. Similarly, the L-type, voltage-operated Ca2+ channel (VOCC) may not be an important component in the NMDA/DHPG phosphorylation of ERK1/2 because the VOCC inhibitor nifedipine, at a concentration effective to block the VOCC activator FPL64176-stimulated ERK1/2 phosphorylation, did not change the hyperphosphorylation of ERK1/2 induced by NMDA/DHPG (Fig. 2B).
Figure 2.

Effects of the AMPA receptor antagonist GYKI52466 (A), the VOCC blocker nifedipine (B), the Na+ channel blocker TTX (C), and the GABAA receptor antagonist bicuculline and GABAB receptor antagonist CGP 55845 (D) on basal and NMDA/DHPG (N/D)-induced ERK1/2 phosphorylation in cultured rat striatal neurons. GYKI52466 (100 μm), nifedipine (20 μm), bicuculline (20 μm), or CGP 55845 (1 μm) was incubated 30 min before and during a 5 min treatment with AMPA (50 μm), the VOCC activator FPL64176 (20 μm), or a mixture of NMDA (3 μm) and DHPG (3 μm). TTX (1 μm) was incubated 45 min before and during a 5 min treatment with a mixture of NMDA (3 μm) and DHPG (3 μm). Note that GYKI52466 and nifedipine completely blocked AMPA- and FPL64176-induced ERK1/2 phosphorylation, respectively. However, GYKI52466 and nifedipine did not affect the ERK1/2 phosphorylation induced by NMDA/DHPG. In the presence of TTX, bicuculline, or CGP 55845, NMDA/DHPG increased ERK1/2 phosphorylation similar to that in the absence of those blockers. Representative immunoblots are shown left of the quantified data of pERK2 (mean ± SEM; n = 4-6). *p < 0.05 versus basal levels; +p < 0.05 versus the corresponding agonist.
Cell firing induced by NMDA and/or DHPG could cause a release of an assortment of transmitters and modulators in the culture, leading to activation of receptors other than NMDARs/group I mGluRs to account for the observed changes in ERK1/2 phosphorylation after NMDA/DHPG application. However, this possibility seems less likely because blockade of action potential firing by pretreatment with TTX (1 μm), a Na+ channel blocker, did not effect NMDA/DHPG-induced ERK1/2 phosphorylation (Fig. 2C).
The striatal culture system used in this study contains predominantly GABAergic neurons. To test the possible contribution of GABAergic transmission to the synergistic effect of NMDA/DHPG, effects of coapplication of NMDA and DHPG on ERK phosphorylation were examined in the presence of a GABAA or GABAB receptor antagonist. From Figure 2D, the basal and NMDA/DHPG-induced ERK1/2 phosphorylation was not changed by the GABAA receptor antagonist bicuculline (20 μm) or the GABAB receptor antagonist CGP 55845 (1 μm). Thus, the GABAergic transmission plays an insignificant role in the NMDA/DHPG effect.
NMDA/DHPG-induced ERK1/2 phosphorylation is independent on Ca2+ and Na+ influx through NMDA channels
As ligand-gated Ca2+ and Na+ channels, NMDARs, after activation, lead to Ca2+ and Na+ influx, which in turn activate a variety of intracelluar signaling cascades. To determine the contribution of NMDAR Ca2+/Na+ channel activity to NMDA/DHPG phosphorylation of ERK1/2, a series of the following experiments were conducted in cultured neurons. Pretreatment with a competitive NMDAR antagonist, AP-5 (50 or 100 μm), blocked the ERK1/2 phosphorylation induced by NMDA and DHPG (3 μm for both drugs) (Fig. 3A), indicating that the response requires the activation of AP-5-sensitive NMDARs. However, in the parallel experiments, we found that a noncompetitive open-channel blocker, MK801 (10 μm), and an ion channel blocker, Mg2+ (5 mm), that were able to completely block NMDA (50 μm)-induced increases in [Ca2+]i concentrations as measured by fura-2 fluorescence (Fig. 3F) did not significantly inhibit NMDA/DHPG (3 μm)-induced ERK1/2 phosphorylation (Fig. 3B). Similarly, the cell-permeable Ca2+ chelators BAPTA-AM (30 μm) and Calcium Green-1/AM (30 μm) that inhibited Ca2+ responses to 50 μm NMDA (Fig. 3F) failed to inhibit the ERK1/2 phosphorylation (Fig. 3C). Furthermore, NMDA/DHPG (3 μm) continued the ability to increase pERK1/2 levels in the extracellular Ca2+-free solution (Fig. 3D) in which NMDA (50 μm) could not induce a Ca2+ rise (Fig. 3F). NMDA (3 μm) and DHPG (3 μm) alone did not change [Ca2+]i levels, whereas the two drugs together at 3 μm induced a small but significant Ca2+ rise in most neurons surveyed, probably because of their additive effects (Fig. 3E). In the presence of MK801, the Ca2+ responses to NMDA/DHPG did not reach a significant level (Fig. 3E). To determine the role of Na+ ions, we found that NMDA/DHPG (3 μm) phosphorylated ERK1/2 in Na+-free medium to the extent comparable with that obtained in Na+-containing medium (data not shown). Together, these results suggest that NMDA and DHPG activate ERK1/2 through an NMDAR-sensitive mechanism but without involving NMDAR-mediated Ca2+/Na+ channel activities.
Figure 3.

Ca2+ independence of NMDAR-mediated ERK1/2 phosphorylation. A, Pretreatment of cultured striatal neurons with the competitive NMDAR antagonist AP-5 blocked NMDA/DHPG (N/D)-induced ERK1/2 phosphorylation. Representative immunoblots are shown left of the quantified data of pERK2 (mean ± SEM; n = 6). B, The open-channel blockers MK801 and Mg2+ did not inhibit NMDA/DHPG-induced ERK1/2 phosphorylation. Representative immunoblots are shown left of the quantified data of pERK2 (mean ± SEM; n = 5). C, The Ca2+ chelator BAPTA-AM did not inhibit ERK2 phosphorylation (mean ± SEM; n = 4). AP-5 (50 or 100 μm), MK801 (10 μm), or BAPTA-AM (30 μm) was incubated 30 min before and during a 5 min treatment with a mixture of NMDA (3 μm) and DHPG (3 μm). D, NMDA and DHPG (3 μm; 5 min) increased ERK2 phosphorylation in Ca2+-free medium (mean ± SEM; n = 4). E, Application of NMDA and DHPG alone at 3 μm induced an insignificant [Ca2+]i rise, whereas NMDA plus DHPG (3 μm) induced an MK801 (10 μm)-sensitive Ca2+ rise. F, Inhibition of NMDA (50 μm)-stimulated Ca2+ responses by AP-5 (50 μm), MK801 (10 μm), BAPTA-AM (30 μm), and Mg2+ (5 mm) and the removal of extracellular Ca2+ ions. The values in E and F are expressed as mean fold changes of basal levels in terms of the peak amplitude of Ca2+ responses measured within 1 min after the start of drug treatment from 17-28 neurons. *p < 0.05 versus basal levels; +p < 0.05 versus NMDA plus DHPG.
