

Abstract
Visual and auditory information is encoded by sensory neurons that tonically release neurotransmitter at high rates. The synaptic ribbon is an essential organelle in nerve terminals of these neurons. Its precise function is unknown, but if the ribbon could be visualized in a living terminal, both its own dynamics and its relation to calcium and vesicle dynamics could be studied. We designed a short fluorescent peptide with affinity for a known binding domain of RIBEYE, a protein unique to the ribbon. When introduced via a whole-cell patch pipette, the peptide labeled structures at the presynaptic plasma membrane of ribbon-type terminals. The fluorescent spots match in size, location, number, and distribution the known features of synaptic ribbons. Furthermore, fluorescent spots mapped by confocal microscopy directly match the ribbons identified by electron microscopy in the same cell. Clearly the peptide binds to the synaptic ribbon, but even at saturating concentrations it affects neither the morphology of the ribbon nor its tethering of synaptic vesicles. It also does not inhibit exocytosis. Using the peptide label, we observed that the ribbon is immobile over minutes and that calcium influx is concentrated at the ribbon. Finally, we find that each ribbon in a retinal bipolar cell contains ∼4000 molecules of RIBEYE, indicating that it is the major component of the synaptic ribbon.
Keywords: synaptic ribbon, exocytosis, photoreceptor, synaptic vesicle, evanescent field microscopy, retina
Introduction
Visual and auditory sensory neurons encode changes in stimulus intensity by changes in tonic rate of vesicle release from their synaptic terminals. Such a system allows these neurons to transmit more information than conventional neurons, which encode information by changes in rate of action potentials (de Ruyter van Steveninck and Laughlin, 1996). Correlated with this capacity for tonic vesicle release, synapses of auditory hair cells and retinal photoreceptors and bipolar cells express a “ribbon” that tethers a dense cluster of vesicles near the active zone. Loss of an anchoring protein, bassoon, causes ribbons to detach from the active zone and also blocks synaptic transmission (Dick et al., 2003), suggesting that the ribbon is required for release. However, it remains to be established exactly what the ribbon is made of, what it does, and how it works.
Fluorescent synaptic vesicles have been directly visualized in these terminals as they move to the membrane and fuse (Zenisek et al., 2000), but in those experiments it was difficult to distinguish vesicles on a ribbon from those free in the cytoplasm. Thus, if both the vesicles and the ribbon could be labeled in vivo, their interactions might be followed in real time, especially in relation to voltage-gated calcium influx.
Several proteins have been identified within the ribbon complex (Wang et al., 1997; Muresan et al., 1999; Schmitz et al., 2000; Dick et al., 2001, 2003), but the only one that localizes exclusively to the ribbon is RIBEYE, which is a splice variant of the transcription factor C-terminal binding protein 2 (CtBP-2). RIBEYE consists of an A-domain without significant homology to any known protein and a B-domain that is nearly identical to CtBP-2 (Schmitz et al., 2000).
The CtBP proteins were initially identified through their binding of the COOH terminus of adenovirus E1A (Boyd et al., 1993; Schaeper et al., 1995) (for review, see Chinnadurai, 2002). The binding site of E1A mapped in part to a stretch of five amino acids containing the sequence PXDLS. We reasoned that RIBEYE might bind the same sequence. Indeed, PXDLS or similar sequences were found in 68% of all clones identified by a two-hybrid screen with CtBP as bait (Vo et al., 2001). Accordingly, we prepared a fluorescent peptide containing this amino acid sequence and dialyzed it into live cells. We report here that the peptide does label synaptic ribbons in live terminals, that each bipolar cell ribbon contains ∼4000 RIBEYE molecules, that the ribbon moves slowly on the membrane, and that calcium influx through voltage-gated channels localizes precisely at the ribbon.
Materials and Methods
Cell and tissue preparation. Goldfish bipolar cells were prepared as previously described (Heidelberger and Matthews, 1992). Goldfish were decapitated, and their eyes were removed and hemisected. After removal of lens, eyecups were incubated for 20 min in an oxygenated HEPES-buffered saline solution containing (in mm): 10 HEPES, 120 NaCl, 0.5 CaCl2, 2.5 KCl, 1.0 MgCl2, 10 glucose, and 1100 U/ml hyaluronidase type V with pH adjusted to 7.4 using NaOH. Next, eyecups were removed and rinsed in a saline solution lacking hyaluronidase. Retinas were removed and cut into 6-10 pieces and placed at room temperature in a papain digestion solution for 30-40 min for isolation of bipolar cells, and 10-20 min for isolation of cones. The papain digestion solution contained (in mm): 10 HEPES, 120 NaCl, 0.5 CaCl2, 2.5 KCl, 1.0 MgCl2, 10 glucose, 0.33 mg/ml cysteine, and 35 U/ml papain (Fluka, Neu-Ulm, Germany) with pH adjusted to 7.4 using NaOH. After papain incubation, pieces of retina were removed and rinsed with saline solution containing (in mm): 10 HEPES, 120 NaCl, 0.5 CaCl2, 2.5 KCl, and 1.0 MgCl2 and stored at 8-12°C. Bipolar cells, horizontal cells, and cones were isolated from pieces of tissue by mechanical trituration using a fire-polished Pasteur pipette into a solution containing (in mm): 10 HEPES, 120 NaCl, 2.5 CaCl2, 2.5 KCl, 1.0 MgCl2, and 10 glucose with pH adjusted to 7.4 using NaOH. Cultured hippocampal neurons from neonatal rats were kindly provided by Gregorio Valdez (State University of New York, Stony Brook, NY) and were used for experiments after 7 d in culture on poly-l-lysine-coated coverslips.
