

Abstract
ATP-gated P2X2 channels undergo permeability changes through a process that is incompletely understood. In the present study, we used fluorescence resonance energy transfer (FRET) and electrophysiology to measure cytosolic gating motions in P2X2 channels as they enter a state with increased permeability. P2X2 channels underwent permeability changes with a time course that was similar to decreases in FRET between cyan fluorescent protein and yellow fluorescent protein attached to the cytosolic domain of P2X2 channels. Wild-type and mutant channels that did not undergo permeability changes also did not show evidence of cytosolic gating motions. Moreover, immobilizing the cytosolic domain by tethering it to the plasma membrane prevented the switch in permeability and impaired the cytosolic gating motions. Both of these phenotypes were restored when the immobilizing tether was cleaved. The data provide a time-resolved measure of state-specific gating motions and suggest how a cytosolic domain may control ion channel permeability.
Keywords: channel, fluorescence, gating, permeability, purinergic, receptor, P2X, FRET, TIRF
Introduction
Transmitter-gated ion channels are key synaptic proteins that bind neurotransmitter and convert this presynaptic chemical signal into a postsynaptic signaling event (Hille, 2001). A large family of transmitter-gated ion channels are the ATP-gated P2X channels (Khakh, 2001; North, 2002). There is strong evidence for the physiological role of distinct P2X channels throughout the body, including in the nervous system, from a combination of physiological, molecular, and behavioral approaches (Khakh, 2001; North, 2002). P2X subunits (P2X1-P2X7) are the products of seven distinct genes and form at least 11 heteromeric channels (Torres et al., 1999). P2X channels are likely trimeric (Nicke et al., 1998; Stoop et al., 1999; Jiang et al., 2003), and each subunit possesses two transmembrane domains. The cytosolic domain of the protein consists of N and C termini. P2X channels are cation selective, but aspects of their ion permeability are dynamic, such as their ability to change ion permeability in an activation-dependent manner on a time scale of seconds (Surprenant et al., 1996; Khakh and Lester, 1999; Khakh et al., 1999, 2001; Virginio et al., 1999a; MacKenzie et al., 2001; Schlichter et al., 2002).
In the case of heterologous expression experiments, permeability changes were first reported for P2X7 channels and subsequently for neuronal P2X2 and P2X4 channels, but there is evidence for variability between studies (Surprenant et al., 1996; Petrou et al., 1997; Khakh et al., 1999; Virginio et al., 1999a; Eickhorst et al., 2002; Smart et al., 2003; Fujiwara and Kubo, 2004). P2X2 permeability changes were measured for heterologously expressed channels as well as for natively expressed P2X2 subunit-containing channels in sympathetic and sensory neurons (Khakh et al., 1999; Virginio et al., 1999a). These studies provided a molecular explanation for early observations on pheochromocytoma 12 cells, in which ATP evoked inward currents when bathed in extracellular solutions comprising organic cations, whereas ACh acting on nicotinic channels did not (Nakazawa et al., 1991). More recently, evidence has also emerged that permeability changes may occur at P2X-expressing dorsal horn neurons during ATP synaptic transmission (Schlichter et al., 2002).
Activation-dependent changes in ionic selectivity are not unique to P2X channels and have been documented for several ion channels, including those gated by voltage, second messengers, and neurotransmitters (Khakh and Lester, 1999; Hille, 2001). Collectively, these data suggest that the selectivity filter in some channels is dynamic and undergoes conformational rearrangements to switch its preference between ions (Khakh and Lester, 1999; Hille, 2001). To address the mechanisms involved, we sought to measure conformational changes in P2X2 channels as they move from a state with high ionic selectivity (I1) to a state (I2) that discriminates poorly between ions (I2) (Khakh et al., 1999; Virginio et al., 1999a). We used electrophysiology combined with fluorescence resonance energy transfer (FRET) (Kim et al., 2003; Riven et al., 2003) between fluorophore-labeled P2X2 subunits and a genetically encoded tag to measure conformational changes in the cytosolic domain of P2X2 channels.
Materials and Methods
Molecular biology. The starting vectors were wild-type (wt) P2X2 and P2X2-green fluorescent protein (GFP) in HindIII (5′) and XbaI(3′) sites of pcDNA3.1 (Khakh et al., 2001): XhoI and XbaI flanked GFP. By PCR (PCR Core kit; Qiagen, Crawley, UK), the P2X2 stop codon was removed and an XhoI site was inserted to generate P2X2XhoI. We generated P2X2-XFP by amplification of cyan fluorescent protein (CFP) and yellow fluorescent protein (YFP) from pECFP-1 and pEYFP-1 (Clontech, Oxford, UK), with in-frame 5′ XhoI sites and 3′ XbaI sites. These were cloned into XhoI and XbaI sites of P2X2-GFP. P2X1-C, P2X1-Y, P2X2-pleckstrin homology (PH), P2X2-PH-C, and P2X2-PH-Y were generated using similar methods (where C equals CFP, and Y equals YFP). Site-directed mutagenesis was performed on the cDNAs using synthetic oligonucleotides to generate T18A mutants (Quickchange; Stratagene, La Jolla, CA). cDNAs were propagated in DH5α Escherichia coli, and plasmids were purified using standard techniques. For transient expression in human embryonic kidney (HEK) 293 cells, we used 0.5-1 μg of plasmid cDNA (Effectene; Qiagen).
Electrophysiology. HEK 293 cells were used for recordings 24-48 hr after transfection, gently mechanically dispersed, and plated onto glass coverslips 2-12 hr before use. We included this step (Egan et al., 1998) to ensure adequate voltage clamp during reversal potential measurements from single spherical cells. The extracellular recording solution was composed of (in mm): 150 NaCl, 1 MgCl2, 1 (or 0.1) CaCl2, 10 HEPES, and 10 glucose, pH 7.4, and the pipette solution was composed of (in mm): 154 NaCl (or CsCl or CsF), 11 EGTA, and 10 HEPES. For some experiments, where stated, we replaced Na+ with equimolar substitutions of N-methyl-d-glucamine (NMDG)+ (Khakh et al., 1999; Virginio et al., 1999a). Whole-cell voltage-clamp recordings were made with 5 MΩ borosilicate glass electrodes (World Precision Instruments, Stevenage, UK), using an Axopatch-1D amplifier controlled by a computer running pClamp 8.1 software via a Digidata 1366 interface (Axon Instruments, Foster City, CA). Data were filtered at 0.5-2 kHz and digitized at 1-5 kHz. Drugs were applied to single cells using a Picospritzer (Intracell, Royston, UK) or in the bathing medium, which flowed at 2-3 ml/min. For pressure application (∼20 psi), we used 2-4 MΩ pipettes; solution exchange occurred in ∼10 msec. We have previously used similar conditions to measure ATP-evoked currents from rapidly desensitizing T18A mutants and natively expressed P2X3 channels in sensory neurons (Khakh et al., 2001). Solution exchange with bath application occurred in ∼10-20 sec, and the only agonist applied in this way was UTP for the experiments shown in Figures 10 and 11; all other experiments used rapid pressure applications. Voltage ramps (0.5 sec duration) were usually applied every 0.5-1 sec, but in the case of T18A and P2X1, 0.1 sec duration ramps every 0.1 sec were also tested. On the basis of initial experiments, the voltage range was adjusted to be ∼400-60 mV either side of the reversal potential for each construct. We performed extensive controls on wt P2X2 channels and found that the speed of the ramps (0.1-1 sec) or their frequency (0.2-10 Hz) did not affect the extent of permeability changes. In the case of channels that desensitize quickly, it is probable that even the first current-voltage relationship missed the peak response, but this is unlikely to affect our interpretation because the pNMDG+/pNa+ of these channels is fixed and almost identical to the I1 state of wt P2X2 (see Table 1) (Khakh et al., 1999) (i.e., there is no evidence that these channels show any enhanced permeability for the first current-voltage relationship we could record or subsequent ones). We never-recorded permeability changes for T18A P2X2 mutants and P2X1 channels regardless of whether we used single or repetitive pulses of ATP (100 μm).
Figure 10.

FRET e for P2X2 channels tagged with the PH domains suggests a splayed cytosolic domain. A, B, Donor (A) and acceptor (B) emission images of an HEK cell expressing P2X2-PH-C/P2X2-PH-Y channels before and after photodestruction of the YFP. C, A graph of the donor and acceptor intensity over time during acceptor photodestruction for cells expressing P2X2-PH-C/P2X2-PH-Y. The donor emission intensity increases concomitantly with the decrease in the acceptor emission. D, A plot of donor fluorescence recovery versus acceptor fluorescence loss. E, Diagrammatic representation of PH-YFP bound to plasma membrane PIP2 in the resting state and cytosolic PH-YFP after PIP2 has been cleaved. F, Photomicrographs of a cell expressing PH-YFP and hP2Y4 receptors before, during, and after UTP (50 μm). G, The intensity of a region of interest from the edge of the cell shown in F and the intensity of an equally sized region from the cytosol area. H, Summary of 10 experiments like those illustrated in F and G with baseline fluorescence normalized to 100% for both the membrane and cytosolic ROI (n = 10). Error bars represent SEM.
Figure 11.

