

Abstract
Adaptive, long-term alterations of excitability have been reported in dendrites and presynaptic terminals but not along axons. Persistent enhancement of axonal excitability has been described in proximal nerve stumps at sites of nerve section in mammals, but this hyperexcitability is considered a pathological derangement important only as a cause of neuropathic pain. Identified neurons in Aplysia were used to test the hypothesis that either axonal injury or the focal depolarization that accompanies axonal injury can trigger a local decrease in action potential threshold [long-term hyperexcitability (LTH)] having memory-like properties. Nociceptive tail sensory neurons and a giant secretomotor neuron, R2, exhibited localized axonal LTH lasting 24 hr after a crush of the nerve or connective that severed the tested axons. Axons of tail sensory neurons and tail motor neurons, but not R2, displayed similar localized LTH after peripheral depolarization produced by 2 min exposure to elevated extracellular [K+]. Neither the induction nor expression of either form of LTH was blocked by saline containing 1% normal [Ca2+] during treatment or testing. However, both were prevented by local application of the protein synthesis inhibitors anisomycin or rapamycin. The features of (1) long-lasting alteration by localized depolarization, (2) restriction of alterations to intensely depolarized regions, and (3) dependence of the alterations on local, rapamycin-sensitive protein synthesis are shared with synaptic mechanisms considered important for memory formation. This commonality suggests that relatively simple, accessible axons may offer an opportunity to define fundamental plasticity mechanisms that were important in the evolution of memory.
Keywords: hyperexcitability, memory, action potential threshold, sensitization, protein synthesis, axotomy, neuropathic pain
Introduction
Synapses between neurons display persistent, activity-dependent alterations in chemical transmission that are thought to be mechanisms for memory (S. J. Martin et al., 2000; Kandel, 2001). Similar synaptic alterations contribute to information storage and behavioral plasticity not only in regions of the mammalian brain specialized for memory function but also in many other neural structures, including invertebrate nervous systems (Krasne and Glanzman, 1995; Burrell and Sahley, 2001; Waddell and Quinn, 2001; Roberts and Glanzman, 2003) and the vertebrate spinal cord, in which they contribute to phenomena such as nociceptive plasticity (Ji et al., 2003). Although synaptic alterations have attracted far more attention, activity-dependent or learning-induced hyperexcitability has now been shown in somata, dendrites, and presynaptic terminals close to altered synapses in many mammalian and invertebrate systems (for review, see Daoudal and Debanne, 2003; Zhang and Linden, 2003). Axons, however, have always been viewed as simple conducting cables lacking any memory capability, and we are unaware of any reports of adaptive, long-term enhancement of excitability in intact axons.
Among the features that appear important for memory storage in various synaptic models are long-lasting alteration of synapses by localized depolarization, restriction of the alterations to intensely depolarized synaptic regions, and dependence of the alterations on local protein synthesis (K. C. Martin et al., 2000; S. J. Martin et al., 2000; Kandel, 2001; Jiang and Schuman, 2002). If some or all of these features can be found in axons, then these much simpler structures might be used to identify fundamental mechanisms that may have important, previously unrecognized axonal functions and might also contribute to memory storage in complex synaptic regions. Under normal conditions, axons have not been reported to exhibit long-term, adaptive increases in excitability. However, mammalian sensory axons can display persistent hyperexcitability at sites of nerve injury (Zimmermann, 2001) and inflammation (Tal and Eliav, 1996; Eliav et al., 2001; Bove et al., 2003).
Using identified neurons in Aplysia, we show that axons of sensory and motor neurons exhibit long-term hyperexcitability (LTH) induced by either injury or transient, localized depolarization of peripheral nerves in the absence of injury. The surprising finding of LTH in peripheral axons that depends on local, rapamycin-sensitive protein synthesis suggests that a common basic mechanism may contribute to at least two different functions: adaptive regulation of conduction in peripheral axons, and memory formation in central synaptic regions. Mechanisms and functions of axonal LTH may provide clues about the early evolution of fundamental mechanisms of memory.
Materials and Methods
General. Aplysia californica (50-300 gm) were anesthetized by injecting isotonic MgCl2 solution (337 mm). The pedal-pleural and abdominal ganglion were excised and placed in chambers with an attached nerve or connective threaded through a series of smaller, 6 mm wells connected by 2 mm slots (Fig. 1) (see also Fig. 4). Ganglia were surgically desheathed in a 1:1 mixture of artificial seawater (ASW) and isotonic MgCl2. In most experiments, the nerve wells and ganglion well were filled with buffered low-Ca saline (1% normal [Ca2+]) containing (in mm): 460 NaCl, 0.1 CaCl2, 10 KCl, 66 MgCl2, and 10 Tris buffer, pH 7.6. In some experiments, buffered ASW was used, containing (in mm): 460 NaCl, 11 CaCl2, 10 KCl, 55 MgCl2, and 10 Tris buffer, pH 7.6. After pretests and treatment on the first day, excised ganglia and nerves were stored overnight at 16°C in a 1:1 mixture of L15 medium and filtered hemolymph or in L15 medium alone. Reagents and drugs (glutaraldehyde, anisomycin, rapamycin, and emetine) were obtained from Sigma (St. Louis, MO).
Figure 1.

Preparations used to examine axonal LTH. p9 or RPAC was threaded through a series of wells. Only the wells containing the stimulated segments of the nerves are shown. The cut nerve ends were always at least 1 cm from the nearest test well. A, Pleural-pedal ganglia with short-segment stimulation configuration illustrated on nerve p9. Intracellular electrodes were used to record evoked action potentials conducted to the somata of tail sensory neurons (SNs) in the pleural ganglion or tail motor neurons (MNs) in the pedal ganglion. The sharp arrow indicates the slot between the wells containing the 2-mm-long tube used to focus test currents flowing from one well to the other on a short nerve segment. The blunt arrow indicates the site of nerve crush, just distal to this test slot. During depolarizing treatment, high-K/low-Ca saline filled the well (6 mm diameter) on each side of the test slot, so that ∼14 mm of the nerve was exposed to the depolarizing solution. Diffusion to other parts of the nerve was prevented by sealing the remaining slots in these two wells with silicone grease. B, Abdominal ganglion with long-segment stimulation configuration illustrated on the RPAC. Evoked spikes were recorded in the soma of giant secretomotor neuron R2 in the abdominal ganglion while applying test currents to a 6 mm segment of the RPAC within a single well, which contained electrodes on each side of the RPAC. In this case, silicone grease sealed both slots of the stimulated well. Note that, in separate experiments, both the short- and long-segment stimulation configurations were used in experiments on tail sensory neurons, tail motor neurons, and R2.
Figure 4.
