

Abstract
Glycogen synthase kinase-3β (GSK-3β) is a critical activator of neuronal apoptosis induced by a diverse array of neurotoxic insults. However, the downstream substrates of GSK-3β that ultimately induce neuronal death are unknown. Here, we show that GSK-3β phosphorylates and regulates the activity of Bax, a pro-apoptotic Bcl-2 family member that stimulates the intrinsic (mitochondrial) death pathway by eliciting cytochrome c release from mitochondria. In cerebellar granule neurons undergoing apoptosis, inhibition of GSK-3β suppressed both the mitochondrial translocation of an expressed green fluorescent protein (GFP)-Baxα fusion protein and the conformational activation of endogenous Bax. GSK-3β directly phosphorylated Baxα on Ser163, a residue found within a species-conserved, putative GSK-3β phosphorylation motif. Coexpression of GFP-Baxα with a constitutively active mutant of GSK-3β, GSK-3β(Ser9Ala), enhanced the in vivo phosphorylation of wild-type Baxα, but not a Ser163Ala mutant of Baxα, in transfected human embryonic kidney 293 (HEK293) cells. Moreover, cotransfection with constitutively active GSK-3β promoted the localization of Baxα to mitochondria and induced apoptosis in both transfected HEK293 cells and cerebellar granule neurons. In contrast, neither a Ser163Ala point mutant of Baxα nor a naturally occurring splice variant that lacks 13 amino acids encompassing Ser163 (Baxσ) were driven to mitochondria in HEK293 cells coexpressing constitutively active GSK-3β. In a similar manner, either mutation or deletion of the identified GSK-3β phosphorylation motif prevented the localization of Bax to mitochondria in cerebellar granule neurons undergoing apoptosis. Our results indicate that GSK-3β exerts some of its pro-apoptotic effects in neurons by regulating the mitochondrial localization of Bax, a key component of the intrinsic apoptotic cascade.
Keywords: glycogen synthase kinase, cerebellar granule neuron, apoptosis, mitochondria, Bax, phosphorylation
Introduction
Aberrant neuronal apoptosis contributes to the progression of several neurodegenerative disorders (Vila and Przedborski, 2003). Protein kinases are key regulators of neuronal cell fate and can be broadly categorized into anti-apoptotic and pro-apoptotic groups. Anti-apoptotic kinases, including AKT and ERK (extracellular signal-regulated kinase), are activated downstream of neurotrophic factor receptors (Fukunaga and Miyamoto, 1998; Brunet et al., 2001). In contrast, protein kinases that play proapoptotic roles in neurons, including JNK (c-Jun NH2-terminal kinase) and glycogen synthase kinase-3β (GSK-3β), are activated by a variety of neuronal insults (Mielke and Herdegen, 2000; Kaytor and Orr, 2002). In particular, GSK-3β is a critical activator of cell death in numerous models of neuronal apoptosis (Hetman et al., 2000; Li et al., 2000; Perez et al., 2003; Phiel et al., 2003).
Identification of substrates of GSK-3β may provide insight into the cellular mechanisms that underlie neurodegeneration. To date, the three most studied substrates of GSK-3β include the metabolic enzyme glycogen synthase, the microtubule bundling protein tau, and a mediator of the Wnt signaling pathway and cell-cell adhesion, β-catenin (Doble and Woodgett, 2003). Although hyperphosphorylation of tau by GSK-3β has been implicated in the pathogenesis of Alzheimer's disease (Kaytor and Orr, 2002), the contribution of this phosphorylation event to neuronal death in other models of neurodegeneration is less well established. Moreover, although the GSK-3β-mediated phosphorylation of β-catenin targets this protein for degradation, phosphorylation-site mutants of β-catenin fail to protect neurons from apoptosis under conditions in which GSK-3β inhibition is neuroprotective (Hetman et al., 2000). These findings suggest that additional unidentified substrates of GSK-3β exist that mediate its pro-apoptotic action in neurons.
Neuronal apoptosis often occurs via an intrinsic apoptotic cascade triggered by the translocation of Bax, a pro-apoptotic Bcl-2 family member, to mitochondria. In response to signals generated by BH3-only Bcl-2 family members, such as Bim and Bid, Bax oligomerizes at the outer mitochondrial membrane and forms a pore that releases cytochrome c from the mitochondria (Zong et al., 2001). Cytosolic cytochrome c then interacts with Apaf-1 and pro-caspase-9 to form a functional apoptosome that ultimately activates downstream executioner caspases (Zou et al., 1999). Many models of neuronal apoptosis occur via this Bax-dependent mitochondrial pathway (Cregan et al., 1999; Putcha et al., 1999; Selimi et al., 2000; Vila et al., 2001). Yet despite the prevalence of Bax involvement in neuronal apoptosis, the cellular mechanisms that regulate this Bcl-2 family member, particularly the role of phosphorylation, have not been clearly defined.
In the current study, we used primary cultures of cerebellar granule neurons (CGNs) isolated from postnatal rats to investigate the role of GSK-3β in the regulation of Bax function. CGNs require serum and depolarizing extracellular potassium for their survival in vitro and die via a mitochondrial apoptotic cascade when deprived of this trophic support (D'Mello et al., 1993; Linseman et al., 2002). CGN apoptosis is dependent on both Bax translocation to mitochondria and activation of GSK-3β (Li et al., 2000; Putcha et al., 2002). Thus, this is an ideal cell model in which to examine the interaction of Bax and GSK-3β during neuronal apoptosis.
Materials and Methods
Reagents. A plasmid encoding an N-terminal green fluorescent protein (GFP) fusion protein of human Baxα was kindly provided by Dr. R. J. Youle (National Institute of Neurological Disorders and Stroke, National Institutes of Health, Bethesda, MD). Enhanced GFP (pEGFP) vector, monoclonal antibody for immunoprecipitation of GFP, and polyclonal living colors antibody for immunoblotting of GFP were from BD Biosciences Clontech (Palo Alto, CA). Tetramethylrhodamine ethyl ester (TMRE) dye and an antibody to cytochrome c oxidase subunit IV (COX IV) were from Molecular Probes (Eugene, OR). Monoclonal antibody to the hemagglutinin (HA) epitope tag and polyclonal antibodies to phospho-GSK-3β (Ser9) and total GSK-3β were obtained from Cell Signaling Technologies (Beverly, MA). Insulin-like growth factor I (IGF-I), LiCl, Hoecsht dye 33258, and 4′,6-diamidino-2-phenylindole (DAPI) were from Sigma (St. Louis, MO). GSK-3β inhibitor II and a specific peptide inhibitor of GSK-3β were from Calbiochem (San Diego, CA). Monoclonal antibody to the active conformation of Bax (clone 6A7) was purchased from Alexis Biochemicals (San Diego, CA). Recombinant, active GSK-3β was from Upstate Biotechnology (Charlottesville, VA). [γ-32P]ATP (3000 Ci/mmol), 32P as orthophosphate (10 mCi/ml), horseradish peroxidase-linked secondary antibodies, and reagents for enhanced chemiluminescence detection were obtained from Amersham Biosciences (Piscataway, NJ). Empty pcDNA3.1 vector was obtained from Invitrogen (Carlsbad, CA). A plasmid encoding HA-tagged GSK-3β(Ser9Ala) was provided by Dr. M. J. Birnbaum (University of Pennsylvania, Philadelphia, PA). A plasmid encoding Baxσ was provided by Dr. R. Bertrand (University of Montreal, Montreal, Quebec, Canada). FITC- and cyanine 3 (Cy3)-conjugated secondary antibodies for immunofluorescence were from Jackson ImmunoResearch (West Grove, PA).
Cell culture. Rat CGNs were isolated from 7-d-old Sprague Dawley rat pups (15-19 gm), as described previously (Li et al., 2000). Briefly, neurons were plated on 35-mm-diameter plastic dishes coated with poly-l-lysine at a density of 2.0 × 106 cells/ml in basal modified Eagle's medium containing 10% fetal bovine serum, 25 mm KCl, 2 mm l-glutamine, and penicillin (100 U/ml)-streptomycin (100 μg/ml) (Invitrogen). Cytosine arabinoside (10 μm) was added to the culture medium 24 hr after plating to limit the growth of non-neuronal cells. Using this protocol, the cultures were ∼95% pure for granule neurons. In general, experiments were performed after 6-7 d in culture. Human embryonic kidney 293 (HEK293) cells were maintained in standard DMEM containing 10% fetal calf serum and routinely passaged every 3-4 d.
