

Abstract
Striatal cholinergic interneurons are critical elements of the striatal circuitry controlling motor planning, movement, and associative learning. Intrastriatal release of dopamine and inhibition of interneuron activity is thought to be a critical link between behaviorally relevant events, such as reward, and alterations in striatal function. However, the mechanisms mediating this modulation are unclear. Using a combination of electrophysiological, molecular, and computational approaches, the studies reported here show that D2 dopamine receptor modulation of Na+ currents underlying autonomous spiking contributes to a slowing of discharge rate, such as that seen in vivo. Four lines of evidence support this conclusion. First, D2 receptor stimulation in tissue slices reduced the autonomous spiking in the presence of synaptic blockers. Second, in acutely isolated neurons, D2 receptor activation led to a reduction in Na+ currents underlying pacemaking. The modulation was mediated by a protein kinase C-dependent enhancement of channel entry into a slow-inactivated state at depolarized potentials. Third, the sodium channel blocker TTX mimicked the effects of D2 receptor agonists on pacemaking. Fourth, simulation of cholinergic interneuron pacemaking revealed that a modest increase in the entry of Na+ channels into the slow-inactivated state was sufficient to account for the slowing of pacemaker discharge. These studies establish a cellular mechanism linking dopamine and the reduction in striatal cholinergic interneuron activity seen in the initial stages of associative learning.
Keywords: sodium channel, dopamine, striatum, scRT-PCR, neuromodulation, voltage clamp, slice, Parkinson's disease
Introduction
Cholinergic interneurons are critical elements of the striatal circuitry controlling motor planning, movement, and associative learning (Graybiel et al., 1994). Their central role was first inferred from clinical observations that striatal cholinergic tone was elevated in Parkinson's disease and that the restoration of a balance with the dopaminergic afferent system was important in alleviating the motor symptoms of the disease (Hornykiewicz, 1976). Although they appear to receive a sparse dopaminergic innervation (Smith et al., 1994), cholinergic interneurons express both D2 and D5 dopamine receptors, making them responsive to volume-transmitted dopamine (Levey et al., 1993; Bergson et al., 1995; Yan et al., 1997). The best characterized effect of dopamine on cholinergic interneurons is mediated by the activation of D2 receptors, which inhibit N-type Ca2+ channel opening and the Ca2+-dependent release of acetylcholine (Lehmann and Langer, 1983; Bertorelli et al., 1992; Stoof et al., 1992; DeBoer et al., 1993; Di Chiara et al., 1994; Yan et al., 1997).
There are reasons to believe that dopamine acts to shape cholinergic interneuron function in other ways as well. Perhaps the most intriguing evidence for additional mechanisms comes from studies of behaving monkeys. Unconditioned reinforcers that are known to activate dopaminergic neurons (Schultz, 2002) inhibit the activity of tonically active neurons (TANs) that have been identified as cholinergic interneurons (Aosaki et al., 1994; Graybiel et al., 1994; Bennett and Wilson, 1999). In the early stages of associative learning paradigms, the activity of dopaminergic neurons and the reduction of TAN activity become bound to conditioned stimuli. Lesioning dopaminergic neurons disrupt conditioning and the linkage between TAN activity and behaviorally relevant stimuli (Aosaki et al., 1994). These findings make a strong case that dopamine not only reduces the release of acetylcholine but also reduces cholinergic interneuron spiking; but how?
Electrophysiological studies of cholinergic interneurons in slices have shown that they are autonomous pacemakers: they are capable of maintained spike discharge in the absence of synaptic input (Bennett and Wilson, 1999). This finding argues that the pause in cholinergic interneuronal discharge must come from either synaptic inhibition or reduction of the intrinsic mechanisms underlying the autonomous activity. D5 dopamine receptor activation does enhance cholinergic interneuronal GABAA receptor currents (Yan and Surmeier, 1997; Suzuki et al., 2001) and spike afterhyperpolarization (Bennett and Wilson, 1998), but studies in vivo suggest that the cholinergic interneuronal pause is dependent primarily on D2 receptors (Watanabe and Kimura, 1998). If this is the case, there are no known synaptic mechanisms involving ionotropic receptors by which dopamine could induce a pause.
Could D2 receptors reduce the pacemaking mechanism? Pacemaking in cholinergic interneurons is dependent on voltage-dependent Na+ channels (Bennett et al., 2000). Phosphorylation of Na+ channel α-subunits by protein kinase C (PKC) is a particularly potent means of reducing Na+ channel currents (Cantrell and Catterall, 2001; Carr et al., 2003). As a consequence, G-protein-coupled receptors that activate PKC are potential regulators of autonomous activity. Because D2 receptors fall into this class (Hernandez-Lopez et al., 2000), it is possible that they are capable of reducing Na+ channel currents and slowing autonomous activity in cholinergic interneurons. The studies reported here provide support for this view.
Materials and Methods
Tissue preparation. C57BL/6 mice (3-5 weeks of age) (Harlan, Indianapolis, IN) were used in all of the experiments presented. A few initial experiments were performed on Sprague Dawley rats (Harlan Sprague Dawley); there were no obvious differences in the physiological properties of the rat neurons. Animals were anesthetized with isofluorane and decapitated in accord with Northwestern University Animal Care and Use Committee guidelines for the care and use of animals. Brains were quickly removed, blocked, and sectioned (300-350 μm; with a VT1000 slicer; Leica, Nussloch, Germany) in an ice-cold sucrose solution containing the following (in mm): 250 sucrose, 11 glucose, 15 HEPES, 4 MgSO4, 1 NaH2PO4, 2.5 KCl, 1 kynurenic acid, 0.1 N-nitro-l-arginine, and 0.005 glutathione, pH 7.4 (300-305 mOsm/l). Unless noted otherwise, all chemicals were obtained from Sigma (St. Louis, MO). Coronal slices were collected in a low-Ca2+ buffer containing 140 mm Naisethionate, 23 mm glucose, 15 mm HEPES, 2 mm KCl, 4 mm MgCl2, 0.2 mm CaCl2, 1 mm kynurenic acid, 0.1 mm N-nitro-l-arginine, and 0.005 mm glutathione, pH 7.4, 300-305 mOsm/l before being incubated for 1-5 hr in sodium bicarbonate-buffered Earle's balanced salt solution (EBSS) bubbled with 95% O2 and 5% CO2. EBSS also contained the following (in mm): 23 glucose, 1 kynurenic acid, 0.1 N-nitro-l-arginine, and 0.005 glutathione.
For acute dissociation, individual slices were transferred to the low-Ca2+ buffer, and the striatum was dissected. Striata were then incubated at 35°C for 25 min in oxygenated HBSS containing 11 mm HEPES, 4 mm MgCl2, 1 mm CaCl2, 1 mm pyruvic acid, 1 mm kynurenic acid, 0.1 mm N-nitro-l-arginine, 0.005 mm glutathione, and 1 mg/ml protease XIV, pH 7.4 (300-305 mOsm/l). After this enzyme incubation, the tissue was transferred to the low-Ca2+ HEPES-buffered saline, rinsed, and mechanically dissociated using fire-polished Pasteur pipettes. The resulting cell suspension was plated onto a 35 mm Petri dish mounted on an inverted microscope. During the course of the experiment, nonrecorded cells were constantly perfused with a background solution containing the following (in mm): 140 NaCl, 23 glucose, 15 HEPES, 2 KCl, 2 MgCl2, and 1 CaCl2, pH 7.4 (300-305 mOsm/l). Although protease treatment may partially degrade surface proteins, reducing the responsiveness of G-protein-coupled receptors, there is no viable alternative to extracting cholinergic interneurons from tissue slices. There is no evidence that protease alters the properties of Na+ channels.
Whole-cell voltage-clamp recording of acutely isolated neurons. Whole-cell recordings were performed using electrodes pulled from Corning (Corning, NY) 7052 glass (1.2 mm outer diameter), coated with R-6101 (Corning), and fire polished immediately before use. Electrodes typically had resistances of 1.5-2.5 MΩ in the bath. Recordings were obtained at room temperature (20-22°C) with an Axopatch 200A amplifier (Axon Instruments, Foster City, CA) interfaced to a Macintosh computer (Apple Computers, Cupertino, CA) running Pulse software (HEKA Elektronik, Lambrecht, Germany). After the gigaohm seal was formed and the cell membrane ruptured, series resistance was compensated (75-80%) and frequently monitored. The intracellular recording solution contained the following (in mm): 70 N-methyl-d-glucamine, 20 HEPES, 50 Cs2SO4, 2 MgCl2, 0.5 Na2SO4, 12 phosphocreatine, 2 Mg-ATP, 0.7 Na2-GTP, and 0.1 leupeptin, pH 7.25, with H2SO4 (265-270 mOsm/l). During recording, cells were bathed in extracellular solutions applied via a gravity-fed capillary perfusion array positioned several hundred micrometers away from the cell under study. Solutions were changed by adjusting the position of the array using a DC motor (Newport, Irvine, CA). Solution changes were complete within <1 sec. For recording rapidly inactivating Na+ currents, the external solution contained the following (in mm): 10 NaCl, 110 tetraethylammonium (TEA) chloride, 10 HEPES, 10 CsCl, 0.3 CdCl2, 1 MgCl2, and 2 BaCl2, pH 7.4 (300-305 mOsm/l). For recording persistent Na+ currents, the external solution contained the following (in mm): 115 NaCl, 45 TEA chloride, 10 HEPES, 0.3 CdCl2, 1 MgCl2, and 2 BaCl2, pH 7.4 (300-305 mOsm/l). The liquid junction potential (<2 mV) was not compensated for.