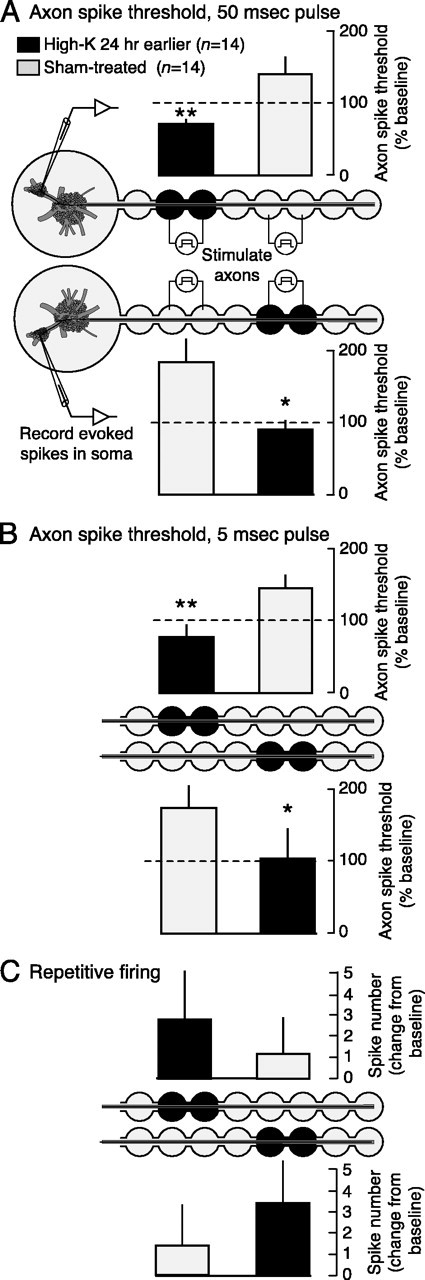
Sensory neuron axons display localized LTH after high-K treatment of a peripheral nerve segment. A, Localization of axonal LTH to the site of depolarization. Test shocks (50 msec) were delivered between the indicated electrodes set up on nerve p9 in the dual-well configuration for stimulating short nerve segments (∼2 mm) between pairs of wells. Tests were delivered before (pretest) and 24 hr after high-K or sham treatment in the test wells. Asterisks indicate significant differences between ipsilateral test segments revealed by paired t tests. Similar differences were also found between contralateral segments (see Results). B, Test pulses (5 msec) also reveal localized axonal LTH. C, Repetitive firing responses are highly variable after high-K treatment and do not differ significantly from responses in control axons. In B and C, the preparations were the same as in A, but the ganglia and electrodes have been omitted from the figure.
Test stimulation. Extracellular electrical test stimuli were applied to segments of the posterior pedal nerve (p9) or the right pleural-abdominal connective (RPAC) while using intracellular electrodes to record action potentials conducted to the soma of a tail sensory neuron in the pleural ganglion (Walters et al., 1983, 2004), a tail motor neuron in the pedal ganglion (Walters et al., 1983), or neuron R2 in the abdominal ganglion (Rayport et al., 1983). Two types of extracellular electrode configuration were used. For localized stimulation of a short segment, test currents were passed through a narrow slot 2 mm long between contiguous wells (Fig. 1 A). Current density was increased by longitudinally bisecting a 2 mm length of tubing, placing the resulting “Quonset hut” over the nerve, and covering the tubing with silicone grease. The tubing diameter was slightly greater than that of the nerve so that fluid and gas exchange were not impeded. In some studies, test stimuli were applied at two different sites on the same nerve that were separated by several intervening wells (see Fig. 4). For stimulation of a longer segment (∼ 6 mm), test currents were delivered through 1 mm slots across a single 6 mm well, aligned perpendicular to the length of the nerve (Fig. 1 B). This configuration allowed more consistent stimulation of the region around an injury site. Axon spike thresholds were tested with ascending series of 5 and 50 msec pulses for tail sensory neurons and tail motor neurons and 10 and 100 msec pulses for R2 (because of its longer time constant). Repetitive firing was tested with 1 sec pulses using a test current one or two times the magnitude of the 50 or 100 msec threshold current (approximately twice rheobase). All pretests and 24 hr tests were made with the nerve and ganglia bathed in low-Ca saline (introduced at least 10 min before the test). Intracellular recordings were made from neuronal somata at room temperature (19-22°C) using standard methods (Walters et al., 1983, 2004).
Nerve crush. Nerve p9 or the RPAC was crushed using fine forceps 0.5-4 mm distal to the test slot in the configuration shown in Figure 1 A or in the middle of the test well in the configuration shown in Figure 1 B (Gunstream et al., 1995). The distance from the end of the slot to the crush site was measured after the 24 hr test. Although nerve crush does not sever the nerve, it transects all the axons in the nerve, and regeneration of these axons takes weeks (Steffensen et al., 1995). Consequently, we did not apply test stimuli distal to the crush site. Crush was made in either normal saline or low-Ca saline (with pretests and 24 hr tests always performed in low-Ca saline). Exposure to protein synthesis inhibitors was interrupted for 5 min (30 min into the 3 hr exposure) while the nerve was crushed in normal saline. In the corresponding uncrushed sham controls, vehicle was temporarily replaced with normal saline for 5 min.
High-K treatment. Nerve segments were depolarized by rapidly withdrawing low-Ca saline from silicone-grease-sealed test wells (Fig. 1) and replacing it immediately with high-K/low-Ca saline, containing (in mm): 0 NaCl, 0.1 CaCl2, 470 KCl, 61 MgCl2, and 10 Tris buffer, pH 7.6. Note that this saline also contained 1% normal [Ca2+]. Intracellular recordings from sensory neuron somata and the giant axon of R2 indicated that membrane potential approached 0 mV in high-K/low-Ca saline. Except when indicated, all changes of solution in nerve wells (i.e., between high-K/low-Ca and low-Ca salines, or between low-Ca saline and culture medium) caused the nerve sheath to be exposed to air for ∼1 sec. To control for possible effects of mechanical agitation or air exposure during solution changes, sham controls received identical treatment, except that low-Ca saline was reintroduced rather than introducing high-K/low-Ca saline. Except when noted, the high-K/low-Ca remained for 2 min before removal and replacement with low-Ca saline.
Data analysis. Data are reported as means ± SEM, except for Figure 7E (see below). Comparisons between two different segments within a nerve or on opposite sides of the same animal used paired t tests. Comparisons across animals used one-way ANOVA, followed by Dunnett's post hoc tests. In each figure, the primary group for comparison is the leftmost column within connected sets of columns. When a group contained subsets of data that were paired and subsets that were unpaired, separate paired t tests and ANOVA-Dunnett's tests were run on different subsets of data. Statistically significant differences are indicated in each figure (⋆p < 0.05; ⋆⋆p < 0.01; ⋆⋆⋆p < 0.001). In one experiment that revealed striking variability in response (see Fig. 7E), the total range of values is presented, with interquartile ranges and medians indicated, and a Mann-Whitney U test was used to compare two unpaired groups. In this case, the Mann-Whitney U test and an unpaired t test yielded similar p values (p < 0.1). Unless otherwise noted, the n values in all experiments indicate the numbers of ganglia tested in each condition. In studies on sensory neurons, 2-10 cells were sampled and averaged to yield a single data point per ganglion. For tail motor neurons, one to three cells were averaged per ganglion. R2 data came from the single cell in each animal.
Figure 7.

Secretomotor neuron R2 displays LTH of its giant axon after nerve crush but not high-K treatment. A, Axonal LTH (monitored as a decrease in spike threshold during 100 msec test pulses) is not observed 24 hr after either 2 or 4 min depolarization of a segment of the RPAC with high-K saline containing 1% normal [Ca2+]. B, LTH of the axon of R2 is produced by crushing the RPAC in the low-Ca solution. C, Crushing the RPAC in normal saline induces LTH of the axon of R2 that requires local protein synthesis. All tests were performed in low-Ca saline. Anisomycin (10 μm; Aniso) or rapamycin (20 nm; Rapa) blocked crush-induced LTH. Asterisks indicate differences relative to the crush (Cr) group. Anisomycin and rapamycin also significantly increased spike threshold compared with the thresholds in uncrushed (Uncr), sham-treated axons (see Results). D, Example of repetitive firing recorded in the soma of R2 during weak 1 sec test pulses (one times the 100 msec pretest threshold) in low-Ca saline before and 1 d after RPAC crush in the test well. E, Repetitive firing in axons expressing LTH (as shown by decreased axonal spike threshold) is highly variable. Each column shows the entire range of repetitive firing responses for each group before and 24 hr after crush of the RPAC in the test well. The top of each box indicates the 75th quartile, the bottom of the box indicates the 25th quartile, and the horizontal line in the middle is the median response. The ends of the vertical lines projecting above and below each box show the maximum and minimum responses in each group.