Transfection of primary CGNs. CGNs were transiently transfected using the Helios Gene-Gun system (Bio-Rad, Hercules, CA). Briefly, 60 μg of plasmid DNA was precipitated onto 30 mg of 0.6-μm-diameter gold beads in a CaCl2-spermidine mixture. The gold/DNA precipitates were washed three times in 100% ethanol and resuspended in 3 ml of ethanol containing 0.05 mg/ml polyvinylpyrrolidone. After thoroughly resuspending the gold/DNA precipitate, it was drawn into ∼74 cm of Tefzel tubing, and the beads were allowed to settle to the bottom of the tubing. After 5 min, the ethanol was slowly drawn off while the beads adhered to the tubing. The tubing was dried under nitrogen for an additional 5 min and then cut into ∼1.3 cm pieces. These pieces can be stored desiccated at 4°C for ∼6 weeks. CGNs to be transfected were seeded at a density of 8 × 105 cells/well on polyethyleneimine-coated glass coverslips in 24-well plates (Corning, Corning, NY). After 5 d in culture, the medium was removed from the wells, and the 1.3 cm lengths of tubing containing the DNA-bound beads were loaded into the Gene-Gun and shot with a burst of ∼100 psi helium through a 40 μm nylon cell strainer placed over the well. The medium was replaced immediately, and cells were grown an additional 48 hr before image analysis. For experiments in which GFPBaxα was cotransfected with either pcDNA3.1 or HA-GSK-3β(S9A), the ratio of plasmids coprecipitated onto the gold beads was 60:1 for empty vector or GSK-3β to the Bax fusion protein.
Transfection of HEK293 cells. HEK293 cells were grown to ∼90% confluency in 35-mm-diameter tissue culture plates. Aliquots (500 ng) of the various GFP-Bax plasmids and 4 μg of either constitutively active HA-GSK3β Ser9Ala or the empty vector control plasmid (pCDNA3.1) were transiently transfected using the Lipofectamine 2000 reagent (Invitrogen) according to the manufacturer's protocol. Cells were grown for 24 hr, after which they were fixed for microscopic imaging analysis of GFP-Bax localization. For morphological assessment of apoptosis, transfected cells were grown for 48 hr before quantitation of apoptosis.
Quantitation of GFP-Bax localization. CGNs or HEK293 cells, which were transfected with various GFP-Bax plasmids, were washed once with PBS and fixed for 20 min in 4% paraformaldehyde. GFP-Bax localization was assessed in HEK293 cells using a 63× water immersion objective. Localization in CGNs was performed under 63× oil after adhering the coverslips to glass slides. Cells were examined for GFP fluorescence and were scored as either diffuse (if the staining observed in the FITC channel was diffuse throughout the cell) or mitochondrial (if the staining observed in the FITC channel localized to discrete subcellular locations within the cytoplasm and around the nucleus). Results for HEK293 cells are averages of blinded quantitations from at least three independent experiments per group in which ∼200 transfected cells were counted per group per experiment. Data for CGNs represent quantitative analysis of at least three separate experiments in which ∼200 transfected CGNs were analyzed per fusion protein under both control and apoptotic conditions.
Immunocytochemistry. CGNs were cultured on polyethyleneimine-coated glass coverslips at a density of ∼2.5 × 105 cells per coverslip. After transfection and incubation as described in Results, cells were fixed in 4% paraformaldehyde and were then permeabilized and blocked in PBS, pH 7.4, containing 0.2% Triton X-100 and 5% BSA. Cells were then incubated for ∼16 hr at 4°C with primary antibody diluted in PBS containing 0.2% Triton X-100 and 2% BSA. The primary antibody was aspirated, and the cells were washed five times with PBS. The cells were then incubated with either Cy3-conjugated or FITC-conjugated secondary antibodies and DAPI for 1 hr at room temperature. CGNs were then washed five more times with PBS, and coverslips were adhered to glass slides in mounting medium (0.1% p-phenylenediamine in 75% glycerol in PBS). Fluorescent images were captured using a 63× oil immersion objective on an Axioplan 2 microscope (Zeiss, Oberkochen, Germany) equipped with a Cooke Sensicam deep-cooled CCD camera and a Slidebook software analysis program for digital deconvolution (Intelligent Imaging Innovations, Denver, CO). Quantitation of active Bax immunoreactivity was performed by determining the percentage of CGNs that showed positive punctate staining with the 6A7 monoclonal antibody to the active conformation of Bax. In general, HEK293 cells were stained using an identical protocol, except that they were cultured on 35-mm-diameter plastic culture dishes and images were captured using a 63× water immersion objective.
Live cell imaging of TMRE fluorescence. HEK293 cells were cotransfected with GFP-Baxα and either pcDNA3.1 empty vector or HA-GSK-3βS9A. At 24 hr after transfection, TMRE (500 nm) and Hoechst dye were added for 30 min, and living cells were imaged using a Cy3 filter to detect TMRE fluorescence.
Preparation of cell extracts. After incubation, the culture medium was aspirated and cells were washed once with 2 ml of ice-cold PBS, pH 7.4, placed on ice, and scraped into lysis buffer (200 μl/35 mm well) containing 20 mm HEPES, pH 7.4, 1% Triton X-100, 50 mm NaCl, 1 mm EGTA, 5 mm β-glycerophosphate, 30 mm sodium pyrophosphate, 100 μm sodium orthovanadate, 1 mm phenylmethylsulfonyl fluoride, 10 μg/ml leupeptin, and 10 μg/ml aprotinin. Cell debris was removed by centrifugation at 6000 × g for 3 min, and the protein concentration of the supernatant was determined using a commercially available protein assay kit (Pierce, Rockford, IL). Aliquots (∼150 μg) of supernatant protein were diluted to a final concentration of 1× SDS-PAGE sample buffer, boiled for 5 min, and electrophoresed through 10% polyacrylamide gels. Proteins were transferred to polyvinylidene difluoride (PVDF) membranes (Millipore, Bedford, MA) and processed for immunoblot analysis.
Immunoblotting. Nonspecific binding sites were blocked in PBS, pH 7.4, containing 0.1% Tween 20 (PBS-T) and 1% BSA for 1 hr at room temperature. Primary antibodies were diluted in blocking solution and incubated with the membranes for 1 hr. Excess primary antibody was removed by washing the membranes three times in PBS-T. The blots were then incubated with the appropriate horseradish peroxidase-conjugated secondary antibody diluted in PBS-T for 1 hr and were subsequently washed three times in PBS-T. Immunoreactive proteins were detected by enhanced chemiluminescence. The autoluminograms shown are generally representative of at least three independent experiments.
Immunoprecipitation of GFP-Bax. At 16-24 hr after transfection, HEK293 cell lysates were prepared in lysis buffer containing 1% Triton X-100, as described above. Ten microliters of monoclonal antibody against GFP were added to 500 μl of lysate (∼1 μg/μl HEK293 cell protein concentration), and samples were mixed for 4 hr at 4°C by continuous inversion. Agarose-conjugated protein A/G (50 μl) was added, and samples were mixed for an additional 2 hr. Immune complexes were pelleted and washed for incubation in the in vitro kinase assay.
In vitro kinase assay. Pelleted GFP-Bax immune complexes were washed twice with 500 μl of cell lysis buffer, followed by two more washes in 500 μl of kinase reaction buffer [8 mm 4-morpholinepropanesulfonic acid (MOPS), pH 7.2, 0.2 mm EDTA, and 10 mm magnesium acetate]. The beads were then resuspended in 20 μl of kinase buffer, and 50 ng (in a total volume of 10 μl) of recombinant active GSK-3β was added, followed by 10 μCi of [γ-32P]ATP (previously diluted in ATP dilution buffer containing 20 mm MOPS, pH 7.2, 75 mm MgCl2, 25 mm β-glycerophosphate, 5 mm EGTA, 1 mm sodium orthovanadate, 1 mm DTT, and 500 μm cold ATP). The kinase reaction mixtures were incubated at 30°C for 30 min with continuous mixing at 1400 rpm. Reactions were stopped by adding 50 μl of 2× SDS-PAGE sample buffer and boiling for 5 min. Samples were then resolved on 10% polyacrylamide gels, and proteins were transferred to PVDF membranes and exposed to film for autoradiography. Subsequently, the membranes were immunoblotted with a polyclonal antibody to GFP to assess the immunoprecipitation efficiencies of the various GFP-Bax variants.
In vivo phosphorylation of GFP-Baxα by constitutively active GSK-3βS9A. HEK293 cell cultures were washed three times in phosphate-free DMEM culture medium and then labeled for 6 hr with 32P-orthophosphate (final concentration, 1.5 mCi/ml). Unincorporated phosphate was removed, and cells were washed three more times in phosphate-free DMEM. HEK293 cells were then cotransfected with wild-type GFP-Baxα, the single Ser163Ala mutant, or the Ser163Ala/Thr167Ala double mutant and either empty vector or constitutively active GSK-3βS9A. Approximately 16 hr after transfection, the GFP-Bax variants were immunoprecipitated using a GFP monoclonal antibody, as described above. Immune complexes were resolved by SDS-PAGE on a 10% polyacrylamide gel and transferred to PVDF, and phosphorylation was assessed by autoradiography.
