

Abstract
The syntrophins are modular adapter proteins that function by recruiting signaling molecules to the cytoskeleton via their direct association with proteins of the dystrophin protein family. We investigated the physiological function of β2-syntrophin by generating a line of mice lacking this syntrophin isoform. The β2-syntrophin null mice show no overt phenotype, or muscular dystrophy, and form structurally normal neuromuscular junctions (NMJs). To determine whether physiological consequences caused by the lack of β2-syntrophin were masked by compensation from the α-syntrophin isoform, we crossed these mice with our previously described α-syntrophin null mice to produce mice lacking both isoforms. The α/β2-syntrophin null mice have NMJs that are structurally more aberrant than those lacking only α-syntrophin. The NMJs of the α/β2-syntrophin null mice have fewer junctional folds than either parent strain, and the remaining folds are abnormally shaped with few openings to the synaptic space. The levels of acetylcholine receptors are reduced to 23% of wild type in mice lacking both syntrophin isoforms. Furthermore, the α/β2-syntrophin null mice ran significantly shorter distances on voluntary exercise wheels despite having normal neuromuscular junction transmission as determined by micro-electrode recording of endplate potentials. We conclude that both α-syntrophin and β2-syntrophin play distinct roles in forming and maintaining NMJ structure and that each syntrophin can partially compensate for the loss of the other.
Keywords: dystrophin, acetylcholine receptor, nNOS, junctional folds, scaffold, neuromuscular
Introduction
The syntrophins are a widely expressed family of adapter proteins that associate with dystrophin and the dystrophin-related proteins utrophin and dystrobrevin. The domain structure of the syntrophins suggests that they function as adapter proteins that recruit signaling proteins, channels, and transporters to the transmembrane dystrophin protein complex. Each of the five syntrophins (α, β1, β2, γ1, γ2) consists of two pleckstrin homology (PH) domains, a postsynaptic density-95/Discs large/zona occludens (PDZ) domain, and a syntrophin unique (SU) region (Ahn et al., 1994; Adams et al., 1995; Piluso et al., 2000). The second PH domain and the SU region form the smallest fragment known to bind to utrophin (Kachinsky et al., 1999), dystrophin, and the dystrobrevins (Ahn and Kunkel, 1995). Many proteins have been identified as syntrophin PDZ domain ligands, including neuronal nitric oxide synthase (nNOS) (Brenman et al., 1996), serine/threonine protein kinases (Hasegawa et al., 1999; Lumeng et al., 1999), diacylglycerol kinase ζ (Hogan et al., 2001), the receptor tyrosine phosphatase-like pancreatic islet protein ICA512 (Ort et al., 2000), the voltage-gated sodium channels Nav1.4 and Nav1.5 (Gee et al., 1998; Ou et al., 2003), aquaporin-4 (Neely et al., 2001), the inward rectifier potassium channels Kir 2.2 and Kir 4.1 (Connors et al., 2004; Leonoudakis et al., 2004), and the ATP-binding cassette transporter A1 (Buechler et al., 2002).
One example of the physiological function of syntrophin-ligand interactions occurs in skeletal muscle where the α-syntrophin PDZ domain is required for localization of nNOS to the sarcolemma (Kameya et al., 1999; Adams et al., 2000, 2001). Disruption of the dystrophin/α-syntrophin/nNOS complex by deletion of dystrophin (Thomas et al., 1998), α-syntrophin (Thomas et al., 2003), or just the α-syntrophin PDZ domain (Thomas et al., 2003) results in the failure of skeletal muscle to attenuate α-adrenergic stimulated vasoconstriction during contraction.
Although all five syntrophins have a similar domain structure and often bind the same proteins in vitro, they do not necessarily do so in vivo. For example, α- and β-syntrophins interact similarly with nNOS in vitro; however, despite their highly conserved PDZ domain primary structure, neither β1 nor β2 syntrophin functionally binds nNOS in skeletal muscle (Adams et al., 2000). These results suggest that each syntrophin has a unique set of preferred PDZ ligands in vivo. Furthermore, cells often express more than one syntrophin isoform. In skeletal muscle, four of the five syntrophin isoforms (α, β1, β2, γ2) are expressed (Adams et al., 1993; Ahn et al., 1994; Piluso et al., 2000), with both α- and β2-syntrophin present at the postsynaptic neuromuscular junction (NMJ) (Peters et al., 1994; Kramarcy and Sealock, 2000).
To investigate the functions of syntrophin in vivo, we previously generated and characterized mice lacking α-syntrophin (Adams et al., 2000). In addition to the loss of nNOS and aquaporin-4 in muscle, these mice have aberrant neuromuscular junctions with sharply reduced levels of acetylcholine receptors (AChRs) and acetylcholine esterase, undetectable postsynaptic utrophin, and altered morphology; however, the α-syntrophin null mice show no sign of muscular dystrophy and appear to function normally under basal laboratory conditions. We therefore postulated that another syntrophin is partially compensating for the loss of α-syntrophin.
In this study, we describe the generation of β2-syntrophin null mice and characterize their neuromuscular junction pheno-type. We also crossed this mouse line with α-syntrophin null mice and used the α/β2-syntrophin double-null mice to investigate the extent to which one syntrophin can compensate for the loss of the other. We show that mice lacking both syntrophin isoforms have a more severe phenotype than mice lacking only one syntrophin.
Materials and Methods
Generation of β2-syntrophin null mice. We have previously characterized the gene encoding mouse β2-syntrophin isolated from a genomic library derived from 129Sv DNA (Adams et al., 1995). A targeting vector was constructed by cloning a 3 kb EcoRI/Ec 47 restriction fragment from the region 5′ of exon 1 and a 3 kb XhoI fragment from intron 1 into the plasmid JNS2 (Dombrowicz et al., 1993) (see Fig. 1). The resulting recombinant gene is missing the entire first exon (amino acids 1-173, which encode half of the first PH domain and the entire PDZ domain), 0.5 kb of 5′ flanking sequence, and 0.3 kb of the first intron. The targeting vector was linearized with NotI and transferred to embryonic day 14 embryonic stem (ES) cells by electroporation. After selection with G418 and gancyclovir (a gift from Roche Biosciences, Palo Alto, CA), homologous recombinant ES cells were identified by Southern blotting and used for C57BL/6 blastocyst injection performed by the University of North Carolina Embryonic Stem Cell Facility. Chimeras were bred with C57BL/6 females, and germline transmission was confirmed by Southern blot analysis. The mice used in this study either were first generation with a mixed 129Sv/C57BL/6 background or have been bred back against C57BL/6 for a minimum of five generations. Mice with either background show similar phenotypes.