PSD-95 contributes to NMDA/DHPG-regulated ERK1/2 phosphorylation
A prominent organizing protein to NMDARs is PSD-95, which binds the C terminus of NMDAR NR2 subunits (Sheng, 2001; Sheng and Kim, 2002). To probe the importance of PSD-95 in converging NMDAR signals to ERK1/2, we chose a strategy to transduce cultured neurons with peptides that disrupt the association of NMDARs with PSD-95 without affecting NMDAR ion channel activity (Aarts et al., 2002). The peptides include the membrane-permeable PSD-95-binding (decoy) peptide Tat-NR2Bct and two control peptides, Tat-NR2Baa with the incapability of binding PSD-95 because of a double point mutation and Tat38-48 with the incapability of transducing into neurons because of its peptide sequence selected from the nontransduction domain. We first detected whether Tat-NR2Bct would transduce into neurons. After bath application of fluorescein-conjugated Tat-NR2Bct (5 μm), living neurons exhibited intense fluorescence in their cytoplasm and processes, demonstrating intracellular peptide uptake (Fig. 4A, left). In contrast, treatment with fluorescein-conjugated Tat38-48 (5 μm) showed no peptide uptake (Fig. 4A, right). Tat-NR2Bct-fluorescein started to accumulate in neurons 10-15 min after application, peaked in the next hour, and remained 3-5 hr after being washed from the cultures.
Figure 4.

Influence of dissociation of NMDAR-PSD-95 interaction by Tat peptides on the NMDA/DHPG-induced ERK1/2 phosphorylation. A, Confocal images visualizing intracellular accumulation of Tat-NR2Bct-fluorescein (5 μm; left) but not of Tat38-48-fluorescein (5 μm; right) 1 hr after the addition to striatal cultures (representative of 5 experiments). B, Coimmunoprecipitation of PSD-95 with NMDAR NR2B subunits in cultured rat striatal neurons treated with Tat peptides (5 μm; 1 hr). The coimmunoprecipitation of PSD-95 in terms of the optical density (O.D.) ratio of PSD-95/NR2B was significantly reduced by Tat-NR2Bct but not by Tat38-48 and Tat-NR2Baa. Left, Representative gels. Right, Means ± SEM of six to eight experiments. *p < 0.05 versus basal levels. C, D, Effects of Tat-NR2Bct and its mutant control (Tat-NR2Baa) on ERK1/2 phosphorylation induced by NMDA/DHPG (N/D). The Tat peptides (0.5 or 5 μm) were incubated 1 hr before and during a 5 min treatment with a mixture of NMDA (3 μm) and DHPG (3 μm). Tat-NR2Bct (C), but not Tat-NR2Baa (D), reduced NMDA/DHPG-induced ERK1/2 phosphorylation. Representative immunoblots are shown left to the quantified data of pERK2 (mean ± SEM; n = 4). *p < 0.05 versus basal levels; +p < 0.05 versus NMDA plus DHPG.
It is assumed that Tat-NR2Bct could disturb NMDAR-PSD-95 binding. To verify this assumption, effects of the peptide on this binding were examined by testing the coimmunoprecipitation of PSD-95 with NR2 subunits. Incubation of cultures with either of the two control peptides, Tat-NR2Baa and Tat38-48 (5 μm; 1 hr), did not reduce the coimmunoprecipitation of PSD-95 with NR2B (Fig. 4B). However, the coimmunoprecipitation of PSD-95 with NR2B was reduced by Tat-NR2Bct incubation (5 μm for 1 hr) (Fig. 4B). The coimmunoprecipitation of PSD-95 with NR2A was unchanged by Tat-NR2Bct (data not shown). Thus, Tat-NR2Bct possesses the ability to selectively perturb PSD-95-NR2B binding. The failure of Tat-NR2Bct to interfere with PSD-95-NR2A binding is consistent with a previous report (Aarts et al., 2002) indicating that NR2A may be more tightly bound to PSD-95 or that Tat-NR2Bct, because of its incomplete homology for the C terminus of NR2A, was less effective in perturbing PSD-95-NR2A binding (Aarts et al., 2002).
After the demonstration of the selective perturbation of NR2B association with PSD-95 by Tat-NR2Bct, we then examined the effects of the peptide on NMDA/DHPG-stimulated ERK1/2 phosphorylation. We found that Tat-NR2Bct (0.5 and 5 μm; 1 hr) reduced the ERK1/2 phosphorylation induced by NMDA/DHPG at 3 μm (Fig. 4C). On the contrary, the control peptide Tat-NR2Baa did not change ERK1/2 phosphorylation (Fig. 4D). These data support the notion that the association of NR2B with PSD-95 is required for activating ERK1/2 by coactivation of NMDARs and group I mGluRs. In addition, Tat-NR2Bct itself had no effect on basal levels of pERK1/2 (Fig. 4C), indicating that the peptide, by simply replacing NR2B binding to PSD-95, had no functional roles in activating the downstream effector, leading to ERK1/2 phosphorylation. Similarly, Tat-NR2Bct (5 μm; 1 hr) did not change Ca2+ responses to NMDA (10-100 μm; data not shown), consistent with previous observations that NMDAR channel activity is unaffected by disrupting PSD-95 (Migaud et al., 1998; Sattler et al., 1999; Aarts et al., 2002).
mGluR5, but not mGluR1, mediates NMDA/DHPG-induced ERK1/2 phosphorylation
A series of studies were performed to evaluate the relative importance of the two subtypes of group I mGluRs, mGluR1 and mGluR5, in mediating NMDA/DHPG-induced ERK1/2 phosphorylation. The noncompetitive mGluR5-selective antagonist MPEP at low doses of 0.04 and 0.2 μm blocked the ERK1/2 phosphorylation induced by NMDA/DHPG (3 μm) (Fig. 5A). In contrast, the noncompetitive mGluR1-selective antagonist CPCCOet at high doses of 5 and 25 μm did not change ERK1/2 phosphorylation (Fig. 5B). The mGluR5-selective agonist CHPG at 10-20 μm did not increase ERK1/2 phosphorylation, but when coincubated with NMDA (3 μm), CHPG induced an increase in pERK1/2 that was sensitive to MPEP blockade (0.2 μm) but not CPCCOet blockade (25 μm) (data not shown). Immunoblots prepared with ERK1/2 antibody showed again that changes in ERK1/2 phosphorylation were not attributable to changes in total ERK1/2 protein (Fig. 5 A, B). Together, these data suggest that selective activation of mGluR5 rather than mGluR1 mediates NMDA/DHPG-induced ERK1/2 phosphorylation.
Figure 5.
