

Abstract
Several proteins are expressed in both immune and nervous systems. However, their putative nonimmune functions in the brain remain poorly understood. KARAP/DAP12 is a transmembrane polypeptide associated with cell-surface receptors in hematopoeitic cells. Its mutation in humans induces Nasu-Hakola disease, characterized by presenile dementia and demyelinization. However, alteration of white matter occurs months after the onset of neuropsychiatric symptoms, suggesting that other neuronal alterations occur in the early phases of the disease. We hypothesized that KARAP/DAP12 may impact synaptic function. In mice deficient for KARAP/DAP12 function, long-term potentiation was enhanced and was partly NMDA receptor (NMDAR) independent. This effect was accompanied by changes in synaptic glutamate receptor content, as detected by the increased rectification of AMPA receptor EPSCs and increased sensitivity of NMDAR EPSCs to ifenprodil. Biochemical analysis of synaptic proteins confirmed these electrophysiological data. In mutants, the AMPA receptor GluR2 subunit expression was decreased only in the postsynaptic densities but not in the whole membrane fraction, demonstrating specific impairment of synaptic receptor accumulation. Alteration of the BNDF-tyrosine kinase receptor B (TrkB) signaling in the mutant was demonstrated by the dramatic decrease of synaptic TrkB with no change in other regulatory or scaffolding proteins. Finally, KARAP/DAP12 was detected only in microglia but not in neurons, astrocytes, or oligodendrocytes. KARAP/DAP12 may thus alter microglial physiology and subsequently synaptic function and plasticity through a novel microglia-neuron interaction.
Keywords: synapse, microglia, LTP, glutamate receptor, BDNF, hippocampus, postsynaptic density
Introduction
Several proteins thought to be specific for the immune system have been detected in the brain. For example, expression of the class-I major histocompatibility complex (MHC-I), the β2 microglobulin, the transporter associated with antigen processing (TAP-1) (Neumann et al., 1995), CD3ζ, a signaling polypeptide associated with the T-cell receptor (TCR) (Corriveau et al., 1998), and the TCR-β mRNA (Syken and Shatz, 2003) have been detected in the nervous system. Yet, the precise identity of cells expressing these proteins and mRNAs remains controversial (Neumann et al., 1995; Corriveau et al., 1998; Linda et al., 1999). Even more rudimentary is the understanding of their putative nonimmune function as well as the relationship between the immune response and the synaptic function (Corriveau et al., 1998; Huh et al., 2000).
Expression of KARAP/DAP12, a 12 kDa transmembrane polypeptide structurally and functionally related to CD3ζ (Olcese et al., 1997; Lanier et al., 1998), has recently been described in the brain and in primary cultures of microglia and oligodendrocytes (Kaifu et al., 2003). In the immune system, KARAP/DAP12 has been detected in lymphoid cells and in cells derived from the myeloid lineage (Bouchon et al., 2000; Lucas et al., 2002). KARAP/DAP12 and CD3ζ bear an intracytoplasmic immunotyrosine-based activation motif (ITAM) and associate with a variety of cell-surface receptors, including MHC-I receptors (Lanier, 2001).
Human mutations of KARAP/DAP12 induce polycystic lipomembranous osteodysplasia with sclerosing leukoencephalopathy (PLOSL; Nasu-Hakola disease; OMIM 604142), characterized by bone cysts and psychotic symptoms rapidly progressing to presenile dementia (Paloneva et al., 2000, 2001). A strong demyelinization is observed in PLOSL patients (Paloneva et al., 2001) as well as in the thalamus of KARAP/DAP12-deficient adult mice (Kaifu et al., 2003). In humans, white-matter alterations can only be detected several months or years after the onset of presenile dementia symptoms (Bianchin et al., 2004), likely reflecting more subtle alterations associated with earlier phases of the disease. Synaptic defects have been proposed to account for the early onset of some dementias (Terry and Katzman, 2001; Selkoe, 2002). Synaptic defects resulting from KARAP/DAP12 deficiency might thus be partly responsible for the early PLOSL symptoms. Interestingly, CD3ζ-deficient mice exhibit an alteration of synaptic plasticity (Huh et al., 2000).
The role of KARAP/DAP12 in synaptic function and plasticity was investigated here at the Schaffer collateral (SC) synapses onto CA1 pyramidal neurons synapses in mice bearing a loss-of-function mutation in the KARAP/DAP12 encoding gene (referred to as KΔ75) (Tomasello et al., 2000). We report that, although expression of KARAP/DAP12 is restricted to perinatal microglia, its loss of function leads to important alterations in synaptic function and plasticity with a dramatic decrease in synaptic expression of the BDNF receptor tyrosine kinase receptor B (TrkB). These results demonstrate a novel regulation of synaptic function through a microglia-neuron interaction.
Materials and Methods
Animals. We used KΔ75 mice, in which the mutated KARAP/DAP12 protein lacks the Y75 residue and wild-type (WT) C-terminus amino acids (Tomasello et al., 2000). The resulting KΔ75 polypeptide is expressed but not functional. KΔ75 mice were backcrossed seven or eight times with C57BL/6J/Rj mice. Another KARAP/DAP12 mutant mouse line also lacking functional protein has been described previously (Kaifu et al., 2003). It is worth noting that, as observed in this mouse line, KΔ75 mice display osteopetrosis and thalamic hypomyelinosis in old animals (data not shown and P. Jurdic and S. Nataf, personal communication)
Electrophysiology. Hippocampal slices were prepared from 18- to 25-d-old WT (n = 14) or KΔ75 (n = 22) mice as described previously (Poncer and Malinow, 2001). Whole-cell recordings were obtained from CA1 pyramidal cells using borosilicate glass microelectrodes (2-5 MΩ) containing (in mm) 115 CsMeSO3, 20 CsCl, 10 HEPES, 0.1 EGTA, 4 Mg-ATP, 0.4 Na3-GTP, and 10 Na-phosphocreatine. When inward rectification of AMPA receptor (AMPAR) currents was examined, 100 μm spermine was added to the internal solution. EPSCs were evoked by extracellular stimulation of Schaffer collateral afferents while holding cells at either -60 or +40 mV. EPSCs were isolated by addition of bicuculline methochloride (20 μm) after a cut was made between CA3 and CA1 areas. Long-term potentiation (LTP) was induced by pairing 200 stimuli delivered at 2 Hz while holding cells at 0 mV, as described previously (Poncer and Malinow, 2001). Signals were filtered at 1 kHz, sampled at 10 kHz, and analyzed using the pClamp8 software suite (Axon Instruments, Union City, CA). For display, stimulation artifacts were digitally subtracted. Values are expressed as mean ± SEM. Statistical significance was estimated using Wilcoxon (paired) or Mann-Whitney (unpaired) rank-sum tests performed under SPSS (Chicago, IL) Sigma-Stat software.