Figure 1.

Generation of β2-syntrophin null mice. A, Schematic diagram of the construct used for targeted recombination of the β2-syntrophin gene. The first exon, 0.5 kb of 5′ flanking region, and 336 base pairs of intron 1 were deleted and replaced with the neomycin resistance gene. B, Southern blot analysis of mice homozygous for wild type (BB), heterozygous (Bb), and homozygous null for β2-syntrophin (bb). Genomic DNA was digested with restriction enzyme XbaI and probed with DNA from the area indicated in A. C, Immunoblot analysis of muscle extracts from wild-type (BB) and β2-syntrophin null mice (bb) using syntrophin isoform-specific antibodies.
Southern analysis. Genomic DNA was isolated from mouse tail biopsies using a QIAmp tissue kit (Qiagen, Valencia, CA), digested with XbaI, and resolved on a 1% agarose gel. GenScreen (DuPont NEN, Boston, MA) replicas were incubated with radiolabeled probe as described previously (Adams et al., 1995).
Antibodies. We have previously characterized the following antibodies: pan-specific syntrophin monoclonal antibody (mAb) SYN1351 (Froehner et al., 1987); isoform specific syntrophin polyclonal antibodies (pAbs) SYN17 (α-syntrophin), SYN28 (β2-syntrophin), and SYN37 (β1-syntrophin) (Peters et al., 1997); pAb 1862 (utrophin) (Kramarcy et al., 1994); pAb 2723 (C terminus of dystrophin) (Kramarcy et al., 1994); dystrobrevin Abs 638 (α-dystrobrevin-1), and DB2 (α-dystrobrevin-2) (Peters et al., 1998). The sodium channel antibody was a gift from S. Rock Levinson (University of Colorado Health Sciences Center). The polyclonal antibody to nNOS was purchased from Diasorin Inc. (Minneapolis, MN), and the neurofilament antibody was from Sigma-Aldrich (St. Louis, MO).
Immunoblotting. Muscle protein extracts were prepared as described previously (Peters et al., 1997). Syntrophin complex was isolated by incubating the extract with the pan syntrophin mAb 1351 followed by precipitation with protein G-coated beads (Sigma). The resulting pellets were resuspended in SDS-PAGE sample buffer, subjected to electrophoresis on a 10% polyacrylamide tricine-buffered gel, transferred to Immobilon-P membrane (Millipore, Bedford, MA), and incubated with the syntrophin isoform-specific antibodies as described previously (Peters et al., 1997).
Fluorescence microscopy. Immunofluorescence labeling of cross sections was performed on unfixed quadriceps muscle using 8 μm cryosections as described (Peters et al., 1998).
For en face views, 40 μm cryosections of sternomastoid muscle were incubated with Alexa 488 α-bungarotoxin (Molecular Probes, Eugene, OR) to visualize AChRs. Confocal microscopy was performed using a Leica TCS NT microscope either at the University of North Carolina (Chapel Hill, NC) or at the W. M. Keck Center for Advanced Studies in Neural Signaling (University of Washington, Seattle, WA). A Zeiss Axioskop with Axiocam was used to capture images for determining fluorescence intensity and for imaging the hematoxylin-eosin-stained tissue.
Fluorescence intensity quantification of labeled AChRs. Sternomastoid muscle AChRs were labeled with Alexa 488 α-bungarotoxin, and NMJs lying fully in the 40 μm section were digitally captured using identical settings for control and syntrophin null samples. Variations in section labeling were minimized by simultaneously labeling control and syntrophin null samples. Variations in excitation lamp intensity were minimized by capturing images of each syntrophin null sample and corresponding control sample on the same day. After the digital image of each NMJ was circumscribed using Adobe Photoshop, the difference between the average pixel intensity in the circumscribed area and average background pixel intensity of the corresponding muscle fiber was determined. The product of this difference and the number of circumscribed pixels gave a total AChR specific intensity measure for each NMJ.
Electron microscopy. Sternomastoid muscles were prepared for electron microscopy, and the nerve-muscle contacts in every NMJ encountered were imaged in only a single plane as described (Adams et al., 2000). Each nerve contact was analyzed by counting the number of junctional fold openings to the synaptic cleft and dividing by the total length of presynaptic membrane immediately apposed to the muscle cell (openings per micrometer). Virtually identical results were obtained by averaging this parameter over all contacts, regardless of their lengths, and by using a length-weighted average to avoid possible skewing of the result by the numerous small, possibly immature contacts.
Mouse running wheels. Equal numbers of wild-type and null mice (four each for α-syntrophin null; six each for β2-syntrophin null and α/β2-syntrophin null) were individually caged with access to a running wheel (Nalgene). The distance run voluntarily was monitored using standard bicycle speedometers (Specialized, Morgan Hill, CA) calibrated for the wheel size. Each set of mice was monitored for 8-10 weeks. For each syntrophin null mouse, the distance run per week was divided by the mean distance run by the control mice over the same week and multiplied by 100 to obtain the percentage of control.
Microelectrode recording. Endplate potentials (EPPs) and miniature endplate potentials (MEPPs) were recorded from diaphragm muscles of three control and syntrophin null mice at 30°C and corrected for a membrane potential of -80 mV. Miniature endplate currents (MEPCs) were recorded at room temperature with the membrane potential clamped at -80 mV (Engel et al., 1993). The quantal content of the EPP was estimated with correction for nonlinear summation and non-Poisson release. Stimulation was at 0.5 Hz. The initial 50 stimuli were discarded, and the next 64 were analyzed by the variance method (Nagel et al., 1990).
Results
Disruption of the α-syntrophin gene produced mice exhibiting structurally abnormal neuromuscular junctions with reduced levels of acetylcholine receptors and acetylcholine esterase and complete absence of detectable postsynaptic utrophin (Adams et al., 2000). Because β2-syntrophin has >43% amino acid identity to α-syntrophin and is highly localized at the NMJ postsynaptic membrane where, like α-syntrophin, it is present both at the AChR-rich crest and in the junctional folds (Peters et al., 1994; Kramarcy and Sealock, 2000), we hypothesized that this isoform may compensate to some extent for the loss of α-syntrophin. To examine this possibility, we used gene disruption technology to generate mice lacking β2-syntrophin (Fig. 1).