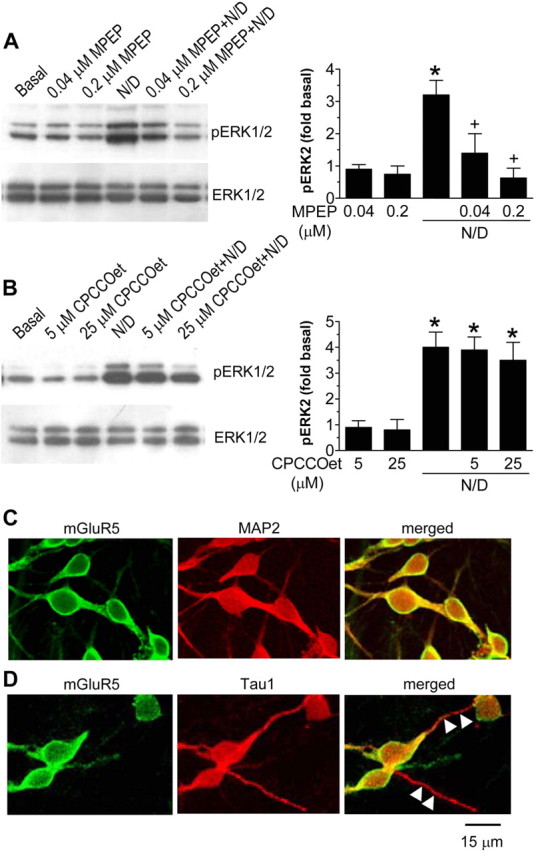
mGluR5-mediated ERK1/2 phosphorylation after NMDA and DHPG application in cultured rat striatal neurons. A, Pretreatment of striatal neurons with the mGluR5-selective antagonist MPEP completely blocked the ERK1/2 phosphorylation induced by NMDA/DHPG (N/D). B, Pretreatment with the mGluR1-selective antagonist CPCCOet failed to change ERK1/2 phosphorylation. MPEP (0.04 or 0.2 μm) or CPCCOet (5 or 25 μm) was incubated 30 min before and during a 5 min treatment with a mixture of NMDA (3 μm) and DHPG (3 μm). Representative immunoblots are shown left to the quantified data of pERK2 (mean ± SEM; n = 4-5). C, D, Confocal immunofluorescent images illustrating subcellular distributions of mGluR5 in cultured rat striatal neurons. Note the colocalization of mGluR5 immunostaining with the MAP2-positive soma and neurites (C) but not with the Tau1-positive neurites (D; white arrowheads). *p < 0.05 versus basal levels; +p < 0.05 versus NMDA plus DHPG.
To evaluate the presynaptic and postsynaptic mechanisms for the mGluR5′ role, we examined the distribution of presynaptic versus postsynaptic mGluR5 in this culture system. The mGluR5 immunolabeling was colocalized with the somatodendritic marker MAP2 (28 of 28 neurites) (Fig. 5C) but not with the axonal marker Tau1 (0 of 30 neurites) (Fig. 5D), a similar result seen in cultured cerebellar granule neurons with transfected mGluR5 and Homer1b (Ango et al., 2000). Thus, regardless of the possible presence of presynaptic mGluR5 in the adult brain (Romano et al., 1995; Shigemoto et al., 1997; Lujan et al., 1997; Ango et al., 2000), endogenous mGluR5 was located in both cell bodies and dendrites but not axons of cultured striatal neurons. This narrows the possibility for a presynaptic mechanism to underlie the mGluR5 participation in NMDA/DHPG-stimulated ERK1/2 phosphorylation.
NMDA/DHPG-induced ERK1/2 phosphorylation is independent on mGluR5-mediated PLCβ1 activation and Ca2+ release
Stimulation of Gαq protein-coupled mGluR5 activates a predominant downstream effector, PLCβ1. Active PLCβ1 then increases PI hydrolysis to produce IP3, which releases Ca2+ from intracellular Ca2+ stores (Nakanishi, 1994; Conn and Pin, 1997). The above finding that demonstrates the mGluR5 contribution to NMDA/DHPG action raises the possibility that this response is mediated by the activation of PLCβ1 and its downstream [Ca2+]i mobilization. To test this possibility, an amino steroid inhibitor of PLCβ1, U73122, was used in this study. We found that U73122 at 40 μm did not change NMDA/DHPG (3 μm)-induced ERK2 phosphorylation (Fig. 6A). Consistent with the ability to inhibit PLCβ1, U73122 (40 μm) totally blocked the DHPG (100 μm)-induced increases in PI hydrolysis (Fig. 6B). It is noted that DHPG (1-3 μm) did not induce significant increases in PI hydrolysis (Fig. 6B). To test the involvement of Ca2+ release, a [Ca2+]i-depleting agent, thapsigargin, was used for 1 hr before application of NMDA/DHPG to discharge internal Ca2+ stores. We found that thapsigargin (2 μm) failed to inhibit NMDA/DHPG (3 μm)-induced ERK2 phosphorylation (Fig. 6C), although the agent was effective in inhibiting the [Ca2+]i rise induced by DHPG (100 μm) (Fig. 6D). Furthermore, the small Ca2+ rise induced by NMDA/DHPG (3 μm) was reduced or completely blocked by adding thapsigargin (2 μm) alone or thapsigargin (2 μm) plus MK801 (10 μm), respectively (Fig. 6D). In the presence of both thapsigargin (2 μm) and MK801 (10 μm), NMDA/DHPG (3 μm) remained to increase ERK1/2 phosphorylation to the extent similar to that seen in the absence of the two drugs (data not shown). These results indicate the lack of the conventional mGluR5-derived second-messenger system (PLCβ1/IP3/Ca2+) in a signaling cascade to ERK1/2 in response to coactivation of NMDARs and mGluR5. The mGluR5-mediated PLCβ1 activation also increases DCG, a stimulator of PKC. To determine the involvement of the DCG-PKC pathway, we examined the NMDA/DHPG effect in the presence of PKC inhibitors. We found that the two PKC inhibitors, Ro-31-8220 and Gö6983 (1 μm for both inhibitors; 30 min), blocked the PKC activator PMA (0.1 μm; 5 min)-stimulated ERK1/2 phosphorylation, sparing the ERK1/2 phosphorylation induced by NMDA/DHPG (3 μm for 5 min; data not shown). Thus, the DCG-PKC pathway is insignificant in mediating the NMDA/DHPG effect.
Figure 6.

The contribution of the mGluR5-associative signaling pathway to ERK2 phosphorylation induced by NMDA/DHPG in cultured rat striatal neurons. A, Pretreatment of striatal neurons with the PLCβ1 inhibitor U73122 did not change the ERK2 phosphorylation induced by NMDA/DHPG (N/D; n = 4). U73122 (40 μm) was incubated 30 min before and during a 5 min treatment with NMDA (3 μm) and DHPG (3 μm). B, DHPG at 1-3 μm did not elevate PI hydrolysis, and DHPG at 100 μm induced a U73122-sensitive increase in PI hydrolysis (n = 3-5). U73122 (40 μm) was incubated 30 min before and during a 5 min treatment with DHPG. C, Thapsigargin (Thap.) did not change the ERK2 phosphorylation induced by NMDA/DHPG (n = 4). Thapsigargin (2 μm) was incubated 1 hr before and during a 5 min treatment with NMDA (3 μm) and DHPG (3 μm). D, Thapsigargin completely blocked intracellular Ca2+ rises induced by DHPG (100 μm). Thapsigargin plus MK801 blocked Ca2+ responses to NMDA/DHPG application. Thapsigargin (2 μm) and/or MK801 (10 μm) were incubated 1 hr before and during a 5 min treatment with NMDA (3 μm) and DHPG (3 μm). The values are expressed as mean fold changes of basal levels in terms of the peak amplitude of Ca2+ responses measured within 1 min after the start of drug treatment from 14-26 neurons. *p < 0.05 versus basal levels; +p < 0.05 versus DHPG (100 μm); #p < 0.05 versus N/D.
