

Abstract
The neuronal response to a Ca2+ stimulus is a complex process involving direct Ca2+/calmodulin (CaM) actions as well as secondary activation of multiple signaling pathways such as cAMP and ERK (extracellular signal-regulated kinase). These signals can act in both the cytoplasm and the nucleus to control gene expression. To dissect the role of nuclear from cytoplasmic Ca2+/CaM signaling in memory formation, we generated transgenic mice that express a dominant inhibitor of Ca2+/CaM selectively in the nuclei of forebrain neurons and only after the animals reach adulthood. These mice showed diminished neuronal activity-induced phosphorylation of cAMP response element-binding protein, reduced expression of activity-induced genes, altered maximum levels of hippocampal long-term potentiation, and severely impaired formation of long-term, but not short-term, memory. Our results demonstrate that nuclear Ca2+/CaM signaling plays a critical role in memory consolidation in the mouse.
Keywords: calcium/calmodulin, memory, consolidation, transgenic, CREB, tetracycline
Introduction
Genetic studies have implicated cAMP response element-binding protein (CREB) in the transcriptional signaling required for consolidation of short-term to long-term synaptic plasticity and memory in Drosophila (Yin et al., 1994, 1995), Aplysia (Kandel, 2001), and mice (Bourtchuladze et al., 1994; Kida et al., 2002; Pittenger et al., 2002). Although the critical role of CREB family transcription factors in long-term memory (LTM) seems to be conserved across species, the second messenger signaling pathways required are less clear. In Aplysia, the formation of LTM is mediated by serotonin stimulation of cAMP and direct CREB activation by protein kinase A (Kandel, 2001). In Drosophila, genetic screens for learning and memory identified mutants, which also implicate direct cAMP-mediated effects in the formation of LTM (Waddell and Quinn, 2001). These data suggest that the primary intracellular signaling for the consolidation of short-term memory (STM) into LTM in invertebrates is mediated by the direct activation of cAMP and phosphorylation of CREB by cAMP-dependent protein kinase (Kandel, 2001). In mammals, a direct role for cAMP-based nuclear signaling in the conversion of STM to LTM is less clear.
Excitatory activity in neurons leads to alterations in gene expression that are mediated by increases in intracellular Ca2+ (Bading, 2000; West et al., 2001). This activity-dependent gene expression is thought to play a critical role in stabilizing forms of neuronal plasticity that may underlie the formation of LTM. Ca2+ signaling to the nucleus has been studied extensively in neurons in culture where it is clear that a primary Ca2+ signal can act both directly through calmodulin (CaM) in the nucleus (Hardingham et al., 1997, 2001; Deisseroth et al., 1998) as well as indirectly through the activation of other second messenger pathways such as cAMP (Chetkovich et al., 1991) and MAP kinase (MAPK) (Dolmetsch et al., 2001; Hardingham et al., 2001). Each of these three signaling pathways can regulate CREB (Shaywitz and Greenberg, 1999), and it has been difficult to determine the relative contribution of each separate pathway to the transcriptional events required for stabilization of behavioral memory.
Extensive pharmacological and genetic data demonstrate behavioral effects from altering both the MAP kinase and cAMP pathways (Abel et al., 1997; Wong et al., 1999; Selcher et al., 2001; Sweatt, 2001; Mazzucchelli et al., 2002). However, in these experiments, it has not been possible to dissociate nuclear from cytoplasmic and acute from developmental roles of these complex signaling pathways. In addition, in most cases, the memory impairments seen in genetic experiments do not parallel the effects of either protein synthesis inhibitors or CREB inhibition, suggesting that these pathways alone do not account for the activation of the transcriptional events required for the stabilization of LTM.
The direct nuclear Ca2+ signaling pathway has been studied less extensively. To examine the role of direct nuclear Ca2+/CaM signaling in a whole animal model, we generated transgenic mice expressing a competitive inhibitor of CaM only in neuronal nuclei. We found that, although these mice had intact STM, their LTM was severely impaired across multiple behavioral tasks. Moreover, we demonstrate that these phenotypes reflect deficits in the encoding of LTM that cannot be attributed to alterations in neuronal development or performance variables required for the tasks.
Materials and Methods
Generation of CaM binding polypeptide transgenic mice
The CaM binding polypeptide (CaMBP) gene from the vector pSVLCaMBP4 (gift from Dr. J. Dedman, University of Cincinnati, Cincinnati, OH) (Wang et al., 1996)) was subcloned into the EcoRV site of pMM400 (Mayford et al., 1996) to create pKL107. In pKL107, the CaMBP gene is downstream of the tetO promoter and flanked by artificial introns and a 3′-polyadenylation signal. The CaMBP transgene was purified away from vector sequences and microinjected into B6/D2 F2 embryos. Founder animals were crossed to mice carrying the tetracycline transactivator (tTA) under control of the calcium-calmodulin-dependent protein kinase IIα (CaMKIIα) promoter (Mayford et al., 1996). Double transgenic males (carrying both the tetO-CaMBP and CaMKIIα-tTA transgenes) were backcrossed to C57BL/6 females to generate the animals used in the experiments. Experiments used 10- to 24-week-old mice for behavior studies and 8- to 16-week-old mice for electrophysiology. All mice were kept on a doxycycline (Dox) diet (40 μg of Dox per 1 gm of mouse chow) during development and until they reached at least 4 weeks of age. To activate gene expression, Dox was removed from the diet 2-4 weeks before starting an experiment.
In situ hybridization
Brains were dissected and rapidly frozen in OCT embedding compound (Sakura, Tokyo, Japan). Parasagittal cryostat sections (20 μm) were mounted onto microscope slides, fixed in 4% paraformaldehyde (PFA) for 10 min, and frozen at -80°C until use. Slices were hybridized as described previously (Mayford et al., 1995) to an 35S-labeled antisense DNA probe corresponding to a 30 bp sequence in the untranslated region of the CaMBP transcript (Abel et al., 1997). The sections were washed twice for 10 min at room temperature in 2× SSC and then twice for 60 min at 60°C in 0.2× SSC. Slides were exposed to autoradiographic film (Biomax MR; Kodak, Rochester, NY) for 2-4 weeks.
Protein analysis
Hippocampi were dissected and homogenized on ice. Nuclear and cytoplasmic fractions were obtained with the NE-PER and TE-PER extraction reagents (Pierce, Rockford, IL), and protease inhibitors were included as recommended by the manufacturer. Protein concentrations were quantified using Pierce Micro BCA protein assay reagent and then diluted to 1 μg/μl with PBS containing protease inhibitors. The samples were aliquoted and stored at -80°C until use. For Western blots, proteins were separated by SDS-PAGE (12% polyacrylamide; Amresco, Solon, OH) and blotted onto nitrocellulose membranes (Protran; Schleicher & Schuell, Dassel, Germany) with a semidry electrophoretic transfer apparatus (Trans-Blot SD; Bio-Rad, Hercules, CA). Membranes were stained with Ponceau S solution, rinsed and incubated in blocking solution (TBS, 0.1% Tween 20, and 5% dry milk) for 1 hr, rinsed briefly, and incubated in primary antibody in blocking solution overnight at 4°C. All primary antibodies were used at the dilution of 1:1000 except for anti-phospho-MAPK (1:500), anti-Fos (1:300), and anti-αTubulin (1:20,000). Monoclonal anti-CaM and polyclonal anti-Fos were obtained from Upstate Biotechnology (Lake Placid, NY); polyclonal anti-, total-, and phospho-MAPK were from Cell Signaling Technology (Beverly, MA); the nuclear loading control monoclonal anti-neuronal nuclei was from Chemicon (Temecula, CA); the cytoplasmic control polyclonal anti-Syntaxin 13 was from Synaptic Systems (Goettingen, Germany); and monoclonal anti-αTubulin and anti-βActin were from Sigma (Milwaukee, WI). The secondary antibodies were a goat anti-mouse and goat anti-rabbit horseradish peroxidase-conjugated IgG (1:5000 and 1:10,000, respectively; Pierce). Signal detection by chemiluminescence was performed with Super Signal according to the instructions of the manufacturer (Pierce).