To ensure adequate voltage control, several steps were taken. Only cells with relatively short (25-50 μm) processes were selected for recording. In experiments designed to examine rapidly inactivating currents, the external Na+ concentration was lowered to 10 mm, and internal Na+ was held near 4 mm; this kept currents relatively small and minimized any residual series resistance errors. In those cases in which these processes retracted during the recording without granulation or a change in leak currents, there was rarely a noticeable change in the quality of the clamp, suggesting that the short processes were adequately controlled. In each cell, current activation plots were generated, and any evidence of loss of voltage control (discontinuities in the current-voltage relationship that would yield slope factors <5 mV) resulted in the cell being discarded. Also, variation in the activation kinetics of test pulse currents evoked in inactivation protocols was taken as evidence of bad space clamp. In several experiments, reversal potentials were examined. These invariably fell within a few millivolts of the prediction based on the Goldman-Hodgkin-Katz equation, suggesting that the transmembrane voltage was adequately controlled. In the ramp experiments, in which external Na+ was near physiological levels, discontinuities in the rising phase of the currents were taken as evidence of bad control; in the worst case, this was manifested as spiking. Where there was ambiguity, the Na+ current driving force was reduced, and the experiments were repeated.
Whole-cell and cell-attached recordings in slices. Slices were obtained as described above and placed for >1 hr into an artificial CSF (ACSF) containing the following (in mm): 125 NaCl, 2.5-3.5 KCl, 25 NaHCO3, 1.25 Na2HPO4, 1 MgCl2, 2 CaCl2, and 25 glucose, pH 7.3 (300 mOsm/l, saturated with 95% CO2 and 5% O2). Thereafter, slices were transferred to a recording chamber and superfused with ACSF at a rate of 3-4 ml/min. Current-clamp recordings were performed on visually identified cholinergic interneurons with an infrared video microscopy system. The pipette solution consisted of the following (in mm): 119 KMeSO4, 1 MgCl2, 0.1 CaCl2, 10 HEPES, 1 EGTA, 12 phosphocreatine, 2 Na2ATP, and 0.7 Na2GTP, pH 7.2-7.3, with KOH (280-300 mOsm/l). Whole-cell recordings were obtained at 32°C; cell-attached recordings were obtained at room temperature (22-25°C). Dopaminergic agonists and antagonists were applied to visually identified cholinergic interneurons with a local puffer pipette.
Pharmacology. Drugs were dissolved as stock solutions in either water or DMSO. Calphostin C and 1-oleoyl-2-acetyl-sn-glycerol (OAG) were obtained from Calbiochem (La Jolla, CA). Quinpirole, R(-)propylnorapomorphine (NPA), and (-)-sulpiride were obtained from Sigma. Stock solutions were dissolved in 0.1% sodium metabisulfite to prevent oxidation. When drugs were dissolved in DMSO or sodium metabisulfite, equivalent amounts were added to all internal or external solutions as controls. BAPTA was obtained from Molecular Probes (Eugene, OR). β-Adrenergic receptor kinase 1 C-terminal peptide (βARK-Cp) comprises residues 548-671 of the rat homolog of βARK. βARK-Cp (4.9 mg/ml) was dialyzed against the recording internal solution by Dr. Heidi Hamm (Vanderbilt University, Nashville, TN). This solution was diluted in the recording internal solution for a final concentration of 1 mg/ml.
Data analysis. Data were plotted and analyzed with IgorPro (Wave-Metrics, Lake Oswego, OR) or Mathematica (Wolfram Research, Champaign, IL). Na+ currents evoked by depolarizing steps were fit with a modified Hodgkin-Huxley (HH) formalism of the following form: g = gmaxm3(V,t)h(V,t) (V - Vrev), where g is the conductance, gmax is the maximal conductance, V is transmembrane voltage, t is time, Vrev is the Na+ reversal potential, m(V,t) = α(1 - exp(-t/τm)), h(V,t) = β(exp(-t/τh1)) + (1 - β - γ) (exp(-t/τh2)) + γ, where α is a scalar, 0 ≤ β < 1 (the component of inactivation that decays with a τh1 time constant), γ is scalar representing the component of the current that is persistent (typically 0.01-0.05), τm is the activation time constant, and τh1 and τh2 are the fast- and slow-inactivation time constants. The development of inactivation between -60 and -40 mV was estimated by stepping into this voltage range for a variable period before delivering a test step to assay for deinactivated channels. Inactivation kinetics were determined by fitting measurements of peak current as a function of prepulse duration. Deactivation kinetics were estimated by briefly depolarizing the membrane to open channels and then repolarizing to hyperpolarized membrane potentials. These tail currents were fit with simple monoexponential or biexponential functions. Nominally steady-state conductance-voltage and inactivation-voltage curves were fit with a Boltzmann function of the following form: g(V) = 1/(1 + exp((V - Vh)/k))C, where Vh is the half-activation or inactivation voltage, and k is the slope factor. Activation data were fit with a third-order (c = 3), and inactivation was fit with a first-order (c = 1) Boltzmann function. Activation and deactivation time constants were plotted as a function of voltage and fit with an equation of the following form: c1 + c2/(α 1 exp(-(V - α2)/α3) + β 1 exp((V + β2)/β3)), where V is transmembrane voltage, and α1-α3, β1-β3, c1, and c2 are fitted constants. This equation is derived from the HH formalism and assumes a single voltage-dependent state transition.
Slow-inactivation voltage curves were fit with a modified Boltzmann equation of the form: I/Imax = (1 - Iresid)/((1 + exp(-(V - Vh)/k)) + Iresid, where Iresid is the residual (noninactivating) fraction of the current, and k is the slope factor. Time constants for the entry into the slow-inactivated state were reasonably fit with a single-exponential function; exit from the slow-inactivated state required a double-exponential fit.
Statistical analyses were performed using SYSTAT (SPSS, Chicago, IL). Sample statistics are given as mean ± SEM for samples ≥10 and as a median (interquartile range) for smaller samples. In data presented as box plots in the figures, the central line represents the median, the edges of the box represent the interquartiles, and the “whisker lines” show the extent of the overall distribution, excluding outliers (points >1.5× interquartile range), which are shown as circles or asterisks in the figures.
Computer simulation. Although the HH formalism yielded accurate fits to the data generated by voltage steps, it does not account for a number of gating properties, including the voltage dependence of persistent Na+ currents. Therefore, a Markov model of channel gating was fit to the data (Raman and Bean, 2001; Taddese and Bean, 2002; Carr et al., 2003) using NEURON (version 5.5) (Hines and Carnevale, 2001). Using the MultiRun Fitter in NEURON, the Markov model was constrained by the experimental data as abstracted by the HH description. To fit the biexponential decay of recorded currents, currents were modeled as the sum of two channels (this was consistent with the molecular profiling; see below); models were constructed with fast and slow kinetics (as well as slow inactivation) (Carr et al., 2003). The fast current had the following parameters: α = 37 exp((V - 45)/40) msec-1, β = 10 exp(-(V + 50)/10) msec-1, γ = 40 msec-1, δ = 30 msec-1, Con = 0.001 msec-1, Coff = 0.1 msec-1, Oon = 1.6 msec-1, Ooff = 0.01 msec-1, a = ((Coff/Con)/) Ooff/Oon), aS1 = 0.0025, aS2 = 0.0002, and bS = 0.00017. The slow channel parameters were as follows: α = 37 exp((V - 45)/40) msec-1, β = 10 exp(-(V + 50)/20) msec-1, γ = 40 msec-1, δ = 30 msec-1, Con = 0.001 msec-1, Coff = 0.1 msec-1, Oon = 0.7 msec-1, Ooff = 0.01 msec-1, and a = ((Coff/Con)/(Ooff/Oon), slow time constants. The ratio of the slow-to-fast channel density was adjusted to 0.4 to match macroscopic currents.
To simulate the pacemaking of cholinergic interneurons, a simplified model was constructed in NEURON using a spherical soma (30 μm) and two dendrites (200 μm long, 2 μm diameter) invested with ionic conductances known to participate in pacemaking based on published (Song et al., 1998; Bennett et al., 2000) and unpublished studies. Membrane resistance was 1 KΩ/cm2, capacitance was 1 μF/cm2, and axial resistivity was 70 Ω/cm. In addition to the Na+ channels described above, somatic currents included voltage-dependent K+ channels (Kv4, Kv2, and KCNQ), Ca2+-dependent K+ channels [BK (large conductance) and SK (small conductance)], and voltage-dependent Ca2+ channels (Cav1.3 and Cav2.1); these channels and voltage-dependent cAMP-modulated cationic channels [HCN (hyperpolarization-activated cyclic-nucleotide-gated channel)] also were placed in the dendrites, albeit at different densities. Intracellular Ca2+ handling was as described by Lazarewicz et al. (2002). The model of SK currents was as described by Lazarewicz et al. (2002), and the model of BK channels was taken from Khaliq et al. (2003). Mod files for the other conductances (Kv4, Kv2, KCNQ, Cav1.3, Cav2.1, HCN1, and HCN2) were generated from experimental data as described above for the Na+ currents. The kinetic models for HCN1 and HCN2 channels were adapted from Wang et al. (2001). Relative densities of the channels were adjusted to yield a pacemaking waveform and rate similar to that found in whole-cell recordings from cholinergic interneurons in the slice (Bennett et al., 2000). The densities in the soma were as follows (in mS/cm2): 1.0 fast Na, 0.4 slow Na, 0.003 Cav1.3, 0.05 Cav2.1, 0.4 Kv4, 0.1 Kv2, 0.002 KCNQ, 1 BK, and 0.003 SK. The densities in the dendrites were as follows (in mS/cm2): 0.25 fast Na, 0.1 slow Na, 0.003 Cav1.3, 0 Cav2.1, 0.4 Kv4, 0.1 Kv2, 0.01 HCN1, 0.03 HCN2, 1 BK, and 0.003 SK. These mod files can be downloaded from the NeuronDB website (http://senselab.med.yale.edu/senselab/ModelDB/).
Single-cell reverse transcription-PCR. These procedures were as described previously (Yan et al., 1997). Briefly, dissociated, individual cholinergic neurons were aspirated into sterile glass micropipettes containing diethylpyrocarbonate-treated water and 1.5 U/μl SUPERase-In (Ambion, Austin, TX). The contents of the pipette were transferred to thin-walled PCR tubes containing dNTPs (1 μl, 10 mm), BSA (0.7 μl, 143 μg/μl), random hexamers or oligo-dT (2.6 μl, 50 ng/μl), and SUPERase-In (0.7 μl, 40 U/μl). All reverse transcription (RT) reagents were obtained from Invitrogen (Carlsbad, CA). This mixture was heated to 65°C for 5 min to linearize mRNA and then placed on ice for 2 min. The following was added to each tube: 10× RT buffer (1 μl), MgCl2 (2 μl, 25 mm), DTT (1 μl, 0.1 m), RNase Out (0.5 μl), and 200U SuperScript II reverse transcriptase. cDNA transcription was performed by heating the reaction mixture to 25°C for 10 min and 42°C for 50 min. The reaction was terminated by incubation at 70°C for 15 min and then placed on ice. RNA was then removed by adding 0.5 μl of RNaseH to each tube and incubating for 20 min at 37°C.
