

Abstract
N-type calcium channels are modulated by acute and chronic ethanol exposure in vitro at concentrations known to affect humans, but it is not known whether N-type channels are important for behavioral responses to ethanol in vivo. Here, we show that in mice lacking functional N-type calcium channels, voluntary ethanol consumption is reduced and place preference is developed only at a low dose of ethanol. The hypnotic effects of ethanol are also substantially diminished, whereas ethanol-induced ataxia is mildly increased. These results demonstrate that N-type calcium channels modulate acute responses to ethanol and are important mediators of ethanol reward and preference.
Keywords: alcoholism, calcium channel, conotoxin, conditioned place preference, ethanol preference, knock-out
Introduction
Voltage-gated calcium channels (VGCCs) are multimeric protein complexes that mediate Ca2+ entry into neurons in response to changes in membrane potential and regulate neuronal excitability, neurotransmitter release, and gene expression (Dunlap et al., 1995; Ghosh and Greenberg, 1995). They have been classified as L, N, P/Q, R, and T type according to their electrophysiological and pharmacological properties (Dunlap et al., 1995). The principal defined role for N-type calcium channels, along with P/Q-type channels, is in the regulation of evoked neurotransmitter release (Reid et al., 2003). The function of N-type channels is inhibited by βγ subunits of heterotrimeric G-proteins (Feng et al., 2001). Accordingly, N-type channels are regulated by several neuropeptides, neuromodulators, and neurotransmitters that bind to G-protein-coupled receptors, such as opioids, cannabinoids, and dopamine (Twitchell et al., 1997; Momiyama and Koga, 2001; Kitamura et al., 2002).
There has been extensive research into the role of L-type calcium channels in mediating the behavioral effects of ethanol, driven primarily by the widespread availability of high-affinity antagonists for these channels (Little, 1995). For example, dihydropyridine nimodipine inhibits both the acquisition and expression of ethanol self-administration (De Beun et al., 1996; Kuzmin et al., 1999) and prevents the inhibitory effects of ethanol on memory in radial arm maze and object recognition tests (Brooks et al., 2002). L-type channel antagonists also reduce alcohol withdrawal seizures in rodents (Watson and Little, 1999).
In addition to effects on L-type channels, ethanol inhibits N- and T-type calcium channels in neural cells (Walter and Messing, 1999). In PC12 cells, ethanol inhibits N-type channels (Solem et al., 1997), and chronic ethanol exposure increases the density of binding sites for the N-type channel antagonist ω-conotoxin GVIA in PC12 cells and in the frontal cortex and hippocampus of mice (McMahon et al., 2000).
Despite the cellular and molecular data indicating that ethanol modulates N-type calcium channels, it is not known whether this interaction contributes to the behavioral effects of ethanol. Currently, available peptide inhibitors of N-type calcium channels, such as ω-conotoxin GVIA, have poor blood-brain barrier permeability and produce tremors and motor impairment when injected intrathecally into mice, limiting their use in behavioral experiments (Olivera et al., 1994). Thus, we chose a genetic approach by studying ethanol responses in mice that carry a null mutation in the calcium channel subunit Cav2.2 and therefore lack functional N-type calcium channels (Kim et al., 2001). Cav2.2 null mice develop normally with no overt phenotypic abnormalities. In dorsal root ganglion cultures from Cav2.2 null mice, the level of L-, P/Q-, and R-type calcium channel currents are unchanged compared with wild-type neurons, indicating a selective absence of N-type channel activity that does not alter the function of remaining VGCCs (Kim et al., 2001).
Here, we report that mice lacking N-type calcium channels show altered ethanol conditioned place preference and taste aversion, reduced ethanol consumption, and altered ataxic and hypnotic responses to acute ethanol administration. Our findings indicate an important role for N-type channels in the reinforcing and rewarding properties of ethanol.
Materials and Methods
Mice. The generation of mice lacking Cav2.2 by homologous recombination has been described previously (Kim et al., 2001). Briefly, a targeting vector was created by replacing part of the IS3 region of the mouse Cav2.2 gene with a green fluorescent protein-Neo cassette. J1 embryonic stem cells containing the mutation were identified and injected into C57BL/6J host blastocysts to generate chimeras, which were then bred with C57BL/6J females to detect germ-line transmission of the null allele. Chimeras were crossed with 129/SvJae mice to generate an inbred line of mice carrying the null mutation. Chimeras were also backcrossed for 10 generations with C57BL/6J mice to generate a line of N10 C57BL/6 mice carrying the null mutation. Wild-type and null mice used in the experiments described here were F1 generation hybrids (50% C57BL/6J and 50%129/SvJae). The genetic background of these mice was homogenous because they were generated by crossing heterozygous inbred 129/SvJae mice with heterozygous N10 C57BL/6 mice. The phenotype we observed with these hybrid mice was confirmed in some experiments using mutant and wild-type mice on the inbred 129/SvJae background, because the phenotype of mutant mice can vary with genetic background (Bowers et al., 1999).
Genotyping of pups was performed by PCR from a tail sample at 8-12 d of age (Kim et al., 2001). Mutant and wild-type mice were housed together in standard Plexiglas cages with food and water available ad libitum. The colony room was maintained on a 12 hr light/dark cycle with lights on at 6:00 A.M. Mice were between the ages of 2 and 6 months at the time of testing. Animal care and handling procedures were in accordance with institutional and National Institutes of Health guidelines. All experiments were performed with male mice. Naive mice were used for two-bottle choice drinking, place preference, taste aversion, and rotarod tests. Mice for loss-of-righting reflex assays were the same as those used in voluntary ethanol drinking experiments, with a 3 week gap between their final day of ethanol consumption and loss-of-righting reflex testing.
Loss of righting reflex. Mice were given an intraperitoneal injection of ethanol (3.6 gm/kg in 0.9 w/v saline) and then assessed for loss of the righting reflex, which was defined as a mouse being unable to right itself three times within 30 sec. After losing the righting reflex, mice were placed on their backs, and the time taken for them to regain the righting reflex was measured. A mouse was said to have regained the righting reflex when it was able to right itself three times within 30 sec. A blood sample was taken from the tail of each mouse when it regained the righting reflex to determine blood ethanol concentration. Blood was centrifuged at 10,000 × g, and serum blood alcohol concentration was determined using an Analox AM-1 analyzer (Analox Instruments, Luneburg, MA).