Results
Saline containing 1% normal [Ca2+] blocks nerve contraction and modulatory effects on sensory neurons produced by nerve shock
To test the excitability of sensory neuron axons, it was necessary to apply repeated and prolonged test stimuli to a peripheral nerve while recording action potentials conducted to the cell soma in the CNS (Fig. 1). However, nerve shock can evoke contractile responses of the nerve (Umitsu et al., 1987) that might change effective test current density, and it can alter the number of action potentials conducted to the central soma by causing the release of neuromodulators in the CNS (Clatworthy and Walters, 1993b) and perhaps the nerve. Because contraction of smooth muscle cells and exocytosis of neuroactive substances are often highly dependent on extracellular Ca2+, we attempted to minimize these complications by delivering nerve test stimuli in low-Ca saline containing 1% normal extracellular [Ca2+]. High-magnification microscopic examination indicated that evoked movements of the nerve, which in normal saline were quite prominent, were completely blocked by this low-Ca saline (n = 4 nerves). Using strong stimulation of nerve p9 as a test stimulus, we also found that low-Ca saline blocked the increase in excitability of the sensory neuron soma observed 1-2 min after p9 shock (n = 10; p = 0.01; paired t test) (Fig. 2A). In addition, hyperpolarizing responses to p9 shock, which can block conduction of sensory neuron spikes (Clatworthy and Walters, 1993b), were completely blocked by low-Ca saline in every sensory neuron tested (n = 6) (Fig. 2B). The blockade of these modulatory effects might be attributable to block of Ca2+-dependent exocytosis. As one indicator of exocytosis, we examined excitatory and inhibitory synaptic responses in pedal motor neurons evoked by p9 shock. All evidence of postsynaptic potentials (PSPs) in these neurons was abolished by the low-Ca saline in all cells examined (n = 40) (Fig. 2C). Low-Ca saline also abolished all evidence of PSPs evoked by RPAC stimulation in giant neuron, R2 (data not shown; n = 65). We also tested synaptic responses in unidentified neurons of various sizes in the left and right pleural ganglia and found that PSPs were abolished in 16 of 17 cells tested. One cell exhibited an EPSP that, when tested in 1% normal extracellular [Ca2+], decreased to ∼5% of its normal amplitude (data not shown). This residual EPSP was then abolished in saline containing ∼0% [Ca2+], suggesting that it was a chemical rather than electrical synapse. Together, only 1 of 122 postsynaptic neurons tested showed any evidence of chemical transmission in low-Ca saline. These results show that the 1% [Ca2+] saline produces a nearly complete block of the release of neurotransmitters and neuromodulators in the nervous system, and it effectively blocks evoked contractions of the nerve.
Figure 2.
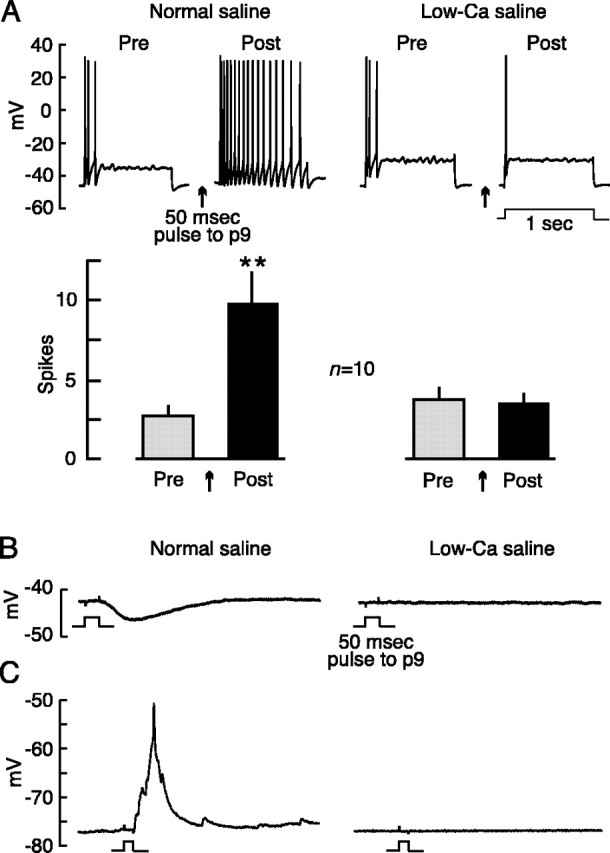
Low-Casaline containing 1% normal [Ca2+] blocks release of neuromodulators in the nervous system. A, Low-Ca saline eliminates hyperexcitability of the soma of pleural sensory neurons observed 1-2 min after p9 shock. Top, Example of responses from a single sensory neuron tested before (Pre) and 1 min after (Post) p9 shock in normal saline and in low-Ca saline. Bottom, Summary of soma excitability effects from 10 sensory neurons tested in both normal and low-Ca saline. **p < 0.05; paired t test. In this figure and the others, error bars indicate SEM (unless otherwise indicated). B, Low-Ca saline eliminates hyperpolarizing responses of pleural sensory neurons evoked by the 50 msec pulse to nerve p9. No evoked slow responses (hyperpolarizing or depolarizing) were detected in any sensory neurons tested in low-Ca saline (n = 6). C, Low-Ca saline (right column) eliminates detectable EPSPs in a pedal motor neuron evoked by a 50 msec pulse to nerve p9. No evoked EPSPs were detected in any motor neurons tested in low-Ca saline (n = 40).
LTH of sensory neuron axons is induced by nerve injury
To see whether long-term changes in axon excitability are induced near a site of nerve injury, we used nociceptive tail sensory neurons in the pleural ganglia. Action potentials (Fig. 3A) recorded intracellularly in the cell body were evoked by test pulses applied to nerve p9, which contains the axons of the tail sensory neurons (Walters et al., 1983, 2004). One of the two pleural-pedal ganglia with nerve p9 attached was used as a control for the other. Electrical test stimuli were delivered focally to short nerve segments (Fig. 1A) in low-Ca saline before and 24 hr after the nerve was crushed either 0.5-1.5 mm (“close”) or 2-4 mm (“far”) distal to the end of the test slot in normal saline. The crush procedure transected all the axons without severing the nerve (Steffensen et al., 1995). Twenty-four hours after nerve crush, repetitive firing increased in 5 of 7 cells tested with stimuli delivered close to the crush site (Fig. 3A), whereas only 5 of 12 uncrushed cells and 3 of 6 cells tested far from the crush site showed an increase in repetitive firing. The changes in repetitive firing showed great variability within each group, and the differences among the groups were not statistically significant. In contrast, action potential (spike) threshold was significantly decreased when the test was close to the crush site, although not when it was far from the crush site (F(2,19) = 4.30; p < 0.05; for 50 msec test pulse, close vs uncrushed, p = 0.039; far vs uncrushed, p = 0.326; Dunnett's tests) (Fig. 3B). Similar differences were found with 5 msec test pulses (data not shown).
Figure 3.

Sensory neuron axons display localized LTH after peripheral nerve injury, and this LTH depends on local protein synthesis. A, Responses recorded in the soma while testing axon spike threshold and repetitive firing before and 24 hr after crushing nerve p9. B, Nerve crush in normal saline significantly decreased axon spike threshold for 50 msec test pulses delivered close to (0.5-1.5 mm away) but not distant from (2-4 mm) the crushed (Cr) site. Asterisk indicates significant difference (Dunnett's test) relative to uncrushed (Uncr) control group. C, Nerve crush in low-Ca saline also decreased spike threshold. D, Crush-induced (in low-Ca saline) decrease in spike threshold was prevented by local application during the crush of protein synthesis inhibitors, anisomycin (10 μm; Aniso) or rapamycin (20 nm; Rapa). In both C and D, asterisks indicate comparisons with the crushed (Cr) group in C (Dunnett's tests).