Amplification cDNAs for 67 kDa isoform of glutamic acid decarboxylase (GAD67), choline acetyltransferase (ChAT), D2 dopamine receptor, and Na+ channel α-subunit cDNA (Nav1.1, Nav1.2, Nav1.5, and Nav1.6) was performed as described previously (Yan et al., 1997; Maurice et al., 2001). The PCR primers for Na+ channel β-subunits (Naβ1, Naβ2, and Naβ3) were developed from GenBank sequences using OLIGO software (National Biosciences, Plymouth, MN). The primers for Naβ1 cDNA (Gen-Bank accession number M91808) were AGAAGGGCACAGAGGAATTTGTCA (position 401) and GACGCTGGTGTTGTGCTCGTAAT (position 611). The predicted product length was 233. The primers for Naβ2 cDNA (GenBank accession number U37026) were CTGCCCTGTACCTTCAACTCCTG (position 308) and CCATCCGTCTTGCCTTCCTC (position 764). The predicted product length was 476. The primers for Naβ3 cDNA (GenBank accession number AJ243395) were TGAGGGCGGTAAAGATTTCCTT (position 560) and CTTCGGCCTTAGAGACCTTTCTGT (position 904). The predicted product length was 368.
Negative controls for extraneous and genomic DNA contamination were run for each experiment. To verify that genomic DNA was not being amplified, reverse transcriptase was omitted during the RT reaction, and the resulting reaction mixture was processed for PCR amplification as described above. Extraneous contamination during the PCR amplification was examined by replacing the cDNA template with buffer solution. If either control was found to be positive, the material from that experiment was discarded.
Results
D2 receptor stimulation reduces evoked and autonomous spiking
Cholinergic interneurons were identified in tissue slices by their large somal diameter and autonomous activity (Bennett and Wilson, 1999). D2 receptor-class agonist NPA (10 μm) consistently reduced spiking evoked by somatic current injection in cholinergic interneurons recorded in tissue slices without changing resting membrane potential (Fig. 1A,B). On average, NPA reduced the number of spikes evoked by near rheobase current injection by nearly one-half (Fig. 1C) (n = 4; p < 0.05;Kruskal-Wallis). To gain a better idea of how this modulation would affect the relationship between discharge frequency and injected current, slow current ramps were delivered (Fig. 1D,E). NPA reduced discharge frequency across a range of current intensities. The frequency-intensity plots were shifted toward higher currents and down on the frequency scale (Fig. 1F). A similar alteration was seen in all of the cholinergic interneurons tested (n = 3).
Figure 1.

D2 receptor activation reduced evoked activity. A, Repetitive spiking evoked in a cholinergic interneuron by intrasomatic current injection during a recording in a tissue slice. B, The same neuron after bath application of NPA (10 μm). Note the reduction in evoked discharge. C, Statistical summary of NPA-induced reduction in evoked spiking. D, Repetitive activity evoked by somatic injection of a current ramp. E, The response to the same stimulus was reduced after application of NPA (10 μm). F, Instantaneous discharge frequency-current injection plot for the neuron shown in D and E. Data were fit with a polynomial function: control, -206.8 + 2.03i -0.0046i2; NPA, -287.4 + 2.74i -0.0062i2, where i is current. Similar results were obtained in two other neurons tested. Triangles indicate NPA application.
As discussed above, cholinergic interneurons are capable of sustained autonomous pacemaking. In the tissue slice, this pacemaking is completely independent of synaptic input, unaltered by blockade of either glutamatergic or GABAergic receptors (Bennett and Wilson, 1999). To determine whether D2 receptor stimulation could influence autonomous pacemaking, cholinergic interneurons were recorded in cell-attached patches in tissue slices. Application of the D2 receptor-class agonist quinpirole (10 μm) slowed cholinergic interneuron discharge (Fig. 2A). The average behavior of our sample (n = 6) can be seen in Figure 2B, in which the discharge rate was normalized, and mean discharge rate (across the sample) was plotted as a function of time after quinpirole application. The median reduction of discharge rate was >20% (Fig. 2C) (n = 10; p < 0.05; Mann-Whitney). Blockade of ionotropic GABA and glutamate receptors had no effect on the modulation (n = 4; p > 0.05; Mann-Whitney). Because discharge rate slowed after receptor activation, the variability of the discharge also increased. One measure of irregularity in interspike intervals is the coefficient of variation (CV); this measure increased significantly after receptor activation (control median CV, 0.3; quinpirole median CV, 0.6; n = 6; p < 0.05; Wilcoxon). In the presence of ionotropic receptor antagonists, coapplication of the D2 receptor antagonist sulpiride (5 μm) blocked the effects of D2 receptor agonists and typically increased the discharge rate of interneurons above the control rate, suggesting that there was an ambient D2 receptor tone (Fig. 2C) (n = 4; p > 0.05; Mann-Whitney). Single-spike pacemaking in cholinergic interneurons is dependent on voltage-dependent Na+ channels (Bennett et al., 2000), suggesting that D2 receptors may be acting to reduce these currents. As a first step toward testing this hypothesis, the properties of the Na+ channels underlying pacemaking were studied.
Figure 2.

D2 receptor activation reduced autonomous spiking. A, Somatic cell-attached recording of autonomous action potential firing in a cholinergic interneuron under control conditions and in the presence of quinpirole (10 μm). The discharge rate was slowed (control, 4.7 Hz; quinpirole, 1.8 Hz), and the discharge coefficient of variation was increased (control, 0.66; quinpirole, 1.47). B, An average time course of the action of quinpirole on the autonomous firing rate of cholinergic interneurons. Inset, The activity of the neuron that was shown in A. C, A summary of the change in firing rate in cell-attached patch recordings after local application of quinpirole (10 μm; n = 10) and after coapplication of the D2 receptor antagonist sulpiride (5 μm) and quinpirole (10 μm; n = 4). Circles are the data points. Note that quinpirole significantly reduced the discharge rate but that coapplication of the D2 receptor antagonist blocked the effect, leading to an increase in discharge rate in some cells.
Na+ channels in cholinergic interneurons are heterogeneous and have features that support pacemaking
Acutely isolated murine cholinergic interneurons were identified by their large somatic diameter (Fig. 3A). As in previous studies (Yan et al., 1997), single-cell (sc) RT-PCR profiling of these neurons revealed that they express ChAT and D2 receptor mRNAs but not mRNA for GAD67 (Fig. 3B). To determine the molecular identity of the Na+ channel subunits expressed by cholinergic interneurons, single neurons were profiled for mRNAs contributing to Na+ channels. Single-cell RT-PCR profiling was performed for three β-subunit mRNAs (Naβ1- Naβ3; n = 10) (Isom et al., 1992, 1995); all three were consistently coexpressed in murine cholinergic interneurons (Fig. 3E,F). Subsequently, profiling was performed for the most commonly expressed pore-forming α-subunit mRNAs in the adult brain: Nav1.1, Nav1.2, and Nav1.6 (Goldin, 1999). Nav1.1 mRNA was detected in all cholinergic interneurons examined (n = 16); Nav1.6 mRNA was detected in a large subset of the sample (12 of 16), whereas Nav1.2 mRNA was detected in only one-half (8 of 16), and Nav1.5 was rarely detected (1 of 6) (Fig. 3C,D). This detection profile differs from that previously reported in cortical pyramidal neurons only in the frequency of Nav1.2 mRNA detection (Maurice et al., 2001). The most parsimonious interpretation of these results is that cholinergic interneurons coexpress significant levels of Nav1.1, Nav1.2, and Nav1.6 α-subunit mRNAs, but Nav1.2 mRNA abundance is relatively low. The relatively high level of Nav1.6 subunits in cholinergic interneurons may lead to relatively larger persistent Na+ currents (Raman et al., 1997; Maurice et al., 2001) and an enhancement of pacemaking capacity.
Figure 3.
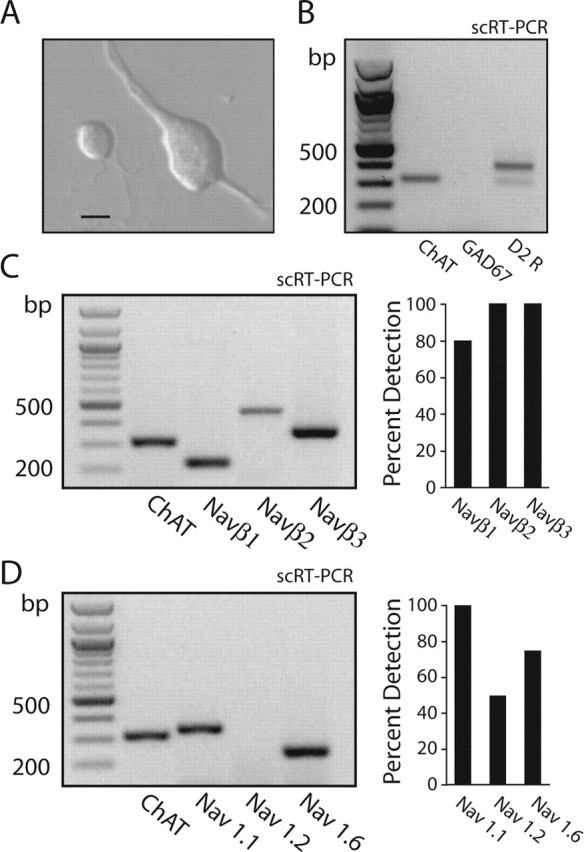
Identified cholinergic interneurons coexpress Na+ channel α- and β-subunit mRNAs. A, Photomicrograph of an acutely isolated cholinergic interneuron and a neuron with an appearance resembling that of a medium spiny neuron. Scale bar, 8 μm. B, Gel showing scRT-PCR amplicons for ChAT, GAD67, and D2 receptor mRNAs. Note that the neuron expressed ChAT and D2 receptor mRNAs but not GAD67. C, Gel showing scRT-PCR amplicons from a ChAT-expressing neuron. The neuron coexpressed Naβ1-Naβ3 mRNAs. Right, A summary from 10 neurons. D, Gel showing scRT-PCR amplicons from a ChAT-expressing neuron. Both Nav1.1 and Nav1.6 mRNAs were detected. Right, A detection summary for Nav1.1, Nav1.2, and Nav1.6 mRNAs in a sample of 16 neurons.