Immunostaining
For CaMBP, mice were perfused transcardially with 4% PFA. The brains were removed and postfixed in PFA for 6 hr and then placed in 20% sucrose solution overnight; on the following day, the brains were frozen in OCT, and coronal sections (6 μm) were mounted on slides. Sections were digested with trypsin for 30 min to recover antigenicity, blocked for 1 hr with 5% normal goat serum in PBS, and then incubated overnight with a rabbit anti-CaMBP antibody (1:25; the anti-CaMBP antibody was a gift from M. Kaetzel and J. Dedman, University of Cincinnati) (Wang et al., 1995). Sections were subsequently washed and incubated with a cyanine 3 (Cy3)-conjugated goat anti-rabbit IgG (1:500; Jackson ImmunoResearch, West Grove, PA). Slides were rinsed and counterstained with 2 μm Syto13 (Molecular Probes, Eugene, OR), rinsed again, and mounted in Slowfade Light antifade mounting medium (Molecular Probes). Fluorescent images were taken with a confocal laser-scanning microscope (Olympus, Tokyo, Japan). For CaM staining, we prepared 5-μm-thick paraffin-embedded brain sections. Antigen retrieval was performed at 95°C for 30 min in a Tris-EDTA buffer. The immunostaining protocol was similar to the CaMBP protocol, except that the rabbit anti-CaM antibody (Zymed Laboratories, San Francisco, CA) was used at 1:100 dilution and the secondary antibody was Cy2 conjugated. Nuclear counterstaining was performed with 10 μm BoPro (Molecular Probes). For CaMBP immunostaining in cell culture, COS-7 cells were transiently transfected with CaMBP and green fluorescent protein (GFP). Cells were fixed in methanol, stained with anti-CaMBP antibody (1:25) followed by incubation with Cy3-coniugated goat anti-rabbit IgG, and counter-stained with 4′,6′-diamidino-2-phenylindole (DAPI) 48 hr after transfection (Wang et al., 1995).
For CREB immunostaining as well as for MAPK and Fos Western blotting, seizure was induced by intraperitoneal injection of pentylenetetrazole (50 mg/kg body weight) (Morgan et al., 1987). Mice were taken from their home cages, injected, and placed into an empty mouse cage for observation. Mice were killed 10 min after seizure onset. Only mice that developed generalized tonic-clonic seizure were used in the present study. Brains were dissected and prepared as described. Coronal sections (12 μm) were taken on a cryostat, mounted on slides, fixed for 30 min in 4% PFA, and washed in PBS. Slides were incubated in blocking solution (PBS, 0.1% Triton X-100, and 10% normal goat serum) for 1 hr and then incubated in primary antibody overnight at 4°C (polyclonal anti-CREB and anti-phospho-CREB, 1:500; Upstate Biotechnology). Slides were rinsed and incubated in Cy3-conjugated goat anti-rabbit IgG (1:1000; Jackson ImmunoResearch) for 2-3 hr. Slides were rinsed again, mounted in Slowfade Light antifade mounting medium (Jackson ImmunoResearch), and cover-slipped. Sections were visualized on a confocal microscope, and the signal was quantified using the MetaMorph software.
Electrophysiology
All electrophysiology was performed in the CA1 region of 400 μm transverse hippocampal slices essentially as described previously (Mayford et al., 1995). Recording was in an interface chamber at 30°C with constant perfusion of artificial CSF (ACSF) (in mm: 124 NaCl, 4.4 KCl, 1.2 Na2HPO4, 25 NaHCO3, 2.5 CaCl2, 1.3 MgCl2, 10 glucose) aerated with a 95% O2/5% CO2 mixture. Slices were permitted to recover for at least 90 min before recording began. Field EPSPs (fEPSPs) were recorded using a bipolar stimulating electrode and low-resistance glass recording electrode (7.5-15 MΩ filled with ACSF) both placed in the stratum radiatum of CA1. Stimuli were delivered at intensities that evoked an fEPSP slope equal to 30% of the maximum in each slice. Test stimuli were delivered at 0.02 Hz. The baseline synaptic response was collected for 30 min before the tetanus. Long-term potentiation (LTP) was induced by one train of 100 Hz for 1 sec or by three trains of 100 Hz for 1 sec at a 5 min interstimulus interval. The same stimulus intensities were used for tetanization and test responses. For statistical comparisons, an unpaired t test was used with the numbers of tested animals as n.
Behavioral analysis
Morris water maze. The water-maze apparatus consisted of a fiberglass tank that was 122 cm in diameter and 61 cm high with white sides and filled with water at 24°C at a level of ∼42 cm. A clear escape platform (10 cm diameter) was hidden ∼1 cm beneath the water surface. The water tank was surrounded by a circular white plastic curtain 70 cm from the edge of the tank. Four visual cues (50 × 76 cm) consisting of simple black-and-white geometric patterns were evenly spaced on the curtain and were 50 cm above the rim on the tank at their lower edge. For the visible platform task, the platform was marked with a red flag and its position was changed for each trial. Every mouse received three trials each day for 4 d, in which the average time to reach the platform was measured. For the hidden platform task, the flag was removed and the platform remained in the same position throughout training for each mouse. In a training trial, a mouse was placed into the water at one of four randomly determined start locations (W, West; S, South; E, East; N, North). The latency to find the hidden platform was recorded. Mice were given a total of 60 sec in which to find the platform, and once there, were allowed 15 sec to rest on the platform. Training consisted of six training trials per day for 6 d with an intertrial interval of 1 hr. Memory for the location of the target platform was assessed by probe trials in which the platform was removed, and the percentage of time spent swimming in the former platform quadrant was recorded. One probe trial was given every morning before the training session (days 1-6) and then 24 hr (day 7) and 2 weeks after the last training trial. The 24 hr and 2 week probe trials are shown in Figure 5.
Figure 5.
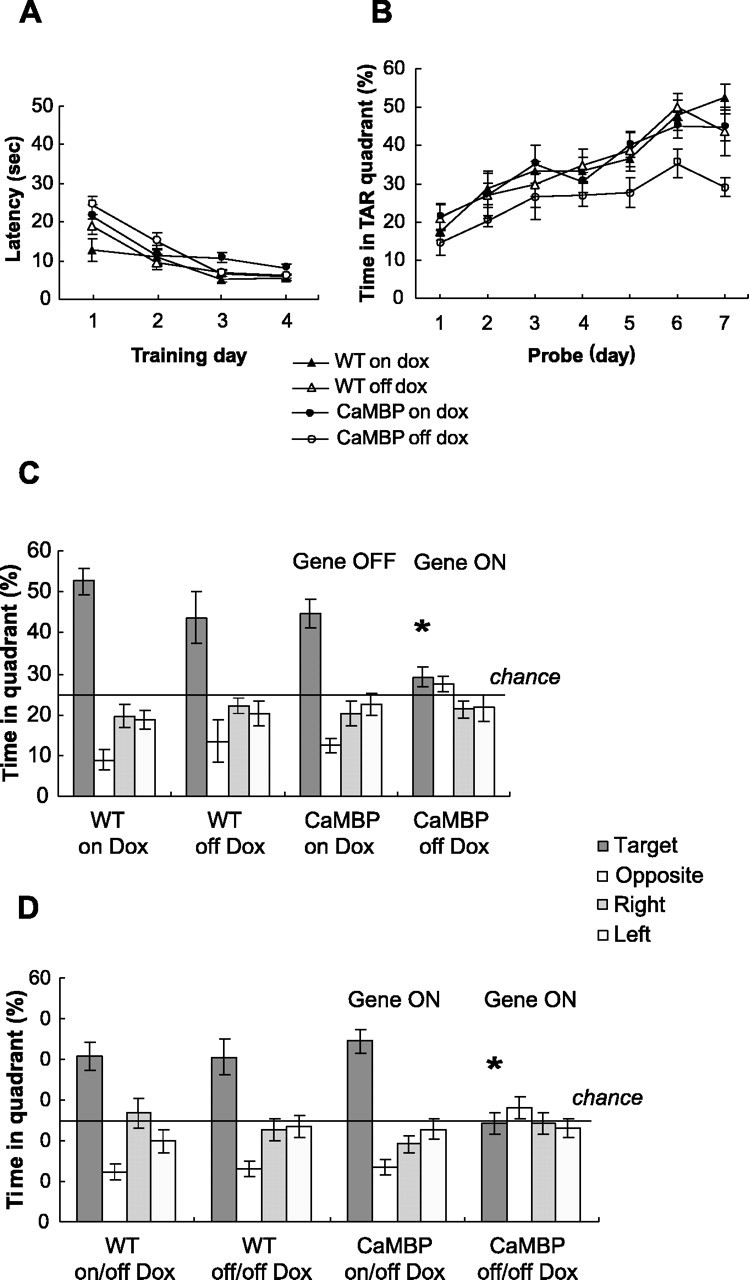
CaMBP transgene disrupts spatial learning and memory formation in the water-maze task. A, Four groups of mice were tested: CaMBP transgenics (n = 15), CaMBP transgenics on Dox (n = 21), wild types (WT) (n = 11), and wild types on Dox (n = 14). They showed normal visible platform learning. The average time to reach the platform was calculated for each day, and values represent group means ± SEM. A two-way ANOVA with repeated measures between CaMBP and wild-type mice off Dox showed no significant effect of genotype. B, CaMBP transgenics are impaired during hidden platform training. A probe trial was given on every training day before starting the training session (days 1-6) and 24 hr after completing training (day 7). Comparison of mean ± SEM percentage time spent in target (TAR) quadrant is shown. Three-way ANOVA with repeated measures revealed a significant two-way interaction (genotype by Dox) (F(1,57) = 4.38; p < 0.05). Planned comparison confirmed that CaMBP mice off Dox spent significantly less time than controls in the target quadrant on the last 3 d (days 5-7) (p < 0.05). C, CaMBP transgenics show impaired spatial memory in the 24 hr probe trial. Bars represent the percentage of time spent swimming in the target, opposite, right, and left of target quadrants. A two-way ANOVA revealed a significant effect of genotype (F(1,57) = 7.82; p < 0.01) and Dox (F(1,57) = 9.13; p < 0.01). Planned comparisons between the four animal groups confirmed a significant difference between CaMBP transgenic mice off Dox and all control groups (*p < 0.02). All three control groups spent significantly more time in the target quadrant than in each of the other quadrants (t test; p < 0.02 for all groups). D, Transgene expression was activated by withdrawal of Dox in the group of CaMBP transgenic mice that had acquired the water-maze task with the transgene suppressed. The mice were placed off Dox immediately after completing the training and the 24 hr probe trial and then retested 2 weeks later; transgene activation after spatial memory acquisition had no effect on recall performance. A two-way ANOVA revealed a significant two-way interaction (genotype by Dox) (F(1,56) = 7.33; p < 0.01). Planned comparisons between the four animal groups confirmed a significant difference between CaMBP mice trained with the gene activated and the three control groups (*p < 0.01).