Glial culture. Hippocampi were dissected from newborn [postnatal day 2 (P2)] Swiss mice and dissociated by incubation in trypsin followed by trituration through a fire-constricted Pasteur pipette in DNase. Cells were plated onto glass poly-ornithine-coated coverslips in MEM with 10% fetal calf serum (PAA Laboratories, Linz, Austria) and grown until apparition of microglial cells and oligodendrocytes among the astrocytes.
Histochemistry. Cultured cells: coverslips were fixed in 4% paraformaldehyde (PFA) with 4% sucrose, permeabilized in 0.1% Triton X-100 plus 0.25% gelatin, and then immunostained. In situ: brains from embryonic and newborn mice were fixed by immersion overnight (o/n) in 4% PFA. Brains from P8 and P15 mice were obtained by intracardiac perfusion with 4% PFA and 4 hr postfixation. Then, they were incubated o/n in PBS plus 20% sucrose (4°C) before rapid freezing. Cryostat sections (12 μm) were incubated o/n at 4°C with primary antibodies in PBS plus Triton X-100 and gelatin. Secondary antibodies were applied sequentially for 1 hr each. When other fixation and labeling protocols were used, nonspecific labeling (not displaced by purified protein) was consistently observed, particularly in neurons. Sections were stained with thionine for Nissl staining. Primary antibodies were: goat anti-DAP12 A20 (0.1 μg/ml; Santa Cruz Biotechnology, Santa Cruz, CA) [comparable results with a rabbit anti-DAP12 (Aoki et al., 2000)], rabbit anti-GFAP (1:500; Sigma, St. Louis, MO), rabbit anti-ionized calcium-binding adaptor molecule 1 (Iba1) (1:500) (Ito et al., 1998) and anti-NG2 (1:600; Chemicon, Temecula, CA), mouse anti-galactocerebroside (GalC; 1:10), rat anti-F4/80 IgG2b (1:100; Serotec, Oxford, UK). Secondary antibodies were: donkey anti-sheep-cyanine 3 (1:400), anti-goat-Texas Red (1:100), or anti-rabbit-FITC (1:400) (Jackson Laboratories, West Grove, PA), horse anti-mouse-FITC (1:100; Vector Laboratories, Burlingame, CA), goat anti-rabbit-Alexa 488 (1:200; Molecular Probes, Eugene, OR). Images were acquired on a Leica (Nussloch, Germany) DMRD microscope equipped with a MicroMax camera and the MetaMorph Image Analysis System (Princeton Instruments, Trenton, NJ).
Preparation of postsynaptic densities. Preparation and analysis of the postsynaptic density (PSD) fraction were performed essentially as described previously (Smalla et al., 2000). Briefly, cerebral cortices and hippocampi from 30-d-old WT or KΔ75 littermates (n = 5 for each genotype) were homogenized in homogenization buffer (5 mm HEPES, pH 7.4, 320 mm sucrose) containing a protease inhibitor mixture (Roche Applied Science, Meylan, France). Nuclei were removed by 1000 × g centrifugation. The supernatant was spun at 100,000 × g for 30 min at 4°C. The pellet (considered as the crude membrane fraction) was resuspended and solubilized for 1 hr in 5 mm Tris, pH 8.1, 0.5% Triton X-100, 320 mm sucrose (buffer A) and then centrifuged at 100,000 × g for 1 h rat 4°C. The resulting detergent-resistant pellet was resuspended in buffer A and spun at 100,000 × g for 1 hr at 4°C. The final pellet represents a protein fraction particularly enriched in PSD proteins and is hereafter referred to as the PSD fraction.
Immunoblots. Equal amounts of the PSD fraction and membrane fraction were applied onto 8% SDS-PAGE and transferred on to polyvinylidene difluoride membrane. Antibodies against the following antigens were used: synapse-associated protein-90 (SAP-90)/PSD-95 (1:2000; Transduction Laboratories, Lexington, KY), neuronal nitric oxide synthase (nNOS; 1:1000; Transduction Laboratories), Capon (1:200; Santa Cruz Biotechnology), TrkB (1:1000; Transduction Laboratories), NR1 (1:1600; Chemicon), NR2A (1:1000; Upstate Biotechnology, Milton Keynes, UK), NR2B (1:250; Upstate Biotechnology), glutamate receptor (GluR) 1 (1:100; Chemicon), and GluR2 (1:100; Chemicon). Horseradish peroxydase-labeled anti-rabbit or anti-mouse antibodies (1:5000) were used as secondary antibodies. Immunoblots were processed with the ECL system (Amersham Biosciences, Orsay, France). Western blots were analyzed using Image Gauge software (Fujifilm, Paris, France). The mean densitometric value and the SEM were calculated from five or three animals of each genotype for the PSD fraction and the membrane fraction, respectively. Statistical analysis was performed with a one-tailed paired Student's t test.
Results
Enhanced hippocampal LTP in KΔ75 mice
To study the role of KARAP/DAP12 in synaptic function, we used KΔ75 mice, in which KARAP/DAP12 protein lacks the Y75 residue and WT C-terminus amino acids (Tomasello et al., 2000). The resulting KΔ75 polypeptide is expressed but not functional (Tomasello et al., 2000). No obvious neurological symptoms could be detected in KΔ75 mice up to the fourth week of postnatal life, and the gross anatomy of the hippocampal formation was unaltered compared with wild-type mice (Fig. 1A). Previous work revealed enhanced LTP in glutamatergic SC inputs onto hippocampal CA1 pyramidal neurons in mice genetically deficient for the ITAM-bearing KARAP/DAP12-related polypeptide CD3ζ (Huh et al., 2000). To evaluate the role of KARAP/DAP12 in synaptic function, we analyzed the properties of SC synapses onto CA1 neurons in KΔ75 mice.
Figure 1.

Enhanced hippocampal LTP in KΔ75 mice. A, Nissl staining of sagittal sections of hippocampus from WT and KΔ75 mice at P20. Scale bar, 200 μm. B, LTP of Schaffer collateral inputs onto CA1 pyramidal cells. A potentiation of 204 ± 13% of the EPSC was induced in WT mice (open symbols). In KΔ75 (filled symbols), the same pairing protocol induced a significantly larger potentiation of 398 ± 23% (p < 0.05). Top, Original traces from individual experiment (average of 15 consecutive EPSCs). Bottom, Averaged data from 9 and 14 recordings, respectively. Each point represents the mean amplitude of five consecutive EPSCs. C, In the presence of the NMDAR antagonist d-AP5 (50 μm), no potentiation was induced by the same pairing protocol in slices from WT mice (103 ± 26%; n = 5; p = 0.81). In contrast, a potentiation of 195 ± 13% was induced in slices from KΔ75 mice (n = 9; p < 0.01). Inset, The EPSC recorded in the presence of d-AP5 was entirely suppressed by the AMPAR antagonist 2,3-dihydroxy-6-nitro-7-sulfonyl-benzo[f]quinoxaline (NBQX; 20 μm). D, Compared effects of 200 stimulations delivered at 2 Hz (pairing) in CA1 cells from KΔ75 mice while holding the postsynaptic cell at 0 mV (circles), -60 mV (squares), or 0 mV (triangles) in the presence of 1 mm BAPTA and 10 mm EGTA in the patch-pipette solution (n = 9, 6, and 7 cells, respectively). Error bars represent SEM.