Subsequently, the biophysical properties of these Na+ channels were evaluated using voltage-clamp techniques. Why? Typically, the ambient membrane potential between spikes in this low-frequency discharge mode is between -60 and -55 mV. In this potential range, Na+ channels undergo a conformational change called fast inactivation, resulting in an inability of Na+ to pass through the channel with additional depolarization (Hille, 2001). If the biophysical properties of Na+ channels in these neurons were similar to those in a number of regular-spiking neurons (e.g., cortical pyramidal neurons) (Maurice et al., 2001), then only ∼20% of the Na+ channels would be available to participate in pacemaking because of high levels of “resting” channel fast inactivation. This would make Na+ channel-dependent pacemaking very inefficient.
To determine whether there was a high level of Na+ channel inactivation during cholinergic interneuron pacemaking, whole-cell voltage-clamp experiments were performed. To obtain an accurate biophysical characterization, Na+ currents were kept small by recording in a low (10 mm) external Na+ concentration and with the internal Na+ concentration near 4 mm. This modest concentration gradient ensured good voltage control and minimized series resistance errors. TTX-sensitive currents evoked by depolarizing voltage steps of increasing amplitude had kinetic features that were voltage dependent (Fig. 4A). These currents were fit with a modified HH model (see Materials and Methods) to generate estimates of maximum conductance as a function of membrane voltage (Fig. 4C). These conductance estimates were well fit with a third-order Boltzmann function, having a half-activation voltage near -40 mV (mean, -39.8 ± 0.9 mV; n = 8) and a slope factor near 8 mV (7.6 ± 0.2 mV) (Fig. 4D). As can be seen by inspection of this plot, the macroscopic conductance was half-maximal at approximately -30 mV. These parameters are similar to those obtained from a variety of other neurons, including those that lack pacemaking ability, and are regular spiking (Magistretti and Alonso, 1999; Maurice et al., 2001). Activation kinetics at depolarized potentials (more than -40 mV) were extracted from HH fits to the currents shown. Deactivation (the reversal of activation) kinetics were estimated by briefly depolarizing the membrane to activate channels and then repolarizing the membrane quickly to generate deactivation tail currents (Fig. 4E). These tail currents were well fit with a double-exponential function; the fast component was taken as deactivation, whereas the slow component was attributable to inactivation development. Pooled kinetic estimates were plotted and fit with a function derived from the HH formalism assuming a single voltage-dependent state transition (Fig. 4F) (see Materials and Methods).
Figure 4.

Cholinergic interneurons express Na+ channel currents with distinctive properties. A, Currents evoked in a cholinergic interneuron by a series of depolarizing steps from -65 to +10 mV from a holding potential of -70 mV. B, Current evoked by a test step to -20 mV from increasingly more depolarized prepulse potentials. Prepulse was 200 msec, and the holding potential was -70 mV. C, Hodgkin-Huxley model fits to the data shown in A. Maximum conductance estimates from these fits were used to generate the activation plot in D. D, Steady-state inactivation and activation data from a sample of neurons (n = 8) are plotted and fit with Boltzmann functions of either first order (inactivation) or third order (activation). Gray lines are from individual neurons. Also shown is the steady-state inactivation plot derived from a sample of cortical pyramidal neurons. E, Deactivation tail currents generated by briefly stepping to -20 mV and then repolarizing to potentials between -110 and -40 mV. F, Plot of activation (n = 8) and deactivation (n = 4) time constants for a sample of neurons. Data were fit with a function of the following form: (7.8e - 5) + (6.1e - 3)/((2.9e - 6) exp(-(V - 204.6)/16.4) + (4.9e2) exp((V - 63.9)/28.6)), where V is membrane voltage. Also shown is the activation plot from D. Circles are the sample mean at each voltage.
The voltage dependence of Na+ channel fast inactivation in cholinergic interneurons was studied with a combination of approaches. Using conditioning pulses of sufficient duration to reach equilibrium (200 msec), the voltage dependence of fast-inactivation gating was extracted from the amplitude of currents evoked by a test voltage step to -20 mV (Fig. 4B). The amplitude of the current evoked by the test step was plotted as a function of conditioning voltage and fit with a first-order Boltzmann function (Fig. 4 D). Half-inactivation voltages were near -55 mV (Vh = -52.9 ± 0.6 mV; n = 8), and slope factors were near 5 mV (k = 5.1 ± 0.2 mV). The half-inactivation voltages are significantly more depolarized than those found in regular-spiking cortical pyramidal neurons recorded under identical circumstances (Vh = -63 ± 1.3 mV; n = 10; p < 0.05; Kruskal-Wallis).
The development of fast inactivation at depolarized potentials (more than -30 mV) was taken from the modified HH fits to currents activated by voltage steps, as shown above. In this potential range, current inactivation was voltage dependent and invariably biexponential (Fig. 5A, semilog plot), with the fast component being the dominant component (although this fraction varied as a function of membrane potential) (Fig. 5A, inset). At membrane potentials near threshold (-70 to -40 mV), inactivation rates were measured with a prepulse protocol (Fig. 5B). Deinactivation of Na+ channels was measured by hyperpolarizing the membrane for variable durations after a depolarizing step (Fig. 5C). Data derived from these protocols were well fit only with a biexponential function, as with inactivation development. Data from all three protocols were pooled and then fit (Fig. 5A) using a single-state transition model.
Figure 5.
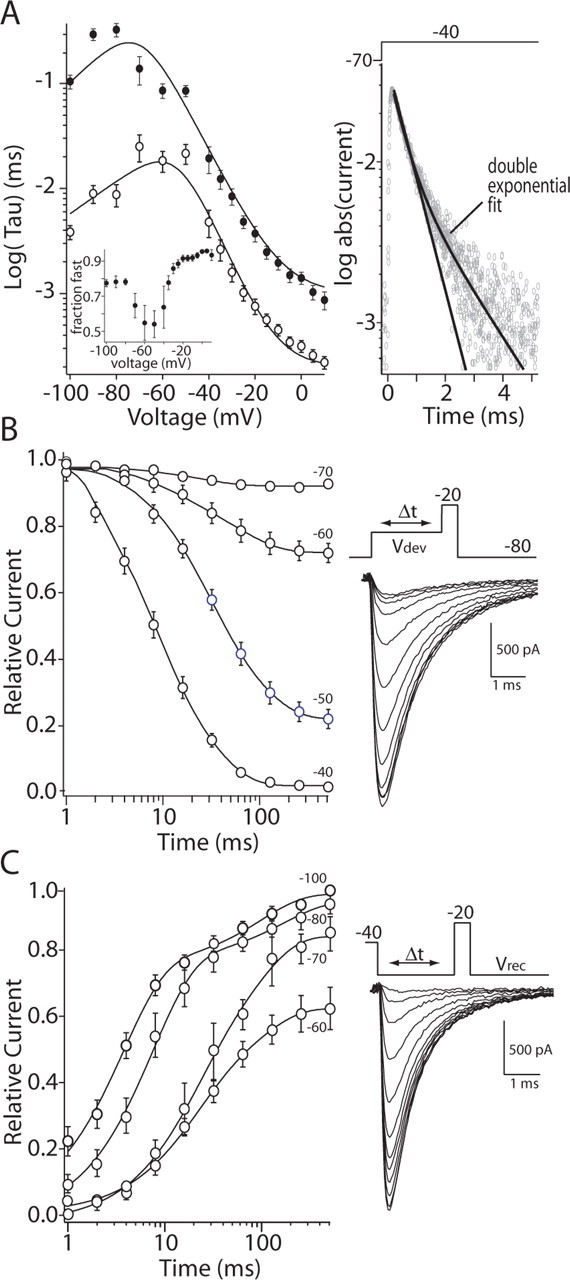
Inactivation kinetics of Na+ currents were biexponential and voltage dependent. A, Summary showing fast-inactivation development and recovery kinetics over a range of membrane potentials between +10 and -100 mV. Data points above -40 mV were derived from HH fits to currents evoked by depolarizing steps (Fig. 2). The decay of these currents was typically biexponential, as shown in the semilog plot at the right. Here, currents evoked by a step to -40 mV are plotted after conversion to absolute values. Data were fit with a function, as in Figure 2 F: fast time constant, (2e - 3) + (5.5e - 3)/((1e - 6) exp(-(V - 180)/20) + (4e3) exp((V - 40)/10)); slow time constant, (7e-4) + (5.5e - 3)/((6e - 8) exp(-(V - 140)/18) + (4e4) exp((V - 90)/11)). Inset, A plot of the relative amplitude of the fast component as a function of membrane potential. B, The development of fast inactivation at potentials between -70 and -40 mV was estimated by plotting the amplitude of the current evoked by a test step to -20 mV that followed a prepulse of variable duration (n = 8). Data points were fit with a biexponential function. C, The recovery from fast inactivation at potentials between -60 and -100 mV was determined by using a similar strategy to that shown in B. Peak current plots for a sample of neurons are shown along with biexponential fits (n = 11). The parameters derived from B and C are plotted in A. Unfilled circles are sample means of the fast time constant fitted to the Na+ current inactivation at the indicated voltages; filled circles are the sample means of the slow time constant.
In summary, these experiments show that (1) the somatic Na+ channel currents in cholinergic interneurons have significantly different inactivation properties from those found in regular-spiking neurons, and (2) the macroscopic currents are likely to arise from at least two kinetically distinguishable channel populations, an inference consistent with the molecular profiling results that revealed coexpression of Na+ channel α- and β-subunits.
