

Abstract
Endocannabinoids are crucial for the extinction of aversive memories, a process that considerably involves the amygdala. Here, we show that low-frequency stimulation of afferents in the lateral amygdala with 100 pulses at 1 Hz releases endocannabinoids postsynaptically from neurons of the basolateral amygdala of mice in vitro and thereby induces a long-term depression of inhibitory GABAergic synaptic transmission (LTDi) via a presynaptic mechanism. Lowering inhibitory synaptic transmission significantly increases the amplitude of excitatory synaptic currents in principal neurons of the central nucleus, which is the main output site of the amygdala. LTDi involves a selective mGluR1 (metabotropic glutamate receptor 1)-mediated calcium-independent mechanism and the activation of the adenylyl cyclase-protein kinase A pathway. LTDi is abolished by the cannabinoid type 1 (CB1) receptor antagonist SR141716A and cannot be evoked in CB1 receptor-deficient animals. LTDi is significantly enhanced in mice lacking the anandamide-degrading enzyme fatty acid amide hydrolase. The present findings show for the first time that mGluR activation induces a retrograde endocannabinoid signaling via activation of the adenylyl cyclase-protein kinase A pathway and the release of anandamide. Furthermore, the results indicate that anandamide decreases the activity of inhibitory interneurons in the amygdala. This disinhibition increases the activity of common output neurons and could provide a prerequisite for extinction by formation of new memory.
Keywords: endocannabinoids, amygdala, long-term depression, mGluR, anandamide, synaptic plasticity
Introduction
Cannabinoid type 1 (CB1) receptors have been shown to play an important role in the control of synaptic inhibition: CB1 receptor activation reduces GABAergic synaptic transmission in the hippocampus (Hoffman and Lupica, 2000), the nucleus accumbens (Hoffman and Lupica, 2001; Manzoni and Bockaert, 2001), and the amygdala (Katona et al., 2001; Azad et al., 2003). Furthermore, endocannabinoids induce a depolarization-induced suppression of inhibition (DSI) in the cerebellum (Kreitzer et al., 2002) and in hippocampal neurons (Ohno-Shosaku et al., 2001; Wilson and Nicoll, 2001). A reduction of transmitter release from GABAergic terminals will eventually reduce an inhibitory tonus exerted on principal neurons (Giuffrida and Piomelli, 2000; Freund et al., 2003). This disinhibition might support mechanisms that are critical for learning and memory, such as long-term potentiation (LTP). It has recently been shown that an endocannabinoid-mediated reduction of GABA release in the hippocampus enhances the excitability of CA1 pyramidal neurons (Carlson et al., 2002; Chevaleyre and Castillo, 2003).
The amygdala expresses high levels of CB1 receptors (Marsicano and Lutz, 1999; Katona et al., 2001; McDonald and Mascagni, 2001). The endocannabinoid system in this brain region has been shown to be crucially involved in the control of nociception (Martin et al., 1999; Manning et al., 2001; Pertwee, 2001) and extinction of aversive memories (Marsicano et al., 2002). However, the cellular mechanisms underlying endocannabinoid action in this brain region are still unclear. Recently, we showed that low-frequency stimulation of afferents in the lateral amygdala (LA) with 100 pulses at 1 Hz (LFSi) induces a significant long-term depression of inhibitory GABAergic synaptic transmission (LTDi) in the basolateral amygdala (BLA) in a CB1 receptor-dependent manner (Marsicano et al., 2002), suggesting an important role of endocannabinoids in long-term synaptic plasticity in this brain region. In this investigation, endocannabinoid levels increased in the amygdala during the extinction of conditioned fear. Therefore, endocannabinoids might facilitate extinction of aversive memories by selectively modulating local inhibitory networks in the amygdala. The present study explores the cellular mechanisms underlying LTDi and its possible role on glutamatergic synaptic transmission involved in long-term associative neuronal plasticity.
Materials and Methods
The present investigations were performed in slices of male C57BL/6J OlaHsd mice (42-63 d old; Harlan Winkelmann, Borchen, Germany). In addition, male mice (50-68 d old) lacking CB1 receptors (CB1-/-) or male mice (52-67 d old) lacking the anandamide-degrading enzyme fatty acid amide hydrolase FAAH (FAAH-/-), as well as their respective wild-type littermates (CB1+/+ and FAAH+/+), were used. CB1-/- mice and FAAH-/- mice were generated as described by Marsicano et al. (2002) and Cravatt et al. (2001), respectively. All electrophysiological experiments were performed in accordance with the guidelines of the Ethical Committee on the Use and Care of Animals (Government of Bavaria, Germany). Slices were prepared during the light phase. For slice preparation, the animals were anesthetized with isoflurane and decapitated. The brains were rapidly removed and placed in ice-cold artificial CSF (ACSF) containing (in mm) 125 NaCl, 2.5 KCl, 25 NaHCO3, 2 CaCl2, 1 MgCl2, 25 d-glucose, and 1.25 NaH2PO4, pH 7.4, and bubbled with a 95% O2/5% CO2 mixture. Coronal slices of the amygdala (400 μm thick) were prepared using a vibroslicer (FTB, Weinheim, Germany). After incubation in a holding chamber with ACSF (22-25°C) for at least 60 min, the slices were placed in the recording chamber and superfused with ACSF at a flow rate of 1.5 ml/min.
Square pulse stimuli (0.066 Hz, 5-12 mA, 200 μsec) were delivered via bipolar concentric tungsten electrodes positioned in the LA to evoke EPSCs, IPSCs, or field potentials (FPs) recorded in the BLA or the central amygdala (CE). The stimulus intensities were adjusted to produce half-maximal responses.
All experiments were performed at room temperature (22-25°C). For whole-cell patch-clamp recordings, principal neurons of the BLA/CE were visualized using infrared video microscopy and the gradient contrast system (Zeiss, Oberkochen, Germany). Principal neurons were distinguished from interneurons according to their morphological and electrophysiological properties (Washburn and Moises, 1992). For recording of EPSCs, glass electrodes (4.5-5 MΩ) contained (in mm) 130 K-d-gluconate, 5 KCl, 2 Mg-ATP, 10 d-glucose, 0.5 EGTA, and 10 HEPES, pH 7.4. Currents were recorded using a switched voltage-clamp amplifier (SEC-10L; npi Electronics, Tamm, Germany) with switching frequencies of 75-80 kHz (25% duty cycle). Series resistance was monitored continuously and compensated in bridge mode. Cells with >10% change in series resistance during the experiment were excluded from the analysis. Neuronal input resistance was monitored by injecting hyperpolarizing current pulses (300 msec, -10 mV, 0.066 Hz) through the patch electrode. All patch-clamp experiments were performed at a holding potential of -70 mV.
Isolation of GABAA receptor-mediated synaptic transmission and induction of LTDi were performed as described previously (Marsicano et al., 2002). Evoked GABAA receptor-mediated IPSCs were recorded in the presence of 50 μm d-(-)-2-amino-5-phosphono-pentanoic acid (d-AP-5) and 5 μm 2,3-dioxo-6-nitro-1,2,3,4-tetrahydro-benzo[f] quinoxaline-7-sulfonamide (NBQX). The pipettes were filled with a solution containing (in mm) 2 Mg-ATP, 100 CsCH3SO3, 60 CsCl, 0.2 EGTA, 10 HEPES, 1 MgCl2, 5 QX314, and and 0.3 Na3GTP, pH 7.3. Under these conditions, the reversal potential for Cl- ions was -28 mV. The amplitudes and frequencies of spontaneous IPSCs (sIPSCs) were studied by continuous recording over 300 sec. The peak amplitudes of the sIPSCs were measured off-line automatically using a customized event detection macro (Igor 4.0; Wave Metrics, Lake Oswego, OR) with an adjustable amplitude threshold. Frequencies were calculated by dividing the total number of sIPSCs by the total time sampled.
FPs were recorded using glass microelectrodes (1-2 MΩ) filled with ACSF. The stimulus intensities were adjusted to produce half-maximal responses. The voltage differences between the sharp negative onset and the negative peak (a), and between the negative peak and the succeeding positive peak (b), were measured, and the amplitudes of the FPs were calculated as (a + b)/2.