Electrophysiology. Bipolar cells, cones, and horizontal cells were recognized by their distinct morphologies. To record whole-cell currents and to introduce labeled peptides and calcium indicators into cells, a high-resistance seal was formed using a glass pipette and the patch of membrane beneath the pipette was ruptured using gentle suction. Cells were voltage-clamped using either an EPC-10 or EPC-9 amplifier (Heka Elektronik, Lambrecht/Pfalz, Germany) controlled by Pulse (InstruTech, Port Washington, NY) acquisition and recording software. For most experiments, the pipette solution contained (in mm): 120 Cs-glutamate, 4 Na2ATP, 0.5 GTP, 4 MgCl2, 10 TEA-Cl, and 0.5 EGTA, pH 7.2 with NaOH. For experiments imaging calcium entry sites, EGTA concentration was raised to 10 mm, and 100 μm Fluo-5F was added to the pipette solution. Capacitance recordings were performed on synaptic terminals, which had become isolated from the soma during the cell isolation procedure. Capacitance jumps from synaptic terminals were included in the average only if calcium currents were >75 pA and leak current at -60 mV was <30 pA.
Peptide synthesis. Peptides were synthesized and purified at the W. M. Keck facility at Yale University. Purity of synthesized peptide was confirmed by mass spectroscopy. All fluorescein- and rhodamine-labeled peptides were purified to >99.9% purity as verified by mass spectroscopy. To generate Alexa 488-labeled peptides, peptides were synthesized with an N-terminal and a C-terminal cysteine (see Fig. 1 A), and cysteines were labeled with Alexa-488 maleimide (Molecular Probes, Eugene, OR). Labeling was accomplished using a procedure modified from the Molecular Probes “Thiol-reactive probes data sheet.” Specifically, 100 μm cysteine-containing peptide was incubated in our patch pipette solution containing 1 mm 3,3′,3′′-phosphinidyne-triproprionic acid hydrochloride (to reduce cysteines) and 2 mm Alexa 488 maleimide (first prepared as 20 mm stock solution in DMSO) for 1 hr at room temperature followed by overnight at 4°C. Labeled peptides were purified by G-25 sephadex gel filtration column.
Figure 1.

Specific peptide labeling of structures at the surface of retinal bipolar cell terminals imaged by TIRFM. A, Sequences of fluorescent peptides used in this study. Bold indicates CtBP binding domain binding. B, Images of four retinal bipolar cell terminals loaded with 27.7 μm fluorescein-labeled peptide and imaged using TIRFM. C, Epifluorescence image of a bipolar cell terminal loaded with the peptide.
Measuring Kd. Assuming first order binding kinetics (Molloy et al., 1998), peptide binding is governed by the equation:
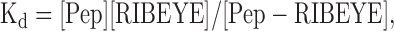 |
where [Pep] is the concentration of peptide, [RIBEYE] is the concentration of RIBEYE molecules, and [Pep-RIBEYE] is the concentration of peptide-RIBEYE complexes. Because the total number of RIBEYE molecules is likely constant over the time course of this experiment, we can rearrange Equation 1 to:
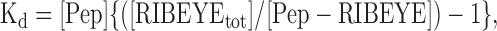 |
where [RIBEYEtot] is the total concentration of RIBEYE available for binding to the peptide. Dialyzing the cell with saturating concentrations of the peptide (415 μm), we assume that the maximum spot fluorescence equates to nearly all RIBEYE molecules bound to the peptide. This assumption seems reasonable because spot fluorescence reached maximum before cytoplasmic fluorescence. Making this assumption, Equation 2 can be rearranged to:
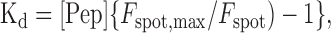 |
where Fspot is the fluorescence of the spot at concentration [Pep], and Fspot, max is the maximum spot fluorescence. This can be rearranged to:
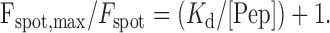 |
Equation 4 indicates that a plot of Fspot, max/Fspot as a function of [Pep]-1 should be linear with a slope equal to the Kd.
[Pep] was measured by monitoring the cytoplasmic fluorescence and assuming that the maximum fluorescence represents the concentration in the pipette. Spot fluorescence was measured as the total fluorescence of a 1 μm diameter circular region encompassing the spot, minus the local background taken from a representative nearby region. Possibly because of photobleaching, spot fluorescence was often observed to plateau and then decrease in intensity during the time course of our experiments. Fspot, max was taken as the maximum spot fluorescence measured from any particular spot.
Total internal reflection fluorescence microscopy. Total internal reflection fluorescence microscopy (TIRFM) was performed as previously described (Zenisek et al., 2002). Briefly, light from the 488 line of an argon-ion laser (MWK, San Bernadino, CA) was passed through a spatial filter and focused to the periphery of the back focal plane of a high numerical aperture (NA) objective (NA = 1.65; Apo X100 0 HR; Olympus, Tokyo, Japan) on an inverted microscope (IX-71; Olympus). Light traveling through the objective passes through immersion oil (Cargille) and coverslip (Plan Optik, Elsoff, Germany) of refractive index n488 = 1.80 where it suffers total internal reflection at the glass-cell interface. From previous measurements of the exit angle from a similar system (Zenisek et al., 2002), we expect an evanescent field with an exponential depth constant of 41-43 nm. Images were acquired using either a front-illuminated CCD camera (Orca ER 1394; Hamamatsu, Bridgewater, NJ) or a back-illuminated, on-chip amplified CCD camera (Cascade 512B; Roper Scientific) controlled by MetaMorph (Universal Imaging, West Chester, PA) acquisition and analysis software.