D2 receptor stimulation reduces Na+ currents at depolarized potentials
As shown above, cholinergic interneurons express mRNA for the D2 dopamine receptor. In most cholinergic interneurons tested (12 of 13), the application of the D2-class receptor agonist NPA (10 μm) rapidly and reversibly reduced the rapidly inactivating Na+ currents evoked by a step from -70 mV (mean reduction, 10.5 ± 1.2%; n = 12; p < 0.05; Kruskal-Wallis) (Fig. 6A,E). The D2-class receptor antagonist sulpiride (10 μm) significantly reduced this modulation (median reduction, 2%; interquartile (25-75%) range, 0-5%; n = 5; p < 0.05; Kruskal-Wallis); lower concentrations of sulpiride (1 μm) gave partial block of the NPA (10 μm) modulation (n = 3; range, 20-80% of block), possibly because of alterations in the receptor during enzymatic dissociation of the tissue. Because cholinergic interneurons do not express detectable levels of mRNA for other members of the D2 receptor class (D3, D4) that bind NPA and sulpiride with high affinity (Yan et al., 1997), these results argue that the modulation observed is mediated by D2 receptors. Dialysis with the Gβγ-subunit scavenger βARK-Cp (Koch et al., 1994) also attenuated the modulation (median reduction, 4%; interquartile range, 1-5%; n = 4). In agreement with this inference that the signaling was mediated by Gβγ-subunits and the demonstration that striatal D2 receptors couple via these subunits to phospholipase C (PLC) β isoforms (Hernandez-Lopez et al., 2000), application of the PKC activator OAG (2 μm) mimicked the effect of D2 receptor agonists, rapidly and reversibly reducing peak Na+ currents evoked by a step from -70 mV (median reduction, 16%; interquartile range, 13-20%; n = 7; p < 0.05; Kruskal-Wallis; data not shown). Dialysis with the PKC inhibitor calphostin C (1 μm) eliminated the effects of D2 receptor agonist application (median modulation, 2%; interquartile range, 0-4%; n = 4; p > 0.05; Kruskal-Wallis; data not shown).
Figure 6.

D2 receptor activation reduced Na+ currents in a voltage-dependent manner. A, Application of the D2 receptor-class agonist NPA (10 μm) reversibly reduced Na+ currents evoked by a test step to -20 mV from a holding potential of -70 mV. B, Currents evoked before and after application of NPA by a fast-inactivation protocol. Peak current data derived from these traces are shown at the right. C, Data were normalized to the peak current in control records and fit with Boltzmann functions (see Materials and Methods); although peak conductance was reduced, there was no change in voltage dependence. Unfilled circles indicate control data; filled circles indicate NPA application. D, In another neuron, NPA had a smaller effect when holding at -70 mV; changing the holding potential to -55 mV increased the magnitude of the modulation. E, Time course showing the increase in modulation at more positive holding potentials. This modulation reversed very slowly at positive potentials; however, the modulation was reversed by a brief step to -90 mV.
The D2 receptor-mediated modulation of Na+ channel currents resulted in an apparent reduction in maximal conductance without prominently shifting the voltage dependence of fast inactivation (Fig. 6B,C) (control, Vh = -55 mV; interquartile range, 53-56 mV; NPA, Vh = -56 mV; interquartile range, 54-58 mV; n = 6). Slope factors were unaltered by NPA as well (control, k = 5.5 mV; interquartile range, 5.2-5.8 mV; NPA, Vh = 5.8 mV; interquartile range, 5.4-6.1 mV; n = 6). Direct activation of PKC with OAG had very similar effects (control, Vh = -54 mV; interquartile range, 52-55 mV; OAG, Vh = -56 mV; interquartile range, 54-58 mV; n = 6). Slope factors were unaltered by OAG (control, k = 5.4 mV; interquartile range, 5.1-5.7 mV; OAG, Vh = 5.4 mV; interquartile range, 5.3-5.6 mV; n = 6).
Although D2 receptor stimulation consistently reduced Na+ currents evoked from -70 mV, the magnitude of the modulation was modest. Holding the membrane potential at more negative membrane potentials (approximately -90 mV) further reduced the modulation of current amplitudes (data not shown). In contrast, when cholinergic interneurons were held at -55 mV (near their normal resting membrane potential) and the D2 receptors were activated, the reduction in current amplitude nearly doubled from that seen in the same cell at -70 mV (Fig. 6D,E). Similar results were obtained in every neuron examined (median increase in modulation at -55 mV was 342%; range, 168-680%; n = 18). However, at depolarized membrane potentials, unlike the situation at -70 mV, reversal of the modulation was typically very slow after washing off the agonist (Fig. 6E). To test whether this sustained reduction was attributable to a voltage-dependent reversible process, neurons were hyperpolarized to -90 mV for 30 sec after agonist application, and then the membrane potential returned to -55 mV. In all of the neurons examined with this protocol (n = 7), brief hyperpolarization reversed the modulation (Fig. 6E). The hyperpolarization was not accompanied by any discernible change in input or series resistance. As shown below, this reversal was presumably accomplished by promoting the exit of Na+ channels from a slow-inactivated state.
D2 receptor stimulation reduces persistent Na+ currents
Activation of D2 receptors with NPA (10 μm) also reversibly diminished persistent Na+ currents evoked by a slow voltage ramp from -80 to 0 mV (8 of 10 neurons tested) (Fig. 7A). At -25 mV, the peak current was reduced by ∼30% (median, 29%; interquartile range, 25-39%; n = 8). This was substantially larger than the reduction in transient Na+ current seen even at a holding potential of -55 mV (Fig. 6). This modulation was mimicked by application of OAG (2 μm), which reduced ramp currents at -25 mV by a similar amount (median, 30%; interquartile range, 21-34%; n = 6). To estimate changes in conductance, currents were converted using the assumption that currents were ohmic, and driving force was determined by the Nernst equation (Hille, 2001). The median reduction in the peak persistent conductance was larger than that for current, which was near 41% (interquartile range, 28-62%; n = 5).
Figure 7.

D2 receptor activation reduced persistent Na+ currents. A, Current-voltage plot of TTX-sensitive persistent Na+ currents evoked by a slow voltage ramp (4 sec) from -80 to 0 mV in an acutely isolated cholinergic interneuron. B, NPA (10 μm) reversibly reduced persistent Na+ currents. The current-voltage relationship of the TTX-sensitive currents before and after NPA application. C, Conversion of the current-voltage plots in A to conductance estimates were fit using an HH model.
D2 receptor stimulation reduces Na+ currents by enhancing a process resembling slow inactivation
What type of mechanism could explain the voltage dependence of the Na+ channel modulation? Recent work has shown that PKC- and PKA-mediated modulation of Na+ channel currents in cortical pyramidal neurons and heterologous expression systems is produced by enhancing channel entry into a slow-inactivated state (Carr et al., 2003). To test whether a similar process was at work here, the voltage dependence and kinetics of slow inactivation were examined in the presence and absence of D2 receptor-class agonists. Na+ channel currents in cholinergic interneurons exhibited slow inactivation, but the extent of slow inactivation was less than that seen in cortical pyramidal neurons (Carr et al., 2003) or medium spiny neurons (J. Mercer and D. J. Surmeier, unpublished observations). In pyramidal neurons, a 5 sec depolarization to -20 mV forces ∼60% of the somatic Na+ channels into a slow-inactivated state, whereas the same protocol converted only ∼20% of the cholinergic interneuronal channels into this state. In this protocol, channels are allowed to recover from fast inactivation by holding the membrane potential at -80 mV for 1 sec before delivering a test pulse (Fig. 8A, inset). Any reduction in current amplitude with this protocol is attributable to slow inactivation. NPA (10 μm) significantly increased the extent of slow inactivation measured using 5 sec voltage steps to -20 mV (mean control slow inactivation, 19 ± 1%; slow inactivation in NPA, 30 ± 2%; n = 18; p < 0.05; Kruskal-Wallis). D2 receptor activation did not discernibly change the voltage dependence of slow inactivation (Fig. 8A) or the rate of entry into the slow-inactivated state produced by a voltage step to -20 mV (Fig. 8 B). Entry in the slow-inactivated state was monoexponential in both cases, with a time constant near 5 sec (Fig. 8B). D2 receptor activation did not alter the rate of recovery from slow inactivation either (Fig. 8C); recovery was biexponential in both cases, with a fast time constant of 1.9 sec and a slow time constant of 13.5 sec. However, D2 receptor activation modestly increased the fraction of the current that recovered rapidly (control, 63%; NPA, 72%; n = 8; p > 0.05; Kruskal-Wallis).
Figure 8.