Ethanol-induced loss-of-righting reflex threshold. We determined the threshold concentration of ethanol required to induce a loss of the righting reflex using the “up and down” method (Dixon, 1965; Findlay et al., 2002). A mouse was given an intraperitoneal injection of 10% v/v ethanol in saline and 5 min later tested for a loss of righting reflex >1 min in duration. If the first mouse lost the righting reflex, then the next mouse was given a lower ethanol dose. If the first mouse did not lose the righting reflex, then the next mouse was given a higher dose. The log dose interval was 0.0138, corresponding to an ∼0.1 gm/kg difference between doses. The ED50 value was determined as described previously (Findlay et al., 2002), with 95% confidence intervals determined by the following equation: 95%CI = dosing increment × square root of (2/n) × 1.96, where n is the last n trials (6) and 1.96 reflects the 0.05 α level (Dixon, 1965).
Alcohol clearance. Mice were given intraperitoneal injections of 4 gm/kg ethanol in 0.9% (w/v) saline. A 20 μl blood sample was taken from the tail at 10, 30, 60, 120, and 180 min after the ethanol injection to determine blood ethanol concentrations.
Rotarod. Mice were trained to remain for 180 sec on a rotarod apparatus (Ugo Basile Biological Research, Varese, Italy) rotating at a fixed rate of 20 rpm. The first test session was performed the following day. During a test session, each mouse was placed on the rotarod, and the time taken for the animal to fall off the rod was recorded. Mice were then given intraperitoneal injections of ethanol and retested every 15 min for 1 hr. Mice were tested on a fixed-rate rotarod after 1.5 gm/kg ethanol in the first test session, then 2 gm/kg ethanol 3 d later. After an additional 3 d, they were tested on an accelerating rotarod (4-40 rpm over 300 sec) after administration of 2 gm/kg ethanol.
Preference drinking. Baseline fluid consumption was monitored by allowing the animals access to two bottles, both containing water only, for 4 d before the experiment.
Mice were then allowed ad libitum access to two drinking bottles, one containing ethanol solution and the other containing water. Bottles and mice were weighed every 2 d to monitor consumption. The side of the cage on which the ethanol bottle was presented (left vs right) was switched every 2 d. Ethanol concentration was increased every 4 d. Data were compared by two-way ANOVA with a repeated measure for ethanol concentration and a factor for genotype. To control for possible differences in taste preference, the same protocol was followed for saccharin and quinine solutions, except that mice were allowed 2 d access to each concentration, and the side on which the tastant was presented was switched daily. The same mice that had been used to assess voluntary ethanol consumption were used in this control experiment. Ethanol, quinine, and saccharin solutions were made in tap water.
Place conditioning. Conditioning was performed using an apparatus consisting of different colored (black vs white) test chambers separated by a central gray access chamber (Med Associates, St. Albans, VT). Floor textures (black side with rod floor vs white side with grid floor) were also different between test chambers. Before conditioning, mice were habituated to the apparatus by allowing them ad libitum access to all chambers for 30 min. Both wild-type and CaV2.2 null mice spent more time in the black chamber compared with the white chamber. Of the total time spent in the two conditioning chambers, wild-type mice spent 57% and null mice spent 53% in the black side during the habituation session. Because the magnitude of this preference for the black side was small and was not significantly different between the two genotypes (p = 0.089; two-tailed t test), we used an unbiased protocol for conditioning (Risinger et al., 2001; Dickinson et al., 2003). Mice were conditioned to ethanol (1.2, 2.0, or 2.8 gm/kg v/v in 0.9% saline, i.p.) in one test chamber and saline in the other. The ethanol-paired chamber was assigned randomly across subjects. Animals were trained with ethanol every other day (4 d ethanol, 4 d saline), with two rest days halfway through training. Individual training sessions were for 5 min. The day after their final training day, mice were tested for conditioned place preference by allowing them 30 min of ad libitum access to all chambers. Data were analyzed separately for each genotype and dose, with the time spent in the ethanol-paired chamber compared with the time spent in the saline-paired chamber, by a Wilcoxon signed rank test. Conditioned place preference was defined as spending significantly more time in the ethanol-paired chamber compared with the saline-paired chamber on the test day.
Conditioned taste aversion. This procedure was performed according to established protocols (Risinger and Boyce, 2002; Chester et al., 2003). Animals were housed singly and acclimatized to limited water access by allowing them access to water for decreasing periods of time (10, 8, 6, and 4 hr per day on subsequent days), followed by 2 hr daily access for 1 week. After acclimatization, they were placed on the conditioning schedule consisting of 1 hr access (1:00 to 2:00 P.M.) to the conditioned stimulus [CS; a solution of 1.2% (w/v) saline], followed immediately by a saline (0.9% w/v) injection. Later that same day (7:00 to 8:00 P.M.) they were allowed access to drinking water. The following day they were allowed 2 hr of access to drinking water (1:00 to 3:00 P.M.). This 2 d procedure constituted a single trial and was repeated seven times to establish baseline consumption levels of 1.2% NaCl. Seven additional trials were then conducted using a 2 gm/kg ethanol injection in place of saline. Because consumption did not change during the saline trials, results were averaged for all eight saline trials and treated as a single value for statistical comparison with the seven ethanol trials by two-way ANOVA with factors for trial and genotype.
Results
Reduced response to the hypnotic effect of ethanol in Cav2.2 null mice
To measure responses to acute ethanol administration, we measured loss of the righting reflex induced by ethanol (Fig. 1A). Cav2.2 null mice lost their righting reflex for a significantly shorter duration when compared with wild-type littermates (p < 0.05; two-tailed t test). Blood alcohol levels when mice regained the righting reflex (Fig. 1B) were significantly higher in Cav2.2 null mice compared with wild-type mice (p < 0.05; two-tailed t test). Differences in loss of righting reflex duration may reflect altered initial sensitivity or altered development of acute tolerance to ethanol. To specifically assay for differences in initial sensitivity, we determined the threshold dose of ethanol required to produce a loss of the righting reflex in the two genotypes. The threshold dose in Cav2.2 null mice was significantly higher than in wild-type mice (Fig. 1C). There was no difference between the genotypes in the clearance of ethanol from the blood after injection of 4 gm/kg ethanol, demonstrating that Cav2.2 null mice do not show altered ethanol metabolism (Fig. 1D). These results show that the hypnotic effect of ethanol is reduced in Cav2.2 null mice.