Confocal microscopy. Laser-scanning confocal microscopy of living neurons (bipolar cells, cones, horizontal cells, hippocampal neurons) was performed on the stage of an upright microscope (Olympus BX61WI) equipped with an Olympus FV300 confocal system. Laser scanning and image acquisition were controlled via Olympus FluoView software. Cells were loaded with fluorescent peptide via a whole-cell patch pipette. For each cell, a series of optical sections was taken at 0.5 μm intervals through the entire cell for later three-dimensional representation of the image of the cell.
Electron microscopy. A bipolar neuron was attached to an Aclar film and, following confocal imaging, was fixed with 2.5% paraformaldehyde plus 2.5% glutaraldehyde in 0.1 m phosphate buffer. A rectangular mark was etched into the Aclar around the imaged cell, and the dish was placed at 4°C overnight. Cells were then rinsed and further fixed in 1% OsO4 plus 1.5% K ferrocyanide in 0.1 m phosphate buffer, dehydrated, and embedded in Embed 812. The embedded sample was peeled from the Aclar film, glued to an Epon blank, and sectioned serially at ∼80 nm. The rectangular mark etched into the Aclar left a corresponding mark in the cured block to guide sectioning. Localization of the cell was also aided by a reference grid printed onto the Aclar substrate using a laser printer. Sequences of five to ten sections were mounted on formvar-coated slot grids, stained in methanolic uranyl acetate, and then photographed in an electron microscope (120 kV) at 10,000×.
Measuring number of peptide binding sites per ribbon. Bipolar cells were whole-cell voltage-clamped and dialyzed with an intracellular solution containing 27 μm fluoresceinated peptide for 5 min. After loading, the patch pipette was removed, and the bipolar cell terminal was imaged using confocal microscopy. Analysis was restricted to the 14 ribbons within a single cell that were spatially separated from other synaptic ribbons. To measure spot intensity, fluorescence was measured in a 6 × 7 voxel region encompassing the ribbon and in three of four regions immediately adjacent to the ribbon, which were used to estimate the fluorescence intensity of the local background within the terminal. To convert the fluorescence units into the number of molecules, we needed to measure the fluorescence at a known dye concentration over a known volume. To do this, we measured the fluorescence within a patch pipette containing 27 μm fluoresceinated peptide at a location where the pipette diameter approximated that of a bipolar cell terminal (diameter, 11 μm). At this location, a 6 × 7 voxel rectangular region had a fluorescence of 155,500 U. To measure the volume of this region, we needed to estimate the thickness of an optical section under our conditions. To estimate this, we took a series of z-sections through a 27 nm FluoSphere bead (Molecular Probes) and monitored the fluorescence as a function of vertical distance. Because this bead is much smaller than the expected thickness of a single section, we used this bead to map out the section thickness. This gave an average section thickness of 1.6 μm. In the x-y plane, our region sampled an area of 0.73 μm2; hence, our region recorded from a volume of 1.168 μm3. At 27 μm, a 1.168 μm3 region contains 19,500 molecules.
Results
Fluorescent peptide labels spots in live synaptic terminals
In vitro, the affinity of CtBP for short peptides containing the PXDLS motif has a dissociation constant between 1-100 μm, depending on the exact sequence (Molloy et al., 1998, 2001). If this affinity were retained by RIBEYE, it might be exploited to label ribbons in live cells. Accordingly, we synthesized a 14 amino acid peptide containing the CtBP-binding sequence and labeled it with fluorescein, Alexa-488, or rhodamine (Fig. 1) (see Materials and Methods). The specific sequence chosen has an affinity of 1.5 μm for CtBP (Molloy et al., 1998). We introduced the peptide into a bipolar cell through a patch pipette and then imaged the cell by TIRFM. This method images the region of the cell membrane nearest a glass-water interface.
Figure 1B shows examples from four different cells loaded with the fluorescein-labeled peptide and imaged by TIRFM. As expected for ribbon labeling, fluorescent spots were readily observable in the synaptic terminal. Across 21 cells, spots in the synaptic terminal were seen at an average density of 0.12 ± 0.03 μm-2, similar to the density of calcium entry sites (0.11 ± 0.02 μm-2) and to the density of synaptic ribbons measured with immunostaining for CtBP (0.10 ± 0.01 μm-2) (Zenisek et al., 2003). It should be noted that 0.12 spots/μm2 predicts 38 ribbons per 10-μm-diameter terminal, somewhat less than the number of ribbons observed in EM serial reconstructions of retinal bipolar cell terminals (∼40-60) (von Gersdorff et al., 1996; Holt et al., 2004), likely because of the difficulty in resolving closely spaced synaptic ribbons at the light level (Zenisek et al., 2002; see also below).
The bipolar cell soma dialyzed with CtBP-binding peptide and imaged with TIRFM exhibited no spots (n = 6; see also below), nor did bipolar terminals dialyzed with a scrambled peptide (n = 4; data not shown). Furthermore, when imaged by epifluorescence, spots tended to be at the cell surface (Fig. 1C). In most bipolar cells dialyzed with the ribbon-binding peptide and imaged by TIRFM, spots remained visible while the cell remained alive. Thus, most spots remain near the plasma membrane. We tracked the movements parallel to the coverslip for 23 spots across 12 cells. Figure 2A plots the lateral displacement of these spots as a function of time, with each spot represented by a different color and symbol. The spots, although not perfectly stationary, were relatively immobile. To estimate the speed of movement, a straight line was fitted to the displacement of each spot by a least-squares criterion, and the slope of the line was taken as the average speed of the spot. The speed of movement ranged from 0.35 to 13.5 nm/sec (Fig. 2B). The median and mean speeds were 0.711 and 1.34 nm/sec, respectively. Although most spots were relatively immobile, two spots were observed to disappear from the membrane during the time course of the experiment, likely representing the displacement of the spot from the surface membrane into the interior of the cell. One such event is shown in Figure 2, C and D (supplemental material, available at www.jneurosci.org).
