

Abstract
Premature death from seizures afflicts gene-targeted mice expressing the Q/R site-unedited glutamate receptor subunit GluR-B(Q) of AMPA receptors in central neurons. Early seizure-related death has now been circumvented by a genetic switch that restricts GluR-B(Q) expression to forebrain principal neurons from postnatal stages onward, prominently in hippocampus and striatum and less so in cortex and amygdala. When switched on, functional receptor incorporation of GluR-B(Q) could be demonstrated by imaging evoked AMPA channel-mediated spinous Ca2+ transients in CA1 pyramidal cells. Sustained GluR-B(Q) expression in adult mice led to smaller excitatory postsynaptic responses in the CA1 region with unchanged presynaptic fiber excitability. Notably, despite the smaller excitatory response, the CA1 cells exhibited a reduced population spike threshold, which might underlie the spontaneous manifestations of epilepsy, including myocloni and generalized seizures with limbic components, observed by synchronous video monitoring and electroencephalographic recordings. No neuropathological symptoms developed when GluR-B(Q) expression was restricted to only hippocampal neurons. Our results show that seizure susceptibility is triggered by GluR-B(Q) expression also in the adult brain and that circuit hyperexcitability is not an immediate consequence of GluR-B(Q) but requires yet unknown downstream events, likely to be induced by non-Hebbian plasticity from Ca2+-permeable AMPA channels in principal neurons.
Keywords: temporal Cre regulation, RNA editing, spinous calcium transients, altered AMPA receptors, hippocampus, population spike
Introduction
In mice, a neurological syndrome with spontaneous and recurrent epileptic seizures starting at postnatal day 14 (P14) and ending with death at approximately P20-P25 can be induced by the inhibition of RNA editing at a single codon position for the AMPA glutamate receptor GluR-B (GluR2) (Brusa et al., 1995; Higuchi et al., 2000). Although presence and levels of unedited GluR-B correlate with the seizure attacks, the molecular and cellular mechanisms underlying the seizure susceptibility remain unknown, and contribution from disturbed development resulting in altered neuronal connectivity could not be excluded.
Among the four AMPA receptor subunits GluR-A to GluR-D (GluR1-GluR4), which constitute heterodimeric, AMPA receptor channels of tetrameric stoichiometry, the GluR-B subunit is of unique importance. GluR-B carries in its pore-forming domain an arginine (R) residue, resulting from RNA editing of a genomically encrypted glutamine (Q) codon (Sommer et al., 1991). This arginine residue is functionally dominant in heteromeric AMPA receptor subunit assemblies, because it determines gating kinetics, unitary channel conductance, voltage-independent gating, Ca2+ permeability, and even channel assembly (Burnashev et al., 1992; Geiger et al., 1995; Swanson et al., 1997; Feldmeyer et al., 1999; Greger et al., 2003). Because >99% of the GluR-B subunits are edited at this Q/R site (Sommer et al., 1991), most neurons express Q/R site-edited GluR-B and therefore contain AMPA receptors with low unitary channel conductance, low Ca2+ permeability, and voltage-independent gating. If editing of the Q codon is abolished by knock-out of the editing enzyme ADAR2 (adenosine deaminases that act on RNA) (Higuchi et al., 2000) or by removing the cis-acting exon complementary sequence (ECS) in intron 11 of the GluR-B gene (Brusa et al., 1995), a substantial portion of the AMPA channels switch from low to high Ca2+ permeability and exhibit a double-rectifying instead of linear current-voltage relationship. If this switch occurs already in the embryo, as it does in GluR-B+/ΔECS and ADAR2-/- mice, it ultimately engenders, by unknown downstream events, a severely compromised phenotype with growth retardation, spontaneous seizures, and premature death.
We have now used genetic regulation to induce the GluR-B(R)-to-GluR-B(Q) AMPA channel switch during juvenile development to investigate whether GluR-B(Q)-containing AMPA receptors when expressed postnatally will still cause an epileptic phenotype. For this, we used the expression-attenuated GluR-Bneo allele (Feldmeyer et al., 1999), which can be converted to the dominant-negative GluR-BΔECS allele by Cre recombinase-mediated removal of the attenuating sequences engineered into intron 11 of the GluR-Bneo allele. Thus, the AMPA receptor switch was tightly controlled by Cre expression. Furthermore, we restricted the GluR-B(R)-to-GluR-B(Q) switch to different principal neuron populations of the forebrain to delineate brain regions responsible for seizure activity. We could show that vulnerability to GluR-B(Q) is not confined to early postnatal life, because seizures were also triggered in the adult mouse from delayed GluR-B(Q) expression. The AMPA receptor switch was accompanied by increased excitability of hippocampal neurons, suggesting a dominant role in seizure induction by the hippocampus. Long-term electroencephalographic (EEG) and video analyses showed that the forebrain-specific AMPA receptor switch ultimately led to myoclonic jerks, followed by secondary generalized seizures. When GluR-B(Q) expression was restricted to only hippocampal neurons, all neuropathological symptoms except occasional spontaneous EEG activities were abolished, indicating that, in addition to hippocampus, other brain regions need to become hyperactive for generalized epileptic activity.
Our EEG analyses and classification of seizures revealed similarities to human temporal lobe epilepsy. However, a role of Q/R site editing of AMPA or kainate receptors suggested by murine models (Sailer et al., 1999; Vissel et al., 2001; this study) remains speculative and has been neither confirmed nor refuted by recent investigations into the editing status of GluR transcripts in brain tissue excised from patients with temporal lobe epilepsy (Grigorenko et al., 1998; Kortenbruck et al., 2001).
Materials and Methods
Mouse lines. The RosaR26 mouse line (Soriano, 1999) was used as Cre reporter. Transgenic TgCre4/GluR-B+/neo mice were of mixed C57BL/6 and NMRI genetic background and generated from TgCre4 (Mantamadiotis et al., 2002) and GluR-B+/neo (Feldmeyer et al., 1999) mice. Tgα-CaMKII-tTA/LC1/GluR-B+/neo mice, also of mixed C57BL/6 and NMRI background, were generated by breeding of lines Tgα-CaMKII-tTA (Mayford et al., 1996), TgLC1 (Schonig et al., 2002), and GluR-B+/neo (Brusa et al., 1995). TgCN10-itTA mice were generated by pronucleus injection of a ∼11 kb minigene containing (5′ to 3′) a 8.5 kb α-CaMKII (calcium/calmodulin-dependent kinase II) promoter fragment (Mayford et al., 1996), 1.0 kb of the NR2C (NMDA receptor subunit 2C) gene with exons 1-3, including a neuron-restrictive silencer element (NRSE) (Suchanek et al., 1997), and an “improved” tTA (itTA) gene with a polyadenylation signal from the human growth hormone gene (Chen et al., 1989). The itTA sequence, generated by assembly PCR (Stemmer et al., 1995), encodes three F-domains (PADALDDFDLDML) in place of VP16 (Baron et al., 1997) and contains numerous nucleotide substitutions that eliminate from the prokaryotic coding sequence (Gossen and Bujard, 1992) potential splice sites and CG dinucleotide sequences and introduces eukaryotic codon usage and a Kozak consensus sequence (Kozak, 1984) for efficient translational initiation (Kim, 2001).
Genotyping. The gene-targeted and transgenic mouse lines were genotyped from tail DNA by PCR with the specific primers listed, generating the following approximately sized DNA bands: TgRosa26R, primers lac5′ (5′-TTACGATGCGCCCATCTACAC-3′) and lac3′ (5′-TTACCCGTAGGTAGTCACGCA-3′), 500 bp; GluR-B+/neo, primers MH53a (5′-GAATGTTGATCATGTGTTTCCCTG-3′), MH117 (neo) (5′-GTTCGAATTCGCCAATGACAAGACG-3′), and MH65 (5′-CAATACAATTGGTGATTTGTGAC-3′), 500 and 400 bp for the GluR-B+ and mutant GluR-Bneo alleles, respectively; Tgα-CaMKII-Cre (“TgCre4”) and TgLC1, primers rspCre1 (5′-ACCAGGTTCGTTCACTCATGG-3′) and rspCre2 (5′-AGGCTA AGTGCCTTCTCTACAC-3′), 200 bp DNA; Tgα-CaMKII-tTA, primers tTA1 (5′-GTGATTAACAGCGCATTAGAGC-3′) and tTA3 (5′-CGCCGTCTAAGTGGAGCTCGTCC-3′), 750 bp; and TgCN10-itTA, htTA1 (5′-AGAGCAAAGTCATCAACTCTGCC-3′) and htTA2 (5′-GTGAGAGCCAGACTCACATTTCA-3′), 600 bp.
Regulation with doxycycline and phenobarbital treatment. Cre recombinase and luciferase expression were suppressed from conception until P0, P5, P10, or P15 with doxycycline (Dox) (50 mg/l; Sigma, Taufkirchen, Germany) in the drinking water of pregnant and nursing females and subsequently induced by transferring pups to Dox-naive foster mothers (Krestel et al., 2001). Phenobarbital (Luminal; 200 mg/ml per injection; Desitin, Hamburg, Germany) was added to 1% sucrose in drinking water of nursing mothers in increasing concentrations (final concentration, 100 mg/kg body weight), assuming that ∼25% of maternal drug blood serum levels are transmitted with milk to pups (data sheet of the manufacturer). Phenobarbital levels were tapered out over 5 d beginning at P50.