Figure 1.
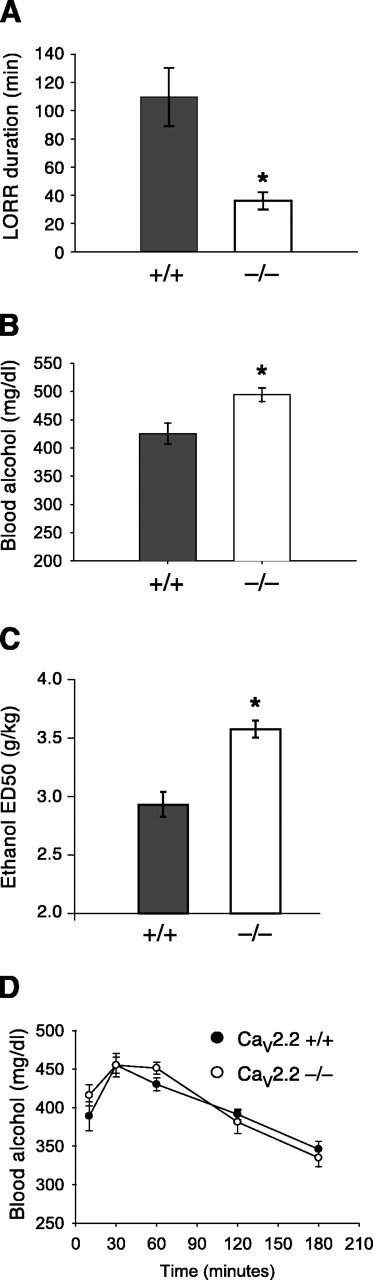
Decreased sensitivity to the hypnotic effect of ethanol in mice lacking Cav2.2. A, Loss of the righting reflex duration in mice given 3.6 gm/kg ethanol intraperitoneally (n = 11 wild-type and n = 12 CaV2.2 null mice). B, Mean blood alcohol levels at the time mice regained the righting reflex (n = 11 wild-type and n = 12 CaV2.2 null mice). C, The threshold dose of ethanol required to induce loss of righting reflex (values are mean ± 95% confidence interval; n = 6 per genotype). D, Blood alcohol clearance after an intraperitoneal injection of 4 gm/kg ethanol (n = 6 per genotype).
Increased ethanol-induced ataxia in Cav2.2 null mice
Studies using inbred mouse lines have shown that the level of response to hypnotic and ataxic doses of ethanol are not directly correlated (Dudek and Phillips, 1990; Pucilowski et al., 1996). We measured the response to lower, ataxia-inducing doses of ethanol using a rotarod apparatus. All mice were successfully trained to stay on a fixed-rate (20 rpm) rotarod for 180 sec after two to three trials. Mice of both genotypes showed a similar decrease in latency to fall from the fixed-rate rotarod after injection of either 1.5 or 2 gm/kg ethanol (Fig. 2A,B). To increase the difficulty of the test, we also examined mice placed on an accelerating rotarod after injection of 2 gm/kg ethanol. Before injection, all mice were able to remain on the accelerating rotarod, but after ethanol injection, Cav2.2 null mice showed a shorter latency to fall compared with wild-type mice (Fig. 2C). A two-way ANOVA with a between-subjects factor for genotype and a repeated measure for time showed an effect of genotype (F(1,68) = 8.32; p < 0.01) and time (F(4,68) = 10.594; p < 0.001), with an interaction between these factors (F(4,68) = 4.25; p = 0.004). Post hoc Tukey tests showed a significant difference (*p < 0.05) in latency to fall between wild-type and CaV2.2 null mice at 15 and 30 min after injection.
Figure 2.

Ethanol-induced ataxia in Cav2.2 null mice. There was no difference between genotypes in the latency to fall from a rotarod rotating at a fixed rate of 20 rpm after an intraperitoneal injection of 1.5 gm/kg (A) or 2 gm/kg (B) ethanol. C, On the accelerating rotarod, Cav2,2 null mice showed a significantly increased latency to fall after 2 gm/kg ethanol. n = 10 per genotype for A-C.
Ethanol consumption and preference are reduced in Cav2.2 null mice
Because antagonists of L-type calcium channels reduce ethanol consumption in several animal species (Rezvani and Janowsky, 1990; Rezvani et al., 1991; Fadda et al., 1992; De Beun et al., 1996), we wanted to assess ethanol self-administration in mice that lack N-type channels. We assayed voluntary ethanol self-administration using a two-bottle choice paradigm in which mice are allowed ad libitum access to ethanol and water presented in separate bottles concomitantly. Cav2.2 null mice drank less ethanol than their wild-type littermates (Fig. 3A). Two-way ANOVA with a between-subjects factor for genotype and a repeated measure for ethanol concentration showed main effects of genotype (F(1,136) = 5.35; p = 0.027) and ethanol concentration (F(4,136) = 7.33; p < 0.001). Cav2.2 null mice also showed reduced ethanol preference (Fig. 3B). Analysis of ethanol preference by two-way ANOVA with a between-subjects factor for genotype and a repeated measure for ethanol concentration also showed an effect of genotype (F(1,136) = 4.23; p = 0.047) and ethanol concentration (F(4,136) = 43.20; p < 0.001). Consumption of bitter (quinine) or sweet (saccharin) solutions did not differ between genotypes, indicating that these results are specific for ethanol (Fig. 3C). We also monitored baseline 24 hr water consumption by allowing mice access to two water bottles. No genotypic differences were seen (data not shown), consistent with a previous report that water and food consumption are normal in Cav2.2 null mice (Beuckmann et al., 2003).
Figure 3.
