

Abstract
Angiotensin II (Ang II) elicits Ang II type 1 receptor (AT1-R)-mediated increases in neuronal firing within the hypothalamus and brainstem that are ultimately responsible for physiological actions such as increased blood pressure and fluid intake. Although there is a growing literature on the intracellular mechanisms that mediate the actions of Ang II via AT1-R in neurons, little is known about the mechanisms that diminish or “switch-off” the neuronal chronotropic action of Ang II. In the present study, we identified macrophage migration inhibitory factor (MIF) as an intracellular inhibitor of the actions of Ang II in neurons. The evidence is as follows. First, Ang II, acting via AT1-R, increases the intracellular levels of MIF in neurons cultured from rat hypothalamus and brainstem. Second, elevation of intracellular MIF by Ang II prevents further chronotropic actions of this peptide. Third, intracellular application of exogenous recombinant MIF abolishes the Ang II-induced chronotropic action in neurons. Finally, intracellular application of the MIF peptide fragment MIF-(50-65), which harbors the thiol oxidoreductase property of the MIF molecule, mimics the inhibitory actions of MIF on Ang II-stimulated neuronal firing. Thus, this study is the first to demonstrate the existence of an intracellular negative regulator of Ang II-induced actions in neurons and indicates that MIF may act as a physiological brake for the chronotropic effects of Ang II in rat neurons.
Keywords: angiotensin II, macrophage migration inhibitory factor, neuron, firing rate, thiol-protein oxidoreductase, reactive oxygen species
Introduction
Angiotensin II (Ang II) acts within the brain to elicit a variety of physiological effects such as increases in water intake and blood pressure, which contribute to its overall role in fluid balance and cardiovascular regulation (Muratani et al., 1996; Averill and Diz, 2000; Ferguson et al., 2001). The importance of Ang II actions in the CNS to blood pressure control is underscored by spontaneously hypertensive rats, which exhibit an overactive brain angiotensin system (Gyurko et al., 1993; Oparil et al., 1994; Han and Sim, 1998). These physiological actions of Ang II begin with stimulation of neuronal Ang II type 1 receptors (AT1-Rs) at circumventricular, hypothalamic, and brainstem sites (de Gasparo et al., 2000; Culman et al., 2002) and involve activation of specific neuronal circuitry within the brain (Jensen et al., 1992; Dampney et al., 2002). To fully understand the physiological actions of Ang II in the brain and how these actions are altered in disease states, it is essential to determine the intracellular mechanisms by which Ang II alters neuronal activity.
In previous studies, we used neurons cultured from newborn rat hypothalamus and brainstem as a model system to study AT1-R signaling (Sumners et al., 2002). We determined that Ang II elicits a chronotropic action (Wang et al., 1997; Sun et al., 2002) that is mediated by activation of both protein kinase C (PKC) and calcium/calmodulin-dependent kinase II and generation of reactive oxygen species (ROS) (Sumners et al., 2002; Sun et al., 2002, 2004). However, it is not known whether there are specific intracellular mechanisms that diminish or suppress the stimulatory actions of Ang II on neuronal firing. Thus, we attempted to identify factors for which neuronal expression is increased by Ang II and that may feed back and influence neuronal firing. To this end, we demonstrated that Ang II increases the expression of macrophage migration inhibitory factor (MIF) in neuronal cultures (Busche et al., 2001). MIF is a cytokine that is widely expressed in immune tissues and in the brain, has established roles in the innate and adaptive immune responses (for review, see Baugh and Bucala, 2002; Calandra and Roger, 2003; Donn and Ray, 2004), and may be a neuroendocrine modulator of inflammation (Fingerle-Rowson and Bucala, 2001). However, there is no evidence to date that MIF can influence neuronal activity or Ang II responses. Interestingly, in addition to the plasma membrane receptor-mediated cytokine actions of MIF (Leng et al., 2003), it has been reported that MIF can exert intracellular actions via association with cytosolic proteins (Kleemann et al., 2000) or potentially via tautomerase or thiol-oxidoreductase properties of the MIF molecule (Rosengren et al., 1996; Nguyen et al., 2003a). Based on the potential of MIF to scavenge ROS via its thioloxidoreductase activity (Nguyen et al., 2003b), we investigated whether it can serve as an intracellular regulator of the chronotropic actions of Ang II in neurons.
Materials and Methods
Materials. Newborn Sprague Dawley (SD) and Wistar-Kyoto (WKY) rats were obtained from our breeding colony, which originated from Charles River Laboratories (Wilmington, MA). DMEM was obtained from Invitrogen (Grand Island, NY). Crystallized trypsin was from Worthington Biochemicals (Freehold, NJ). α-Internexin and glial fibrillary acidic protein (GFAP) monoclonal antibodies were kindly provided by Dr. Gerry Shaw (University of Florida, Gainesville, FL). Rabbit anti-rat MIF antibody was purchased from Torrey Pines Biolabs (Houston, TX). Alexa Fluor 488 goat anti-rabbit IgG, Alexa Fluor 594 goat anti-mouse IgG, and dihydroethidium (DHE) were purchased from Molecular Probes (Eugene, OR). Rat-mouse MIF ELISA kits and monoclonal anti-neuronal-specific nuclear protein (NeuN) antibody were purchased from Chemicon International (Temecula, CA). Protein assay dye, 10% Tris-HCl criterion gels, and nitrocellulose were purchased from Bio-Rad (Hercules, CA). Western Lightning Chemiluminescence Reagent Plus was purchased from PerkinElmer Life Sciences (Boston, MA). RNAeasy kits and OneStep reverse transcription (RT)-PCR kits were obtained from Qiagen (Valencia, CA). DNase I was from Ambion (Austin, TX). TA cloning kits were obtained from Invitrogen (Carlsbad, CA). Adeno-X Rapid Titer kits were purchased from BD Biosciences (Palo Alto, CA). Mouse recombinant MIF (rMIF) was prepared as the native protein from an Escherichia coli expression system and purified free of endotoxin by C8 chromatography as described previously (Bernhagen et al., 1994). Mouse MIF differs from rat MIF by a single amino acid substitution (mMIF: asn54, rMIF:ser54) that, to date, has not been found to affect the influence of the bioactivity or immunoreactivity of the protein in different murine bio-assays (Bernhagen et al., 1994; Bacher et al., 1997). d-dopachrome tautomerase (27% sequence identity with MIF) was prepared by PCR cloning from mouse cDNA, and its structural fidelity was established by DNA sequencing. The recombinant protein was purified from an E. coli expression system by sequential Mono-Q and C8 column FPLC followed by slow refolding from DTT/urea. These procedures closely followed those described previously for recombinant MIF (Bernhagen et al., 1994). The peptide fragments of rat MIF (Sakai et al., 1994), MIF-(50-65), and C57S/C60S-MIF-(50-65) were synthesized by the Tufts University Core Facility (Boston, MA). These peptides were unmodified at the N and C termini. Structures are as follows. MIF-(50-65): [H]-F-S-G-T-S-D-P-C-A-L-C-S-L-H-S-I-[OH]. C57S/C60S-MIF-(50-65): [H]-F-S-G-T-S-D-P-S-A-L-S-S-L-H-S-I-[OH]. pMH4-I-synapsin 1 (SYN)-enhanced green fluorescent protein (EGFP) was kindly provided by Dr. Sebastien Kugler (University of Göttingen, Göttingen, Germany). Ang II, l-(-)-norepinephrine, carbachol, and all other chemicals were purchased from Sigma-Aldrich (St. Louis, MO).