NMDA/DHPG activates ERK1/2 via an mGluR5-Homer1b/c signaling cascade
Recent studies indicate that synaptic scaffolding/signaling proteins regulate glutamate receptor signaling and, specifically, that the Homer family of proteins contributes to mGluR5 signaling activity (Brakeman et al., 1997; Kato et al., 1998; Tu et al., 1998, 1999). Among the constitutively expressed long forms of Homer proteins, Homer1b/c proteins are most abundant, whereas Homer3 proteins are lacking, in striatal neurons in vivo (Shiraishi et al., 2004) and in vitro (Ango et al., 2000). Thus, the importance of Homer1b/c in mediating mGluR5 signals to ERK was evaluated by an approach using siRNAs. We first wanted to explore whether a functional RNA interference (RNAi) system exists in our cultured striatal neurons. Figure 7A shows that the cells transfected with a construct expressing EGFP (0.5 μg/well) showed strong expression of EGFP. The expressed EGFP was not affected by cotransfected control siRNAs (0.3-0.9 μg/well). In contrast, the cells cotransfected with the GFP siRNA (0.3-0.9 μg/well) showed no EGFP expression (Fig. 7A), indicating an existence of an effective RNAi apparatus in striatal neurons. There was no significant difference in cell viability as detected by the double fluorescein diacetate-propidium iodide staining between control and GFP siRNA-treated cultures.
Figure 7.

The contribution of the mGluR5-Homer1b/c signaling pathway to ERK2 phosphorylation induced by NMDA/DHPG in cultured rat striatal neurons. A, Expression of transfected EGFP in striatal neurons and depletion of EGFP expression by exposure to GFP siRNAs. Striatal neurons were cotransfected with EGFP-expressing vectors (0.5 μg/well) and control or GFP siRNAs (0.9 μg/well; 2-4 hr). One day after cotransfection, cultures were visualized by confocal microscopy for EGFP (green; left). The nucleus was visualized using the DNA dye DAPI (blue; right). The evenly stained, round morphology is typical of a healthy (nonapoptotic) nucleus. B, Homer1b/c siRNAs, but not control siRNAs, selectively reduced cellular Homer1b/c, but not Homer2a/b, protein levels. Control or Homer1b/c siRNAs were incubated for 2-4 hr at 1 μg/well, and cultures were lysed 24 hr later. Representative immunoblots are shown left of the quantified data (mean± SEM; n = 6). Lane 1, Basal; lane 2, control siRNAs; lane 3, Homer1b/c siRNAs. C, Control and Homer1b/c siRNAs did not affect basal and DHPG-stimulated PI hydrolysis. siRNAs (1 μg/well) were incubated for 2-4 hr, and effects of DHPG (100 μm; 5 min) on PI hydrolysis were tested 24 hr later (n = 4). D, Homer1b/c siRNAs reduced the NMDA/DHPG (N+D)-induced ERK1/2 phosphorylation. Control or Homer1b/c siRNAs were incubated for 2-4 hr at 1 μg/well, and NMDA (3 μm) and DHPG (3 μm) were then incubated alone or in combination 24 hr later for 5 min. Representative immunoblots are shown left of the quantified data (mean ± SEM; n = 6-7). *p < 0.05 versus basal levels; +p < 0.05 versus NMDA plus DHPG treatment after exposure to control siRNAs.
We next attempted to determine whether the Homer1b/c siRNA can selectively reduce the cellular levels of its targets. As shown in Figure 7B, a high basal level of Homer1b/c was exhibited in the untreated cultures. Cultures transfected with the control siRNAs (1 μg/well) showed no significant changes in total Homer1b/c levels. In contrast, cultures, after exposure to Homer1b/c siRNAs (1 μg/well), showed a reduced level of Homer1b/c by 63%. The effect of Homer1b/c siRNAs was evident 24 hr after transfection and persisted for 2 d. The suppression was only partial by 4 d safter transfection. To control the selectivity of Homer1b/c siRNAs, effects of the siRNAs on cellular levels of a closely related Homer family member Homer2a/b were tested. No significant effects of Homer1b/c siRNAs were detected on immunoreactivity of Homer2a/b (Fig. 7B). To evaluate the influence of Homer1b/c knockdown on mGluR5-mediated PI signaling, effects of Homer1b/c siRNAs on PI hydrolysis were examined in cultured neurons. Both basal and DHPG-stimulated (100 μm) or CHPG-stimulated (1 mm) PI hydrolysis was not changed in cultures treated with either control or Homer1b/c siRNAs (Fig. 7C), indicating that the siRNA knockdown of Homer1b/c does not affect the mGluR5-mediated PI hydrolysis. In separate studies, Homer2a/b siRNAs (1 μg/well; 24 hr) reduced cellular levels of Homer2a/b, but not Homer1b/c, by ∼40-50%.
After the demonstration of the effective and selective knockdown of Homer1b/c and Homer2a/b, we then detected the ability of NMDA/DHPG to phosphorylate ERK1/2 in Homer1b/c or Homer2a/b siRNA-treated cultures. In cultures treated with control siRNAs (1 μg/well), NMDA/DHPG (3 μm) induced a marked increase in ERK1/2 phosphorylation (Fig. 7D). In contrast, NMDA/DHPG (3 μm) failed to induce a significant increase in ERK1/2 phosphorylation in cultures exposed to 1 μg of Homer1b/c siRNAs at 24 hr (Fig. 7D). On the fourth day, NMDA/DHPG (3 μm) continued to increase pERK1/2 levels to a lesser extent compared with that shown in control siRNA-treated cultures (data not shown), indicating the reversible nature of the inhibition of the NMDA/DHPG effect by Homer1b/c siRNAs. Both control and Homer1b/c siRNAs did not change total ERK1/2 proteins (Fig. 7D). These results support that Homer1b/c proteins are essential components in the post-mGluR5 signaling complex mediating mGluR5 signals to ERK1/2. In contrast to Homer1b/c siRNAs, Homer2a/b siRNAs (1 μg/well; 24 hr) did not change the NMDA/DHPG (3 μm)-induced increase in pERK1/2 (data not shown), indicating an insignificant role of Homer2a/b in the event.