β-Galactosidase staining. Briefly, β-galactosidase was visualized by incubating vibratome-cut brain slices in X-gal solution [5 mm K4Fe(CN)6, 5 mm K3Fe(CN)6, 2 mm MgCl2, and 2 mg/ml 5-bromo-4-chloro-3-indolyl β-d-galactopyranoside (X-gal) in PBS] in the dark at room temperature overnight (Krestel et al., 2001). All chemicals were from Sigma (Taufkirchen, Germany).
Immunohistochemistry and quantification of immunolabeled cells by confocal laser-scanning microscopy. Polyclonal anti-Cre recombinase IgG (1:5000; Covance, Richmond, CA) and monoclonal anti-neuronal nuclei IgG (NeuN) (1:1000; Chemicon, Temecula, CA) were used for immunohistochemical detection of Cre and NeuN as described previously (Krestel et al., 2001). Secondary antibodies were Texas Red-conjugated donkey anti-rabbit IgG (1:100; Jackson ImmunoResearch, West Grove, PA) and FITC-conjugated donkey anti-mouse IgG (1:100; Jackson ImmunoResearch). Coronal brain slices from three prenatally Dox-treated and three Dox-naive Tgα-CaMKII-tTA/LC1/Rosa+/R26R mice at age P30 were analyzed for coexpression of Cre and the neuronal marker NeuN. Quantification was by determining the ratio of anti-NeuN-FITC-labeled and anti-Cre-Texas Red neurons in confocal images of CA1, CA3, dentate gyrus (DG), striatum, and somatosensory cortex layers 5/6. Between 100 and 400 neurons were analyzed per image.
Video and EEG monitoring of mice. All animal procedures were conducted in accordance with a European Communities Council Directive (86/609/EEC). Mice were monitored with a video camera (CCD video camera WV-CL920 by Panasonic; Deininger, Weingarten, Germany) attached to a digital video recorder (Eneo, DLR-116/80NW; Deininger) continuously for 5-14 d. An infrared (IR) light-emitting diode spotlight (Videor Technical, Göttingen, Germany) permitted observation in the dark.
Telemetric EEG recording was performed after surgical implantation of telemetric transmitters (TA10EA-F20; DataSciences International, St. Paul, MN) with permission of the Veterinarian State Department. Shortly, skin overlaying skull and neck was incised, and a subcutaneous skin pouch was created by blunt dissection underneath skin on the back of the animal. Telemetric implants were placed into the pouch, and flexible leads were connected to stainless steel screws (length 2.6 mm; thread diameter, 1 mm) positioned epidurally over the frontal and parietal cortex (1.5 mm lateral to sagittal suture and rostral to bregma; second screw was positioned on the contralateral side 1.5 mm lateral to sagittal suture and caudal to bregma). The neck skin incision was sutured, and the remaining gap on the skull with screws and attached leads was covered with dental cement. EEG data were recorded with Dataquest A.R.T. 2.2 software (DataSciences International). Off-line interictal spike sorting and frequency analysis was by Spike2 software version 4.08 (Cambridge Electronics Design, Cambridge, UK). To discriminate abnormal interictal spikes from physiologically occurring ones in epidural one-channel electroencephalograms, only those spikes were counted manually that were followed by a wave. Transmitters were implanted into Dox-naive Tgα-CaMKII-tTA/LC1/GluR-B+/neo mice on average at P47, in mice on Dox until birth on average at P92, and into TgCN10-itTA/LC1/GluR-B+/neo mice on average at P96. Mice showed the first seizure attack ∼6 d after implantation.
Multiple depth-electrode EEG recordings were conducted on six mice (male and female, 25-35 gm). Mice were placed in a mouse-adapted stereotaxic frame (Cunningham mouse frame; Stoelting, Wood Dale, IL) in a flat skull position. Bipolar electrodes made of two twisted enamel isolated wires (0.17 mm in diameter) were implanted in the right dorsal hippocampus (anteroposterior, 2.0 mm; mediolateral, 1.5 mm; dorsoventral, 1.9 mm) with the bregma as reference (Franklin and Paxinos, 1997). The animals were also equipped with monopolar cortical electrodes made of tungsten rods (0.25 mm in diameter; Phymep, Paris, France) onto the left fronto-parietal cortex, and the right frontal cortex. Hippocampal and cortical electrodes were soldered on male pins (Farnell, Paris, France) and secured onto the skull with acrylic dental cement. EEG activities were recorded with a computer-based digital acquisition system (Coherence; Deltamed, Paris, France) in animals freely moving in a Faraday cage. A referential setup was used in which cortical and hippocampal electrodes were referenced with an electrode placed over the cerebellum. This setup allowed visualizing the EEG recording in different derivations after the acquisition. Each animal was EEG monitored for several hours. After completion of the experiments, animals were injected with a lethal dose of Nembutal (100 mg/kg, i.p.). Brains were removed, frozen, and cut in 20 μm sections on a cryostat. Histological analysis was performed after cresyl violet staining to verify the location of the hippocampal and cortical electrodes.
Two-photon imaging. Acute transverse hippocampal slices (250 μm thick) were prepared from P28-P33 mice as described previously (Stuart et al., 1993). Whole-cell patch-clamp recordings were performed with an EPC-9 amplifier, filtered at 3 kHz, and digitized with 10 kHz. A galvanometer scanning unit (TCS NT; Leica) was adapted to an upright microscope (DMLFS; Leica) equipped with a 63× objective (harmonic components X apochromatic glass W63× ultraviolet index; numerical aperture, 0.9; Leica). For two-photon excitation (Denk et al., 1990), short pulses of 170-200 fsec at 76 MHz from a titanium/sapphire laser (MIRA 900F; Coherent, Santa Clara, CA) pumped by a solid-state laser (Verdi 5W; Coherent) at 870-890 nm were used. Line-scan imaging was performed using both scan directions at highest magnification, resulting in a high-duty cycle with high temporal resolution (2.2 msec/line). External non-descanned detection behind the objective and the condenser was used for signal collection, yielding high signal collection efficiency. Transmission and epifluorescence signals were recorded by photomultiplier tubes (R6357; Hamamatsu Photonics, Herrsching, Germany) selected for high quantum efficiency and summed off-line. Simultaneously to fluorescence image acquisition, IR-scanning gradient contrast images were recorded. The transmitted IR laser light was imaged through a gradient-contrast tube (Rathenberg et al., 2003) onto a photomultiplier. Fluorescence line-scan images were analyzed using custom software macros based on the Leica confocal software. A line was scanned every 2.2 msec and two lines were averaged off-line, resulting in a temporal resolution of 4.4 msec. The fluorescence for each time point was averaged for regions of interest enclosing the spine examined. Stimulation protocols began 150 msec after the start of the line scan. Before stimulation, fluorescence was averaged for 100 msec to obtain the basal fluorescence F0. A region distant to any indicator-containing structure was chosen to determine the background fluorescence FB. Relative fluorescence changes were calculated as ΔF/F(t) = (F(t) - F0)/(F0 - FB) and were fitted with a single exponential using a least-square fit routine (Igor; WaveMetrics, Lake Oswego, OR). This fit yielded the peak amplitude of the fluorescence increase and the decay time constant τ.
Hippocampal field recordings and analyses. Mice (P60-P90) were killed with halothane. The brain was removed, and transverse slices (400 μm) were cut from each hippocampus with a vibroslicer in artificial CSF (ACSF) (4°C, bubbled with 95% O2-5% CO2, pH 7.4) containing the following (in mm): 124 NaCl, 2 KCl, 1.25 KH2PO4, 2 MgSO4, 1 CaCl2,26 NaHCO3, and 12 glucose. Slices were placed in a humidified interface chamber at 28-32°C and perfused with ACSF containing 2 mm CaCl2. Orthodromic synaptic stimuli (<290 μA, 0.1 Hz) were delivered through a tungsten electrode placed in the stratum radiatum. A glass electrode (filled with ACSF) was placed in the synaptic layer (stratum radiatum) and recorded the presynaptic volley and the field EPSP (fEPSP), whereas another electrode in the pyramidal cell body layer (stratum pyramidale) monitored the population spike. After obtaining stable responses, we stimulated the afferent fibers with increasing strength (increasing the stimulus duration in steps of 10 μsec from 0 to 150 μsec, five consecutive stimulations at each step). A similar approach was used to elicit paired-pulse responses (50 msec interstimulus interval, the two stimuli being equal in strength).
To assess synaptic excitability, we measured for different stimulation strengths the amplitude of the presynaptic volley and the amplitude of the fEPSP. In the pyramidal layer recording, the population spike amplitude was obtained by measuring the maximal negative deflection of the population spike onto a line drawn between the maximum prespike and postspike fEPSP positivity. To pool data from the paired-pulse experiments, we selected responses to stimulation strength just below the threshold for eliciting a population spike on the second fEPSP. Data are pooled across mice of the same genotype and are presented as mean ± SEM. Statistical significance was evaluated by Student's t test.