We first compared LTP in WT and KΔ75 mutant mice. In WT mice, a pairing protocol associating 2 Hz afferent stimulation with postsynaptic depolarization to 0 mV induced a robust LTP (276 ± 15% of control) with a moderate decay to 204 ± 13% after the first 15 min of induction (n = 9) (Fig. 1B). In KΔ75 mice, however, the same induction protocol produced a significantly larger potentiation (435 ± 19%; n = 14; p < 0.05) with a smaller decay than in WT after 15 min of induction (398 ± 23%; p < 0.05). The increased LTP in KΔ75 mice was not associated with a difference in glutamate release probability at Schaffer collateral inputs, as reflected by the similar paired-pulse ratio values in WT and KΔ75 mice (1.57 ± 0.13 and 1.71 ± 0.16, respectively, for an interstimulus interval of 50 msec; p = 0.69).
Induction of LTP requires postsynaptic Ca2+ influx, usually through NMDA receptors (NMDARs) (Malenka and Nicoll, 1993). We thus examined the NMDAR dependence of LTP in WT and KΔ75 mice (Fig. 1C). A pairing protocol delivered in the presence of the specific NMDAR antagonist d-2-amino-5-phosphonopentanoate (d-AP5; 50 μm) failed to produce persistent changes in the amplitude of evoked EPSCs in WT mice (94 ± 15% of control immediately after pairing and 103 ± 26% of control 15 min after pairing; n = 5; p = 0.69 and 0.81, respectively). In contrast, pairing delivered in the presence of d-AP5 in KΔ75 mutants caused a slowly developing yet significant potentiation of EPSCs (134 ± 6% of control immediately after pairing and 195 ± 13% after 15 min; n = 9; p < 0.01). We conclude that LTP at SC synapses onto CA1 pyramidal neurons is increased in KΔ75 mutants compared with WT and that this increase partly reflects a NMDAR-independent component of LTP. Because LTP in KΔ75 was approximately threefold larger than in WT mice and NMDAR-independent LTP was increased by ∼100%, we suggest that this component contributes to the enhanced LTP in KΔ75 by ∼50%. KARAP/DAP12 disruption thus impacts on hippocampal synaptic plasticity.
Impaired properties of excitatory synapses in KΔ75 mice
The existence of an NMDAR-independent LTP in KΔ75 mice suggests that repetitive synaptic stimulations may lead to postsynaptic calcium influx independent of NMDAR activation. We first tested this hypothesis in recordings made with internal solution containing the calcium chelators EGTA (10 mm) and BAPTA (1 mm). In these conditions, no potentiation was induced by a pairing protocol delivered in the presence of d-AP5 (90 ± 0.5% of control immediately after pairing and 94 ± 0.5% after 15 min; n = 7) (Fig. 1D), demonstrating that the NMDAR-independent potentiation in mutant mice indeed depends on postsynaptic calcium. A form of Ca2+-dependent, NMDAR-independent LTP has been described in mice lacking the GluR2 subunit of AMPARs (Jia et al., 1996). In these mice, postsynaptic Ca2+ influx was carried by Ca2+-permeable, GluR2-lacking AMPARs (Burnashev et al., 1992). If NMDAR-independent LTP in KΔ75 mice was caused by Ca2+ influx through GluR2-lacking AMPARs, it should persist when the cell is held at -60 mV during afferent stimulation. We indeed observed significant LTP when “pairing” was delivered while holding the postsynaptic cell at -60 mV (150 ± 7% of control immediately after pairing and 142 ± 5% after 15 min; n = 6) (Fig. 1D). However, the later phase of LTP was reduced compared with that obtained while holding the cell at 0 mV (142 ± 5 vs 195 ± 13% of control) suggesting that other voltage-dependent sources of Ca2+ may be recruited during pairing in KΔ75 mice. We did not, however, pursue this issue further.
Ca2+ permeability and the current-voltage (I-V) relationship of AMPARs are both determined by GluR2 subunits: whereas AMPARs containing GluR2 are Ca2+ impermeable and show linear I-V relationships, AMPARs lacking GluR2 are Ca2+ permeable and show complete inward rectification (Verdoorn et al., 1991; Burnashev et al., 1992). To test whether SC synapses onto CA1 neurons express Ca2+-permeable AMPARs in KΔ75 mice, we compared the rectification of AMPAR-mediated EPSCs in CA1 pyramidal cells in WT and KΔ75 mice (Fig. 2A). We recorded EPSCs at -60 and +40 mV, and the ratio of these amplitudes was used as a rectification index (Hayashi et al., 2000). In WT mice, this index was 2.0 ± 0.3 (n = 6), as described previously (Hayashi et al., 2000). In KΔ75 mice, however, this ratio was significantly higher (3.9 ± 0.7; n = 11; p < 0.04), a value comparable with that observed in neurons overexpressing GluR2-lacking receptors (Hayashi et al., 2000). This demonstrates that SC→CA1 synapses in KΔ75 mice express Ca2+-permeable AMPARs, most probably lacking the GluR2 subunit.
Figure 2.

Altered AMPAR- and NMDAR-mediated EPSCs in KΔ75 mice. A, AMPAR-mediated EPSCs recorded at -60 and + 40 mV in the presence of bicuculline and d-AP5. Left, Sample traces showing more prominent inward rectification of EPSCs recorded in CA1 pyramidal cells from KΔ75 than from WT mice. The amplitude ratio of the EPSC recorded at -60 and +40 mV was used to compute a rectification index. Right, Averaged data from 11 KΔ75 and six WT cells, respectively. The rectification index of AMPAR-mediated EPSCs was 2.0 ± 0.3 in WT mice and 3.9 ± 0.7 in KΔ75 mice (p < 0.03). B, NMDAR-mediated EPSCs recorded at +40 mV in the presence of bicuculline and 2,3-dihydroxy-6-nitro-7-sulfonyl-benzo[f]quinoxaline (NBQX; 20 μm). The effect of the NR2B-specific antagonist ifenprodil (5 μm) was much more pronounced in cells from KΔ75 than wild-type mice (-33 ± 8 and -5 ± 6% of control, respectively; n = 5 for each group; p < 0.01). Error bars represent SEM.