Preparation of neuronal and astroglial cultures. Neuronal cultures (∼95% neurons/5% astroglia), mixed neuronal-astrocyte cultures (∼50% neurons, 50% astroglia), and pure astroglial cultures were prepared from the hypothalami and brain stems or from the cerebral cortex of newborn SD or WKY rats, as detailed previously (Sumners et al., 1991). Cultures were grown in 35 mm Nunc (Naperville, IL) plastic tissue culture dishes or 24-well plates, precoated with poly-l-lysine, and were used at 12-16 d in vitro.
Analysis of MIF protein. For Western blotting, proteins were isolated from control or drug-treated neuronal cultures, and the expression of MIF protein was assessed via Western immunoblot as detailed by us previously (Busche et al., 2001). MIF protein expression was quantified by densitometry using a calibrated imaging densitometer (model GS710; Bio-Rad). For ELISA assays, growth media were removed from control or drug-treated neuronal cultures and reserved for analysis of extracellular MIF. Cultures were washed once with ice-cold PBS, pH 7.4, and then were incubated with sterile water (0.3 ml/dish) on ice for 15 min to lyse the cells. Cells were scraped from the dishes, vortexed vigorously, and centrifuged to remove cells debris, and the supernatants were used to analyze the intracellular levels of MIF. Intracellular and extracellular MIF were quantified using an ELISA kit according to the instructions provided (Chemicon International).
Real-time RT-PCR. MIF mRNA was analyzed via quantitative real-time RT-PCR. Oligonucleotide primers and TaqMan probes specific for rat MIF were obtained from Applied Biosystems (Foster City, CA). Total RNA was isolated from control or Ang II-treated neuronal cultures using an RNAeasy kit. Isolated RNA underwent DNase I treatment to remove genomic DNA, and 25 ng of the purified RNA was used to perform RT-PCR in an Applied Biosystems PRISM 7000 sequence detection system, with the use of the one-step RT-PCR Master Mix reagents. Data were normalized to 18S RNA.
Immunocytochemistry. Neuronal cultures from WKY rats were washed briefly with Dulbecco's PBS and then fixed for 10 min with PBS containing 0.1% Tween 20 (PBS/Tween) and 4% formaldehyde solution. Dishes were then washed briefly with PBS/Tween. Goat serum (10%) in PBS/Tween was added to the dish for 30 min at 37°C to reduce nonspecific binding, followed by an additional wash with PBS/Tween. Primary antibodies (monoclonal α-internexin or GFAP; 1:50) and rabbit anti-rat MIF antibody (1:500), diluted in a 1 ml total volume of PBS/Tween, were added to the dish and incubated overnight at 4°C. After two 30 min washes with PBS/Tween, the neurons were incubated with secondary antibodies (Alexa Fluor 488 goat anti-rabbit IgG and Alexa Fluor 594 goat anti-mouse IgG; 1:1000), washed twice for 30 min each time with PBS/Tween, and mounted with anti-bleaching medium and a glass coverslip. The slides were detected using a fluorescence confocal microscope equipped with a three-channel acquisition system.
Electrophysiology. Spontaneous action potentials (APs) in neurons in culture were recorded using whole-cell voltage-clamp methods in the current-clamp mode, exactly as detailed by us previously (Wang et al., 1997; Zhu et al., 2001; Sun et al., 2002). The resting membrane potential was defined as the potential within a 1 sec time period during which there was no spontaneously firing AP. The neuronal firing rate was measured as the number of fully developed APs per second (in Hertz). Ang II was applied to the neurons in the superfusate (modified Tyrode's) solution. Intracellular application of rMIF, MIF peptides, or anti-MIF antibodies was achieved by injection through the patch pipette as detailed by us previously (Zhu et al., 2000). In brief, a sidearm pipette holder was attached to the head stage of the Axopatch. One side arm was used to supply suction for seal formation, and a second side arm was used to advance a very fine polyethylene catheter (PE-50) down the inside of the patch pipette. Control measurements of firing rate were made 5 min after the whole-cell configuration was established in a given neuron. After this, the rMIF, MIF peptides, or anti-MIF antibodies were injected into the tip of the recording electrode via the PE-50 tube. From the pipette tip, the rMIF, MIF peptides, anti-MIF antibodies, or d-dopachrome tautomerase (DCT) were allowed to diffuse into the neuron, and measurements of firing rate made 4 min later, at which time a stable peak response was obtained. Care was taken not to overperfuse the neuron, and this is monitored electrically via the Axopatch and on the television monitor. Thus, the concentrations of rMIF, MIF peptides, or anti-MIF antibodies that are given in the results refer to the amounts that are injected at the pipette tip and, thus, are higher than the actual amounts that reach the intracellular site of action.
Determination of intracellular ROS. Intracellular ROS were detected in neuronal cultures using the oxidant-sensitive fluorogenic probe DHE, as described previously (Zimmerman et al., 2002). DHE is oxidized to fluorescent ethidium by superoxide radicals. After loading with DHE (100 nm) for 30 min at 37°C, cultures were treated with Ang II (100 nm) or control solution (PBS) for 5 min. Each treatment condition was run in triplicate within experiments, and each set of experiments was performed three separate times. After these treatments, cultures were washed once with PBS, and ethidium-positive cells were detected using a fluorescence microscope (Nikon, Melville, NY) equipped with a digital camera. Fluorescence intensity of individual cells was analyzed using Quantity One software (Bio-Rad).
Construction and transduction of recombinant adenoviral constructs. Total RNA was prepared from SD rat cerebral cortical neuronal cultures using an RNeasy kit. Rat MIF cDNA containing the full coding sequences was generated by RT-PCR using a OneStep RT-PCR kit using the following primers: 5′ CTGACGAATTCCCACCATGCCTATGTTCATCGT 3′ (upstream); 5′ AGTATGCGGCCGCTCAAGCGAAGGTGGAACCGTT 3′(downstream).
Single PCR products of the expected size were purified by agarose gel electrophoresis and cloned into pCR2.1 vector using TA cloning kits. The recombinant plasmids were prepared and sequenced (both strands) to ensure the rat MIF inserts were correct. The AdEasy Vector System (He et al., 1998) was used for construction of the rat MIF adenoviral vector. The transfer vector pShuttle-SYN-MIF was constructed using a standard method (Sambrook et al., 1989). The vector contains MIF cDNA driven by the neuronal specific SYN gene promoter. The synapsin promoter, chimeric intron, and SV40 polyadenylation sequence are derived from pMH4-I-SYN-EGFP (Kugler et al., 2001). The pShuttle-SYN-MIF was linearized with PmeI and separately cotransformed into competent E. coli strain BJ5183 together with pAdeasy-1, the viral DNA plasmid. The recombinant clones (pAd5-SYN-MIF) were identified by restriction enzyme analysis. As a control, pAd5-SYN-EGFP containing the EGFP gene driven by the synapsin promoter was constructed using the same procedure described above.