Site-directed mutagenesis of GFP-Baxα. Site-directed mutagenesis of potential GSK3β phosphorylation sites within Baxα was performed on the pEGFP-Baxα plasmid using the Quikchange kit from Stratagene (La Jolla, CA). The following primers and annealing temperatures were used (underlined nucleotides indicate mutated site): Ser163Ala: upper primer 5′-GGTTGGGACGGCCTCCTCGCCTACTTTGGGACGCC-3′, lower primer 5′-ACCCTGCCGGAGGAGCGGATGAAACCCTGC-3′, 69°C annealing temperature (5% v/v DMSO was added to the reaction mix); Thr167Ala: upper primer 5′-TCCTCTCCTACTTTGGGGCGCCACGT-3′, lower primer 5′-GAGAGGATGAAACCCCGCGGGTGCA-3′, 78.6°C annealing temperature. The double mutant Ser163Ala/Thr167Ala was constructed using the Ser163Ala pEGFP-Baxα plasmid and the Thr167 upper and lower primers at an annealing temperature of 70°C.
All reactions were performed with the following program: 95°C for 30 sec, appropriate annealing temperature for 1 min, and 72°C for 5 min for 18 cycles.
Quantitation of apoptosis. HEK293 cells were transfected with variants of GFP-Bax in the absence or presence of constitutively active GSK-3β. At 48 hr after transfection, Hoechst dye was added directly to the wells to stain nuclei and cells were scraped into the culture medium so that all cells were harvested, including those that had detached from the dish. The cells were then pelleted, washed once with PBS, and resuspended in PBS. Nuclear morphology of GFP-positive cells was assessed by fluorescence microscopy. Cells containing chromatin that was condensed and/or fragmented were scored as apoptotic. Approximately 150 transfected cells were quantitated per well, and each construct was transfected into duplicate wells in a total of two separate experiments.
Data analysis. The results shown represent the means ± SEM for the number (n) of independent experiments that were performed. Statistical differences between the means of unpaired sets of data were evaluated by one-way ANOVA, followed by a post hoc Dunnett's or Tukey's test. A p value of <0.01 was considered statistically significant.
Results
GSK-3β inhibitors suppress Bax translocation to mitochondria and Bax conformational activation in trophic factor-deprived CGNs
CGNs were transfected with plasmids expressing either GFP or an NH2-terminal GFP fusion protein of human Bax α (GFP-Baxα) (Wolter et al., 1997). Neurons were transfected using gold beads coated with the precipitated plasmid DNA via particle-mediated gene transfer by a Helios Gene-Gun (Wellmann et al., 1999). At 48 hr after transfection, cells were either maintained in control medium containing serum and 25 mm KCl (25K+Ser) or switched to apoptotic medium lacking serum and containing 5 mm KCl (5K-Ser). After an additional 4 hr incubation, neurons were fixed in paraformaldehyde, and nuclei were stained with Hoechst dye. The localization of GFP or GFP-Baxα and nuclear morphology were examined by fluorescence microscopy (Fig. 1). GFP displayed a diffuse distribution over the entire cell body in CGNs cultured in either control or apoptotic medium (Fig. 1A, top). In contrast, the localization of GFP-Baxα changed from a diffuse pattern in control medium to a punctate distribution in apoptotic medium. The punctate localization of GFP-Baxα in apoptotic medium coincided with condensation and fragmentation of the chromatin in CGNs (Fig. 1A, bottom). To confirm that the punctate distribution of GFP-Baxα corresponded to its translocation to mitochondria, CGNs were stained for the integral mitochondrial membrane protein COX IV. In control medium, there was little specific overlap between the localization of GFP-Baxα and COX IV (Fig. 1B, top). While in apoptotic medium, GFP-Baxα showed substantial colocalization with COX IV (Fig. 1B, bottom). These data show that removal of serum and depolarizing extracellular potassium (trophic factor withdrawal) triggers translocation of GFP-Baxα to mitochondria in CGNs undergoing apoptosis.
Figure 1.
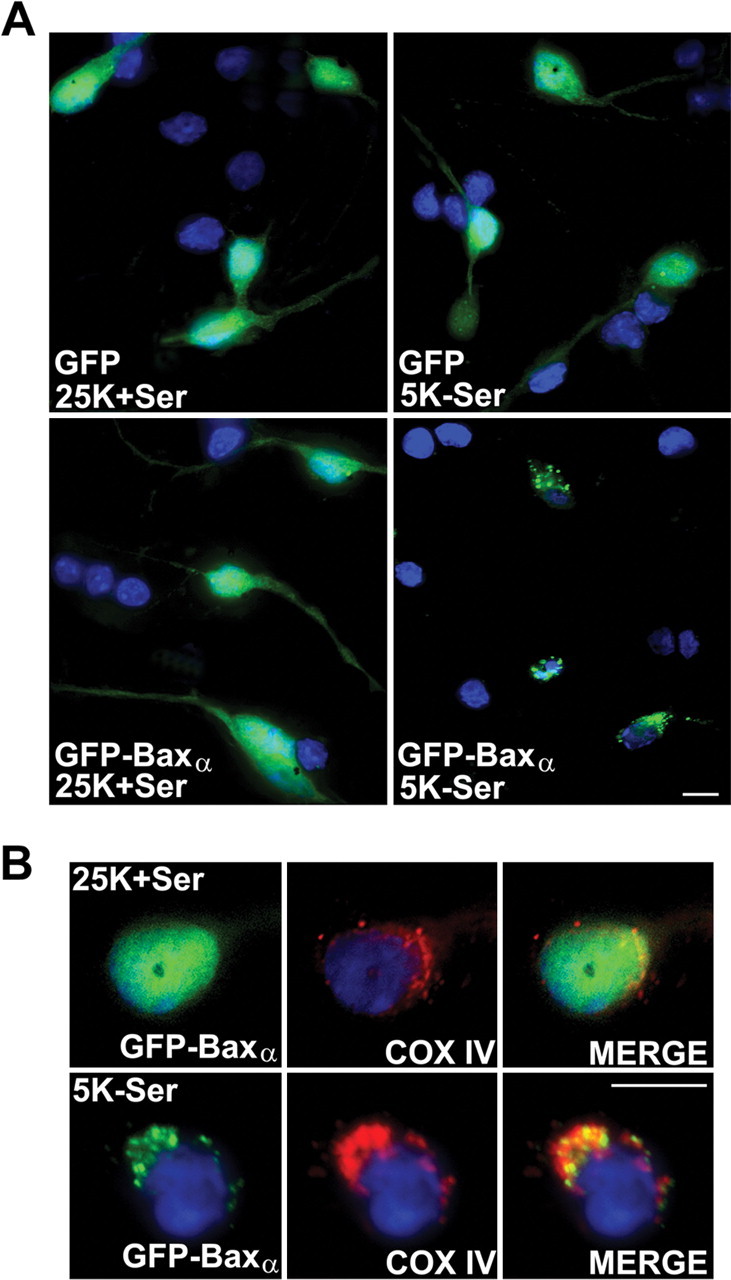
An expressed GFP-Baxα fusion protein translocates to mitochondria in CGNs undergoing apoptosis. A, Representative images of CGNs transfected with either GFP or GFP-Baxα and subsequently incubated for 4 hr in either control medium (25 mm KCl with serum; 25K+Ser) or apoptotic medium (5 mm KCl without serum; 5K-Ser). Images show the localization of GFP or GFP-Baxα and nuclear morphology after staining with Hoechst dye (shown in blue). B, Neurons transfected with GFP-Baxα were incubated as described in A, followed by immunostaining for COX IV to identify mitochondria. Scale bars, 10 μm.
Examination of the kinetics of GFP-Baxα translocation revealed a rapid movement of the expressed fusion protein to mitochondria after induction of apoptosis (Fig. 2A). In control medium, <30% of the CGNs expressing GFP-Baxα showed a mitochondrial localization of the fusion protein. Within 1 hr of trophic factor deprivation, a significant increase in mitochondrial GFP-Baxα was detectable. After 4 hr of incubation in apoptotic medium, ∼70% of the transfected CGNs demonstrated a mitochondrial distribution of GFP-Baxα. Measurement of GSK-3β activation in CGNs undergoing apoptosis revealed kinetics that closely paralleled the mitochondrial translocation of GFP-Baxα. When dephosphorylation of the inhibitory residue Ser9 was used as an index of GSK-3β activation, significant stimulation was observed within 1 hr, and nearly complete dephosphorylation occurred after 4 hr of incubation in apoptotic medium (Fig. 2B). Thus, the activation of GSK-3β is temporally correlated with the movement of Bax to mitochondria in CGNs undergoing apoptosis.
Figure 2.