Permeability changes and FY/FC changes at PH-tagged P2X2 channels in the absence and presence of UTP to cleave PIP2. A, B, Experiments in the absence of UTP for PH-tagged P2X2 channels. A, Experiments with NMDG+ replacing Na+. Top, Steady-statecurrents at -60 mV in the absence of UTP (100 μm ATP). Bottom, The reversal potential of the ATP-evoked response shown in the top panel, starting at the peak of the outward current over time, up to 8 sec into ATP. B, Top, ATP-evoked inward current in Na+ solutions (30 μm ATP); bottom, the FY/FC ratio over time. C, D, Experiments in the presence of UTP for PH-tagged P2X2 channels. C, Experiments with NMDG+ replacing Na+. Top, Steady-state currents at -60 mV (100 μm ATP) in the presence of UTP. Bottom, The reversal potential of the ATP-evoked response shown in the top panel, starting at the peak of the outward current over time, up to 8 sec into ATP. D, Top, ATP-evoked current (30 μm ATP) in Na+ solutions; bottom, the FY/FC ratio over time, after a 20 sec pulse of UTP to cleave PIP2 (see Results and Table 1 for average data). Bottom, The traces for P2X2 channels are superimposed in gray for comparison (see Fig. 6). Error bars represent SEM.
Table 1.
Comparison of permeability changes, ΔFy/Fc, and desensitization kinetics in physiological solutions for wt and engineered P2X channels tagged with CFP and YFP
|
|
wt P2X2 |
T18A P2X2 |
wt P2X1 |
P2X2-PH |
P2X2-PH + UTP |
n |
|---|---|---|---|---|---|---|
| Permeability | ||||||
| I1 NMDG+ Erev (mV) | −64.9 ± 1.0 | −62.1 ± 1.5 | −58.7 ± 3.2 | −64.2 ± 1.1 | −75.1 ± 1.7c | |
| I1 pNMDG+/pNa | 0.08 ± 0.003 | 0.08 ± 0.005 | 0.10 ± 0.01 | 0.08 ± 0.004 | 0.05 ± 0.004 | 5-23 |
| Erev I1 to I2 (mV) | +23.9 ± 2.7 | <2 | <2 | +4.0 ± 0.9 | +19.5 ± 5.5 | |
| Fold change in pNMDG+/pNa+ | 2.8 ± 0.4 | <0.1 | <0.1 | 0.2 ± 0.04 | 2.8 ± 0.8 | |
| I2 | Yes | No | No | No | Yes | 5-23 |
| FRET | ||||||
| Basal FRET e (%) | 35.4 ± 5.0 | 26.3 ± 6.2 | 18.9 ± 4.3 | 17.2 ± 2.0 | ND | 6-23 |
| ΔFy/Fc (%) | −14.1 ± 2.0 | −1.7 ± 2.2a | +4.5 ± 3.4a | −1.9 ± 1.8a | −9.3 ± 1.8 | 9-23 |
| Desensitization | ||||||
| τfast (sec) | 74.5 ± 14.4 | 0.22 ± 0.04 | 0.058 ± 0.008 | 8.8 ± 1.6 | 35.3 ± 6.7 | |
| Amplitudefast (%) | 54.0 ± 5.3 | 84.0 ± 3.0 | 93.7 ± 2.0 | 29.5 ± 3.6 | 65.1 ± 5.5 | 5-14 |
| τslow (sec) | b | 2.9 ± 0.8 | b | 91.4 ± 16.5 | b | |
| Amplitudeslow (%) | b | 12.3 ± 1.5 | b | 61.1 ± 4.9 | b | 5-13 |
| Plateau current (%) |
46 |
4 |
6 |
9 |
35 |
|
ND, Not determined.
These values were not significant compared with the control period (Fig. 8).
These desensitization data were best fit by a single exponential (Fig. 5).
This value was significantly different from the others for I1 NMDG + Erev (see Results for discussion). The ATP concentration was 30-100 μm.
FRET microscopy. We used an Olympus (Southall, UK) BX50 scope equipped with an Imago cooled CCD camera, lamp, and condenser (all from TILL Photonics, Munich, Germany) and an Image Splitter (Cairn Research Ltd., Faversham, Kent, UK) with an adjustable pinhole. The control of excitation and image acquisition was achieved using TIllVision software, hardware, and drivers (TILL Photonics). We used the following filters for acquiring CFP, YFP, or FRET images (order is exciter, dichroic, emitter in nanometers; Glen Spectra, Stanmore, UK): CFP (440AF21, 455DRLP, 480AF30), YFP (500AF25, 525DRLP, 545AF35), and FRET (440AF21, 455DRLP, 545AF26). For the time-resolved measurements of FRET, we used 440 nm light from a monochromator, a 440AF21 and 455DRLP filter in the rotating sextuplet of the microscope, a DQ505LP dichroic mirror in the Image Splitter, and 480/30 and 535/25 filters in the emission paths of the CFP and YFP images, respectively. The Image Splitter ensures that CFP and YFP images are captured in the same frame on the CCD chip. To photo destroy the YFP, we used 525 nm light from the monochromator and a 525 DRLP dichroic. Three-cube FRET imaging was performed essentially as described previously (Erickson et al., 2001). Occasionally we came across cells that expressed both fluorophores but paradoxically showed no FRET or showed no change in FY/FC. For our most complete data set (P2X2 channels), this amounted to 7 of 55 and 6 of 26 cells, respectively, which were not studied further. We do not have a complete explanation for this but suggest it may represent cells that preferentially assembled a mixture of channels with just donors and acceptors, which we expect from previous work to not support FRET (Cha et al., 1999). If assembly of trimeric channels has no specificity and follows a binomial distribution, we expect 50% of channels to contain at least one donor and one acceptor and thus support FRET (see Results).
Total internal reflection fluorescence microscopy. Briefly, we used an Olympus IX70 scope equipped with a Princeton Instruments (Trenton, NJ) cooled I-PentaMAX camera with High Blue Gen III Intensifier (Roper Scientific, Trenton, NJ). The control of excitation and image acquisition was achieved using MetaMorph software and drivers (Universal Imaging, Marlow, UK) and shutters, filter wheels, and a Proscan II control box (Prior Scientific, Cambridge, UK). The beams of 488/515 nm argon (150 mW) and 442 nm helium cadmium (12.5 mW) lasers (Melles Griot, Carlsbad, CA) at <5% power (constant for all experiments reported) were combined and controlled with an IX2-COMB (Olympus), Uniblitz Shutters (Prior Scientific) and acoustoptical tunable filter and controller (AOTF; AA Optoelectronics, Les Chevreuse, France) and fed into a fiber optic (FV5-FUR; Olympus) for entry into the total internal reflection fluorescence (TIRF) condenser (IX2-RFAEVA-2; Olympus). Cells were plated onto glass-bottom Petri dishes (170 nm thick; Willco Wells BV, Amsterdam, The Netherlands) 24-48 hr before imaging and viewed with a 60× oil immersion objective lens with a numerical aperture of 1.45 (Olympus). FRET was determined with three-cube methods and donor dequenching, as described below. The gain was adjusted for maximum signal-to-noise for each cell and kept constant for all image acquisitions.
Confocal microscopy. We used confocal microscopy to measure changes in the distribution of PH-YFP during addition of UTP (50 μm). Cells plated on glass-bottom Petri dishes (MatTek, Ashland, MA) were placed on the stage of a Nikon (Tokyo, Japan) Eclipse TE300 scope equipped with a Bio-Rad (Hemel Hempstead, UK) Radiance 2000 confocal imaging setup and computer running Laser Sharp 2000 software (Bio-Rad).
Analysis. For donor dequenching experiments, the FRET efficiency was calculated as follows:
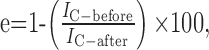 |
where IC-before is the donor fluorescence intensity before photodestruction, and IC-after is the intensity after photodestruction. The photodestruction of the YFP should proceed with a rate equivalent to the dequenching of the donor, and plotting the photorecovery versus photodestruction yields a linear plot (see Fig. 1 F). We routinely used such linear plots and extrapolated to 100% acceptor photodestruction to calculate the maximum donor dequenching for epifluorescence microscopy (see Fig. 1 F); FRET efficiency (e) is given by the y-axis intercept. For TIRF microscopy, we estimated FRET e from a single time point when the acceptor was >90% destroyed (<1 min with the 515 nm laser line at 100%) and extrapolated to 100% destruction by constraining the linear regression to pass through the origin. To approximate distances, we used the Forster equation:
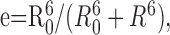 |
Figure 1.