At a variety of central synapses (Pickard et al., 2000; Kumar et al., 2002), GluR2 content is low in immature synapses and progressively increases as synapses mature. This raised the intriguing possibility that KARAP/DAP12 mutation might interfere with glutamatergic synapse development. During development, the NMDAR subunit NR2B is progressively replaced by the NR2A subunit (Monyer at al., 1994). The NR2B subunit confers NMDAR with distinctive biophysical and pharmacological properties, including higher sensitivity to the antagonist ifenprodil (Williams et al., 1993). To evaluate the contribution of NR2B subunit to the composition of NMDARs, we compared the sensibility of NMDAR EPSCs to ifenprodil in WT and mutant mice. Figure 2B shows that evoked NMDAR currents in neurons from WT mice were not significantly affected by ifenprodil, as expected at this developmental stage (18-25 d; 95 ± 6% of control; n = 5; p = 0.44). In contrast, ifenprodil significantly depressed NMDAR currents in neurons from KΔ75 mice to 67 ± 8% of control (n = 5; p < 0.01). Therefore, hippocampal synapses of mutant mice display altered functional properties with a decreased AMPAR GluR2 content and probably an increased NMDAR NR2B content.
Decreased synaptic AMPARs and TrkB in KΔ75 mice
To further substantiate the characterization of KΔ75 mutant synapses, we undertook biochemical analysis of synaptic proteins in mutant and WT mice. Synaptic glutamate receptors are organized by the PSD, and we first examined the amount of glutamate receptors subunits in the PSD fractions (Fig. 3A,B). Levels of both GluR1 and GluR2 subunits were significantly reduced in the PSDs of the mutant mice compared with WT (GluR2, 61 ± 1%; GluR1, 80 ± 3%; n = 5; p < 0.005) (Fig. 3B). In agreement with the higher rectification of AMPARs observed in mutant mice, GluR2 amount in PSD decreased 1.3-fold more than GluR1 amount (n = 5; p < 0.05). Interestingly, no significant change in the AMPAR subunit levels was visible in the whole membrane fraction (GluR1, 90 ± 18% of control; GluR2, 85 ± 14% of control; n = 3; p = 0.85 and 0.94, respectively) (Fig. 3B). These results suggest that the KARAP/DAP12 loss-of-function mutation affects synaptic accumulation of GluR1 and GluR2 without interfering with their cellular expression levels. Despite the higher ifenprodil sensitivity of NMDARs in mutant mice, suggestive of an increased NR2B content, we could not detect significant changes in the amounts of NR1, NR2A, and NR2B subunits in the PSDs of WT and mutant animals (Fig. 3A,D).
Figure 3.
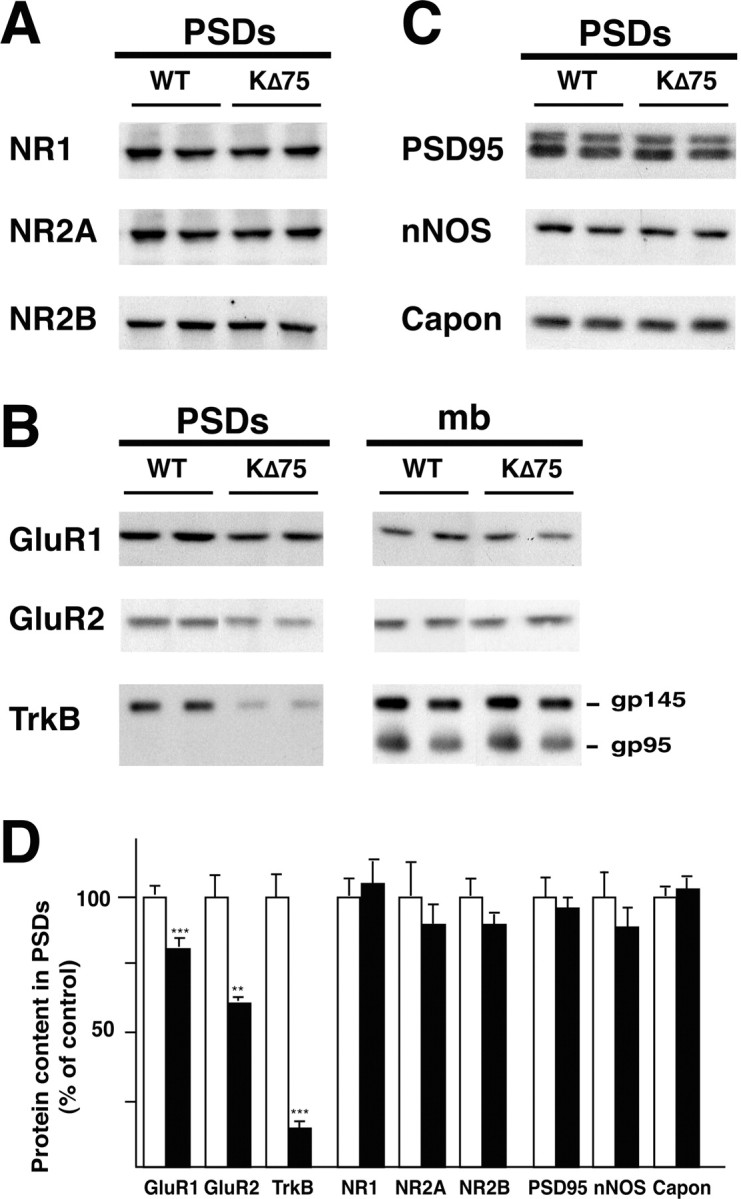
Specific alteration in protein content of PSD fraction in KΔ75 mice demonstrated by Western blot analysis. A, Synaptic NMDA receptor subunits are not modified. B, Decreased AMPA receptor subunits and TrkB (gp145) in immunoblots of PSD. Proportions are not modified in the whole membrane fractions (mb). Note that truncated TrkB (gp95) is detected in the whole membrane fractions but not in the PSDs, confirming the purity of the PSD preparation (Wu el al., 1996). C, Unaltered synaptic contents of the signaling protein nNOS and Capon and of PSD-95/SAP-90 in KΔ75 mice. D, Changes in the PSD fraction quantified by densitometric quantification. Data are expressed as percentage of control (mean ± SEM). n = 5 animals for each genotype; **p < 0.01; ***p < 0.001; Student's t test.
Neurotrophins are known to modulate synaptic transmission and LTP (Klintsova and Greenough, 1999). In particular, BDNF affects NMDAR function through phosphorylation of NR1 and NR2B subunits (Levine et al., 1998; Levine and Kolb, 2000) and promotes AMPAR trafficking to synaptic sites (Itami et al., 2003). We thus hypothesized that impairment of BDNF signaling might be responsible for the synaptic phenotype observed in KΔ75 mice. We tested this hypothesis by analyzing the synaptic expression of the BDNF receptor TrkB (Fig. 3B), which revealed a dramatic decrease of the full-length TrkB isoform (145 kDa) content in PSDs from mutant mice (15 ± 2% of control; n = 5; p < 0.003) (Fig. 3B) with no significant change in the whole membrane fraction (107 ± 7% of control; n = 5; p = 0.65). Note that the truncated isoform of TrkB (95 kDa) was not detected in the PSD fraction, confirming the purity of this preparation (Wu et al., 1996), and that it was unchanged in the membrane fraction (102 ± 6% of control; n = 5; p = 0.85) (Fig. 3B).