Generation and characterization of β2-syntrophin null mice
The targeted gene disruption removed the entire first exon, 0.5 kb of 5′ flanking sequence and 0.3 kb of the first intron (Fig. 1A). Genomic DNA from the first generation of mice was analyzed by Southern blot and gave bands of the predicted size, demonstrating that the β2-syntrophin gene was disrupted (Fig. 1B). Offspring of the founding heterozygotes were genotyped and found to occur at the expected Mendelian ratio of 1:2:1 (wild type/heterozygote/null) and therefore showed no evidence of embryonic lethality.
Using antibodies known to bind well downstream of exon I-encoded sequence (Peters et al., 1997; Adams et al., 2001), we confirmed the absence of β2-syntrophin at the protein level by performing immunoblots on muscle preparations enriched for syntrophins (Fig. 1C). Immunofluorescence studies of β2-syntrophin null skeletal muscle confirmed that β2-syntrophin is absent from the postsynaptic membrane, its predominant site of expression in muscle cells (Fig. 2). The loss of β2-syntrophin does not appear to affect the levels or distribution of postsynaptic α-syntrophin or β1-syntrophin at the NMJ. α-Syntrophin is present at both the crest and depths of the junctional folds, a distribution similar to that seen in wild-type mice. β1-syntrophin remains presynaptic at the β2-syntrophin null NMJ, as it is in wild-type mice, and is not upregulated postsynaptically in these adult mice (Fig. 2).
Figure 2.

β2-syntrophin is absent from the postsynaptic neuromuscular junction. Immunofluorescence of the neuromuscular junction of β2-syntrophin null mice shows absence of β2-syntrophin but no substantial upregulation of α- or β1-syntrophin. Scale bar, 5 μm.
The β2-syntrophin null mice have no overt phenotype. They are motile and fertile and live up to 2 years under laboratory conditions. Because β2-syntrophin associates directly with members of the dystrophin protein family, we examined the muscle for any dystrophic phenotype. Overall muscle structure as assessed by hematoxylin-eosin staining did not appear different from that of wild-type controls (Fig. 3). We saw no sign of significant muscle fibrosis or regeneration. Numbers of central nuclei were similar for controls and the β2-syntrophin null mice (data not shown).
Figure 3.

Muscle histology. Hematoxylin-eosin staining of mouse quadriceps muscle shows no evidence of muscle pathology in mice lacking β2-syntrophin (bb) or both α- and β2-syntrophin (aabb). Scale bar, 50 μm.
Generation and characterization of mice lacking both α-syntrophin and β2-syntrophin
To investigate the possibility that α-syntrophin and β2-syntrophin could compensate for each other when one is absent, we bred the α-syntrophin null mouse with the β2-syntrophin null to generate α/β2-syntrophin double-null mice. Like the parent strains, the α/β2-syntrophin null mice are motile and fertile and show no overt phenotype. Furthermore, hematoxylin-eosin staining of the α/β2-syntrophin null muscle revealed no indications of muscular dystrophy (Fig. 3).
Molecular characterization of NMJs
In α-syntrophin null muscle, junctional AChR levels are reduced to ∼35% of normal levels, and the AChR is distributed over abnormally shallow synaptic gutters, with unusual fingers of AChR extending outside the gutters (Adams et al., 2000). We used en face images of α-bungarotoxin-labeled NMJs to assess the β2- and α/β2-syntrophin null mice with respect to these characteristics.
The β2-syntrophin null NMJs have AChR levels slightly higher than those of control C57BL/6 mice (mean, 115% of wild type; p < 0.02). The observed increase in fluorescence intensity of the β2-syntrophin null NMJs appears to be caused mainly by a brighter labeling of the lateral edges, suggesting a slightly deeper neural groove. The distribution of AChR was normal (Fig. 4). Mice lacking both syntrophin isoforms express levels of AChRs significantly lower (p < 0.001) than those of the α-syntrophin null (mean, 23% of wild type), and the derangement of the AChR distribution is profound (Fig. 5), ranging from at least as significant as in the α-syntrophin null mouse to even more striking. In the latter case, broad fields of AChR organized in dots, short streaks, and fingers, with little hint of a synaptic gutter, and smaller, poorly formed patches of AChR that vary greatly in size and staining intensity are common. Most, but not all, of the small AChR accumulations are innervated (Fig. 5). In common with the most deranged regions of α-syntrophin null NMJs, all of these AChR distributions label weakly or not at all with the lectin Vicia villosa-B4, a marker for junctional folds (data not shown). These observations suggest the presence of immature contacts and possibly a high rate of synaptic remodeling.
Figure 4.
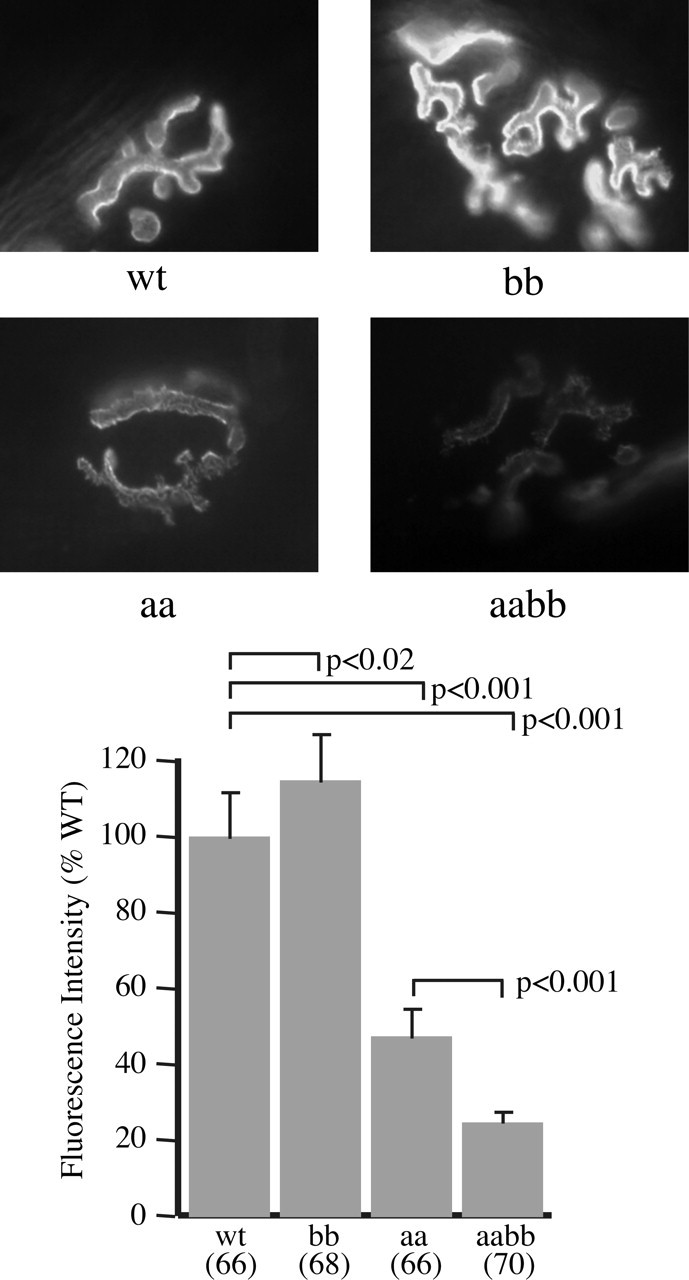
Acetylcholine receptor levels in syntrophin null mice. Images of sternomastoid muscle NMJs labeled with fluorescent α-bungarotoxin were captured under identical conditions using a 40× objective and used to determine the relative levels of AChRs in control and syntrophin null mice. Error bars indicate SEM.