Novel object recognition. Mice were trained and tested in the home cages. During training each mouse was allowed to explore an object (A), randomly selected from a group of seven different toys, for 2 hr. Three retention test trials were performed at 2 min, 1 hr, and 24 hr after removing the familiar object from the cage. During testing, mice were presented with both the familiar object (A) and a novel object (B), and the time spent exploring the familiar object (TA) and the novel object (TB) was recorded for 2 min by stopwatch. Memory for the familiar object is associated with increased exploration of the novel object, and the result is expressed as a recognition index, which is the ratio (TB × 100)/(TA + TB) (Dodart et al., 1997).
Fear conditioning. Two different mouse operant chambers were used (San Diego Instruments, San Diego, CA). The training and context conditioning test took place in the training chamber, a clear acrylic box (22 × 14 × 15 inches) with a metal grid floor. Cued conditioning was tested in the tone chamber, another acrylic box, which differed from the training chamber in color (walls covered by black paper), floor structure (no grid), light intensity, and scent. On the training day, mice were placed inside the conditioning chamber for 3 min and then a 20 sec tone (77 dB, 2.8 kHz) was presented. During the last 1.5 sec of the tone, a 0.75 mA footshock was delivered through the grid floor. Two more tone-shock pairings were given with 1 min intertrial intervals. At the completion of training, mice were returned to their home cages. LTM was assessed 24 hr after training for both the context-shock and the tone-shock associations, and STM was evaluated 1 hr after training for context only. For the context test, mice were placed back into the conditioning chamber and freezing was measured for 3 min by stopwatch. Freezing was defined as a total lack of movement with the exception of respiration. For the cued test, mice were placed into the tone chamber for 3 min of baseline locomotor activity measurement, and then three 20 sec, 77 dB tones were presented, separated by 1 min intervals. Freezing was measured only during the tone presentations.
Conditioned taste aversion. Mice were trained to drink from two different water bottles, one containing 0.005 m saccharin and one containing plain water. The experiment was performed 2 weeks later, and in this period, they were not exposed to saccharin. During 2 consecutive conditioning days, mice were water deprived and presented with saccharin instead of water for 30 min. Ten minutes after removing the saccharin solution, they were injected intraperitoneally with 0.15 m LiCl (2% of body weight) as the malaise-inducing agent. On the testing days, mice were presented with two bottles, one containing water and one containing saccharin, and the consumption of liquids was measured for 30 min (Masugi et al., 1999).
Sensory-motor tests. Performance of the CaMBP mice in a series of simple sensory-motor tests (Crawley and Paylor, 1997) was assessed, and no significant differences were found. In addition, mice were tested in an open field and a clear plastic box (50 × 34 × 30 cm), and a video tracking system (Poly-Track; San Diego Instruments) was used to monitor the activity of the mouse. Locomotor activity was measured as a function of speed and distance traveled, whereas anxiety level was scored by the relative time spent in the central area and by the amount of defecation, urination, and freezing.
Results
Generation and characterization of CaMBP-expressing transgenic mice
Depolarization of neurons leads to elevations in nuclear Ca2+ levels and the translocation of the CaM from the cytoplasm to the nucleus (Deisseroth et al., 1998; Hardingham et al., 2001). To investigate the role of this direct nuclear Ca2+/CaM pathway in a whole animal model, we generated transgenic mice in which nuclear CaM signaling was specifically impaired. We took advantage of a synthetic CaMBP composed of tandem reiterated CaM binding domains from the rabbit skeletal muscle myosin light chain kinase (Wang et al., 1995). Because CaMBP contains multiple nuclear localization sequences, the peptide acts as a competitive inhibitor of CaM signaling specifically in the nucleus. We expressed the transgene in a spatially and temporally regulated manner using the tetracycline system (Mayford et al., 1996). We studied mice carrying both a tetO-CaMBP transgene and a CaMKIIα promoter-driven tetracycline transactivator transgene. In double transgenic mice, the CaMKIIα promoter limits expression to forebrain projection neurons, and this expression can be fully suppressed by administration of Dox (Fig. 1A). Distribution of the CaMBP transgene in the CNS was examined by in situ hybridization, which showed expression throughout the forebrain, including the neocortex, striatum, and hippocampus (Fig. 1B). CaMBP expression could be completely suppressed by oral administration of Dox (Fig. 1B). Eosin/hematoxylin, Nissl, and Golgi-stained brain sections from wild-type and transgenic mice were indistinguishable when compared for changes in the anatomy of the brain for the neuronal number in regions such as CA1 or for defects in the gross cell structure (data not shown).
Figure 1.