A reduced synaptic expression of TrkB may reflect a more general alteration of the postsynaptic density. To test this possibility, we examined the expression level of other proteins important for glutamate receptor function and/or known to be regulated by synaptic activity in the PSD (Wyneken et al., 2001). As shown in Figure 3, we observed no change in the relative content of SAP-90/PSD-95, a major scaffolding protein thought to interact with NMDARs (Kornau et al., 1995) and regulate the availability of AMPARs in synapses via stargazin (El-Husseini et al., 2002). Similarly, no change was observed in the expression of the signaling enzyme nNOS or its interacting protein, Capon (Jaffrey et al., 1998). These results demonstrate that the KARAP/DAP12 mutation specifically affects the postsynaptic targeting of TrkB.
KARAP/DAP12 is only expressed in microglia
We have shown that deficient KARAP/DAP12 function affects synaptic properties in the absence of any morphological defect, raising the question of the identity of the cell type expressing this protein in the hippocampus. Detection of KARAP/DAP12 in myeloid cells suggests that it might be expressed in microglia (Bakker et al., 2000). This was confirmed recently in purified and cultured cells by reverse transcription PCR (RT-PCR), Northern blots, and Western blots (Kaifu et al., 2003). This study also revealed expression of KARAP/DAP12 in oligodendrocytes. However, these results were not confirmed at the histochemical level.
In mixed glial culture (Fig. 4), KARAP/DAP12 immunoreactivity (IR) was only detected in cells positive for the microglial-specific marker Iba1 (Fig. 4A) (Ito et al., 1998), demonstrating KARAP/DAP12 expression in microglia. Lower KARAP/DAP12-IR could be observed in more ramified, differentiated microglial cells (data not shown) but not in astrocytes characterized by GFAP expression (Fig. 4B), in mature oligodendrocytes characterized by GalC-IR (Fig. 4C) (Ranscht et al., 1982), or in oligodendrocytes at earlier stages of differentiation characterized by O4-IR or A2B5-IR (data not shown). We conclude that, in our experimental conditions, KARAP/DAP12 expression is found only in microglia.
Figure 4.

Detection of KARAP/DAP12 in microglial cells but not in astrocytes or oligodendrocytes. Cultures were labeled with anti-DAP12 antibody (red; A1, B1, C1) and with anti-Iba1 (A2), anti-GFAP (B2), or anti-GalC (C2) antibodies to identify microglia, astrocytes, and oligodendrocytes, respectively. A3, B3, C3, Merges of DAPI (blue), KARAP/DAP12 (red), and Iba1, GFAP, or GalC (green) stainings. Arrowheads, KARAP/DAP12-positive cells; arrows, nuclei of KARAP/DAP12-negative cells. Scale bars (in A3, B3, C3), 5 μm.
We next investigated the expression pattern of KARAP/DAP12 in the developing hippocampus. At embryonic day 17 (E17), a few amoeboid microglia identified by their Iba1-IR had started to invade the hippocampus and were mainly located next to the pial surface and in the fimbria (Fig. 5A1, arrows). At this stage, only a few microglial cells displayed KARAP/DAP12-IR (Fig. 5A2,A3, arrowheads). At P0, the density of microglial cells increased (Fig. 5B1, arrows). They were distributed mainly close to the pial surface, in the hippocampal fissure, but also in the parenchyma in the strata radiatum and oriens. Most, but not all, amoeboid microglia displayed KARAP/DAP12-IR (Fig. 5B3, arrowheads), and in accordance with the in vitro observations, more differentiated primitive ramified microglia expressed a lower level of KARAP/DAP12 (data not shown). Kaifu et al. (2003) detected KARAP/DAP12 transcripts and protein at all stages of oligodendrocytes development in culture. We thus examined KARAP/DAP12 expression in oligodendrocytes in situ. At P0, NG2-positive cells were numerous, in particular in the fimbria (Fig. 5B2, arrows), but none of them displayed KARAP/DAP12-IR (Fig. 5B2,B3). This shows that NG2-positive oligodendrocytes (Ong and Levine, 1999) do not express KARAP/DAP12. At a later stage (P17), microglial cells were mostly ramified and distributed all over the hippocampus (Fig. 5C1, arrowheads), but KARAP/DAP12-immunopositive cells could not be detected (Fig. 5C2). At all stages examined, the anatomically identified neurons of the pyramidal layer were never stained (Fig. 5C). Similarly, the CD3ζ protein and corresponding mRNA were not found in neurons by immunohistochemistry or RT-PCR (data not shown). We therefore conclude that KARAP/DAP12 is expressed in microglia but not in neurons, astrocytes, or oligodendrocytes.
Figure 5.

Detection of KARAP/DAP12 in the hippocampus during development. A1, B1, Distribution of Iba1-positive microglia (arrows) in the hippocampus at E17 (A1) and P0 (B1). A2, A3, C1, C2, Iba1-positive microglia (green; A2, C1) display KARAP/DAP12-IR at E17 (red; A3, arrowheads) but not at P17 (C2, arrows). B2, B3, At P0, NG2-positive oligodendrocytes (B2, arrows) are not KARAP/DAP12 positive (B3, arrowheads). Scale bars: A1, B1, 50 μm; (in A2, B2) A2, A3, B2, B3, 10 μm; (in C2) C1, C2, 50 μm. AH, Ammon's horn; cp, choroid plexus; Fi, fimbria; HiF, hippocampal fissure; lv, lateral ventricle; py, pyramidal cell layer; pia, pia mater.
Discussion
KARAP/DAP12 is an ITAM-bearing polypeptide first described in cells of the innate immune system (Olcese et al., 1997; Lanier, 2001). In this report, we investigated its synaptic function in brain, based on its involvement in a human neurological disease (Paloneva et al., 2000, 2001) and its structural and functional similarity with CD3ζ (Tomasello et al., 2000). We show that KARAP/DAP12 expression in the hippocampus is restricted to microglial cells and that its loss of function leads to modifications in glutamatergic transmission and synaptic plasticity and to major changes in the composition of the PSDs.
KARAP/DAP12 mutation induces a synaptic phenotype in mutant mice
We have shown that the function of both NMDARs and AMPARs at synapses formed onto CA1 pyramidal cells was altered by the functional inactivation of KARAP/DAP12. The physiological properties of AMPAR EPSCs recorded in CA1 neurons are consistent with the biochemical analysis of the PSD fractions. We observed an increased inward rectification of AMPAR EPSCs in KARAP/DAP12 mutants, suggestive of a reduced synaptic GluR2 content (Geiger et al., 1995). Accordingly, a reduced synaptic recruitment of GluR2 was observed in KΔ75 mice. The greater calcium permeability of AMPARs lacking the GluR2 subunit has important functional consequences. In mice deficient for GluR2 (Jia et al., 1996), a form of NMDAR-independent LTP was observed. Consistent with this observation, we also observed NMDAR-independent LTP in KΔ75 mice. However, this form of LTP shows a slower onset than that observed in GluR2-deficient mice and is not entirely conserved during pairing at -60 mV. These results suggest that NMDAR-independent LTP in KΔ75 may not entirely rely on Ca2+-permeable AMPARs. Instead, this form of LTP might arise from a voltage-dependent synaptic source of calcium elevation or a potentiation mechanism acting downstream of Ca2+ influx. Finally, such a slowly developing, NMDAR-independent form of LTP in KΔ75 may partly contribute to enhanced LTP observed in these mice. It might also be responsible for the lesser decay of LTP observed in mutant animals (-8 vs -26% within the first 15 min after induction). These mechanisms and their relative contribution to the facilitation of LTP in KΔ75 remain to be clarified further.