Figure 5.
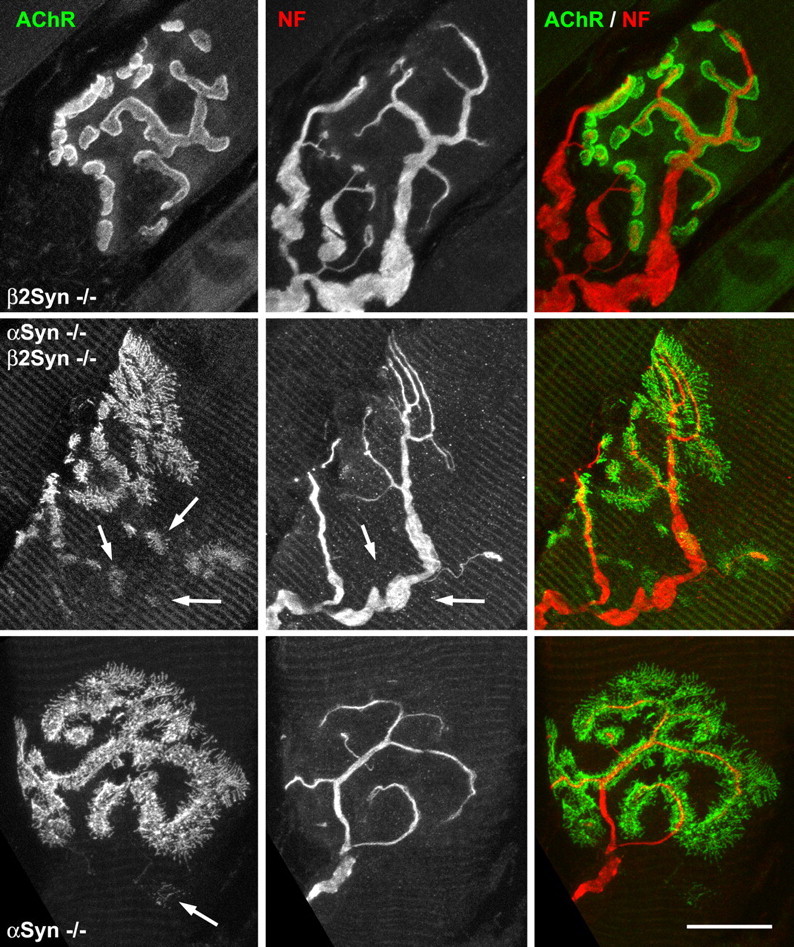
AChR distributions and innervation. NMJs in sternomastoid muscle labeled for AChR (green) and neurofilament (NF) (red). Top row, NMJs in β2-syntrophin null mice (AAbb) are essentially identical to wild-type controls except as noted in Figure 4. Middle row, α/β2-syntrophin double-null (aabb) NMJ showing large fields of AChR organized in dots, streaks, and fingers, plus several smaller patches of variable, lower-density AChR (left panel, arrows). Nerve branches going to such patches are of ten not apparent (middle panel, arrows). Bottom row, NMJs in the α-syntrophin null mouse (aaBB) appear to be less severely altered than in the double-null mice. Microscope parameters were adjusted to give similar maximum intensities of AChR image, despite the much lower AChR densities in the α-syntrophin null NMJs. Scale bars: top row, 13.9 μm; middle row, 15 μm; bottom row, 12.5 μm.
Both α- and β2- syntrophin are known to bind to members of the dystrophin protein family present at the NMJ, and, in principle, these associations could be essential for incorporation of these proteins at the NMJ. This idea was confirmed in the α-syntrophin null mouse, where the absence of α-syntrophin entrains elimination of utrophin from NMJs, without change in dystrophin at the NMJ or on the sarcolemma (Adams et al., 2000). We therefore investigated the distributions of proteins that interact with syntrophins in the NMJs of the β2- and α/β2-syntrophin null mice. In the β2-syntrophin null mice, dystrophin, utrophin, and dystrobrevin show distributions similar to those of control mice (Fig. 6). In particular, utrophin is present at normal levels and colocalized precisely with AChRs. Similarly, localization of dystrophin and dystrobrevin remain essentially unperturbed in mice lacking both α- and β2-syntrophin. Observed minor differences are likely caused by the structural alterations discussed below. Utrophin is absent from the postsynaptic NMJ in the α/β2-syntrophin double-null mice (Fig. 6A).
Figure 6.