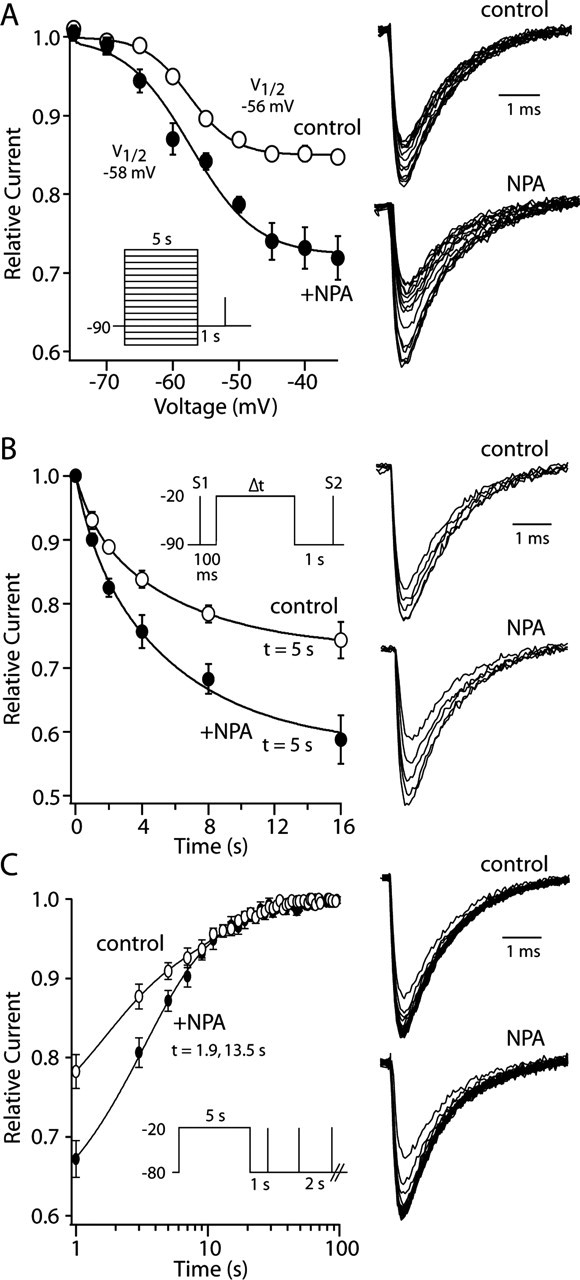
D2 receptor activation enhanced slow inactivation of Na+ currents. A, NPA (10 μm) increased the extent of slow inactivation evoked by a 5 sec voltage step to potentials between -35 and -60 mV but did not change the apparent voltage dependence of the process. Boltzmann fits to normalized data are shown for control (unfilled circles) and NPA (filled circles) data (n = 4). Representative current traces are shown at the right. B, The rate of entry into the slow-inactivated state was unchanged by NPA (10 μm). Single-exponential fits to control (unfilled circles) and NPA (filled circles) data are shown (n = 5). Both data were well fit with a single exponential having a time constant near 5 sec. The extent of slow inactivation was greater in NPA, however. Representative current traces are shown on the right; inset shows protocol. C, NPA did not significantly alter the rate of recovery from slow inactivation at -80 mV. Double-exponential fits to control (unfilled circles) and NPA (filled circles) data are shown (n = 8). Both data sets were well fit with a double exponential having a time constant of 1.9 and 13.5 sec. Representative current traces are shown at the right. Protocol is shown in the inset.
Distinguishing features of the D2 receptor modulation of Na+ channels are slow kinetics and voltage dependence. Long-lasting but not short hyperpolarization of the membrane should reverse the modulation. To test this conjecture, the effect of D2 receptor activation on “postanode break” spiking, such as that triggered by termination of somatic GABAergic inhibitory input, was examined. This postanode break depolarization is generated by slowly deactivating HCN channels in cholinergic interneurons (Bennett and Wilson, 1999). With strong enough HCN activation, the rebound depolarization is accompanied by spiking (Fig. 9A). NPA (10 μm) had no discernible effect on HCN channel activation, as judged by the sag in membrane potential with membrane hyperpolarization, but it did reduce spiking during the rebound depolarization. With a 1 sec hyperpolarization to approximately -80 mV (Fig. 9B), similar results were obtained in other neurons (median reduction in rebound spike number, 63%; n = 5; p < 0.05; Kruskal-Wallis) (Fig. 9C). In the presence of TTX, the rebound depolarization produced by HCN activation was unaffected by NPA (data not shown; n = 4). If the reduction of rebound discharge was a consequence of enhanced slow inactivation of Na+ channels, increasing the duration of the hyperpolarizing pulses should reverse the effects of NPA. To test this hypothesis, the current steps were lengthened to 10 sec, a period that at 35°C should deinactivate ∼90% of the Na+ channel population. In this situation, NPA did not have a significant impact on rebound spiking (Fig. 9D-F) (n = 5; p > 0.05; Kruskal-Wallis).
Figure 9.

D2 receptor activation reduced rebound spiking. A, Hyperpolarizing current pulse (1 sec) induces rebound discharge in a cholinergic interneuron. B, Application of the D2 receptor agonist NPA reduces the rebound discharge (dark line); control data from A is shown in the background. C, Statistical summary of the inhibition of rebound discharge after application of NPA (n = 5). D, Rebound discharge after a longer hyperpolarizing prepulse (10 sec). E, NPA failed to noticeably alter the rebound discharge. F, Statistical summary showing that NPA produced only a modest inhibition of discharge with the long prepulse, in contrast to the short prepulse (n = 5).
Is the D2 receptor-mediated reduction of pacemaking attributable to modulation of Na+ channels?
The data presented here are consistent with the hypothesis that the effects of D2 receptor activation on evoked activity and pacemaking are attributable to an enhanced slow inactivation of voltage-dependent Na+ channels. Is this plausible? The reduction in the transient Na+ current produced by D2 receptor activation was modest: 15-20%. The reduction of persistent Na+ current was twice as large: 30-40%. However, is this sufficient to explain the change in pacemaker rate? If so, then simply blocking 10-30% of the Na+ channels with TTX should reproduce the effects of D2 receptor agonists (recall that interneuron spiking in the slice is not affected by blocking synaptic input). To determine which concentration of TTX would suffice to test this hypothesis, the affinity of interneuron Na+ channels for TTX was determined using whole-cell voltage-clamp recordings. The data were well fit with a Langmuir isotherm, with an IC50 value of ∼5 nm (Fig. 10A), a value very close to that found in other preparations (Goldin, 2001). This data suggest that the application of 1 nm TTX should block 10% of the interneuron Na+ channels. Doing so slowed interneuronal discharge rate in the slice preparation by >20% on average (Fig. 10B,C). These data argue that even a modest reduction of Na+ channel currents can have significant effects on pacemaking.
Figure 10.
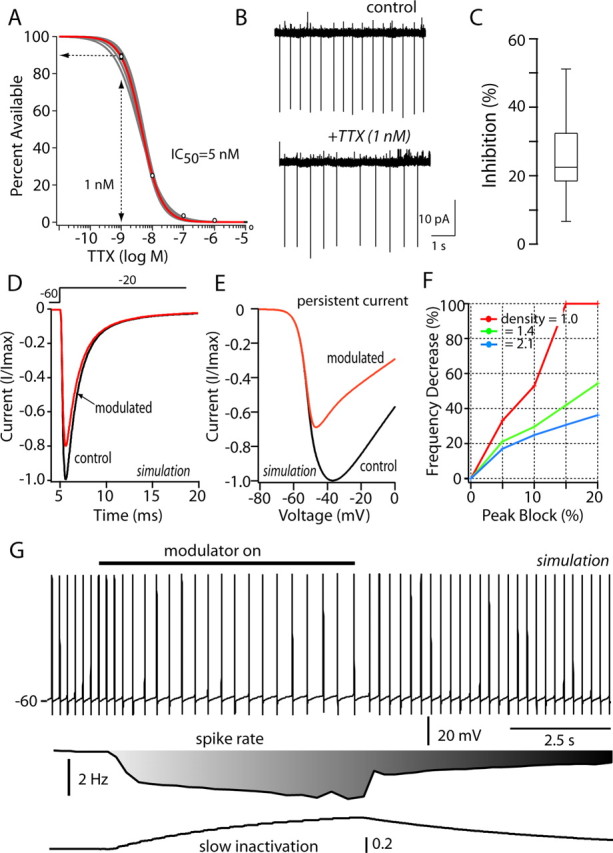
Modest reduction of Na+ channel currents mimics D2 receptor effects on pacemaking. A, Dose-response for relationship for TTX in acutely isolated cholinergic interneurons (n = 5). Peak transient Na+ current, evoked by a step to -20 mV from a holding potential of -80 mV as a function of TTX dose, was fit with a Langmuir isotherm, having a slope of one and an IC50 of 5 nm. B, Cell-attached patch recording in tissue slices before and after bath application of 1 nm TTX. C, The median reduction in discharge rate was just >20% (n = 5); data are shown in box plot format. D, Simulation of Na+ channel currents evoked by a step to -20 mV before and after enhancing entry into the slow-inactivated state from the fast-inactivated state (Carr et al., 2003) (aS2 = 0.0002 → 0.005); the modulation was restricted to the kinetically fast current. Total (fast + slow) Na+ current evoked by the step was reduced by 20% as in experimental observations. E, The same change produced a larger reduction (∼40%) in the total current evoked by a voltage ramp. F, The reduction in autonomous discharge rate increased monotonically as the percentage reduction in transient current was increased from 5 to 20%. Channel densities from 1.0 to 2.1 mS/cm2 yielded qualitatively similar relationships. G, In a simulation of pacemaking (Na+ channel density of 1.4 mS/cm2), mimicking the D2 receptor modulation by selectively increasing Na+ channel entry into a slow-inactivated state (as in D, E) slowed the discharge rate by >40%. Restoration of the control values for this rate constant restored the discharge rate. The bar depicts the onset and offset of the change in slow inactivation. At the bottom, the change in discharge rate and slow inactivation in the affected channel population are plotted.
To provide an additional test, a computational approach was used that allowed selective manipulation of Na+ channels. Using the program NEURON (Hines and Carnevale, 2001), a simulation was constructed using a soma, two dendrites (Fig. 3A), and a complement of channels known to contribute to pacemaking and repetitive discharge in cholinergic interneurons (see Materials and Methods). To match the biexponential kinetic features of macroscopic currents, the Na+ channel population was broken into fast and slow channel types, an assumption that was consistent with the molecular heterogeneity of Na+ channel subunit expression (see above and Materials and Methods). The cell model accurately reproduced the autonomous discharge of interneurons and yielded membrane trajectories resembling those found in whole-cell recordings (Fig. 10G). Enhancing entry of the faster channel population into the slow-inactivated state (Carr et al., 2003) accurately reproduced the macroscopic modulation, decreasing peak transient current by 20% and persistent Na+ current by ∼40% (Fig. 10D,E). This modulation approximately halved the rate of autonomous discharge (Fig. 10G). Alteration in no other channel was necessary to produce this change in discharge rate. Grading the modulation to produce transient current reductions of 5-20% produced a graded reduction in pacemaker rate over a wide range of Na+ channel densities (Fig. 10F). At lower channel densities, a modulation sufficient to produce a 20% reduction in transient Na+ current almost completely stopped spiking, producing a >90% reduction in spike rate. At intermediate channel densities, a modest reduction of Na+ currents led to a reduction of discharge rates, such as that seen experimentally (20-40%) with the application of D2 receptor agonists. These findings, together with those derived from TTX application, provide direct support for the conclusion that modulation of Na+ channel currents in cholinergic interneurons is a major component of the D2 receptor-mediated modulation of interneuron excitability and the reduction in autonomous spiking.