LTDi was induced by 100 stimuli at 1 Hz (LFSi). For high-frequency stimulation (HFS), two trains of 100 pulses with 50 Hz and an interstimulus interval of 10 sec were applied in the LA. All recordings were amplified, filtered (3 kHz), and digitized (9 kHz). The digitized data were stored to disk on a Power Macintosh G3 computer by a data acquisition and evaluation program (Pulse version 8.5; Heka Electronic, Lambrecht, Germany).
Statistical analysis was performed using the Student's paired t test to compare the values of 10 min of a stable baseline (control) and after application of LFSi, HFS, or substances, respectively. The latter were obtained by choosing the values as soon as LFSi, HFS, or substances led to stable effects for at least 10 min. The Kolmogorov-Smirnov test (Stat-View 5.0; SAS Institute, Cary, NC) was used for comparison of the amplitude distributions of sIPSCs. In both tests, p < 0.05 was considered as a significant difference. Data are presented as mean ± SEM.
Drugs were applied via the superfusion system or added to the recording pipette. In all experiments, in which the different enzymes were blocked postsynaptically, the respective substances [1-(6-[(17β]-3-methoxyestra-1,3,5[10]-trien-17-yl)amino]hexyl)-1H-pyrrole-2,5-dione (U73122), Rp-cAMP, 9-(tetrahydro-2-furanyl)-9-H-purin-6-amine (SQ22,536), GTPγ-S, and BAPTA] were allowed to diffuse to the postsynaptic site after whole-cell access for at least 20 min (if they did not affect the IPSC amplitude) or as long as they still changed the IPSC amplitude (BAPTA, GTPγ-S, and Rp-cAMP slightly). After this time, the stable control values were recorded for 15 min before inducing LTDi. Only the last 10 min of the control are depicted in all graphs.
The following pharmacological compounds were used: WIN55,212-2, SQ22,536, Rp-cAMP, NBQX, d-AP-5, BAPTA, 7-hydroxyiminocyclopropan[b]-chromen-1a-carboxylic acid ethyl ester (CPCCOEt), 2-methyl-6-(phenylethynyl)-pyridine (MPEP), U73122, 1,6-bis(cyclohexyloximino-carbonylamino)hexane (RHC-80267), and GTPγ-S (Sigma/RBI, Natick, MA); (S)-3,5-dihydroxyphenylglycine (DHPG) (Tocris, Biotrend, Köln, Germany); and N-(piperidin-1-yl)-5-(4-chlorophenyl)-4-methyl-1H-pyrazole-3-carboxamide (SR141716A; National Institute of Mental Health Chemical Synthesis and Drug Supply Program, Bethesda, MD).
Stock solutions of WIN55,212-2 (10 mm) and SR141716A (10 mm) were prepared in DMSO and stored at -20°C. Final DMSO concentrations were ≤0.05%. Before all experiments, fatty acid-free bovine serum albumin (1 mg/ml) was rinsed through the system to avoid binding of WIN55,212-2 and SR141716A to the walls of the tubing.
Results
CB1 receptor-dependent LTDi in the BLA does not require postsynaptic Ca2+ influx
To investigate the phenomenon of LTDi in the amygdala in detail, inhibitory GABAA receptor-mediated currents (IPSCs) in the BLA were evoked by stimulating afferents in the LA in the presence of the NMDA receptor antagonist d-AP-5 (50 μm) and the AMPA/kainate receptor blocker NBQX (5 μm). LFSi (100 pulses/1 Hz) of afferents from the LA induced a LTDi in the BLA of C57BL/6JOlaHsd mice (control, 100%; LFSi, 78 ± 5%; n = 6; p < 0.05) (Fig. 1A). Because LTDi is inducible in wild-type mice (CB1+/+), but not in animals lacking the CB1 receptor (CB1-/-), nor in slices of wild-type mice pretreated with the CB1 receptor antagonist SR141716A (Marsicano et al., 2002), we postulated that LTDi is mediated by endocannabinoids. We therefore investigated whether LTDi can be occluded by application of the CB1 receptor agonist WIN55,212-2. Pretreatment of slices from C57BL/6JOlaHsd mice with WIN55,212-2 (5 μm) decreased the amplitude of isolated IPSCs (control, 100%; WIN55,212-2, 56 ± 9%; n = 5; p < 0.05) recorded in the BLA, and inhibited the induction of LTDi (control plus WIN55,212-2, 100%; LFSi, 104 ± 10%; n = 5; p > 0.05) (Fig. 1B). These results suggest that LTDi involves the release of endocannabinoids and the activation of CB1 receptors.
Figure 1.
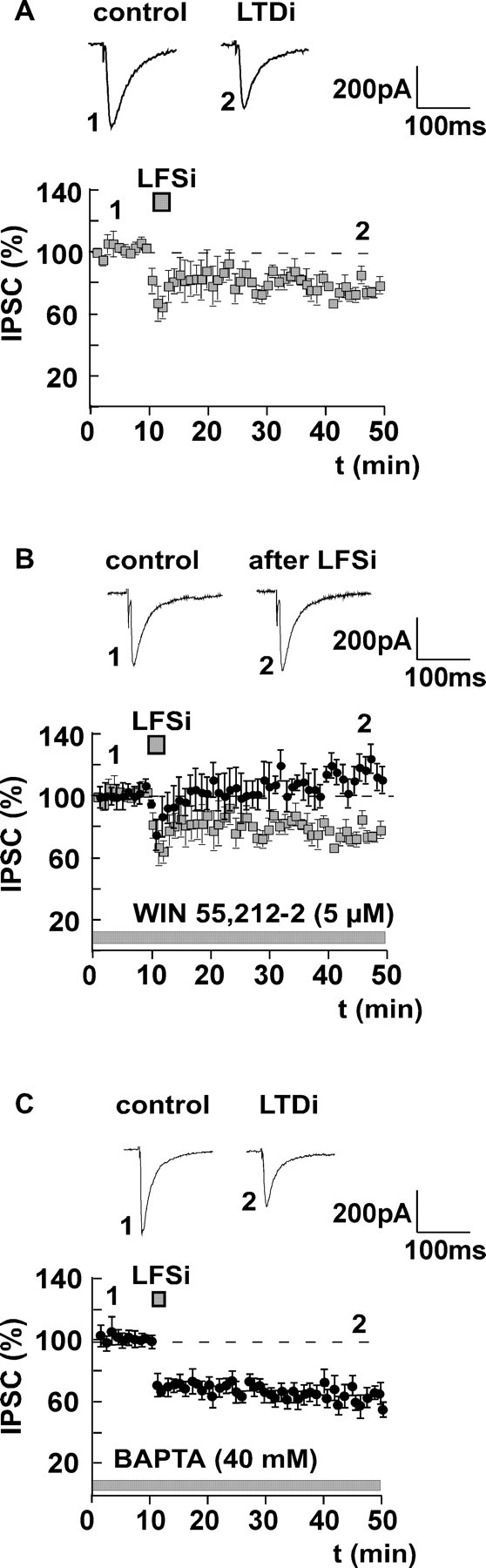
LTDi is mediated by endocannabinoids and is independent of postsynaptic Ca2+ influx. A, LFSi of afferents in the LA (100 pulses/1 Hz) leads to a significant long-term depression of IPSCs recorded in principal BLA neurons (LTDi) to 78 ± 5% of control (n = 6; p < 0.05). B, Pretreatment of the slices with the CB1 receptor agonist WIN55,212-2 (5 μm) occludes the induction of LTDi (104 ± 10% of control; n = 5; p > 0.05). LTDi without WIN55,212-2 (data from A) is shown for comparison (squares). C, LTDi is not inhibited when BAPTA (40 mm) is applied to the postsynaptic cell (67 ± 7% of control; n = 7; p < 0.05). Representative traces of IPSCs before (1) and after (2) LFSi are shown. The experiments were performed in C57BL/6JOlaHsd mice.
DSI is an endocannabinoid-mediated phenomenon that can be blocked by Ca2+ chelators (Pitler and Alger, 1992; Lenz and Alger, 1999), indicating that it requires postsynaptic Ca2+ influx. To determine whether a rise in postsynaptic Ca2+ is also required for LTDi induction, neurons in the BLA were recorded with BAPTA-filled (40 mm) patch pipettes. After whole-cell access, BAPTA was allowed to diffuse to the postsynaptic cell for at least 30 min. Afterward, control values were recorded for 15 min before LFSi was applied. The postsynaptic application of BAPTA did not affect the induction of LTDi (control plus BAPTA, 100%; LFSi, 67 ± 7%; n = 7; p < 0.05) (Fig. 1C), indicating that, in contrast to DSI, LTDi induction in the amygdala is independent of postsynaptic Ca2+ influx.