We then asked whether axonal LTH could be induced if the nerve crush was made while the nerve segment was bathed in low-Ca saline. In this and subsequent studies on crush-induced plasticity, we used a nerve test configuration that stimulated a longer segment containing the crush site (Fig. 1B) to reduce variability attributable to inadvertent differences among preparations in the distance of the crush site from the test site. Crushing nerve p9 in low-Ca saline caused a long-term decrease in axonal spike threshold tested in low-Ca saline near the crush site (Fig. 3C). In this study, the uncrushed axons displayed a significant increase in spike threshold compared with their baseline responses 24 hr earlier (p = 0.04; paired t test; n = 10 sensory neurons in 5 animals). Increases in threshold were also found 24 hr after baseline tests in every other uncrushed or untreated control group in this report, although the increases in threshold were not always statistically significant (Fig. 3B). The pattern of increased thresholds in uncrushed sensory neuron axons 24 hr after the pretest indicates that some aspect of the long-term ex vivo preparation reduces axonal excitability. In contrast, the long-term decrease in spike threshold in injured nerve segments shows that nerve injury induces strong LTH in sensory axons near the injury site, and its induction in low-Ca saline suggests that the LTH induction does not depend on local or central release of neurotransmitters.
Injury-induced LTH of sensory axons requires local protein synthesis
A feature of long-term synaptic plasticity that is attracting increasing attention from memory researchers is a contribution of local protein synthesis at modifiable synapses (K. C. Martin et al., 2000; Jiang and Schuman, 2002). To see whether injury-induced LTH in sensory neuron axons depends on local synthesis, we applied a protein synthesis inhibitor to the nerve segment for 3 hr, beginning 30 min before nerve crush. Two inhibitors were used that have been shown to be effective in Aplysia: anisomycin (10 μm), an inhibitor of general protein synthesis in Aplysia neurons (Schwartz et al., 1971; Greenberg et al., 1987), and rapamycin (20 nm), which selectively blocks a component of protein synthesis (in Aplysia and mammals) whose initiation can be regulated through the 5′ untranslated region of mRNAs (Yanow et al., 1998; Khan et al., 2001). Rapamycin-sensitive protein synthesis is required for marking activated synapses and stabilizing synaptic growth after prolonged 5-HT treatment in Aplysia (Casadio et al., 1999). Injury-induced axonal LTH was blocked by local application of either anisomycin or rapamycin (comparisons of all unpaired subgroups, F(2,14) = 6.00, p = 0.013; crushed vs crushed plus anisomycin, p < 0.01; crushed vs crushed plus rapamycin, p < 0.05, Dunnett's tests; crushed vs uncrushed, p = 0.0002, paired t test) (Fig. 3D). Axons treated with either anisomycin or rapamycin showed significant increases in spike threshold relative to their own pretests (p < 0.05 in each case; paired t tests). Although these increases appeared greater than that produced in uncrushed control axons, the differences were not statistically significant in this study. We also found that application to the injury site of another protein synthesis inhibitor, emetine (100 μm), blocked crush-induced LTH in three of three animals treated.
High-K treatment induces localized axonal LTH
Our nerve crush procedure results in axonal transection at the crush site (Steffensen et al., 1995) and thus rupture of the axonal membrane. Axonal transection in vitro produces an abrupt depolarization, centered at the injury site, which can last minutes (Berdan et al., 1993; Spira et al., 1993; Ziv and Spira, 1993; Krause et al., 1994). We asked whether similar depolarization, in the absence of other effects of nerve injury, is sufficient to induce LTH of sensory neuron axons. To mimic transection-induced depolarization, the well on each side of a test slot (Fig. 1A), either ∼2 or ∼4 cm from the ganglion, was bathed for 2 min with saline containing elevated [K+] and 1% normal [Ca2+]. This treatment depolarized resting membrane potential to ∼0 mV and usually evoked firing recorded in the sensory neuron soma that lasted a few seconds (Lin et al., 2003). Firing ranged from 0 to 37 spikes, and lasted 60 sec in one case. No correlation was found between the number of spikes evoked during the high-K treatment and subsequent LTH. Excitability tests in low-Ca saline were delivered focally to the treated test slot as well as to the control test slot on the same nerve (Fig. 4). If the proximal site was treated, the corresponding site on the contralateral side was used as a control and the distal site on the contralateral side was treated. Thus, each treated site was compared with ipsilateral and contralateral controls.
Twenty-four hours after high-K treatment, planned comparisons using paired t tests showed that sensory axons in the treated segments displayed lower action potential thresholds to our standard 50 msec test pulses than did sham-treated axons in either the same or contralateral nerves (high-K proximal vs sham distal, p = 0.001; high-K proximal vs sham proximal, p = 0.009; high-K distal vs sham proximal, p = 0.025; high-K distal vs sham distal, p = 0.050; n = 14 for each group) (Fig. 4A). This experiment also confirmed that the high-K treatment of a proximal segment did not block subsequent conduction from the distal segment, which suggests that it did not injure the treated segment. To see whether the long duration of the test pulse (50 msec) was important for revealing LTH, we also tested with a brief, 5 msec test pulse, which revealed the same depression of spike threshold (high-K proximal vs sham distal, p = 0.005; high-K proximal vs sham proximal, p = 0.004; high-K distal vs sham proximal, p = 0.004; high-K distal vs sham distal, p = 0.016; n = 14 for each group) (Fig. 4B). No difference was found with either long or short test pulses between excitability of untreated nerves and untreated segments of treated nerves (data not shown). As found after nerve crush, the number of spikes during repetitive firing evoked by standard 1 sec test pulses did not show a statistically significant increase, although a tendency to increase was observed (Fig. 4C). Because effects on spike number were more variable than effects on spike threshold, and no difference was found between LTH measured by short and long test pulses, our primary measure of axonal LTH in the remainder of this report is a decrease in threshold current for evoking an axon spike with a long test pulse.
LTH induced by high-K treatment is attributable to depolarization and not to nerve contraction
Because LTH occurred when both the test stimuli and high-K treatment were applied in low-Ca saline that blocked observable contractions of the nerve, the decreases in spike threshold observed were unlikely to be caused by changes in current density secondary to movements of the nerve. Additional evidence that nerve contraction was not responsible for the decreases in spike threshold came from fixing the sheath with glutaraldehyde (0.5% for 30 sec) before the experiment. This pretreatment, like the low-Ca solution, blocked observable movements of the nerve. In addition, when applied alone to the entire preparation, both glutaraldehyde and low-Ca saline greatly increased the stability of intracellular impalements of tail sensory and motor neurons, further supporting the conclusion that these treatments prevented contractions. Glutaraldehyde pretreatment failed to prevent LTH (high-K, 60 ± 12%; sham, 177 ± 31%; p = 0.011; paired t test; n = 5).
Although high-K treatment strongly depolarized the neurons, it remained possible that other potential effects of this treatment were responsible for inducing LTH. To obtain additional evidence that depolarization itself can induce LTH, instead of high-K treatment, we substituted a 2 min current pulse applied across a test slot, using a level twice the 50 msec current for spike threshold. During this prolonged DC stimulus, the sensory neurons displayed an immediate, high-frequency burst of spikes that usually accommodated rapidly. However, in one case, firing persisted for 52 sec (producing 320 spikes in the soma). This depolarizing treatment of the nerve also caused a 24 hr reduction in axon spike threshold (shocked, 38 ± 9%; unshocked, 127 ± 24%; p = 0.039; paired t test; n = 4).