Discussion
D2 receptor activation reduces cholinergic interneuronal pacemaking
Dopaminergic inhibition of cholinergic interneuron function was first inferred from clinical observations that striatal cholinergic tone was elevated in Parkinson's disease (Hornykiewicz, 1976). A major component of the dopaminergic regulation of cholinergic function is mediated by D2 receptor inhibition of spike-driven Ca2+ influx and acetylcholine release (Lehmann and Langer, 1983; Bertorelli et al., 1992; Stoof et al., 1992; DeBoer et al., 1993; Di Chiara et al., 1994; Yan et al., 1997). However, there are compelling reasons to believe there is more to this story. In associative learning paradigms, the ongoing autonomous spiking of primate cholinergic interneurons (or TANs) is reduced by the presentation of unconditioned positive stimuli and by learned conditioned stimuli (Aosaki et al., 1994; Graybiel et al., 1994; Raz et al., 1996). In the initial stages of learning, the pause in interneuronal spiking is dependent on stimulus-linked elevations in dopamine cell activity and dopamine release in the striatum (Aosaki et al., 1994). Work in vivo has shown that the pause in cholinergic interneuronal discharge is primarily mediated by D2 receptors (Watanabe and Kimura, 1998). In agreement with this conclusion, the application of D2 receptor agonists to cholinergic interneurons in the slice preparation reduced autonomous spiking. In this preparation, activity is independent of synaptic connectivity (Bennett and Wilson, 1999), arguing that the D2 receptors expressed by the cholinergic interneurons themselves are critical to the response. The experiments reported here provide a direct linkage between these interneuronal D2 receptors and the ionic mechanism underlying their autonomous activity.
Na+ channel currents in cholinergic interneurons are well suited to a central role in pacemaking
Striatal cholinergic interneurons are autonomous pacemakers; that is, they rhythmically spike in the absence of synaptic input (Bennett and Wilson, 1999). This intrinsically generated activity relies on voltage-dependent Na+ channel currents that appear tailored for this role. Unlike channels in regular-spiking neurons, the Na+ channels in cholinergic interneurons have relatively depolarized fast-inactivation voltage dependence. The half-inactivation voltage is 8-10 mV more depolarized in cholinergic interneurons than in neighboring, regular-spiking medium spiny neurons or cortical pyramidal neurons. Slow inactivation of interneuronal Na+ channels is also modest. This distinctive feature of cholinergic interneuronal Na+ channels was not an obvious consequence of the rate of entry into the slow-inactivated state or the voltage dependence of the process. It may be that basal serine-threonine kinase phosphorylation of Na+ channels in cholinergic interneurons is low (Carr et al., 2003).
Na+ channel currents in cholinergic interneurons also display a prominent persistence. In many cell types, persistent currents are key determinants of the rate and regularity of spiking (Raman et al., 1997; Taddese and Bean, 2002). After spike-repolarizing currents wane, these Na+ currents provide the inward depolarizing force necessary to bring the membrane potential to spike threshold again. The persistence of these currents is critical to slow pacemakers, such as cholinergic interneurons, in which the interspike interval typically ranges from 200 to 1000 msec.
D2 receptor activation decreases Na+ channel currents
In isolated neurons, activation of postsynaptic D2 receptors reduced the Na+ currents underlying pacemaking. The reduction did not depend on an alteration in the voltage dependence of channel opening or fast inactivation. Rather, the reduction in Na+ channel current was brought about by an enhanced entry into a slow-inactivated state (Carr et al., 2003). This endowed the modulation with profound voltage dependence. Holding the membrane potential at -55 mV, rather than -70 mV, tripled the magnitude of the modulation of peak Na+ current, whereas holding at -90 mV virtually eliminated the development of the modulation or quickly reversed it. This feature should maximize the impact of the D2 receptor modulation on pacemaking, in which the modal membrane potential of cholinergic interneurons is near -55 mV. On the other hand, the modulation should have much less of an impact on rhythmic bursting, in which slow inactivation of Na+ channels should be less prominent (Bennett and Wilson, 1999).
The D2 receptor modulation appeared to be accomplished via a Gβγ signaling pathway that activated PKC. This inference is based on (1) the ability of a Gβγ scavenger (βARK-Cp) to reduce the modulation, (2) the mimicry of the modulation by PKC activators (OAG), and (3) the blockade of the modulation by intracellular dialysis with a PKC inhibitor (calphostin C). Although at variance with the conventional model of D2 receptor coupling (Stoof et al., 1992), this signaling linkage aligns with several recent lines of study. For example, D2 receptors have been shown to preferentially couple via Gβγ-subunits associated with Goα to activate PLC isoforms (Hernandez-Lopez et al., 2000; Jiang et al., 2001). PKC activation is a well characterized consequence of PLC stimulation, resulting from the generation of diacylglycerol and mobilization of Ca2+ from inositol triphosphate-sensitive intracellular stores. This linkage is also consistent with the recent demonstration that phosphorylation of Na+ channels enhances entry into a slow-inactivated state (Carr et al., 2003).
Is the reduction of Na+ channel currents sufficient to explain D2 receptor-mediated changes in pacemaking?
Although it is clear that activation of D2 receptors led to a reduction of Na+ channel currents, the modulation of transient Na+ currents was modest. Although within the range reported in previous studies (Cantrell and Catterall, 2001), peak transient current at -55 mV was reduced by only ∼15-20% by D2 receptor agonists or PKC activators. The maximal persistent Na+ conductance was more substantially reduced (∼30-40%) by the same manipulations, but is this sufficient to account for the reduction in pacemaking seen in tissue slices? There are two lines of evidence that suggest so. First, application of the specific Na+ channel blocker TTX at an adequate concentration to block only 10% of the Na+ channel population (with full penetration of the tissue) reduced pacemaking by >20% on average. Because pacemaking is not dependent on synaptic input (Bennett and Wilson, 1999), this reduction is directly attributable to the blockade of the Na+ channels underlying pacemaking. Second, computer simulation of the pacemaking process in cholinergic interneurons revealed that a modest enhancement of Na+ channel slow inactivation, sufficient to bring about reductions in transient and persistent current, such as those seen experimentally, had profound effects on pacemaking. At intermediate channel densities that gave discharge rates such as those seen in vivo (3-6 Hz), reduction of the transient Na+ channel current by 15% and the persistent current by ∼30% brought about a 30-50% reduction in spiking rate. The simulations also showed that the relationship between channel inactivation and discharge rate was monotonic over a wide range, providing a potential readout of the intensity of D2 receptor stimulation. The sensitivity of pacemaking to Na+ channel modulation is not surprising, given that the slope of the current-voltage relationship in cholinergic interneurons is very shallow in the voltage range in which Na+ channels begin to open (Bennett et al., 2000), allowing small changes in current to significantly change the trajectory of the membrane potential to the next spike. This appears to be a common feature of Na+ channel-dependent pacemakers (Raman and Bean, 1999; Taddese and Bean, 2002; Do and Bean, 2003).
There are undoubtedly collateral mechanisms via which dopamine might act directly to inhibit the activity of cholinergic interneurons. The best characterized of these is the D5 receptor-mediated augmentation of spike afterhyperpolarization, which should slow discharge (Bennett and Wilson, 1998). This modulation could account, in part, for conjectured involvement of D1-class receptors in generation of the pause (Watanabe and Kimura, 1998). These alterations in intrinsic properties governing autonomous spiking complement the direct D2 receptor-mediated inhibition of acetylcholine release (Lehmann and Langer, 1983; Bertorelli et al., 1992; Stoof et al., 1992; DeBoer et al., 1993; Di Chiara et al., 1994; Yan et al., 1997), further reducing the impact of cholinergic interneurons on striatal circuitry.
Although dopamine acts directly on interneurons to reduce autonomous discharge, it may also sculpt synaptic inputs to cholinergic interneurons that are of significance in vivo, contributing to the generation of pauses. D1/D5 dopamine receptor activation enhances the activity of GABAergic interneurons (Bracci et al., 2002) and potentiates interneuronal extrasynaptic GABAA receptor function (Yan and Surmeier, 1997). D1/D5 receptor-dependent long-term potentiation of this input (Suzuki et al., 2001) could be particularly important in enabling pause generation with overtraining in associative learning paradigms when the pause in interneuron activity appears to become independent of striatal dopamine release. The facilitation of this synaptic linkage also could help explain the role of thalamic structures in the regulation of cholinergic interneuron activity (Matsumoto et al., 2001).
Conclusions
The studies reported here show that D2 dopamine receptor activation reduces evoked activity and autonomous pacemaking in identified cholinergic interneurons. These studies establish a mechanistic framework in which the pause of cholinergic interneurons in associative and instrumental learning paradigms can be understood. They also provide a complement to our existing understanding of how dopamine depletion leads to an elevation in striatal cholinergic tone and dysfunction in Parkinson's disease.
Footnotes
This work was supported by National Institutes of Health Grants NS34696 and NS47085 (D.J.S.). We thank Karen Burnell for expert technical support and Dr. Heidi Hamm for βARK-Cp.
Correspondence should be addressed to D. James Surmeier, Department of Physiology, Northwestern University Institute for Neuroscience, 320 East Superior Street, Searle 5-447, Feinberg School of Medicine, Northwestern University, Chicago, IL 60611. E-mail: j-surmeier@northwestern.edu.
N. Maurice's present address: Institut National de la Santé et de la Recherche Médicale, U.114, Chaire de Neuropharmacologie, Collège de France, 11, place Marcelin Berthelot, 75231 Paris Cedex 05, France.