The pAdeasy-1 is E1 and E3 deleted; its E1 function can be complemented in 293A cells. The recombinant adenoviral constructs pAd5-SYN-MIF and pAd5-SYN-EGFP were then cleaved with PacI to expose their inverted terminal repeats and transfected into 293A cells to produce viral particles. The recombinant viruses were identified with PCR, RT-PCR, and Western blot. The recombinant viruses were purified through two cesium chloride gradients and then purified virus was desalted by dialysis at 4°C against 10 mm Tris-hydrochloric acid buffer with 10% glycerol and stored in aliquots in liquid nitrogen. The titer of virus preparations was determined using an Adeno-X Rapid Titer kit. Expression of MIF in vitro was determined by incubating cerebral cortical neuronal cultures with purified viral particles (Ad5-SYN-MIF, 4.4 × 105, 2 × 106, or 1 × 107 infectious U/ml of media; Ad5-SYN-EGFP, 1 × 107 infectious U/ml of media) for 3 d. After this, the expression of MIF was determined by Western blot as above.
Data analysis. Results are expressed as means ± SEM. Statistical significance was evaluated with the use of a one-way ANOVA, followed by a Newman-Keuls test to compare individual means. Differences were considered significant at p < 0.05.
Results
Effects of Ang II on MIF expression in WKY rat neuronal cultures
In previous studies, we determined that Ang II elicits an AT1-R-mediated and time-dependent increase in the expression of MIF mRNA and protein in hypothalamus-brainstem neuronal cultures from SD rats (Busche et al., 2001). The initial goal of the present study was to establish whether Ang II alters intracellular levels of MIF, MIF release, or both. Incubation of WKY rat hypothalamus-brainstem neuronal cultures with Ang II (100 nm) for 5 hr resulted in significant increases in MIF mRNA and protein expression, similar to the effects of this peptide in SD rat neurons (Fig. 1A). ELISA analyses revealed that Ang II increased the intracellular levels of MIF in these neuronal cultures but did not alter MIF levels in the growth media, implying that Ang II does not increase MIF secretion from these cells (Fig. 1B).
Figure 1.

Effects of angiotensin II on MIF expression in WKY rat neuronal cultures. A, Neuronal cultures from WKY rats were incubated with control (Con) solution (PBS) or Ang II (100 nm) for 5 hr at 37°C followed by analysis of MIF mRNA and protein levels as described in Materials and Methods. Left, Bar graphs are real-time PCR data showing the mean levels of MIF mRNA (±SEM) under each treatment condition (n = 7 experiments). Right, Top, Representative blot showing the levels of MIF protein under each treatment condition; Bottom, bar graphs showing the mean levels of MIF protein (±SEM; n = 5 experiments). *p < 0.01 versus respective control. B, WKY rat neuronal cultures were treated as in A, followed by analysis of intracellular and extracellular (media) MIF by ELISA as described in Materials and Methods. Bar graphs show means ± SEM (n = 4 experiments). *p < 0.01 versus respective control. C, WKY rat neuronal cultures were incubated with control solution (PBS) or Ang II (100 nm) for 0.5, 1, 3, 6, 12, or 24 hr, cells were lysed, and intracellular MIF was analyzed by ELISA. Line graphs are levels of intracellular MIF in PBS- and Ang II-treated cells at each time point and are means ± SEM (n = 3 experiments). *p < 0.005 versus controls. D, WKY rat neuronal cultures were incubated with control solution (PBS), NE (10 μm), or carbachol (Carb; 100 μm) for 5 hr at 37°C, followed by analysis of MIF levels by ELISA. Bar graphs show means ± SEM of intracellular MIF (left) and extracellular MIF levels (right) (n = 3 experiments).
To determine the time optimum for Ang II action in these cells, neuronal cultures from WKY rats were incubated with Ang II (100 nm) for 0.5, 1, 3, 6, 12, or 24 hr, followed by ELISA analysis of intracellular MIF. The results indicate that in WKY rat neurons, Ang II begins to produce an increase in MIF expression within 30 min, an effect that is statistically significant by 1 hr and that reaches a maximum by 3 hr. This action of Ang II persists through 24 hr (Fig. 1C). To investigate the specificity of this Ang II induction of MIF expression, we tested the effects of other agents that act via G-protein-coupled receptors and stimulate intracellular calcium-dependent processes, similar to Ang II activation of AT1-R. Incubation of neuronal cultures with α1-adrenergic receptor agonist norepinephrine (NE) (10 μm; 5 hr) or with the muscarinic agonist carbachol (100 μm; 5 hr) resulted in no changes in MIF levels, either intracellular or extracellular (Fig. 1D).
It is also important to note that the stimulatory effects of Ang II on MIF levels were not attributable to effects on the astroglia, which account for ∼5% of the cell population of the neuronal cultures used here. The micrographs in Figure 2 demonstrate that MIF is primarily localized in neurons in culture, and incubation of pure WKY rat astroglial cultures with Ang II (100 nm; 5 hr) produced no significant changes in the levels of intracellular MIF (7.4 ± 0.6 ng/mg protein in controls vs 7.8 ± 0.8 ng/mg protein in Ang II-treated neurons).
Figure 2.

MIF immunostaining within neural cells in culture. A-C are confocal images taken from the same field of WKY rat neuronal cultures stained for the neuronal marker α-internexin (red channel) (A), MIF (green channel) (B), and overlap between α-internexin and MIF staining (yellow) (C). D-F are confocal images taken from the same field of WKY rat mixed neuronal-glial cultures stained for the astroglial-specific marker GFAP (red channel) (D) and MIF (green channel) (E); F demonstrates little overlap between MIF and GFAP staining. Scale bar, 50 μm.
MIF decreases neuronal firing
Ang II produces an AT1-R-mediated increase in firing rate in normotensive WKY and SD rat neurons in culture via a ROS-dependent mechanism (Wang et al., 1997; Sun et al., 2004) and increases MIF expression in both cases (Busche et al., 2001) (Fig. 1). In addition, the MIF molecule exhibits a thiol-oxidoreductase activity with the potential to scavenge ROS (Nguyen et al., 2003a,b). From this, we hypothesized that increased expression of MIF may negatively regulate Ang II-induced neuronal firing in normotensive rat neurons via a thiol-oxidoreductase-ROS scavenging mechanism. This hypothesis was examined in the following series of studies.
We first examined the effects of rMIF on the basal firing rate of WKY and SD rat neurons. The data shown in Figure 3, A and B, indicate that intracellular application of rMIF (80 nm ≡ 1.0 ng/μl) produces a significant decrease in the basal firing of neurons prepared from WKY or SD rats. The concentration dependency of this MIF effect was tested by intracellular application of the recombinant protein (0.8-800 nm) into WKY rat neurons. The results demonstrate that a significant reduction of basal neuronal firing was obtained with 8 nm rMIF, and that the response is maximal between 80 and 800 nm (Fig. 3C). This effect of rMIF was abolished by denaturing the protein through boiling for 20 min (data not shown). In addition, the reduction of basal firing produced by rMIF was abolished by simultaneous administration of anti-MIF antibodies (aMIF; 1:500) (Fig. 3D). Extracellular application of rMIF (1000 nm) in the superfusate solution produced no changes in neuronal firing (0.87 ± 0.14 Hz in controls vs 0.95 ± 0.18 Hz in rMIF-treated neurons; n = 4 neurons).