Figure 2.

Ribbon displacement in the synaptic terminal. A, Ribbons were imaged using TRFM microscopy and tracked using a software tracking algorithm (MetaMorph; Universal Imaging) and their displacement from the location where they were first visible plotted as a function of time. In general, displacement increased with time. B, To estimate the speed of ribbon movement, the plot displacement as a function of time was best least-squares fit to a straight line through the origin, and the slopes of those lines are plotted as a histogram. Although ribbons were relatively immobile on the time scale of our experiments, ribbons were twice observed to make comparatively rapid movements in bipolar cells. C, An example of a bipolar cell with a ribbon undergoing a rapid movement. Images are consecutive frames taken in 30 sec intervals. D, Same as in C, except at higher magnification.
We next estimated the affinity of the peptide for these spots. Dialyzing a synaptic terminal with a saturating concentration of the fluorescent peptide via a patch pipette placed on the synaptic terminal, we measured the fluorescence of the spot as the peptide gradually diffused into the cell. After “break-in,” cytoplasmic fluorescence rose over several minutes (Fig. 3A, inset). Assuming that the asymptotic cytoplasmic fluorescence intensity was equivalent to the patch pipette concentration, we estimated the intracellular peptide concentration (Fig. 3, [peptide]) at any given time point by comparing the cytoplasmic fluorescence at that time to the asymptotic fluorescence (Fig. 3A, inset). To estimate the Kd for peptide binding to the ribbon, we used the relationship in Equation 4 (see Materials and Methods), which indicates that the ratio of spot fluorescence at saturation (Fspot, max) to the spot fluorescence (Fspot) should be linearly related to the inverse of peptide concentration ([Pep]-1) with slope equal to the Kd. Figure 3A shows Fspot, max/Fmax versus peptide concentration plotted for six cells loaded with 415 μm fluoresceinated peptide. From Figure 3A, it is apparent that this concentration sufficed to saturate binding to the spots. Figure 3B shows [Pep]-1 as a function of Fspot, max/Fspot. From this plot, we measured a Kd of 27 μm.
Figure 3.

Measurements of peptide affinity for spots. Bipolar cells were loaded with 415 μm fluorescein-labeled peptide and imaged using TIRFM. The fluorescence intensity of spots were measured (Fspot) and normalized to the maximum fluorescence for that spot (Fspot, max) over the time course of cell loading. The fluorescence of the cytoplasm outside the fluorescent spots was taken as an index of cytosolic peptide concentration. A, Fspot/Fspot, max is plotted as a function of peptide concentration ([peptide]) within the cell. Inset shows one example of a plot of fluorescence as a function of time for a spot (open circles) and in the cytoplasm (filled squares). The right axis in the inset shows the conversion of cytoplasmic fluorescence to peptide concentration, assuming that the asymptotic concentration is the same as the pipette concentration. Note that fluorescence of the spot saturates sooner than the cytoplasmic fluorescence. B, Plot of Fspot, max/Fspot versus [peptide]-1. First-order binding predicts a linear relationship with a slope equal to the Kd (see Materials and Methods). Slope of linear least-squares fit indicates a Kd of 27 μm.
This calculation assumes that binding to the ribbon equilibrates quickly relative to the speed of peptide loading, which seems reasonable given the slow rise of cytoplasmic fluorescence (Fig. 3A, inset). However, if this assumption were false, then our estimate of Kd should be sensitive to loading speed. To test for this possibility, we loaded three cells with the peptide using a patch pipette placed on the cell body, which retards loading of the terminal because of the interposed axon. Using this configuration, we estimated Kd = 32 μm, which is similar to the affinity obtained with the pipette placed on the terminal (27 μm). So, we conclude that binding of the peptide to the ribbon is rapid on the relatively slow time scale of dialysis.
Peptide-labeled structures are confined to ribbon-type synaptic terminals
If the labeled structures in the bipolar cell actually represent ribbons, then they should be present only in the terminal but not the soma. Also, fluorescent spots should be present only in cells that make ribbon synapses, such as photoreceptors and bipolar neurons, but not in cells that lack ribbons, such as horizontal cells. To test this, we again dialyzed with fluorescent peptide, but switched from TIRFM, which reveals only the region where a cell contacts the substrate, to laser-scanning confocal microscopy, which provides an overview of labeling throughout the cell.
The confocal results were congruent with results from TIRFM: bright spots were observed in giant synaptic terminals of bipolar cells (Fig. 4A), which contain ∼40-60 separate ribbons, based on direct counts in three terminals reconstructed from serial electron micrographs (two in von Gersdorff et al., 1996; one in this paper, see below) and on an estimate from ribbon density in individual thin sections (Holt et al., 2004). In agreement with the known number of ribbons, we counted 31-75 distinct spots in the synaptic terminals of ten bipolar cells dialyzed with the peptide (51 ± 13; mean ± SD), but no labeled structures were observed in the cell body or axon. So, the location and number of labeled structures is consistent with specific labeling of bipolar cell ribbons by the peptide.
Figure 4.