Copyright © 2004 Society for Neuroscience 0270-6474/04/2410289-13$15.00/0
References
- Aosaki T, Graybiel AM, Kimura M (1994) Effect of the nigrostriatal dopamine system on acquired neural responses in the striatum of behaving monkeys. Science 265: 412-415. [DOI] [PubMed] [Google Scholar]
- Bennett BD, Wilson CJ (1998) Synaptic regulation of action potential timing in neostriatal cholinergic interneurons. J Neurosci 18: 8539-8549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennett BD, Wilson CJ (1999) Spontaneous activity of neostriatal cholinergic interneurons in vitro J Neurosci 19: 5586-5596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennett BD, Callaway JC, Wilson CJ (2000) Intrinsic membrane properties underlying spontaneous tonic firing in neostriatal cholinergic interneurons. J Neurosci 20: 8493-8503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bergson C, Mrzljak L, Smiley JF, Pappy M, Levenson R, Goldman-Rakic PS (1995) Regional, cellular and subcellular variation in the distribution of D1 and D5 dopamine receptors in primate brain. J Neurosci 15: 7821-7836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bertorelli R, Zambelli M, Di Chiara G, Consolo S (1992) Dopamine depletion preferentially impairs D1-over D2-receptor regulation of striatal in vivo acetylcholine release. J Neurochem 59: 353-357. [DOI] [PubMed] [Google Scholar]
- Bracci E, Centonze D, Bernardi G, Calabresi P (2002) Dopamine excites fast-spiking interneurons in the striatum. J Neurophysiol 87: 2190-2194. [DOI] [PubMed] [Google Scholar]
- Cantrell AR, Catterall WA (2001) Neuromodulation of Na+ channels: an unexpected form of cellular plasticity. Nat Rev Neurosci 2: 397-407. [DOI] [PubMed] [Google Scholar]
- Carr DB, Cantrell AR, Day M, Catterall WA, Scheuer T, Surmeier DJ (2003) Transmitter modulation of sodium channel availability endows neurons with a novel form of cellular plasticity. Neuron 39: 793-806. [DOI] [PubMed] [Google Scholar]
- DeBoer P, Abercrombie ED, Heeringa M, Westerink BH (1993) Differential effect of systemic administration of bromocriptine and l-dopa on the release of acetylcholine from striatum of intact and 6-OHDA-treated rats. Brain Res 608: 198-203. [DOI] [PubMed] [Google Scholar]
- Di Chiara G, Morelli M, Consolo S (1994) Modulatory functions of neurotransmitters in the striatum: ACh/dopamine/NMDA interactions. Trends Neurosci 17: 228-233. [DOI] [PubMed] [Google Scholar]
- Do MT, Bean BP (2003) Subthreshold sodium currents and pacemaking of subthalamic neurons: modulation by slow inactivation. Neuron 39: 109-120. [DOI] [PubMed] [Google Scholar]
- Goldin AL (1999) Diversity of mammalian voltage-gated sodium channels. Ann NY Acad Sci 868: 38-50. [DOI] [PubMed] [Google Scholar]
- Goldin AL (2001) Resurgence of sodium channel research. Annu Rev Physiol 63: 871-894. [DOI] [PubMed] [Google Scholar]
- Graybiel AM, Aosaki T, Flaherty AW, Kimura M (1994) The basal ganglia and adaptive motor control. Science 265: 1826-1831. [DOI] [PubMed] [Google Scholar]
- Hernandez-Lopez S, Tkatch T, Perez-Garci E, Galarraga E, Bargas J, Hamm H, Surmeier DJ (2000) D2 dopamine receptors in striatal medium spiny neurons reduce L-type Ca2+ currents and excitability via a novel PLC[β]1-IP3-calcineurin-signaling cascade. J Neurosci 20: 8987-8995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hille B (2001) Gating: voltage sensing and inactivation. In: Ion channels of excitable membranes, pp 603-634. Sunderland, MA: Sinauer.
- Hines ML, Carnevale NT (2001) NEURON: a tool for neuroscientists. The Neuroscientist 7: 123-135. [DOI] [PubMed] [Google Scholar]
- Hornykiewicz O (1976) Neurohumoral interactions and basal ganglia function and dysfunction. Res Publ Assoc Res Nerv Ment Dis 55: 269-280. [PubMed] [Google Scholar]
- Isom LL, De Jongh KS, Patton DE, Reber BF, Offord J, Charbonneau H, Walsh K, Goldin AL, Catterall WA (1992) Primary structure and functional expression of the beta 1 subunit of the rat brain sodium channel. Science 256: 839-842. [DOI] [PubMed] [Google Scholar]
- Isom LL, Ragsdale DS, De Jongh KS, Westenbroek RE, Reber BF, Scheuer T, Catterall WA (1995) Structure and function of the beta 2 subunit of brain sodium channels, a transmembrane glycoprotein with a CAM motif. Cell 83: 433-442. [DOI] [PubMed] [Google Scholar]
- Jiang M, Spicher K, Boulay G, Wang Y, Birnbaumer L (2001) Most central nervous system D2 dopamine receptors are coupled to their effectors by Go. Proc Natl Acad Sci USA 98: 3577-3582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khaliq ZM, Gouwens NW, Raman IM (2003) The contribution of resurgent sodium current to high-frequency firing in Purkinje neurons: an experimental and modeling study. J Neurosci 23: 4899-4912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koch WJ, Hawes BE, Inglese J, Luttrell LM, Lefkowitz RJ (1994) Cellular expression of the carboxyl terminus of a G protein-coupled receptor kinase attenuates G beta gamma-mediated signaling. J Biol Chem 269: 6193-6197. [PubMed] [Google Scholar]
- Lazarewicz MT, Migliore M, Ascoli GA (2002) A new bursting model of CA3 pyramidal cell physiology suggests multiple locations for spike initiation. Biosystems 67: 129-137. [DOI] [PubMed] [Google Scholar]
- Lehmann J, Langer SZ (1983) The striatal cholinergic interneuron: synaptic target of dopaminergic terminals? Neuroscience 10: 1105-1120. [DOI] [PubMed] [Google Scholar]
- Levey AI, Hersch SM, Rye DB, Sunahara RK, Niznik HB, Kitt CA, Price DL, Maggio R, Brann MR, Ciliax BJ (1993) Localization of D1 and D2 dopamine receptors in brain with subtype-specific antibodies. Proc Natl Acad Sci USA 90: 8861-8865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magistretti J, Alonso A (1999) Biophysical properties and slow voltage-dependent inactivation of a sustained sodium current in entorhinal cortex layer-II principal neurons: a whole-cell and single-channel study. J Gen Physiol 114: 491-509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsumoto N, Minamimoto T, Graybiel AM, Kimura M (2001) Neurons in the thalamic CM-Pf complex supply striatal neurons with information about behaviorally significant sensory events. J Neurophysiol 85: 960-976. [DOI] [PubMed] [Google Scholar]
- Maurice N, Tkatch T, Meisler M, Sprunger LK, Surmeier DJ (2001) D1/D5 dopamine receptor activation differentially modulates rapidly inactivating and persistent sodium currents in prefrontal cortex pyramidal neurons. J Neurosci 21: 2268-2277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raman IM, Bean BP (1999) Ionic currents underlying spontaneous action potentials in isolated cerebellar Purkinje neurons. J Neurosci 19: 1663-1674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raman IM, Bean BP (2001) Inactivation and recovery of sodium currents in cerebellar Purkinje neurons: evidence for two mechanisms. Biophys J 80: 729-737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raman IM, Sprunger LK, Meisler MH, Bean BP (1997) Altered subthreshold sodium currents and disrupted firing patterns in Purkinje neurons of Scn8a mutant mice. Neuron 19: 881-891. [DOI] [PubMed] [Google Scholar]
- Raz A, Feingold A, Zelanskaya V, Vaadia E, Bergman H (1996) Neuronal synchronization of tonically active neurons in the striatum of normal and parkinsonian primates. J Neurophysiol 76: 2083-2088. [DOI] [PubMed] [Google Scholar]
- Schultz W (2002) Getting formal with dopamine and reward. Neuron 36: 241-263. [DOI] [PubMed] [Google Scholar]
- Smith Y, Bennett BD, Bolam JP, Parent A, Sadikot AF (1994) Synaptic relationships between dopaminergic afferents and cortical or thalamic input in the sensorimotor territory of the striatum in monkey. J Comp Neurol 344: 1-19. [DOI] [PubMed] [Google Scholar]
- Song WJ, Tkatch T, Baranauskas G, Ichinohe N, Kitai ST, Surmeier DJ (1998) Somatodendritic depolarization-activated potassium currents in rat neostriatal cholinergic interneurons are predominantly of the A type and attributable to coexpression of Kv4.2 and Kv4.1 subunits. J Neurosci 18: 3124-3137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stoof JC, Drukarch B, de Boer P, Westerink BH, Groenewegen HJ (1992) Regulation of the activity of striatal cholinergic neurons by dopamine. Neuroscience 47: 755-770. [DOI] [PubMed] [Google Scholar]
- Suzuki T, Miura M, Nishimura K, Aosaki T (2001) Dopamine-dependent synaptic plasticity in the striatal cholinergic interneurons. J Neurosci 21: 6492-6501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taddese A, Bean BP (2002) Subthreshold sodium current from rapidly inactivating sodium channels drives spontaneous firing of tuberomammillary neurons. Neuron 33: 587-600. [DOI] [PubMed] [Google Scholar]
- Wang J, Chen S, Siegelbaum SA (2001) Regulation of hyperpolarization-activated HCN channel gating and cAMP modulation due to interactions of COOH terminus and core transmembrane regions. J Gen Physiol 118: 237-250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe K, Kimura M (1998) Dopamine receptor-mediated mechanisms involved in the expression of learned activity of primate striatal neurons. J Neurophysiol 79: 2568-2580. [DOI] [PubMed] [Google Scholar]
- Yan Z, Surmeier DJ (1997) D5 dopamine receptors enhance Zn2+-sensitive GABA(A) currents in striatal cholinergic interneurons through a PKA/PP1 cascade. Neuron 19: 1115-1126. [DOI] [PubMed] [Google Scholar]
- Yan Z, Song WJ, Surmeier J (1997) D2 dopamine receptors reduce N-type Ca2+ currents in rat neostriatal cholinergic interneurons through a membrane-delimited, protein-kinase-C-insensitive pathway. J Neurophysiol 77: 1003-1015. [DOI] [PubMed] [Google Scholar]