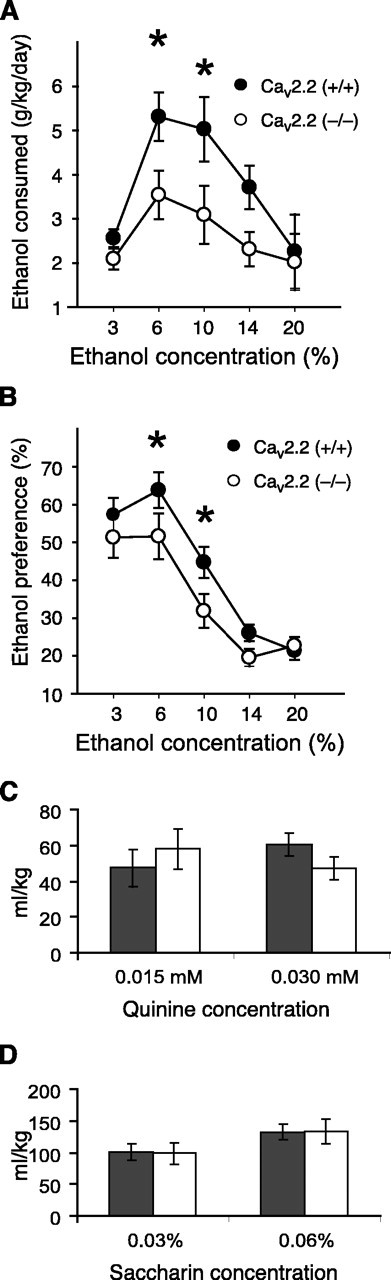
Decreased voluntary ethanol drinking in Cav2.2 null mice. A, Cav2.2 null mice (n = 15) showed decreased ethanol consumption when compared with wild-type mice (n = 21) (*p < 0.05 compared with CaV2.2 null mice given the same concentration of ethanol). B, Ethanol preference (volume of ethanol consumed/total volume of fluid consumed) was also lower for Cav2.2 null mice (*p < 0.05 compared with CaV2.2 null mice at the same concentration of ethanol). There was no difference between Cav2.2 null (□) and wild-type (▪) mice in the consumption of quinine (C) or saccharin (D) solutions (n = 12 for both genotypes).
Conditioned place preference for ethanol is altered in Cav2.2 null mice
Cav2.2 null mice may consume less ethanol than wild-type mice because they find it less rewarding and are thus not motivated to consume it or, conversely, because they find it more rewarding at a lower dose and thus require less ethanol to achieve satiety. To investigate these possibilities, we examined ethanol-induced place conditioning (Fig. 4A,B), which measures an animal's preference for a context associated with administration of a drug (Tzschentke, 1998). We trained animals with 2 gm/kg ethanol, a dose that establishes strong place preference in mice (Risinger et al., 2001; Dickinson et al., 2003). Wild-type mice showed a robust place preference for ethanol (p = 0.0093; Wilcoxon signed rank test) that was completely absent in Cav2.2 null mice (p > 0.05). However, Cav2.2 null mice did develop place preference at a lower dose of ethanol (1.2 gm/kg) that did not produce place preference in wild-type mice (p = 0.0068 for CaV2.2 null mice and p > 0.05 for wild-type mice; Wilcoxon signed rank tests). Neither genotype developed place preference at a higher dose of ethanol (2.8 gm/kg).
Figure 4.
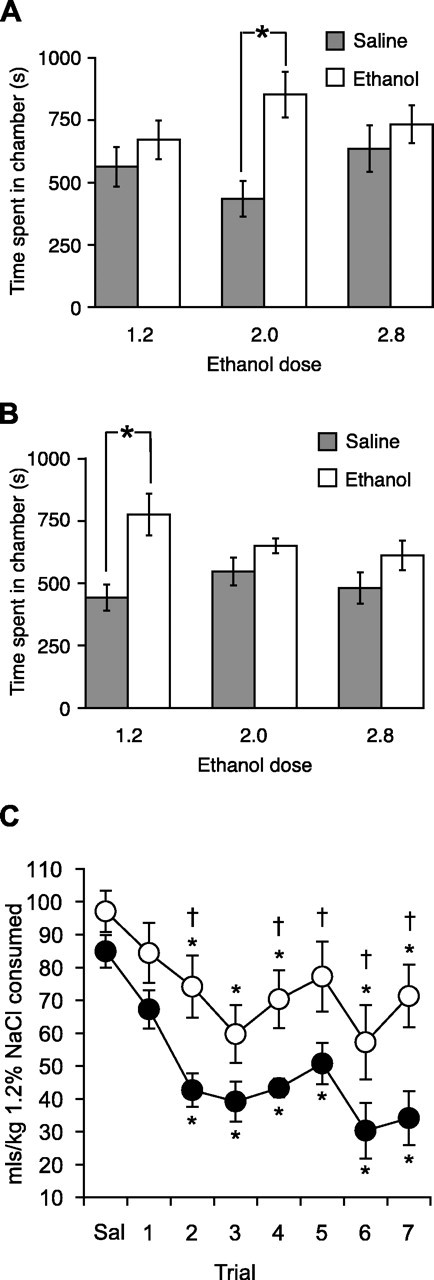
Altered motivational effects of ethanol in Cav2.2 null mice. A, Wild-type mice showed robust conditioned place preference for 2 gm/kg ethanol but not for higher (2.8 gm/kg) or lower (1.2 gm/kg) doses, whereas Cav2.2 null mice (B) showed place preference for 1.2 gm/kg ethanol but not for higher doses (n = 10-18 per genotype at each dose). C, Ethanol-conditioned taste aversion was reduced in Cav 2.2 null mice compared with wild-type mice. Data shown are mean daily consumption of 1.2% NaCl for 7 d after injection of saline and then individual trials after ethanol (2 gm/kg) injection (*p < 0.05 compared with saline; †p < 0.05 compared with wild type; n = 10 per genotype).