It has been proposed that LTP results from the delivery of functional AMPARs into “NMDAR-only” silent synapses (Malinow and Malenka, 2002). The reduced expression of synaptic AMPARs observed in KΔ75 mice is compatible with a greater proportion of silent synapses in these mice. In addition, the TrkB ligand BDNF converts silent synapses into functional ones by activating the delivery of AMPAR to synaptic sites (Itami et al., 2003). Accordingly, we found a strikingly lower expression of TrkB in PSD fractions from mutant mice with the overall levels of AMPARs and TrkB unchanged, suggesting that a larger pool of these receptors might be available for rapid synaptic recruitment. Although we did not specifically address this issue at the physiological and electron microscopy levels, we suggest that a larger fraction of synapses may remain silent in KΔ75 mice, and LTP might thus be enhanced as the consequence of a greater conversion of silent synapses into functional ones.
Some reports have suggested that genetic background may influence LTP induction and expression (Nguyen et al., 2000). This influence is unlikely in the KΔ75 mice, because they were backcrossed seven times to C57BL/6 mice. In addition, mutations most often abolished, and not increased, LTP (Sanes and Lichtman, 1999), and the modifications of the synaptic parameters observed in KΔ75 mice are all compatible with an increased LTP. Finally, LTP was also enhanced in mice genetically deficient for the KARAP/DAP12-related CD3ζ (Huh et al., 2000). It is thus likely that the increased LTP in KΔ75 mice results from a dys-function of KARAP/DAP12 rather than from unspecific genetic differences.
In the CD3ζ mice, the mechanisms of increased LTP were not investigated in detail. However, no NMDAR-independent LTP was reported, in contrast to the present study. Therefore, CD3ζ and KARAP/DAP12, although they share structural and functional similarities, may affect synaptic function through partially distinct mechanisms. This may reflect different expression patterns. Whereas KARAP/DAP12 was only detected in developing microglia, transcripts encoding CD3ζ have been detected in lateral geniculate nucleus (Corriveau et al., 1998; Huh et al., 2000) and in adult hippocampus (Corriveau et al., 1998).
Synaptic NMDARs were more sensitive to ifenprodil in mutant than in WT mice, although NMDAR subunit expression was unchanged. The higher sensitivity of NMDARs to ifenprodil is thus unlikely to reflect a higher NR2B content. Instead, two non-exclusive hypotheses might explain this apparent discrepancy. First, the splicing of NR1 exon 5 was shown to affect the sensitivity of NR1/NR2B to ifenprodil (Kashiwagi et al., 1996). KARAP/DAP12 mutation might thus promote the synaptic insertion of specific NR1 splice variants that could not be discriminated in our Western blot analysis of the PSD. Alternatively, by homology with the decreased sensitivity of NR2A to its specific antagonist Zn2+ by phosphorylation (Zheng et al., 1998), it may be hypothesized that the decreased TrkB content prevented phosphorylation of NR2B in mutant mice, thus inducing a higher sensitivity to ifenprodil (Lin et al., 1999).
KARAP/DAP12 expression is restricted to microglia
Our immunohistochemical analysis showed that during development, KARAP/DAP12 is only expressed in amoeboid microglial cells but not in neurons or oligodendrocytes. KARAP/DAP12 is expressed in multiple cell types derived from the myeloid lineage (Bouchon et al., 2000), but its presence in microglia was only mentioned as unpublished results (Bakker et al., 2000; Paloneva et al., 2000). Recently, Kaifu et al. (2003) detected KARAP/DAP12 in immunopurified or cultured microglia but also in oligodendrocytes by RT-PCR and Western blots. This is in apparent contradiction with our results. However, microglia and oligodendrocytes both express Fc receptors (Vedeler et al., 1994; Nakahara et al., 2003) and are thus susceptible to bind nonspecifically the Fc portion of IgGs used to purify or label these cell types. Therefore, immunopurified cells are likely to be contaminated by microglia, and expression data obtained from in-mass analysis of immunopurified cells without appropriate purity controls should be interpreted carefully. In our study, we have consistently controlled the specificity of our immunostainings by using normal, nonimmune sera or irrelevant antibodies (data not shown). It could be that KARAP/DAP12 expression in oligodendrocytes is too low to be detected in our conditions. Alternatively, it might be restricted to an uncharacterized population of oligodendrocytes derived from the myeloid lineage that was not detected in our study; however, such a lineage has never been demonstrated (Vitry et al., 2003). In macrophages, KARAP/DAP12 expression depends on the cell activation state and/or differentiation stage (Aoki et al., 2000). This is in agreement with the differential expression observed between amoeboid and ramified microglia. We also found that only a fraction of amoeboid microglial cells expressed KARAP/DAP12. This suggests that the expression level of KARAP/DAP12 in the hippocampus might be variable and that we may not have detected low-expressing cells. Alternatively, KARAP/DAP12 expression by microglia might be transient.
A link between microglial and synaptic functions
KARAP/DAP12 is detected in microglia around birth, and disruption of its expression suffices to induce a persistent effect on the function and plasticity of glutamatergic synapses. This demonstrates that the changes in synaptic properties observed in the mutant mice are not based on a cell-autonomous mechanism but rather involve a novel regulatory link between microglial function and neurons. Yet, it remains unclear whether regulation occurred directly or indirectly as a consequence of microglial alterations induced by the mutation of KARAP/DAP12. Microglia express a large repertoire of secreted factors (Cuadros and Navascues, 1998), and some of them, such as BDNF or NT-3, are known to regulate synapse maturation and plasticity (Klintsova and Greenough, 1999). Although hippocampal BDNF content around birth was not different in mutant and WT mice (data not shown), KARAP/DAP12 loss-of-function mutation might result in reduced secretion of other cytokines that may lead to the observed synaptic phenotype (Mason et al., 2000). Our results are thus compatible with a model in which microglia control synaptic TrkB expression, the synaptic recruitment of GluR2-containing AMPARs, and phosphorylation of synaptic NMDARs.