Depolarization-induced LTH of sensory axons requires local protein synthesis
Like LTH induced by nerve crush, high-K-induced LTH was prevented by pretreatment with either anisomycin or rapamycin (comparisons of all unpaired subgroups, F(4,93) = 12.36, p < 0.0001; high-K vs untreated, p < 0.01; high-K vs high-K plus anisomycin, p < 0.01; high-K vs untreated plus anisomycin, p < 0.05; high-K vs high-K plus rapamycin, p < 0.01, Dunnett's tests; high-K vs sham, p = 0.0001, paired t test; n = 22) (Fig. 5). Although the axon spike thresholds of the high-K plus anisomycin and high-K plus rapamycin groups appeared greater than those of the sham-treated and untreated groups, these differences were not statistically significant in this study. Application of anisomycin to untreated control nerves produced no significant long-term effects on axonal excitability. Neither protein synthesis inhibitor affected axonal excitability 5-20 min after high-K treatment (data not shown).
Figure 5.

LTH of sensory neuron axons induced by peripheral high-K treatment requires local protein synthesis. Local application of the protein synthesis inhibitors anisomycin (10 μm; Aniso) or rapamycin (20 nm; Rapa) blocked the 24 hr decrease in axonal spike threshold. Asterisks indicate statistically significant differences from the high-K-treated group. Untreated nerves did not receive the sham wash with ASW.
Tail motor neuron axons display LTH after high-K treatment
Is depolarization-induced axonal LTH unique to sensory neurons? We next examined axons of identified tail motor neurons, which, like tail sensory neurons, project their axons into peripheral nerve p9 (Walters et al., 1983) and display LTH in the cell body after axotomy (Walters et al., 1991; Gunstream et al., 1995; Ambron et al., 1996). Moreover, several studies indicate that motor neurons in Aplysia may be sites of short- and long-term memory storage (Trudeau and Castellucci, 1995; Zhu et al., 1997; Roberts and Glanzman, 2003). We found that tail motor neuron axons, like the sensory neuron axons, displayed localized axonal LTH 24 hr after high-K treatment, and this LTH was blocked by anisomycin (comparisons of all unpaired subgroups, F(3,22) = 7.70, p = 0.001; high-K vs untreated, p < 0.05; high-K vs high-K plus anisomycin, p < 0.01; high-K vs untreated plus anisomycin, p < 0.05, Dunnett's tests; high-K vs sham, p = 0.0002, paired t test) (Fig. 6). The axon spike thresholds of the high-K plus anisomycin group appeared greater than those of the sham-treated and untreated groups, but these differences were not statistically significant.
Figure 6.

Tail motor neuron axons display localized LTH after high-K treatment, which depends on local protein synthesis. During high-K treatment and all excitability tests, the nerve segment was bathed in low-Ca saline. Local application of anisomycin (10 μm; Aniso) blocked the 24 hr decrease in spike threshold of the motor axons. Asterisks indicate statistically significant differences from the high-K-treated group.
Giant axon of secretomotor neuron R2 displays LTH after connective crush but not after high-K treatment
R2, one of the largest cells known, is a secretomotor neuron that innervates mucus glands throughout the body (Rayport et al., 1983) and has a giant axon in the RPAC, which interconnects two central ganglia. High-K treatment (in 1% normal [Ca2+]) applied for either 2 or 4 min to the RPAC failed to alter axon excitability 24 hr later (Fig. 7A). This shows that localized depolarization is not sufficient to induce LTH in all axons of Aplysia. In contrast, crushing the connective in low-Ca solution did induce LTH of the axon of R2 (p = 0.0025; paired t test) (Fig. 7B). Crushing the connective in normal saline also caused a significant long-term decrease in spike threshold in the axon of R2, and this injury-induced LTH was prevented by local application of either anisomycin or rapamycin (F(5,37) = 12.23, p < 0.0001; crushed vs uncrushed sham, p < 0.05; crushed vs crushed plus anisomycin, p < 0.01; crushed vs uncrushed plus anisomycin, p < 0.05; crushed vs crushed plus rapamycin, p < 0.01; crushed vs un-crushed plus rapamycin, p < 0.01) (Fig. 7C). These results indicate that mechanisms similar to those underlying depolarization-induced LTH can be present in an axon and activated by injury, even if they are not activated by depolarization alone.
Interestingly, post hoc comparisons with Newman-Keuls tests revealed significant increases in axon spike threshold of the crushed plus anisomycin and crushed plus rapamycin groups compared with the uncrushed sham group (p < 0.01 in each case). Thus, nerve injury causes a long-term decrease in axon excitability if local protein synthesis is blocked at the time of injury. This decrease in excitability is probably superimposed on a separate decrease that often occurs with time in uncrushed and untreated axons in the ex vivo preparation (Figs. 3, 4, 5, 6).
Repetitive firing (Fig. 7D) evoked by 1 sec test pulses to the axon of R2 was highly variable after nerve crush when evoked in the axon of R2 by a weak test stimulus (a 1 sec pulse at one times the threshold current for evoking a spike with a 100 msec pulse in the pretest). Indeed, the number of evoked spikes observed 24 hr after crush ranged from 1 to 42 (Fig. 7E). Nonetheless, there was a trend for more spikes to be conducted to the soma of R2 from crushed axons than from axons of sham controls (p = 0.09; Mann-Whitney U test). In contrast, when a test current two times threshold was used, no differences between crushed and control axons were apparent. Thus, the axon of R2, like the other axons we examined, expresses LTH primarily as a decrease in spike threshold rather than as an increase in repetitive firing.
Discussion
Axonal LTH after nerve crush
Axonal LTH was expressed 1 d after nerve crush in tail sensory neurons and giant secretomotor neuron, R2. In each case, LTH was expressed reliably as a decrease in axonal spike threshold. We failed to find consistent increases in repetitive firing, although firing usually tended to increase (Figs. 3A, 7E). Because axonal spikes were monitored in the soma, far from the treated region, an apparent lack of effect on axonal spike number should be interpreted cautiously. For example, axonal LTH might increase depolarizing responses of the axon in the tested region to a level at which substantial inactivation of Na+ channels occurs, which could reduce firing in hyperexcitable axons. The observation that, after nerve crush, repetitive firing in the axon of R2 tended to increase when tested with weak but not strong depolarizing stimuli (Fig. 7E) is consistent with this possibility.
In mammalian somatic sensory neurons, hyperexcitability of axon stumps occurs within the neuroma that forms at a site of nerve transection (Devor and Govrin-Lippmann, 1983; Papir-Kricheli and Devor, 1988; Chen and Devor, 1998), and this hyperexcitability contributes to neuropathic pain (Zimmermann, 2001). Hyperexcitability of axon stumps has been considered to be a pathological (rather than adaptive) consequence of severing the axon, caused by accumulation of Na+ channels en route to sensory terminals but obstructed at the site of transection (Devor et al., 1993) or by changes in the relative expression of different Na+ channel isoforms (Waxman et al., 1999). Alterations of these and other conductances potentially underlying axonal LTH may be amenable to direct examination in the giant axon of R2 (Horn, 1978). The signals responsible for producing LTH in mammalian neuromas are not yet known, but inflammatory mediators, cytokines, and perhaps neurotransmitter release in the neuroma may contribute to electrophysiological and behavioral alterations after nerve section (Zimmermann, 2001).