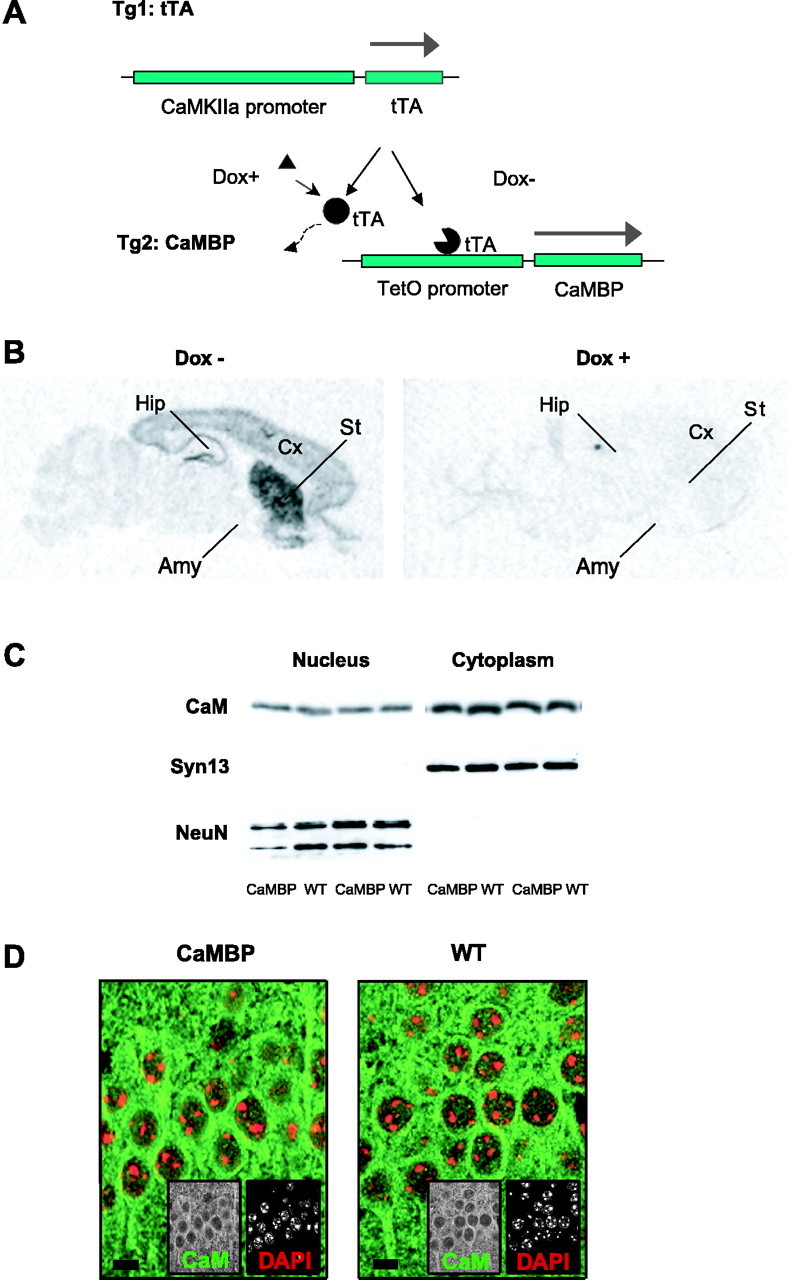
Generation and characterization of the CaMBP mice. A, Transgenic mice expressing the tTA with the CaMKIIα promoter were crossed with mice carrying the tetO promoter linked to the CaMBP gene. The CaMBP is expressed only in double transgenic mice in the absence of Dox. B, Regional distribution of CaMBP mRNA was analyzed by in situ hybridization. CaMBP expression was detected in the cortex (Cx), hippocampus (Hip), and striatum (St). Amy, Amygdala. Dox suppresses CaMBP expression. C, Western blot analysis of hippocampal nuclear and cytoplasmic fractions was performed to evaluate CaM distribution. No difference was detected between wild-type (WT) and CaMBP transgenic mice in either nuclear or cytoplasmic CaM levels. A nuclear antigen [neuronal-specific nuclear protein (NeuN)] and a cytoplasmic antigen [Syntaxin 13 (Syn13)] were used as controls. D, Immunostaining of brain sections confirmed that CaM distribution is not altered in CaMBP transgenic mice. The CA1 region of the hippocampus is shown for a wild-type and a transgenic mouse. DAPI staining was used to label nuclei. Scale bar, 5 μm.
To establish the cellular localization and the brain distribution of CaMBP, we stained cell cultures expressing CaMBP and brains from wild-type and CaMBP transgenic mice with a CaMBP-specific antibody (Wang et al., 1995). In cell culture, CaMBP immunoreactivity was limited only to the nuclei of cells transfected with the full-length CaMBP construct (Fig. 2A), whereas in brain sections, CaMBP immunoreactivity was also exclusively nuclear (Fig. 2B) and corresponded to the neuroanatomical pattern of CaMBP expression observed by in situ hybridization. Brains from wild-type mice showed no CaMBP immunoreactivity. To test for potential CaMBP-induced changes in CaM distribution, we examined nuclear and cytoplasmic extracts from the hippocampus of transgenic and wild-type animals and found no difference in the levels of cytoplasmic and nuclear CaM (Fig. 1C). In addition, brain sections from wild-type and CaMBP transgenic mice were stained for CaM, corresponding immunoreactivities were quantified, and cytoplasmic/nuclear CaM ratios were calculated (1.8 ± 0.13 and 1.82 ± 0.07 U for wild-type and transgenic mice, respectively; n = 5 animals each; NS) and indicated that expression of CaMBP in the nuclei did not affect the cellular distribution of endogenous CaM (Fig. 1D). Moreover, to exclude possible unobserved developmental effects, in all subsequent experiments, the transgene was suppressed during development and activated only in adulthood before electrophysiological or behavioral testing.
Figure 2.
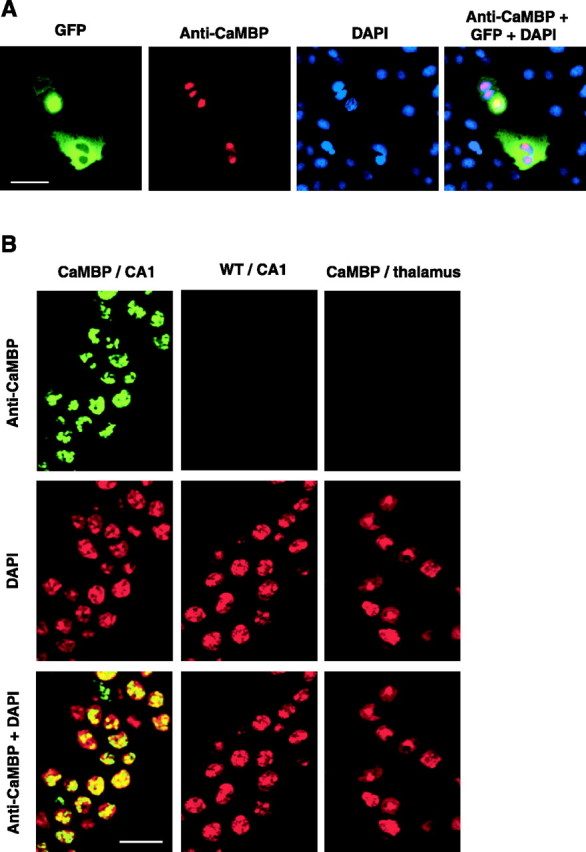
CaMBP immunolocalization. A, CaMBP immunostaining in cell culture. COS-7 cells were transiently transfected with CaMBP and GFP. Cells that are positive for GFP are also positive for CaMBP; nuclear counterstaining confirms CaMBP nuclear localization. Scale bar, 30 μm. B, The nuclear localization of CaMBP was confirmed by staining brain sections. DAPI staining labels nuclei. CaMBP-positive cells were detected only in brain areas in which CaMBP mRNA is expressed and was exclusively nuclear. Immunostaining is shown for the CA1 region of the hippocampus and thalamus of CaMBP transgenic mice and in the CA1 region of the hippocampus of wild-type (WT) animals. Scale bar, 25 μm.
Altered S133-CREB phosphorylation and gene expression
Previous studies in neuronal cell culture have demonstrated that CaMBP expression inhibits the rapid component of depolarization-induced S133 phosphorylation of CREB (Deisseroth et al., 1998), which is required for CREB-mediated gene expression (Shaywitz and Greenberg, 1999). To test whether expression of CaMBP also affects activity-induced S133 phosphorylation of CREB in adult neurons, we induced seizures with pentylenetetrazole in wild-type and transgenic mice and examined tissue at 10 min after the onset of generalized clonic seizures (Fig. 3). Brain sections were stained for total and phosphorylated CREB and imaged using confocal microscopy. Quantitative analysis of the nuclear signals obtained from the CA1 region of the hippocampus revealed no differences in baseline levels of CREB or phospho-S133-CREB. However, after seizure, the wild-type mice showed an increase in CREB phosphorylation that was blocked in the CaMBP transgenic animals (Fig. 3A-C). This demonstrates that in vivo nuclear CaM signaling participates in activity-induced S133 CREB phosphorylation but does not contribute to the significant baseline level of its phosphorylation; similar results were obtained by Western blotting (our unpublished data). The ability of extracellular signal-regulated kinase 1/2 (ERK1/2) MAPKs to activate nuclear CREB has been shown previously (Impey et al., 1998; Roberson et al., 1999). However, cytoplasmic but not nuclear Ca2+/CaM signaling induces the Ras-ERK1/2 pathway, which therefore should not be affected by CaMBP expression (Hardingham et al., 2001). We used ERK1/2 phosphorylation as a measure of MAPK activation and found no difference in levels of phospho-MAPK between wild-type and mutant mice in baseline conditions or after seizures (Fig. 3C). Collectively, these data suggest that inhibition of CREB phosphorylation by CaMBP occurs by a direct nuclear action rather than by blocking secondary signaling pathways in the cytoplasm.
Figure 3.