Selective peptide-labeling of structures in the synaptic terminal of ribbon-containing neurons. Each picture shows planar projections of confocal optical sections through the entire depth of the cell. A, Bipolar cell. B, Cone photoreceptor. C, Horizontal cell. D, Cultured hippocampal neuron. The cell body of the labeled hippocampal neuron was above the field of view, and a labeled process followed the principal process of a nearby unlabeled cell, the position of which is indicated by the outline.
The terminals of cone photoreceptors also contain synaptic ribbons, which are larger and fewer than in bipolar cells. Figure 4B shows a labeled cone with one large, bright structure in the synaptic terminal but only uniform fluorescence throughout the rest of the cell (axon, cell body, and outer segment). Thus, the labeling in cones is again consistent with concentration of the peptide specifically at ribbons. Movies showing three-dimensional labeling patterns in cone and bipolar cell terminals are available as supplemental materials (supplemental material, available at www.jneurosci.org).
Horizontal cells are retinal interneurons that lack ribbon-type active zones and therefore should not exhibit bright fluorescent spots. Indeed, as shown in Figure 4C, horizontal cells exhibited only diffuse, dim fluorescence after dialysis with the fluorescent peptide, as expected if the peptide is specific for ribbons. But, horizontal cells also lack conventional synapses, so to establish if spots of bright fluorescence would be observed at conventional synapses, we dialyzed cultured hippocampal neurons with fluorescent peptide and examined boutons at contact points with other nearby cultured neurons. Figure 4D shows that the processes and en passant boutons of hippocampal neurons were uniformly labeled with the fluorescent peptide, like horizontal cells. The presence of intensely labeled spots in bipolar and photoreceptor terminals and their absence in horizontal cells and hippocampal neurons further confirms the specificity of the peptide for synaptic ribbons.
Electron microscopy confirms that labeled spots correspond to synaptic ribbons
To directly determine if labeled spots observed in living cells actually correspond to synaptic ribbons, we turned to electron microscopy. A bipolar cell was filled with labeled peptide via a whole-cell patch pipette, and confocal optical sections were collected covering the entire depth of the cell. The cell was then fixed, and ultrathin sections were taken serially parallel to the substrate (i.e., in nearly the same plane as the confocal optical sections). We used landmarks such as the contact point with the substrate and the insertion point of the axon to aid in establishing correspondence between the optical sections and the electron micrographs. Figure 5 shows examples of spatial correspondence between labeled structures in the living synaptic terminal and synaptic ribbons in electron micrographs from the same terminal.
Figure 5.
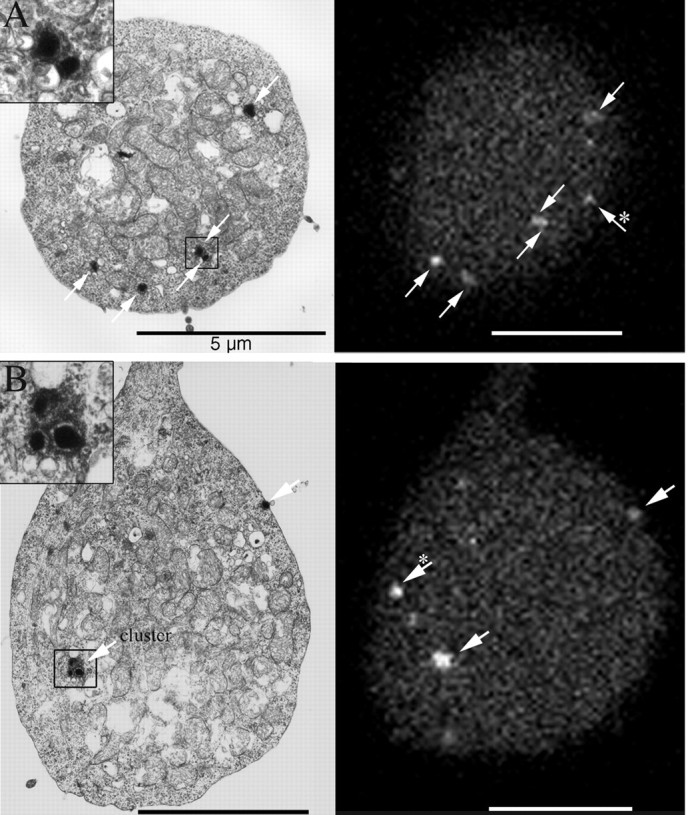
Spatial correspondence of labeled structures in confocal optical sections with ribbons in electron micrographs of the same synaptic terminal. Arrows indicate fluorescent spots and corresponding ribbons. The insets show close-up views of the ribbon clusters indicated by boxes. Clusters were rare and were only observed within the interior of the terminal of the one cell shown here. Asterisks indicate fluorescent spots that corresponded to ribbons in nearby semithin sections.
In one synaptic terminal that was completely reconstructed, electron microscopy revealed 68 synaptic ribbons. Most ribbons were spatially isolated from other ribbons, but 21 ribbons found within the interior of the terminal were grouped in eight clusters, each containing two to five ribbons (Fig. 5). Because the clusters were counted as single spots in the optical sections, the electron microscopy data led us to expect 55 labeled spots in the fluorescence images. In that terminal, confocal microscopy of the living cell showed 47 labeled spots, somewhat fewer than expected. The difference may reflect the signal-to-noise ratio of the fluorescence images. In two other synaptic terminals that were partially reconstructed, there were 40 and 17 ribbons, none of which occurred in clusters. In confocal optical sections, the corresponding regions of these terminals exhibited 39 and 16 labeled spots, in good agreement with the expected numbers.