Our finding that LTH was expressed in tests in low-Ca saline and our observation that this solution blocks chemical transmission suggest that release of signals by Ca2+-dependent exocytosis may not be required for the expression of axonal LTH in Aplysia (although such signals may contribute under some conditions). Because LTH was induced by nerve crush in low-Ca saline, the same is true for the induction of axonal LTH. The induction and expression of axonal LTH in low-Ca saline made it highly unlikely that the LTH was attributable to two potential artifacts of nerve crush: (1) contraction or movement of the tested nerves (Umitsu et al., 1987), or (2) alterations in spike conduction produced by central release of neuromodulators (Clatworthy and Walters, 1993a,b). LTH of the sensory neuron axons was restricted to a region <2 mm from the crush site (Fig. 3B). This suggests a link to signals closely associated with a site of injury, such as the localized depolarization that persists for minutes or longer after axonal transection (Spira et al., 1993; Fishman and Bittner, 2003). The induction of LTH in low-Ca saline does not mean that the induction mechanisms are independent of Ca2+. Even when reduced to 10-4 m, substantial concentration and electrical gradients remain to drive extracellular Ca2+ into the cell, and induction might also involve Ca2+ release from intracellular stores.
Depolarization-induced LTH of intact axon segments
Our demonstration of LTH of sensory and motor axons after treatment with either high-K saline or prolonged shock indicates that focal depolarization of an axon for 2 min is sufficient to induce localized LTH. This suggests that axonal depolarization acts as a primary signal for inducing local LTH during nerve injury. This report is the first to describe long-lasting hyperexcitability produced in intact axons by localized depolarization. Previous reports focused on decreases rather than increases in excitability, produced by either high-frequency activation of an axon (Debanne, 2004) or axon injury (Titmus and Faber, 1990). Although enhancement of action potential conduction (or reflection) at branch points or in neuronal processes prone to conduction failure has been described, reported examples of enhancement barely outlast the duration of axonal firing or neuromodulatory input responsible for the enhancement (Mar and Drapeau, 1996; Baccus et al., 2000; Evans et al., 2003). A long-term, localized decrease in spike threshold triggered by depolarization during axon injury should counteract the increased probability of conduction failure that occurs in an injured region, e.g., by impedance mismatch when spikes invade normal segments after propagating through axon segments narrowed by injury (Titmus and Faber, 1990) or through thin regenerating neurites (Steffensen et al., 1995). The observation of depolarization-induced LTH in intact axons shows that axonal LTH can be produced by mechanisms other than damming of Na+ channels at the proximal stump of a severed axon (Devor and Govrin-Lippmann, 1983; Devor et al., 1993). The distance of the treated nerve segment from the neuronal soma (2-4 cm) and the blockade of synaptic transmission in the ganglion during induction and testing of LTH, coupled with reported rates of axonal transport in Aplysia (∼1.5 mm/hr) (Ambron et al., 1992; Gunstream et al., 1995; Sung et al., 2004), suggests that the axonal LTH seen 24 hr after treatment does not depend on transport of retrograde signals that produce transcriptional or translational effects in the soma (Lin et al., 2003; Sung et al., 2004) and subsequent anterograde transport of plasticity-mediating molecules to the axonal test site.
Dependence of axonal LTH on local protein synthesis
Axonal LTH induced by both nerve crush and high-K treatment was abolished in sensory and motor axons by 3 hr local exposure to protein synthesis inhibitors, including rapamycin, which selectively blocks a regulated form of synthesis considered important for memory formation (Casadio et al., 1999; Khan et al., 2001; Tang et al., 2002). Interestingly, axon spike thresholds usually increased after crush or high-K treatment in the presence of the inhibitors (Figs. 3, 5, 6, 7), whereas thresholds of axons in un-injured or untreated nerves were unaffected by the inhibitors. This indicates that depolarization or injury would cause a long-lasting decrease in excitability if protein synthesis were absent at the time of treatment, suggesting that the LTH has compensatory as well as sensitizing functions in regulating excitability. The sites of protein synthesis required for LTH might be extra-axonal (e.g., in glia), but evidence in mammalian and invertebrate preparations has shown that protein synthesis can occur in adult axons (Giuditta et al., 2002), including axons of dissociated Aplysia sensory neurons (Martin et al., 1997). Nerve injury in mammals can induce local protein synthesis within ∼12 hr, which is independent of the cell soma (Tobias and Koenig, 1975a,b), and axonal regeneration can be slowed by inhibiting local protein synthesis (Edbladh et al., 1994; Gaete et al., 1998; Zheng et al., 2001). Intraaxonal protein synthesis might be important for both regeneration and adaptive hyperexcitability of axons.
Axonal LTH may represent a primitive memory mechanism
LTH of sensory neuron axons in Aplysia displays features implicated in memory storage in central synapses, including long-lasting alteration by localized depolarization, restriction of the alterations to intensely depolarized regions, and dependence of the alterations on local, rapamycin-sensitive protein synthesis (Casadio et al., 1999; K. C. Martin et al., 2000; S. J. Martin et al., 2000; Kandel, 2001; Jiang and Schuman, 2002; Tang et al., 2002). Thus, the present findings add to increasing evidence for a link between injury responses and memory mechanisms (Walters, 1991, 1994; Lin et al., 2003; Cohen-Armon et al., 2004). In the ancient, soft-bodied animals that were ancestral to existing animal phyla, peripheral axons and sensory terminals would have been highly vulnerable to injury, and local, long-lasting decreases in spike threshold could have helped to counteract the increased probability of conduction failure after trauma. Selection pressures for localized repair and long-term sensory compensation mechanisms after peripheral injury might have driven the early evolution of forms of plasticity, such as localized LTH, that could later be used in other regions for other purposes. For example, mechanisms of local protein synthesis involved in producing axonal LTH might also stabilize long-term alterations in “tagged” synapses during learning (Frey and Morris, 1997; Martin et al., 1997; Casadio et al., 1999; K. C. Martin et al., 2000; Jiang and Schuman, 2002; Steward and Worley, 2002).
Hyperexcitability itself might contribute to memory. Neuronal LTH was one of the first correlates of learning reported in invertebrates and vertebrates (Woollacott and Hoyle, 1977; Brons and Woody, 1980; Crow and Alkon, 1980; Disterhoft et al., 1986; Scholz and Byrne, 1987; Walters, 1987). Dendritic and somatic hyperexcitability have recently been correlated with behaviorally expressed learning or long-term potentiation (LTP) in numerous preparations (for review, see Daoudal and Debanne, 2003; Zhang and Linden, 2003). An intriguing finding is that LTP of synapses onto hippocampal neurons is accompanied by localized dendritic hyperexcitability, and both this hyperexcitability and LTP are blocked by NMDA receptor antagonists (Frick et al., 2004). It will be interesting to see whether localized dendritic hyperexcitability persists and involves local protein synthesis (Frey and Morris, 1997; Tang et al., 2002; Bradshaw et al., 2003). If LTH near mammalian synapses and LTH in Aplysia axons involve homologous mechanisms, then favorable axons (such as the giant axon of R2) can be used to define fundamental plasticity mechanisms that are difficult to isolate in complex synaptic regions specialized for memory formation.
Footnotes
This work was supported by National Institutes of Health Grant NS35979 (E.T.W.) and Training Grant T32-NS07467 (which provided partial support for R.M.S.W.). We thank D. Englot, whose finding of short-term axonal hyperexcitability induced by nerve depolarization led to this study, and C. Brou for technical assistance.
Correspondence should be addressed to Dr. Edgar T. Walters, Department of Integrative Biology and Pharmacology, University of Texas-Houston Medical School, 6431 Fannin Boulevard, MSB 4.116, Houston, TX 77030. E-mail: edgar.t.walters@uth.tmc.edu.