Physiological roles of NMDAR/mGluR5-sensitive ERK1/2 signaling in regulating gene expression
One of the particularly noticeable roles that active ERK1/2 plays is to facilitate gene expression via phosphorylating nuclear transcription factors (Wang et al., 2004). Among potential factor targets of active ERK1/2, we tested Elk-1 (Sgambato et al., 1998) and cAMP response element-binding protein (CREB). In addition, an immediate-early gene, c-Fos, was tested as a reporter of inducible gene expression downstream to Elk-1 and CREB to determine a final output of gene expression. We found that NMDA/DHPG (3 μm; 15 min) was effective in elevating both Elk-1 and CREB phosphorylation as opposed to no changes in phosphorylation of the two factors when the two drugs were given alone (data not shown). More dramatically, NMDA/DHPG at 3 μm induced rapid and transient phosphorylation of Elk-1 (Fig. 8A) and CREB (Fig. 8B) and induction of c-Fos expression (Fig. 8C). All three events together with the dynamic ERK1/2 phosphorylation (Fig. 1B) kinetically corresponded well into sequential responses: ERK1/2 phosphorylation precedes Elk-1/CREB phosphorylation followed by c-Fos induction. In contrast to the elevated pElk-1 and pCREB levels, cellular levels of Elk-1 and CREB were not changed by NMDA/DHPG (Fig. 8A,B).
Figure 8.

Coactivation of NMDARs and mGluR5 phosphorylates Elk-1 and CREB and increases c-Fos expression in cultured rat striatal neurons. Note that NMDA/DHPG induced a rapid and transient increase in Elk-1 (A) and CREB (B) phosphorylation, followed by an increase in c-Fos protein levels (C). Total Elk-1 and CREB proteins remained unchanged for all the time points surveyed (A, B). NMDA and DHPG were coincubated at 3 μm for different durations. Representative immunoblots are shown left to the quantified data (mean ± SEM; n = 4-5). *p < 0.05 versus basal levels.
To determine whether ERK1/2 mediates Elk-1/CREB phosphorylation and c-Fos expression, a mitogen-activated protein kinase kinase (MEK)-selective inhibitor, U0126, was used to inhibit the ERK1/2 pathway in response to NMDA/DHPG stimulation. The inhibition was confirmed (Fig. 9A) in which U0126 (1 μm) completely blocked the ERK1/2 phosphorylation induced by NMDA/DHPG (3 μm). Consistent with the ability to block ERK1/2 phosphorylation, U0126 blocked the NMDA/DHPG (3 μm)-induced Elk-1 and CREB phosphorylation (Fig. 9B,C). U0126 also essentially reduced the c-Fos response to NMDA/DHPG (Fig. 9D). No significant differences were found in total ERK1/2, Elk-1, and CREB proteins after the U0126 treatment (Fig. 9A-C). These results suggest that the MEK-mediated ERK1/2 activation leads to the subsequent phosphorylation of the two transcription factors and c-Fos expression after coactivation of NMDARs and mGluR5.
Figure 9.

Effects of the MEK inhibitor U0126 on the NMDA/DHPG (N/D)-induced phosphorylation of ERK1/2, Elk-1, and CREB and c-Fos expression in cultured rat striatal neurons. Note that U0126 blocked the NMDA/DHPG-induced phosphorylation of ERK1/2 (A), Elk-1 (B), and CREB (C) and c-Fos expression (D), without changing cellular levels of the three proteins. U0126 (1 μm) was incubated 30 min before and during treatment with NMDA (3 μm) and DHPG (3 μm) for 5 min (pERK1/2), 15 min (pElk-1 and pCREB), or 30 min (c-Fos). Representative immunoblots are shown left to the quantified data (mean ± SEM; n = 4-5). *p < 0.05 versus basal levels; +p < 0.05 versus NMDA plus DHPG.
Discussion
The present study investigated the interaction between NMDARs and mGluR5 in the activation of ERK1/2 in neurons. We found that coactivation of NMDARs and mGluR5 by their selective agonists at a low dose range triggered a PSD-95- and Homer1b/c-sensitive signaling mechanism in the PSD. This signaling is independent of Ca2+ signals derived from NMDAR-mediated Ca2+ influx and mGluR5-mediated Ca2+ release and converges on ERK1/2 to upregulate its phosphorylation. The ERK1/2 activated by this pathway possesses the ability to phosphorylate the two transcription factors, Elk-1 and CREB, resulting in c-Fos expression. These results demonstrate an integral signaling complex in the PSD that couples signals from concurrent activation of NMDARs and mGluR5 to ERK1/2 for regulating gene expression.
Although separate stimulation of NMDARs and group I mGluRs activates ERK, impact of concurrent stimulation of the two receptors on ERK activity is poorly understood. In neurons, this study found that costimulation of the two receptors produced a significant left-shift of the dose-response curve of ERK1/2 activation. Particularly at the low dose range, the agonists together induced a significant synergy. Thus, NMDARs and group I mGluRs, after concurrent stimulation around threshold and subthreshold levels, converge the excitatory synaptic inputs to ERK. Because the ERK response was only sensitive to the mGluR5, but not the mGluR1, antagonist, mGluR5 is believed to be the subtype of group I mGluRs that is linked to ERK. Noticeably, the NMDA/DHPG effect was not mediated via a conventional prime second messenger, Ca2+ signals, derived from either NMDAR or mGluR5 stimulation. Although coapplication of NMDA and DHPG at a low dose (3 μm) induced a small Ca2+ rise, blockade of the Ca2+ rise by the NMDA open-channel blockers or the internal Ca2+-depleting agent unaffected the NMDA/DHPG-induced ERK phosphorylation. Removal of extracellular Ca2+ or loading the Ca2+ chelators did not change the NMDA/DHPG action. Thus, neither the NMDA-mediated Ca2+ influx nor the mGluR5-mediated Ca2+ release participates in coupling the two receptor signals to ERK. The results from this study provide additional evidence supporting a model in which certain synaptic NMDAR signaling can be achieved in a use (ligand)-dependent manner, even when the ion flux is not sufficiently activated (Vissel et al., 2001).
In additional efforts to dissect signaling mechanisms, we identified two PSD proteins that organize the receptor signals to ERK. PSD-95 that binds the C-terminus tSXV motif of NMDAR NR2 subunits (Kornau et al., 1995) shows the linkages to other downstream proteins in the PSD. In this study, we found that the peptide that disrupts the interaction of NMDARs with PSD-95 attenuated the ERK phosphorylation induced by coactivation of NMDARs and mGluR5. Thus, PSD-95 is likely a protein coupling Ca2+-independent signals derived from NMDARs to ERK. Parallel with PSD-95, mGluR5-related Homer1b/c seemingly serves as another essential synaptic protein organizing the mGluR5 interaction with signals from NMDARs. Homer1b/c proteins are constitutively expressed in cultured rat striatal neurons at a high level (present study). Its C-terminal coiled-coil domain facilitates the formation of homomeric multivalent complexes, such as Homer dimers and tetramers (Brakeman et al., 1997; Xiao et al., 1998), whereas the highly conserved N-terminal EVH (Enabled/VASP homology) domain forms heteromeric binding to a proline-rich motif in the C terminus of mGluR5 (Gertler et al., 1996; Kato et al., 1998; Tu et al., 1998) as well as other synaptic proteins. Homer1b/c proteins are enriched in the central as well as lateral region of the PSD (Xiao et al., 1998). The mGluR5 is also present at the PSD with a higher concentration at the lateral margin (Baude et al., 1993; Nusser et al., 1994; Lujan et al., 1997). This unique ultrastructural arrangement allows Homer1b/c to form a physical tether linking membrane mGluR5 with other PSD proteins. Indeed, the reduction of Homer1b/c by an RNAi approach attenuated the ERK phosphorylation induced by NMDA/DHPG stimulation, indicating that Homer1b/c proteins transduce mGluR5 signals to converge with signals from NMDAR/PSD-95 to activate ERK. This finding also supports the notion that proteins in a given signaling cascade needs to be physically coupled, just like the mitogen-activated protein kinase cascade in which all members form a complex of cytosolic kinases with their specific substrates.