FRET in closed P2X2 channels. Ai, Diagram representing the presently understood membrane topology of a P2X subunit, with a fluorophore attached to the C-terminal tail. The photomicrographs show YFP and CFP images of two cells expressing P2X2-Y/P2X2-C channels, acquired with the confocal aperture sufficiently open to allow good signal-to-noise for the entire cell. Aii, Bar graph of fluorescence intensity for ROI on the perimeter of the cell, the subperimeter, and from intracellular areas; the values are normalized to 100% at the perimeter. iii, iv, Examples of confocal sections (left) and surface plots (right) used to generate the graph in ii from cells expressing P2X2-Y. B, Diagram (i-iv) representations of wt, P2X2-Y, P2X2-C, and P2X2-C/P2X2-Y channels; only two subunits are represented. Traces show normalized ATP-evoked currents. C, D, Donor (C) and acceptor (D) emission images of an HEK cell expressing P2X2-C/P2X2-Y channels before and after photodestruction of the YFP. E, Donor emission intensity increases concomitantly (τ, 8.7 ± 1.5 min) with the decrease in the acceptor emission (τ, 8.3 ± 0.4 min). F, Plot of donor recovery versus acceptor loss for P2X channels at the cell perimeter (plasma membrane) and across the entire cell. Error bars represent SEM.
where R0 includes terms for quantum efficiency, the solvent refractive index, the orientation factor (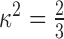 ), and the overlap of the donor emission and acceptor absorption spectra. R0 is the distance at which e is 50% for any given fluorophore pair, at 5 nm for CFP and YFP. We corrected the calculated distance R for the center-to-center of fluorophore distance by subtraction (Michalet et al., 2003). In the case of GFP, which contains the fluorophore buried in the center of a β-can structure, this distance is equal to two radii, at ∼30 Å (Tsien, 1998). We also used the three-cube method to determine the FRET ratio (FR). This nondestructive method uses three filter cubes and a set of predetermined constants. The methods used are essentially identical to those used previously (Erickson et al., 2001) and are reproduced here only for completeness. FR, the value representative of the increase in acceptor emission caused by FRET from a sample containing both donor and acceptor fluorophores, is given by the following:
), and the overlap of the donor emission and acceptor absorption spectra. R0 is the distance at which e is 50% for any given fluorophore pair, at 5 nm for CFP and YFP. We corrected the calculated distance R for the center-to-center of fluorophore distance by subtraction (Michalet et al., 2003). In the case of GFP, which contains the fluorophore buried in the center of a β-can structure, this distance is equal to two radii, at ∼30 Å (Tsien, 1998). We also used the three-cube method to determine the FRET ratio (FR). This nondestructive method uses three filter cubes and a set of predetermined constants. The methods used are essentially identical to those used previously (Erickson et al., 2001) and are reproduced here only for completeness. FR, the value representative of the increase in acceptor emission caused by FRET from a sample containing both donor and acceptor fluorophores, is given by the following:
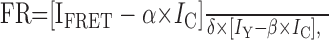 |
where IFRET is the fluorescence intensity measured using the FRET filter cube, IC is the fluorescence intensity measured using the CFP filter cube, and IY is the fluorescence intensity measured using the YFP filter cube. This equation contains the following experimentally determined constants:
 |
where α is the intensity ratio of CFP fluorescence passing through the FRET and CFP filter cubes with data collected from cells that express just donor, β is the fluorescence intensity ratio of CFP through the YFP and CFP filter cubes from cells expressing just donor, and δ is the fluorescence intensity ratio of YFP through the FRET and YFP filter cubes from cells expressing just acceptor. We determined constants for α, β, and δ in cells expressing P2X2 tagged with CFP or YFP as well as for cytosolic CFP or YFP. These were similar to those reported by Erickson et al. (2001) at 0.345 ± 0.011 (n = 51), 0.014 ± 0.001 (n = 51), and 0.064 ± 0.002 (n = 61) for α, β, and δ, respectively, for CFP and YFP. The values for P2X2-C and P2X2-Y were 0.465 ± 0.005, 0.005 ± 0.001, and 0.04 ± 0.001 (n = 6-8), and for T18A P2X2-C and T18A P2X2-Y they were 0.461 ± 0.005, 0.015 ± 0.011, and 0.034 ± 0.002 (n = 6-9) for α, β, and δ, respectively. For TIRF microscopy, the values for P2X2-C and P2X2-Y were 0.359 ± 0.03, 0.012 ± 0.003, and 0.066 ± 0.009 (n = 12-17), for α, β, and δ, respectively. Effective FRET efficiency (Eeff) can be estimated as follows:
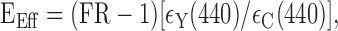 |
where ϵY(440)/ϵc(440) = 0.094 as the ratio of YFP and CFP molar extinction coefficients (Erickson et al., 2001). When comparing donor dequenching and three-cube methods, we back-calculated FR using the above function from estimates of e from linear plots such as those shown in Figure 1. For the FRET experiments, we chose regions of interest (ROI) post hoc. Our criteria were that ROI: (1) were bright in both CFP and YFP images, (2) were located at the edge, presumably the plasma membrane of the cell, (3) did not change or drift in intensity during the control period, (4) did not go out of focus during the entire experiment, and (5) did not move when ATP was applied.
Ion permeability ratios were calculated from shifts in reversal potentials using the following function:
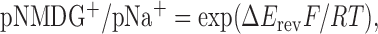 |
where pNMDG+ is the permeability to NMDG+, pNa+ is the permeability to Na+, ΔErev is the shift in reversal potential, and F, R, and T have their usual meaning (Eickhorst et al., 2002). In some of the electrophysiological traces, we used adjacent point averaging. Data in the text and graphs are shown as mean ± SEM from n determinations, as indicated. All ramps shown in the figures are leak subtracted.
Results
This study has three main parts. First, we show that fluorophore-labeled P2X2 channels are functional and undergo FRET. Second, we present evidence for cytosolic conformational changes correlated with permeability changes. Third, we use a genetically encoded tag to reversibly hinder permeability changes and cytosolic conformational changes.
Fluorophore-labeled P2X2 channels are functional and located predominantly in the plasma membrane
Our previous work mainly concerned P2X4 (Khakh et al., 1999), but in the present study, we focused on P2X2, because they are stably expressed in the plasma membrane and do not undergo endocytosis in response to ATP (Khakh et al., 2001; Bobanovic et al., 2002). CFP and YFP undergo FRET if they are within ∼70 Å. Fusion of CFP and YFP (Tsien, 1998) onto the cytosolic C-terminal domain of P2X2 subunits (Khakh et al., 2001) (Fig. 1A) to generate P2X2-C and P2X2-Y, respectively, resulted in functional channels (Fig. 1B; supplemental material, available at www.jneurosci.org). HEK 293 cells expressing P2X2-C and P2X2-Y channels fluoresced blue and yellowish-green when appropriately excited (Fig. 1A). The highest-intensity fluorescence was in the perimeter, which we interpret as plasma membrane, with little in the subperimeter regions and lower but significant levels inside cells, which may represent endoplasmic reticulum (Fig. 1Aii-iv). ATP EC50 values for P2X2-C and P2X2-Y were 7.4 ± 2.7 and 7.1 ± 1.5 μm (n = 6 and 7, respectively), which are similar to wt P2X2 (Khakh et al., 2001). Moreover, the ATP-evoked currents (100 μm for 1-30 sec) at P2X2, P2X2-C, P2X2-Y, and P2X2-C/P2X2-Y channels in Na+ and NMDG+ solutions were indistinguishable (Fig. 1B; supplemental material, available at www.jneurosci.org).
FRET in closed P2X2 channels
We used the donor dequenching method (Michalet et al., 2003) to estimate FRET e for channels expressed in HEK 293 cells (Fig. 1C-F). We measured a marked decrease in P2X2-Y fluorescence intensity with a time course that followed that of the P2X2-C increase (Fig. 1C-E) when the acceptor was optically destroyed. Maximum P2X2-C photorecovery when P2X2-Y was 100% photodestroyed (Fig. 1F) corresponds to FRET e, which in the case of P2X2-C/P2X2-Y channels near the cell perimeter was 35.4 ± 5.0% (n = 14), whereas for the whole cell, e was 12.3 ± 2.7% (n = 7). We suggest that FRET near the perimeter of the cell represents functional channels in the plasma membrane, whereas intracellular FRET may represent partially assembled channels as well, perhaps located in the endoplasmic reticulum (Nashmi et al., 2003). In subsequent studies, we restricted our measurements to fluorescence near the cell edges.
With TIRF microscopy, we determined FRET e within ∼100 nm of the plasma membrane (Steyer and Almers, 2001; Riven et al., 2003). FRET e was quantified with donor dequenching and with a linear unmixing three-cube method (Erickson et al., 2001) for ROI that covered the area of the cell in close apposition to glass within the evanescent field of illumination, an area we call the footprint. The intensity of the footprint was 13.4 ± 3.3% (n = 13) of that for the whole cell imaged with epifluorescence illumination (Fig. 2A,B), and the footprint represented ∼16-35% of the total cell surface area at 376 ± 54 μm2 (n = 13; with cell capacitance at 10-20 pF) (Fig. 2C-F). This corresponds to ∼600-1200 channels within the footprint, or on average approximately two to three channels per square micrometer (Khakh et al., 2001). The FRET e for channels in the footprint determined by donor dequenching and three-cube FRET was 33.2 ± 4.1 and 32.5 ± 5.4%, respectively (FR, 4.5 ± 0.6; n = 18). This compares well with our determination of FRET e for membrane ROI with epifluorescence imaging at 35.4 ± 5.0% (n = 14) (Fig. 1). There was a strong correlation (Fig. 3) between FRET estimates determined with donor dequenching and three-cube methods for both epifluorescence and TIRF microscopy (Erickson et al., 2001). With the assumption that P2X2 channels are trimeric (Nicke et al., 1998; Stoop et al., 1999; Jiang et al., 2003), the fluorophores attached to the P2X2 channel cytosolic domain tips may sit at the corners of a triangle of sides ∼25 Å in length. This estimate is within the range of sizes reported for cytosolic domains of channels of known shape or structure (Chang et al., 1998; Miyazawa et al., 1999; Sokolova et al., 2001; Higgins et al., 2002; Jiang et al., 2002; da Fonseca et al., 2003; Kuo et al., 2003).
Figure 2.

FRET for P2X2 channels in the plasma membrane determined with TIRF microscopy. A, The diagram illustrates the principle of TIRF (Steyer and Almers, 2001). Laser light was focused onto the back focal plane of the objective lens, and its position was adjusted so that it emerged into the immersion oil at an angle shallower than the critical angle. This created an evanescent field of illumination of depth ∼100 nm into the cell, which was adhered to the coverslip. B, The top panel shows an epifluorescence image of two HEK cells expressing P2X2-C/P2X2-Y channels, and the bottom panel shows the footprint of these cells onto the glass coverslip viewed with TIRF microscopy. The intensity of the footprint was  that of the entire cell (see Results), and so for presentation, the images are artificially scaled. C-F, Donor (E, F) and acceptor (C, D) emission images of an HEK cell footprint expressing P2X2-C/P2X2-Y channels before and after photodestruction of the YFP.
that of the entire cell (see Results), and so for presentation, the images are artificially scaled. C-F, Donor (E, F) and acceptor (C, D) emission images of an HEK cell footprint expressing P2X2-C/P2X2-Y channels before and after photodestruction of the YFP.