Figure 3.

MIF reduces the basal firing of SD and WKY rat neurons. A, Recordings of APs from representative SD and WKY rat neurons under the following conditions. Superfusion of control (Con) solution (PBS) followed by intracellular application of rMIF (80 nm) as detailed in Materials and Methods. B, Bar graphs show means ± SEM basal firing rate values under each treatment condition in SD and WKY rat neurons. Numbers of neurons are indicated in parentheses. *p < 0.05 versus respective control. C, rMIF was applied intracellularly at the indicated concentrations, and basal firing was recorded. Note that one concentration of rMIF was tested in one neuron. Shown here is the percentage inhibition of 1 firing (mean ± SEM; n = 5-7 neurons) produced by each concentration of rMIF, compared with controls (no rMIF). *p < 0.01 versus control. D, rMIF (80 nm) was applied intracellularly in the presence of anti-MIF antibodies (aMIF; 1:500), and basal firing was recorded. Bar graphs show means ± SEM basal firing rate values under each treatment condition (n = 5 neurons).
Having established that MIF alone can alter basal neuronal firing, we investigated whether MIF could influence Ang II-induced increases in firing and used two different approaches. In the first approach, we determined whether endogenous MIF, produced in response to Ang II treatment, can influence the Ang II-induced chronotropic action. WKY rat neuronal cultures were pretreated with either control solution (PBS) or Ang II (100 nm) for 5 hr, the latter treatment producing an increase in intracellular MIF levels (Fig. 2). In the neurons pretreated with PBS, superfusion of Ang II (100 nm) produced a significant increase in firing rate (Fig. 4A). This chronotropic action of Ang II was not affected by simultaneous intracellular application of MIF-neutralizing antibody (aMIF; 1:500) (Fig. 4A). Intracellular application of aMIF alone into PBS-pretreated neurons produced no changes in firing rate (Fig. 4A). The response profile of the Ang II-pretreated neurons was very different. In these cells, superfusion of Ang II (100 nm) failed to alter firing rate (Fig. 4B). However, in the presence of intracellular application of aMIF, similar superfusion of Ang II produced a significant chronotropic action (Fig. 4B). Intracellular application of aMIF alone into the Ang II-pretreated neurons also produced a significant chronotropic action (Fig. 4B). In control experiments, we determined that intracellular application of normal rabbit serum (IgG; 1:500) into PBS or Ang II pretreated cultures elicited no significant changes in neuronal firing rate (Fig. 4A,B). Collectively, these studies suggest that the increased levels of MIF produced by Ang II pretreatment act to suppress subsequent effects of Ang II on neuronal firing.
Figure 4.
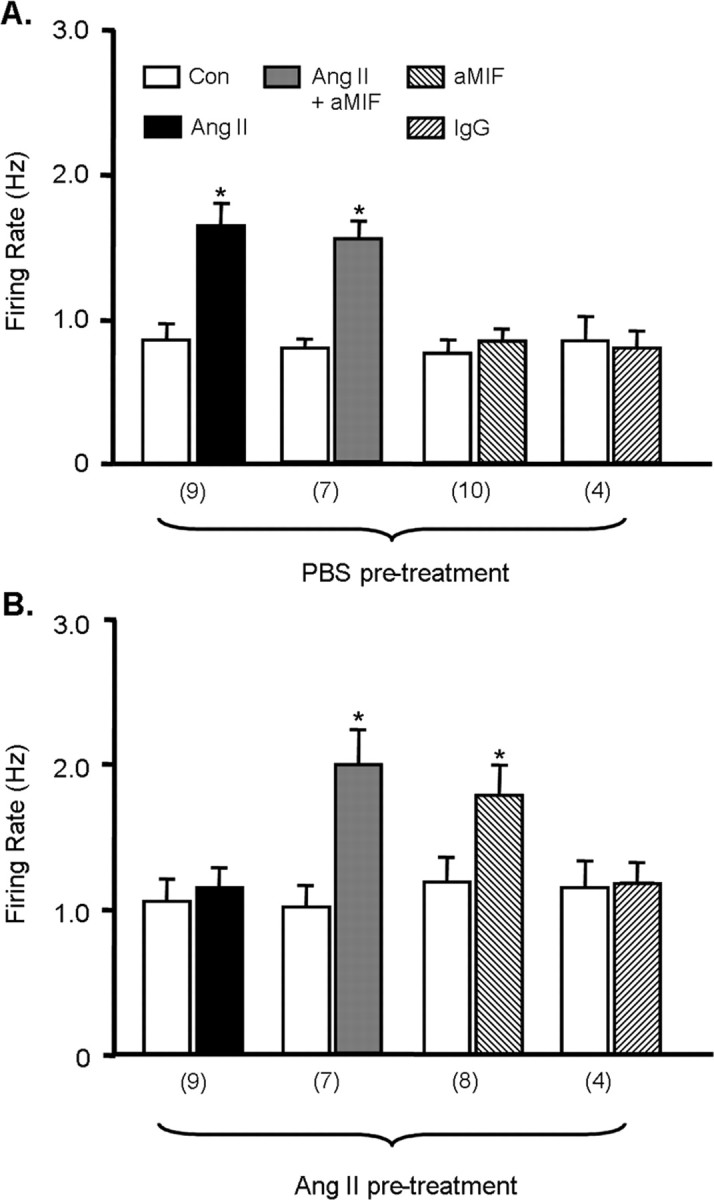
Intracellular MIF modulates Ang II-induced increases in neuronal firing. A, B, WKY rat neurons were pretreated with control (Con) solution (PBS; A) or 100 nm Ang II (B) for 5 hr at 37°C and then neuronal firing was recorded, as described in Materials and Methods, under the following treatment conditions: superfusion with control solution (PBS), superfusion with 100 nm Ang II, superfusion with 100 nm Ang II plus intracellular application of anti-MIF antibodies (aMIF; 1:500), intracellular application of anti-MIF antibodies (aMIF; 1:500), and intracellular application of normal rabbit serum (IgG; 1:500). Data are mean ± SEM firing rate values in each treatment condition. Numbers of neurons tested for each group are indicated in parentheses below the x-axis. *p < 0.05 versus respective control cultures.