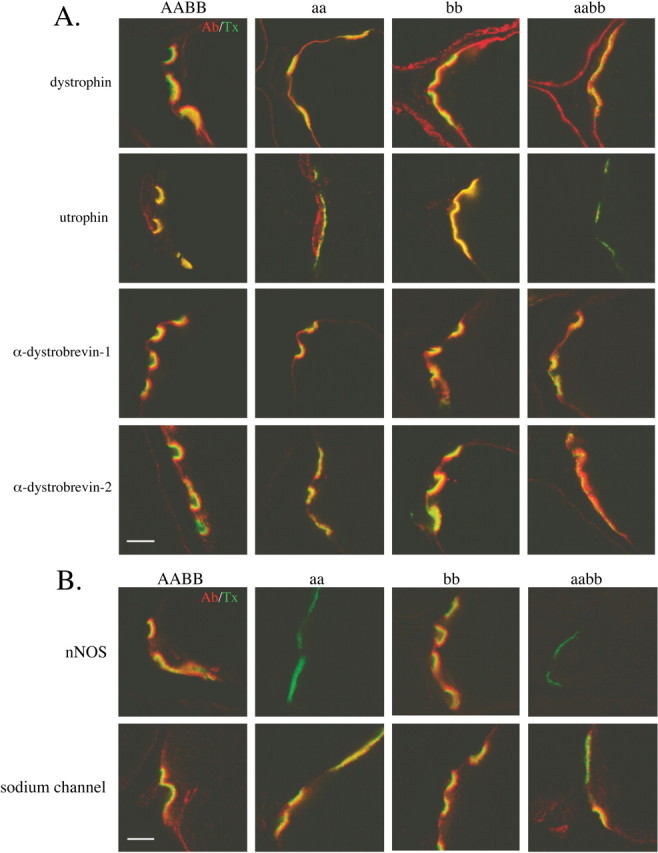
Syntrophin-associated proteins at the neuromuscular junction of syntrophin null mice. Composite images of the indicated dystrophin family proteins (red) and AChR (green). Note that utrophin is lost from the postsynaptic membrane only when α-syntrophin is absent. The absence of β2-syntrophin does not affect the distribution of the dystrophin family proteins. B, Composite images of nNOS or sodium channels (NaCh) (red) and AChR (green). nNOS remains present at the NMJ of the β2-syntrophin null mice but is lost when no α-syntrophin is present. Sodium channel distribution is unaffected by the loss of α-syntrophin, β2-syntrophin, or both syntrophin isoforms. Scale bar, 5 μm.
In vitro, the PDZ domains of both α- and β2-syntrophin bind to nNOS and the voltage-gated sodium channel, two signaling proteins present at the NMJ (Brenman et al., 1996; Gee et al., 1998). Mice lacking α-syntrophin lose the ability to localize nNOS at the sarcolemma and the NMJ despite the presence of β2-syntrophin (Adams et al., 2000). As expected, nNOS is also absent in the α/β2-syntrophin double-null mice, but in the absence of only β2-syntrophin it remains concentrated on the postsynaptic membrane (Fig. 6B). The distribution of the voltage-gated sodium channels, Nav 1.4 and Nav1.5, appears unaffected by the absence of either or both syntrophin isoforms (Fig. 6B).
Postsynaptic folds of syntrophin null mice
In wild-type NMJs, the junctional folds are basal lamina-lined folds of the postsynaptic membrane that are oriented more or less radially relative to the synaptic cleft and open to the cleft through most of their extent (Fig. 7B). In the α-syntrophin null mouse, the folds are plentiful but often misoriented relative to the surface and sometimes fragmented (Adams et al., 2000). Even those oriented toward the synaptic cleft are often not open to the cleft in any single image. A quantitative indication of this disorganization was given by the number of junctional fold openings to the synaptic cleft per micrometer of nerve-muscle contact; this parameter was reduced by 70% in the α-syntrophin null mouse relative to littermate controls (Adams et al., 2000). In the β2-syntrophin null mouse, the appearance of the folds was normal (Fig. 7A), and the number of openings in randomly selected single images of nerve-muscle contacts was not different from control (average of four mice = 2.31 openings per micrometer compared with 2.34 for one littermate wild-type mouse and 2.35 for a C57BL/6 control). An effect was apparent, however, when the β2-syntrophin null condition was placed on the α-syntrophin null background. The deficit of openings, even when numerous folds were present, was striking (Fig. 7C,D), and openings per micrometer were reduced by 90%, from 2.00 and 2.19 in two control mice to 0.19 and 0.16 in two α/β2-syntrophin null mice. Results of a qualitative examination of a third mouse were similar, i.e., the majority of nerve-muscle contacts showed no fold openings. The quantitative data for the three genotypes are summarized in Figure 7E. As in the α-syntrophin null and wild-type mice, all junctional folds in the α/β2-syntrophin null mice contained basal lamina (Fig. 7), and staining with tannic acid during fixation showed that they communicate with the extracellular space (data not shown). The location at which that communication takes place is not visible in most sections.
Figure 7.
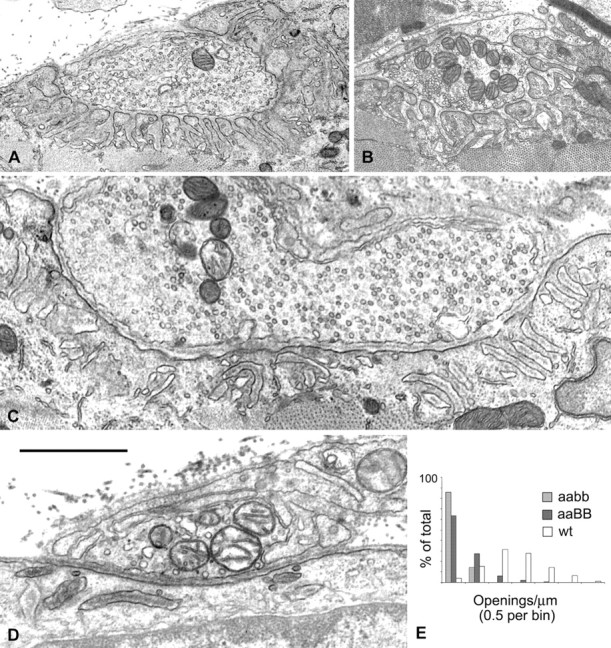
Electron microscopy of junctional folds. A, Nerve-muscle contacts in a β2-syntrophin null mouse and its wild-type littermate (B) show numerous, well organized junctional folds and associated openings to the synaptic cleft. C, D, Nerve-muscle contacts from α/β2-syntrophin null mice. C, A nerve-muscle contact of mature appearance. Approximately 20 fold-like structures are shown. Many are disorganized (left two-thirds of the image), but even where they are reasonably organized (right one-third), there are no openings to the synaptic cleft in the section shown. D, A contact of immature appearance. Disorganized fold structures are apparent just below the muscle cell surface. The central dark stripes within the fold structures in C and D are basal lamina. Scale bar: A, 2.5 μm; B, 1.8 μm; C, 1.2 μm; D, 1.0 μm. E, The values for openings to the synaptic cleft per micrometer of presynaptic membrane for all nerve-muscle contacts are grouped by bins of 0.5 for two α/β2-syntrophin null mice (this study) and α-syntrophin null mice and their wild-type littermates (Adams et al., 2000).