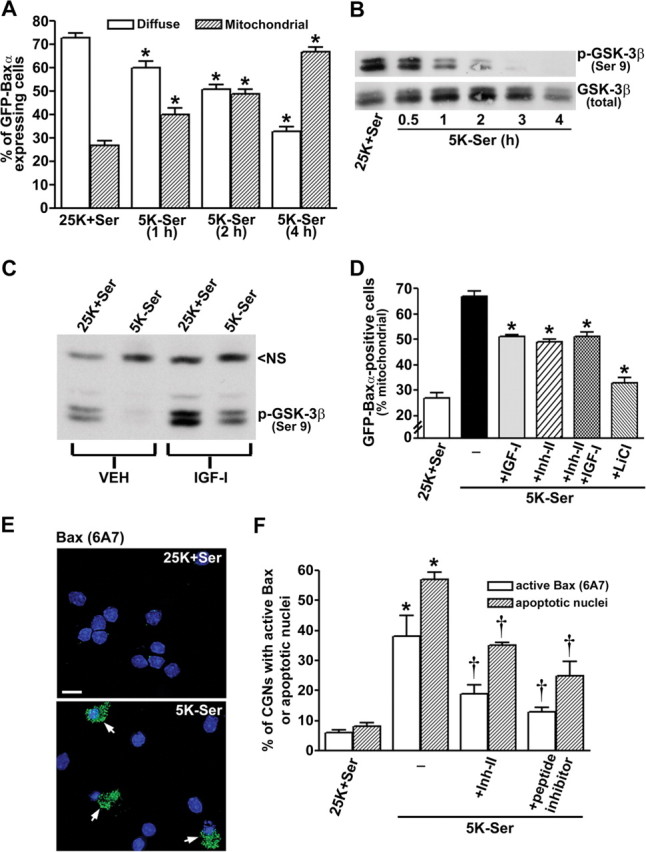
GSK-3β activity regulates Bax translocation to mitochondria and Bax conformational activation in apoptotic CGNs. A, Quantitation of the percentage of GFP-Baxα-expressing cells that demonstrated either a diffuse or mitochondrial localization of the fusion protein. The data shown are from three independent experiments in which ∼200 transfected CGNs were counted per experiment (*p < 0.01 compared with the 25K+Ser control). B, Time course of GSK-3β activation after trophic factor withdrawal in CGNs was measured by dephosphorylation at Ser9 (top blot). Total GSK-3β was not significantly altered over the duration of the experiment (bottom blot). All subsequent experiments involved a 4 hr incubation in 5K-Ser medium, unless noted otherwise. C, Effects of dilute acetic acid vehicle (VEH) or IGF-I (200 ng/ml) on GSK-3β activation (dephosphorylation) in trophic factor-deprived CGNs. NS, Nonspecific protein detected by the phospho-GSK-3β antibody, shown as a loading control. D, Quantitation of GFP-Baxα-positive CGNs that displayed mitochondrial localization of the fusion protein in either control or apoptotic medium containing IGF-I (200 ng/ml), GSK-3β inhibitor II (10 μm), or LiCl (20 mm). The results shown are from three separate experiments in which ∼200 transfected CGNs were counted per experiment (*p < 0.01 compared with 5K-Ser). E, Representative images of the activation of endogenous Bax detected immunocytochemically in trophic factor-deprived CGNs using an active conformation-specific monoclonal antibody (clone 6A7). Nuclei are stained with DAPI (shown in blue). Scale bar, 10μm. F, Quantitation of CGN apoptosis and immunoreactivity for active Bax in cells incubated for 16 hr in control or apoptotic medium containing either GSK-3β inhibitor II (10 μm) or a specific peptide inhibitor of GSK-3β (50 μm). The data represent the results from three independent experiments in which apoptosis and active Bax immunoreactivity were quantitated in ∼300 cells per condition per experiment (*p < 0.01 compared with 25K+Ser; †p < 0.01 compared with 5K-Ser).
To determine whether GSK-3β activity is required to trigger Bax translocation to mitochondria in CGNs, we evaluated the effects of several distinct GSK-3β inhibitors on the movement of GFP-Baxα. IGF-I is a known survival factor for CGNs that signals through phosphatidylinositol 3-kinase to activate the anti-apoptotic kinase AKT (D'Mello et al., 1993; Linseman et al., 2002). AKT subsequently phosphorylates and inactivates several pro-apoptotic molecules including Bad, caspase-9, GSK-3β, and the forkhead transcription factor FKHRL1 (Lawlor and Alessi, 2001). In the case of GSK-3β, AKT phosphorylates the inhibitory residue Ser9. As shown in Figure 2C, IGF-I significantly attenuated the dephosphorylation of GSK-3β on Ser9 in CGNs switched to apoptotic medium. In addition to IGF-I, we also used a substituted oxadiazole GSK-3β inhibitor, GSK-3β inhibitor II (Inh-II; Calbiochem) (Naerum et al., 2002), and the monovalent cation lithium, which is a noncompetitive inhibitor of GSK-3β that is protective in many models of neuronal apoptosis including CGNs (Linseman et al., 2003). Inclusion of either IGF-I or Inh-II resulted in an ∼40% decrease in the (5K-Ser)-induced translocation of GFP-Baxα to mitochondria (Fig. 2D). Moreover, the combination of IGF-I and Inh-II did not produce an additive inhibitory effect on GFP-Baxα translocation, suggesting that each of these agents blocks the same signal for Bax movement (i.e., GSK-3β activation) (Fig. 2D). Finally, the noncompetitive GSK-3β inhibitor lithium was most effective at blocking GFPBaxα translocation in apoptotic medium (Fig. 2D).
We next analyzed the activation of the endogenous Bax protein by immunostaining CGNs with a monoclonal antibody that specifically recognizes the active Bax conformation (clone 6A7) (Hsu and Youle, 1997). CGNs maintained in control medium showed little to no detectable 6A7 staining, whereas neurons in apoptotic medium showed active Bax immunoreactivity (Fig. 2E). Moreover, the 6A7 staining often coincided with condensed and/or fragmented chromatin. Inclusion of either the GSK-3β inhibitor II or a specific phosphopeptide inhibitor of GSK-3β (Plotkin et al., 2003) significantly inhibited both the conformational activation of endogenous Bax and CGN apoptosis (Fig. 2F). In a similar manner, the peptide inhibitor of GSK-3β also blunted GFP-Baxα translocation to mitochondria in transfected CGNs incubated in apoptotic medium (data not shown). Collectively, these results demonstrate that GSK-3β activity is necessary to induce the conformational change and mitochondrial translocation of Bax in trophic factor-deprived CGNs.
GSK-3β directly phosphorylates Bax on Ser163 in vitro and in vivo
To determine whether Bax is a direct substrate of GSK-3β, we examined the amino acid sequences of several species variants of Baxα to establish whether they contain any conserved GSK-3β consensus phosphorylation motifs (Fig. 3A). GSK-3β phosphorylates the consensus site S*/T*XXXS(P)/T(P), where (P) indicates a previously phosphorylated or “primed” serine or threonine residue and the asterisk denotes the site targeted by GSK-3β (Fiol et al., 1987). Baxα contains a SXXXT motif (residues Ser163 through Thr167) that is conserved in human, rat, mouse, and bovine Baxα (Fig. 3A). To ascertain whether Ser163 is phosphorylated by GSK-3β, we first analyzed the ability of purified GSK-3β to phosphorylate GFP-Baxα in vitro. GFP-Baxα was immunoprecipitated from HEK293 cells at 24 hr after transfection using a monoclonal antibody to GFP. Equivalent amounts of immunoprecipitated GFP-Baxα were then subjected to in vitro kinase assays using purified GSK-3β in the absence or presence of lithium. A small amount of phosphorylation occurred on GFPBaxα in vitro in the absence of GSK-3β, and this increased markedly when GSK-3β was added to the kinase reaction (Fig. 3B, top). Autophosphorylation of GSK-3β was also detected during the kinase reaction (Fig. 3B, bottom). Both the autophosphorylation of GSK-3β and the enhanced phosphorylation of GFP-Baxα were blocked by inclusion of lithium. Furthermore, when GFP-Baxα was immunoprecipitated from HEK293 cells that had been cotransfected with a constitutively active mutant of GSK-3β(S9A), it was less efficiently phosphorylated by GSK-3β in vitro compared with GFP-Baxα immunoprecipitated from cells cotransfected with empty vector (Fig. 3C). This latter result suggests that fewer Baxα sites were available for in vitro phosphorylation after previous exposure to constitutively active GSK-3βS9A. Collectively, these findings indicate that GFP-Baxα is a substrate for GSK-3β both in vitro and in intact cells.
Figure 3.