Figure 3.

Comparison of FR determined by donor dequenching and three-cube FRET for epifluorescence and TIRF microscopy. Our measurements in closed channels (see Results) rely on determination of e with donor dequenching. To be confident of our data, we compared our values of FRET determined with donor dequenching with FR determined with a recently introduced unmixing method, called the three-cube method. This latter method extracts values for FR (see Materials and Methods) using an algorithm that is described briefly in the data analysis section but more extensively by Erickson et al. (2001). To make this comparison possible, we converted our measurement of FRET e determined with donor dequenching to FR by back-calculation (see Materials and Methods) and compared these values with FR determined with the three-cube method. We performed this analysis for fluorescence from whole cells expressing cytosolic CFP/YFP, YC2.1, P2X2-C/P2X2-Y, and T18A P2X2-C/T18A P2X2-Y. We repeated the comparison for TIRF experiments with ROI encompassing whole footprints (n = 18). Note that the FR value for P2X2-C/P2X2-Y channels is higher in the membrane determined by TIRF rather than in whole cells and not significantly different from the calculated value of FRET e for membrane ROI using the EPI scope (see Results). For both TIRF and epifluorescence FRET microscopy, there was good agreement between FR determined by the three-cube and donor dequenching methods. Moreover, our values of FR for cytosolic CFP/YFP and YC2.1 are almost identical to those determined previously with both donor dequenching and three-cube methods. Collectively, these data provide strong evidence to indicate that our measurements of FRET are (1) consistent with past data and (2) consistent between two independent methods. Error bars represent SEM.
We performed experiments with cytosolic proteins and Yellow cameleon 2.1 to be confident that our instrumentation and measurements of e were reliable and consistent with past data (Erickson et al., 2001). With these controls in hand (Fig. 3), we suggest that FRET e is 35.4 ± 5.0% in closed P2X2 channels.
Slow entry into and slow recovery from the P2X2 channel I2 state
Previous work on rat P2X2 channels shows that ATP- and time-dependent permeability changes occurred in 40% of HEK cells (Virginio et al., 1999a). However, consistent with previous work on Xenopus oocytes (Eickhorst et al., 2002), we recorded permeability changes in all HEK cells with 100 μm ATP (56 of 56 cells from 13 transfections). Variability in expression levels (Fujiwara and Kubo, 2004) may explain these differences between our work and that already published (Virginio et al., 1999a). Given these differences, we considered it important to examine the essential features of permeability changes in HEK cells (Fig. 4). The channels initially opened to the I1 state with a reversal potential near -60 mV (Fig. 4A,B), representing a low permeability to NMDG+, and with time they entered the I2 state with a reversal potential ∼20-40 mV more positive (Fig. 4A,B), representing a higher permeability to NMDG+. The channels opened to I1 in <100 msec, whereas the transition from I1 to I2 occurred with a 10-90% rise time of ∼13 sec (rate, 0.17 ± 0.06 sec-1; n = 11) (Fig. 4D). Overall, these data are consistent with those presented previously for a subpopulation of HEK 293 cells (Virginio et al., 1999a) and Xenopus oocytes (Eickhorst et al., 2002). Interestingly, once the channels had opened to I2 with a ∼20 sec pulse of ATP, they also readily opened to I2 with a subsequent brief puff of ATP (100 μm; 100 msec). This has not been reported previously (Figs. 4A,C,D) and suggests the existence of a long-lived nonconducting state that is permissive for I2. P2X2 channels recovered from this permissive state in ∼200 sec (τ, 62 ± 12 sec; n = 11) (Fig. 4A,C,D).
Figure 4.

Permeability changes at wt P2X2 channels. A, Steady-state current at -60 mV from an HEK cell expressing P2X2 channels in extracellular buffer containing NMDG+ instead of Na+ (154 mm). The ATP was applied for the periods indicated by the solid bars above the graph, initially for ∼20 sec and then periodically for <100 msec (100 μm ATP). B, Current-voltage relationships during the response evoked by the initial 20 sec pulse of ATP, starting at the peak outward current (I1) and finishing at the peak inward current (I2). C, Current-voltage relationships during the response evoked by brief 100 msec puffs of ATP (at the times indicated) after the initial priming 20 sec pulse of ATP. D, A graph of average data from 11 cells showing the time course of the shift in the reversal potential in NMDG+ during the development of I2 and during its reversal over time after ATP washout. Error bars represent SEM.
Some P2X channels undergoing permeability changes display biphasic or sustained inward Na+ currents (Khakh and Lester, 1999; Khakh et al., 1999; North, 2002), with the sustained or slower current being attributable to the I2 state. Thus, a rapidly desensitizing current may indicate opening to only I1 in physiological solutions, whereas a sustained current may represent opening to I2 (Khakh et al., 1999; North, 2002). All P2X2 channel-expressing cells displayed slowly desensitizing ATP-evoked currents (τ, 74.5 ± 14.4 sec; n = 11 of 11 cells; 100 μm ATP) and a sustained plateau current in the presence of ATP (∼46% of peak) and recovered from desensitization in ∼200 sec. In contrast, wt P2X1 channels and mutant T18A P2X2 channels desensitized quickly (<100 msec; 100 μm ATP) (Fig. 5, Table 1), displayed minimal plateau currents, and did not undergo increases in NMDG+ permeability (Table 1). Our experiments do not reveal the mechanism of desensitization, but we suggest that in physiological solutions, the P2X2 I1 state is followed by the I2 state, which can be seen as a plateau current that desensitizes slowly over tens of seconds, whereas the lack of the I2 state may result in rapidly desensitizing currents such as those measured for T18A P2X2 and wt P2X1 channels.
Figure 5.

ATP-evoked currents for P2X channels in physiological solutions. The recordings show ATP-evoked currents at -60 mV with 30 μm ATP applied for the times indicates by the solid bars.
Slow entry into and recovery from a conformational change in the cytosolic domain
The seconds timescale I1-I2 transition is appropriate for FRET experiments (Michalet et al., 2003). Our experiments in NMDG+ solutions to track I2 at the same time as measuring the YFP to CFP ratio (FY/FC) were thwarted because NMDG+, or a component of this buffer (154 mm NMDG+), quenched the fluorophores as it flowed into the cell during entry into I2. To circumvent these limitations, we measured and plotted FY/FC over time, from cells in which we also measured ATP-evoked membrane currents in Na+ solutions at the same time (30 μm ATP) (Fig. 6A-C) and used the presence of a slowly desensitizing plateau current as a reflection of entry to the I2 state in physiological solutions (see above section, Fig. 5). For 20 sec applications of ATP (30 μm ATP), we recorded inward currents of -5836 ± 660 pA (Fig. 6A) and a 38.4 ± 3.6% (n = 11) decrease in FY/FC, implying a decrease in FRET (Fig. 6A,B). There was a trend for the 10-90% rise time for the decrease in FY/FC (18.0 ± 4.1 sec; n = 11) to be longer than the experimentally determined 10-90% rise time for the increase in permeability to NMDG+ at 10.9 ± 4.2 sec (Fig. 4D), but this did not reach statistical significance (the calculated 10-90% rise time of ∼13 sec for a single exponential process with a rate constant of 0.17 ± 0.06 sec-1 is within the error limits of the experimental data). Next, we puffed ATP for only 100 msec, briefer than that required to trigger permeability changes, monitored membrane current and FY/FC, and recorded a significant change in FY/FC of only 14.1 ± 2.0% and an inward current of -4805 ± 988 pA (n = 9; 30 μm ATP) (Fig. 6). The change occurred with a 10-90% rise time (12.5 ± 3.2 sec; n = 9) that was also not significantly different from that for permeability changes (10.9 ± 4.9 sec) or for the change in FY/FC for 20 sec ATP applications. There was little correlation between the magnitudes of ΔFY/FC and peak ATP-evoked currents (r = 0.2). The change in FY/FC decayed back to baseline slowly, and in some cases incompletely, compared with the ATP-evoked current (Fig. 6B). The slow reversal of the FY/FC (τ, 52 ± 7 sec for 100 msec puffs and 77 ± 12 sec for 20 sec pulses of ATP; n = 6 and 3) was similar to the recovery from the I2 permissive state (62 ± 12 sec; n = 11) (Fig. 4C,D). The incomplete nature of FY/FC recovery in some cells has been noted previously in FRET experiments and may additionally represent long-lived dark states of the fluorophores (Zheng et al., 2003). These data imply that the change in FY/FC has the same time course as the I1 to I2 transition and that its return to baseline follows the time course of recovery from the I2 permissive state. Thus, FRET-based measures provide evidence for a state in which the cytosolic domain persists in a conformation that is permissive for I2, even when ATP is removed and the pore is closed (Figs. 4, 6). The cytosolic domain can also be recruited to this state by brief puffs of ATP, possibly explaining the history dependence of permeability changes in P2X channels (Khakh et al., 1999) (Fig. 4A-D).
Figure 6.