Copyright © 2004 Society for Neuroscience 0270-6474/04/2410393-09$15.00/0
References
- Ambron RT, Schmied R, Huang CC, Smedman M (1992) A signal sequence mediates the retrograde transport of proteins from the axon periphery to the cell body and then into the nucleus. J Neurosci 12: 2813-2818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ambron RT, Zhang X-P, Gunstream JD, Povelones M, Walters ET (1996) Intrinsic injury signals enhance growth, survival, and excitability of Aplysia neurons. J Neurosci 16: 7469-7477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baccus SA, Burrell BD, Sahley CL, Muller KJ (2000) Action potential reflection and failure at axon branch points cause stepwise changes in EPSPs in a neuron essential for learning. J Neurophysiol 83: 1693-1700. [DOI] [PubMed] [Google Scholar]
- Berdan RC, Easaw JC, Wang R (1993) Alterations in membrane potential after axotomy at different distances from the soma of an identified neuron and the effect of depolarization on neurite outgrowth and calcium channel expression. J Neurophysiol 69: 151-164. [DOI] [PubMed] [Google Scholar]
- Bove GM, Ransil BJ, Lin HC, Leem JG (2003) Inflammation induces ectopic mechanical sensitivity in axons of nociceptors innervating deep tissues. J Neurophysiol 90: 1949-1955. [DOI] [PubMed] [Google Scholar]
- Bradshaw KD, Emptage NJ, Bliss TV (2003) A role for dendritic protein synthesis in hippocampal late LTP. Eur J Neurosci 18: 3150-3152. [DOI] [PubMed] [Google Scholar]
- Brons JF, Woody CD (1980) Long-term changes in excitability of cortical neurons after Pavlovian conditioning and extinction. J Neurophysiol 44: 605-615. [DOI] [PubMed] [Google Scholar]
- Burrell BD, Sahley CL (2001) Learning in simple systems. Curr Opin Neurobiol 11: 757-764. [DOI] [PubMed] [Google Scholar]
- Casadio A, Martin KC, Giustetto M, Zhu H, Chen M, Bartsch D, Bailey CH, Kandel ER (1999) A transient, neuron-wide form of CREB-mediated long-term facilitation can be stabilized at specific synapses by local protein synthesis. Cell 99: 221-237. [DOI] [PubMed] [Google Scholar]
- Chen Y, Devor M (1998) Ectopic mechanosensitivity in injured sensory axons arises from the site of spontaneous electrogenesis. Eur J Pain 2: 165-178. [DOI] [PubMed] [Google Scholar]
- Clatworthy AL, Walters ET (1993a) Rapid amplification and facilitation of mechanosensory discharge in Aplysia by noxious stimulation. J Neurophysiol 70: 1181-1194. [DOI] [PubMed] [Google Scholar]
- Clatworthy AL, Walters ET (1993b) Activity-dependent depression of mechanosensory discharge and excitability in Aplysia J Neurophysiol 70: 1195-1209. [DOI] [PubMed] [Google Scholar]
- Cohen-Armon M, Visochek L, Katzoff A, Levitan D, Susswein AJ, Klein R, Valbrun M, Schwartz JH (2004) Long-term memory requires polyADP-ribosylation. Science 304: 1820-1822. [DOI] [PubMed] [Google Scholar]
- Crow TJ, Alkon DL (1980) Associative behavioral modification in Hermissenda: cellular correlates. Science 209: 412-414. [DOI] [PubMed] [Google Scholar]
- Daoudal G, Debanne D (2003) Long-term plasticity of intrinsic excitability: learning rules and mechanisms. Learn Mem 10: 456-465. [DOI] [PubMed] [Google Scholar]
- Debanne D (2004) Information processing in the axon. Nat Rev Neurosci 5: 304-316. [DOI] [PubMed] [Google Scholar]
- Devor M, Govrin-Lippmann R (1983) Axoplasmic transport block reduces ectopic impulse generation in injured peripheral nerves. Pain 16: 73-85. [DOI] [PubMed] [Google Scholar]
- Devor M, Govrin-Lippmann R, Angelides K (1993) Na+ channel immunolocalization in peripheral mammalian axons and changes following nerve injury and neuroma formation. J Neurosci 13: 1976-1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Disterhoft JF, Coulter DA, Alkon DL (1986) Conditioning-specific membrane changes of rabbit hippocampal neurons measured in vitro. Proc Natl Acad Sci USA 83: 2733-2737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edbladh M, Tonge D, Golding J, Ekstrom AR, Edstrom A (1994) Early regeneration in vitro of adult mouse sciatic axons is dependent on local protein synthesis but may not involve neurotrophins. Neurosci Lett 168: 37-40. [DOI] [PubMed] [Google Scholar]
- Eliav E, Benoliel R, Tal M (2001) Inflammation with no axonal damage of the rat saphenous nerve trunk induces ectopic discharge and mechanosensitivity in myelinated axons. Neurosci Lett 311: 49-52. [DOI] [PubMed] [Google Scholar]
- Evans CG, Jing J, Rosen SC, Cropper EC (2003) Regulation of spike initiation and propagation in an Aplysia sensory neuron: gating-in via central depolarization. J Neurosci 23: 2920-2931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fishman HM, Bittner GD (2003) Vesicle-mediated restoration of a plasmalemmal barrier in severed axons. News Physiol Sci 18: 115-118. [DOI] [PubMed] [Google Scholar]
- Frey U, Morris RG (1997) Synaptic tagging and long-term potentiation. Nature 385: 533-536. [DOI] [PubMed] [Google Scholar]
- Frick A, Magee J, Johnston D (2004) LTP is accompanied by an enhanced local excitability of pyramidal neuron dendrites. Nat Neurosci 7: 126-135. [DOI] [PubMed] [Google Scholar]
- Gaete J, Kameid G, Alvarez J (1998) Regenerating axons of the rat require a local source of proteins. Neurosci Lett 251: 197-200. [DOI] [PubMed] [Google Scholar]
- Giuditta A, Kaplan BB, van Minnen J, Alvarez J, Koenig E (2002) Axonal and presynaptic protein synthesis: new insights into the biology of the neuron. Trends Neurosci 25: 400-404. [DOI] [PubMed] [Google Scholar]
- Greenberg SM, Castellucci VF, Bayley H, Schwartz JH (1987) A molecular mechanism for long-term sensitization in Aplysia Nature 329: 62-65. [DOI] [PubMed] [Google Scholar]
- Gunstream JD, Castro GA, Walters ET (1995) Retrograde transport of plasticity signals in Aplysia sensory neurons following axonal injury. J Neurosci 15: 439-448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horn R (1978) Propagating calcium spikes in an axon of Aplysia J Physiol (Lond) 281: 513-534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ji RR, Kohno T, Moore KA, Woolf CJ (2003) Central sensitization and LTP: do pain and memory share similar mechanisms? Trends Neurosci 26: 696-705. [DOI] [PubMed] [Google Scholar]
- Jiang C, Schuman EM (2002) Regulation and function of local protein synthesis in neuronal dendrites. Trends Biochem Sci 27: 506-513. [DOI] [PubMed] [Google Scholar]
- Kandel ER (2001) The molecular biology of memory storage: a dialogue between genes and synapses. Science 294: 1030-1038. [DOI] [PubMed] [Google Scholar]
- Khan A, Pepio AM, Sossin WS (2001) Serotonin activates S6 kinase in a rapamycin-sensitive manner in Aplysia synaptosomes. J Neurosci 21: 382-391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krasne FB, Glanzman DL (1995) What we can learn from invertebrate learning. Annu Rev Psychol 46: 585-624. [Google Scholar]
- Krause TL, Fishman HM, Ballinger ML, Bittner GD (1994) Extent and mechanism of sealing in transected giant axons of squid and earthworms. J Neurosci 14: 6638-6651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin H, Bao J, Ju-Sung Y, Walters ET, Ambron RT (2003) Rapid electrical and delayed molecular signals regulate the serum response element after nerve injury: convergence of injury and learning signals. J Neurobiol 57: 204-220. [DOI] [PubMed] [Google Scholar]