Conditioned taste aversion is attenuated in Cav2.2 null mice
In addition to its rewarding properties, ethanol has aversive effects, which can also be assayed using classical conditioning paradigms (Cunningham and Henderson, 2000). Having established that the rewarding effects of ethanol are altered in Cav2.2 null mice, we next investigated the aversive properties of ethanol using a conditioned taste aversion assay with ethanol as the unconditioned stimulus and 1.2% NaCl as the CS. Using 1.2% NaCl as the CS is thought to measure genuine aversion to ethanol, rather than reward, which may be measured using saccharin as the CS (Grigson, 1997; Risinger and Boyce, 2002; Chester et al., 2003). After commencement of ethanol treatment, both genotypes rapidly developed conditioned taste aversion. Analysis of these data by two-way repeated ANOVA showed significant effects of trial (F(7,126) = 18.406; p < 0.001) and genotype (F(1,126) = 7.582; p = 0.013), with the magnitude of conditioned taste aversion being significantly reduced in Cav2.2 null mice compared with their wild-type littermates (Fig. 4C).
Influence of genetic background
The expression of ethanol-related behaviors in mutant mouse lines can be influenced by the genetic background (Bowers et al., 1999). The mice used in these experiments were C57BL/6J × 129/SvJae F1 hybrids. Although the C57BL/6J parents of these mice had been backcrossed into the C57BL/6J lineage for 10 generations, there still remained a slight possibility that the Cav2.2 null phenotype might be attributable to cosegregation of genes near the CaV2.2 locus derived from the 129/SvJae background, and not a consequence of the null mutation. To confirm that the phenotype we had observed with the Cav2.2 null hybrid mice was attributable to absence of the Cav2.2 gene, we studied inbred 129/SvJae Cav2.2 null and wild-type mice because these mice are genetically identical, except for the Cav2.2 allele. We used a measure of acute response: loss of the righting reflex. Inbred 129/SvJae mice showed the same phenotype as their F1 hybrid counterparts; Cav2.2 null showed decreased duration of ethanol-induced loss of the righting reflex compared with wild-type littermates (mean duration ± SEM: wild type, 128 ± 19.3; null mice, 52.56 ± 7.3; p < 0.05 by two-tailed t test).
Discussion
Our study set out to investigate the behavioral responses to ethanol in mice lacking functional N-type calcium channels, based on in vitro data showing that acute ethanol treatment inhibits N-type calcium channels (Solem et al., 1997) and chronic ethanol treatment increases N-type calcium channel density (McMahon et al., 2000). We observed that, compared with their wild-type littermates, Cav2.2 null mice showed a substantially reduced response to hypnotic doses of ethanol but a slightly increased response to a lower dose of ethanol that induces ataxia. Compared with wild-type littermates, these mice also voluntarily consumed less alcohol, developed place preference at a lower dose of ethanol, and were less sensitive to the aversive effects of ethanol. Thus, our results demonstrate for the first time that N-type calcium channels modulate several behavioral responses to ethanol.
Behavioral experiments using a pharmacological manipulation of N-type calcium channels are difficult because currently available inhibitors cause motor impairment when injected into mice (Olivera et al., 1994). Despite this, a previous study investigating the role of N-type calcium channels in hypnotic responses to ethanol showed that intraventricular administration of the N-type calcium channel antagonist ω-conotoxin GVIA, which by itself is not hypnotic, dose dependently prolongs the ethanol-induced loss of the righting reflex in food-deprived rats (Brown et al., 1993). These results, together with previous work demonstrating that acute ethanol exposure inhibits N-type calcium channels (Wang et al., 1991; Solem et al., 1997), suggest that inhibition of N-type calcium channels promotes to the hypnotic effects of ethanol.
If inhibition of N-type calcium channels facilitates ethanol-induced hypnosis, then animals that lack N-type channels should show a reduced hypnotic response to ethanol. Indeed, in the current study, we found that CaV2.2 null mice showed a 70% decrease in the duration of the ethanol-induced loss of the righting reflex and a 22% increase in the threshold ethanol dose that induces a loss of the righting reflex when compared with wild-type mice. The much greater effect of gene deletion on loss-of-righting reflex duration compared with the threshold dose required to induce a loss of the righting reflex suggests that N-type channels may modulate not only to initial sensitivity but also acute tolerance to the hypnotic effect of ethanol.
Ethanol-induced loss of the righting reflex assesses the response to high, anesthetic doses of ethanol. To examine the response to a lower intoxicating dose, we measured ethanol-induced ataxia. Interestingly, Cav2.2 null mice displayed an increase in ethanol-induced ataxia compared with wild-type littermates. This increase was modest, present only when mice were tested on an accelerating rotarod, but not on a fixed-rate apparatus. Ethanol-induced ataxia and ethanol-induced loss of the righting reflex are, like many ethanol-associated behaviors, genetically distinct: the level of response to ethanol in one assay does not necessarily correlate with the level of response in the other. This has been demonstrated using inbred strains of mice (Dudek and Phillips, 1990; Crabbe et al., 1994) and rats (Radcliffe et al., 2000). Our data suggest that N-type channels play a minor role in the effects of ethanol on coordination but a major role in the hypnotic effects of ethanol. These findings demonstrate that the ethanol phenotype of Cav2.2 null mice is not simply attributable to a global, unidirectional change in acute response but is behavior specific.
Alcoholism is a disease with a substantial genetic component, and as a consequence, many different knock-out and transgenic mouse lines have been screened for altered responses to ethanol. The dominant pattern in these studies is an inverse correlation between changes in the acute intoxicating properties of ethanol, usually assayed by loss of the righting reflex, and changes in voluntary ethanol consumption (Thiele et al., 1998, 2000, 2002; Hodge et al., 1999; Weinshenker et al., 2000; Wand et al., 2001; Naassila et al., 2002, 2004; Spanagel et al., 2002). This relationship resembles observations in some children of human alcoholics, in which a low level of subjective response and a reduced level of postural instability induced by a three-drink alcohol challenge correlates with increased likelihood of future alcohol abuse or dependence (Schuckit, 1994).