Simultaneous electrophysiological recording and FY/FC imaging from the same cells suggests a cytosolic conformational change. A-C, Pulses of ATP (20 sec; 30 μm). A, ATP-evoked inward currents from HEK cells expressing P2X2-C/P2X2-Y channels (mean ± SEM; n = 11 of 13; 2 cells showed robust currents but no change in FY/FC). B, Top, Representative trace for FY/FC. Bottom, The average ratio of FY/FC for the same cells as in A. There was variability in the recovery phase, but on average the FY/FC ratio at a time point corresponding to 200-300 sec was not significantly different from baseline before ATP. Superimposed are records from cells (n = 13) in which we imaged the cell but did not puff any ATP. C, The same data superimposed on an expanded time scale. D-F, Brief puffs (100 msec; 30 μm) of ATP. C, ATP-evoked inward currents from HEK cells expressing P2X2-C/P2X2-Y (mean ± SEM; n = 9 of 13; 4 cells showed robust currents but no change in FY/FC). E, Top, Representative trace for FY/FC. Bottom, The ratio of FY/FC for cells in D. F, The same data superimposed on an expanded time scale. In this and other figures, the average current traces are shown with points for SEM at the times indicated. For the FY/FC traces, the underlying traces are averages, but SEMs are shown only for every 10th acquisition point. The asterisks indicate those data points that were significantly different from the control period before ATP as judged by Dunnett's tests with p < 0.05 taken as significant.
The above experiments assume that P2X2 channels actually undergo permeability changes in physiological solutions. There is evidence for this already (Khakh et al., 1999; Virginio et al., 1999a), but we repeated the key experiments to be sure this occurred under our experimental conditions (Fig. 7). Single-cell imaging experiments confirmed that P2X2 channels become permeable to the propidium dye YOPRO-1 in Na+ solutions (with 1 or 0.1 mm Ca2+) for 20 sec applications of 30 μm ATP (n = 5 and 12) (Fig. 7). We also performed a specific set of experiments to determine whether P2X2 channel NMDG+ permeability increased by the same amount in solutions containing either 0.1 or 1 mm Ca2+. The data were clear: the NMDG+ reversal potential shifted equally by +39.8 ± 4.3 and +32.6 ± 2.5 mV for solutions containing 0.1 and 1 mm Ca2+, respectively (from -56.6 ± 3.3 and -54.6 ± 2.3 mV; n = 7 for 0.1 mm and n = 17 for 1 mm Ca2+; p > 0.05). YOPRO-1 permeates only the I2 state (Khakh et al., 1999; Virginio et al., 1999a). Because YOPRO-1 uptake and NMDG+ permeability changes occurred in 1 mm Ca2+, these data support the hypothesis that P2X2 channels undergo permeability changes in the Na+-based physiological solutions used for the dual electrophysiology and imaging experiments (see above, Fig. 7).
Figure 7.

Permeability changes occur in solutions used for electrophysiology and imaging. A, Photomicrographs of an HEK cell bathed in YOPRO-1 (10 μm) containing extracellular solution (0.1 mm Ca2+) before and at various times in ATP (30 μm). Note that clear increases in YOPRO-1 fluorescence can be seen within 6 sec. B, C, Average time course for the increase in YOPRO-1 fluorescence in response to ATP with 1 mm (B; n = 5) or 0.1 mm (C; n = 12) Ca2+ in the extracellular buffer. The peak responses differed by ∼33%. It is problematic to interpret this difference precisely, because the magnitude of the YOPRO-1 signal depends on the number of channels in the membrane, the size of the cells, and the amount of nucleotides per cell and is therefore not directly related to the extent of permeability changes. These experiments simply indicate that YOPRO-1 uptake occurs in extracellular solutions containing Na+ with 0.1 or 1 mm Ca2+. B, C, Images were acquired every second (the line shows the mean), but for clarity error bars are only shown for every 10th point. D, Steady-state current at -60 mV from an HEK cell expressing P2X2 channels in extracellular buffer containing NMDG+ instead of Na+ (154 mm) with 1 mm Ca2+. The ATP was applied for the periods indicated by the solid bars above the graph. The bottom panel plots current-voltage relationships during the ATP-evoked response at the peak outward current (I1) and at the peak inward current (I2). E, Same as in D but with 0.1 mm Ca2+ in the extracellular buffer. See Results for average data. Error bars represent SEM.
Cytosolic conformational changes are absent in channels that lack the I2 state
To test the relationship between the I2 state and changes in FY/FC further, we studied channels that carry a mutation in the cytosolic N terminus (T18A), because they display normal I1 but no I2 (Khakh et al., 2000). ATP-evoked currents (in Na+ solutions; 100 μm ATP) at T18A-P2X2 (Boue-Grabot et al., 2000) and T18AP2X2-C/T18A-P2X2-Y were transient and decayed with a rate constant (5.3 ± 0.7 sec-1; n = 16) 30-fold faster than the transition from I1 to I2 (0.17 ± 0.06 sec-1; n = 4) (Fig. 5). Moreover, no time-dependent increase in NMDG+ permeability could be measured for T18A mutants (Table 1). We next determined FRET e for T18A-P2X2-C/T18A-P2X2-Y channels in the resting state, which was 26.3 ± 6.2% (n = 6) (Fig. 8A,B) and not significantly different from that for P2X2-C/P2X2-Y channels (Figs. 1, 2), implying that the cytosolic domains are similar. At cells expressing T18A-P2X2-C/T18A-P2X2-Y channels, we measured robust ATP-evoked inward currents but no changes in the FY/FC ratio during or after ATP (30 μm ATP) (Fig. 8C,D; traces for wt P2X2 channels are superimposed for comparison). We suggest that the T18A mutant lacks I2 because the permissive conformational change in the C-terminal tail is impaired. Consistent with this hypothesis, previous work has suggested that the N and C termini of P2X2 channels may interact (Boue-Grabot et al., 2000).
Figure 8.

T18A P2X2-C/T18A P2X2-Y and P2X1-C/P2X1-Y channels expressed in HEK cells. A-D, Data for T18A P2X2-C/P2X2-Y channels. A, A graph of the donor and acceptor intensity over time during acceptor photo destruction for cells expressing T18A P2X2-C/T18A P2X2-Y. Note that the donor emission intensity increases with a time course similar to the decrease in the acceptor emission. B, Plot of donor fluorescence recovery versus acceptor loss. The y-axis intercept was 26.3 ± 6.2% (n = 6), which is equal to FRET e for T18A P2X2-C/T18A P2X2-Y channels. C, ATP-evoked inward currents from HEK cells expressing T18A P2X2-C/T18A P2X2-Y channels. D, The ratio of FY/FC plotted on the same time axis for these cells. The ATP was applied for ∼100 msec (30 μm ATP); there was no change in FY/FC. Superimposed for comparison are responses for wt P2X2-C/P2X2-Y channels for 100 msec and 20 sec applications of ATP (see Fig. 6). E-H, Data for P2X1-C/P2X1-Y channels. E, A graph of the donor and acceptor intensity over time during photodestruction of YFP for cells expressing P2X1-C/P2X1-Y. Note that the donor emission intensity increases with a time course similar to the decrease in the acceptor emission. F, Plot of donor fluorescence recovery versus acceptor loss. The y-axis intercept was 18.9 ± 4.3% (n = 8), which is equal to FRET e for P2X1-C/P2X1-Y channels. G, H, ATP-evoked inward currents from an HEK cell expressing P2X1-C/P2X1-Y channels (G), and the ratio of FY/FC plotted on the same time axis for these cells (H). The ATP (30 μm) was applied for ∼100 msec; there was no change in FY/FC. Error bars represent SEM.
In the case of another member of the P2X family, P2X1-C/P2X1-Y channels (Li et al., 2000) were also functional, gave robust transient ATP-evoked currents (-774 ± 174 pA) (Figs. 5, 8G), displayed normal NMDG+ permeability in the I1 state (Table 1), and displayed significant FRET with an e of 18.9 ± 4.3%, lower than P2X2-C/P2X2-Y channels (Table 1, Fig. 8E,F). However, P2X1 channels did not undergo permeability changes in response to ATP (30 μm ATP) (Table 1) and also did not display changes in FY/FC measured from cells in which ATP-evoked currents were concomitantly recorded (Fig. 8G,H). Thus, wt (P2X1) and mutant channels (T18A P2X2) lacking I2 also lack ATP-evoked changes in FY/FC but otherwise function normally. This supports the suggestion that changes in FY/FC are specific for conformational changes leading to the I2 state rather than the I1 state.