Electron microscopy also showed that the NMJs of the α/β2-syntrophin null mice often had numerous small terminals or axon branches in addition to terminals of normal adult appearance (Fig. 7). The interaction of these terminals with the accompanying Schwann cells was often complex, whereas the interaction with the muscle cell surface tended to be simple, with no or only a very shallow synaptic gutter, even when folds were present. These apparently immature contacts reinforce the suggestion that remodeling is a characteristic of these mice.
Voluntary exercise in the syntrophin null mice
As a functional test of whole-animal physiology, mice were caged individually with free access to a running wheel. Wheel revolutions and the calculated distance run were monitored simultaneously for control and syntrophin null mice (Fig. 8). Mice lacking α-syntrophin (Adams et al., 2000) or β2-syntrophin ran distances similar to those of the control mice (Fig. 8); however, mice lacking both α- and β2-syntrophin ran only about half the distance, significantly less than the controls (Fig. 8).
Figure 8.

Voluntary exercise on running wheels in the syntrophin null mice. Mice lacking both α- and β2-syntrophin ran significantly less (p < 0.01) on voluntary running wheels than mice lacking only one of the syntrophin isoforms. Error bars indicate SEM.
Electrophysiological analysis of neuromuscular transmission
To determine whether the running deficit of mice lacking both α- and β2-syntrophin was caused by a functional defect of neuromuscular transmission, we performed electrophysiological comparisons of the mutant and normal mice (Table 1). A small but statistically significant decrease of the MEPP amplitude in both mouse lines lacking α-syntrophin was observed. Furthermore, the MEPC amplitude was significantly decreased in the α/β2-syntrophin null mouse. The double syntrophin null mice also had significantly decreased decay time constants for both MEPPs and MEPCs. The number of transmitter quanta released by nerve impulse in the syntrophin null mice (both single and double mutants) was not significantly different from that of the control mice. Thus, the running deficiency is most likely not caused by defects in neuromuscular junction transmission.
Table 1.
Microelectrode recordings from mouse diaphragm
|
|
MEPP |
MEPC |
Quantal content of EPP |
||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Mouse |
n
|
Amplitude (mV) |
Amplitude (msec) |
n
|
Amplitude (nA) |
Amplitude (msec) |
n
|
|
|||||
| Control (C57BL/6) | 49 | 0.89 ± 0.036 | 1.45 ± 0.041 | 31 | 2.27 ± 0.090 | 0.55 ± 0.017 | 29 | 42.8 ± 2.6 | |||||
| α-Syntrophin null | 32 | 0.69 ± 0.020 (p < 0.001) | 1.31 ± 0.057 (p < 0.05) | 45 | 2.09 ± 0.087 (p, NS) | 0.39 ± 0.023 (p < 0.001) | 28 | 49.4 ± 3.4 (p, NS) | |||||
| α/β2-Syntrophin null |
47 |
0.73 ± 0.025 (p < 0.001) |
1.53 ± 0.056 (p, NS) |
42 |
1.71 ± 0.059 (p < 0.001) |
0.39 ± 0.022 (p < 0.001) |
17 |
39.1 ± 3.9 (p, NS) |
|||||
Values indicate mean ± SE; n indicates the number of endplates examined. NS, Not significant.
Discussion
The syntrophins are modular proteins that recruit signaling molecules to the cytoskeleton via their direct association with proteins of the dystrophin family. We have previously investigated the physiological significance of this function in vivo by generating a line of mice lacking α-syntrophin (Adams et al., 2000). These mice fail to properly localize aquaporin-4 and nNOS to the muscle sarcolemma and utrophin to the postsynaptic membrane of the neuromuscular junction. Furthermore, they have structurally aberrant neuromuscular junctions with a shallow neural groove, radiating fingers of AChR, and reduced levels of both the AChR and acetylcholinesterase (Adams et al., 2000). In this study, we used a similar approach to investigate the in vivo function of β2-syntrophin in skeletal muscle by generating a line of mice lacking this syntrophin isoform. We then investigated the degree to which the α and β2 isoforms of syntrophin can compensate for the loss of the other by generating mice lacking both of these syntrophins.
β2-Syntrophin null mice
The β2-syntrophin null mice showed no apparent muscle phenotype, except possibly elevated AChR number at neuromuscular junctions; however, we cannot exclude the possibility that they may have a non-muscle phenotype. β2-Syntrophin is widely expressed and interacts with various signaling molecules in other tissues. These include the tyrosine phosphatase-like ICA512 in pancreas (Ort et al., 2000), the ATP-binding cassette transporter A1 in liver (Buechler et al., 2002), and the syntrophin-associated serine/threonine kinase in brain (Lumeng et al., 1999). Because the β2-syntrophin null mice live a normal lifespan (at least in the laboratory setting), however, these interactions must not be required for survival.
α/β2-Syntrophin double-null mice
a-Syntrophin and β2-syntrophin have similar protein domain organizations and are both present at the NMJ. Because they share overlapping but not identical distributions, it is plausible that one isoform could partially compensate for the loss of the other. To test this possibility, we crossed the α-syntrophin null mice with the β2-syntrophin null mice to produce α/β2-syntrophin double-null mice. Like their parent strains, the double syntrophin null mice show no indications of muscular dystrophy; however, the double syntrophin null mice run significantly less (∼50%) on voluntary exercise wheels than wild-type mice or either parent strain. Because we observed no damage to the muscle fiber using histology techniques, we investigated the possibility that the double syntrophin null mice run less because of a deficit in neuromuscular transmission. Quantal release by nerve impulse was not significantly different among control (C57BL/6), α-syntrophin null, and α/β2-syntrophin null mice. We did observe, however, a small but significant decrease in the amplitude of MEPP in the α-syntrophin null and the α/β2-syntrophin null mice and a decrease of the MEPC in the α/β2-syntrophin null mice. We also observed a decrease in the MEPP and MEPC decay time constants in both mouse lines lacking α-syntrophin, suggesting either faster channel closure or perhaps faster destruction of acetylcholine by acetylcholine esterase attributable to changes in endplate geometry. These small changes in the MEPP and MEPC are not sufficient to explain the reduced level of voluntary running in the α/β2-syntrophin double-null mice. The running deficit may be caused by a non-muscle defect, perhaps affecting metabolism or neuronal function.