Results
Conditional Cre expression in forebrain
The regional and temporal control of the AMPA receptor switch is central to our studies. Therefore, to optimize the Cre-mediated switch of AMPA receptors, we analyzed the Cre expression and activity patterns in different mouse lines carrying Cre transgenes (Fig. 1A). For this analysis, Cre indicator mice were used because the Cre-mediated AMPA receptor switch itself cannot be monitored by immunohistochemistry or in situ hybridization. In mouse line RosaR26, the Rosa26 locus harbors a lacZ gene silenced with a transcriptional terminator sequence (Soriano, 1999) (Fig. 1B). Cre-mediated removal of the floxed transcriptional terminator sequence activates lacZ gene expression, which can be easily monitored by X-gal staining.
Figure 1.

Schematic view of the genetic elements used for the conditional gene switch. A, Different transgenic lines were used for conditional expression of Cre in the mouse brain. In line Tgα-CaMKII-Cre [line “TgCre4” (Mantamadiotis et al., 2002)] is directly controlled by a 8 kb promoter fragment from the gene for the α-subunit of CaMKII. In line TgLC1 (Schonig et al., 2002), Cre expression requires activation by the transcription factor tTA, which has to be provided by other transgenes, such as from line Tgα-CaMKII-tTA. Dox inhibits DNA binding of tTA, which, without Dox, binds to the bidirectional tTA-dependent promoter (Ptetbi) to induce luciferase and Cre expression from the transgenic LC1 locus. We used the forebrain-specific tTA expression provided again by the 8 kb promoter fragment of the α-CaMKII subunit gene of mouse line Tgα-CaMKII-tTA (Mayford et al., 1996) to direct Cre expression to the forebrain. In addition, we used line TgCN10-itTA with the itTA expressed from the α-CaMKII promoter combined with the NRSE comprising three untranslated exons (e1-e3) of the NR2C gene (Suchanek et al., 1997). B, The different target genes for Cre are the targeted Rosa26 locus harboring a lacZ gene silenced (lacZ-) with a floxed transcriptional terminator sequence (neo pA) (Soriano, 1999) and the GluR-Bneo allele silenced by a floxed thymidine kinase and neomycin gene (TK-neo pA) in intron 11 (Feldmeyer et al., 1999). Exons 10-12, with 11 and 12 encoding membrane-spanning segments M1-M3 of GluR-B, are indicated together with the ECS in intron 11 of the wild-type allele, present in these mice. Cre activity excises the floxed sequences and activates the expression of the newly formed alleles Rosa26Δ (lacZ+) and GluR-BΔECS.
Abundant lacZ gene expression could be induced in forebrain by Cre recombinaseexpressed from a transgenic α-CaMKII promoter fragment [Tgα-CaMKII-Cre (Mantamadiotis et al., 2002)], with striatum, cortex, hippocampus and olfactory bulb showing strong X-gal staining (Fig. 2A). An even more abundant Cre-induced lacZ expression was observed in mice expressing Cre controlled by α-CaMKII promoter-driven tTA (Tgα-CaMKII-tTA/LC1). X-gal staining was obtained from embryonic stages onward and increased in intensity postnatally in major forebrain areas that also showed Cre immunostaining throughout postnatal development (Fig. 2A) (supplemental Fig. 1, available at www.jneurosci.org as supplemental material). However, in this genetic paradigm, the extent of Cre expression could be restricted by Dox. If Dox was applied from conception, no Cre activity was detectable in the brain (supplemental Fig. 1, available at www.jneurosci.org as supplemental material), concordant with previous results (Hasan et al., 2001; Schonig et al., 2002). If Dox was applied from conception until birth (P0) (Fig. 2A) (supplemental Fig. 1, available at www.jneurosci.org as supplemental material), the Cre-specific immunosignal in stratum pyramidale of CA1 (Fig. 2A, Δ) increased over time, reflecting postnatal increase in tTA from the transgenic α-CaMKII promoter, but revealed slow Cre induction after Dox withdrawal (Fig. 2B).
Figure 2.

Regulation of Cre expression in mouse forebrain. A, Cre-immunolabeled (αCre) and corresponding X-gal-stained (lacZ) coronal brain sections and sagittal X-gal-stained cerebellar sections of TgCre4, Dox-naive (no Dox), and prenatally Dox-treated (Dox till P0) Tgα-CaMKII-tTA/LCR and TgCN10-itTA/LC1 mice. All sections were taken between P30 and P60 from mice that carried the Cre-indicator lacZ gene Rosa+/R26R to permit the visualization of Cre activity by X-gal staining. CA1, CA1 stratum pyramidale; pcx, piriform cortex. B, Confocal analysis of Cre- and NeuN-immunolabeled neurons in hippocampal CA1, DG neurons, and striatum in prenatally Dox-treated Tgα-CaMKII-tTA/LC1/Rosa+/R26R mice at P30. C, Ratios of Cre-positive to NeuN-positive neurons, in several brain regions (CA1 and CA3 stratum pyramidale, dentate gyrus, striatum, and somatosensory cortex layer 5/6). Ratios (error bars, mean ± SD) were assessed by confocal analysis (see B) from four sections of three Dox-naive and three prenatally Dox-treated mice. Note that prenatal Dox exposure increases the extent of mosaic expression substantially in some regions.
To quantify the extent of Cre reactivation and to obtain cellular resolution of Cre activity, we evaluated the number of Cre-expressing neurons in striatum, in the CA1, CA3, and DG subfields of the dorsal hippocampus, and in somatosensory cortex of P30 Tgα-CaMKII-tTA/LC1/Rosa+/R26R mice. Optical sections of single confocal scans were used to determine in single cell layers the proportion of Cre-positive neurons, identified as NeuN-immunopositive cells (Fig. 2B). In three Dox-naive mice, 80-90% of the neurons in CA1, DG, cortex, and striatum showed Cre expression, whereas only ∼60% of the CA3 pyramidal cells were positive for Cre. In three mice suppressed by Dox during intrauterine development, high Cre/NeuN ratios were reached by P30 in striatal and CA1 neurons only. However, in CA3 pyramidal cells, DG cells, and neurons of the somatosensory cortex, Cre expression was induced in only a minor subset (Fig. 2C). Further aging of the mice and different time windows for Dox suppression did not significantly improve the Cre induction (data not shown).
An even more restricted pattern of Cre activity was finally obtained, without taking recourse to Dox regulation, by introducing the neuron-restrictive silencer NRSE element of the NR2C promoter (Suchanek et al., 1997) into the α-CaMKII promoter fragment. When analyzing Cre activity on the Rosa26R locus in TgCN10/LC1/Rosa+/R26R mice, we observed a remarkably limited pattern of Cre activity, which appeared to be present selectively in hippocampal CA1 pyramidal and DG granule cells (Fig. 2A, bottom row).
Lethal seizures by forebrain expression of GluR-B(Q)
Next we determined whether a correlation existed between the Cre expression pattern of the different mouse lines and the neurological phenotype mediated by the Cre-induced GluR-B+/neo to GluR-B+/ΔECS switch. GluR-B+/neo mice carrying the transgene of line TgCre4, which expresses Cre recombinase in forebrain from a transgenic α-CaMKII promoter fragment (Mantamadiotis et al., 2002), became seizure prone. These mice, although longer-lived than GluR-B+/ΔECS mice (Brusa et al., 1995), also died prematurely from seizure attacks (Fig. 3), indicating that the AMPA receptor switch occurred in many brain regions early in development. Therefore, we used the transgenic LC1 line in which Cre expression is stringently controlled by the Dox-dependent transcription factor tTA (Hasan et al., 2001; Schonig et al., 2002) (supplemental Fig. 1, available at www.jneurosci.org as supplemental material). Double-transgenic Tgα-CaMKII-tTA/LC1 mice carrying a gene-targeted GluR-Bneo allele (Brusa et al., 1995) died of seizures within 3 weeks of life (Fig. 3, -Dox), much earlier than the TgCre4/GluR-B+/neo genotype, because of earlier embryonic onset of Cre activity (supplemental Fig. 1, available at www.jneurosci.org as supplemental material). However, they survived without signs of phenotypic impairments when given Dox in utero and during subsequent postnatal times (Fig. 3, +Dox) (supplemental Fig. 1, available at www.jneurosci.org as supplemental material). Notably, most Tgα-CaMKII-tTA/LC1/GluR-B+/neo mice (10 of 13) survived when Dox treated in utero until birth (P0) (Fig. 3, DoxP0), and the others died between P15 and P30. This postnatal period may be particularly sensitive for seizure-related death from the dominant-negative GluR-BΔECS allele, the generation of which is suppressed by the Dox in utero regimen until at least 2 weeks after birth (supplemental Fig. 1, available at www.jneurosci.org as supplemental material). Moreover, mice not given Dox in utero had a ∼50% survival chance when Dox was administered during early postnatal life (Fig. 3, DoxP5-30). The higher death rate of mice put on Dox only after P5 is likely to reflect the early postnatal conversion of GluR-Bneo to GluR-BΔECS. Remarkably, when Cre expression was restricted entirely to hippocampus by expressing tTA from the modified α-CaMKII promoter of transgenic TgCN10-itTA mice (Figs. 1, 2A), Dox-naive TgCN10-itTA/LC1/GluR-B+/neo mice manifested no overt seizures but developed rhythmic spike activity in epidural electroencephalograms (see below). Thus, additional forebrain regions need to express GluR-B(Q) and be recruited for behavioral seizure manifestation.