Homer binds the proline-rich motif in IP3 receptors (Tu et al., 1998), and the putative Homer ligand motif presents in ryanodine receptors (Feng et al., 2002; Hwang et al., 2003). Thus, Homer proteins, by linking to these receptors on internal Ca2+ stores, could actively regulate Ca2+ signaling. However, this regulation may not play a significant role in the ERK phosphorylation seen in this study because of a Ca2+-independent nature of the ERK activation. Other PSD proteins that may participate in the organization of Homer and PSD-95 signals to ERK include the Shank/ProSAP family (Tu et al., 1999; Sala et al., 2001). Shank/ProSAP proteins bind Homer and are also linked to NMDARs via inner PSD proteins of the synapse-associated protein 90/PSD-95-associated protein/guanylate kinase-associated protein and PSD-95 (Naisbitt et al., 1999; Boechers et al., 1999). Thus, through the above scaffolding/signaling molecules in the PSD, NMDARs and mGluR5 could jointly form an effective signaling complex. This complex could allow the sophisticated interaction between the two receptors after concomitant stimulation (Tu et al., 1998; Sheng and Kim, 2000; Thomas, 2002). The results from this study confirm the existence of a synergistic type of interaction in the complex that results in activation of ERK. The detailed mechanisms underlying the signaling transmission among the NMDA/mGluR5 receptors and their synaptic proteins that eventually leads to ERK activation are unclear at present. More studies need to be conducted to address this issue.
TTX-insensitive spontaneous release of glutamate from presynaptic terminals substrates activity-independent miniature EPSPs (mEPSPs) in postsynaptic neurons. Because the frequency rather than the amplitude of mEPSPs is dependent on transmitter release probability (Fatt and Katz, 1952; Redman, 1990), a selective evaluation of the presynaptic regulation of excitatory synaptic transmission can be performed by measuring changes in the frequency of mEPSPs. Through this approach, two articles reported an increase in the frequency but not the amplitude of mEPSPs after mGluR1/5 agonist application in the rat spinal cord or reticulospinal neurons (Schwartz and Alford, 2000; Park et al., 2004). This indicates the presence of presynaptic facilitation of spontaneous glutamate release by mGluR1/5. However, this presynaptic mechanism is less likely involved in mediating the NMDAR-mGluR5 interaction observed in this study. This is because the presence of presynaptic mGluR5 was not found in this culture system. In addition, the NMDAR antagonist MK801 and extracellular Mg2+, which can block the NMDAR-dependent mEPSPs, and the AMPA receptor antagonists GYKI52466 and DNQX, which can block the AMPA receptor-dependent mEPSPs (Araque et al., 1998; Schwartz and Alford, 2000), had no effect on the synergistic effect of NMDA/DHPG. Thus, glutamatergic mEPSPs through NMDA and AMPA channels are not related to the NMDAR-mGluR5 interaction. Finally, group I mGluR facilitation of presynaptic glutamate release is Ca2+ dependent (Schwartz and Alford, 2000). The Ca2+-independent nature of the NMDAR-mGluR interaction narrows the participation of a Ca2+-dependent presynaptic mechanism in this event.
Given the existence of a Ca2+-dependent ERK pathway to Elk-1 and CREB in response to glutamate stimulation (Rajadhyaksha et al., 1999; Vanhoutte et al., 1999; Zanassi et al., 2001; Perkinton et al., 2002; Mao and Wang, 2003b,c), it is intriguing to know whether the new Ca2+-independent ERK pathway identified in this study possesses the similar ability. We found that NMDA/DHPG-induced ERK phosphorylation kinetically corresponded well with increased Elk-1 and CREB phosphorylation and c-Fos expression. Blockade of ERK phosphorylation prevented Elk-1 and CREB activation and c-Fos induction. Thus, the novel Ca2+-independent ERK pathway, like the Ca2+-dependent ERK pathway, possesses the ability to facilitate Elk-1- and CREB-sensitive gene expression. However, it is assumed that subtle differences in regulating gene expression may exist among multiple glutamate signaling pathways. These differences could translate various synaptic signals into precise responses of DNA transcription.
In summary, the present work has identified a novel Ca2+-independent signaling pathway mediating synapse-to-nucleus communication. This pathway is activated by concurrent stimulation of NMDARs and mGluR5, and signals from the two receptors are then coordinated by PSD-95 and Homer1b/c. The coordinated signals could synergistically converge to ERK to phosphorylate transcription factors and facilitate gene expression.
Footnotes
This work was supported by National Institutes of Health Grants DA010355 and MH061469 to J.Q.W.
Correspondence should be addressed to Dr. John Q. Wang, Department of Basic Medical Science, University of Missouri-Kansas City, School of Medicine, 2411 Holmes Street, Kansas City, MO 64108. E-mail: wangjq@umkc.edu.