Point mutation in human KARAP/DAP12, as well as in its associated receptor TREM-2 (triggering receptor expressed on myeloid cells-2), induces PLOSL (Paloneva et al., 2000, 2002). Patients display bone cysts as well as severe neurological symptoms including progressive frontal-type dementia. At the histological level, demyelinization and microglial activation are observed, although no obvious neuronal loss is detected in the neocortex or hippocampus (Paloneva et al., 2001). Our results suggest that higher functions may be perturbed as a result of synaptic defects. More generally, inflammatory reaction with microglial activation is a common feature of neurodegenerative diseases (Wyss-Coray and Mucke, 2002). Because synaptic disorders often precede the dementia in these diseases (Terry and Katzman, 2001; Selkoe, 2002), the demonstration of a relationship between microglia and synaptic function is thus of potentially high diagnostic and therapeutic interest.
Footnotes
This work was supported by grants from the European Commission (SYNAPTOGENET, QLG3-CT-2001-01181), the Association Française Contre les Myopathies, and the GIS-Institut des Maladies Rares. J.-C.P. was supported by Centre National de la Recherche Scientifique (CNRS) and Institut National de la Santé et de la Recherche Médicale (INSERM). E.V. and E.T. were supported by institutional grants from INSERM, CNRS, the Ministère de l'Enseignement Supérieur et de la Recherche, and specific grants from the Ligue Nationale Contre le Cancer (Equipe labelisée La Ligue). We thank Dr. Imaïfor the anti-IbaI antibody, Dr. Aoki for the anti-DAP12 antibody, Dr. Zalc for the anti-GalC, anti-O4, and anti-A2B5 antibodies, Dr. Jaillard for expert advices on oligodendrocytes, and Drs. J.-L. Bessereau, R. Miles, and P. Paoletti for helpful comments on this manuscript.
Correspondence should be addressed to Alain Bessis, INSERM U497, Ecole Normale Supérieure, 46 rue d'ulm, 75005 Paris, France. E-mail: abessis@ens.fr.
Copyright © 2004 Society for Neuroscience 0270-6474/04/2411421-08$15.00/0
A.R., C.B., and J.-C.P. contributed equally to this work.
References
- Aoki N, Kimura S, Takiyama Y, Atsuta Y, Abe A, Sato K, Katagiri M (2000) The role of the DAP12 signal in mouse myeloid differentiation. J Immunol 165: 3790-3796. [DOI] [PubMed] [Google Scholar]
- Bakker AB, Wu J, Phillips JH, Lanier LL (2000) NK cell activation: distinct stimulatory pathways counterbalancing inhibitory signals. Hum Immunol 61: 18-27. [DOI] [PubMed] [Google Scholar]
- Bianchin MM, Capella HM, Chaves DL, Steindel M, Grisard EC, Ganev GG, da Silva Jr JP, Neto Evaldo S, Poffo MA, Walz R, Carlotti Jr CG, Sakamoto AC (2004) Nasu-Hakola disease (polycystic lipomembranous osteodysplasia with sclerosing leukoencephalopathy-PLOSL): a dementia associated with bone cystic lesions. From clinical to genetic and molecular aspects. Cell Mol Neurobiol 24: 1-24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouchon A, Dietrich J, Colonna M (2000) Cutting edge: inflammatory responses can be triggered by TREM-1, a novel receptor expressed on neutrophils and monocytes. J Immunol 164: 4991-4995. [DOI] [PubMed] [Google Scholar]
- Burnashev N, Monyer H, Seeburg PH, Sakmann B (1992) Divalent ion permeability of AMPA receptor channels is dominated by the edited form of a single subunit. Neuron 8: 189-198. [DOI] [PubMed] [Google Scholar]
- Corriveau RA, Huh GS, Shatz CJ (1998) Regulation of class I MHC gene expression in the developing and mature CNS by neural activity. Neuron 21: 505-520. [DOI] [PubMed] [Google Scholar]
- Cuadros MA, Navascues J (1998) The origin and differentiation of microglial cells during development. Prog Neurobiol 56: 173-189. [DOI] [PubMed] [Google Scholar]
- El-Husseini Ael-D, Schnell E, Dakoji S, Sweeney N, Zhou Q, Prange O, Gauthier-Campbell C, Aguilera-Moreno A, Nicoll RA, Bredt DS (2002) Synaptic strength regulated by palmitate cycling on PSD-95. Cell 108: 849-863. [DOI] [PubMed] [Google Scholar]
- Geiger JR, Melcher T, Koh DS, Sakmann B, Seeburg PH, Jonas P, Monyer H (1995) Relative abundance of subunit mRNAs determines gating and Ca2+ permeability of AMPA receptors in principal neurons and interneurons in rat CNS. Neuron 15: 193-204. [DOI] [PubMed] [Google Scholar]
- Hayashi Y, Shi SH, Esteban JA, Piccini A, Poncer JC, Malinow R (2000) Driving AMPA receptors into synapses by LTP and CaMKII: requirement for GluR1 and PDZ domain interaction. Science 287: 2262-2267. [DOI] [PubMed] [Google Scholar]
- Huh GS, Boulanger LM, Du H, Riquelme PA, Brotz TM, Shatz CJ (2000) Functional requirement for class I MHC in CNS development and plasticity. Science 290: 2155-2159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Itami C, Kimura F, Kohno T, Matsuoka M, Ichikawa M, Tsumoto T, Nakamura S (2003) Brain-derived neurotrophic factor-dependent unmasking of “silent” synapses in the developing mouse barrel cortex. Proc Natl Acad Sci USA 100: 13069-13074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito D, Imai Y, Ohsawa K, Nakajima K, Fukuuchi Y, Kohsaka S (1998) Microglia-specific localisation of a novel calcium binding protein, Iba1. Brain Res Mol Brain Res 57: 1-9. [DOI] [PubMed] [Google Scholar]
- Jaffrey SR, Snowman AM, Eliasson MJ, Cohen NA, Snyder SH (1998) CA PON: a protein associated with neuronal nitric oxide synthase that regulates its interactions with PSD95. Neuron 20: 115-124. [DOI] [PubMed] [Google Scholar]
- Jia Z, Agopyan N, Miu P, Xiong Z, Henderson J, Gerlai R, Taverna FA, Velumian A, MacDonald J, Carlen P, Abramow-Newerly W, Roder J (1996) Enhanced LTP in mice deficient in the AMPA receptor GluR2. Neuron 17: 945-956. [DOI] [PubMed] [Google Scholar]
- Kaifu T, Nakahara J, Inui M, Mishima K, Momiyama T, Kaji M, Sugahara A, Koito H, Ujike-Asai A, Nakamura A, Kanazawa K, Tan-Takeuchi K, Iwasaki K, Yokoyama WM, Kudo A, Fujiwara M, Asou H, Takai T (2003) Osteopetrosis and thalamic hypomyelinosis with synaptic degeneration in DAP12-deficient mice. J Clin Invest 111: 323-332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kashiwagi K, Fukuchi J, Chao J, Igarashi K, Williams K (1996) An aspartate residue in the extracellular loop of the N-methyl-d-aspartate receptor controls sensitivity to spermine and protons. Mol Pharmacol 49: 1131-1141. [PubMed] [Google Scholar]