Induction of LTDi involves the activation of metabotropic glutamate receptor 1 but not of metabotropic glutamate receptor 5
Besides a rise in postsynaptic Ca2+, the synthesis and release of endocannabinoids can also be triggered by activation of group I metabotropic glutamate receptors (mGluRs) in the cerebellum (Maejima et al., 2001; Galante and Diana, 2004) and the hippocampus (Varma et al., 2001; Chevaleyre and Castillo, 2003). There are two subtypes of group I mGluRs: mGluR1 and mGluR5 (Bordi and Ugolini, 1999; Valenti et al., 2002). Evidence exists that both receptor subtypes are expressed in the amygdala (Rodrigues et al., 2002; Lavreysen et al., 2003, 2004; Li and Neugebauer, 2004). We therefore investigated whether a similar mechanism is involved in the induction of LTDi. We found that bath application of the unselective group I mGluR agonist DHPG (50 μm) evoked a suppression of GABAergic currents similar to LTDi (control, 100%; DHPG, 72 ± 5%; n = 8; p < 0.05) (Fig. 2A) in CB1+/+ mice. DHPG had no effect in CB1-/- mice (control, 100%; DHPG, 101 ± 3%; n = 5; p > 0.05) (Fig. 2A). Furthermore, application of DHPG occluded LTDi (control plus DHPG, 100%; LFSi, 106 ± 13%; n = 6; p > 0.05) (Fig. 2B). To mimick the short-lasting activation of mGluRs during LFSi, we also performed experiments in which DHPG was applied only transiently. In these experiments, DHPG (50 μm) also induced a long-lasting decrease of the IPSC amplitude to 64 ± 3% (n = 8; p < 0.05) (Fig. 2C). This effect of DHPG was occluded by a preceding induction of LTDi (control, 100%; LFSi, 66 ± 8%; n = 5; p < 0.05; DHPG, 75 ± 4%; n = 5; p > 0.05) (Fig. 2D). Together, these results underline the close relationship between group I mGluR activation and the release of endocannabinoids in the amygdala. To further identify the mGluR subtype involved in LTDi, LFSi was applied in the presence of the selective mGluR1 antagonist CPCCOEt (50 μm) and the mGluR5 antagonist MPEP (10 μm), respectively. In accordance with the results observed in other brain regions (Battaglia et al., 2001; Mannaioni et al., 2001), neither application of CPCCOEt nor MPEP (two different batches; n = 2 and n = 3) per se had any significant effect on basal GABAergic transmission (control, 100%; CPCCOEt, 98 ± 9%; p > 0.05; control, 100%; MPEP, 90 ± 15%; p > 0.05). Bath application of CPCCOEt abolished the effect of LFSi on GABAergic currents and inhibited the induction of LTDi (control plus CPCCOEt, 100%; LFSi, 105 ± 4%; n = 6; p > 0.05) (Fig. 3A), whereas bath application of the mGluR5 antagonist MPEP had no effect on LTDi induction (control plus MPEP, 100%; LFSi, 63 ± 10%; n = 5; p < 0.05) (Fig. 3B).
Figure 2.

Induction of LTDi involves activation of group I mGluRs. A, The unselective group I mGluR agonist DHPG (50 μm) leads to a long-lasting suppression of GABAergic currents similar to LTDi in wild-type mice (CB1+/+; effect of DHPG, 72 ± 5% of control; n = 8; p < 0.05; •) but not in mice lacking CB1 receptors (CB1-/-; effect of DHPG, 101 ± 3%; n = 5; p > 0.05; ○). B, Application of DHPG (50 μm) occludes the induction of LTDi by LFSi (effect of LFSi in the presence of DHPG, 106 ± 13% of control; n = 6; p > 0.05). LTDi without DHPG (data from Fig. 1 A) is shown for comparison (squares). C, The transient application of DHPG (50 μm) also induces a long-lasting decrease of the IPSC amplitude to 64 ± 3% (n = 8; p < 0.05). D, This effect of DHPG is occluded by a preceding induction of LTDi (control, 100%; LFSi, 66 ± 8%; n = 5; p < 0.05; DHPG, 75 ± 4%; n = 5; p > 0.05). Representative traces of IPSCs are shown. The experiments depicted in B-D were performed in C57BL/6JOlaHsd mice.
Figure 3.
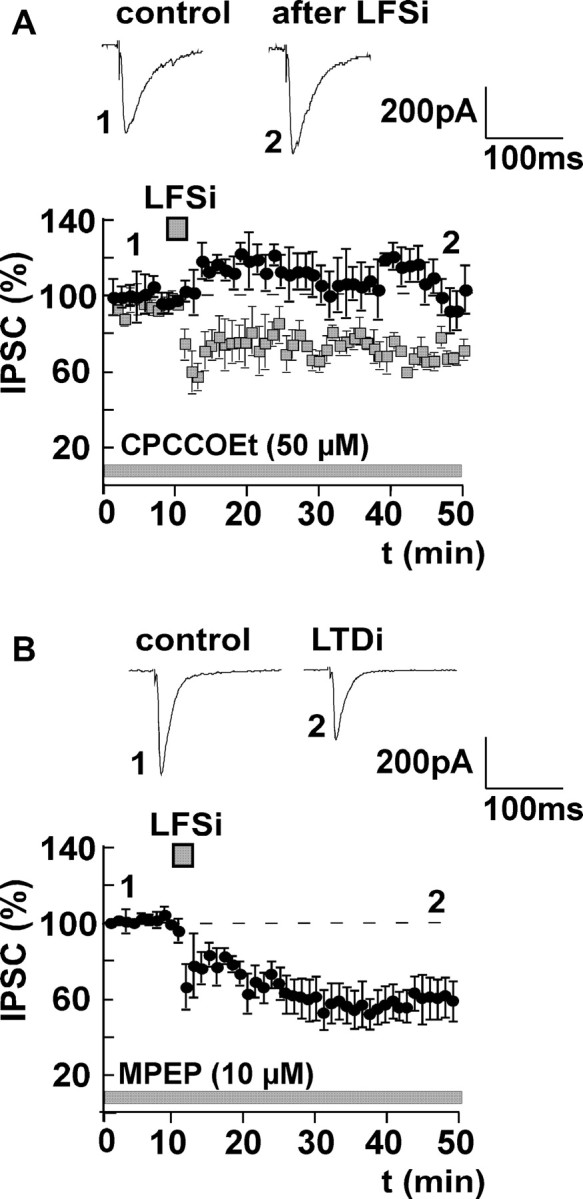
Induction of LTDi involves activation of mGluR1, but not of mGluR5. A, LTDi induction is completely inhibited by bath application of the mGluR1 antagonist CPCCOEt (50 μm; effect of LFSi, 105 ± 4% of control; n = 6; p > 0.05). LTDi without CPCCOEt (data from Fig. 1 A) is shown for comparison (squares). B, Preincubation of the slices with the mGluR5 antagonist MPEP (10 μm) has no effect on the induction of LTDi by LFSi (effect of LFSi in the presence of MPEP, 63 ± 10% of control; n = 5; p < 0.05). Representative traces of IPSCs before (1) and after (2) LFSi are shown. The experiments were performed in C57BL/6JOlaHsd mice.
LTDi involves the presynaptic inhibition of GABA release and the postsynaptic activation of G-proteins
During LTDi induction, the paired-pulse ratio is increased (Marsicano et al., 2002), suggesting a decrease of presynaptic transmitter release (Manabe et al., 1993). To confirm this finding, we investigated the effect of LFSi on the frequency and amplitude of sIPSCs. LFSi significantly reduced the frequency of sIPSCs from 6.4 ± 0.6 Hz to 2.7 ± 0.5 Hz (n = 5; p < 0.05), without affecting their amplitude (control, 7.4 ± 1.1 pA; LFSi, 6.9 ± 0.9 pA; n = 5; p > 0.05) (Fig. 4A), indicating that LTDi involves presynaptic mechanisms.
Figure 4.