Figure 3.

Survival curves of GluR-B(Q)-expressing mice. TgCre4/GluR-B+/neo mice showed high mortality, and, by P60, 60% died from seizures (gray line, open squares). Tgα-CaMKII-tTA/LC1/GluR-B+/neo mice (dark lines) lived seemingly normal lives with life-long Dox administration (filled circles). Without Dox (open triangles), the mice did not survive P20. When Dox exposure was confined to prenatal stages (filled squares), periodic mild seizure attacks were observed, but most mice survived. Postnatal Dox administration (open circles) prevented seizure-related death in less than half of the mice until P60. In Dox-naive mice, therapeutic phenobarbital (Pb) administration (open stars) prolonged survival, and mice surviving P50 died from seizures within 10 d after drug removal. The number of mice (n) observed for each treatment and genotype is indicated on the right.
Seizures can be controlled by pharmacological means, which might also suppress the seizure-related lethality resulting from forebrain expression of GluR-B(Q). Hence, we tested whether barbiturates, which are effective anticonvulsants, suppress overt seizures in Dox-naive Tgα-CaMKII-tTA/LC1/GluR-B+/neo mice, and administered phenobarbital from P5 to P50 (1 mg/ml drinking water) (Fig. 3, +Pb). This intervention was only partly successful, because 8 of 14 mice died from seizures by P30. Withdrawal of phenobarbital at P50 caused seizure relapse in the survivors, which died within 10 d. These observations emphasized the need for genetic control of GluR-B(Q) expression.
Spinous AMPA channel-mediated Ca2+ transients in CA1 pyramidal cells
To indicate altered AMPA receptor properties after the Cre-induced AMPA receptor switch, we monitored the Q/R codon ratio for GluR-B transcripts as a measure of Cre activity at the GluR-Bneo locus in hippocampi of Tgα-CaMKII-itTA/LC1/GluR-B+/neo mice and recorded spinous Ca2+ transients in CA1 pyramidal cells (Fig. 4A,B).
Figure 4.
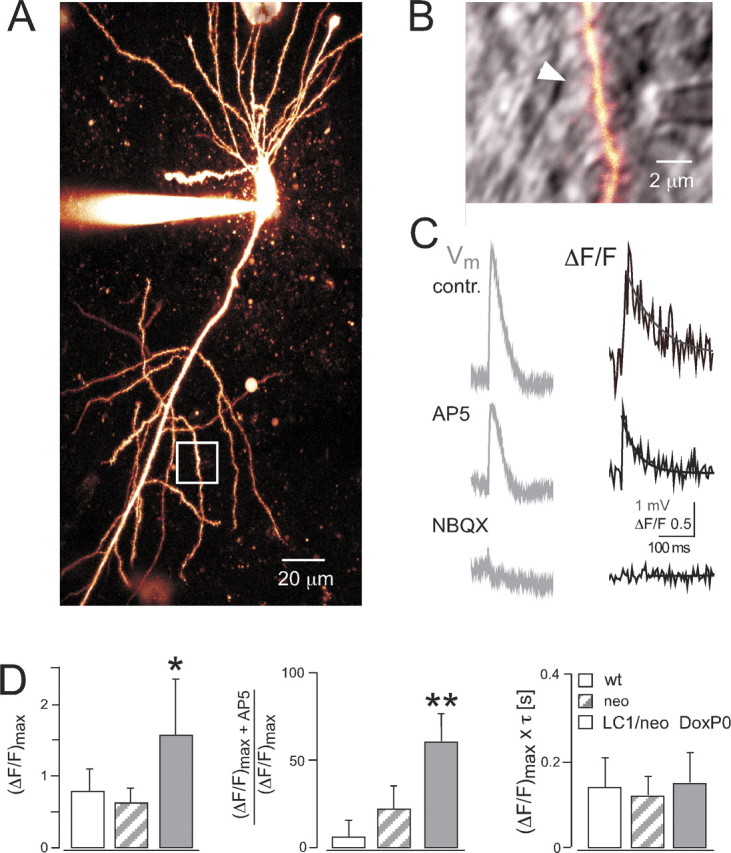
Spinous Ca2+ transients mediated by altered AMPA channels. A, Two-photon excitation image of CA1 pyramidal neuron filled with Oregon Green BAPTA-1 from prenatally Dox-treated P30 Tgα-CaMKII-tTA/LC1/GluR-B+/neo mouse. B, Enlarged view of dendritic region indicated by white box in A. The fluorescence channel is overlaid with the infrared-scanning grading contrast channel to show position of bipolar stimulation pipette close to dendrite. The white arrow indicates spine with clear Ca2+ transient after subthreshold synaptic stimulation. C, Somatic voltage recordings (Vm, membrane potential) and fluorescence changes (ΔF/F) in spine head indicated in B. Bath application of the NMDA receptor blocker AP-5 (25 μm) decreased Ca2+ influx (AP5), which could be abolished by additional application of the AMPA receptor blocker NBQX (10 μm, bottom trace). Ca2+ transients were fitted with single exponential (solid line), yielding the peak amplitude (ΔF/F)max and decay time τ. D, Left, Averaged peak amplitudes of Ca2+ transients. *p<0.05 compared with wild type (wt) and neo. Middle, Reduction of peak Ca2+ transient amplitudes by bath application of AP-5. **p < 0.005 compared with wild type (wt) and neo. Right, Averaged time-integrated spinous Ca2+, calculated by multiplication of peak amplitude with decay time. Values represent mean ± SD.
Q/R ratios for GluR-B cDNA were determined at P30 in mice, continuously given Dox, exposed to Dox during prenatal stages, or raised without Dox. Q/R ratios are <0.1% in wild type (Feldmeyer et al., 1999) but were 8.5 and 8.3% in two mice continuously on Dox, in good correspondence to the ratio determined previously for GluR-B+/neo mice (Feldmeyer et al., 1999). The Q/R ratio increased to 14 ± 0.8% (mean ± SD; n = 4) for Tgα-CaMKII-itTA/LC1/GluR-B+/neo mice, on Dox until P0. In two Dox-naive mice, analyzed at P19 because they would not survive 3 weeks of age (Fig. 3) (see also below), the ratio further increased to 23%. This value is somewhat lower than the 25-28% determined for P15-P18 GluR-B+/ΔECS mice (Brusa et al., 1995; Feldmeyer et al., 1999), probably reflecting the mosaic expression of Cre protein in Dox-naive Tgα-CaMK-itTA/LC1 mice (supplemental Fig. 1, available at www.jneurosci.org as supplemental material).
The increased Q/R ratio of GluR-B should result in spinous Ca2+ influx through the functionally altered AMPA channels, which can be visualized by a transient increase in fluorescence of Ca2+-sensitive dyes during two-photon laser excitation. Indeed, the maximal Ca2+ transient amplitude [(ΔF/F)max], evoked by subthreshold synaptic stimulation of Schaffer collateral fibers, in spine heads of CA1 pyramidal cells was much larger in prenatally Dox-treated Tgα-CaMKII-itTA/LC1/GluR-Bneo mice at P30 [(ΔF/F)max = 1.6 ± 0.8; n = 8 spines] than in age-matched GluR-B+/neo [(ΔF/F)max = 0.6 ± 0.2; n = 9 spines; p < 0.01; one-sided, unpaired t test] and wild-type [(ΔF/F)max = 0.8 ± 0.3; n = 9 spines; p < 0.05] mice (Fig. 4C, contr., D, left diagram). Bath application of the NMDA channel blocker AP-5 (25 μm) resulted on average in a reduction of the peak amplitude to 60 ± 16% (n = 6 spines) in Tgα-CaMKII-itTA/LC1/GluR-B+/neomice, to 22 ± 13% (n = 6 spines) in GluR-B+/neo mice, and 6 ± 8% (n = 5 spines) in wild-type mice (Fig. 4C, AP5, D, middle diagram). Additional bath application of the AMPA receptor blocker 2,3-dihydroxy-6-nitro-7-sulfonyl-benzo[f]quinoxaline (NBQX) (10 μm; n = 4 spines) abolished the residual Ca2+ transient in Tgα-CaMKII-tTA/LC1/GluR-B+/neo mice, indicating that spinous Ca2+ influx indeed occurred via Ca2+-permeable AMPA receptors (Fig. 4C, NBQX).
Notably, the decay time constant of spinous Ca2+ transients in Tgα-CaMK-tTA/LC1/GluR-B+/neo mice (τ = 100 ± 30 msec; n = 8 spines) was significantly smaller than in GluR-B+/neo (τ = 200 ± 50 msec; n = 9 spines; p < 0.0005) and wild-type (τ = 180 ± 50 msec; n = 9 spines; p < 0.005) mice, which generated equal time-integrated Ca2+ influx in these genotypes (Fig. 3D, right diagram).