In the second approach, we tested the idea of MIF as an intracellular regulator of Ang II action in a more direct manner by examining whether intracellular application of rMIF inhibits the Ang II-induced increase in firing. For these experiments, we chose a concentration of MIF (0.8 nm) that produced no significant change in basal firing (Fig. 3C). Superfusion of WKY rat neurons in culture with Ang II (100 nm) elicited a significant chronotropic effect, as expected (Fig. 5A,B). This effect of Ang II was significantly depressed by intracellular application of rMIF (0.8 nm) (Fig. 5A,B). The inhibitory action of rMIF was abolished by denaturing the protein by boiling (data not shown). To examine the specificity of this MIF action, we tested the effects of a control protein, DCT, on the Ang II-induced chronotropic action. DCT was chosen because it shares ∼27% amino acid identity with MIF, at 118 amino acids is similar in size, and, like MIF, exhibits tautomerase activity (Zhang et al., 1995). Of importance, DCT also lacks one of the two homologous cysteines (cys60) that mediate the oxido-reductase activity of MIF. The data shown in Figure 5, C and D, clearly indicate that intracellular application of DCT (0.8 nm) produces no inhibitory effects on the Ang II-induced chronotropic action in WKY rat neurons in culture.
Figure 5.

rMIF inhibits the chronotropic action of Ang II in WKY rat neurons. A, Recordings of APs from a representative WKY rat neuron under the following sequential treatment conditions: superfusion of control (Con) solution (PBS), superfusion of Ang II (100 nm), wash with superfusate solution, intracellular application of rMIF (0.8 nm), and intracellular application of rMIF plus superfusion of Ang II. B, Bar graphs are means ± SEM of firing rate recorded in each treatment situation in A and are from five neurons. *p < 0.01 versus respective control treatment; #p < 0.05 versus Ang II treatment. C, Recordings of APs from a representative WKY rat neuron under the following sequential treatment conditions: superfusion of control solution (PBS), superfusion of Ang II (100 nm), wash with superfusate solution, intracellular application of DCT (0.8 nm), and intracellular application of DCT plus superfusion of Ang II. D, Bar graphs are means ± SEM of firing rate recorded in each treatment situation in C and are from seven neurons. *p < 0.01 versus respective control treatment.
MIF-induced decrease in neuronal firing: role of the MIF oxidoreductase activity and ROS.
Amino acid residues C57 and C60 of the MIF molecule exert a thiol-protein oxidoreductase property that is responsible for certain cellular actions of MIF (Lue et al., 2002). A recent study has indicated that a 16 amino acid peptide fragment of MIF [MIF-(50-65)] containing these residues mimics the thiol-protein oxidoreductase property and has MIF-like biological activity (Nguyen et al., 2003a). Thus, we determined the effects of rat MIF-(50-65) on neuronal firing to determine whether this peptide mimics the MIF effects. The data presented in Figure 6 clearly show that intracellular application of MIF-(50-65) (0.8 nm) produces complete inhibition of the chronotropic action of Ang II, similar to rMIF. In contrast, a mutant peptide in which the C57 and C60 residues have been replaced by serines and lacks the thiol-protein oxidoreductase property of MIF fails to affect the chronotropic action of Ang II after intracellular application (Fig. 6). These data suggest that the inhibitory action of rMIF on neuronal firing may involve the C57 and C60 residues and the thiol-protein oxidoreductase property of the MIF molecule.
Figure 6.

MIF-(50-65) inhibits the chronotropic action of Ang II in WKY rat neurons. A, Recordings of APs from a representative WKY rat neuron under the following sequential treatment conditions: superfusion of control (Con) solution (PBS), superfusion of Ang II (100 nm), wash with superfusate solution, intracellular application of MIF-(50-65) (0.8 nm), and intracellular application of MIF-(50-65) plus superfusion of Ang II. B, Bar graphs are means ± SEM of firing rate recorded in each treatment situation described in A and in similar experiments where C57S/C60S-MIF-(50-65) was substituted for MIF-(50-65). Data are from five neurons in each case. *p < 0.01 versus respective control treatment; #p < 0.05 versus Ang II treatment.
In other studies, we determined that the AT1-R-mediated chronotropic action of Ang II is mediated by generation of ROS (Sun et al., 2004). For example, the increase in neuronal firing produced by 100 nm Ang II (0.63 ± 0.15 Hz in controls vs 1.68 ± 0.36 Hz in Ang II-treated neurons; n = 7; p < 0.05) was significantly blunted by simultaneous treatment with 1 mm tempol, an ROS scavenger (0.81 ± 0.14 Hz in Ang II/tempol-treated neurons and 0.6 ± 0.1 Hz in neurons with tempol alone; n = 7). Thus, one way in which the C57 and C60 sulfhydryl residues of MIF may be involved in blunting the chronotropic action of Ang II is by scavenging ROS. To provide support for this idea, we determined whether overexpressing MIF in neurons could alter the Ang II-induced ROS generation. For this, we developed an adenoviral construct containing MIF cDNA driven by the neuron-specific SYN gene promoter (pAd5-SYN-MIF) and an associated control vector pAd5-SYN-EGFP (Fig. 7A). Incubation of WKY rat neuronal cultures with the Ad5-SYN-MIF construct (2 × 106 or 1 × 107 infectious units (ifu)/ml) caused a large increase in MIF protein expression (Fig. 7B). Incubation of neuronal cultures with pAd5-SYN-EGFP (1 × 107 ifu/ml) resulted in ∼95% of the neurons within the culture dish displaying green fluorescence (Fig. 7C-E). When neuronal cultures were incubated with Ang II (100 nm; 2 min), there was an increase in ROS generation in cultures that had been pretreated with Ad5-SYN-EGFP (1 × 107 ifu/ml) (Fig. 7F-H,L). However, in the cultures preincubated with Ad5-SYN-MIF (1 × 107 ifu/ml), the Ang II did not increase ROS levels (Fig. 7I-K,L). These data demonstrate that Ang II fails to increase ROS levels in neurons where MIF has been overexpressed.
Figure 7.

Effect of overexpression of MIF in WKY rat neurons on the Ang II-stimulated increases in ROS levels. A, Ad5-SYN-MIF and Ad5-SYN-EGFP constructs. B, Western blot showing that MIF expression in WKY rat cerebral cortex neuronal cultures is increased by Ad5-SYN-MIF. Lane 1, Marker; lane 2, control neurons; lane 3, Ad5-SYN-EGFP (1 × 107 ifu); lanes 4-6, Ad5-SYN-MIF (4.4 × 105, 2 × 106, and 1 × 107 ifu, respectively). C-E, Fluorescence micrographs of a neuron infected with Ad5-SYN-EGFP (1 × 107 ifu) for 3 d. C, EGFP-positive cells. D, NeuN (red) immunostaining of the same field of cells. E, Overlap (yellow-orange) between EGFP and NeuN-positive cells. F-H, Representative fluorescence micrographs of a neuron infected with Ad5-SYN-EGFP (1 × 107 ifu) for 3 d. F, Transduced neuron exhibiting green fluorescence. G, The same transduced neuron as in F, exhibiting ethidium fluorescence under control conditions. Scale bar, 50 μm. H, The same transduced neuron as in F, exhibiting ethidium fluorescence (arrow) after Ang II treatment (100 nm; 2 min). I-K, Micrographs of a neuron infected with Ad5-SYN-MIF (1 × 107 if u) for 3 d. I, Transduced neuron under phase. J, Thesame transduced neuron as in I, exhibiting ethidium fluorescence under control conditions. K, The same transduced neuron as in I, exhibiting ethidium fluorescence after Ang II treatment (100 nm; 2 min). L, Bar graphs are means ± SEM of ethidium fluorescence intensity from Ad5-SYN-EGFP and Ad5-SYN-MIF-infected neurons after treatment with control solution or Ang II (n = 21 neurons). *p < 0.01 versus respective control treatment. LITR, Left inverted terminal repeats; RITR, right inverted terminal repeats.