Number of peptide binding sites per ribbon
By measuring the intensity of the spots visualized using confocal microscopy and taking into account the affinity of the peptide for its binding site, we could estimate the number of peptide binding sites per ribbon and thus, the number of RIBEYE molecules. We restricted the analysis to ribbons that were spatially separated from other spots and measured the fluorescent spot intensity after subtracting the local cytoplasmic background. In a bipolar cell dialyzed with 27 μm fluoresceinated peptide, 14 synaptic ribbons had an average fluorescence of 16,000 ± 1090 U above background. An 11 μm section of a patch pipette loaded with the same concentration of fluorescent peptide yielded an average fluorescence intensity of 132,700 U/μm3 (see Materials and Methods), a volume containing ∼16,700 peptide molecules; hence, we determined that a ribbon bound 2000 ± 130 peptide molecules to RIBEYE. From our measured Kd for ribbon-peptide binding, we expect ∼50% of the binding sites to be occupied in the presence of 27 μm peptide; thus, a bipolar cell ribbon expresses ∼4000 binding sites. Electron microscopic evaluation of ribbons in the same cell indicated that they were approximately spherical with a diameter of 133 ± 4 nm (n = 41) or a volume of 764,000 nm3. Because crystallography indicates that each CtBP molecule binds one peptide (Nardini et al., 2003), each binding site measured here likely represents one RIBEYE molecule. Therefore, RIBEYE is found at a density of 0.005 molecules/nm3 or 549 Da/nm3. By comparison, if the ribbon were pure RIBEYE its concentration would be 790-860 Da/nm3 (Harpaz et al., 1994). Hence, RIBEYE is a major constituent of the ribbon.
Peptide labeling does not affect ribbon function
Close inspection of electron micrographs showed that ribbons exposed to saturating concentrations of the peptide were densely studded with synaptic vesicles (Fig. 5A,B, insets). Therefore, peptide binding did not prevent vesicles from attaching to the ribbon. Furthermore, capacitance responses and calcium current evoked by depolarization were normal. In seven dissociated terminals dialyzed with 415 μm peptide, depolarizing to 0 mV for 10 and 250 msec elicited average capacitance jumps of 41 ± 11 and 114 ± 30 fF, respectively, 1-2 min after achieving the whole-cell configuration. We also assayed the degree of recovery from paired-pulse depression. In terminals dialyzed with the peptide, when the membrane was twice depolarized to 0 mV for 250 msec, with five seconds intervening, the second capacitance jump was 48.0 ± 9% as large as the first. Similarly, when the membrane was twice depolarized to 0 mV for 10 msec, the second capacitance jump was 43.7 ± 8.6% as large as the first. These results resemble previous reports (von Gersdorff and Matthews, 1994; Mennerick and Matthews, 1996; Neves and Lagnado, 1999).
Peptide labeling colocalizes with calcium entry sites
When a terminal is loaded with Fluo-5f calcium indicator, plus a high concentration of EGTA, and then depolarized, TIRFM can image the calcium entry sites (Zenisek et al., 2003). These sites are predicted to colocalize with ribbons because they show similar distributions (see also Roberts et al., 1990) and also because immunostaining for calcium channels colocalizes with ribbon markers (Berntson et al., 2003; Claes et al., 2004). The calcium indicator itself does not generate fluorescent spots when calcium channels are closed (Zenisek et al., 2003) as found for certain hair cells (Issa and Hudspeth, 1996). Consequently, we could visualize ribbons (with the peptide) and calcium entry sites (with indicator dye) to directly compare their locations in the living terminal.
Figure 6, A and C, shows fluorescent spots at resting potential. Control experiments with peptide alone showed no fluorescence rise during voltage steps (n = 15 cells; data not shown). Thus, the fluorescence at rest marks the location of ribbons. During the voltage step, fluorescence rose at the calcium channel clusters because of binding with the indicator (Fig. 6B). Figure 6D shows the average fluorescence rise during depolarization. This figure was generated by subtracting the average fluorescence during a step to 0 mV (Fig. 6B) from the average fluorescence before and after the depolarization (Fig. 6A,C). Thus, Figure 6D indicates the locations of calcium entry. Figure 6E, showing fluorescence versus time for this cell (in the same region as Fig. 6A), illustrates that fluorescence changes rapidly near the ribbons, but not beyond them.
Figure 6.

Ribbons colocalize with calcium entry sites. Bipolar cells were loaded with the fluorescent CtBP-binding peptide (28 μm), Fluo-5f (100 μm), and 10 mm EGTA and imaged using TIRFM. TIRFM images the region of the cell that is adherent to the glass, and in this example the cell adhered in two locations. A, Fluorescence collected and averaged for 250 msec just before a depolarization to 0 mV. B, Fluorescence collected and averaged during a 250 msec depolarization to 0 mV. C, Fluorescence collected for 250 msec immediately after return of the membrane potential to its resting potential of -60 mV. D, Image of B subtracted by the average of the images in A and C. E, Top shows fluorescence in regions marked in A. Note the rapid change in fluorescence at locations near spots seen at rest but not at other locations within the terminal.
In eight cells with calcium currents >50 pA, 21 of the 23 fluorescence spots observed at resting membrane potential colocalized with calcium entry sites. Conversely, 21 of the 22 spots that were identified as calcium entry sites colocalized with fluorescent spots. Thus, in living bipolar terminals, calcium entry sites closely colocalize with synaptic ribbons.