Thus, increased AMPA receptor-mediated Ca2+ entry can be detected in postsynaptic locations in GluR-B(Q)-expressing mice. However, a compensatory faster removal of free Ca2+ diminishes the potentially higher Ca2+ signal in these mice, strengthening the hypothesis that Ca2+ signaling by the modified AMPA channels is not a primary cause for seizures in GluR-B(Q)-expressing mice (Feldmeyer et al., 1999).
Increased synaptic excitability but reduced excitatory transmission in hippocampus by sustained GluR-B(Q) expression
To unmask excitability changes, which may be related to the epileptic phenotype ultimately resulting from sustained GluR-B(Q) expression, we recorded simultaneously in the apical dendritic and soma layers in the CA1 region of hippocampal slices (P60-P90 mice). We monitored the presynaptic fiber volley, the fEPSP, and the population spike at different stimulation intensities. Stratum radiatum responses from an “input-output” series are shown by the top traces in Figure 5A, and soma layer recordings in response to a paired-pulse paradigm are shown by the bottom traces.
Figure 5.

Increased synaptic excitability in hippocampal slices from GluR-B(Q)-expressing mice. A, Top traces, Means of five consecutive synaptic responses elicited by different stimulation strengths recorded in stratum radiatum in slices from wild-type (wt, left) and GluR-B(Q)-expressing (right) mice. The prevolleys preceding the fEPSPs are indicated by a filled circle. Bottom traces, Recordings from stratum pyramidale elicited by paired-pulse stimulation (50 msec interstimulus interval). Arrowheads indicate population spike threshold, and arrows indicate the appearance of multiple population spikes. B, Stimulation strength (in nano-coulombs) necessary to elicit a prevolley of a given amplitude (0.5, 1.0, and 1.5 mV) and fEPSP amplitudes as a function of prevolley amplitude. C, Paired-pulse facilitation ratio (PPF) in the two genotypes at an interstimulus interval of 50 msec. D, fEPSP amplitude necessary to elicit a just detectable population spike (1) and a population spike of 2 mV amplitude (2). Data are shown as mean ± SEM. *p < 0.01. Number of experiments ranged from 23 to 48.
The fiber excitability appeared unchanged by GluR-B(Q) expression, judging from the similar stimulation strengths necessary to elicit fiber volleys with different amplitudes in the two genotypes (Fig. 5B, left diagram). Furthermore, comparison between the two genotypes revealed no statistical difference in the stimulation strength or the fiber volley amplitude necessary to elicit a just-above-threshold population spike (p = 0.73 and p = 0.35, respectively). However, the evoked fEPSPs were significantly lowered by 24, 29, and 30% for presynaptic fiber volleys at 0.5, 1.0, and 1.5 mV in GluR-B(Q)-expressing mice (Fig. 5B, right diagram), which indicates that excitatory synaptic transmission is smaller in GluR-B(Q)-expressing mice than wild-type mice. This reduced synaptic transmission seems to be caused by a weaker response of the postsynaptic cell because paired-pulse facilitation (Fig. 5C) and the excitability of the presynaptic fibers (Fig. 5B, left) were not altered. However, despite the reduced excitatory synaptic transmission in GluR-B(Q)-expressing mice, the threshold for generating a population spike was lowered by 25%. Similarly, the fEPSP size necessary to elicit a population spike of 2 mV was 22% smaller than in wild type (EPSP spike potentiation) (Fig. 5D). Increased excitability was further indicated by the multiple population spikes during the paired-pulse facilitation experiments (Fig. 5A, bottom traces). Notably, multiple spikes were only observed in response to the second stimulus and occurred in 10 of 35 experiments in slices of GluR-B(Q)-expressing mice but only in 1 of 47 of wild type.
Thus, despite the smaller excitatory response, the CA1 cells exhibited a reduced population spike threshold, which might underlie the spontaneous manifestations of epilepsy.
Seizure phenotypes depend on regional expression of altered AMPA receptors
Depth-electrode and single-channel epidural long-term EEG recording combined with video monitoring (5-14 d) were used for detailed analysis of seizure activity in adult mice. Tgα-CaMKII-tTA/LC1/GluR-B+/neo mice given Dox until birth and surviving until P30 had mild seizure attacks on average every 4 d. A similar frequency of seizures was recorded in GluR-B(Q)-expressing TgCre4/GluR-B+/neo mice. Most attacks occurred at night, during the active diurnal phase. Seizure relapses monitored in two Dox-naive Tgα-CaMKII-tTA/LC1/GluR-B+/neo mice after phenobarbital withdrawal occurred in clusters at night, were very severe, and eventually lethal.
In general, the EEG analysis showed interictal EEG patterns consisting of spikes, spike series, polyspikes, and 8-10 Hz rhythmic sharp activity (Fig. 6A), which were rarely accompanied by behavioral abnormalities. Spike series usually appeared 1 or 2 d before seizure attacks, and individual spikes consistently increased in number, days before seizures, in both mice given Dox prenatally (Fig. 6B, LC1/neo DoxP0) and Dox-naive mice after phenobarbital treatment (Fig. 6B, LC1/neo + Pb). However, the number of spikes did not correlate with seizure severity. Mice previously on phenobarbital showed fewer spikes per day but had more severe and eventually lethal seizure attacks than prenatally Dox-treated mice. Interictal spikes were also observed in GluR-B+/neo mice, and, in one of these mice, the number of spikes surged for 1 d without seizure incidence (Fig. 6B, neo). In GluR-B+/neo mice, the GluR-Bneo allele is incompletely silenced (Feldmeyer et al., 1999), and the low levels of GluR-B(Q) from this allele seem to be sufficient for the appearance of interictal EEG patterns.
Figure 6.
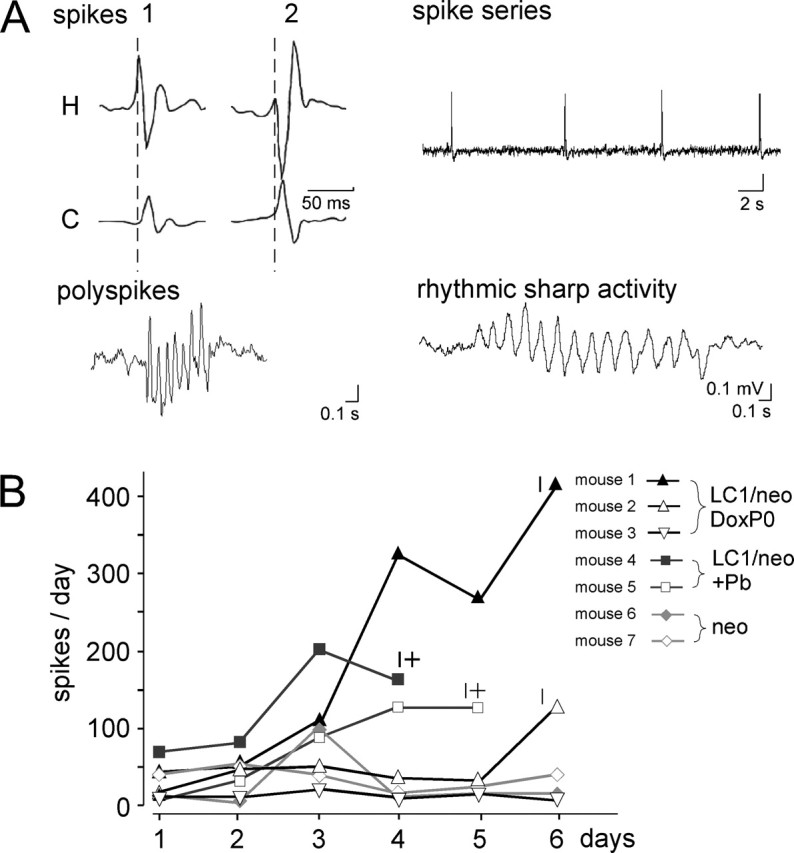
Abnormal EEG patterns by Q/R site-unedited GluR-B. A, Spikes (example taken from depth-electrode recordings), spike series, polyspikes, and sharp waves characterize interictal patterns in epidural electroencephalogram of Tgα-CaMKII-tTA/LC1/GluR-B+/neo mice treated with phenobarbital or prenatally with Dox. Interictal spikes in two prenatally Dox-treated mice (1, 2) started in hippocampus (H) and propagated to cortex (C) with constant propagation time of 12 msec (see delayed cortical peaks in relation to hippocampal peaks, calibrated by broken lines). B, Number of spikes per day, counted in epidural EEG recordings of three experimental groups (neo, GluR-B+/neo; LC1/neo +Pb, Tgα-CaMKII-tTA/LC1/GluR-B+/neo mice withdrawn from phenobarbital; and LC1/neo DoxP0, prenatally Dox-treated Tgα-CaMKII-tTA/LC1/GluR-B+/neo mice) indicated by similar symbols, with the individual mice shown in different shades of gray. Note that the number of spikes increased 1-3 days before seizure attacks but that this increase does not correlate with seizure severity.