LTDi is expressed presynaptically and induced postsynaptically via activation of G-proteins. A, Induction of LTDi is accompanied by a reduction of the frequency of sIPSCs from 6.4 ± 0.6 Hz to 2.7 ± 0.5 Hz (n = 5; p < 0.05), whereas the amplitude of sIPSC remains unaltered (control, 7.4 ± 1.1 pA; LFSi, 6.9 ± 0.9 pA; n = 5; p > 0.05). Representative traces of sIPSCs and the cumulative probability of the sIPSC amplitude before and after LFSi are shown. B, Postsynaptic application of the non-hydrolizable GTP analog GTPγ-S (600 μm) via the patch pipette blocks LTDi induction (effect of LFSi in the presence of GTPγ-S, 110 ± 9%; n = 4; p > 0.05). LTDi without GTPγ-S (data from Fig. 1 A) is shown for comparison (squares). Representative traces of IPSCs before (1) and after (2) LFSi are shown. The experiments were performed in C57BL/6JOlaHsd mice.
However, because endocannabinoids are assumed to act as retrograde transmitters, it remained to be proven that, during LTDi, endocannabinoids are released from the postsynaptic site. We therefore induced LTDi while blocking the postsynaptic G-proteins by the non-hydrolizable GTP analog GTPγ-S. When the neurons were loaded with GTPγ-S (600 μm) via the patch pipette, IPSC amplitudes increased to 130 ± 11% during the equilibration (20-30 min) after whole-cell access. After the amplitudes had stabilized, control values were recorded for 10 min, before LFSi was applied. GTPγ-S completely blocked LTDi induction (control plus GTPγ-S, 100%; LFSi, 110 ± 9%; n = 4; p > 0.05) (Fig. 4B), indicating that LTDi depends on the postsynaptic activation of G-proteins.
LTDi does not involve the activation of phospholipase C and diacylglycerol lipase
Group I mGluRs activate phospholipase C (PLC) via GTP-binding proteins and enhance the release of Ca2+ from intracellular stores via an increase of intracellular inositol-(1,4,5)triphosphate (Lee and Boden, 1997; Masgrau et al., 2001). This mechanism is considered to be involved in the synthesis and release of 2-arachidonoyl-glycerol (2-AG) (Stella et al., 1997; Di Marzo et al., 1998). Because we found that group I mGluRs play a major role in the induction of LTDi, we focused on the role of the PLC in LTDi. As described previously (Chevaleyre and Castillo, 2003), PLC was blocked by preincubation of the slices with the specific inhibitor U73122 (5 μm) for at least 60 min. During recording, 5 μm of the inhibitor was added to the bath solution. Unexpectedly, inhibition of the PLC did not affect LTDi (control plus U73122, 100%; LFSi, 67 ± 6%; n = 7; p < 0.05) (Fig. 5A), indicating that activation of the PLC is not required for the induction of LTDi in the amygdala. Because this is in clear contrast to previous investigations on endocannabinoid signaling, we also tested whether an inhibition of the diacylglycerol (DAG) lipase by RHC-80267 has any effect on the induction of LTDi. The DAG lipase is required for the synthesis of 2-AG (Stella et al., 1997; Di Marzo et al., 1998). After incubation of the slices with 100 μm RHC-80267 for at least 60 min, control values were recorded in the presence of 50 μm of this substance. We found that RHC-80267 did not affect the induction of LTDi (control plus RHC-80267, 100%; LFSi, 68 ± 4%; n = 8; p < 0.05) (Fig. 5B).
Figure 5.
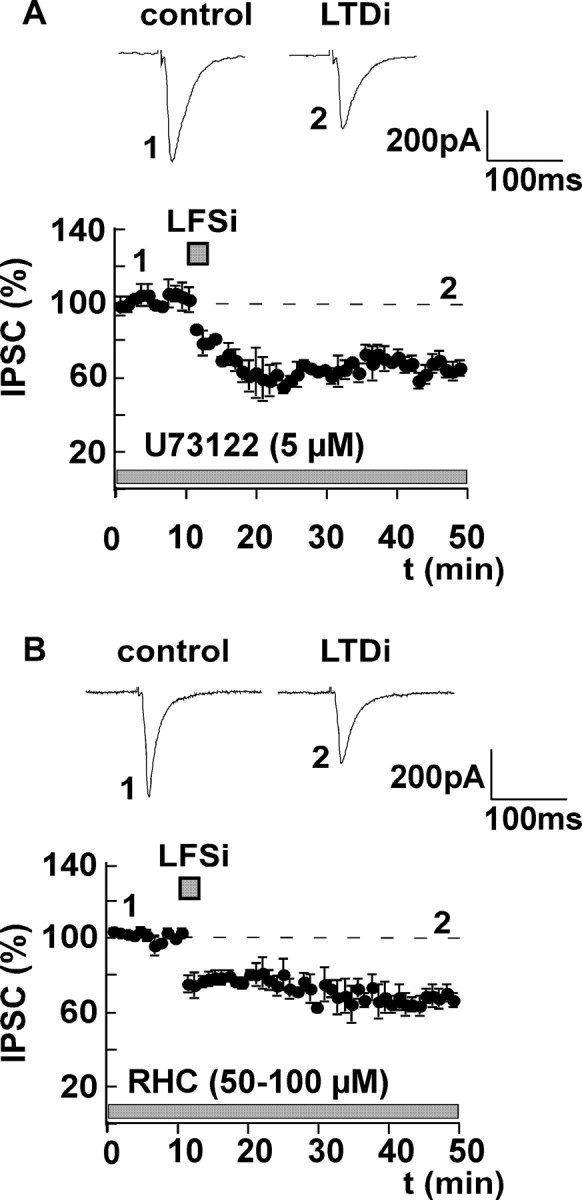
LTDi induction does not require activation of PLC and DAG lipase. A, After preincubation of the slices with the specific PLC inhibitor U73122 (5 μm) for at least 60 min, IPSCs were recorded in the presence of 5 μm U73122. Inhibition of PLC does not inhibit the induction of LTDi (effect of LFSi in the presence of U73122, 67 ± 6% of control; n = 7; p < 0.05). B, To block the DAG lipase, slices were preincubated with 100 μm RHC-80267 for at least 60 min. Afterward, control values were recorded in the presence of 50 μm RHC-80267. The DAG lipase inhibitor has no effect on the induction of LTDi (control plus RHC-80267, 100%; LFSi, 68 ± 4%; n = 8; p < 0.05). Representative traces of IPSCs before (1) and after (2) LFSi are shown. The experiments were performed in C57BL/6JOlaHsd mice.
LTDi involves the activation of the adenylyl cyclase-protein kinase A pathway and the release of anandamide
The above results indicate that, in the BLA, differently from the hippocampus (Chevaleyre and Castillo, 2003), the endocannabinoid 2-AG does not play a central role in CB1 receptor-dependent long-term synaptic plasticity of GABAergic transmission. To investigate the involvement of the other major endocannabinoid, anandamide, we evoked LTDi in mice lacking the anandamide-degrading enzyme FAAH (FAAH-/-) and in the respective wild-type littermates (FAAH+/+). In FAAH-/- mice, the degradation of the endocannabinoid anandamide is severely impaired (Cravatt et al., 2001). LTDi was significantly more pronounced in FAAH-/- mice compared with FAAH+/+ mice (FAAH+/+: control, 100%; LFSi, 78 ± 6%; n = 9; p < 0.05; FAAH-/-: control, 100%; LFSi, 48 ± 9%; n = 6; p < 0.05; LTDi FAAH+/+ vs FAAH-/-: p < 0.05) (Fig. 6A).
Figure 6.

LTDi involves the activation of the AC-PKA pathway and the release of anandamide. A, LTDi is significantly enhanced in mice lacking the anandamide-degrading enzyme FAAH (FAAH-/- mice, 48 ± 9% of control; n = 6; p < 0.05; ○) compared with the wild-type littermates (FAAH+/+ mice, 78 ± 6% of control; n = 9; p < 0.05; •; LTDi in FAAH+/+ vs LTDi in FAAH-/-, p < 0.05). B, LTDi induction is abolished when AC is blocked postsynaptically by 200 μm SQ22,536 (effect of LFSi in the presence of SQ22,536, 103 ± 15% of control; n = 5; p > 0.05). C, LTDi is also inhibited by the postsynaptic application of the PKA inhibitor Rp-cAMP (25 μm; effect of LFSi in the presence of Rp-cAMP, 101 ± 8% of control; n = 6; p > 0.05). LTDi without SQ22,536 or Rp-cAMP (data from Fig. 1 A) is shown for comparison (squares) in B and C, respectively. Representative traces of IPSCs before (1) and after (2) LFSi are shown. The experiments depicted in B and C were performed in C57BL/6JOlaHsd mice.