GSK-3β phosphorylates Baxα on the species-conserved residue Ser163. A, Amino acid sequence alignment of Baxα from human (GenBank accession number NP_620116), rat (Q63690), mouse (AAA03622), and bovine (AAC48806). A conserved GSK-3β consensus phosphorylation motif is highlighted in bold lettering at residues Ser163 to Thr167. B, HEK293 cells were transfected with GFP-Baxα as described in Materials and Methods. At 24 hr after transfection, the fusion protein was immunoprecipitated from cell lysates using a GFP monoclonal antibody. Immune complexes were then subjected to in vitro kinase assay by incubating for 30 min at 30°C with purified GSK-3β (50 ng) and [γ-32P]ATP (10μCi), in either the absence or the presence of LiCl (20 mm). 32P incorporation into GFP-Baxα (top) and GSK-3β (bottom) was detected by autoradiography. C, GFP-Baxα was immunoprecipitated from HEK293 cells that were cotransfected (Co-Tfx) with either empty vector (pcDNA3.1) or constitutively active GSK-3β(S9A). Immunecomplexes were subjected to GSK-3β invitro kinase assay, and 32P incorporation into GFP-Baxα was detected by autoradiography (top). The membrane was later immunoblotted (IB) for GFP-Baxα using a polyclonal antibody to GFP to demonstrate transfection and immunoprecipitation efficiences (bottom). D, HEK293 cells were transfected with either wild-type GFP-Baxα, the site-directed mutants Ser163Ala or Thr167Ala, or the Ser163Ala/Thr167Ala double mutant (SerThrDM). At 24 hr after transfection, the fusion proteins were immunoprecipitated using a GFP monoclonal antibody, and immune complexes were subjected to GSK-3β in vitro kinase assay. The amount of 32P incorporation into each of the fusion proteins (top) was normalized to the densitometric signal of the GFP immunoblot (bottom) to correct for differences in either expression or immunoprecipitation efficiences. The normalized ratio for wild-type GFP-Baxα in the absence of GSK-3β was set to 1.0, and all other values are relative to that control. E, GFP-Baxα (wild type, the single Ser163Ala mutant, or the Ser163Ala/Thr167Ala double mutant) was immunoprecipitated from 32P-orthophosphate-labeled HEK293 cells that were cotransfected (Co-Tfx) for ∼16 hr with either empty vector (pcDNA3.1) or constitutively active GSK-3β(S9A). Immune complexes were resolved by SDS-PAGE, and 32P incorporation into GFP-Baxα was detected by autoradiography.
Next, we analyzed the ability of GSK-3β to phosphorylate several site-directed mutants of GFP-Baxα in vitro. When the 32P incorporation into the fusion protein was normalized to the amount of GFP-Bax immunoprecipitated from transfected HEK293 cells, the addition of GSK-3β induced an ∼12-fold increase in the phosphorylation of wild-type GFP-Baxα (Fig. 3D, compare lanes 1 and 2). A Ser163Ala mutant of GFP-Baxα was not significantly phosphorylated by GSK-3β in vitro, consistent with Ser163 being a major phosphorylation site (Fig. 3D, compare lanes 2 and 3). In contrast, GFP-Baxα mutated at the putative priming site, Thr167Ala, was efficiently phosphorylated by GSK-3β in vitro (Fig. 3D, lane 4), whereas the double mutant Ser163Ala/Thr167Ala showed substantially decreased phosphorylation relative to wild-type Baxα (Fig. 3D, lane 5). Overall, these data indicate that the species-conserved residue Ser163 of Baxα is a substrate for GSK-3β-mediated phosphorylation in vitro. However, phosphorylation of the putative priming residue Thr167 is apparently not necessary for GSK-3β to target Ser163 in vitro.
Finally, to demonstrate that Ser163 of Bax is a substrate for GSK-3β in vivo, we cotransfected 32P-orthophosphate-labeled HEK293 cells with wild-type GFP-Baxα, the single Ser163Ala mutant, or the Ser163Ala/Thr167Ala double mutant in combination with either empty vector or constitutively active GSK-3βS9A. Approximately 16 hr after transfection, the GFP-Bax variants were immunoprecipitated, immune complexes were resolved by SDS-PAGE, and phosphorylation was assessed by autoradiography. Cotransfection with GSK-3βS9A markedly increased the phosphorylation of wild-type GFP-Baxα by approximately fourfold (Fig. 3E, compare lanes 1 and 2). In contrast, coexpression with constitutively active GSK-3β did not enhance 32P incorporation into either the single Ser163Ala mutant or the double mutant of GFP-Baxα (Fig. 3E, lanes 3-6). These data strongly suggest that Ser163 of Bax is a principal substrate for GSK-3β in vivo in intact cells.
Constitutively active GSK-3β drives GFP-Baxα to mitochondria in transfected HEK293 cells and CGNs
To assess the functional consequences of phosphorylation by GSK-3β, we examined the subcellular distribution of GFPBaxα in HEK293 cells cotransfected with either empty vector or HA-tagged, constitutively active GSK-3βS9A. The majority of cells cotransfected with empty vector showed a GFP-Baxα distribution that was diffuse over the entire cell (Fig. 4A, left; B, top). In contrast, cells cotransfected with constitutively active GSK-3βS9A showed a marked redistribution of GFP-Baxα to organelles that were structurally identifiable as mitochondria (Fig. 4A, right; B, bottom). These structures were confirmed to be mitochondria by colabeling live HEK293 cells with the mitochondrial marker TMRE. Cells cotransfected with GSK-3βS9A showed significant colocalization of GFP-Baxα with TMRE-stained mitochondria (Fig. 4C). These data indicate that activation of GSK-3β is sufficient to trigger localization of Bax to mitochondria in transfected HEK293 cells.
Figure 4.

Coexpression of GFP-Baxα with a constitutively active mutant of GSK-3β(S9A) induces translocation of the Bax fusion protein to mitochondria in transfected HEK293 cells. A, HEK293 cells were cotransfected with GFP-Baxα and either empty vector or HA-tagged GSK-3βS9A. At 24 hr after transfection, cotransfected cells were visualized by staining for the epitope tag using an HA polyclonal antibody and a Cy3-conjugated secondary. B, The areas demarcated by the boxes in A were enlarged 300% to show fine structure. C, HEK293 cells were cotransfected with GFP-Baxα and HA-tagged GSK-3βS9A. At 24 hr after transfection, cells were incubated for 30 min with Hoechst dye (to stain nuclei) and TMRE to label mitochondria. Live cells were then visualized with a 63× water immersion objective, and digitally deconvolved images were obtained using an Axioplan 2 microscope (Zeiss) equipped with a Cooke Sensicam deep-cooled CCD camera and a Slidebook software analysis program for digital deconvolution (Intelligent Imaging Innovations). Cotransfection with GSK-3βS9A induced a marked redistribution of GFP-Baxα that showed significant colocalization with the mitochondrial marker TMRE. The images shown are representative of data obtained in two independent experiments. Scale bars, 10 μm.
Next, we examined the effects of constitutively active GSK-3β on the localization of GFP-Baxα and induction of apoptosis in CGNs. As described in Figure 1, CGNs cotransfected with GFP-Baxα and empty vector (maintained in control medium) showed a diffuse distribution of the Bax fusion protein and no nuclear condensation (Fig. 5A,B, left). In contrast, CGNs coexpressing GFP-Baxα and HA-GSK-3βS9A (maintained in control medium) displayed a punctate localization of the Bax fusion protein and demonstrated significant nuclear condensation and fragmentation (Fig. 5A,B, right). Thus, constitutive activation of GSK-3β promotes Bax localization to mitochondria and induces CGN apoptosis even in the presence of survival medium.
Figure 5.
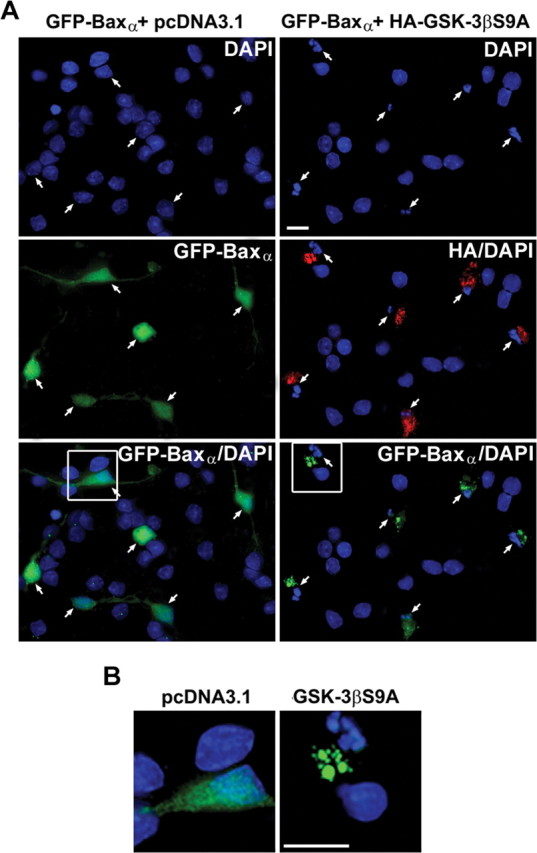
Cotransfection of CGNs with GFP-Baxα and a constitutively active mutant of GSK-3β(S9A) induces translocation of the Bax fusion protein to mitochondria and triggers CGN apoptosis. A, CGNs were cotransfected with GFP-Baxα and either empty vector or HA-tagged GSK-3βS9A using the Helios Gene-Gun system, as described in Materials and Methods. At 48 hr after transfection, cotransfected cells were maintained in control medium and were visualized by staining for the epitope tag using an HA polyclonal antibody and a Cy3-conjugated secondary antibody. The images shown are composites of four to six fields captured with a 63× oil objective to give a representative view of the localization of the Bax fusion protein in the absence or presence of constitutively active GSK-3β. B, The areas demarcated by the boxes in A were enlarged 300% to show fine structure. Scale bars, 10 μm.