Discussion
In this study, we investigated the interactions between Ang II and MIF in neurons cultured from newborn rat hypothalamus and brainstem. The major findings are as follows. First, Ang II increases the intracellular levels of MIF in neurons but does not alter MIF release. Ang II does not alter MIF expression or release in cultured astroglia. Second, increased levels of intracellular MIF inhibit the stimulatory actions of Ang II on neuronal firing. Third, the data suggest that these inhibitory actions of MIF are mediated via a thiol-oxidoreductase activity of the MIF molecule and subsequent scavenging of ROS. Collectively, these findings are consistent with the idea that MIF is an intracellular inhibitor of Ang II-induced increase in neuronal activity. As such, the data are the first to demonstrate the existence of a negative regulator of the actions of Ang II in neurons. However, many questions remain unanswered.
One set of questions concerns the nature of the intracellular signaling mechanisms by which Ang II increases MIF expression in normotensive rat neurons. The MIF gene promoter region, at least in mouse and humans, contains several DNA-binding consensus sequences for transcription factors, including activator protein-1 (AP-1) and cAMP response element binding protein (Mitchell et al., 1995; Calandra and Roger, 2003). Previously, we determined that Ang II elicits an AT1-R-mediated increase in PKC activity and induces AP-1 DNA binding in normotensive rat neuronal cultures (Sumners et al., 1996; Fleegal and Sumners, 2003). In addition, we determined that the Ang II-induced increase in neuronal MIF expression is calcium and PKC dependent (Busche et al., 2001). Thus, it is reasonable to conclude that the Ang II-induction of MIF expression involves a PKC/AP-1 mechanism. However, the situation may not be that simple. For example, we have demonstrated that the Ang II-induced increase in PKC activity in neurons causes a subsequent increase in protein kinase A (PKA) activity and cAMP levels (S. Busche, S. Gallinat, and C. Sumners, unpublished observations). In addition, treatment of neuronal cultures with PKC or PKA stimulatory agents elicits a large increase in MIF expression (C. Sun, M. K. Raizada, and C. Sumners, unpublished observations). Thus, it is possible that the Ang II induction of MIF also involves PKA activation. The above ideas are clearly speculative, however, and will only be supported by additional studies.
Another major question that arises from our studies concerns the mechanisms that are involved in the MIF regulation of neuronal firing. Clearly, MIF can exert effects on cellular function via binding to a cell surface CD74 binding protein (Leng et al., 2003). In addition, intracellular actions of MIF have been proposed. For example, studies indicated that MIF can be internalized from the extracellular milieu and interact with c-Jun activity-binding protein (JAB-1) and consequently inhibit JAB-1 function (Kleemann et al., 2000, 2002). However, our studies indicate that extracellular application of rMIF to neuronal cultures produces no changes in neuronal firing. Furthermore, our data indicate that Ang II increases MIF intracellularly, and it is the increase in the intracellular level of MIF that can alter neuronal firing. Thus, we propose that MIF remains within the intracellular compartment and exerts an influence on Ang II-induced neuronal firing via an enzymatic activity of the MIF molecule. Specifically, it has been shown that MIF can act as a thiol-oxidoreductase by virtue of a cysteine-alanine-leucine-cysteine (CALC) motif at residues 57-60 of the MIF molecule (Kleemann et al., 1998). Our data demonstrate that intracellular application of a peptide that contains this CALC motif mimics the inhibitory action of MIF on Ang II-induced chronotropic actions. Therefore, the data suggest that the actions of MIF on neuronal firing are attributable to the thiol-oxidoreductase activity of MIF. One potential consequence of increases in thiol-oxidoreductase activity is scavenging of ROS and blockade of oxidant-mediated intracellular actions (Nguyen et al., 2003b). Considering that Ang II signals via ROS in neurons (Zimmerman et al., 2002; Sun et al., 2004), and our data (Fig. 7) indicate that Ang II fails to increase ROS production in neurons where MIF has been overexpressed, it is possible that the MIF inhibition of Ang II-stimulated neuronal firing involves a ROS-scavenging mechanism. However, more studies are required to prove this idea and to test other possibilities such as an inhibitory effect of MIF, via thiol-oxidoreductase, at the level of the AT1-R. It is also well known that the MIF molecule exhibits a tautomerase activity (Rosengren et al., 1996). Our studies indicate that intracellular application of the protein, d-dopachrome tautomerase, which shares with MIF an N-terminal tautomerase but not oxido-reductase activity, did not affect the Ang II-induced increase in neuronal firing. These data, together with the MIF peptide results, are consistent with the conclusion that the oxidoreductase activity of MIF regulates neuronal firing in response to Ang-II. Additional structure-function studies, in genetically defined mouse strains for instance (Bozza et al., 1999), will help to better define the action of MIF in neuronal physiology.
An interesting point that arose from the current studies is that MIF can depress baseline neuronal firing, albeit at higher concentrations than were needed for the inhibition of chronotropic actions of Ang II. Although the significance of this finding is not clear, it may mean that MIF can exert a general depressive action on neuronal firing if its intracellular levels are stimulated beyond a certain level.
Although our studies have so far focused on MIF and Ang II interactions in vitro using primary neuronal cultures, it is clear that additional studies must also address this interaction in vivo. In other words, the question is whether increased MIF expression within the brain can exert inhibitory effects on any of the Ang II-induced physiological actions such as increases in blood pressure and drinking. Our previous studies demonstrate that intracerebroventricular injection of Ang II increases MIF expression in SD rat hypothalamus, similar to its effects in neuronal cultures (Busche et al., 2001). Thus, it will be important to identify the particular loci within the hypothalamus where Ang II increases MIF levels and to modulate the neuronal expression of MIF at those sites and determine the effects on Ang II-induced physiological actions. One possible way of achieving this might be to use mutant mice (MIF-/-) that are devoid of MIF (Bozza et al., 1999). Thus far, there have been no reports of studies on Ang II receptors or responses in those animals. However, the MIF knock-out mice may not be the best system to use for a number of reasons. First, they represent a global knock-out of MIF, and it is likely that the changes in MIF in the brain produced by Ang II are highly localized to certain nuclei. Second, based on the present studies, it is important that MIF expression is altered in neurons only, rather than in all brain cell types, as would be the case with the MIF-/- animals. In addition, it is also clear that Ang II can increase MIF expression in tissues outside the CNS, such as the kidney (Rice et al., 2003). Therefore, it is important that we focus on modulatory MIF expression in neurons at the sites of Ang II action in the CNS, in the absence of changes in MIF expression at other sites or tissues. Overexpression may be achieved with the Ad-Syn-MIF construct used in the present study via microinjection into specific brain nuclei. Depleting of MIF at these sites will be more difficult but may be possible through the use of specific MIF RNAi. Once the means for specific overexpression or depletion of MIF in the brain have been achieved, we will be able to better address the subject of MIF interactions with Ang II in vivo.