Because there is evidence that activation of group I mGluRs may also lead to an increase in cAMP formation (Balazs et al., 1998), we investigated whether the adenylyl cyclase (AC)-protein kinase A (PKA) pathway is involved in LTDi. Interestingly, LTDi was abolished when either AC or PKA was blocked postsynaptically by SQ22,536 (200 μm; control plus SQ22,536, 100%; LFSi, 103 ± 15%; n = 5; p > 0.05) (Fig. 6B) and Rp-cAMP (25 μm; control plus Rp-cAMP, 100%; LFSi, 101 ± 8%; n = 6; p > 0.05) (Fig. 6C), respectively. During the equilibration process after whole-cell access (30 min), baseline IPSC amplitudes remained stable in the presence of SQ22,536 (97 ± 5%; n = 5) and Rp-cAMP (113 ± 7%; n = 6; p > 0.05). Together, these findings suggest that LTDi involves the release of anandamide via an activation of the AC-PKA pathway.
LFSi enhances HFS-induced LTP in the BLA and increases excitatory synaptic transmission in the central nucleus
It has previously been reported that a reduction of inhibitory synaptic transmission enhances LTP in the amygdala (Watanabe et al., 1995; Huang and Kandel, 1998; Rammes et al., 2000). Provided that LFSi induces an endocannabinoid-mediated decrease of GABA release, it should increase the excitability of principal neurons in this structure. We therefore investigated the effect of LFSi on synaptic transmission and synaptic plasticity in the amygdala. We found that HFS of afferents in the LA with two trains of 100 pulses (50 Hz) and an interstimulus interval of 10 sec induced a LTP of extracellularly recorded FPs in the BLA (control, 100%; HFS, 115 ± 3%; n = 6; p < 0.05) (Fig. 7A). When LFSi was applied 10 min before HFS, the resulting LTP was significantly enhanced to 143 ± 7% (n = 7; p < 0.05; HFS without LFSi vs HFS with LFSi, p < 0.05) (Fig. 7A). Application of the CB1 antagonist SR141716A (5 μm) for at least 40 min did not affect HFS-induced LTP (control plus SR141716A, 100%; HFS, 118 ± 5%; n = 5; p < 0.05) (Fig. 7B) but abolished the enhancing effect of LFSi on LTP (LFSi plus HFS with SR141716A, 122 ± 7%; n = 6; p < 0.05; LFSi plus HFS vs LFSi plus HFS plus SR141716A, p < 0.05) (Fig. 7B). We also wanted to know whether the disinhibition induced by LFSi has any effect on excitatory synaptic transmission in the central nucleus, which is the main output site of the amygdala (LeDoux, 2000). We therefore recorded EPSCs in the central nucleus while stimulating in the LA. We found that LFSi delivered to the LA significantly enhanced the amplitude of EPSCs in the central nucleus (control, 100%; LFSi, 125 ± 10%; n = 6; p < 0.05) (Fig. 7C). This effect was abolished by application of the CB1 receptor antagonist SR141716A (control plus SR141716A, 100%; LFSi, 101 ± 5%; n = 6; p > 0.05; LFSi vs LFSi plus SR141716A, p < 0.05) (Fig. 7C), which per se did not affect EPSC amplitudes (control, 100%; SR141716A, 104 ± 4%; n = 6; p > 0.05).
Figure 7.
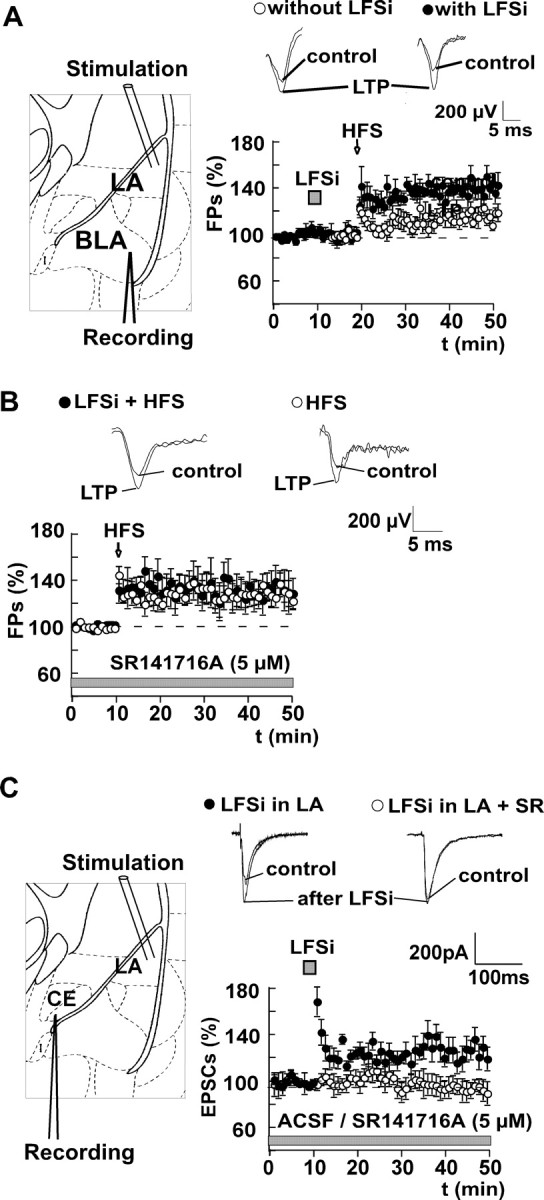
LFSi enhances HFS-induced LTP in the BLA and increases excitatory synaptic transmission in the central nucleus via activation of CB1 receptors. A, HFS of afferents in the LA (2 trains of 100 pulses with 50 Hz and an interstimulus interval of 10 sec) induces an LTP of FPs in the BLA (effect of HFS, 115 ± 3% of control; n = 6; p < 0.05; ○). When LFSi is applied 10 min before HFS, the resulting LTP is significantly enhanced (HFS after LFSi, 143 ± 7%; n = 7; p < 0.05; •; HFS vs HFS plus LFSi, p < 0.05). B, Application of the CB1 antagonist SR141716A (5 μm) for at least 40 min does not affect HFS-induced LTP (control plus SR141716A, 100%; HFS, 118 ± 5%; n = 5; p < 0.05; ○) but abolishes the enhancing effect of LFSi on LTP (LFSi plus HFS with SR141716A, 122 ± 7%; n = 6; p < 0.05; LFSi plus HFS vs LFSi plus HFS plus SR141716A, p < 0.05; •). C, LFSi of afferents in the LA increases the amplitude of EPSCs recorded from principal neurons in the central nucleus (CE; 125 ± 10% of control; n = 6; p < 0.05; •). This effect is abolished by preincubation of the slices with the CB1 receptor antagonist SR141716A (SR; 5 μm; effect of LFSi, 101 ± 5% of control; n = 6; p > 0.05; ○; LFSi vs LFSi plus SR141716A, p < 0.05). Schemes on the left depict the respective positions of stimulation and recording electrodes. Representative traces are shown. The experiments were performed in C57BL/6JOlaHsd mice.
Discussion
The present findings suggest that LFSi releases glutamate from afferents in the LA and thereby induces an mGluR1-mediated on-demand release of endocannabinoids, presumably mainly anandamide, in the BLA. This endocannabinoid acts as a retrograde transmitter and enhances excitability in distinct intraamygdaloid pathways by reducing the inhibitory synaptic transmission of GABAergic interneurons. LTDi cannot be evoked in CB1-/- mice, is blocked by the CB1 receptor antagonist SR141716A (Marsicano et al., 2002), and is occluded by application of the CB1 receptor agonist WIN55,212-2. A specific role of anandamide in LTDi is underscored by the findings that LTDi is significantly enhanced in mice lacking the enzyme FAAH and that the release requires the activation of the AC-PKA pathway but not of the PLC-DAG lipase cascade.