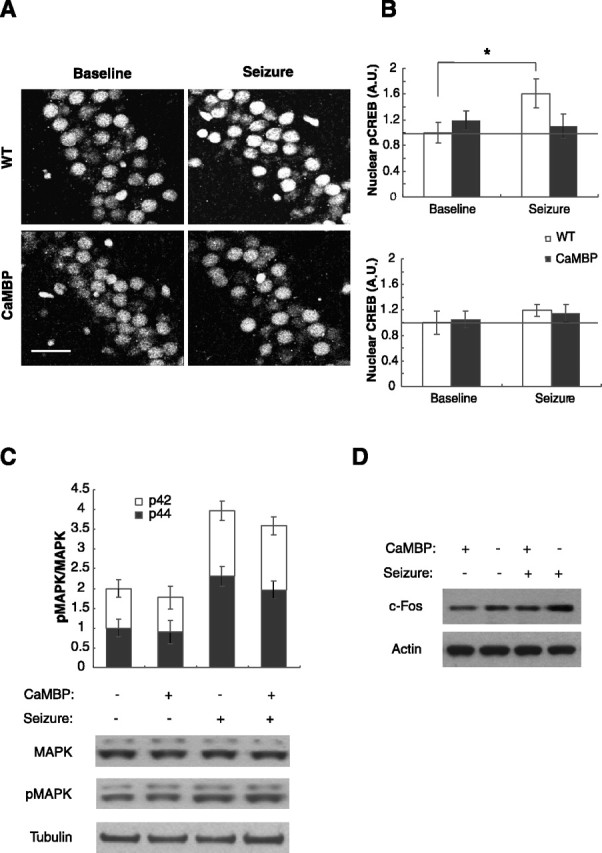
Altered S133-CREB phosphorylation and c-Fos levels in CaMBP mice after seizure induction. A, CREB phosphorylation was detected by immunofluorescence with a phospho-S133-specific antibody in wild-type (WT) and CaMBP transgenics at baseline or 10 min after seizure onset. Representative images from the CA1 hippocampal subregion are shown. Scale bar, 50 μm. B, Quantification of phospho-S133 CREB and total CREB in the CA1 region of hippocampus. Values represent group mean intensity of phospho-S133 CREB or CREB immunoreactivity and have been normalized to baseline of the wild-type group. Seizure induced CREB phosphorylation in wild-type mice (t test; *p < 0.05; n = 7) but not in CaMBP transgenics. There was no significant difference in baseline pCREB levels between wild types and CaMBP mice (CaMBP, 119 ± 14% of wild type). Total CREB levels were not affected by seizures and did not differ between wild types and transgenics (CaMBP, 105.1 ± 12% of wild-type; n = 4). C, Western blot quantification of phospho-MAPK in the hippocampus. Values represent the ratio of phosphorylated to total p42 and p44 MAPK levels in whole-cell hippocampal extracts from wild-type and mutant mice with and without seizure (n = 4). Seizure caused an increase in p42 and p44 phosphorylation, but no difference was detected between wild-type and CaMBP transgenic mice. D, Protein levels of c-fos in the striatum. Seizure caused an increase in c-fos in the wild-type but not in the transgenic mice. See Results for quantification.
Because we established a link between nuclear CaMBP and the inhibition of activity-dependent CREB phosphorylation, we next asked whether CaMBP affects gene expression 1 hr after seizure. The c-fos gene is a classic example of a neuronal activity-induced immediate early gene (Kiessling and Gass, 1993; Curran and Morgan, 1995). We analyzed the level of c-fos expression in the striatum by Western blotting and found a 40% increase in the levels of c-fos at 1 hr after seizure in wild-type animals, whereas the levels remained unchanged in the CaMBP mice (Fig. 3D). The ratios between seizure and baseline conditions were 1.39 ± 0.071 and 1.01 ± 0.055 for wild-type and CaMBP mice, respectively (n = 5 animals each; unpaired two-tailed Student's t test, p < 0.001).
Reduced maximal LTP in CaMBP transgenic mice
The maintenance of late-phase LTP (L-LTP) produced by repeated high-frequency stimulation of the hippocampus is disrupted by inhibitors of protein and RNA synthesis and in mutant mice carrying a hypomorphic CREB allele (Bourtchuladze et al., 1994; Nguyen et al., 1994) but not in mice carrying a CREB dominant-negative transgene (Pittenger et al., 2002). We characterized baseline synaptic transmission, paired-pulse facilitation (PPF), and LTP induced by a single high-frequency train (E-LTP) in the Schafer collateral pathway and found them to be indistinguishable between CaMBP and wild-type mice (Fig. 4A-C). Both PPF and E-LTP have been reported to be impaired by general blockers of CaM function, confirming that the transgene does not alter cytoplasmic CaM-based signaling (Reymann et al., 1988; Malenka et al., 1989; Wang and Kelly, 1996). When we examined L-LTP induced by three spaced high-frequency trains, we found a deficit in the CaMBP mice (Fig. 4 D). However, the deficit occurs shortly after stimulation, suggesting that it does not reflect a block of the RNA and protein synthesis-dependent phase of L-LTP but rather a decrease in the maximal levels of LTP that can be achieved.
Figure 4.

CaMBP mice exhibit LTP deficits. A, Analysis of input-output curves plotting the fEPSP slopes and their corresponding presynaptic fiber volley amplitudes of wild-type (WT) (n = 5) and CaMBP transgenic (n = 5) mice shows no significant differences. B, Paired-pulse facilitation in slices from wild-type (n = 8) and CaMBP transgenic mice (n = 9). Percentage of facilitation, measured by taking a ratio of the second fEPSP slope over the first fEPSP slope between 50 and 250 msec, was quantified. Paired-pulse facilitation in the CaMBP transgenic mice does not differ from wild type. C, LTP induced by one-train tetanic stimulation (100 Hz for 1 sec) in wild-type mice (n = 7) and CaMBP transgenic mice (n = 7). The level and time course of potentiation in the two groups was indistinguishable. D, Late-phase LTP induced by three-train tetanic stimulation was decreased in CaMBP transgenic mice. Potentiation at 3 hr after tetanus: wild type, 220 ± 22%, n = 8; CaMBP, 150 ± 29%, n = 11 (t test; p < 0.05).
CaMBP expression impairs formation, but not retrieval, of spatial memories
To evaluate the role of nuclear Ca2+ signaling on spatial learning, we tested CaMBP transgenic and wild-type littermates treated either with or without Dox on the Morris water-maze task (Morris et al., 1982). In the visible platform version of the water maze, mice expressing the transgene performed comparably to the three control groups in escape latency and swim speed, indicating that expression of the transgene did not disrupt the perceptual, motor, or motivational parameters required to perform the water-maze task (Fig. 5A). However, when we trained mice in the hidden version of the water maze, all control groups showed a spatially localized search, whereas the CaMBP-expressing group was impaired (Fig. 5B,C). This could result from a deficit in memory encoding, the disruption of processes required for recall, or sensorimotor-motivational abilities specific to the spatial version of the task. To distinguish between these possibilities, we activated the transgene in mice that had previously learned the task and assessed their performance with the transgene active. As shown (Fig. 5D), expression of the transgene had no effect on the performance of animals that had previously acquired the task with the transgene suppressed. Thus, expression of the nuclear localized CaM inhibitor blocks encoding of spatial memories without disrupting memory retrieval or the performance variables required for the task.
CaMBP expression leads to defect in LTM but not STM
Acquisition of spatial memory in the water maze requires prolonged training over multiple days and does not allow a clear distinction between STM and LTM. To achieve this distinction, we used a visual paired comparisons task to test both short- and long-term recognition memory (Tang et al., 1999; Clark et al., 2000). In this task, animals are allowed to explore two objects during the training phase. After a defined delay period, the memory test consists of a probe trial in which the animals are exposed to one of the familiar objects and a novel object. Animals tend to explore a novel object, and this increased preference is used to infer a memory for the previously explored familiar object. This is an explicit memory test that is impaired by hippocampal lesions in rats, monkeys, and humans (Squire and Zola, 1996; Clark et al., 2000; Zola et al., 2000). All groups spent more time exploring the novel object when tested immediately or 1 hr after training (Fig. 6A), demonstrating that expression of the CaMBP transgene did not disrupt learning or memory at short time delays. When tested at a 24 hr delay, all of the control groups still showed a preference for the novel object, whereas the group expressing the CaMBP transgene performed at chance levels.
Figure 6.