Copyright © 2004 Society for Neuroscience 0270-6474/04/2410846-12$15.00/0
References
- Aarts M, Liu Y, Liu L, Besshoh S, Arundine M, Gurd JW, Wang YT, Salter MW, Tymianski M (2002) Treatment of ischemic brain damage by perturbing NMDA receptor-PSD-95 protein interactions. Science 298: 846-850. [DOI] [PubMed] [Google Scholar]
- Ango F, Pin JP, Tu JC, Xiao B, Worley PF, Bockaert J, Fagni L (2000) Dendritic and axonal targeting of type 5 metabotropic glutamate receptor is regulated by Homer1 proteins and neuronal excitation. J Neurosci 20: 8710-9716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Araque A, Sanzgiri RP, Parpura V, Haydon PG (1998) Calcium elevation in astrocytes causes an NMDA receptor-dependent increase in the frequency of miniature synaptic currents in cultured hippocampal neurons. J Neurosci 18: 6822-6829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baude A, Nusser Z, Roberts JD, Mulvihill E, Mcllhinney RA, Somogyi P (1993) The metabotropic glutamate receptor (mGluR1α) is concentrated at perisynaptic membrane of neuronal subpopulations as detected by immunogold reaction. Neuron 11: 771-787. [DOI] [PubMed] [Google Scholar]
- Boechers TM, Winter C, Smalla KH, Kreutz MR, Bokmann J, Seidenbecher C, Garner CC, Gundelfinger ED (1999) Proline-rich synapse-associated proteins ProSAP1 and ProSAP2 interact with synaptic proteins of the SAPAP/GKAP family. Biochem Biophys Res Commun 264: 247-252. [DOI] [PubMed] [Google Scholar]
- Brakeman PR, Lanahan AA, O'Brien R, Roche K, Barnes CA, Huganir RL, Worley PF (1997) Homer: a protein that selectively binds metabotropic glutamate receptors. Nature 386: 284-288. [DOI] [PubMed] [Google Scholar]
- Caplen NJ, Parrish S, Imani F, Fire A, Morgan RA (2001) Specific inhibition of gene expression by small double-stranded RNAs in invertebrate and vertebrate systems. Proc Natl Acad Sci USA 98: 9742-9747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conn PU, Pin JP (1997) Pharmacology and function of metabotropic glutamate receptors. Annu Rev Pharmacol Toxicol 37: 205-237. [DOI] [PubMed] [Google Scholar]
- Dingledine R, Borges K, Bowie D, Traynelis SF (1999) The glutamate receptor ion channels. Pharmacol Rev 51: 7-61. [PubMed] [Google Scholar]
- Fatt P, Katz B (1952) Spontaneous subthreshold activity at motor nerve endings. J Physiol (Lond) 117: 109-128. [PMC free article] [PubMed] [Google Scholar]
- Feng W, Tu J, Yang T, Vernon PS, Allen PD, Worley PF, Pessah IN (2002) Homer regulates gain of ryanodine receptor type 1 channel complex. J Biol Chem 277: 44722-44730. [DOI] [PubMed] [Google Scholar]
- Gertler FB, Niebuhr K, Reinhard M, Wehland J, Soriano P (1996) Mena, a relative of VASP and Drosophila Enabled, is implicated in the control of microfilament dynamics. Cell 87: 227-239. [DOI] [PubMed] [Google Scholar]
- Hwang SY, Wei J, Westhoff JH, Duncan RS, Ozawa F, Volpe P, Inokuchi K, Koulen P (2003) Differential functional interaction of two Vesl/Homer protein isoforms with ryanodine receptor type 1: a novel mechanism for control of intracellular calcium signaling. Cell Calcium 34: 177-184. [DOI] [PubMed] [Google Scholar]
- Jones KH, Senft JA (1985) An improved method to determine cell viability by simultaneous staining with fluorescein diacetate-propidium iodide. J Histochem Cytochem 33: 77-79. [DOI] [PubMed] [Google Scholar]
- Kato A, Ozawa F, Saitoh Y, Fukazawa Y, Sugiyama H, Inoduchi K (1998) Novel members of the Vesl/Homer family of PDZ proteins that bind metabotropic glutamate receptors. J Biol Chem 273: 23969-23975. [DOI] [PubMed] [Google Scholar]
- Kornau HC, Schenker LT, Kennedy MB, Seeburg PH (1995) Domain interaction between NMDA receptor subunits and the postsynaptic density protein PSD-95. Science 269: 1737-1740. [DOI] [PubMed] [Google Scholar]
- Lujan R, Roberts JD, Shigemoto R, Ohishi H, Somogyi P (1997) Differential plasma membrane distribution of metabotropic glutamate receptors mGluR1α, mGluR2 and mGluR5, relative to neurotransmitter release sites. J Chem Neuroanat 13: 219-241. [DOI] [PubMed] [Google Scholar]
- Mann DA, Frankel AD (1991) Endocytosis and targeting of exogenous HIV-1 Tat protein. EMBO J 10: 1733-1739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mao L, Wang JQ (2002) Glutamate cascade to cAMP response element-binding protein phosphorylation in cultured striatal neurons through calcium-coupled group I mGluRs. Mol Pharmacol 62: 473-484. [DOI] [PubMed] [Google Scholar]
- Mao L, Wang JQ (2003a) Group I metabotropic glutamate receptor-mediated calcium signaling and immediate early gene expression in cultured rat striatal neurons. Eur J Neurosci 17: 741-750. [DOI] [PubMed] [Google Scholar]
- Mao L, Wang JQ (2003b) Phosphorylation of cAMP response element-binding protein in cultured striatal neurons by metabotropic glutamate receptor subtype 5. J Neurochem 84: 233-243. [DOI] [PubMed] [Google Scholar]
- Mao L, Wang JQ (2003c) Metabotropic glutamate receptor subtype 5-regulated Elk-1 phosphorylation and immediate early gene expression in striatal neurons. J Neurochem 85: 1006-1017. [DOI] [PubMed] [Google Scholar]
- Mao L, Tang Q, Samdani S, Liu Z, Wang JQ (2004) Regulation of MAPK/ERK phosphorylation via ionotropic glutamate receptors in cultured rat striatal neurons. Eur J Neurosci 19: 1207-1216. [DOI] [PubMed] [Google Scholar]
- McBain CJ, Mayer ML (1994) N-methyl-d-aspartate structure and function. Physiol Rev 74: 723-760. [DOI] [PubMed] [Google Scholar]
- Migaud M, Charlesworth P, Dempster M, Webster LC, Watabe AM, Makhinson M, He Y, Ramsay MF, Morris RG, Morrison JH, O'Dell TJ, Grant SG (1998) Enhanced long-term potentiation and impaired learning in mice with mutant postsynaptic density-95 protein. Nature 396: 433-439. [DOI] [PubMed] [Google Scholar]
- Naisbitt S, Kim E, Tu JC, Xiao B, Sala C, Valtschanoff J, Weinberg RJ, Worley PF, Sheng M (1999) Shank, a novel family of posysynaptic density proteins that bind to the NMDA receptor/PSD-95/GKAP complex and cortactin. Neuron 23: 569-582. [DOI] [PubMed] [Google Scholar]
- Nakanishi S (1994) Metabotropic glutamate receptors: synaptic transmission, modulation, and plasticity. Neuron 13: 1031-1037. [DOI] [PubMed] [Google Scholar]
- Nusser Z, Mulvihill E, Streit P, Somogyi P (1994) Subsynaptic segregation of metabotropic and ionotropic glutamate receptors as revealed by immunogold localication. Neuroscience 61: 421-427. [DOI] [PubMed] [Google Scholar]