Endocannabinoids released from postsynaptic sites influence the presynaptic release of excitatory and inhibitory transmitters (Ohno-Shosaku et al., 2001; Alger, 2002; Wilson and Nicoll, 2002; Melis et al., 2004) and induce long-term changes of neuronal excitability in various brain regions (Gerdeman et al., 2002; Robbe et al., 2002; Chevaleyre and Castillo, 2003; Gerdeman and Lovinger, 2003). Endocannabinoids were also shown to mediate a timing-dependent form of LTD in the neocortex (Sjöström et al., 2003).
In the present study, the CB1 receptor-dependent LTDi involves the activation of group I mGluRs. In line with this finding, the application of the group I mGluR agonist DHPG induced a long-lasting reduction of inhibitory GABAergic currents similar to LTDi in wild-type mice but not in CB1-/- mice. It has previously been shown that the release of endocannabinoids can be triggered by activation of mGluR5 or by activation of both mGluR1 and mGluR5 in various brain regions (Maejima et al., 2001; Ohno-Shosaku et al., 2002; Robbe et al., 2002; Chevaleyre and Castillo, 2003). Interestingly, endocannabinoid-mediated LTDi in the BLA seems to require mainly the activation of mGluR1.
The release of endocannabinoids during DSI and depolarization-induced suppression of excitation are initiated by voltage-dependent postsynaptic Ca2+ influx (Lenz and Alger, 1999; Kreitzer and Regehr, 2001). Also, endocannabinoid-mediated LTD of excitatory synaptic currents in the striatum (Gerdeman et al., 2002) and the nucleus accumbens (Robbe et al., 2002) depends on a postsynaptic rise of Ca2+. However, LTDi in the BLA does not require a postsynaptic Ca2+ influx, thus resembling group I mGluR-mediated endocannabinoid release in the cerebellum (Maejima et al., 2001) and the hippocampus (Ohno-Shosaku et al., 2002; Chevaleyre and Castillo, 2003).
Metabotropic GluR1/5 receptors couple to the phosphoinositide-Ca2+ cascade via an activation of PLC (Abe et al., 1992). This mechanism is involved in the mGluR1/5-mediated endocannabinoid release in the nucleus accumbens (Robbe et al., 2002) and the hippocampus (Chevaleyre and Castillo, 2003). Unexpectedly, in the present study, LTDi was independent of the activation of PLC or DAG lipase but required the activation of the AC-PKA signaling cascade. We propose that LTDi in the BLA involves the postsynaptic activation of mGluR1a and a subsequent coupling to the AC-PKA pathway. The mGluR1a receptor subtype may activate both PLC and AC via different G-proteins. Whereas mGluR1a-mediated phosphoinositide hydrolysis is clearly reduced in the absence of Ca2+ ions, mGluR1a-induced stimulation of cAMP formation appears to be independent of extracellular Ca2+ concentration (Thomsen, 1996). In the present study, neither the postsynaptic inhibition of PLC nor the postsynaptic application of Ca2+ chelators affected mGluR1-mediated LTDi. There is evidence that activation of PLC and DAG lipase preferentially affects the formation of the endocannabinoid 2-AG (Stella et al., 1997; Di Marzo et al., 1998; Mechoulam et al., 1998; Bisogno et al., 2003), whereas anandamide formation involves N-acyltransferase (NAT) and phospholipase D as key enzymes (Di Marzo et al., 1994; Sugiura et al., 2002; Piomelli, 2003). The AC-PKA pathway may be important for the potentiation of NAT activity (Cadas et al., 1996). Our finding that the AC-PKA pathway, but not the PLC pathway, is involved in LTDi favors the assumption that LTDi in the amygdala is mediated preferentially by anandamide. This view is strongly supported by the observation that LTDi is significantly enhanced in FAAH-/- mice. FAAH also degrades other bioactive fatty acid amides such as oleamide and palmitoylethanolamide (Cravatt et al., 1995, 2001). However, the fact that LTDi depends on CB1 receptor activation (Marsicano et al., 2002), together with the basically abolished degradation of anandamide in FAAH-/- mice (Cravatt et al., 2001), suggests that anandamide is responsible for the specific enhancement of LFSi-induced LTDi in these animals. The 15-fold increased basal level of anandamide in FAAH-/- mice (Cravatt et al., 2001) may lead to an occupation of CB1 receptors already under basal conditions and may induce compensatory processes that alter the functionality of the endogenous cannabinoid system. However, in FAAH-/- mice, LTDi induction is not occluded but rather significantly increased compared with FAAH+/+ animals. Moreover, CB1 receptor levels are not changed in FAAH-/- mice (Lichtman et al., 2002), excluding a direct compensation of enhanced anandamide signaling by a downregulation of CB1 receptors. Therefore, the present data suggest that LFSi induces a dynamically regulated release of anandamide and that the observed enhancement of LTDi in mutant mice is primarily attributable to an increased amount of anandamide available at specific GABAergic synapses containing CB1 receptors.
Provided that endocannabinoids reduce transmitter release from GABAergic terminals exerting an inhibitory tonus on principal neurons (Giuffrida and Piomelli, 2000), mechanisms critical for learning and memory such as LTP (Watanabe et al., 1995; Huang and Kandel, 1998; Rammes et al., 2000; Chevaleyre and Castillo, 2003) will be favored. LTP of excitatory glutamatergic transmission in the BLA, induced by HFS in the LA, was enhanced by a preceding LFSi. Furthermore, LFSi per se markedly enhanced excitatory synaptic transmission in the central nucleus. The disinhibitory effects on the excitability of principal neurons was blocked by application of SR141716A, suggesting an LFSi-induced endocannabinoid release as the main underlying mechanism.
The amygdala plays a key role in fear conditioning and anxiety (McKernan and Shinnick-Gallagher, 1997; LeDoux, 2000; Walker and Davis, 2002). In a previous investigation, we found that mice lacking CB1 receptors show strongly impaired extinction but unaffected acquisition and consolidation of aversive memories. We also found that tone presentation during extinction trials resulted in elevated levels of endocannabinoids in the BLA (Marsicano et al., 2002). Compared with acquisition and consolidation of memories, the information available about mechanisms underlying extinction is rather scant and less clear (McGaugh et al., 1990; Falls et al., 1992; Harris and Westbrook, 1998; Milad and Quirk, 2002; Myers and Davis, 2002). Based on our previous findings, we assumed that the release of endocannabinoids in the BLA during LTDi might be related to extinction, rather than to acquisition and consolidation of aversive memories. LTDi leads to an enhancement of excitatory synaptic transmission in the CE, which is the main output site of the amygdala. Recent theories suggest that extinction is at least partially based on an active relearning process, in which the prolonged reexposure to the tone in the absence of the footshock triggers the formation of a new memory trace encoding the dissociation between the tone and the footshock (Quirk et al., 2000; Myers and Davis, 2002; Suzuki et al., 2004). In this context, CB1 receptor-dependent LTDi in the BLA might selectively enhance the excitability in specific neuronal circuits, which encode for this novel extinction memory trace. LTDi might therefore underlie an active mnemonic process involved in the extinction of fear memories on a cellular level.
Footnotes
This work was supported by a grant from the Claussen-Simon Stiftung des Stifterverbandes der Deutschen Wissenschaft (S.C.A.), by the Deutsche Forschungsgemeinschaft (SFB 391 to W.Z., LU755/1-3 to B.L.), and by a scholarship from the Hertie Foundation (B.L.). We thank Christine Hilf, Barbara Wölfel, and Barbara Fackelmeier for excellent technical assistance and Carola Hetzel for secretarial work. S.C.A. is also grateful to Dr. h. c. K. Peter, Dr. A. Beyer, and Prof. P. Schöps for supporting her research.
Correspondence should be addressed to Dr. Shahnaz Christina Azad, Max-Planck-Institute of Psychiatry, Clinical Neuropharmacology, Kraepelinstrasse 2, 80804 München, Germany. E-mail: azad@mpipsykl.mpg.de.