Discussion
Our key finding is that a short peptide with affinity for a binding domain in the CtBP subunit of RIBEYE labels synaptic ribbons in living cells. The evidence for this interaction is as follows: (1) peptide concentrates in ribbon-type synapses, showing bright puncta beneath the plasma membrane; (2) scrambled peptide does not show puncta, nor does the functional peptide show them in cell types that lack ribbons; (3) puncta at the resemble in size, location, number, and overall distribution the ribbons described at the light level in previous studies; (4) puncta mapped in the live terminal by confocal microscopy were subsequently identified as ribbons by electron microscopy of serial ultrathin sections from the same cell.
Implications for ribbon function
Because the PXDLS motif is widely used by proteins that interact with CtBP, it might mediate tethering of vesicles to the ribbon (Schmitz et al., 2000). If so, a saturating concentration of peptide containing this motif should compete with the endogenous binding partners of RIBEYE and block tethering. Similar experiments in vitro show that peptides containing PXDLS effectively compete with adenovirus E1A protein (Molloy et al., 1998). However, in our experiments, binding of peptide to ribbons did not affect the concentration of vesicles at the ribbon and did not noticeably alter either the amount of exocytosis or recovery from paired-pulse depression. Thus, the PXDLS binding motif of CtBP likely has some role at the ribbon other than vesicle tethering and mobilization.
Because the ribbon was proposed as an organelle for vesicle priming, a key question has been whether it is required for exocytosis. Recent studies using TIRFM to visualize synaptic vesicles in the bipolar terminal identify specific active zones, where most fusion events occur, and “outlier” fusion events beyond the active zones (Zenisek et al., 2000, 2003). Although this implied that some vesicles fuse without the ribbons, it seemed possible that outlier fusions arose from mobile ribbons. The present results show that ribbons are virtually stationary over the time scale of those experiments (<5 min), indicating that outlier fusions probably occur without help from a ribbon.
Ribbons and calcium entry
The colocalization between ribbons and sites of voltage-gated calcium entry has been demonstrated in live hair cells (Issa and Hudspeth, 1996). However, evidence for this relationship at live ribbon-type synapses of the retina has been indirect: (1) bipolar cell exocytosis requires calcium concentrations that are only achieved very near the calcium channels (Heidelberger et al., 1994), implying that they should colocalize; (2) the number of calcium entry sites identified in live cells strikingly resemble the number of active zones and ribbons (Zenisek et al., 2003); (3) cell-attached recordings of membrane capacitance demonstrate a tight relationship between calcium entry, calcium-activated potassium channels, and vesicle release sites (Llobet et al., 2003). Our results now confirm these findings by directly demonstrating in live bipolar neurons that, to submicron resolution, calcium channels and ribbon sites colocalize.
RIBEYE number in the bipolar cell ribbon
The fluorescent peptide allowed us to estimate that a ribbon contains ∼4000 PXDLS binding sites. Assuming the specific volume of RIBEYE is similar to that of a typical globular protein (0.73 ml/gm; Harpaz et al., 1994), 4000 RIBEYE molecules (molecular weight = 120 kDa; Schmitz et al., 2000) contribute an estimated 67% of the total ribbon volume. This value may represent an underestimate of the number of RIBEYE molecules per ribbon, because (1) we cannot be certain that the peptide concentration within the cell reached the pipette concentration during the time course of our experiments; (2) some RIBEYE molecules may have their binding sites obscured by other endogenous proteins possibly containing the consensus sequence for RIBEYE binding, and (3) confocal serial sections may have caused some photobleaching of ribbon-bound peptide. Nevertheless, our results indicate that RIBEYE is the major structural component of the synaptic ribbon.
Footnotes
This work was supported by National Institutes of Health Grants EY014990 (D.Z., C.M.), EY00828 (P.S.), and EY03821 (G.M.) and scholar awards from the McKnight and Kinship Foundations (D.Z.). We thank Jian Li for electron microscopy. We thank Elizabeth Prescott and Michael Coggins for critical reading of this manuscript.
Correspondence should be addressed to David Zenisek, Department of Cellular and Molecular Physiology, Yale University School of Medicine, 333 Cedar Street, New Haven, CT 06520. E-mail: david.zenisek@yale.edu.