The α/β2-syntrophin null mice have neuromuscular junction structural defects similar in nature but more severe than those observed in the α-syntrophin null mice. En face views of the α/β2-syntrophin null NMJs show more extensive branching of the radiating fingers of AChRs than those of the α-syntrophin null (Fig. 5). Furthermore, the synaptic levels of receptor are even lower in the α/β2-syntrophin null mice (23% of wild type) compared with the α-syntrophin null mice (35% of wild type) (Fig. 4). The junctional folds are also more aberrant in the α/β2-syntrophin null mice, with fewer synaptic cleft openings per micrometer of nerve contact (Fig. 7). These abnormalities in NMJ structure occur despite the presence of normal levels of dystrophin, dystrobrevin, and sodium channels.
Syntrophin isoform compensation
We observed here that NMJ defects in the α/β2-syntrophin double-null mice are more severe than in the α-syntrophin null mice and that deficiencies in voluntary running appear only when both α- and β2-syntrophin are absent. These observations suggest that these two syntrophins can compensate for each other in at least some activities at the NMJ. This compensation could be expected, given the high degree of sequence similarity in this family and similar behaviors in some biochemical measures. On the other hand, mechanisms in which compensation does not occur are known. For example, the β2-syntrophin PDZ domain binds syntrophin-associated serine/threonine kinase and microtubule-associated serine/threonine kinase in vitro, but the α-syntrophin PDZ domain does not (Lumeng et al., 1999). nNOS is absent from synapses in α-syntrophin null muscle, despite the presence of β2-syntrophin (Adams et al., 2000), and the loss of β2-syntrophin alone has no detectable effect on the level of synaptic nNOS. The same applies to β1-syntrophin on the sarcolemma. In the absence of α-syntrophin,β1-syntrophin cannot retain nNOS (or aquaporin-4) on the membrane, even when present at higher than normal amounts (Adams et al., 2001). Sarcolemmal nNOS retention is known to depend critically on interaction of the nNOS and α-syntrophin PDZ domains (Adams et al., 2001) but may also require other, less clearly defined interactions within the dystrophin complex (Rafael et al., 1994; Chao et al., 1996). Because the PDZ domains of all three syntrophins bind the nNOS PDZ domain in vitro, the requirement for only α-syntrophin presumably reflects in vivo regulatory processes that are absent in vitro.
Clearly, both α- and β2-syntrophin play an important role in NMJ synapse formation and/or maintenance. Furthermore, our results indicate that these two syntrophins can only partially compensate for the loss of the other isoform. Finally, the running wheel data coupled with the neuromuscular transmission experiments suggest that the syntrophins have important roles outside of the neuromuscular junction. Studies of α-syntrophin in the brain, aided by the availability of the α-syntrophin null mouse, have revealed a critical role in the expression and regulation of aquaporin-4 in astroglial end feet. Further investigation of the β2-syntrophin null mouse may prove similarly informative.
Footnotes
This work was supported by National Institutes of Health Grants NS6277 (A.G.E.), NS44211 (R.S.), and NS33145 (S.C.F.) and the Muscular Dystrophy Association (M.E.A., A.G.E., R.S., S.C.F.). We thank Stuart Krall and Heather Mueller (University of North Carolina) for technical support. We thank S. Rock Levinson (University of Colorado Health Science Center) for the generous gift of sodium channel antibody.
Correspondence should be addressed to Marvin E. Adams, Department of Physiology and Biophysics, University of Washington, Seattle, WA 98195. E-mail: marva@u.washington.edu.
Copyright © 2004 Society for Neuroscience 0270-6474/04/2410302-08$15.00/0
References
- Adams ME, Butler MH, Dwyer TM, Peters MF, Murnane AA, Froehner SC (1993) Two forms of mouse syntrophin, a 58 kd dystrophin-associated protein, differ in primary structure and tissue distribution. Neuron 11: 531-540. [DOI] [PubMed] [Google Scholar]
- Adams ME, Dwyer TM, Dowler LL, White RA, Froehner SC (1995) Mouse alpha 1- and beta 2-syntrophin gene structure, chromosome localization, and homology with a discs large domain. J Biol Chem 270: 25859-25865. [DOI] [PubMed] [Google Scholar]
- Adams ME, Kramarcy N, Krall SP, Rossi SG, Rotundo RL, Sealock R, Froehner SC (2000) Absence of alpha-syntrophin leads to structurally aberrant neuromuscular synapses deficient in utrophin. J Cell Biol 150: 1385-1398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adams ME, Mueller HA, Froehner SC (2001) In vivo requirement of the alpha-syntrophin PDZ domain for the sarcolemmal localization of nNOS and aquaporin-4. J Cell Biol 155: 113-122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahn AH, Kunkel LM (1995) Syntrophin binds to an alternatively spliced exon of dystrophin. J Cell Biol 128: 363-371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahn AH, Yoshida M, Anderson MS, Feener CA, Selig S, Hagiwara Y, Ozawa E, Kunkel LM (1994) Cloning of human basic A1, a distinct 59-kDa dystrophin-associated protein encoded on chromosome 8q23-24. Proc Natl Acad Sci USA 91: 4446-4450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brenman JE, Chao DS, Gee SH, McGee AW, Craven SE, Santillano DR, Wu Z, Huang F, Xia H, Peters MF, Froehner SC, Bredt DS (1996) Interaction of nitric oxide synthase with the postsynaptic density protein PSD-95 and alpha1-syntrophin mediated by PDZ domains. Cell 84: 757-767. [DOI] [PubMed] [Google Scholar]
- Buechler C, Boettcher A, Bared SM, Probst MC, Schmitz G (2002) The carboxy terminus of the ATP-binding cassette transporter A1 interacts with a beta2-syntrophin/utrophin complex. Biochem Biophys Res Commun 293: 759-765. [DOI] [PubMed] [Google Scholar]