LTM is impaired in the CaMBP transgenic mice. A, Object recognition. Four groups of mice were tested: wild types (WT) off (n = 17) and on (n = 16) Dox and CaMBP transgenics off (n = 20) and on (n = 19) Dox. Values represent group means ± SEM. All mice spent more time exploring the novel object at the immediate and 1 hr delay tests. When tested 24 hr after training, the control mice spent significantly more time exploring the novel object compared with the mutants off Dox. A three-way ANOVA with repeated measures revealed a significant effect of genotype (F(1,68) = 8.22; p < 0.01). Planned comparisons for the CaMBP mice off Dox showed a significant difference between the 24 hr test and both the immediate and 1 hr tests (p < 0.01) and in 24 hr memory from each of the control groups (p < 0.02 in all cases). There was no difference in total exploration time between the groups (WT off Dox, 16.6 ± 1.8 sec; CaMBP off Dox, 19.1 ± 2.7 sec; WT on Dox, 20.1 ± 3.3 sec; CaMBP on Dox, 23.8 ± 2.5 sec). B, Time spent freezing during 1 hr contextual, 24 hr contextual, and 24 hr cued fear conditioning. There was no difference in 1 hr context memory between CaMBP transgenic mice off Dox (n = 22) and wild-type mice off Dox (n = 17). Four groups (CaMBP off Dox, n = 29; WT off Dox, n = 26; CaMBP on Dox, n = 17; WT on Dox, n = 17) were trained and tested for 24 hr contextual and cued memory. A two-way ANOVA for context freezing revealed a significant effect of genotype (F(1,85) = 5.89; p < 0.02) and Dox (F(1,85) = 6.76; p < 0.02). Post hoc Scheffe showed that the CaMBP transgenic mice off Dox froze less than all of the control groups (*p < 0.025 in all cases). There was no significant effect of genotype in 24 hr cued conditioning. C, CaMBP transgenic (n = 5) and wild-type (n = 5) mice were assayed for conditioned taste aversion using saccharin/water discrimination. Both groups preferred saccharin to water before conditioning. Mice showed a marked aversion to saccharin both 48 hr and 8 d after conditioning. A two-way ANOVA with repeated measures showed a significant effect of conditioning (F(2,20) = 28.7; p < 0.0001) but no effect of genotype. There was no difference in the total water intake between the two groups (CaMBP, 1.98 ± 0.3; WT, 1.72 ± 0.1).
To explore further the possibility that expression of CaMBP produces a selective defect in LTM, without affecting STM, the mice were tested in contextual and cued fear conditioning (Schafe et al., 2001). Memory encoding in these tasks involves different neuroanatomical substrates; contextual conditioning requires the function of hippocampus and amygdala, whereas cued conditioning appears to depend primarily on the amygdala. Transgenic and wild-type mice showed similar levels of conditioning during training and in a 1 hr memory test (Fig. 6B). However, when tested 24 hr after training, CaMBP transgenic mice exhibited a significant reduction in contextual, but not cued, fear conditioning. The lack of a cued conditioning deficit is consistent with the lack of transgene expression in the amygdala and suggests that the context deficit arises from a disruption of hippocampal function.
In addition to the three learning and memory tasks discussed above, we tested mice in conditioned taste aversion and several sensory-motor control tests. We found no significant difference between wild types and mice expressing the CaMBP transgene in any of these tests. In conditioned taste aversion, the mice were conditioned to saccharin and tested 48 hr and 8 d after training. Controls and mutants showed a marked aversion to saccharin in both tests (Fig. 6C). Thus, the only behavioral deficits associated with expression of the CaMBP transgene are in tasks that depend on the hippocampus, and in those tasks, the deficit is only in 24 hr memory with 1 hr memory remaining intact. These deficits cannot be attributable to developmental abnormalities and are specific to LTM encoding as opposed to recall.
Discussion
Inhibitors of protein synthesis, administered globally or to specific brain regions, produce a selective deficit in 24 hr memory but fail to disrupt STM measured at 1 hr (Davis and Squire, 1984). This phenomenon has been seen in many species using several different behavioral paradigms and has generally been interpreted as indicating that the stabilization of long-term memories requires new gene expression. This raises the critical question of what transcriptional signaling pathways are required to stabilize LTM. Studies in invertebrates have implicated CREB family transcription factors as critical mediators of this LTM consolidation (Yin et al., 1994; Kandel, 2001). Analysis of hypomorphic CREB alleles suggests a similar role in mammals (Bourtchuladze et al., 1994). Although the severity of the deficits in CREB hypomorphs appears to depend on genetic background, two recent studies using inducible dominant-negative inhibitors of CREB have clearly established a role for acute CREB-based transcriptional signaling in the adult in the consolidation of short-term 1 hr memory into 24 hr LTM (Kida et al., 2002; Pittenger et al., 2002).
The critical second messenger signaling pathways that stimulate the transcription needed for LTM consolidation are less clear. Although Ca2+ is the primary signal for the activation in gene expression in excitatory neurons by synaptic activity, a primary Ca2+ signal can activate many different second messenger pathways, including cAMP and MAP kinase, both of which can stimulate CREB-mediated transcription (Shaywitz and Greenberg, 1999). Deletion of the AC1 and AC8 adenyl cyclase isoforms yields mice lacking any detectable calcium-stimulated adenyl cyclase (Wong et al., 1999). The memory deficits in these mice did not parallel the time course seen with inhibitors of protein synthesis or CREB dominant-negative expression. Examination of transgenic mice expressing a dominant inhibitor of cAMP-stimulated A-kinase activity produces memory deficits in several tasks that paralleled deficits produced by protein synthesis inhibitors (Abel et al., 1997). However, these studies could not distinguish nuclear from cytoplasmic and acute from developmental effects of the transgene. The MAP kinase signaling pathway is complex and contains many parallel pathways that are difficult to segregate experimentally in a whole animal model. Pharmacological studies have indicated a role for MAP kinase signaling in memory (Schafe et al., 2000; Ohno et al., 2001; Hebert and Dash, 2002). Surprisingly, deletion of the gene encoding one of the major MAP kinase isoforms in the brain (ERK1) produces no memory deficit or enhanced memory in certain tasks (Selcher et al., 2001; Mazzucchelli et al., 2002). Thus, although cAMP and MAP kinase are clearly important modulators of memory in mice, they may not be the primary mediators of the critical transcriptional events required for LTM consolidation.
We have used the tetracycline system for transgene regulation to express a dominant-negative inhibitor of CaM signaling specifically in the nucleus of excitatory forebrain neurons. The specificity of the nuclear inhibition is supported by immunofluorescence showing nuclear localization of the inhibitor polypeptide, biochemical data showing no change in the nuclear and cytoplasmic distribution of CaM, and electrophysiological data showing that two processes sensitive to cytoplasmic but not nuclear CaM, E-LTP and PPF, are not altered. We found robust deficits in 24 hr memory in three different commonly used behavioral tasks, whereas STM at 1 hr is spared. These deficits cannot be attributable to alterations in neuronal development, because the transgene was activated only in adult mice. Most importantly, expression of the transgene affects encoding of LTM without impacting memory recall or performance variables. These memory defects parallel the time course of impairments seen in many organisms after inhibition of protein synthesis during behavioral training. This suggests that direct nuclear Ca2+ signaling is a major second messenger pathway used in stimulating the transcriptional changes required for LTM consolidation.
One likely target of nuclear CaM signaling is CaMKIV, which is a major Ca2+-dependent activator of CREB in the nucleus (Soderling, 1999). Several studies examining the function of CaMKIV using conventional knock-out and transgenic mice have yielded conflicting results in learning and memory tests. In one study, CaMKIV null mice showed normal spatial learning but impaired fear conditioning when measured at 24 hr (Ho et al., 2000; Wei et al., 2002). In the second study, using a dominant-negative inhibitor of CaMKIV, spatial memory was impaired but fear conditioning at 24 hr was unaltered (Kang et al., 2001). The discrepancies could arise from differences in the developmental timing of CaMKIV inhibition in the two animal models. Although these results demonstrate a role for CaMKIV in normal behavioral responses, they do not distinguish between an acute versus a developmental role for the enzyme. Moreover, because the behavioral phenotypes do not parallel phenotypes seen with protein synthesis inhibitors, the results suggest that CaMKIV alone cannot be the sole target for the transcriptional signaling necessary for LTM consolidation. In contrast, the behavioral phenotype of mice carrying a dominant-negative inhibitor of CREB family transcription factors is consistent with the one observed in the CaMBP mice (Kida et al., 2002; Pittenger et al., 2002). This suggests that although CREB is a critical transcription factor target in memory consolidation, it is the coordinated action of multiple nuclear CaM targets that is necessary for its appropriate activation. Recent data showing that CREB phosphorylation at sites acted on by CaMKII but not CaMKIV can alter the transcriptional response suggest nuclear isoforms of this kinase as another potential CaM target (Kornhauser et al., 2002). In addition, there are likely to be other targets (for example, the CREB binding protein) (Alarcon et al., 2004; Korzus et al., 2004) or Ca2+-dependent protein phosphatases, that are critical for the coordinated transcriptional response to increases in nuclear Ca2+ (Agell et al., 1998; Kitani et al., 2003). Finally, it is likely that nuclear CaM also exerts effects via CREB-independent pathways.