Ser163 is required for GSK-3β to induce Bax translocation and apoptosis in HEK293 cells
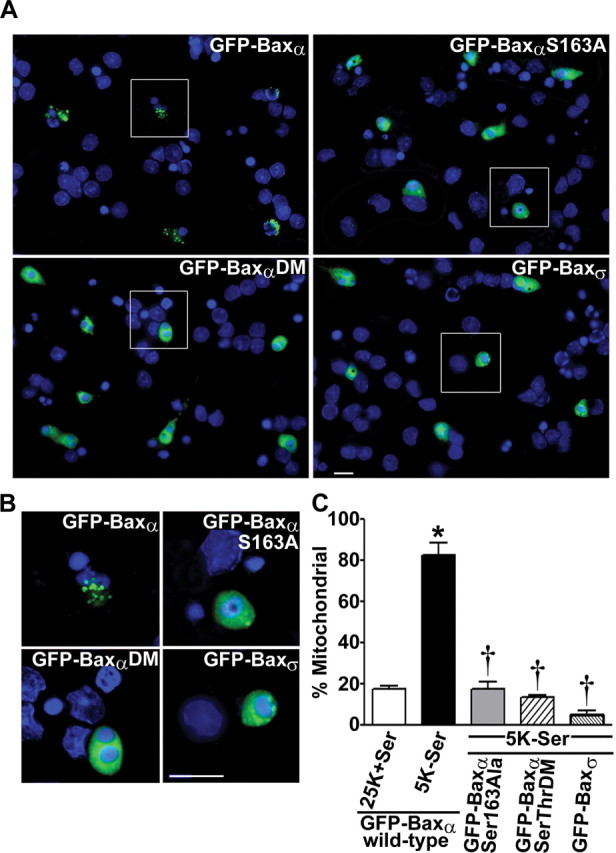
To confirm that the putative GSK-3β phosphorylation site Ser163 is necessary for Bax localization to mitochondria, we evaluated the ability of constitutively active GSK-3β to affect the subcellular distribution of various Bax mutants in cotransfected HEK293 cells. Consistent with its relative inefficiency as a GSK-3β substrate (Fig. 3D,E), the Ser163Ala/Thr167Ala double mutant of GFP-Baxα failed to localize to mitochondria when cotransfected with GSK-3βS9A (Fig. 6A). Quantitative analysis of the mitochondrial localization of various GFP-Baxα mutants is shown in Figure 6B. Cotransfection of wild-type GFP-Baxα with GSK-3βS9A induced a statistically significant increase in the mitochondrial localization of the fusion protein. In contrast, the single alanine substitution mutants at Ser163 and Thr167, as well as the Ser163/Thr167 double Ala mutant, were predominantly cytosolic regardless of the presence of GSK-3βS9A.
Figure 6.

The ability of constitutively active GSK-3β to promote the mitochondrial translocation of GFP-Baxα in transfected HEK293 cells is dependent on Ser163. A, HEK293 cells were cotransfected with the Ser163Ala/Thr167Ala double mutant of GFP-Baxα and either empty vector or HA-tagged GSK-3βS9A. Cotransfected cells were visualized by staining for the HA epitope tag. B, Quantitation of the percentage of transfected HEK293 cells demonstrating a mitochondrial distribution of the GFP-Bax variants [wild-typeα, Ser163Alaα, Thr167Alaα, Ser163Ala/Thr167Alaα (SerThrDM), and Baxσ (GenBank accession number AAF71267)] after cotransfection with either empty vector or HA-GSK-3βS9A. Data represent the means ± SEM of at least three separate experiments for each Bax variant in which ∼200 transfected cells were counted per experiment (*p < 0.01 compared with cotransfection of the same fusion protein with empty vector). C, Partial sequence alignment of human Baxα and human Baxσ including amino acids 121-180. Representative images of HEK293 cells cotransfected with GFP-Baxσ (lacking Baxα residues 159-171) and HA-GSK-3βS9A are shown. D, Quantitation of HEK293 cell apoptosis at 48 hr after transfection with the indicated constructs. The results shown are the means ± SEM (n = 4 transfected wells per group from a total of 2 separate experiments). *p < 0.01, GFP-Baxα significantly different from GFP; †p < 0.01, GFP-Baxα + GSK-3βS9A significantly different from GFP-Baxα alone; ‡p < 0.01, significantly different from GFP-Baxα (wild type) + GSK-3βS9A. DM, Ser163Ala/Thr167Ala double mutant. Scale bars: A, C, 10 μm.
The above results parallel the phosphorylation data shown in Figure 3D, except for the single Thr167Ala mutant that was phosphorylated by GSK-3β in vitro but failed to localize to mitochondria in intact cells. This apparent discrepancy may reflect a physiological necessity for priming in order for Bax to be phosphorylated by GSK-3β in vivo. The high concentration of GSK-3β added to the in vitro kinase reaction most likely circumvented this requirement. Alternatively, it is possible that an additional docking site for GSK-3β exists on Bax distinct from the phosphorylated threonine at position (+4) relative to Ser163 (Biondi and Nebreda, 2003).
Additional evidence implicating Ser163 as a critical residue in the regulation of Bax function was acquired from analysis of the various Bax isoforms (Oltvai et al., 1993). In particular, a recently described isoform, human Bax σ (Baxσ) (Schmitt et al., 2000), is nearly identical to Baxα, except it lacks residues 159-171, which encompass the putative GSK-3β-regulated site Ser163 (Fig. 6C). Like the Ser163Ala point mutant of Baxα, HEK293 cells cotransfected with GFP-Baxσ and HA-GSK-3βS9A demonstrated a diffuse localization of the Bax fusion protein with little accumulation in mitochondria (Fig. 6B,C).
Finally, in agreement with the mitochondrial localization of Bax triggering the intrinsic death pathway, the amount of apoptosis induced in transfected HEK293 cells reflected the ability of GSK-3βS9A to drive wild-type Baxα, but not the Ser163Ala Baxα single mutant, the Ser163Ala/Thr167Ala Baxα double mutant, or Baxσ, to mitochondria (Fig. 6D). These data indicate that phosphorylation of Ser163 on Bax by GSK-3β promotes the localization of Bax to mitochondria and induction of apoptosis in transfected HEK293 cells.
Bax translocation to mitochondria in apoptotic CGNs requires Ser163
To determine whether the mechanistic data obtained in transfected HEK293 cells reflects the regulation of Bax in primary neurons, we investigated the localization of Bax variants lacking Ser163 in CGNs undergoing apoptosis. As described in Figure 1, CGNs deprived of trophic support demonstrate a marked translocation of GFP-Baxα from the cytosol to mitochondria and significant nuclear condensation and fragmentation (Fig. 7A,B, top left). In contrast, the Ser163Ala single mutant of GFP-Baxα, the Ser163Ala/Thr167Ala double mutant of GFP-Baxα, or GFP-Baxσ did not localize to mitochondria in trophic factor-deprived CGNs (Fig. 7A,B, top right and bottom). Neurons expressing these Bax mutants were still induced to undergo apoptosis, indicated by their condensed and fragmented nuclei, because of translocation of endogenous Bax (Fig. 7A,B, top right and bottom). Quantitation of the mitochondrial localization of each of these Bax variants is shown in Figure 7C. Trophic factor withdrawal induced a fourfold increase in the percentage of GFP-Baxα-transfected CGNs that displayed a mitochondrial localization of the fusion protein. In contrast, Bax variants lacking Ser163 showed no significant translocation to mitochondria in CGNs undergoing apoptosis. These data suggest that expression of Bax variants that fail to translocate to mitochondria does not impede the activation of endogenous Bax and its ability to subsequently trigger apoptosis in CGNs. Moreover, these results corroborate the data obtained in transfected HEK293 cells and support the conclusion that phosphorylation of Bax on Ser163 by GSK-3β is required for translocation of this proapoptotic protein to mitochondria in neurons undergoing apoptosis.