Without exception, all seizures in GluR-B(Q)-expressing mice started with a 1-min-long series of jerks that coincided with spikes in the electroencephalogram and hence justified the diagnosis of myoclonic jerks (Fig. 7A,B, 1). Jerks were more severe in Dox-naive mice that had undergone previous phenobarbital treatment. In these mice, bursts of jerks corresponded to groups of polyspikes (1-3 sec duration) compared with single myoclonic jerks (spikes ≤500 msec duration) in mice treated prenatally with Dox. Beside the different severity, jerks or groups of jerks occurred on average every 8 sec at a remarkably stable frequency of 0.1-0.2 Hz (Fig. 7A,B, 1). Myoclonic jerks in both Dox-naive and prenatally Dox-treated mice switched into seizure attacks with signs for generalized seizures [EEG, 4-7 Hz (Fig. 7A,B, 2)] and tonic [9-15 Hz (Fig. 7A,B, 3)] and focal seizures with oral automatisms [4-9 Hz (Fig. 7A,B, 4)] and hypermotor fits [7-10 Hz (Fig. 7B, 5)]. Oral automatisms included chewing and repetitive tongue flicking, and hypermotor activity consisted of extensive running fits.
Figure 7.
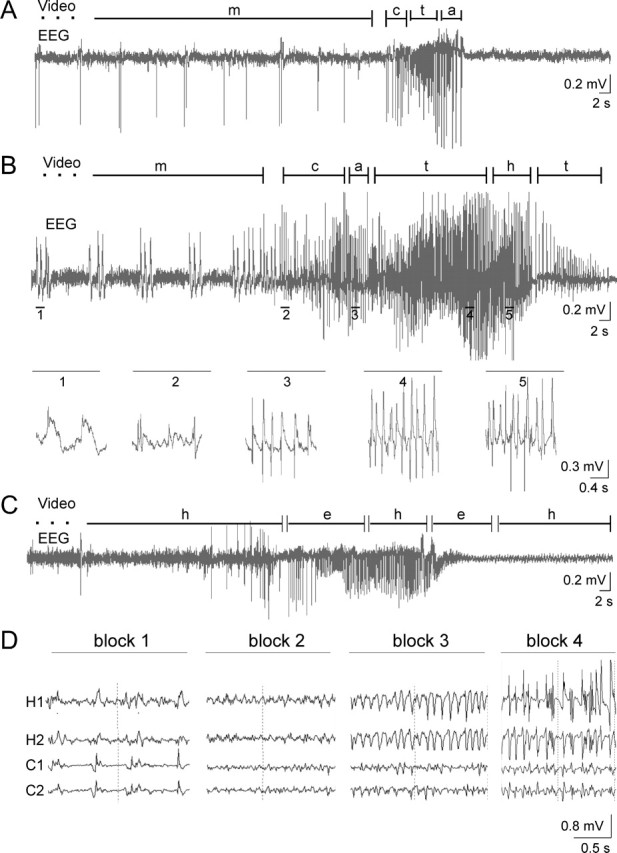
Seizure phenotypes. Long-term EEG recordings by epidural electrodes and synchronous video observation (A-C) monitored seizures in adult Tgα-CaMKII-tTA/LC1/GluR-B+/neo mice, either prenatally Dox-treated (P69; A) or withdrawn from phenobarbital (P54; B), and in adult TgCN10-tTA/LC1/GluR-B+/neo mice (P123; C). Different phenotypes are given (m, myoclonic jerks; c, clonic; t, tonic seizures; a, complex-motor; h, hypermotor behavior; and e, exploratory activity), and durations are indicated by segments. Five fold-expanded EEG traces in B represent spike discharges during the corresponding numbered seizure phenotypes. Clinical seizure phenotypes were most severe in mice withdrawn from phenobarbital (B) were mild and of shorter duration in prenatally Dox-treated mice (A) and absent in TgCN10-tTA/LC1/GluR-B+/neo mice (C). D, EEG recordings with four depth electrodes (H1, H2, hippocampal electrodes; C1, C2, cortical electrodes) in prenatally Dox-treated Tgα-CaMKII-tTA/LC1/GluR-B+/neo mice. A seizure attack in a P92 mouse is traced by EEG recording, shown in four blocks. The seizure started in hippocampus with a 2 Hz spike pattern (block 1), followed by 40 Hz low-amplitude, fast-activity superimposed on basic α-activity (block 2), a 9-10 Hz pattern (block 3), and high-amplitude spike activity (block 4), again pronounced in hippocampus.
Notably, when GluR-B(Q) expression was restricted to hippocampus (TgCN10-itTA/LC1/GluR-B+/neo mice; Fig. 1B) (see also Fig. 2A, bottom panel), the mice still displayed rhythmic spike discharge (5-8 Hz activity) but showed no overt seizure activity in simultaneous video monitoring (Fig. 7C). This may indicate failure of synchronous hippocampal activity to spread to other brain regions to elicit seizure symptoms.
The hippocampus plays a dominant role in seizure episodes
Additional evidence for the hippocampus as location of seizure onset was obtained by use of depth electrodes. Two electrodes were placed into the right hippocampus and two electrodes were placed in deeper layers of parietal cortex of mice expressing GluR-B(Q) after birth (n = 3; P80-P100) (mice 1, 3, two electrodes in left cortex; mouse 2, one electrode in each side of the parietal cortex) (for electrode positions, see supplemental Fig. 2, available at www.jneurosci.org as supplemental material).
For each mouse undergoing seizures, 100 randomly chosen interictal spikes were analyzed for occurrence in hippocampal and/or cortical electrode recordings and relative temporal onset. In all three mice, most interictal spikes (86, 77, and 94%) occurred only in the hippocampal recording or showed hippocampal onset and propagated subsequently to the neocortical electrode recording (Fig. 7D). Moreover, the recorded amplitudes were always larger in hippocampus than cortex. Propagation time from hippocampal to cortical electrode recording was impressively constant at 13 msec in mice 1 and 3 and 11 msec in mouse 2. The remaining 14% of spikes in mouse 1 occurred in only one of the unilateral cortical recordings. For mouse 2, 18% of spikes showed simultaneous onset in cortical and hippocampal recordings, and 5% occurred in only one of the cortical recordings. The remaining 6% of spikes in mouse 3 were again of simultaneous onset in cortical and hippocampal recordings.
As observed in the simultaneous epidural one-channel EEG and video recordings, multiple depth-electrode EEG recordings revealed spike series indicative of myoclonic jerks. In one myoclonic series preceding a seizure episode, spikes occurred simultaneously in hippocampal and cortical recordings, whereas in another series, most hippocampal spikes preceded cortically recorded spikes (data not shown). The myoclonic spike series preceding both seizure attacks shifted to a 2 Hz spike pattern with leading hippocampal activity, followed by a low-amplitude, fast-activity pattern of ∼40 Hz superimposed on basic α-activity predominantly in hippocampus. The generalized seizure evolved into a 9-10 Hz pattern and subsequently in high-amplitude spike and polyspike activities that were again pronounced in the hippocampus (Fig. 7D).
Collectively, these data strongly suggest that seizure activity in our model is triggered in hippocampal circuits, in agreement with the high GluR-B(Q) expression in CA1 pyramidal cells relative to other brain regions (supplemental Fig. 1, available at www.jneurosci.org as supplemental material).
Discussion
To permit the analysis of seizures brought about by functionally altered AMPA receptors in adult mice, we implemented a genetic switch directing the expression of the dominant-negative GluR-B(Q) subunit selectively to postnatal principal forebrain neurons.
The genetic control was achieved by inducing GluR-B(Q) expression via tTA-dependent Cre activity. Prenatal Dox administration suppressed Cre activity in the developing nervous system, and withdrawal from Dox after birth led to a restricted Cre expression pattern in the adult mouse relative to that observed in Dox-naive mice. Cre induction as monitored with the Rosa26R reporter gene was slow and even after months remained restricted to hippocampus, striatum, and amygdala. The reason for the region-specific failure of Cre induction after prenatal Dox administration is enigmatic. One likely explanation is that epigenetic silencing causes “switch-off” of the tTA-dependent promoter Ptetbi, which appears to be slowly reactivated in neuronal populations expressing high tTA levels, such as hippocampal CA1 pyramidal cells.
The AMPA receptor switch when operated already at prenatal stages produced a phenotype characterized by early onset seizures and premature death, similar to that resulting from global GluR-B(Q) expression (Brusa et al., 1995; Higuchi et al., 2000). However, when the AMPA receptor switch was attenuated by inducing it in only few forebrain areas during juvenile development, early death could be avoided, and most mice lived longer than 2 months. As shown by video monitoring and EEG recordings, the myoclonic seizures eventually developing in these mice were less severe and lacked hypermotor running fits. They appeared to originate in the hippocampus, as shown by multiple depth-electrode recordings, but had limbic components with clear amygdala contribution, exemplified by oral automatisms. Collectively, these observations indicate that the expression of the unedited GluR-B(Q) subunit itself suffices in triggering the development of the phenotype and that the seizure attacks in juvenile GluR-B+/ΔECS mice are unlikely to reflect abnormal CNS development.
The hippocampal origin of the seizure attacks mediated by GluR-B(Q) finds support in mice that express GluR-B(Q) exclusively in the postnatal hippocampus. This selective expression was obtained by physically linking the α-CaMKII promoter driving tTA to the neuronal silencer of the NR2C gene of NMDA receptors, which suppresses NR2C gene expression in brain areas other than cerebellum, hippocampus, and some cortical neurons (Suchanek et al., 1997). GluR-B(Q) expression, now even in the absence of Dox confined to hippocampal CA1 pyramidal and DG granule cells, never resulted in clinical seizure symptoms at adult stages. The rhythmic paroxysmal discharge in the electroencephalogram was, however, maintained, indicating that hippocampal expression of functionally switched AMPA receptors is sufficient to generate epileptic activity. Lack of GluR-B(Q) expression in additional brain areas may prevent spread of epileptic excitation to other limbic lobe and motor cortex structures in this clinically mute mouse.