Although LTP has many features that make it an attractive candidate for a cellular mechanism of learning, there have been a number of cases in which severe LTP deficits in mutant mice have failed to strongly impact behavioral learning and memory (Ho et al., 2000), suggesting that the LTP phenotype in the current mice is not directly casually related to the memory phenotype. The current study links nuclear CaM signaling to activity-induced CREB phosphorylation and induction of c-fos. Preliminary gene chip studies suggest that the activity-induced expression of a number of other immediate early genes is also impaired in the mutant mice (our unpublished results). Although the signaling pathway is clear, which of the downstream targets of this regulation are critical and how they alter neuronal function to form the physical basis of a memory trace are unclear.
The cellular and molecular signaling mechanisms of LTM are most thoroughly characterized in invertebrates. In Aplysia, sensitization of the gill withdrawal reflex involves a form of heterosynaptic facilitation of synaptic transmission in which 5-HT acts to directly increase cAMP and activate CREB-dependent transcription (Kandel, 2001). Characterization of learning mutants in Drosophila also suggests a role for direct cAMP-dependent signaling to CREB in pavlovian conditioning (Yin and Tully, 1996; Dubnau et al., 2003). Previous studies in mice demonstrate that the function of CREB family transcription factors in memory consolidation is conserved in the mammalian brain (Bourtchuladze et al., 1994; Kida et al., 2002; Pittenger et al., 2002). However, the current results suggest that the direct action of Ca2+/CaM in the nucleus is a primary signaling pathway in mice. Memory formation in the mammalian brain, particularly hippocampal-dependent forms of explicit memory, presumably requires that the important plasticity captures aspects of complex patterns of excitatory activity in multiple brain regions. Because Ca2+ influx is directly linked to excitatory neuronal activity both through synaptic receptors and through voltage-gated channels, it is well suited to serve as the primary signaling molecule for this task. Thus, whereas the CREB-dependent transcriptional pathways involved in the stabilization of LTM are evolutionarily old and conserved, the use of the direct Ca2+/CaM pathway to signal to CREB may allow for the behaviorally relevant plasticity to be more directly linked to complex patterns of excitatory activity required to capture explicit forms of memory in the mammalian brain.
Footnotes
The work was supported by National Institutes of Health Grant 5R01MH057368-06 and by the McKnight Foundation. We thank James Watson, Elizabeth Gardiner, and Kathy Spencer for their technical help.
Correspondence should be addressed to Mark Mayford, Cell Biology, ICND202, The Scripps Research Institute, La Jolla, CA 92037. E-mail: mmayford@scripps.edu.
Copyright © 2004 Society for Neuroscience 0270-6474/04/2410858-10$15.00/0
References
- Abel T, Nguyen PV, Barad M, Deuel TA, Kandel ER, Bourtchouladze R (1997) Genetic demonstration of a role for PKA in the late phase of LTP and in hippocampus-based long-term memory. Cell 88: 615-626. [DOI] [PubMed] [Google Scholar]
- Agell N, Aligue R, Alemany V, Castro A, Jaime M, Pujol MJ, Rius E, Serratosa J, Taules M, Bachs O (1998) New nuclear functions for calmodulin. Cell Calcium 23: 115-121. [DOI] [PubMed] [Google Scholar]
- Alarcon JM, Malleret G, Touzani K, Vronskaya S, Ishii S, Kandel ER, Barco A (2004) Chromatin acetylation, memory, and LTP are impaired in CBP+/- mice: a model for the cognitive deficit in Rubinstein-Taybi syndrome and its amelioration. Neuron 42: 947-959. [DOI] [PubMed] [Google Scholar]
- Bading H (2000) Transcription-dependent neuronal plasticity the nuclear calcium hypothesis. Eur J Biochem 267: 5280-5283. [DOI] [PubMed] [Google Scholar]
- Bourtchuladze R, Frenguelli B, Blendy J, Cioffi D, Schutz G, Silva AJ (1994) Deficient long-term memory in mice with a targeted mutation of the cAMP-responsive element-binding protein. Cell 79: 59-68. [DOI] [PubMed] [Google Scholar]
- Chetkovich DM, Gray R, Johnston D, Sweatt JD (1991) N-methyl-d-aspartate receptor activation increases cAMP levels and voltage-gated Ca2+ channel activity in area CA1 of hippocampus. Proc Natl Acad Sci USA 88: 6467-6471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark RE, Zola SM, Squire LR (2000) Impaired recognition memory in rats after damage to the hippocampus. J Neurosci 20: 8853-8860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crawley JN, Paylor R (1997) A proposed test battery and constellations of specific behavioral paradigms to investigate the behavioral phenotypes of transgenic and knockout mice. Horm Behav 31: 197-211. [DOI] [PubMed] [Google Scholar]
- Curran T, Morgan JI (1995) Fos: an immediate-early transcription factor in neurons. J Neurobiol 26: 403-412. [DOI] [PubMed] [Google Scholar]
- Davis HP, Squire LR (1984) Protein synthesis and memory: a review. Psychol Bull 96: 518-559. [PubMed] [Google Scholar]
- Deisseroth K, Heist EK, Tsien RW (1998) Translocation of calmodulin to the nucleus supports CREB phosphorylation in hippocampal neurons. Nature 392: 198-202. [DOI] [PubMed] [Google Scholar]
- Dodart JC, Mathis C, Ungerer A (1997) Scopolamine-induced deficits in a two-trial object recognition task in mice. NeuroReport 8: 1173-1178. [DOI] [PubMed] [Google Scholar]
- Dolmetsch RE, Pajvani U, Fife K, Spotts JM, Greenberg ME (2001) Signaling to the nucleus by an L-type calcium channel-calmodulin complex through the MAP kinase pathway. Science 294: 333-339. [DOI] [PubMed] [Google Scholar]
- Dubnau J, Chiang AS, Tully T (2003) Neural substrates of memory: from synapse to system. J Neurobiol 54: 238-253. [DOI] [PubMed] [Google Scholar]
- Hardingham GE, Chawla S, Johnson CM, Bading H (1997) Distinct functions of nuclear and cytoplasmic calcium in the control of gene expression. Nature 385: 260-265. [DOI] [PubMed] [Google Scholar]
- Hardingham GE, Arnold FJ, Bading H (2001) A calcium microdomain near NMDA receptors: on switch for ERK-dependent synapse-to-nucleus communication. Nat Neurosci 4: 565-566. [DOI] [PubMed] [Google Scholar]
- Hebert AE, Dash PK (2002) Extracellular signal-regulated kinase activity in the entorhinal cortex is necessary for long-term spatial memory. Learn Mem 9: 156-166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ho N, Liauw JA, Blaeser F, Wei F, Hanissian S, Muglia LM, Wozniak DF, Nardi A, Arvin KL, Holtzman DM, Linden DJ, Zhuo M, Muglia LJ, Chatila TA (2000) Impaired synaptic plasticity and cAMP response element-binding protein activation in Ca2+/calmodulin-dependent protein kinase type IV/Gr-deficient mice. J Neurosci 20: 6459-6472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Impey S, Obrietan K, Wong ST, Poser S, Yano S, Wayman G, Deloulme JC, Chan G, Storm DR (1998) Cross talk between ERK and PKA is required for Ca2+ stimulation of CREB-dependent transcription and ERK nuclear translocation. Neuron 21: 869-883. [DOI] [PubMed] [Google Scholar]
- Kandel ER (2001) The molecular biology of memory storage: a dialogue between genes and synapses. Science 294: 1030-1038. [DOI] [PubMed] [Google Scholar]
- Kang H, Sun LD, Atkins CM, Soderling TR, Wilson MA, Tonegawa S (2001) An important role of neural activity-dependent CaMKIV signaling in the consolidation of long-term memory. Cell 106: 771-783. [DOI] [PubMed] [Google Scholar]
- Kida S, Josselyn SA, de Ortiz SP, Kogan JH, Chevere I, Masushige S, Silva AJ (2002) CREB required for the stability of new and reactivated fear memories. Nat Neurosci 5: 348-355. [DOI] [PubMed] [Google Scholar]