Copyright © 2004 Society for Neuroscience 0270-6474/04/249953-09$15.00/0
References
- Abe T, Sugihara H, Nawa H, Shigemoto R, Mizuno N, Nakanishi S (1992) Molecular characterization of a novel metabotropic glutamate receptor mGluR5 coupled to inositol phosphate/Ca2+ signal transduction. J Biol Chem 267: 13361-13368. [PubMed] [Google Scholar]
- Alger BE (2002) Retrograde signaling in the regulation of synaptic transmission: focus on endocannabinoids. Prog Neurobiol 68: 247-286. [DOI] [PubMed] [Google Scholar]
- Azad SC, Eder M, Marsicano G, Lutz B, Zieglgänsberger W, Rammes G (2003) Activation of the cannabinoid receptor type 1 decreases glutamatergic and GABAergic synaptic transmission in the lateral amygdala of the mouse. Learn Mem 10: 116-128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balazs R, Miller S, Chun Y, O'Toole J, Cotman CW (1998) Metabotropic glutamate receptor agonists potentiate cyclic AMP formation induced by forskolin or beta-adrenergic receptor activation in cerebral cortical astrocytes in culture. J Neurochem 70: 2446-2458. [DOI] [PubMed] [Google Scholar]
- Battaglia G, Bruno V, Pisani A, Centonze D, Catania MV, Calabresi P, Nicoletti F (2001) Selective blockade of type-1 metabotropic glutamate receptors induces neuroprotection by enhancing gabaergic transmission. Mol Cell Neurosci 17: 1071-1083. [DOI] [PubMed] [Google Scholar]
- Bisogno T, Howell F, Williams G, Minassi A, Cascio MG, Ligresti A, Matias I, Schiano-Moriello A, Paul P, Williams EJ, Gangadharan U, Hobbs C, Di Marzo V, Doherty P (2003) Cloning of the first sn1-DAG lipases points to the spatial and temporal regulation of endocannabinoid signaling in the brain. J Cell Biol 163: 463-468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bordi F, Ugolini A (1999) Group I metabotropic glutamate receptors: implications for brain diseases. Prog Neurobiol 59: 55-79. [DOI] [PubMed] [Google Scholar]
- Cadas H, Gaillet S, Beltramo M, Venance L, Piomelli D (1996) Biosynthesis of an endogenous cannabinoid precursor in neurons and its control by calcium and cAMP. J Neurosci 16: 3934-3942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carlson G, Wang Y, Alger BE (2002) Endocannabinoids facilitate the induction of LTP in the hippocampus. Nat Neurosci 5: 723-724. [DOI] [PubMed] [Google Scholar]
- Chevaleyre V, Castillo PE (2003) Heterosynaptic LTD of hippocampal GABAergic synapses: a novel role of endocannabinoids in regulating excitability. Neuron 38: 461-472. [DOI] [PubMed] [Google Scholar]
- Cravatt BF, Prospero-Garcia O, Siuzdak G, Gilula NB, Henriksen SJ, Boger DL, Lerner RA (1995) Chemical characterization of a family of brain lipids that induce sleep. Science 268: 1506-1509. [DOI] [PubMed] [Google Scholar]
- Cravatt BF, Demarest K, Patricelli MP, Bracey MH, Giang DK, Martin BR, Lichtman AH (2001) Supersensitivity to anandamide and enhanced endogenous cannabinoid signaling in mice lacking fatty acid amide hydrolase. Proc Natl Acad Sci USA 98: 9371-9376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Marzo V, Fontana A, Cadas H, Schinelli S, Cimino G, Schwartz JC, Piomelli D (1994) Formation and inactivation of endogenous cannabinoid anandamide in central neurons. Nature 372: 686-691. [DOI] [PubMed] [Google Scholar]
- Di Marzo V, Melck D, Bisogno T, De Petrocellis L (1998) Endocannabinoids: endogenous cannabinoid receptor ligands with neuromodulatory action. Trends Neurosci 21: 521-528. [DOI] [PubMed] [Google Scholar]
- Falls WA, Miserendino MJ, Davis M (1992) Extinction of fear-potentiated startle: blockade by infusion of an NMDA antagonist into the amygdala. J Neurosci 12: 854-863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freund TF, Katona I, Piomelli D (2003) Role of endogenous cannabinoids in synaptic signaling. Physiol Rev 83: 1017-1066. [DOI] [PubMed] [Google Scholar]
- Galante M, Diana MA (2004) Group I metabotropic glutamate receptors inhibit GABA release at interneuron-Purkinje cell synapses through endocannabinoid production. J Neurosci 24: 4865-4874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerdeman GL, Lovinger DM (2003) Emerging roles for endocannabinoids in long-term synaptic plasticity. Br J Pharmacol 140: 781-789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerdeman GL, Ronesi J, Lovinger DM (2002) Postsynaptic endocannabinoid release is critical to long-term depression in the striatum. Nat Neurosci 5: 446-451. [DOI] [PubMed] [Google Scholar]
- Giuffrida A, Piomelli D (2000) The endocannabinoid system: a physiological perspective on its role in psychomotor control. Chem Phys Lipids 108: 151-158. [DOI] [PubMed] [Google Scholar]
- Harris JA, Westbrook RF (1998) Evidence that GABA transmission mediates context-specific extinction of learned fear. Psychopharmacology 140: 105-115. [DOI] [PubMed] [Google Scholar]
- Hoffman AF, Lupica CR (2000) Mechanisms of cannabinoid inhibition of GABAA synaptic transmission in the hippocampus. J Neurosci 20: 2470-2479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffman AF, Lupica CR (2001) Direct actions of cannabinoids on synaptic transmission in the nucleus accumbens: a comparison with opioids. J Neurophysiol 85: 72-83. [DOI] [PubMed] [Google Scholar]
- Huang YY, Kandel ER (1998) Postsynaptic induction and PKA-dependent expression of LTP in the lateral amygdala. Neuron 21: 169-178. [DOI] [PubMed] [Google Scholar]
- Katona I, Rancz EA, Acsady L, Ledent C, Mackie K, Hajos N, Freund TF (2001) Distribution of CB1 cannabinoid receptors in the amygdala and their role in the control of GABAergic transmission. J Neurosci 21: 9506-9518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kreitzer AC, Regehr WG (2001) Retrograde inhibition of presynaptic calcium influx by endogenous cannabinoids at excitatory synapses onto Purkinje cells. Neuron 29: 717-727. [DOI] [PubMed] [Google Scholar]
- Kreitzer AC, Carter AG, Regehr WG (2002) Inhibition of interneuron firing extends the spread of endocannabinoid signaling in the cerebellum. Neuron 34: 787-796. [DOI] [PubMed] [Google Scholar]
- Lavreysen H, Janssen C, Bischoff F, Langlois X, Leysen JE, Lesage AS (2003) [3H]R214127: a novel high-affinity radioligand for the mGlu1 receptor reveals a common binding site shared by multiple allosteric antagonists. Mol Pharmacol 63: 1082-1093. [DOI] [PubMed] [Google Scholar]
- Lavreysen H, Pereira SN, Leysen JE, Langlois X, Lesage AS (2004) Metabotropic glutamate 1 receptor distribution and occupancy in the rat brain: a quantitative autoradiographic study using [3H]R214127. Neuropharmacology 46: 609-619. [DOI] [PubMed] [Google Scholar]
- LeDoux JE (2000) Emotion circuits in the brain. Annu Rev Neurosci 23: 155-184. [DOI] [PubMed] [Google Scholar]
- Lee K, Boden PR (1997) Characterization of the inward current induced by metabotropic glutamate receptor stimulation in rat ventromedial hypothalamic neurones. J Physiol (Lond) 504: 649-663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lenz RA, Alger BE (1999) Calcium dependence of depolarization-induced suppression of inhibition in rat hippocampal CA1 pyramidal neurons. J Physiol (Lond) 521: 147-157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li W, Neugebauer V (2004) Differential roles of mGluR1 and mGluR5 in brief and prolonged nociceptive processing in central amygdala neurons. J Neurophysiol 91: 13-24. [DOI] [PubMed] [Google Scholar]
- Lichtman AH, Hawkins EG, Griffin G, Cravatt BF (2002) Pharmacological activity of fatty acid amides is regulated, but not mediated, by fatty acid amide hydrolase in vivo. J Pharmacol Exp Ther 302: 73-79. [DOI] [PubMed] [Google Scholar]