Immobilizing the cytosolic domains of P2X2 channels with a genetically encoded tag hinders the I2 state and cytosolic conformational changes
If increases in NMDG+ permeability require a cytosolic conformational change to occur, then it may be possible to prevent permeability changes with an appropriate tag that impairs the cytosolic conformational change. We used the PH domain of phospholipase C δ (PLCδ) to test for this (Ferguson et al., 1995). The PH domain displays micromolar affinity for membrane phosphatidylinositol biphosphate (PIP2) and has been used extensively to recruit proteins such as GFP to the membrane (Ferguson et al., 1995; Kobrinsky et al., 2003). We speculated that the PH tag would tether the tips of the P2X2 cytosolic domain to the plasma membrane. Reproducing the published data, we found that the PH domain can recruit YFP to the membrane (Fig. 9 A, B). We next tested P2X2 channels tagged on their C termini with the PH domain (P2X2-PH, P2X2-PH-C, and P2X2-PH-Y). We imaged (0.5-1 μm) confocal sections of cells expressing P2X2-Y and P2X2-PH-Y and noted very profound membrane fluorescence in cells expressing P2X2-PH-Y (Fig. 9D), whereas in cells expressing P2X2-Y, there was additional intracellular fluorescence (Khakh et al., 2001) (Fig. 9C). Fusion of the PH domain to the cytosolic domain of P2X2 channels increased the current density approximately two- to fivefold (Fig. 9E), suggesting that the PH tag results in increased plasma membrane expression of P2X2 channels. FRET e in cells expressing P2X2-PH-C/P2X2-PH-Y at 17.2 ± 2.0% (n = 5) (Fig. 10 A-D) was significantly lower than that for P2X2-C/P2X2-Y channels at 35% (Figs. 1, 2). This suggests that the PH tag splays the tails, but not to the extent that the distance between them is so great that there is no FRET at all. We suppose that the extent of splaying is a product of the length and flexibility of the C tail and the size of the PH domain itself (Ferguson et al., 1995). We next determined the consequences of the PH tether on permeability changes by testing for any differences between wt P2X2 channels and P2X2-PH channels with respect to increases in pNMDG+/pNa+. As expected, wt channels showed a robust 2.8 ± 0.4-fold increase in pNMDG+/pNa+ (+23.9 ± 2.7 mV shift in Erev; n = 17) and slow desensitization in Na+ solutions (Fig. 5). In marked contrast, P2X2-PH channels showed no significant increase in pNMDG+/pNa+ (0.2 ± 0.05-fold; +4.0 ± 0.9 mV shift in Erev; n = 15) (Fig. 11 A) and displayed significantly faster desensitization (Fig. 5, Table 1). These data support the hypothesis that the physiological correlate of I2 is a slowly desensitizing plateau current in Na+ solutions. Based on our work with wt, T18A mutant, and P2X1 channels (see above), we would expect that P2X2-PH-C/P2X2-PH-Y channels would also not show changes in FY/FC. This was the case; although we measured robust ATP-evoked inward currents, we found no evidence of changes in FY/FC (-1.9 ± 1.8% change; n = 5) (Fig. 11 B). Seemingly, P2X2 channels with immobilized-splayed cytosolic domains do not undergo permeability changes, display a faster component to desensitization, and do not display changes in FY/FC.
Figure 9.

The PH domain recruits YFP to the plasma membrane and increases P2X2 current density in the plasma membrane. A, Photomicrograph of HEK cells expressing cytosolic YFP. Note the cytosolic distribution of fluorescence revealed in the line profile. B, Photomicrograph of two neighboring HEK cells expressing PH-YFP; fluorescence is localized to the plasma membrane. C, Photomicrograph of HEK cells expressing P2X2-Y with membrane and intracellular fluorescence. D, Photomicrograph of HEK cells expressing P2X2-PH-Y. Note the fluorescence localized predominantly to the plasma membrane. E, Summary bar graph showing peak ATP-evoked current densities from HEK cells expressing various cDNAs, as indicated (n = 6-15). Error bars represent SEM.
Freeing the immobilized cytosolic domain restores the I2 state and cytosolic conformational changes
If tethering the cytosolic domain of P2X2 channels leads to impaired permeability changes, is it possible to cleave the tether and restore permeability changes? This experiment is feasible because the PH domain has high affinity for inositol 1,4,5 triphosphate (IP3) (∼2 nm) relative to PIP2 (∼3 μm). In a resting cell, this means that PH-Y is bound to plasma membrane PIP2, but when PIP2 is cleaved and IP3 is generated, the PH-Y molecule is freed and moves in to the cytosol in seconds (Stauffer et al., 1998). We tested for this under our conditions by imaging PH-Y over time in HEK cells expressing hP2Y4 receptors (Filippov et al., 2003) that are expected to cleave PIP2 through activation of PLC. We used the hP2Y4 receptor as the trigger to cleave PIP2 because it is activated by UTP and not by ATP (Filippov et al., 2003). In contrast, ATP and not UTP activates P2X2 channels (North, 2002). After UTP addition to the bathing medium, translocation of PH-Y to the cytosol was observed in ∼10 sec, was maintained for ∼20-30 sec in the presence of UTP, and then reversed such that the fluorescence returned back to the periphery of the cell (n = 10) (Fig. 10E-H). We used these conditions to cleave PIP2 in an attempt to free the cytosolic domain from its nonpermissive conformation in P2X2-PH channels and then applied ATP to voltage-clamped cells to determine NMDG+ permeability (UTP itself did not affect the function of P2X2 channels). Under these conditions, the channels did show an increase in NMDG+ permeability (Fig. 11C). Thus, when we applied UTP in the bathing medium for ∼20 sec and then pulsed ATP for ∼30 sec to measure changes in pNMDG+/pNa+, we recorded a significant 2.8 ± 0.8-fold increase in NMDG+ permeability (+19.1 ± 5.4 mV shift in Erev; n = 11; 100 μm ATP) (Fig. 11C) that was almost identical to wt P2X2 channels but significantly larger than the minor changes recorded from P2X2-PH channels with no UTP applied (Fig. 11A). Interestingly, application of UTP also converted fast desensitization of P2X2-PH channels measured in Na+ solutions to a slower form, which more closely resembled wt P2X2 (Fig. 5, Table 1). Interestingly, for cells expressing P2X2-PH-C/P2X2-PH-Y channels (after a 20 sec pulse of UTP), we measured robust ATP-evoked inward currents and significant changes in FY/FC (-9.3 ± 1.8%; n = 7) (Fig. 11D) that were not significantly different from those observed with P2X2 channels (Fig. 11D, superimposed in gray for comparison; Table 1).
We did not measure significant changes in FY/FC for P2X2-PH-Y/P2X2-PH-C channels after UTP application alone (1.7 ± 3.1% change in FY/FC; n = 6), although UTP application restored permeability changes and ATP-evoked changes in FY/FC (Fig. 11). This suggests that the most distal parts of the C tail may remain splayed even after UTP application, whereas functional domains within the tails that are permissive for permeability changes and cytosolic conformational changes are restored. Additional details must await high-resolution structural information, but our previous work suggests that residues permissive for the I2 state are indeed located ∼20 residues before the last amino acid (Eickhorst et al., 2002). Interestingly, in the presence of UTP, P2X2-PH channels displayed an initial reversal potential (in NMDG+) that was ∼10 mV more negative than either P2X2 or P2X2-PH channels (Table 1). This suggests that after PIP2 cleavage and untethering of the cytosolic tail, P2X2-PH channels adopt a conformation whereby the permeability to NMDG+ is low in the I1 state. However, these channels still show shifts in reversal potential and increases in permeability that are identical to those for wt P2X2 channels and significantly smaller than those for P2X2-PH channels in the presence of ATP (Table 1). We interpret these data to indicate that by untethering the cytosolic domain, the ability to undergo permeability changes and enter the I2 state is restored. Overall, there was good correlative agreement between ATP-evoked increases in NMDG+ permeability and changes in FY/FC in terms of activation time (Table 1) and magnitude (Fig. 12), suggesting that the two measures may report a common-related conformational transition(s) in P2X2 channels.
Figure 12.

Correlative relationship between the magnitude of NMDG+ permeability increases and ΔFY/FC. The scatter graph plots the relationship between the magnitudes of ΔFY/FC and the shift in NMDG+ reversal potential for various wt and engineered rP2X2 channels (raw data are in Table 1). The dotted line represents a linear fit to the data and has the form ΔFY/FC(%) =-0.603 (ΔErev; mV) + 1.221. Error bars represent SEM.
Discussion
The data suggest that the cytosolic domain of P2X2 channels undergoes a conformational change that is correlated with permeability changes and in the simplest case is consistent with an expansion and/or rotational movement within this domain. Our experiments cannot discriminate between changes in interfluorophore distance and relative orientation. Previous work suggested that CFP and YFP adopt random orientations when tagged to the C termini of other proteins, allowing FRET to be interpreted in terms of distance (Riven et al., 2003; Zheng et al., 2003). Regardless, together, our data suggest a link between state-specific cytosolic conformational changes and entry into the I2 state. The most salient findings are summarized in a model available as supplemental material (available at www.jneurosci.org).
In a recent model of G-protein-gated inwardly rectifying K+ channels, a conformational change in the cytosolic domain was proposed to be sufficient to open the pore (Riven et al., 2003). From this work, it was unclear whether the cytosolic conformational change occurred before channel opening or was a consequence of it (Evans and Yue, 2003; Riven et al., 2003), primarily because access of the agonist to the receptors was slow. We have managed to circumvent this limitation, because P2X2 channels are predominantly found in the plasma membrane and are amenable to study with rapid agonist applications. FRET efficiencies for P2X2 channels with epifluorescence and TIRF microscopy were nearly identical, implying that sub-membrane fluorescence contributed little to our measurements. Time-resolved measurements favor the idea that cytosolic conformational changes are not simply a reflection of changes in the pore, because they persist even when the pore is closed.