Compatible with the EEG recordings, the hippocampal slices from GluR-B(Q)-expressing forebrains showed increased synaptic excitability, as judged by a lower threshold for population spikes in the CA1 pyramidal cell layer. The reduced spike threshold and the EPSP spike potentiation, previously described as features of LTP (Andersen et al., 1980; Abraham et al., 1987; Chavez-Noriega et al., 1990; Staff and Spruston, 2003; Frick et al., 2004), may thus also feature in epileptiform circuits. We could not yet demonstrate a reduced spike threshold in single GluR-B(Q)-expressing CA1 pyramidal cells by current injection or evoked EPSPs. We surmise that downstream consequences of GluR-B(Q) expression underlying the enhanced excitability include altered intrinsic neuronal excitability from, for example, acquired dendritic channelopathy (Bernard et al., 2004) and/or alterations in the relative contribution of evoked excitation-inhibition (for review, see Zhang and Linden, 2003). Indeed, a significant reduction in the number of somatostatin-positive interneurons in hippocampal stratum oriens was revealed in a study on the cellular consequences of long-term GluR-B(Q) expression (D. Shimshek, Y. Geng, H. Krestel, P. H. Seeburg, and R. Sprengel, unpublished observations), which could, in part, explain the increased excitability. Because the genetic switch to GluR-B(Q) implemented here by use of the α-CaMKII promoter does not operate in GABAergic interneurons in which this promoter is silent, the reduction in interneuron number may well result from recurrent seizure activity, as reported in other seizure models (Cossart et al., 2001). Thus, the persistent increase in circuit excitability by long-term GluR-B(Q) expression may primarily derive from increased intrinsic neuronal excitability by as yet unknown molecular changes in principal neurons and may be accelerated by seizure-induced death of interneurons.
Although the molecular events causing the CNS dysfunctions by GluR-B(Q) remain to be resolved, our study documents that expression of the Q/R site-unedited form of the GluR-B subunit is sufficient to produce over time pathological synchronized firing in neuronal populations. Efficient RNA editing in the pore-forming membrane loop M2 of GluR-B can thus be viewed as a critical safeguard against epileptiform network activity. This mayimplicate impaired Q/R site editing of GluR-B as a causative factor in clinical epilepsy forms, but, as of today, experimental evidence in support of this hypothesis is lacking. Several important properties of AMPA receptors are determined by Q/R site-edited GluR-B. First, the arginine residue in the pore loop of GluR-B is a signal for retention in endoplasmic reticulum, ensuring the generation of functional heteromeric receptors (Greger et al., 2003). The GluR-B(Q) form would override this assembly checkpoint, and GluR-B(Q) containing channels might exhibit altered synaptic trafficking properties (Shi et al., 2001). Second, the arginine in the pore lop confers to AMPA channels insensitivity to polyamines and reduced Ca2+ permeability and unitary conductance (Burnashev et al., 1992; Swanson et al., 1997). The increased Ca2+-permeability of AMPA channels in CA1 cells expressing GluR-B(Q), previously determined in nucleated patches (Brusa et al., 1995; Feldmeyer et al., 1999), was demonstrated by AMPA channel-mediated spinous Ca2+ transients in the present study. The altered AMPA channel properties in our conditional mouse model were recorded at P30, weeks before seizures became manifest. Thus, circuit hyperexcitability appears not to be an immediate consequence of GluR-B(Q) expression but to require additional molecular and cellular events. Third, the synaptically localized GluR-B(Q)-containing channels can induce NMDA receptor independent (“non-Hebbian”) plasticity (Feldmeyer et al., 1999), which may trigger the postulated additional events. These may determine the faster decrease of free Ca2+ levels in spines of mutant mice, mediated for example by changes in Ca2+ buffering and/or Ca2+ ATPase (pump) activity. Fourth, GluR-B(Q) expression increases the macroscopic AMPA receptor-mediated conductance in nucleated patches of CA1 pyramidal cells (Feldmeyer et al., 1999). However, analysis of synaptic responses reveals that the increased somatic AMPA conductance is not paralleled by a corresponding synaptic increase. Rather, we observed reduced excitatory synaptic transmission, judging from the significantly smaller fEPSPs elicited by prevolleys in GluR-B(Q)-expressing mice. This could be confirmed by recording from single CA1 pyramidal cells after stimulation of Schaffer collaterals in acute slices derived from P40 mice. Wild-type cells showed higher EPSC amplitudes than mutant cells [mean ± SEM of stimulation intensity (in pA/V): wild type, 14.2 ± 1.9 (n = 6); mutant, 8.6 ± 1.1 (n = 10); p = 0.03] (B. Schupp, D. Shimshek, P. H. Seeburg, R. Sprengel, and G. Kohr, unpublished observations), with AMPA currents exhibiting pronounced rectification in the mutant, as expected from the GluR-B(Q) expression. We were unable to detect a presynaptic contribution to the reduced excitatory transmission, as judged from unchanged excitability of presynaptic fibers and unaltered paired-pulse facilitation. Postsynaptic AMPA receptor channel function may be downregulated by sustained GluR-B(Q) expression, perhaps by non-Hebbian synaptic plasticity (Feldmeyer et al., 1999) or by reduced GluR-B gene transcription after seizures (Huang et al., 2002).
It should be noted that the AMPA channel-mediated Ca2+ influx in principal neurons cannot be the sole determinant for triggering the epileptic phenotype of mice expressing GluR-B(Q), because mice with genetically induced expression of AMPA receptors in their maximally Ca2+-permeable form, by either global ablation of GluR-B (Jia et al., 1996) or GluR-B depletion in forebrain principal neurons (Krestel, Shimshek, Sprengel, and Seeburg, unpublished observations), do not manifest this phenotype. Hence, the presence of GluR-B in AMPA receptors appears to be necessary to bring about circuit hyperexcitability. This is best explained by the function of GluR-B in AMPA receptor trafficking. According to studies in virus-infected brain slices, GluR-B is important for stabilizing AMPA receptors for normal synaptic transmission (Shi et al., 2001). In the absence of GluR-B, synaptic AMPA receptors are reduced in number, as can be deduced from the decreased synaptic transmission in GluR-B-deficient mice (Jia et al., 1996; Meng et al., 2003). Thus, Ca2+ signaling by AMPA channels without GluR-B differs from that of AMPA channels with GluR-B(Q), implicating GluR-B itself (in its unedited form) as a critical determinant in generating epileptiform activity. The investigation of downstream mechanisms after GluR-B(Q) expression, aided by use of the genetically controlled mouse model described here, should ultimately help in elucidating the molecular and cellular changes responsible for the epileptic phenotype.
Footnotes
We thank Drs. M. Mayford and P. Soriano for mouse lines; Drs. B. Schupp and G. Kohr for EPSC measurements; A. Herold, J. Kern, and M. Lang for technical assistance; and Dr. P. Andersen for discussions.
Correspondence should be addressed to Peter H. Seeburg, Max-Planck-Institute for Medical Research, Jahnstrasse 29, 69120 Heidelberg, Germany. E-mail: seeburg@mpimf-heidelberg.mpg.de.
J. Kim's present address: National Institute of Child Health and Human Development, Laboratory of Cellular and Synaptic Neurophysiology, 49 Convent Drive, Building 49, Room 5C68, Bethesda, MD 20892. E-mail: kimji@mail.nih.gov.
Y. Geng's present address: Sir Run Run Shaw Hospital, 3 Qing-chun Road East, 310016 Hangzhou, Zhejiang, China. E-mail: gengyun@hzcnc.com.