Figure 7.
Bax variants lacking Ser163 fail to localize to mitochondria in CGNs undergoing apoptosis. A, CGNs were transfected using the Helios Gene-Gun with wild-type GFP-Baxα, the GFP-Baxα Ser163Ala single mutant or the Ser163Ala/Thr167Ala double mutant (DM), or GFP-Baxσ. At 48 hr after transfection, neurons were switched to apoptotic medium (5K-Ser) for 4 hr. CGNs were then fixed, and nuclei were stained with Hoechst dye. The images shown are composites of four to six fields captured with a 63× oil objective to give a representative view of the localization of the various fusion proteins. B, The regions demarcated by the boxes in A were enlarged 250% to magnify the fusion protein distributions and nuclear morphologies. C, Quantitation of the fraction of transfected CGNs showing a mitochondrial distribution of the GFP-Bax variants [wild-type α, Ser163Alaα, Ser163Ala/Thr167Alaα (SerThrDM), and Baxσ] after incubation in either control or apoptotic medium. Data represent the means ± SEM of at least three separate experiments with each Bax variant in which ∼200 transfected CGNs were counted per experiment. *p < 0.01 compared with wild-type α incubated in control medium; †p < 0.01 compared with GFP-Baxα (wild type) in apoptotic medium. Scale bars: A, B, 10 μm.
Discussion
In the present study, we have identified the pro-apoptotic Bcl-2 family member Bax as a novel substrate of GSK-3β in cultured neurons undergoing apoptosis. We present several lines of evidence indicating that phosphorylation by GSK-3β regulates the subcellular localization of Bax during neuronal apoptosis. First, inhibitors of GSK-3β activity blunt the translocation of Bax to mitochondria and suppress the conformational activation of Bax in CGNs undergoing apoptosis. Second, GSK-3β directly phosphorylates Bax on Ser163 within a species-conserved, putative GSK-3β phosphorylation motif, and this phosphorylation occurs both in vitro and in vivo in intact cells. Third, constitutive activation of GSK-3β drives Bax to mitochondria and promotes apoptosis of transfected HEK293 cells and CGNs. Finally, Bax variants lacking Ser163 fail to localize to mitochondria in CGNs undergoing apoptosis. These data are the first to identify a component of the intrinsic apoptotic cascade as a direct target of GSK-3β action. These findings also provide a molecular mechanism for the pro-apoptotic function of GSK-3β in neurons. Finally, these results are the first to demonstrate that Bax function in neurons is regulated by phosphorylation.
The identification of Ser163 as a pivotal residue involved in the translocation of Bax to mitochondria is consistent with recent work by Schinzel et al. (2004), who showed that mutation of Pro168, a residue immediately C terminal to the putative GSK-3β phosphorylation motif (Ser163XXXThr167), similarly prevents Bax localization to mitochondria in fibroblasts exposed to staurosporine. These authors suggest that Pro168 may be the target of a posttranslational modification that modulates the Bax conformational change and subcellular localization (Schinzel et al., 2004). Our results suggest another possibility: that Pro168 may be required to target the pro-directed kinase GSK-3β to phosphorylate Bax at Ser163. Future experiments will determine whether a Pro168 mutant of Bax is, in fact, a poor substrate for GSK-3β.
Our finding that Bax is a direct target of GSK-3β complements a recent study by Watcharasit et al. (2003) showing that GSK-3β indirectly stimulates the transcription of Bax via regulation of p53 activity. Collectively, these results suggest that GSK-3β modulates Bax expression and function at the transcriptional and posttranslational levels, respectively, to promote mitochondrial apoptosis.
Where and how GSK-3β and Bax interact in neurons remains to be investigated. Previous studies examining the interactions of GSK-3β with β-catenin have revealed that the kinase is brought in close proximity to its substrate by their interactions with the axin-adenomatous polyposis coli scaffold complex (Hart et al., 1998). Similarly, GSK-3β and tau interact via their common association with cytosolic scaffolding proteins of the 14-3-3 family (Agarwal-Mawal et al., 2003). Because cytosolic Bax was shown recently to interact with 14-3-3 proteins (Nomura et al., 2003), it is possible that 14-3-3 scaffolds also mediate the interaction of GSK-3β with Bax.
The current finding that GSK-3β phosphorylates and regulates Bax function impacts on several recent reports. First, activation of AKT was reported to inhibit Bax conformational change in B-cells subjected to cytokine withdrawal (Yamaguchi and Wang, 2001) and Bax translocation to mitochondria in COS-1 cells incubated with staurosporine (Tsuruta et al., 2002). Although there is no consensus AKT phosphorylation site on Bax, Gardai et al. (2004) recently reported that Bax is phosphorylated in neutrophils on the C-terminal Ser184 in an AKT-dependent manner. Furthermore, they suggest that this phosphorylation event inhibits the pro-apoptotic action of Bax in neutrophils. Our results indicate that the ability of AKT to modulate Bax conformation and movement in these non-neuronal cells may also involve regulation of GSK-3β activity. Second, Somervaille et al. (2001) reported that lithium prevents the conformational change in Bax in human erythroid progenitor cells deprived of growth factors. Although they related this effect to inhibition of GSK-3β activity, these authors did not show that Bax is a direct substrate of GSK-3β. Given the coincident activation of Bax and GSK-3β in many models of apoptosis, it is likely that the interaction of these two molecules acts as a common trigger for activation of the intrinsic death pathway.
Precisely how phosphorylation of Bax by GSK-3β stimulates Bax movement to mitochondria will require further investigation. There are several potential events that could be influenced by Bax phosphorylation. The phosphorylation may alter the conformation of Bax and facilitate its ability to oligomerize or to incorporate into the outer mitochondrial membrane. Indeed, we found that peptide and non-peptide inhibitors of GSK-3β activity effectively blocked the conformational change of endogenous Bax induced by trophic factor deprivation in CGNs. Alternatively, phosphorylation may decrease the affinity of Bax for cytosolic anchoring proteins like 14-3-3. Conversely, the affinity of Bax for mitochondrial membrane proteins may be enhanced by phosphorylation, thus promoting its targeting to mitochondria. Additional elucidation of the mechanism by which GSK-3β-mediated phosphorylation regulates Bax function may reveal novel targets for inhibiting neuronal apoptosis.
Footnotes
This work was supported by Department of Veterans Affairs Merit Awards (K.A.H. and D.A.L.), Department of Defense Grant USAMRMC 03281009 (K.A.H. and D.A.L.), National Institutes of Health (NIH) Grant NS38619-01A1 (K.A.H.), and the Department of Veterans Affairs Research Enhancement Award Program (K.A.H. and D.A.L.). Molecular Biology Core Services were supported by NIH Diabetes Endocrinology Research Care Grant P30-DK57516. We thank Dr. R. J. Youle (National Institute of Neurological Disorders and Stroke, NIH, Bethesda, MD) for GFP-Baxα, Dr. R. Bertrand (University of Montreal, Montreal, Quebec, Canada) for the Baxσ construct, and Dr. M. J. Birnbaum (University of Pennsylvania, Philadelphia, PA) for HA-GSK-3βS9A.
Correspondence should be addressed to Dr. Kim A. Heidenreich, Department of Pharmacology, University of Colorado Health Sciences Center at Fitzsimons, P.O. Box 6511, Mail Stop 8303, Aurora, CO 80045-0511. E-mail: Kim.Heidenreich@UCHSC.edu.
Copyright © 2004 Society for Neuroscience 0270-6474/04/249993-10$15.00/0
D.A.L. and B.D.B. contributed equally to this work.