How does the disposition and movement of the cytosolic domain relate to changes in the pore? Previous work (Eickhorst et al., 2002) suggested that the narrowest region of the P2X2 pore dilates by >3 Å, resulting in an increase in the area by at least 60-72 Å2. Interestingly, the FRET efficiency for P2X2 channels with long C termini (∼120 residues) was significantly higher than for P2X1 channels with smaller C termini (∼20 residues) at 35 and 19%, respectively (Table 1). This approximates to distances of 25 and 34 Å between the three tips of the C termini in a trimeric channel. Assuming that P2X channels have similar transmembrane domain organization, this implies that the P2X2 cytosolic domain may narrow by ∼9 Å, like an inverted tripod, from near the plasma membrane (supplemental material, available at www.jneurosci.org). The FRET efficiency for P2X2-PH channels (17%) suggests that the cytosolic domain in these channels does not narrow, resulting in a conformation that is nonpermissive for permeability changes (Table 1). Overall, the data indicate that gating motions may involve distances of no more than a couple of angstroms from the resting conformation of the C tail with tips ∼25 Å apart, whereas larger changes of ∼10 Å, as in P2X2-PH channels, result in nonpermissive states. A question that arises from our work is exactly how the binding of ATP to the extracellular surface can cause motions in the cytosolic domain, given that they have to propagate through the transmembrane segments. The atomic scale details of this will have to await a high-resolution model of the whole channel, but we may be able to extend our work to map individual moving residues within single P2X2 ion channels by using small cysteine-reactive fluorophores (Sonnleitner et al., 2002) and to directly discriminate between distance and rotational movements of these residues with patch-clamp photochemistry (Leite et al., 2003). However, there is precedence for small extracellular ligands causing cytosolic changes in transmembrane proteins. For example, the binding of small molecule transmitters to G-protein-coupled receptors can cause conformational changes that ultimately change the conformation of G-proteins and effectors that are intracellularly bound (Hille, 2001). In the case of receptor tyrosine kinases, binding of extracellular ligand triggers activation of cytosolic domain enzymatic activity (Alberts et al., 2002). More recently, there has been FRET-based evidence for cytosolic domain separation for integrins as a result of binding extracellular ligand (Kim et al., 2003). As far as we know, our findings are the first to show that binding of extracellular transmitter can cause state-specific cytosolic motions in an ion channel.
Given that P2X channels discriminate poorly between cations, will permeability changes result in measurable pathological or physiological effects? In the case of P2X7 channels, there is evidence to suggest that permeability changes may be associated with morphological changes, cell death, release of signaling molecules, and vesicle shedding (Surprenant et al., 1996; Rassendren et al., 1997; Virginio et al., 1999b; MacKenzie et al., 2001; North, 2002; Duan et al., 2003). By analogy, a physiological role for P2X2 channel permeability changes may also be expected (Khakh and Lester, 1999; Khakh et al., 1999; Virginio et al., 1999a; Fujiwara and Kubo, 2004). Indeed, broadly consistent with the morphological changes associated with P2X7 channel activation (North, 2002), previous studies have also described ATP-evoked changes in neuronal dendritic morphology because of enhanced ion flow through the P2X2 channel I2 state (Khakh et al., 2001). It is also possible that permeability changes, and the associated pore dilation, may allow intracellular solutes to leave the cell down their concentration gradients, as well as allowing more ions to enter the cell from the extracellular milieu. This was first demonstrated for P2X7-like channels in mast cells (Cockcroft and Gomperts, 1979) and more recently for astrocytes that release glutamate and aspartate into the extracellular space when P2X7 channels undergo permeability changes (Duan et al., 2003). It will be of interest to determine whether P2X2 channels that are found in presynaptic terminals (Khakh et al., 2003) also directly allow the passage of neurotransmitters into the extracellular space. Furthermore, lack of fast desensitization is a defining feature of recombinant and native homomeric (and some heteromeric) P2X2 subunit-containing channels (North, 2002). Our data suggest that the molecular motions correlated with permeability changes may also contribute to sustained slowly desensitizing P2X2 responses, implying that permeability changes contribute to enhanced ion flow in physiological solutions. It is also noteworthy that recent work on P2X2 channels shows that differences in relative ion permeability arise because of specific polar residues in the pore region, perhaps because they are optimally positioned to provide a favorable environment for passing ions (Migita et al., 2001; Egan and Khakh, 2004). Given that the P2X2 channel pore dilates by >3 Å (Eickhorst et al., 2002), it is tempting to speculate that the mechanisms that determine preferential flow of one ion over another may also change. In future work, it will be important to test this hypothesis for Na+, K+, and Ca2+ (Migita et al., 2001; Egan and Khakh, 2004). From the perspective of ATP signaling, cytosolic conformational changes in other P2X channels may also offer a simple interpretation for several physiological responses that are described but incompletely understood. These include how ATP triggers endocytosis of some P2X channels (Li et al., 2000; Bobanovic et al., 2002), P2X7 channel interactions with intracellular proteins (Kim et al., 2001), and activation of intra-cellular signal transduction pathways (North, 2002). Cytosolic gating motions may also allow P2X channels to directly interact with intracellular processes and add a temporal dimension to ATP signaling that could outlast the surge of extracellular ATP. It is interesting that many ion channels possess structured cytosolic domains that sit on the inner aspects of the pore (Chang et al., 1998; Miyazawa et al., 1999; Sokolova et al., 2001; Higgins et al., 2002; Jiang et al., 2002; da Fonseca et al., 2003; Kuo et al., 2003). In the case of serotonin-gated 5-HT3 channels, cytosolic residues have been shown recently to affect channel conductance (Kelley et al., 2003). It may be of interest to determine whether cytosolic gating affects pore properties such as conductance and permeability in these and other channels and allows them to communicate with intracellular processes and therefore impact signaling.
Footnotes
J.A.F. was supported by a Medical Research Council (MRC) studentship, and G.G. was supported by an MRC undergraduate summer vacation award. The Medical Research Council, European Molecular Biology Organization, Human Frontiers Science Program, and National Institutes of Health support our research. We are indebted to Evgeny Kobrinsky, Roger Tsien, and Joseph Simon for PH domain, YC2.1, and hP2Y4 cDNAs, respectively. We thank Henry A. Lester for discussions and encouragement and David N Bowser for comments on this work.
Correspondence should be addressed to Baljit S. Khakh, Medical Research Council Laboratory of Molecular Biology, Hills Road, Cambridge CB2 2QH, UK. E-mail: bsk@mrc-lmb.cam.ac.uk.
Copyright © 2004 Society for Neuroscience 0270-6474/04/2410475-13$15.00/0
References
- Alberts B, Johnson A, Lewis J, Raff M, Roberts K, Walter P (2002) Cell communication. In: Molecular biology of the cell, Ed 4, pp 831-906. New York: Garland Science.
- Bobanovic LK, Royle SJ, Murrell-Lagnado RD (2002) P2X receptor trafficking in neurons is subunit specific. J Neurosci 22: 4814-4824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boue-Grabot E, Archambault V, Seguela P (2000) A protein kinase C site highly conserved in P2X subunits controls the desensitization kinetics of P2X(2) ATP-gated channels. J Biol Chem 275: 10190-10195. [DOI] [PubMed] [Google Scholar]
- Cha A, Snyder GE, Selvin PR, Bezanilla F (1999) Atomic scale movement of the voltage-sensing region in a potassium channel measured via spectroscopy. Nature 402: 809-813. [DOI] [PubMed] [Google Scholar]
- Chang G, Spencer RH, Lee AT, Barclay MT, Rees DC (1998) Structure of the MscL homolog from Mycobacterium tuberculosis: a gated mechanosensitive ion channel. Science 282: 2220-2226. [DOI] [PubMed] [Google Scholar]
- Cockcroft S, Gomperts BD (1979) ATP induces nucleotide permeability in rat mast cells. Nature 279: 541-542. [DOI] [PubMed] [Google Scholar]
- da Fonseca PC, Morris SA, Nerou EP, Taylor CW, Morris EP (2003) Domain organization of the type 1 inositol 1,4,5-trisphosphate receptor as revealed by single-particle analysis. Proc Natl Acad Sci USA 100: 3936-3941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duan S, Anderson CM, Keung EC, Chen Y, Chen Y, Swanson RA (2003) P2X7 receptor-mediated release of excitatory amino acids from astrocytes. J Neurosci 23: 1320-1328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Egan TM, Khakh BS (2004) Contribution of calcium ions to P2X channel responses. J Neurosci 24: 3413-3420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Egan TM, Haines WR, Voigt MM (1998) A domain contributing to the ion channel of ATP-gated P2X2 receptors identified by the substituted cysteine accessibility method. J Neurosci 18: 2350-2359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eickhorst A, Berson A, Cockayne D, Lester HA, Khakh BS (2002) Control of P2X2 channel permeability by the cytosolic domain. J Gen Physiol 120: 119-131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erickson MG, Alseikhan BA, Peterson BZ, Yue DT (2001) Preassociation of calmodulin with voltage-gated Ca(2+) channels revealed by FRET in single living cells. Neuron 31: 973-985. [DOI] [PubMed] [Google Scholar]
- Evans J, Yue DT (2003) New turf for CFP/YFP FRET imaging of membrane signaling molecules. Neuron 38: 145-147. [DOI] [PubMed] [Google Scholar]
- Ferguson KM, Lemmon MA, Schlessinger J, Sigler PB (1995) Structure of the high affinity complex of inositol trisphosphate with a phospholipase C pleckstrin homology domain. Cell 83: 1037-1046. [DOI] [PubMed] [Google Scholar]
- Filippov AK, Simon J, Barnard EA, Brown DA (2003) Coupling of the nucleotide P2Y4 receptor to neuronal ion channels. Br J Pharmacol 138: 400-406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujiwara Y, Kubo Y (2004) Density-dependent changes of the pore properties of the P2X2 receptor channel. J Physiol (Lond) 558: 31-43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Higgins MK, Weitz D, Warne T, Schertler GF, Kaupp UB (2002) Molecular architecture of a retinal cGMP-gated channel: the arrangement of the cytoplasmic domains. EMBO J 21: 2087-2094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hille B (2001) Ion channels of excitable membranes, Ed 3. Sunderland, MA: Sinauer.