A final point of discussion concerns the cellular location of the interactions of Ang II with MIF in neurons. Our studies focus on modulation by MIF of the postsynaptic actions of Ang II. However, it appears from a number of studies that certain AT1-Rs are localized presynaptically, and that Ang II has presynaptic actions in the brain (Huang et al., 2003; Latchford and Ferguson, 2003; Li et al., 2003). Thus, an additional question concerns whether MIF can act as a regulator of the presynaptic actions of Ang II via AT1-R.
In summary, this study provides the first evidence for the existence of a negative regulator of the actions of Ang II in neurons. That MIF may be an important factor in the central control of Ang II function is of high interest, given the recent description of functional alleles of the MIF gene in the human population (Baugh et al., 2002).
Footnotes
This work was supported by National Institutes of Health Grants 2R37 HL-33610 (M.K.R., C.S.) and 2RO1 AIO42310-07 (L.L., R.B.). We thank Fan Lin and Yongxin Gao for preparation of neuronal cultures.
Correspondence should be addressed to Dr. Colin Sumners, Department of Physiology and Functional Genomics, College of Medicine, P.O. Box 100274, 1600 Southwest Archer Road, University of Florida, Gainesville, FL 32610-0274. E-mail: csumners@phys.med.ufl.edu.
Copyright © 2004 Society for Neuroscience 0270-6474/04/249944-09$15.00/0
References
- Averill DB, Diz DI (2000) Angiotensin peptides and baroreflex control of sympathetic outflow: pathways and mechanisms of the medulla oblongata. Brain Res Bull 51: 119-128. [DOI] [PubMed] [Google Scholar]
- Bacher M, Meinhardt A, Lan HY, Mu W, Metz CN, Chesney JA, Calandra T, Gemsa D, Donnelly T, Atkins RC, Bucala R (1997) Migration inhibitory factor expression in experimentally induced endotoxemia. Am J Pathol 150: 235-246. [PMC free article] [PubMed] [Google Scholar]
- Baugh JA, Bucala R (2002) Macrophage migration inhibitory factor. Crit Care Med 30: S27-S35. [PubMed] [Google Scholar]
- Baugh JA, Chitnis S, Donnelly SC, Monteiro J, Lin X, Plant BJ, Wolfe F, Gregersen PK, Bucala R (2002) A functional promoter polymorphism in the macrophage migration inhibitory factor (MIF) gene associated with disease severity in rheumatoid arthritis. Genes Immun 3: 170-176. [DOI] [PubMed] [Google Scholar]
- Bernhagen J, Mitchell RA, Calandra T, Voelter W, Cerami A, Bucala R (1994) Purification, bioactivity, and secondary structure analysis of mouse and human macrophage migration inhibitory factor (MIF). Biochemistry 33: 14144-14155. [DOI] [PubMed] [Google Scholar]
- Bozza M, Satoskar AR, Lin G, Lu B, Humbles AA, Gerard C, David JR (1999) Targeted disruption of migration inhibitory factor gene reveals its critical role in sepsis. J Exp Med 189: 341-346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Busche S, Gallinat S, Fleegal MA, Raizada MK, Sumners C (2001) Novel role of macrophage migration inhibitory factor in angiotensin II regulation of neuromodulation in rat brain. Endocrinology 142: 4623-4630. [DOI] [PubMed] [Google Scholar]
- Calandra T, Roger T (2003) Macrophage migration inhibitory factor: a regulator of innate immunity. Nat Rev Immunol 3: 791-800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Culman J, Blume A, Gohlke P, Unger T (2002) The renin-angiotensin system in the brain: possible therapeutic implications for AT(1)-receptor blockers. J Hum Hypertens 16[Suppl 3]: S64-S70. [DOI] [PubMed] [Google Scholar]
- Dampney RA, Fontes MA, Hirooka Y, Horiuchi J, Potts PD, Tagawa T (2002) Role of angiotensin II receptors in the regulation of vasomotor neurons in the ventrolateral medulla. Clin Exp Pharmacol Physiol 29: 467-472. [DOI] [PubMed] [Google Scholar]
- de Gasparo M, Catt KJ, Inagami T, Wright JW, Unger T (2000) International union of pharmacology. XXIII. The angiotensin II receptors. Pharmacol Rev 52: 415-472. [PubMed] [Google Scholar]
- Donn RP, Ray DW (2004) Macrophage migration inhibitory factor: molecular, cellular and genetic aspects of a key neuroendocrine molecule. J Endocrinol 182: 1-9. [DOI] [PubMed] [Google Scholar]
- Ferguson AV, Washburn DL, Latchford KJ (2001) Hormonal and neuro-transmitter roles for angiotensin in the regulation of central autonomic function. Exp Biol Med (Maywood) 226: 85-96. [DOI] [PubMed] [Google Scholar]
- Fingerle-Rowson GR, Bucala R (2001) Neuroendocrine properties of macrophage migration inhibitory factor (MIF). Immunol Cell Biol 79: 368-375. [DOI] [PubMed] [Google Scholar]
- Fleegal MA, Sumners C (2003) Angiotensin II induction of AP-1 in neurons requires stimulation of PI3-K and JNK. Biochem Biophys Res Commun 310: 470-477. [DOI] [PubMed] [Google Scholar]
- Gyurko R, Wielbo D, Phillips MI (1993) Antisense inhibition of AT1 receptor mRNA and angiotensinogen mRNA in the brain of spontaneously hypertensive rats reduces hypertension of neurogenic origin. Regul Pept 49: 167-174. [DOI] [PubMed] [Google Scholar]
- Han NL, Sim MK (1998) Hypothalamic angiotensin receptor subtypes in normotensive and hypertensive rats. Am J Physiol 275: H703-H709. [DOI] [PubMed] [Google Scholar]
- He TC, Zhou S, da Costa LT, Yu J, Kinzler KW, Vogelstein B (1998) A simplified system for generating recombinant adenoviruses. Proc Natl Acad Sci USA 95: 2509-2514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang J, Hara Y, Anrather J, Speth RC, Iadecola C, Pickel VM (2003) Angiotensin II subtype 1A (AT1A) receptors in the rat sensory vagal complex: subcellular localization and association with endogenous angiotensin. Neuroscience 122: 21-36. [DOI] [PubMed] [Google Scholar]
- Jensen LL, Harding JW, Wright JW (1992) Role of paraventricular nucleus in control of blood pressure and drinking in rats. Am J Physiol 262: F1068-F1075. [DOI] [PubMed] [Google Scholar]
- Kleemann R, Kapurniotu A, Frank RW, Gessner A, Mischke R, Flieger O, Juttner S, Brunner H, Bernhagen J (1998) Disulfide analysis reveals a role for macrophage migration inhibitory factor (MIF) as thiol-protein oxidoreductase. J Mol Biol 280: 85-102. [DOI] [PubMed] [Google Scholar]