Copyright © 2004 Society for Neuroscience 0270-6474/04/249752-08$15.00/0
References
- Berntson A, Taylor WR, Morgans CW (2003) Molecular identity, synaptic localization, and physiology of calcium channels in retinal bipolar cells. J Neurosci Res 71: 146-151. [DOI] [PubMed] [Google Scholar]
- Boyd JM, Subramanian T, Schaeper U, La Regina M, Bayley S, Chinnadurai G (1993) A region in the C-terminus of adenovirus 2/5 E1a protein is required for association with a cellular phosphoprotein and important for the negative modulation of T24-ras mediated transformation, tumorigenesis and metastasis. EMBO J 12: 469-478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chinnadurai G (2002) CtBP, an unconventional transcriptional corepressor in development and oncogenesis. Mol Cell 9: 213-224. [DOI] [PubMed] [Google Scholar]
- Claes E, Seeliger M, Michalakis S, Biel M, Humphries P, Haverkamp S (2004) Morphological characterization of the retina of the CNGA3(-/-)Rho(-/-) mutant mouse lacking functional cones and rods. Invest Ophthalmol Vis Sci 45: 2039-2048. [DOI] [PubMed] [Google Scholar]
- de Ruyter van Steveninck RR, Laughlin SB (1996) (1996) The rate of information transfer at graded-potential synapses. Nature 379: 642-645. [Google Scholar]
- Dick O, Hack I, Altrock WD, Garner CC, Gundelfinger ED, Brandstatter JH (2001) Localization of the presynaptic cytomatrix protein Piccolo at ribbon and conventional synapses in the rat retina: comparison with Bassoon. J Comp Neurol 439: 224-234. [DOI] [PubMed] [Google Scholar]
- Dick O, tom Dieck S, Altrock WD, Ammermuller J, Weiler R, Garner CC, Gundelfinger ED, Brandstatter JH (2003) The presynaptic active zone protein bassoon is essential for photoreceptor ribbon synapse formation in the retina. Neuron 37: 775-786. [DOI] [PubMed] [Google Scholar]
- Harpaz Y, Gerstein M, Chothia C (1994) Volume changes on protein folding. Structure 2: 641-649. [DOI] [PubMed] [Google Scholar]
- Heidelberger R, Matthews G (1992) Calcium influx and calcium current in single synaptic terminals of goldfish retinal bipolar neurons. J Physiol (Lond) 447: 235-256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heidelberger R, Heinemann C, Neher E, Matthews G (1994) Calcium dependence of the rate of exocytosis in a synaptic terminal. Nature 371: 513-515. [DOI] [PubMed] [Google Scholar]
- Holt M, Cooke A, Neef A, Lagnado L (2004) High mobility of vesicles supports continuous exocytosis at a ribbon synapse. Curr Biol 14: 173-183. [DOI] [PubMed] [Google Scholar]
- Issa NP, Hudspeth AJ (1996) Characterization of fluo-3 labelling of dense bodies at the hair cell's presynaptic active zone. J Neurocytol 25: 257-266. [DOI] [PubMed] [Google Scholar]
- Llobet A, Cooke A, Lagnado L (2003) Exocytosis at the ribbon synapse of retinal bipolar cells studied in patches of presynaptic membrane. J Neurosci 23: 2706-2714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mennerick S, Matthews G (1996) Ultrafast exocytosis elicited by calcium current in synaptic terminals of retinal bipolar neurons. Neuron 17: 1241-1249. [DOI] [PubMed] [Google Scholar]
- Molloy DP, Milner AE, Yakub IK, Chinnadurai G, Gallimore PH, Grand RJ (1998) Structural determinants present in the C-terminal binding protein binding site of adenovirus early region 1A proteins. J Biol Chem 273: 20867-20876. [DOI] [PubMed] [Google Scholar]
- Molloy DP, Barral PM, Bremner KH, Gallimore PH, Grand RJ (2001) Structural determinants outside the PXDLS sequence affect the interaction of adenovirus E1A, C-terminal interacting protein and Drosophila repressors with C-terminal binding protein. Biochim Biophys Acta 1546: 55-70. [DOI] [PubMed] [Google Scholar]
- Muresan V, Lyass A, Schnapp BJ (1999) The kinesin motor KIF3A is a component of the presynaptic ribbon in vertebrate photoreceptors. J Neurosci 19: 1027-1037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nardini M, Spano S, Cericola C, Pesce A, Massaro A, Millo E, Luini A, Corda D, Bolognesi M (2003) CtBP/BARS: a dual-function protein involved in transcription co-repression and Golgi membrane fission. EMBO J 22: 3122-3130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neves G, Lagnado L (1999) The kinetics of exocytosis and endocytosis in the synaptic terminal of goldfish retinal bipolar cells. J Physiol (Lond) 515: 181-202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts WM, Jacobs RA, Hudspeth AJ (1990) Colocalization of ion channels involved in frequency selectivity and synaptic transmission at presynaptic active zones of hair cells. J Neurosci 10: 3664-3684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaeper U, Boyd JM, Verma S, Uhlmann E, Subramanian T, Chinnadurai G (1995) Molecular cloning and characterization of a cellular phosphoprotein that interacts with a conserved C-terminal domain of adenovirus E1A involved in negative modulation of oncogenic transformation. Proc Natl Acad Sci USA 92: 10467-10471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmitz F, Konigstorfer A, Sudhof TC (2000) RIBEYE, a component of synaptic ribbons: a protein's journey through evolution provides insight into synaptic ribbon function. Neuron 28: 857-872. [DOI] [PubMed] [Google Scholar]
- Vo N, Fjeld C, Goodman RH (2001) Acetylation of nuclear hormone receptor-interacting protein RIP140 regulates binding of the transcriptional corepressor CtBP. Mol Cell Biol 21: 6181-6188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- von Gersdorff H, Matthews G (1994) Dynamics of synaptic vesicle fusion and membrane retrieval in synaptic terminals. Nature 367: 735-739. [DOI] [PubMed] [Google Scholar]
- von Gersdorff H, Vardi E, Matthews G, Sterling P (1996) Evidence that vesicles on the synaptic ribbon of retinal bipolar neurons can be rapidly released. Neuron 16: 1221-1227. [DOI] [PubMed] [Google Scholar]
- Wang Y, Okamoto M, Schmitz F, Hofmann K, Sudhof TC (1997) Rim is a putative Rab3 effector in regulating synaptic-vesicle fusion. Nature 388: 593-598. [DOI] [PubMed] [Google Scholar]
- Zenisek D, Steyer JA, Almers W (2000) Transport, capture and exocytosis of single synaptic vesicles at active zones. Nature 406: 849-854. [DOI] [PubMed] [Google Scholar]
- Zenisek D, Steyer JA, Feldman ME, Almers W (2002) A membrane marker leaves synaptic vesicles in milliseconds after exocytosis in retinal bipolar cells. Neuron 35: 1085-1097. [DOI] [PubMed] [Google Scholar]
- Zenisek D, Davila V, Wan L, Almers W (2003) Imaging calcium entry sites and ribbon structures in two presynaptic cells. J Neurosci 23: 2538-2548. [DOI] [PMC free article] [PubMed] [Google Scholar]