- Jiang LH, Kim M, Spelta V, Bo X, Surprenant A, North RA (2003) Subunit arrangement in P2X receptors. J Neurosci 23: 8903-8910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang Y, Lee A, Chen J, Cadene M, Chait BT, MacKinnon R (2002) Crystal structure and mechanism of a calcium-gated potassium channel. Nature 417: 515-522. [DOI] [PubMed] [Google Scholar]
- Kelley SP, Dunlop JI, Kirkness EF, Lambert JJ, Peters JA (2003) A cytoplasmic region determines single-channel conductance in 5-HT3 receptors. Nature 424: 321-324. [DOI] [PubMed] [Google Scholar]
- Khakh BS (2001) Molecular physiology of P2X receptors and ATP signalling at synapses. Nat Rev Neurosci 2: 165-174. [DOI] [PubMed] [Google Scholar]
- Khakh BS, Lester HA (1999) Dynamic selectivity filters in ion channels. Neuron 23: 653-658. [DOI] [PubMed] [Google Scholar]
- Khakh BS, Bao X, Labarca C, Lester HA (1999) Neuronal P2X receptor-transmitter-gated cation channels change their ion selectivity in seconds. Nat Neurosci 2: 322-330. [DOI] [PubMed] [Google Scholar]
- Khakh BS, Zhou X, Sydes J, Galligan JJ, Lester HA (2000) State-dependent cross-inhibition between transmitter-gated cation channels. Nature 406: 405-410. [DOI] [PubMed] [Google Scholar]
- Khakh BS, Smith WB, Chiu CS, Ju D, Davidson N, Lester HA (2001) Activation-dependent changes in receptor distribution and dendritic morphology in hippocampal neurons expressing P2X2-green fluorescent protein receptors. Proc Natl Acad Sci USA 98: 5288-5293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khakh BS, Gitterman DP, Cockayne D, Jones AM (2003) ATP modulation of excitatory synapses onto interneurons. J Neurosci 23: 7426-7437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim M, Jiang LH, Wilson HL, North RA, Surprenant A (2001) Proteomic and functional evidence for a P2X7 receptor signalling complex. EMBO J 20: 6347-6358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim M, Carman CV, Springer TA (2003) Bidirectional transmembrane signaling by cytoplasmic domain separation in integrins. Science 301: 1720-1725. [DOI] [PubMed] [Google Scholar]
- Kobrinsky E, Schwartz E, Abernethy DR, Soldatov NM (2003) Voltage-gated mobility of the Ca2+ channel cytoplasmic tails and its regulatory role. J Biol Chem 278: 5021-5028. [DOI] [PubMed] [Google Scholar]
- Kuo A, Gulbis JM, Antcliff JF, Rahman T, Lowe ED, Zimmer J, Cuthbertson J, Ashcroft FM, Ezaki T, Doyle DA (2003) Crystal structure of the potassium channel KirBac1.1 in the closed state. Science 300: 1922-1926. [DOI] [PubMed] [Google Scholar]
- Leite JF, Blanton MP, Shahgholi M, Dougherty DA, Lester HA (2003) Conformation-dependent hydrophobic photolabeling of the nicotinic receptor: electrophysiology-coordinated photochemistry and mass spectrometry. Proc Natl Acad Sci USA 100: 13054-13059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li GH, Lee EM, Blair D, Holding C, Poronnik P, Cook DI, Barden JA, Bennett MR (2000) The distribution of P2X receptor clusters on individual neurons in sympathetic ganglia and their redistribution on agonist activation. J Biol Chem 275: 29107-29112. [DOI] [PubMed] [Google Scholar]
- MacKenzie A, Wilson HL, Kiss-Toth E, Dower SK, North RA, Surprenant A (2001) Rapid secretion of interleukin-1beta by microvesicle shedding. Immunity 15: 825-835. [DOI] [PubMed] [Google Scholar]
- Michalet X, Kapanidis AN, Laurence T, Pinaud F, Doose S, Pflughoefft M, Weiss S (2003) The power and prospects of fluorescence microscopies and spectroscopies. Annu Rev Biophys Biomol Struct 32: 161-182. [DOI] [PubMed] [Google Scholar]
- Migita K, Haines WR, Voigt MM, Egan TM (2001) Polar residues of the second transmembrane domain influence cation permeability of the ATP-gated P2X2 receptor. J Biol Chem 276: 30934-30941. [DOI] [PubMed] [Google Scholar]
- Miyazawa A, Fujiyoshi Y, Stowell M, Unwin N (1999) Nicotinic acetylcholine receptor at 4.6 A resolution: transverse tunnels in the channel wall. J Mol Biol 288: 765-786. [DOI] [PubMed] [Google Scholar]
- Nakazawa K, Fujimori K, Takanaka A, Inoue K (1991) Comparison of adenosine triphosphate- and nicotine-activated inward currents in rat phaeochromocytoma cells. J Physiol (Lond) 434: 647-660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nashmi R, Dickinson ME, McKinney S, Jareb M, Labarca C, Fraser SE, Lester HA (2003) Assembly of α4β2 nicotinic acetylcholine receptors assessed with functional fluorescently labeled subunits: effects of localization, trafficking, and nicotine-induced upregulation in clonal mammalian cells and in cultured midbrain neurons. J Neurosci 23: 11554-11567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicke A, Baumert HG, Rettinger J, Eichele A, Lambrecht G, Mutschler E, Schmalzing G (1998) P2X1 and P2X3 receptors form stable trimers: a novel structural motif of ligand-gated ion channels. EMBO J 17: 3016-3028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- North RA (2002) Molecular physiology of P2X receptors. Physiol Rev 82: 1013-1067. [DOI] [PubMed] [Google Scholar]
- Petrou S, Ugur M, Drummond RM, Singer JJ, Walsh JVJ (1997) P2X7 purinoceptor expression in Xenopus oocytes is not sufficient to produce a pore-forming P2Z-like phenotype. FEBS Lett 411: 339-345. [DOI] [PubMed] [Google Scholar]
- Rassendren F, Buell GN, Virginio C, Collo G, North RA, Surprenant A (1997) The permeabilizing ATP receptor, P2X7 Cloning and expression of a human cDNA. J Biol Chem 272: 5482-5786. [DOI] [PubMed] [Google Scholar]
- Riven I, Kalmanzon E, Segev L, Reuveny E (2003) Conformational rearrangements associated with the gating of the G protein-coupled potassium channel revealed by FRET microscopy. Neuron 38: 225-235. [DOI] [PubMed] [Google Scholar]
- Schlichter R, Rodeau JL, Hugel S (2002) Synaptic release of ATP induces pore dilation at central P2X receptors in dorsal horn neurones of the rat spinal cord. FENS Abstr 1: A081.017. [Google Scholar]
- Smart ML, Gu B, Panchal RG, Wiley J, Cromer B, Williams DA, Petrou S (2003) P2X7 receptor cell surface expression and cytolytic pore formation are regulated by a distal C-terminal region. J Biol Chem 278: 8853-8860. [DOI] [PubMed] [Google Scholar]
- Sokolova O, Kolmakova-Partensky L, Grigorieff N (2001) Three-dimensional structure of a voltage-gated potassium channel at 2.5 nm resolution. Structure 9: 215-220. [DOI] [PubMed] [Google Scholar]
- Sonnleitner A, Mannuzzu LM, Terakawa S, Isacoff EY (2002) Structural rearrangements in single ion channels detected optically in living cells. Proc Natl Acad Sci USA 99: 12759-12764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stauffer TP, Ahn S, Meyer T (1998) Receptor-induced transient reduction in plasma membrane PtdIns(4,5)P2 concentration monitored in living cells. Curr Biol 8: 343-346. [DOI] [PubMed] [Google Scholar]
- Steyer JA, Almers W (2001) A real-time view of life within 100 nm of the plasma membrane. Nat Rev Mol Cell Biol 2: 268-275. [DOI] [PubMed] [Google Scholar]
- Stoop R, Thomas S, Rassendren F, Kawashima E, Buell G, Surprenant A, North R (1999) Contribution of individual subunits to the multimeric P2X2 receptor: estimates based on methanethiosulfonate block at T336C. Mol Pharmacol 56: 973-981. [DOI] [PubMed] [Google Scholar]
- Surprenant A, Rassendren F, Kawashima E, North RA, Buell G (1996) The cytolytic P2Z receptor for extracellular ATP identified as a P2X receptor (P2X7). Science 272: 735-738. [DOI] [PubMed] [Google Scholar]
- Torres G, Egan T, Voigt M (1999) Hetero-oligomeric assembly of P2X receptor subunits. Specificities exist with regard to possible partners. J Biol Chem 274: 6653-6659. [DOI] [PubMed] [Google Scholar]
- Tsien RY (1998) The green fluorescent protein. Annu Rev Biochem 67: 509-544. [DOI] [PubMed] [Google Scholar]
- Virginio C, MacKenzie A, Rassendren FA, North RA, Surprenant A (1999a) Pore dilation of neuronal P2X receptor channels. Nat Neurosci 2: 315-321. [DOI] [PubMed] [Google Scholar]
- Virginio C, MacKenzie A, North RA, Surprenant A (1999b) Kinetics of cell lysis, dye uptake and permeability changes in cells expressing the rat P2X7 receptor. J Physiol (Lond) 519: 335-346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng J, Varnum MD, Zagotta WN (2003) Disruption of an intersubunit interaction underlies Ca2+-calmodulin modulation of cyclic nucleotide-gated channels. J Neurosci 23: 8167-8175. [DOI] [PMC free article] [PubMed] [Google Scholar]