- Kleemann R, Hausser A, Geiger G, Mischke R, Burger-Kentischer A, Flieger O, Johannes FJ, Roger T, Calandra T, Kapurniotu A, Grell M, Finkelmeier D, Brunner H, Bernhagen J (2000) Intracellular action of the cytokine MIF to modulate AP-1 activity and the cell cycle through Jab1. Nature 408: 211-216. [DOI] [PubMed] [Google Scholar]
- Kleemann R, Grell M, Mischke R, Zimmermann G, Bernhagen J (2002) Receptor binding and cellular uptake studies of macrophage migration inhibitory factor (MIF): use of biologically active labeled MIF derivatives. J Interferon Cytokine Res 22: 351-363. [DOI] [PubMed] [Google Scholar]
- Kugler S, Meyn L, Holzmuller H, Gerhardt E, Isenmann S, Schulz JB, Bahr M (2001) Neuron-specific expression of therapeutic proteins: evaluation of different cellular promoters in recombinant adenoviral vectors. Mol Cell Neurosci 17: 78-96. [DOI] [PubMed] [Google Scholar]
- Latchford KJ, Ferguson AV (2003) Angiotensin II activates a nitric-oxide-driven inhibitory feedback in the rat paraventricular nucleus. J Neurophysiol 89: 1238-1244. [DOI] [PubMed] [Google Scholar]
- Leng L, Metz CN, Fang Y, Xu J, Donnelly S, Baugh J, Delohery T, Chen Y, Mitchell RA, Bucala R (2003) MIF signal transduction initiated by binding to CD74. J Exp Med 197: 1467-1476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li DP, Chen SR, Pan HL (2003) Angiotensin II stimulates spinally projecting paraventricular neurons through presynaptic disinhibition. J Neurosci 23: 5041-5049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lue H, Kleemann R, Calandra T, Roger T, Bernhagen J (2002) Macrophage migration inhibitory factor (MIF): mechanisms of action and role in disease. Microbes Infect 4: 449-460. [DOI] [PubMed] [Google Scholar]
- Mitchell R, Bacher M, Bernhagen J, Pushkarskaya T, Seldin MF, Bucala R (1995) Cloning and characterization of the gene for mouse macrophage migration inhibitory factor (MIF). J Immunol 154: 3863-3870. [PubMed] [Google Scholar]
- Muratani H, Teruya H, Sesoko S, Takishita S, Fukiyama K (1996) Brain angiotensin and circulatory control. Clin Exp Pharmacol Physiol 23: 458-464. [DOI] [PubMed] [Google Scholar]
- Nguyen MT, Beck J, Lue H, Funfzig H, Kleemann R, Koolwijk P, Kapurniotu A, Bernhagen J (2003a) A 16-residue peptide fragment of macrophage migration inhibitory factor, MIF-(50-65), exhibits redox activity and has MIF-like biological functions. J Biol Chem 278: 33654-33671. [DOI] [PubMed] [Google Scholar]
- Nguyen MT, Lue H, Kleemann R, Thiele M, Tolle G, Finkelmeier D, Wagner E, Braun A, Bernhagen J (2003b) The cytokine macrophage migration inhibitory factor reduces pro-oxidative stress-induced apoptosis. J Immunol 170: 3337-3347. [DOI] [PubMed] [Google Scholar]
- Oparil S, Yang RH, Jin HG, Chen SJ, Meng QC, Berecek KH, Wyss JM (1994) Role of anterior hypothalamic angiotensin II in the pathogenesis of salt sensitive hypertension in the spontaneously hypertensive rat. Am J Med Sci. 307[Suppl 1]: S26-S37. [PubMed] [Google Scholar]
- Rice EK, Tesch GH, Cao Z, Cooper ME, Metz CN, Bucala R, Atkins RC, Nikolic-Paterson DJ (2003) Induction of MIF synthesis and secretion by tubular epithelial cells: a novel action of angiotensin II. Kidney Int 63: 1265-1275. [DOI] [PubMed] [Google Scholar]
- Rosengren E, Bucala R, Aman P, Jacobsson L, Odh G, Metz CN, Rorsman H (1996) The immunoregulatory mediator macrophage migration inhibitory factor (MIF) catalyzes a tautomerization reaction. Mol Med 2: 143-149. [PMC free article] [PubMed] [Google Scholar]
- Sakai M, Nishihira J, Hibiya Y, Koyama Y, Nishi S (1994) Glutathione binding rat liver 13k protein is the homologue of the macrophage migration inhibitory factor. Biochem Mol Biol Int 33: 439-446. [PubMed] [Google Scholar]
- Sambrook JF, Fritsch EF, Maniatis T (1989) Molecular cloning: a laboratory manual, Chap 1. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory.
- Sumners C, Tang W, Zelezna B, Raizada MK (1991) Angiotensin II receptor subtypes are coupled with distinct signal transduction mechanisms in neurons and astrocytes from rat brain. Proc Natl Acad Sci USA 88: 7567-7571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sumners C, Zhu M, Gelband CH, Posner P (1996) Angiotensin II type 1 receptor modulation of neuronal K+ and Ca2+ currents: intracellular mechanisms. Am J Physiol 271: C154-C163. [DOI] [PubMed] [Google Scholar]
- Sumners C, Fleegal MA, Zhu M (2002) Angiotensin AT1 receptor signalling pathways in neurons. Clin Exp Pharmacol Physiol 29: 483-490. [DOI] [PubMed] [Google Scholar]
- Sun C, Sumners C, Raizada MK (2002) Chronotropic action of angiotensin II in neurons via protein kinase C and CaMKII. Hypertension 39: 562-566. [DOI] [PubMed] [Google Scholar]
- Sun C, Raizada MK, Sumners C (2004) Angiotensin II-induced increases in neuronal firing: role of ROS. FASEB J 18: 834.3. [Google Scholar]
- Wang D, Gelband CH, Sumners C, Posner P (1997) Mechanisms underlying the chronotropic effect of angiotensin II on cultured neurons from rat hypothalamus and brain stem. J Neurophysiol 78: 1013-1020. [DOI] [PubMed] [Google Scholar]
- Zhang M, Aman P, Grubb A, Panagopoulos I, Hindemith A, Rosengren E, Rorsman H (1995) Cloning and sequencing of a cDNA encoding rat d-dopachrome tautomerase. FEBS Lett 373: 203-206. [DOI] [PubMed] [Google Scholar]
- Zhu M, Natarajan R, Nadler JL, Moore JM, Gelband CH, Sumners C (2000) Angiotensin II increases neuronal delayed rectifier K+ current: role of 12-lipoxygenase metabolites of arachidonic acid. J Neurophysiol 84: 2494-2501. [DOI] [PubMed] [Google Scholar]
- Zhu M, Sumners C, Gelband CH, Posner P (2001) Chronotropic effect of angiotensin II via type 2 receptors in rat brain neurons. J Neurophysiol 85: 2177-2183. [DOI] [PubMed] [Google Scholar]
- Zimmerman MC, Lazartigues E, Lang JA, Sinnayah P, Ahmad JM, Spitz DR, Davisson RL (2002) Superoxide mediates the actions of angiotensin II in the central nervous system. Circ Res 91: 1038-1045. [DOI] [PubMed] [Google Scholar]