- Maejima T, Hashimoto K, Yoshida T, Aiba A, Kano M (2001) Presynaptic inhibition caused by retrograde signal from metabotropic glutamate to cannabinoid receptors. Neuron 31: 463-475. [DOI] [PubMed] [Google Scholar]
- Manabe T, Wyllie DJ, Perkel DJ, Nicoll RA (1993) Modulation of synaptic transmission and long-term potentiation: effects on paired pulse facilitation and EPSC variance in the CA1 region of the hippocampus. J Neurophysiol 70: 1451-1459. [DOI] [PubMed] [Google Scholar]
- Mannaioni G, Marino MJ, Valenti O, Traynelis SF, Conn PJ (2001) Metabotropic glutamate receptors 1 and 5 differentially regulate CA1 pyramidal cell function. J Neurosci 21: 5925-5934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manning BH, Merin NM, Meng ID, Amaral DG (2001) Reduction in opioid- and cannabinoid-induced antinociception in rhesus monkeys after bilateral lesions of the amygdaloid complex. J Neurosci 21: 8238-8246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manzoni OJ, Bockaert J (2001) Cannabinoids inhibit GABAergic synaptic transmission in mice nucleus accumbens. Eur J Pharmacol 412: R3-R5. [DOI] [PubMed] [Google Scholar]
- Marsicano G, Lutz B (1999) Expression of the cannabinoid receptor CB1 in distinct neuronal subpopulations in the adult mouse brain. Eur J Neurosci 11: 4213-4225. [DOI] [PubMed] [Google Scholar]
- Marsicano G, Wotjak CT, Azad SC, Bisogno T, Rammes G, Cascio MG, Hermann H, Tang J, Hofmann C, Zieglgänsberger W, Di Marzo V, Lutz B (2002) The endogenous cannabinoid system controls extinction of aversive memories. Nature 418: 530-534. [DOI] [PubMed] [Google Scholar]
- Martin WJ, Coffin PO, Attias E, Balinsky M, Tsou K, Walker JM (1999) Anatomical basis for cannabinoid-induced antinociception as revealed by intracerebral microinjections. Brain Res 822: 237-242. [DOI] [PubMed] [Google Scholar]
- Masgrau R, Servitja JM, Young KW, Pardo R, Sarri E, Nahorski SR, Picatoste F (2001) Characterization of the metabotropic glutamate receptors mediating phospholipase C activation and calcium release in cerebellar granule cells: calcium-dependence of the phospholipase C response. Eur J Neurosci 13: 248-256. [DOI] [PubMed] [Google Scholar]
- McDonald AJ, Mascagni F (2001) Localization of the CB1 type cannabinoid receptor in the rat basolateral amygdala: high concentrations in a sub-population of cholecystokinin-containing interneurons. Neuroscience 107: 641-652. [DOI] [PubMed] [Google Scholar]
- McGaugh JL, Castellano C, Brioni J (1990) Picrotoxin enhances latent extinction of conditioned fear. Behav Behav Neurosci 104: 264-267. [DOI] [PubMed] [Google Scholar]
- McKernan MG, Shinnick-Gallagher P (1997) Fear conditioning induces a lasting potentiation of synaptic currents in vitro. Nature 390: 607-611. [DOI] [PubMed] [Google Scholar]
- Mechoulam R, Fride E, Di Marzo V (1998) Endocannabinoids. Eur J Pharmacol 359: 1-18. [DOI] [PubMed] [Google Scholar]
- Melis M, Pistis M, Perra S, Muntoni AL, Pillolla G, Gessa GL (2004) Endocannabinoids mediate presynaptic inhibition of glutamatergic transmission in rat ventral tegmental area dopamine neurons through activation of CB1 receptors. J Neurosci 24: 53-62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milad MR, Quirk GJ (2002) Neurons in medial prefrontal cortex signal memory for fear extinction. Nature 420: 70-74. [DOI] [PubMed] [Google Scholar]
- Myers KM, Davis M (2002) Behavioral and neural analysis of extinction. Neuron 36: 567-584. [DOI] [PubMed] [Google Scholar]
- Ohno-Shosaku T, Maejima T, Kano M (2001) Endogenous cannabinoids mediate retrograde signals from depolarized postsynaptic neurons to presynaptic terminals. Neuron 29: 729-738. [DOI] [PubMed] [Google Scholar]
- Ohno-Shosaku T, Shosaku J, Tsubokawa H, Kano M (2002) Cooperative endocannabinoid production by neuronal depolarization and group I metabotropic glutamate receptor activation. Eur J Neurosci 15: 953-961. [DOI] [PubMed] [Google Scholar]
- Pertwee RG (2001) Cannabinoid receptors and pain. Prog Neurobiol 63: 569-611. [DOI] [PubMed] [Google Scholar]
- Piomelli D (2003) The molecular logic of endocannabinoid signalling. Nat Rev Neurosci 4: 873-884. [DOI] [PubMed] [Google Scholar]
- Pitler TA, Alger BE (1992) Postsynaptic spike firing reduces synaptic GABAA responses in hippocampal pyramidal cells. J Neurosci 12: 4122-4132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quirk GJ, Russo GK, Barron JL, Lebron K (2000) The role of ventromedial prefrontal cortex in the recovery of extinguished fear. J Neurosci 20: 6225-6231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rammes G, Steckler T, Kresse A, Schütz G, Zieglgänsberger W, Lutz B (2000) Synaptic plasticity in the basolateral amygdala in transgenic mice expressing dominant-negative cAMP response element-binding protein (CREB) in forebrain. Eur J Neurosci 12: 2534-2546. [DOI] [PubMed] [Google Scholar]
- Robbe D, Kopf M, Remaury A, Bockaert J, Manzoni OJ (2002) Endogenous cannabinoids mediate long-term synaptic depression in the nucleus accumbens. Proc Natl Acad Sci USA 99: 8384-8388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodrigues SM, Bauer EP, Farb CR, Schafe GE, LeDoux JE (2002) The group I metabotropic glutamate receptor mGluR5 is required for fear memory formation and long-term potentiation in the lateral amygdala. J Neurosci 22: 5219-5229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sjöström PJ, Turrigiano GG, Nelson SB (2003) Neocortical LTD via coincident activation of presynaptic NMDA and cannabinoid receptors. Neuron 39: 641-654. [DOI] [PubMed] [Google Scholar]
- Stella N, Schweitzer P, Piomelli D (1997) A second endogenous cannabinoid that modulates long-term potentiation. Nature 388: 773-778. [DOI] [PubMed] [Google Scholar]
- Sugiura T, Kobayashi Y, Oka S, Waku K (2002) Biosynthesis and degradation of anandamide and 2-arachidonoylglycerol and their possible physiological significance. Prostaglandins Leukot Essent Fatty Acids 66: 173-192. [DOI] [PubMed] [Google Scholar]
- Suzuki A, Josselyn SA, Frankland PW, Masushige S, Silva AJ, Kida S (2004) Memory reconsolidation and extinction have distinct temporal and biochemical signatures. J Neurosci 24: 4787-4795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomsen C (1996) Metabotropic glutamate receptor subtype 1a activates adenylate cyclase when expressed in baby hamster kidney cells. Prog Neuropsychopharmacol Biol Psychiatry 20: 709-726. [DOI] [PubMed] [Google Scholar]
- Valenti O, Conn PJ, Marino MJ (2002) Distinct physiological roles of the Gq-coupled metabotropic glutamate receptors co-expressed in the same neuronal populations. J Cell Physiol 191: 125-137. [DOI] [PubMed] [Google Scholar]
- Varma N, Carlson GC, Ledent C, Alger BE (2001) Metabotropic glutamate receptors drive the endocannabinoid system in hippocampus. J Neurosci 21: RC188(1-5). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walker DL, Davis M (2002) The role of amygdala glutamate receptors in fear learning, fear-potentiated startle, and extinction. Pharmacol Biochem Behav 71: 379-392. [DOI] [PubMed] [Google Scholar]
- Washburn MS, Moises HC (1992) Electrophysiological and morphological properties of rat basolateral amygdaloid neurons in vitro. J Neurosci 12: 4066-4079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe Y, Ikegaya Y, Saito H, Abe K (1995) Roles of GABAA, NMDA and muscarinic receptors in induction of long-term potentiation in the medial and lateral amygdala in vitro. Neurosci Res 21: 317-322. [DOI] [PubMed] [Google Scholar]
- Wilson RI, Nicoll RA (2001) Endogenous cannabinoids mediate retrograde signalling at hippocampal synapses. Nature 410: 588-592. [DOI] [PubMed] [Google Scholar]
- Wilson RI, Nicoll RA (2002) Endocannabinoid signaling in the brain. Science 296: 678-682. [DOI] [PubMed] [Google Scholar]