References
- Agarwal-Mawal A, Qureshi HY, Cafferty PW, Yuan Z, Han D, Lin R, Paudel HK (2003) 14-3-3 connects glycogen synthase kinase-3 beta to tau within a brain microtubule-associated tau phosphorylation complex. J Biol Chem 278: 12722-12728. [DOI] [PubMed] [Google Scholar]
- Biondi RM, Nebreda AR (2003) Signalling specificity of Ser/Thr protein kinases through docking-site-mediated interactions. Biochem J 372: 1-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brunet A, Datta SR, Greenberg ME (2001) Transcription-dependent and -independent control of neuronal survival by the PI3K-Akt signaling pathway. Curr Opin Neurobiol 11: 297-305. [DOI] [PubMed] [Google Scholar]
- Cregan SP, MacLaurin JG, Craig CG, Robertson GS, Nicholson DW, Park DS, Slack RS (1999) Bax-dependent caspase-3 activation is a key determinant in p53-induced apoptosis in neurons. J Neurosci 19: 7860-7869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D'Mello SR, Galli C, Ciotti T, Calissano P (1993) Induction of apoptosis in cerebellar granule neurons by low potassium: inhibition of death by insulin-like growth factor I and cAMP. Proc Natl Acad Sci USA 90: 10989-10993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doble BW, Woodgett JR (2003) GSK-3: tricks of the trade for a multi-tasking kinase. J Cell Sci 116: 1175-1186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fiol CJ, Mahrenholz AM, Wang Y, Roeske RW, Roach PJ (1987) Formation of protein kinase recognition sites by covalent modification of the substrate. Molecular mechanism for the synergistic action of casein kinase II and glycogen synthase kinase 3. J Biol Chem 262: 14042-14048. [PubMed] [Google Scholar]
- Fukunaga K, Miyamoto E (1998) Role of MAP kinase in neurons. Mol Neurobiol 16: 79-95. [DOI] [PubMed] [Google Scholar]
- Gardai SJ, Hildeman DA, Frankel SK, Whitlock BB, Frasch SC, Borregaard N, Marrack P, Bratton DL, Henson PM (2004) Phosphorylation of Bax Ser184 by Akt regulates its activity and apoptosis in neutrophils. J Biol Chem 279: 21085-21095. [DOI] [PubMed] [Google Scholar]
- Hart MJ, de los Santos R, Albert IN, Rubinfeld B, Polakis P (1998) Downregulation of beta-catenin by human axin and its association with the APC tumor suppressor, beta-catenin and GSK3 beta. Curr Biol 8: 573-581. [DOI] [PubMed] [Google Scholar]
- Hetman M, Cavanaugh JE, Kimelman D, Xia Z (2000) Role of glycogen synthase kinase-3β in neuronal apoptosis induced by trophic withdrawal. J Neurosci 20: 2567-2574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsu YT, Youle RJ (1997) Nonionic detergents induce dimerization among members of the Bcl-2 family. J Biol Chem 272: 13829-13834. [DOI] [PubMed] [Google Scholar]
- Kaytor MD, Orr HT (2002) The GSK3 beta signaling cascade and neurodegenerative disease. Curr Opin Neurobiol 12: 275-278. [DOI] [PubMed] [Google Scholar]
- Lawlor MA, Alessi DR (2001) PKB/Akt: a key mediator of cell proliferation, survival and insulin responses? J Cell Sci 114: 2903-2910. [DOI] [PubMed] [Google Scholar]
- Li M, Wang X, Meintzer MK, Laessig T, Birnbaum MJ, Heidenreich KA (2000) Cyclic AMP promotes neuronal survival by phosphorylation of glycogen synthase kinase 3beta. Mol Cell Biol 20: 9356-9363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linseman DA, Phelps RA, Bouchard RJ, Le SS, Laessig TA, McClure ML, Heidenreich KA (2002) Insulin-like growth factor-I blocks Bcl-2 interacting mediator of cell death (Bim) induction and intrinsic death signaling in cerebellar granule neurons. J Neurosci 22: 9287-9297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linseman DA, Cornejo BJ, Le SS, Meintzer MK, Laessig TA, Bouchard RJ, Heidenreich KA (2003) A myocyte enhancer factor 2D (MEF2D) kinase activated during neuronal apoptosis is a novel target inhibited by lithium. J Neurochem 85: 1488-1499. [DOI] [PubMed] [Google Scholar]
- Mielke K, Herdegen T (2000) JNK and p38 stress kinases-degenerative effectors of signal-transduction-cascades in the nervous system. Prog Neurobiol 61: 45-60. [DOI] [PubMed] [Google Scholar]
- Naerum L, Norskov-Lauritsen L, Olesen PH (2002) Scaffold hopping and optimization towards libraries of glycogen synthase kinase-3 inhibitors. Bioorg Med Chem Lett 12: 1525-1528. [DOI] [PubMed] [Google Scholar]
- Nomura M, Shimizu S, Sugiyama T, Narita M, Ito T, Matsuda H, Tsujimoto Y (2003) 14-3-3 interacts directly with and negatively regulates proapoptotic Bax. J Biol Chem 278: 2058-2065. [DOI] [PubMed] [Google Scholar]
- Oltvai ZN, Milliman CL, Korsmeyer SJ (1993) Bcl-2 heterodimerizes in vivo with a conserved homolog, Bax, that accelerates programmed cell death. Cell 74: 609-619. [DOI] [PubMed] [Google Scholar]
- Perez M, Rojo AI, Wandosell F, Diaz-Nido J, Avila J (2003) Prion peptide induces neuronal cell death through a pathway involving glycogen synthase kinase 3. Biochem J 372: 129-136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phiel CJ, Wilson CA, Lee VM, Klein PS (2003) GSK-3alpha regulates production of Alzheimer's disease amyloid-beta peptides. Nature 423: 435-439. [DOI] [PubMed] [Google Scholar]
- Plotkin B, Kaidanovich O, Talior I, Eldar-Finkelman H (2003) Insulin mimetic action of synthetic phosphorylated peptide inhibitors of glycogen synthase kinase-3. J Pharmacol Exp Ther 305: 974-980. [DOI] [PubMed] [Google Scholar]
- Putcha GV, Deshmukh M, Johnson Jr EM (1999) Bax translocation is a critical event in neuronal apoptosis: regulation by neuroprotectants, Bcl-2, and caspases. J Neurosci 19: 7476-7485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Putcha GV, Harris CA, Moulder KL, Easton RM, Thompson CB, Johnson Jr EM (2002) Intrinsic and extrinsic pathway signaling during neuronal apoptosis: lessons from the analysis of mutant mice. J Cell Biol 157: 441-453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schinzel A, Kaufmann T, Schuler M, Martinalbo J, Grubb D, Borner C (2004) Conformational control of Bax localization and apoptotic activity by Pro168. J Cell Biol 164: 1021-1032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmitt E, Paquet C, Beauchemin M, Dever-Bertrand J, Bertrand R (2000) Characterization of Bax-sigma, a cell death-inducing isoform of Bax. Biochem Biophys Res Commun 270: 868-879. [DOI] [PubMed] [Google Scholar]
- Selimi F, Vogel MW, Mariani J (2000) Bax inactivation in lurcher mutants rescues cerebellar granule cells but not purkinje cells or inferior olivary neurons. J Neurosci 20: 5339-5345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Somervaille TC, Linch DC, Khwaja A (2001) Growth factor withdrawal from primary human erythroid progenitors induces apoptosis through a pathway involving glycogen synthase kinase-3 and Bax. Blood 98: 1374-1381. [DOI] [PubMed] [Google Scholar]
- Tsuruta F, Masuyama N, Gotoh Y (2002) The phosphatidylinositol 3-kinase (PI3K)-Akt pathway suppresses Bax translocation to mitochondria. J Biol Chem 277: 14040-14047. [DOI] [PubMed] [Google Scholar]
- Vila M, Przedborski S (2003) Targeting programmed cell death in neurodegenerative diseases. Nat Rev Neurosci 4: 365-375. [DOI] [PubMed] [Google Scholar]
- Vila M, Jackson-Lewis V, Vukosavic S, Djaldetti R, Liberatore G, Offen D, Korsmeyer SJ, Przedborski S (2001) Bax ablation prevents dopaminergic neurodegeneration in the 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine mouse model of Parkinson's disease. Proc Natl Acad Sci USA 98: 2837-2842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watcharasit P, Bijur GN, Song L, Zhu J, Chen X, Jope RS (2003) Glycogen synthase kinase-3beta (GSK3beta) binds to and promotes the actions of p53. J Biol Chem 278: 48872-48879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wellmann H, Kaltschmidt B, Kaltschmidt C (1999) Optimized protocol for biolistic transfection of brain slices and dissociated cultured neurons with a hand-held gene gun. J Neurosci Methods 92: 55-64. [DOI] [PubMed] [Google Scholar]
- Wolter KG, Hsu YT, Smith CL, Nechushtan A, Xi XG, Youle RJ (1997) Movement of Bax from the cytosol to mitochondria during apoptosis. J Cell Biol 139: 1281-1292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamaguchi H, Wang HG (2001) The protein kinase PKB/Akt regulates cell survival and apoptosis by inhibiting Bax conformational change. Oncogene 20: 7779-7786. [DOI] [PubMed] [Google Scholar]
- Zong WX, Lindsten T, Ross AJ, MacGregor GR, Thompson CB (2001) BH3-only proteins that bind pro-survival Bcl-2 family members fail to induce apoptosis in the absence of Bax and Bak. Genes Dev 15: 1481-1486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zou H, Li Y, Liu X, Wang X (1999) An APAF-1. cytochrome c multimeric complex is a functional apoptosome that activates procaspase-9. J Biol Chem 274: 11549-11556. [DOI] [PubMed] [Google Scholar]