According to these patterns, the higher threshold and shorter duration shown by Cav2.2 null mice in loss-of-righting reflex experiments would predict increased voluntary ethanol consumption in Cav2.2 null mice. Instead, the amount of ethanol consumed by these mice was significantly reduced compared with wild-type animals. This striking result led us to investigate the motivational effects of ethanol in Cav2.2 null mice. We observed that Cav2.2 null mice developed place preference for a low dose of ethanol (1.2 gm/kg) that is not rewarding to wild-type mice but did not develop place preference to 2 gm/kg ethanol, which was rewarding to wild-type littermates and is the standard dose used in mouse ethanol place-conditioning studies (Risinger et al., 2001; Dickinson et al., 2003). In contrast, CaV2.2 null mice developed conditioned taste aversion when administered 2 gm/kg ethanol, although this was reduced in magnitude when compared with wild-type animals. These results suggest that Cav2.2 null mice may consume less ethanol because gene deletion has enhanced ethanol reward, thereby requiring less ethanol to achieve the same effect normally produced in wild-type mice, whereas larger volumes of ethanol produce only aversive effects. These findings also suggest that the level of response to a hypnotic dose of ethanol is not a reliable predictor of alcohol consumption and preference. It would be worthwhile in future studies of ethanol responses in transgenic mouse lines to investigate whether the level of ethanol induced in coordination is a better predictor of drinking behavior, because it appears to be in some humans with a family history of alcoholism.
Our results indicate that N-type channels contribute to multiple aspects of ethanol-induced behavior, suggesting that there may be several mechanisms by which ethanol and N-type calcium channels interact. One appears to involve direct inhibition of N-type channels by ethanol, as we have shown previously in PC12 cells (Solem et al., 1997) and as has been demonstrated recently in a brain slices (Maldve et al., 2004). These findings indicate that N-type calcium channels are an important molecular target for ethanol and predict that some of the behavioral effects of ethanol will be blunted by removing this target, as we observed in Cav2.2 null mice.
Other evidence suggests that N-type calcium channels act indirectly to mediate responses to ethanol. For example, ethanol stimulates release of β-endorphin from primary cultures of hypothalamic neurons, and this is prevented by pretreatment with ω-conotoxin GVIA, which does not alter basal release from these cells (De et al., 1999). This suggests that N-type channels contribute to ethanol activation of the opioid system. N-type calcium channels also regulate the release of neurotransmitters that bind to receptor-gated ion channels modulated by ethanol. For example, N-type calcium channels regulate the release of glutamate (Gruner and Silva, 1994), the actions of which at NMDA receptors are inhibited by ethanol (Allgaier, 2002). This is thought to be important for several different behavioral responses to ethanol (Krystal et al., 2003). N-type calcium channels also modulate the release of GABA (Momiyama and Koga, 2001), and interactions between ethanol and GABA at GABAA receptors contribute to acute behavioral effects of alcohol (Davies, 2003).
N-type channels may also indirectly influence behavioral responses to ethanol through interactions with neural systems in-volved in drug reward and reinforcement. N-type calcium channels are modulated by G-protein-coupled receptors for neuropeptides and neurotransmitters known to play significant roles in ethanol reward, such as opioids (Kitamura et al., 2002; Hjelmstad and Fields, 2003), cannabinoids (Twitchell et al., 1997), and dopamine (Momiyama and Koga, 2001). Consistent with the importance of these receptors in ethanol self-administration is the finding that mice deficient in CB1 cannabinoid (Naassila et al., 2004), μ-opioid (Roberts et al., 2000) or D2 dopamine receptors (Phillips et al., 1998) consume less alcohol than wild-type littermates. Activation of these receptors can inhibit N-type calcium channels through release of βγ subunits (Feng et al., 2001), which appear to be important in promoting ethanol self-administration (Yao et al., 2002).
In summary, deletion of N-type calcium channels altered acute behavioral responses to ethanol, producing a slight increase in ethanol-induced ataxia and a substantial decrease in ethanol-induced hypnosis. Most importantly, deletion of N-type channels caused mice to voluntarily consume less alcohol and experience reward at lower ethanol doses. These findings indicate that the N-type calcium channel may be an attractive pharmacological target for alcoholism, because inhibitors of these N-type channels might reduce ethanol consumption.
Footnotes
This research was supported by funds provided by the National Institutes of Health-National Institute on Alcohol Abuse and Alcoholism to R.O.M. (award numbers R01 AA008117 and R37 AA013588); by funds provided by the State of California for medical research on alcohol and substance abuse through the University of California, San Francisco; and by a grant to H.-S.S. from the Chemoinformatics Program of Korea Institute of Science and Technology. We thank Jackie Connolly and Gerry Brush for technical assistance.
Correspondence should be addressed to Dr. Robert O. Messing, Ernest Gallo Clinic and Research Center, 5858 Horton Street, Suite 200, Emeryville, CA 94608. E-mail: romes@itsa.ucsf.edu.
Copyright © 2004 Society for Neuroscience 0270-6474/04/249862-08$15.00/0
References
- Allgaier C (2002) Ethanol sensitivity of NMDA receptors. Neurochem Int 41: 377-382. [DOI] [PubMed] [Google Scholar]