- Park YK, Galik J, Ryu PD, Randic M (2004) Activation of presynaptic group I metabotropic glutamate receptors enhances glutamate release in the rat spinal cord substantia gelatinosa. Neurosci Lett 361: 220-224. [DOI] [PubMed] [Google Scholar]
- Perkinton MS, Sihra TS, Williams RJ (1999) Ca2+-permeable AMPA receptors induce phosphorylation of cAMP response element-binding protein through a phosphatidylinositol 3-kinase-dependent stimulation of the mitogen-activated protein kinase signaling cascade in neurons. J Neurosci 19: 5861-5874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perkinton MS, Ip JK, Wood GL, Crossthwaite AJ, Williams RJ (2002) Phosphatidylinositol 3-kinase is a central mediator of NMDA receptor signalling to MAP kinase (Erk1/2), Akt/PKB and CREB in striatal neurons. J Neurochem 80: 239-254. [DOI] [PubMed] [Google Scholar]
- Peyssonnaux C, Eychene A (2001) The Raf/MEK/ERK pathway: new concepts of activation. Biol Cell 93: 53-62. [DOI] [PubMed] [Google Scholar]
- Rajadhyaksha A, Barczak A, Macias W, Leveque JC, Lewis SE, Konradi C (1999) L-type Ca2+ channels are essential for glutamate-mediated CREB phosphorylation and c-fos gene expression in striatal neurons. J Neurosci 19: 6348-6359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Redman S (1990) Quantal analysis of synaptic potentials in neurons of central nervous system. Physiol Rev 70: 165-198. [DOI] [PubMed] [Google Scholar]
- Romano C, Sesma MA, MacDonald C, O'Malley K, van den Pol AN, Olney JW (1995) Distribution of metabotropic glutamate receptor mGluR5 immunoreactivity in rat brain. J Comp Neurol 355: 455-469. [DOI] [PubMed] [Google Scholar]
- Sala C, Piech V, Wilson NR, Passafaro M, Liu G, Sheng M (2001) Regulation of dendritic spine morphology and synaptic function by Shank and Homer. Neuron 31: 115-130. [DOI] [PubMed] [Google Scholar]
- Sattler R, Xiong Z, Lu WY, Hafner M, MacDonald JF, Tymianski M (1999) Specific coupling of NMDA receptor activation to nitric oxide neurotoxicity by PSD-95 protein. Science 284: 1845-1848. [DOI] [PubMed] [Google Scholar]
- Schwartz NE, Alford S (2000) Physiological activation of presynaptic metabotropic glutamate receptors increases intracellular calcium and glutamate release. J Neurophysiol 84: 415-427. [DOI] [PubMed] [Google Scholar]
- Schwarze SR, Ho A, Voero-Akbani A, Dowdy SF (1999) In vivo protein transduction: delivery of a biologically active protein into the mouse. Science 285: 1569-1572. [DOI] [PubMed] [Google Scholar]
- Sgambato V, Vanhoutte P, Pages C, Rogard M, Hipskind R, Besson MJ, Caboche J (1998) In vivo expression and regulation of Elk-1, a target of the extracellular-regulated kinase signaling pathway, in the adult rat brain. J Neurosci 18: 214-226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheng M (2001) Molecular organization of the postsynaptic specialization. Proc Natl Acad Sci USA 98: 7058-7061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheng M, Kim E (2000) The Shank family of scaffold proteins. J Cell Sci 113: 1851-1856. [DOI] [PubMed] [Google Scholar]
- Sheng M, Kim MJ (2002) Postsynaptic signaling and plasticity mechanisms. Science 298: 776-780. [DOI] [PubMed] [Google Scholar]
- Shigemoto R, Kinoshita A, Wada E, Nomura S, Ohishi H, Takada M, Flor PJ, Neki A, Abe T, Nakanishi S, Mizuno N (1997) Differential presynaptic localization of metabotropic glutamate receptor subtypes in the rat hippocampus. J Neurosci 17: 7503-7522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shiraishi Y, Mizutani A, Yuasa S, Mikoshiba K, Furuichi T (2004) Differential expression of Homer family proteins in the developing mouse brain. J Comp Neurol 473: 582-599. [DOI] [PubMed] [Google Scholar]
- Takagi N, Logan R, Teves L, Wallace MC, Gurd JW (2000) Altered interaction between PSD-95 and the NMDA receptor following transient global ischemia. J Neurochem 74: 169-178. [DOI] [PubMed] [Google Scholar]
- Thandi S, Blank JL, Challiss RA (2002) Group I metabotropic glutamate receptors, mGluR1a and mGluR5 couple to extracellular signal-regulated kinase (ERK) activation via distinct, but overlapping, signaling pathways. J Neurochem 83: 1139-1153. [DOI] [PubMed] [Google Scholar]
- Thomas U (2002) Modulation of synaptic signaling complexes by Homer proteins. J Neurochem 81: 407-413. [DOI] [PubMed] [Google Scholar]
- Tu J, Xiao B, Naisbitt S, Yuan J, Petralia R, Brakeman P, Aakalu V, Lanahan A, Sheng M, Worley P (1999) mGluR/Homer and PDS-95 complexes are linked by the Shank family of postsynaptic density proteins. Neuron 23: 583-592. [DOI] [PubMed] [Google Scholar]
- Tu JC, Xiao B, Yuan JP, Lanahan AA, Leoffert K, Li M, Linden DJ, Worley PF (1998) Homer binds a novel proline-rich motif and links group 1 metabotropic glutamate receptors with IP3 receptors. Neuron 21: 717-726. [DOI] [PubMed] [Google Scholar]
- Vanhoutte P, Barnier JV, Guibert B, Pages C, Besson MJ, Hipskind RA, Caboche J (1999) Glutamate induces phosphorylation of Elk-1 and CREB, along with c-fos activation, via an extracellular signal-regulated kinase-dependent pathway in brain slices. Mol Cell Biol 19: 136-146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vissel B, Krupp JJ, Heinemann SF, Westbrook GL (2001) A use-dependent tyrosine dephosphorylation of NMDA receptors is independent of ion flux. Nat Neurosci 4: 587-596. [DOI] [PubMed] [Google Scholar]
- Volmat V, Pouyssegur J (2001) Spatiotemporal regulation of the p42/p44 MAPK pathway. Biol Cell 93: 71-79. [DOI] [PubMed] [Google Scholar]
- Wang JQ, Tang Q, Samdani S, Liu Z, Parelkar NK, Choe ES, Yang L, Mao L (2004) Glutamate signaling to Ras-MAPK in striatal neurons: mechanisms for inducible gene expression and plasticity. Mol Neurobiol 29: 1-14. [DOI] [PubMed] [Google Scholar]
- Xiao B, Tu JC, Petralia RS, Yuan JP, Doan A, Breder CD, Ruggiero A, Lanahan AA, Wenthold RJ, Worley PF (1998) Homer regulates the association of group I metabotropic glutamate receptors with multivalent complexes of homer-related, synaptic proteins. Neuron 21: 707-716. [DOI] [PubMed] [Google Scholar]
- Xiao B, Tu JC, Worley PF (2000) Homer: a link between neural activity and glutamate reception. Curr Opin Neurobiol 10: 370-374. [DOI] [PubMed] [Google Scholar]
- Zanassi P, Paolillo M, Feliciello A, Avvedimento EV, Gallo V, Schinelli S (2001) cAMP-dependent protein kinase induces cAMP-response element-binding protein phosphorylation via an intracellular calcium release/ERK-dependent pathway in striatal neurons. J Biol Chem 276: 11487-11495. [DOI] [PubMed] [Google Scholar]
- Ziff EB (1997) Enlightening the postsynaptic density. Neuron 19: 1163-1174. [DOI] [PubMed] [Google Scholar]