- Kiessling M, Gass P (1993) Immediate early gene expression in experimental epilepsy. Brain Pathol 3: 381-393. [DOI] [PubMed] [Google Scholar]
- Kitani T, Okuno S, Takeuchi M, Fujisawa H (2003) Subcellular distributions of rat CaM kinase phosphatase N and other members of the CaM kinase regulatory system. J Neurochem 86: 77-85. [DOI] [PubMed] [Google Scholar]
- Kornhauser JM, Cowan CW, Shaywitz AJ, Dolmetsch RE, Griffith EC, Hu LS, Haddad C, Xia Z, Greenberg ME (2002) CREB transcriptional activity in neurons is regulated by multiple, calcium-specific phosphorylation events. Neuron 34: 221-233. [DOI] [PubMed] [Google Scholar]
- Korzus E, Rosenfeld MG, Mayford M (2004) CBP histone acetyltransferase activity is a critical component of memory consolidation. Neuron 42: 961-972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malenka RC, Kauer JA, Perkel DJ, Mauk MD, Kelly PT, Nicoll RA, Waxham MN (1989) An essential role for postsynaptic calmodulin and protein kinase activity in long-term potentiation. Nature 340: 554-557. [DOI] [PubMed] [Google Scholar]
- Masugi M, Yokoi M, Shigemoto R, Muguruma K, Watanabe Y, Sansig G, van der Putten H, Nakanishi S (1999) Metabotropic glutamate receptor subtype 7 ablation causes deficit in fear response and conditioned taste aversion. J Neurosci 19: 955-963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayford M, Wang J, Kandel ER, O'Dell TJ (1995) CaMKII regulates the frequency-response function of hippocampal synapses for the production of both LTD and LTP. Cell 81: 891-904. [DOI] [PubMed] [Google Scholar]
- Mayford M, Bach ME, Huang YY, Wang L, Hawkins RD, Kandel ER (1996) Control of memory formation through regulated expression of a CaMKII transgene. Science 274: 1678-1683. [DOI] [PubMed] [Google Scholar]
- Mazzucchelli C, Vantaggiato C, Ciamei A, Fasano S, Pakhotin P, Krezel W, Welzl H, Wolfer DP, Pages G, Valverde O, Marowsky A, Porrazzo A, Orban PC, Maldonado R, Ehrengruber MU, Cestari V, Lipp HP, Chapman PF, Pouyssegur J, Brambilla R (2002) Knockout of ERK1 MAP kinase enhances synaptic plasticity in the striatum and facilitates striatal-mediated learning and memory. Neuron 34: 807-820. [DOI] [PubMed] [Google Scholar]
- Morgan JI, Cohen DR, Hempstead JL, Curran T (1987) Mapping patterns of c-fos expression in the central nervous system after seizure. Science 237: 192-197. [DOI] [PubMed] [Google Scholar]
- Morris RG, Garrud P, Rawlins JN, O'Keefe J (1982) Place navigation impaired in rats with hippocampal lesions. Nature 297: 681-683. [DOI] [PubMed] [Google Scholar]
- Nguyen PV, Abel T, Kandel ER (1994) Requirement of a critical period of transcription for induction of a late phase of LTP. Science 265: 1104-1107. [DOI] [PubMed] [Google Scholar]
- Ohno M, Frankland PW, Chen AP, Costa RM, Silva AJ (2001) Inducible, pharmacogenetic approaches to the study of learning and memory. Nat Neurosci 4: 1238-1243. [DOI] [PubMed] [Google Scholar]
- Pittenger C, Huang YY, Paletzki RF, Bourtchouladze R, Scanlin H, Vronskaya S, Kandel ER (2002) Reversible inhibition of CREB/ATF transcription factors in region CA1 of the dorsal hippocampus disrupts hippocampus-dependent spatial memory. Neuron 34: 447-462. [DOI] [PubMed] [Google Scholar]
- Reymann KG, Brodemann R, Kase H, Matthies H (1988) Inhibitors of calmodulin and protein kinase C block different phases of hippocampal long-term potentiation. Brain Res 461: 388-392. [DOI] [PubMed] [Google Scholar]
- Roberson ED, English JD, Adams JP, Selcher JC, Kondratick C, Sweatt JD (1999) The mitogen-activated protein kinase cascade couples PKA and PKC to cAMP response element binding protein phosphorylation in area CA1 of hippocampus. J Neurosci 19: 4337-4348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schafe GE, Atkins CM, Swank MW, Bauer EP, Sweatt JD, LeDoux JE (2000) Activation of ERK/MAP kinase in the amygdala is required for memory consolidation of pavlovian fear conditioning. J Neurosci 20: 8177-8187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schafe GE, Nader K, Blair HT, LeDoux JE (2001) Memory consolidation of Pavlovian fear conditioning: a cellular and molecular perspective. Trends Neurosci 24: 540-546. [DOI] [PubMed] [Google Scholar]
- Selcher JC, Nekrasova T, Paylor R, Landreth GE, Sweatt JD (2001) Mice lacking the ERK1 isoform of MAP kinase are unimpaired in emotional learning. Learn Mem 8: 11-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaywitz AJ, Greenberg ME (1999) CREB: a stimulus-induced transcription factor activated by a diverse array of extracellular signals. Annu Rev Biochem 68: 821-861. [DOI] [PubMed] [Google Scholar]
- Soderling TR (1999) The Ca-calmodulin-dependent protein kinase cascade. Trends Biochem Sci 24: 232-236. [DOI] [PubMed] [Google Scholar]
- Squire LR, Zola SM (1996) Structure and function of declarative and non-declarative memory systems. Proc Natl Acad Sci USA 93: 13515-13522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sweatt JD (2001) The neuronal MAP kinase cascade: a biochemical signal integration system subserving synaptic plasticity and memory. J Neurochem 76: 1-10. [DOI] [PubMed] [Google Scholar]
- Tang YP, Shimizu E, Dube GR, Rampon C, Kerchner GA, Zhuo M, Liu G, Tsien JZ (1999) Genetic enhancement of learning and memory in mice. Nature 401: 63-69. [DOI] [PubMed] [Google Scholar]
- Waddell S, Quinn WG (2001) What can we teach Drosophila? What can they teach us? Trends Genet 17: 719-726. [DOI] [PubMed] [Google Scholar]
- Wang J, Campos B, Jamieson Jr GA, Kaetzel MA, Dedman JR (1995) Functional elimination of calmodulin within the nucleus by targeted expression of an inhibitor peptide. J Biol Chem 270: 30245-30248. [DOI] [PubMed] [Google Scholar]
- Wang J, Moreira KM, Campos B, Kaetzel MA, Dedman JR (1996) Targeted neutralization of calmodulin in the nucleus blocks DNA synthesis and cell cycle progression. Biochim Biophys Acta 1313: 223-228. [DOI] [PubMed] [Google Scholar]
- Wang JH, Kelly PT (1996) Regulation of synaptic facilitation by postsynaptic Ca2+/CaM pathways in hippocampal CA1 neurons. J Neurophysiol 76: 276-286. [DOI] [PubMed] [Google Scholar]
- Wei F, Qiu CS, Liauw J, Robinson DA, Ho N, Chatila T, Zhuo M (2002) Calcium calmodulin-dependent protein kinase IV is required for fear memory. Nat Neurosci 5: 573-579. [DOI] [PubMed] [Google Scholar]
- West AE, Chen WG, Dalva MB, Dolmetsch RE, Kornhauser JM, Shaywitz AJ, Takasu MA, Tao X, Greenberg ME (2001) Calcium regulation of neuronal gene expression. Proc Natl Acad Sci USA 98: 11024-11031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong ST, Athos J, Figueroa XA, Pineda VV, Schaefer ML, Chavkin CC, Muglia LJ, Storm DR (1999) Calcium-stimulated adenylyl cyclase activity is critical for hippocampus-dependent long-term memory and late phase LTP. Neuron 23: 787-798. [DOI] [PubMed] [Google Scholar]
- Yin JC, Tully T (1996) CREB and the formation of long-term memory. Curr Opin Neurobiol 6: 264-268. [DOI] [PubMed] [Google Scholar]
- Yin JC, Wallach JS, Del Vecchio M, Wilder EL, Zhou H, Quinn WG, Tully T (1994) Induction of a dominant negative CREB transgene specifically blocks long-term memory in Drosophila Cell 79: 49-58. [DOI] [PubMed] [Google Scholar]
- Yin JC, Del Vecchio M, Zhou H, Tully T (1995) CREB as a memory modulator: induced expression of a dCREB2 activator isoform enhances long-term memory in Drosophila Cell 81: 107-115. [DOI] [PubMed] [Google Scholar]
- Zola SM, Squire LR, Teng E, Stefanacci L, Buffalo EA, Clark RE (2000) Impaired recognition memory in monkeys after damage limited to the hippocampal region. J Neurosci 20: 451-463. [DOI] [PMC free article] [PubMed] [Google Scholar]