- Mar A, Drapeau P (1996) Modulation of conduction block in leech mechanosensory neurons. J Neurosci 16: 4335-4343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin KC, Casadio A, Zhu H, Yaping E, Rose JC, Chen M, Bailey CH, Kandel ER (1997) Synapse-specific, long-term facilitation of Aplysia sensory to motor synapses: a function for local protein synthesis in memory storage. Cell 91: 927-938. [DOI] [PubMed] [Google Scholar]
- Martin KC, Barad M, Kandel ER (2000) Local protein synthesis and its role in synapse-specific plasticity. Curr Opin Neurobiol 10: 587-592. [DOI] [PubMed] [Google Scholar]
- Martin SJ, Grimwood PD, Morris RG (2000) Synaptic plasticity and memory: an evaluation of the hypothesis. Annu Rev Neurosci 23: 649-711. [DOI] [PubMed] [Google Scholar]
- Papir-Kricheli D, Devor M (1988) Abnormal impulse discharge in primary afferent axons injured in the peripheral versus the central nervous system. Somatosens Mot Res 6: 63-77. [DOI] [PubMed] [Google Scholar]
- Rayport SG, Ambron RT, Babiarz J (1983) Identified cholinergic neurons R2 and LPl1 control mucus release in Aplysia J Neurophysiol 49: 864-876. [DOI] [PubMed] [Google Scholar]
- Roberts AC, Glanzman DL (2003) Learning in Aplysia: looking at synaptic plasticity from both sides. Trends Neurosci 26: 662-670. [DOI] [PubMed] [Google Scholar]
- Scholz KP, Byrne JH (1987) Long-term sensitization in Aplysia: biophysical correlates in tail sensory neurons. Science 235: 685-687. [DOI] [PubMed] [Google Scholar]
- Schwartz JH, Castellucci VF, Kandel ER (1971) Functioning of identified neurons and synapses in abdominal ganglion of Aplysia in absence of protein synthesis. J Neurophysiol 34: 939-953. [DOI] [PubMed] [Google Scholar]
- Spira ME, Benbassat D, Dormann A (1993) Resealing of the proximal and distal cut ends of transected axons: electrophysiological and ultrastructural analysis. J Neurobiol 24: 300-316. [DOI] [PubMed] [Google Scholar]
- Steffensen I, Dulin MF, Walters ET, Morris CE (1995) Peripheral regeneration and central sprouting of sensory neurone axons in Aplysia californica following nerve injury. J Exp Biol 198: 2067-2078. [DOI] [PubMed] [Google Scholar]
- Steward O, Worley P (2002) Local synthesis of proteins at synaptic sites on dendrites: role in synaptic plasticity and memory consolidation? Neurobiol Learn Mem 78: 508-527. [DOI] [PubMed] [Google Scholar]
- Sung YJ, Walters ET, Ambron RT (2004) A neuronal isoform of protein kinase G couples mitogen-activated protein kinase nuclear import to axotomy-induced long-term hyperexcitability in Aplysia sensory neurons. J Neurosci 24: 7583-7595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tal M, Eliav E (1996) Abnormal discharge originates at the site of nerve injury in experimental constriction neuropathy (CCI) in the rat. Pain 64: 511-518. [DOI] [PubMed] [Google Scholar]
- Tang SJ, Reis G, Kang H, Gingras AC, Sonenberg N, Schuman EM (2002) A rapamycin-sensitive signaling pathway contributes to long-term synaptic plasticity in the hippocampus. Proc Natl Acad Sci USA 99: 467-472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Titmus MJ, Faber DS (1990) Axotomy-induced alterations in the electrophysiological characteristics of neurons. Prog Neurobiol 35: 1-51. [DOI] [PubMed] [Google Scholar]
- Tobias GS, Koenig E (1975a) Axonal protein synthesizing activity during the early outgrowth period following neurotomy. Exp Neurol 49: 221-234. [DOI] [PubMed] [Google Scholar]
- Tobias GS, Koenig E (1975b) Influence of nerve cell body and neurolemma cell on local axonal protein synthesis following neurotomy. Exp Neurol 49: 235-245. [DOI] [PubMed] [Google Scholar]
- Trudeau LE, Castellucci VF (1995) Postsynaptic modifications in long-term facilitation in Aplysia: upregulation of excitatory amino acid receptors. J Neurosci 15: 1275-1284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Umitsu Y, Matsumoto H, Koike H (1987) Active contraction of nerve bundle and identification of a nerve-contractor motoneuron in Aplysia J Neurophysiol 58: 1016-1034. [DOI] [PubMed] [Google Scholar]
- Waddell S, Quinn WG (2001) Flies, genes, and learning. Annu Rev Neurosci 24: 1283-1309. [DOI] [PubMed] [Google Scholar]
- Walters ET (1987) Multiple sensory neuronal correlates of site-specific sensitization in Aplysia J Neurosci 7: 408-417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walters ET (1991) A functional, cellular, and evolutionary model of nociceptive plasticity in Aplysia Biol Bull 180: 241-251. [DOI] [PubMed] [Google Scholar]
- Walters ET (1994) Injury-related behavior and neuronal plasticity: an evolutionary perspective on sensitization, hyperalgesia and analgesia. Int Rev Neurobiol 36: 325-427. [DOI] [PubMed] [Google Scholar]
- Walters ET, Byrne JH, Carew TJ, Kandel ER (1983) Mechanoafferent neurons innervating tail of Aplysia I. Response properties and synaptic connections. J Neurophysiol 50: 1522-1542. [DOI] [PubMed] [Google Scholar]
- Walters ET, Alizadeh H, Castro GA (1991) Similar neuronal alterations induced by axonal injury and learning in Aplysia Science 253: 797-799. [DOI] [PubMed] [Google Scholar]
- Walters ET, Bodnarova M, Billy AJ, Dulin MF, Diaz-Rios M, Miller MW, Moroz LL (2004) Somatotopic organization and functional properties of mechanosensory neurons expressing sensorin-A mRNA in Aplysia californica J Comp Neurol 471: 219-240. [DOI] [PubMed] [Google Scholar]
- Waxman SG, Cummins TR, Dib-Hajj S, Fjell J, Black JA (1999) Sodium channels, excitability of primary sensory neurons, and the molecular basis of pain. Muscle Nerve 22: 1177-1187. [DOI] [PubMed] [Google Scholar]
- Woollacott M, Hoyle G (1977) Neural events underlying learning in insects: changes in pacemaker. Proc R Soc Lond B Biol Sci 195: 395-415. [DOI] [PubMed] [Google Scholar]
- Yanow SK, Manseau F, Hislop J, Castellucci VF, Sossin WS (1998) Biochemical pathways by which serotonin regulates translation in the nervous system of Aplysia J Neurochem 70: 572-583. [DOI] [PubMed] [Google Scholar]
- Zhang W, Linden DJ (2003) The other side of the engram: experience-driven changes in neuronal intrinsic excitability. Nat Rev Neurosci 4: 885-900. [DOI] [PubMed] [Google Scholar]
- Zheng JQ, Kelly TK, Chang B, Ryazantsev S, Rajasekaran AK, Martin KC, Twiss JL (2001) A functional role for intra-axonal protein synthesis during axonal regeneration from adult sensory neurons. J Neurosci 21: 9291-9303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu H, Wu F, Schacher S (1997) Site-specific and sensory neuron-dependent increases in postsynaptic glutamate sensitivity accompany serotonin-induced long-term facilitation at Aplysia sensorimotor synapses. J Neurosci 17: 4976-4986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zimmermann M (2001) Pathobiology of neuropathic pain. Eur J Pharmacol 429: 23-37. [DOI] [PubMed] [Google Scholar]
- Ziv NE, Spira ME (1993) Spatiotemporal distribution of Ca2+ following axotomy and throughout the recovery process of cultured Aplysia neurons. Eur J Neurosci 5: 657-668. [DOI] [PubMed] [Google Scholar]