- Beuckmann CT, Sinton CM, Miyamoto N, Ino M, Yanagisawa M (2003) N-type calcium channel alpha1B subunit (Cav2.2) knock-out mice display hyperactivity and vigilance state differences. J Neurosci 23: 6793-6797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bowers BJ, Owen EH, Collins AC, Abeliovich A, Tonegawa S, Wehner JM (1999) Decreased ethanol sensitivity and tolerance development in gamma-protein kinase C null mutant mice is dependent on genetic background. Alcohol Clin Exp Res 23: 387-397. [PubMed] [Google Scholar]
- Brooks SP, Hennebry G, McAlpin GP, Norman G, Little HJ (2002) Nimodipine prevents the effects of ethanol in tests of memory. Neuropharmacology 42: 577-585. [DOI] [PubMed] [Google Scholar]
- Brown LM, Sims JS, Randall P, Wilcox R, Leslie SW (1993) ω-Conotoxin increases sleep time following ethanol injection. Alcohol 10: 159-162. [DOI] [PubMed] [Google Scholar]
- Chester JA, Lumeng L, Li TK, Grahame NJ (2003) High- and low-alcohol-preferring mice show differences in conditioned taste aversion to alcohol. Alcohol Clin Exp Res 27: 12-18. [DOI] [PubMed] [Google Scholar]
- Crabbe JC, Gallaher ES, Phillips TJ, Belknap JK (1994) Genetic determinants of sensitivity to ethanol in inbred mice. Behav Neurosci 108: 186-195. [DOI] [PubMed] [Google Scholar]
- Cunningham CL, Henderson CM (2000) Ethanol-induced conditioned place aversion in mice. Behav Pharmacol 11: 591-602. [DOI] [PubMed] [Google Scholar]
- Davies M (2003) The role of GABAA receptors in mediating the effects of alcohol in the central nervous system. J Psychiatry Neurosci 28: 263-274. [PMC free article] [PubMed] [Google Scholar]
- De A, Boyadjieva NI, Sarkar DK (1999) Effect of voltage-dependent calcium channel blockers on ethanol-induced beta-endorphin release from hypothalamic neurons in primary cultures. Alcohol Clin Exp Res 23: 850-855. [PubMed] [Google Scholar]
- De Beun R, Schneider R, Klein A, Lohmann A, De Vry J (1996) Effects of nimodipine and other calcium channel antagonists in alcohol-preferring AA rats. Alcohol 13: 263-271. [DOI] [PubMed] [Google Scholar]
- Dickinson SD, Lee EL, Rindal K, Cunningham CL (2003) Lack of effect of dopamine receptor blockade on expression of ethanol-induced conditioned place preference in mice. Psychopharmacology (Berl) 165: 238-244. [DOI] [PubMed] [Google Scholar]
- Dixon W (1965) The Up- and -Down Method for Small Samples. J Am Stat Assoc 60: 967-978. [Google Scholar]
- Dudek BC, Phillips TJ (1990) Distinctions among sedative, disinhibitory, and ataxic properties of ethanol in inbred and selectively bred mice. Psychopharmacology (Berl) 101: 93-99. [DOI] [PubMed] [Google Scholar]
- Dunlap K, Luebke JI, Turner TJ (1995) Exocytotic Ca2+ channels in mammalian central neurons. Trends Neurosci 18: 89-98. [PubMed] [Google Scholar]
- Fadda F, Garau B, Colombo G, Gessa GL (1992) Isradipine and other calcium channel antagonists attenuate ethanol consumption in ethanol-preferring rats. Alcohol Clin Exp Res 16: 449-452. [DOI] [PubMed] [Google Scholar]
- Feng ZP, Arnot MI, Doering CJ, Zamponi GW (2001) Calcium channel beta subunits differentially regulate the inhibition of N-type channels by individual Gbeta isoforms. J Biol Chem 276: 45051-45058. [DOI] [PubMed] [Google Scholar]
- Findlay GS, Wick MJ, Mascia MP, Wallace D, Miller GW, Harris RA, Blednov YA (2002) Transgenic expression of a mutant glycine receptor decreases alcohol sensitivity of mice. J Pharmacol Exp Ther 300: 526-534. [DOI] [PubMed] [Google Scholar]
- Ghosh A, Greenberg ME (1995) Calcium signaling in neurons: molecular mechanisms and cellular consequences. Science 268: 239-247. [DOI] [PubMed] [Google Scholar]
- Grigson PS (1997) Conditioned taste aversions and drugs of abuse: a reinterpretation. Behav Neurosci 111: 129-136. [PubMed] [Google Scholar]
- Gruner W, Silva LR (1994) Omega-conotoxin sensitivity and presynaptic inhibition of glutamatergic sensory neurotransmission in vitro. J Neurosci 14: 2800-2808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hjelmstad GO, Fields HL (2003) Kappa opioid receptor activation in the nucleus accumbens inhibits glutamate and GABA release through different mechanisms. J Neurophysiol 89: 2389-2395. [DOI] [PubMed] [Google Scholar]
- Hodge CW, Mehmert KK, Kelley SP, McMahon T, Haywood A, Olive MF, Wang D, Sanchez-Perez AM, Messing RO (1999) Supersensitivity to allosteric GABAA receptor modulators and alcohol in mice lacking PKCϵ. Nat Neurosci 2: 997-1002. [DOI] [PubMed] [Google Scholar]
- Kim C, Jun K, Lee T, Kim SS, McEnery MW, Chin H, Kim HL, Park JM, Kim DK, Jung SJ, Kim J, Shin HS (2001) Altered nociceptive response in mice deficient in the alpha(1B) subunit of the voltage-dependent calcium channel. Mol Cell Neurosci 18: 235-245. [DOI] [PubMed] [Google Scholar]
- Kitamura G, Ohta T, Kai T, Kon Y, Ito S (2002) Inhibitory effects of opioids on voltage-dependent Ca(2+) channels and catecholamine secretion in cultured porcine adrenal chromaffin cells. Brain Res 942: 11-22. [DOI] [PubMed] [Google Scholar]
- Krystal JH, Petrakis IL, Mason G, Trevisan L, D'Souza DC (2003) N-methyl-d-aspartate glutamate receptors and alcoholism: reward, dependence, treatment, and vulnerability. Pharmacol Ther 99: 79-94. [DOI] [PubMed] [Google Scholar]
- Kuzmin A, Semenova S, Zvartau E, De Vry J (1999) Effects of calcium channel blockade on intravenous self-administration of ethanol in rats. Eur Neuropsychopharmacol 9: 197-203. [DOI] [PubMed] [Google Scholar]
- Little HJ (1995) The role of calcium channels in drug dependence. Drug Alcohol Depend 38: 173-194. [DOI] [PubMed] [Google Scholar]
- Maldve RE, Chen X, Zhang TA, Morrisett RA (2004) Ethanol selectively inhibits enhanced vesicular release at excitatory synapses: real-time visualization in intact hippocampal slices. Alcohol Clin Exp Res 28: 143-152. [DOI] [PubMed] [Google Scholar]
- McMahon T, Andersen R, Metten P, Crabbe JC, Messing RO (2000) Protein kinase C epsilon mediates up-regulation of N-type calcium channels by ethanol. Mol Pharmacol 57: 53-58. [PubMed] [Google Scholar]