Copyright © 2004 Society for Neuroscience 0270-6474/04/2410568-11$15.00/0
References
- Abraham WC, Gustafsson B, Wigström H (1987) Long-term potentiation involves enhanced synaptic excitation relative to synaptic inhibition in guinea-pig hippocampus. J Physiol (Lond) 394: 367-380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andersen P, Sundberg SH, Sveen O, Swann JW, Wigström H (1980) Possible mechanisms for long-lasting potentiation of synaptic transmission in hippocampal slices from guinea-pigs. J Physiol (Lond) 302: 463-482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baron U, Gossen M, Bujard H (1997) Tetracycline-controlled transcription in eukaryotes: novel transactivators with graded transactivation potential. Nucleic Acids Res 25: 2723-2729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernard C, Anderson A, Becker A, Poolos NP, Beck H, Johnston D (2004) Acquired dendritic channelopathy in temporal lobe epilepsy. Science 305: 532-535. [DOI] [PubMed] [Google Scholar]
- Brusa R, Zimmermann F, Koh DS, Feldmeyer D, Gass P, Seeburg PH, Sprengel R (1995) Early-onset epilepsy and postnatal lethality associated with an editing-deficient GluR-B allele in mice. Science 270: 1677-1680. [DOI] [PubMed] [Google Scholar]
- Burnashev N, Monyer H, Seeburg PH, Sakmann B (1992) Divalent ion permeability of AMPA receptor channels is dominated by the edited form of a single subunit. Neuron 8: 189-198. [DOI] [PubMed] [Google Scholar]
- Chavez-Noriega LE, Halliwell JV, Bliss TVP (1990) A decrease in firing threshold observed after induction of the EPSP-spike (E-S) component of long-term potentiation in rat hippocampal slices. Exp Brain Res 79: 633-641. [DOI] [PubMed] [Google Scholar]
- Chen EY, Liao YC, Smith DH, Barrera-Saldana HA, Gelinas RE, Seeburg PH (1989) The human growth hormone locus: nucleotide sequence, biology, and evolution. Genomics 4: 479-497. [DOI] [PubMed] [Google Scholar]
- Cossart R, Dinocourt C, Hirsch JC, Merchan-Perez A, De Felipe J, Ben-Ari Y, Esclapez M, Bernard C (2001) Dendritic but not somatic GABAergic inhibition is decreased in experimental epilepsy. Nat Neurosci 4: 52-62. [DOI] [PubMed] [Google Scholar]
- Denk W, Strickler JH, Webb WW (1990) Two-photon laser scanning fluorescence microscopy. Science 248: 73-76. [DOI] [PubMed] [Google Scholar]
- Feldmeyer D, Kask K, Brusa R, Kornau HC, Kolhekar R, Rozov A, Burnashev N, Jensen V, Hvalby O, Sprengel R, Seeburg PH (1999) Neurological dysfunctions in mice expressing different levels of the Q/R site-unedited AMPAR subunit GluR-B. Nat Neurosci 2: 57-64. [DOI] [PubMed] [Google Scholar]
- Franklin K, Paxinos G (1997) The mouse brain in stereotaxic coordinates. New York: Academic.
- Frick A, Magee J, Johnston D (2004) LTP is accompanied by an enhanced local excitability of pyramidal neuron dendrites. Nat Neurosci 7: 126-135. [DOI] [PubMed] [Google Scholar]
- Geiger JR, Melcher T, Koh DS, Sakmann B, Seeburg PH, Jonas P, Monyer H (1995) Relative abundance of subunit mRNAs determines gatting and Ca2+ permeability of AMPA receptors in principal neurons and interneurons in rat CNS. Neuron 15: 193-204. [DOI] [PubMed] [Google Scholar]
- Gossen M, Bujard H (1992) Tight control of gene expression in mammalian cells by tetracycline-responsive promoters. Proc Natl Acad Sci USA 89: 5547-5551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greger IH, Khatri L, Kong X, Ziff EB (2003) AMPA receptor tetramerization is mediated by Q/R editing. Neuron 13: 763-774. [DOI] [PubMed] [Google Scholar]
- Grigorenko EV, Bell WL, Glazier S, Pons T, Deadwyler S (1998) Editing status at the Q/R site of the GluR2 and GluR6 glutamate receptor subunits in the surgically excised hippocampus of patients with refractory epilepsy. NeuroReport 9: 2219-2224. [DOI] [PubMed] [Google Scholar]
- Hasan MT, Schonig K, Berger S, Graewe W, Bujard H (2001) Long-term, noninvasive imaging of regulated gene expression in living mice. Genesis 29: 116-122. [DOI] [PubMed] [Google Scholar]
- Higuchi M, Maas S, Single FN, Hartner J, Rozov A, Burnashev N, Feldmeyer D, Sprengel R, Seeburg PH (2000) Point mutation in an AMPA receptor gene rescues lethality in mice deficient in the RNA-editing enzyme ADAR2. Nature 406: 78-81. [DOI] [PubMed] [Google Scholar]
- Huang Y, Doherty JJ, Dingledine R (2002) Altered histone acetylation at glutamate receptor 2 and brain-derived neurotrophic factor genes is an early event triggered by status epilepticus. J Neurosci 22: 8422-8428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jia Z, Agopyan N, Miu P, Xiong Z, Henderson J, Gerlai R, Taverna FA, Velumian A, MacDonald J, Carlen P, Abramow-Newerly W, Roder J (1996) Enhanced LTP in mice deficient in the AMPA receptor GluR2. Neuron 17: 945-956. [DOI] [PubMed] [Google Scholar]
- Kim J (2001) Improvement and establishment of the +TA-dependent inducible system in the mouse brain. PhD thesis, University of Heidelberg.
- Kortenbruck G, Berger E, Speckmann EJ, Musshoff U (2001) RNA editing at the Q/R site for the glutamate receptor subunits GluR2, GluR5, and GluR6 in hippocampus and temporal cortex from epileptic patients. Neurobiol Dis 8: 459-468. [DOI] [PubMed] [Google Scholar]
- Kozak M (1984) Compilation and analysis of sequences upstream from the translational start site in eukaryotic mRNAs. Nucleic Acids Res 12: 857-872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krestel HE, Mayford M, Seeburg PH, Sprengel R (2001) A GFP-equipped bidirectional expression module well suited for monitoring tetracycline-regulated gene expression in mouse. Nucleic Acids Res 29: E39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mantamadiotis T, Lemberger T, Bleckmann SC, Kern H, Kretz O, Martin Villalba A, Tronche F, Kellendonk C, Gau D, Kapfhammer J, Otto C, Schmid W, Schutz G (2002) Disruption of CREB function in brain leads to neurodegeneration. Nat Genet 31: 47-54. [DOI] [PubMed] [Google Scholar]
- Mayford M, Bach ME, Huang YY, Wang L, Hawkins RD, Kandel ER (1996) Control of memory formation through regulated expression of α-CaMKII transgene. Science 274: 1678-1683. [DOI] [PubMed] [Google Scholar]
- Meng Y, Zhang Y, Jia Z (2003) Synaptic transmission and plasticity in the absence of AMPA glutamate receptor GluR2 and GluR3. Neuron 39: 163-176. [DOI] [PubMed] [Google Scholar]
- Rathenberg J, Nevian T, Witzemann V (2003) High-efficiency transfection of individual neurons using modified electrophysiology techniques. J Neurosci Methods 126: 91-98. [DOI] [PubMed] [Google Scholar]
- Sailer A, Swanson GT, Perez-Otano I, O'Leary L, Malkmus SA, Dyck RH, Dickinson-Anson H, Schiffer HH, Maron C, Yaksh TL, Gage FH, O'Gorman S, Heinemann SF (1999) Generation and analysis of GluR5 (Q636R) kainate receptor mutant mice. J Neurosci 19: 8757-8764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schonig K, Schwenk F, Rajewsky K, Bujard H (2002) Stringent doxycycline dependent control of CRE recombinase in vivo. Nucleic Acids Res 30: e134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi S, Hayashi Y, Esteban JA, Malinow R (2001) Subunit-specific rules governing AMPA receptor trafficking to synapses in hippocampal pyramidal neurons. Cell 105: 331-343. [DOI] [PubMed] [Google Scholar]
- Sommer B, Kohler M, Sprengel R, Seeburg PH (1991) RNA editing in brain controls a determinant of ion flow in glutamate-gated channels. Cell 67: 11-19. [DOI] [PubMed] [Google Scholar]
- Soriano P (1999) Generalized lacZ expression with the ROSA26 Cre reporter strain. Nat Genet 21: 70-71. [DOI] [PubMed] [Google Scholar]
- Staff NP, Spruston N (2003) Intracellular correlate of EPSP-spike potentiation in CA1 pyramdial neurons is controlled by GABAergic modulation. Hippocampus 13: 801-805. [DOI] [PubMed] [Google Scholar]
- Stemmer WP, Crameri A, Ha KD, Brennan TM, Heyneker HL (1995) Single-step assembly of a gene and entire plasmid from large numbers of oligodeoxyribonucleotides. Gene 164: 49-53. [DOI] [PubMed] [Google Scholar]
- Stuart GJ, Dodt HU, Sakmann B (1993) Patch-clamp recordings from the soma and dendrites of neurons in brain slices using infrared video microscopy. Pflügers Arch 423: 511-518. [DOI] [PubMed] [Google Scholar]
- Suchanek B, Seeburg PH, Sprengel R (1997) Tissue specific control regions of the N-methyl-d-aspartate receptor subunit NR2C promoter. Biol Chem 378: 929-934. [PubMed] [Google Scholar]
- Swanson GT, Kamboj SK, Cull-Candy SG ((1997) Single-channel properties of recombinant AMPA receptors depend on RNA editing, splice variation, and subunit composition. J Neurosci 17: 58-69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vissel B, Royle GA, Christie BR, Schiffer HH, Ghetti A, Tritto T, Perez-Otano I, Radcliffe RA, Seamans J, Sejnowski T, Wehner JM, Collins AC, O'Gorman S, Heinemann SF (2001) The role of RNA editing of kainate receptors in synaptic plasticity and seizures. Neuron 29: 217-227. [DOI] [PubMed] [Google Scholar]
- Zhang W, Linden DJ (2003) The other side of the engram: experience-driven changes in neuronal intrinsic excitability. Nat Neurosci Rev 4: 885-900. [DOI] [PubMed] [Google Scholar]