- Chao DS, Gorospe JRM, Brenman JE, Rafael JA, Peters MF, Froehner SC, Hoffman EP, Chamberlain JS, Bredt DS (1996) Selective loss of sarcolemma nitric oxide synthase in Becker muscular dystrophy. J Exp Med 184: 609-618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Connors NC, Adams ME, Froehner SC, Kofuji P (2004) The potassium channel Kir4.1 associates with the dystrophin glycoprotein complex via alpha-syntrophin in glia. J Biol Chem 279: 28387-28392. [DOI] [PubMed] [Google Scholar]
- Dombrowicz D, Flamand V, Brigman KK, Koller BH, Kinet J-P (1993) Abolition of anaphylaxis by targeted gene disruption of the high affinity immunoglobulin E receptor alpha chain gene. Cell 75: 969-976. [DOI] [PubMed] [Google Scholar]
- Engel AG, Nagel A, Walls TJ, Harper CM, Waisburg HA (1993) Congenital myasthenic syndromes: I. Deficiency and short open-time of the acetylcholine receptor. Muscle Nerve 16: 1284-1292. [DOI] [PubMed] [Google Scholar]
- Froehner SC, Murnane AA, Tobler M, Peng HB, Sealock R (1987) A postsynaptic Mr 58,000 (58K) protein concentrated at acetylcholine receptor-rich sites in Torpedo electroplaques and skeletal muscle. J Cell Biol 104: 1633-1646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gee SH, Madhavan R, Levinson SR, Caldwell JH, Sealock R, Froehner SC (1998) Interaction of muscle and brain sodium channels with multiple members of the syntrophin family of dystrophin-associated proteins. J Neurosci 18: 128-137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hasegawa M, Cuenda A, Spillantini MG, Thomas GM, Buee-Scherrer V, Cohen P, Goedert M (1999) Stress-activated protein kinase-3 interacts with the PDZ domain of alpha1-syntrophin. A mechanism for specific substrate recognition. J Biol Chem 274: 12626-12631. [DOI] [PubMed] [Google Scholar]
- Hogan A, Shepherd L, Chabot J, Quenneville S, Prescott SM, Topham MK, Gee SH (2001) Interaction of {gamma}1-syntrophin with diacylglycerol kinase-{zeta}: regulation of nuclear localization by PDZ interactions. J Biol Chem 276: 26526-26533. [DOI] [PubMed] [Google Scholar]
- Kameya S, Miyagoe Y, Nonaka I, Ikemoto T, Endo M, Hanaoka K, Nabeshima Y, Takeda S (1999) Alpha-syntrophin gene disruption results in the absence of neuronal-type nitric oxide synthase at the sarcolemma but does not induce muscle degeneration. J Biol Chem 274: 2193-2200. [DOI] [PubMed] [Google Scholar]
- Kramarcy NR, Sealock R (2000) Syntrophin isoforms at the neuromuscular junction: developmental time course and differential localization. Mol Cell Neurosci 15: 262-274. [DOI] [PubMed] [Google Scholar]
- Kramarcy NR, Vidal A, Froehner SC, Sealock R (1994) Association of utrophin and multiple dystrophin short forms with the mammalian Mr 58,000 dystrophin-associated protein (syntrophin). J Biol Chem 269: 2870-2876. [PubMed] [Google Scholar]
- Leonoudakis D, Conti LR, Anderson S, Radeke CM, McGuire LM, Adams ME, Froehner SC, Yates III JR, Vandenberg CA (2004) Protein trafficking and anchoring complexes revealed by proteomic analysis of inward rectifier potassium channel (Kir2.x)-associated proteins. J Biol Chem 279: 22331-22346. [DOI] [PubMed] [Google Scholar]
- Lumeng C, Phelps S, Crawford GE, Walden PD, Barald K, Chamberlain JS (1999) Interactions between beta2-syntrophin and a family of microtubule-associated serine/threonine kinases. Nat Neurosci 2: 611-617. [DOI] [PubMed] [Google Scholar]
- Nagel A, Lehmann-Horn F, Engel AG (1990) Neuromuscular transmission in the mdx mouse. Muscle Nerve 13: 742-749. [DOI] [PubMed] [Google Scholar]
- Neely JD, Amiry-Moghaddam M, Ottersen OP, Froehner SC, Agre P, Adams ME (2001) Syntrophin-dependent expression and localization of Aquaporin-4 water channel protein. Proc Natl Acad Sci USA 98: 14108-14113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ort T, Maksimova E, Dirkx R, Kachinsky AM, Berghs S, Froehner SC, Solimena M (2000) The receptor tyrosine phosphatase-like protein ICA512 binds the PDZ domains of beta2-syntrophin and nNOS in pancreatic beta-cells. Eur J Cell Biol 79: 621-630. [DOI] [PubMed] [Google Scholar]
- Ou Y, Strege P, Miller SM, Makielski J, Ackerman M, Gibbons SJ, Farrugia G (2003) Syntrophin gamma 2 regulates SCN5A gating by a PDZ domain-mediated interaction. J Biol Chem 278: 1915-1923. [DOI] [PubMed] [Google Scholar]
- Peters MF, Kramarcy NR, Sealock R, Froehner SC (1994) β2-Syntrophin: localization at the neuromuscular junction in skeletal muscle. NeuroReport 5: 1577-1580. [PubMed] [Google Scholar]
- Peters MF, Adams ME, Froehner SC (1997) Differential association of syntrophin pairs with the dystrophin complex. J Cell Biol 138: 81-93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peters MF, Sadoulet-Puccio H, Grady RM, Kramarcy NR, Kunkel LM, Sanes JR, Sealock R, Froehner SC (1998) Differential membrane localization and intermolecular associations of alpha-dystrobrevin isoforms in skeletal muscle. J Cell Biol 142: 1269-1278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piluso G, Mirabella M, Ricci E, Belsito A, Abbondanza C, Servidei S, Puca AA, Tonali P, Puca GA, Nigro V (2000) Gamma1- and gamma2-syntrophins, two novel dystrophin-binding proteins localized in neuronal cells. J Biol Chem 275: 15851-15860. [DOI] [PubMed] [Google Scholar]
- Rafael JA, Sunada Y, Cole NM, Campbell KP, Faulkner JA, Chamberlain JS (1994) Prevention of dystrophic pathology in mdx mice by a truncated dystrophin isoform. Hum Mol Genet 3: 1725-1733. [DOI] [PubMed] [Google Scholar]
- Thomas GD, Sander M, Lau KS, Huang PL, Stull JT, Victor RG (1998) Impaired metabolic modulation of alpha-adrenergic vasoconstriction in dystrophin-deficient skeletal muscle. Proc Natl Acad Sci USA 95: 15090-15095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas GD, Shaul PW, Yuhanna IS, Froehner SC, Adams ME (2003) Vasomodulation by skeletal muscle-derived nitric oxide requires alpha-syntrophin-mediated sarcolemmal localization of neuronal nitric oxide synthase. Circ Res 92: 554-560. [DOI] [PubMed] [Google Scholar]