- Momiyama T, Koga E (2001) Dopamine D(2)-like receptors selectively block N-type Ca(2+) channels to reduce GABA release onto rat striatal cholinergic interneurones. J Physiol (Lond) 533: 479-492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naassila M, Ledent C, Daoust M (2002) Low ethanol sensitivity and increased ethanol consumption in mice lacking adenosine A2A receptors. J Neurosci 22: 10487-10493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naassila M, Pierrefiche O, Ledent C, Daoust M (2004) Decreased alcohol self-administration and increased alcohol sensitivity and withdrawal in CB1 receptor knockout mice. Neuropharmacology 46: 243-253. [DOI] [PubMed] [Google Scholar]
- Olivera BM, Miljanich GP, Ramachandran J, Adams ME (1994) Calcium channel diversity and neurotransmitter release: the ω-conotoxins and ω-agatoxins. Annu Rev Biochem 63: 823-867. [DOI] [PubMed] [Google Scholar]
- Phillips TJ, Brown KJ, Burkhart-Kasch S, Wenger CD, Kelly MA, Rubinstein M, Grandy DK, Low MJ (1998) Alcohol preference and sensitivity are markedly reduced in mice lacking dopamine D2 receptors. Nat Neurosci 1: 610-615. [DOI] [PubMed] [Google Scholar]
- Pucilowski O, Ayensu WK, D'Ercole AJ (1996) Insulin-like growth factor I expression alters acute sensitivity and tolerance to ethanol in transgenic mice. Eur J Pharmacol 305: 57-62. [DOI] [PubMed] [Google Scholar]
- Radcliffe RA, Bohl ML, Lowe MV, Cycowski CS, Wehner JM (2000) Mapping of quantitative trait loci for hypnotic sensitivity to ethanol in crosses derived from the C57BL/6 and DBA/2 mouse strains. Alcohol Clin Exp Res 24: 1335-1342. [PubMed] [Google Scholar]
- Reid CA, Bekkers JM, Clements JD (2003) Presynaptic Ca2+ channels: a functional patchwork. Trends Neurosci 26: 683-687. [DOI] [PubMed] [Google Scholar]
- Rezvani AH, Janowsky DS (1990) Decreased alcohol consumption by verapamil in alcohol preferring rats. Prog Neuro-Psychopharmacol Biol Psychiatr 14: 623-631. [DOI] [PubMed] [Google Scholar]
- Rezvani AH, Grady DR, Janowsky DS (1991) Effect of calcium-channel blockers on alcohol consumption in alcohol-drinking monkeys. Alcohol Alcohol 26: 161-167. [DOI] [PubMed] [Google Scholar]
- Risinger FO, Boyce JM (2002) Conditioning tastant and the acquisition of conditioned taste avoidance to drugs of abuse in DBA/2J mice. Psychopharmacology (Berl) 160: 225-232. [DOI] [PubMed] [Google Scholar]
- Risinger FO, Freeman PA, Greengard P, Fienberg AA (2001) Motivational effects of ethanol in DARPP-32 knock-out mice. J Neurosci 21: 340-348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts AJ, McDonald JS, Heyser CJ, Kieffer BL, Matthes HW, Koob GF, Gold LH (2000) Mu-opioid receptor knockout mice do not self-administer alcohol. J Pharmacol Exp Ther 293: 1002-1008. [PubMed] [Google Scholar]
- Schuckit MA (1994) Low level of response to alcohol as a predictor of future alcoholism. Am J Psychiatry 151: 184-189. [DOI] [PubMed] [Google Scholar]
- Solem M, McMahon T, Messing RO (1997) Protein kinase A regulates inhibition of N- and P/Q-type calcium channels by ethanol in PC12 cells. J Pharmacol Exp Ther 282: 1487-1495. [PubMed] [Google Scholar]
- Spanagel R, Siegmund S, Cowen M, Schroff KC, Schumann G, Fiserova M, Sillaber I, Wellek S, Singer M, Putzke J (2002) The neuronal nitric oxide synthase gene is critically involved in neurobehavioral effects of alcohol. J Neurosci 22: 8676-8683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thiele TE, Marsh DJ, Ste. Marie L, Bernstein IL, Palmiter RD (1998) Ethanol consumption and resistance are inversely related to neuropeptide Y levels. Nature 396: 366-369. [DOI] [PubMed] [Google Scholar]
- Thiele TE, Willis B, Stadler J, Reynolds JG, Bernstein IL, McKnight GS (2000) High ethanol consupmtion and low sensitivity to ethanol-induced sedation in protein kinase A-mutant mice. J Neurosci 20: RC75(1-6). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thiele TE, Koh MT, Pedrazzini T (2002) Voluntary alcohol consumption is controlled via the neuropeptide Y Y1 receptor. J Neurosci 22: RC208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Twitchell W, Brown S, Mackie K (1997) Cannabinoids inhibit N- and P/Q-type calcium channels in cultured rat hippocampal neurons. J Neurophysiol 78: 43-50. [DOI] [PubMed] [Google Scholar]
- Tzschentke TM (1998) Measuring reward with the conditioned place preference paradigm: a comprehensive review of drug effects, recent progress and new issues. Prog Neurobiol 56: 613-672. [DOI] [PubMed] [Google Scholar]
- Walter H, Messing RO (1999) Regulation of neuronal voltage-gated calcium channels by ethanol. Neurochem Int 35: 95-101. [DOI] [PubMed] [Google Scholar]
- Wand G, Levine M, Zweifel L, Schwindinger W, Abel T (2001) The cAMP-protein kinase A signal transduction pathway modulates ethanol consumption and sedative effects of ethanol. J Neurosci 21: 5297-5303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Lemos JR, Dayanithi G, Nordmann JJ, Treistman SN (1991) Ethanol reduces vasopressin release by inhibiting calcium currents in nerve terminals. Brain Res 551: 339-341. [DOI] [PubMed] [Google Scholar]
- Watson WP, Little HJ (1999) Correlation between increases in dihydropyridine binding in vivo and behavioural signs of ethanol withdrawal in mice. Alcohol Alcohol 34: 35-42. [DOI] [PubMed] [Google Scholar]
- Weinshenker D, Rust NC, Miller NS, Palmiter RD (2000) Ethanol-associated behaviors of mice lacking norepinephrine. J Neurosci 20: 3157-3164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao L, Arolfo MP, Dohrman DP, Jiang Z, Fan P, Fuchs S, Janak PH, Gordon AS, Diamond I (2002) Betagamma dimers mediate synergy of dopamine D2 and adenosine A2 receptor-stimulated PKA signaling and regulate ethanol consumption. Cell 109: 733-743. [DOI] [PubMed] [Google Scholar]