- Klintsova AY, Greenough WT (1999) Synaptic plasticity in cortical systems. Curr Opin Neurobiol 9: 203-208. [DOI] [PubMed] [Google Scholar]
- Kornau HC, Schenker LT, Kennedy MB, Seeburg PH (1995) Domain interaction between NMDA receptor subunits and the postsynaptic density protein PSD-95. Science 269: 1737-1740. [DOI] [PubMed] [Google Scholar]
- Kumar SS, Bacci A, Kharazia V, Huguenard JR (2002) A developmental switch of AMPA receptor subunits in neocortical pyramidal neurons. J Neurosci 22: 3005-3015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lanier LL (2001) On guard-activating NK cell receptors. Nat Immunol 2: 23-27. [DOI] [PubMed] [Google Scholar]
- Lanier LL, Corliss BC, Wu J, Leong C, Phillips JH (1998) Immunoreceptor DAP12 bearing a tyrosine-based activation motif is involved in activating NK cells. Nature 391: 703-707. [DOI] [PubMed] [Google Scholar]
- Levine ES, Kolb JE (2000) Brain-derived neurotrophic factor increases activity of NR2B-containing N-methyl-d-aspartate receptors in excised patches from hippocampal neurons. J Neurosci Res 62: 357-362. [DOI] [PubMed] [Google Scholar]
- Levine ES, Crozier RA, Black IB, Plummer MR (1998) Brain-derived neurotrophic factor modulates hippocampal synaptic transmission by increasing N-methyl-d-aspartic acid receptor activity. Proc Natl Acad Sci USA 95: 10235-10239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin SY, Wu K, Len GW, Xu JL, Levine ES, Suen PC, Mount HT, Black IB (1999) Brain-derived neurotrophic factor enhances association of protein tyrosine phosphatase PTP1D with the NMDA receptor subunit NR2B in the cortical postsynaptic density. Brain Res Mol Brain Res 70: 18-25. [DOI] [PubMed] [Google Scholar]
- Linda H, Hammarberg H, Piehl F, Khademi M, Olsson T (1999) Expression of MHC class I heavy chain and beta2-microglobulin in rat brainstem motoneurons and nigral dopaminergic neurons. J Neuroimmunol 101: 76-86. [DOI] [PubMed] [Google Scholar]
- Lucas M, Daniel L, Tomasello E, Guia S, Horschowski N, Aoki N, Figarella-Branger D, Gomez S, Vivier E (2002) Massive inflammatory syndrome and lymphocytic immunodeficiency in KARAP/DAP12-transgenic mice. Eur J Immunol 32: 2653-2663. [DOI] [PubMed] [Google Scholar]
- Malenka RC, Nicoll RA (1993) NMDA-receptor-dependent synaptic plasticity: multiple forms and mechanisms. Trends Neurosci 16: 521-527. [DOI] [PubMed] [Google Scholar]
- Malinow R, Malenka RC (2002) AMPA receptor trafficking and synaptic plasticity. Annu Rev Neurosci 25: 103-126. [DOI] [PubMed] [Google Scholar]
- Mason LH, Willette-Brown J, Mason AT, McVicar D, Ortaldo JR (2000) Interaction of Ly-49D+ NK cells with H-2Dd target cells leads to Dap-12 phosphorylation and IFN-gamma secretion. J Immunol 164: 603-611. [DOI] [PubMed] [Google Scholar]
- Monyer H, Burnashev N, Laurie DJ, Sakmann B, Seeburg PH (1994) Developmental and regional expression in the rat brain and functional properties of four NMDA receptors. Neuron 12: 529-540. [DOI] [PubMed] [Google Scholar]
- Nakahara J, Tan-Takeuchi K, Seiwa C, Gotoh M, Kaifu T, Ujike A, Inui M, Yagi T, Ogawa M, Aiso S, Takai T, Asou H (2003) Signaling via immunoglobulin Fc receptors induces oligodendrocyte precursor cell differentiation. Dev Cell 4: 841-852. [DOI] [PubMed] [Google Scholar]
- Neumann H, Cavalie A, Jenne DE, Wekerle H (1995) Induction of MHC class I genes in neurons. Science 269: 549-552. [DOI] [PubMed] [Google Scholar]
- Nguyen PV, Duffy SN, Young JZ (2000) Differential maintenance and frequency-dependent tuning of LTP at hippocampal synapses of specific strains of inbred mice. J Neurophysiol 84: 2484-2493. [DOI] [PubMed] [Google Scholar]
- Olcese L, Cambiaggi A, Semenzato G, Bottino C, Moretta A, Vivier E (1997) Human killer cell activatory receptors for MHC class I molecules are included in a multimeric complex expressed by natural killer cells. J Immunol 158: 5083-5086. [PubMed] [Google Scholar]
- Ong WY, Levine JM (1999) A light and electron microscopic study of NG2 chondroitin sulfate proteoglycan-positive oligodendrocyte precursor cells in the normal and kainate-lesioned rat hippocampus. Neuroscience 92: 83-95. [DOI] [PubMed] [Google Scholar]
- Paloneva J, Kestila M, Wu J, Salminen A, Bohling T, Ruotsalainen V, Hakola P, Bakker AB, Phillips JH, Pekkarinen P, Lanier LL, Timonen T, Peltonen L (2000) Loss-of-function mutations in TYROBP (DAP12) result in a presenile dementia with bone cysts. Nat Genet 25: 357-361. [DOI] [PubMed] [Google Scholar]
- Paloneva J, Autti T, Raininko R, Partanen J, Salonen O, Puranen M, Hakola P, Haltia M (2001) CNS manifestations of Nasu-Hakola disease: a frontal dementia with bone cysts. Neurology 56: 1552-1558. [DOI] [PubMed] [Google Scholar]
- Paloneva J, Manninen T, Christman G, Hovanes K, Mandelin J, Adolfsson R, Bianchin M, Bird T, Miranda R, Salmaggi A, Tranebjaerg L, Konttinen Y, Peltonen L (2002) Mutations in two genes encoding different subunits of a receptor signaling complex result in an identical disease phenotype. Am J Hum Genet 71: 656-662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pickard L, Noel J, Henley JM, Collingridge GL, Molnar E (2000) Developmental changes in synaptic AMPA and NMDA receptor distribution and AMPA receptor subunit composition in living hippocampal neurons. J Neurosci 20: 7922-7931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poncer JC, Malinow R (2001) Postsynaptic conversion of silent synapses during LTP affects synaptic gain and transmission dynamics. Nat Neurosci 4: 989-996. [DOI] [PubMed] [Google Scholar]
- Ranscht B, Clapshaw PA, Price J, Noble M, Seifert W (1982) Development of oligodendrocytes and Schwann cells studied with a monoclonal antibody against galactocerebroside. Proc Natl Acad Sci USA 79: 2709-2713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanes JR, Lichtman JW (1999) Can molecules explain long-term potentiation? Nat Neurosci 2: 597-604. [DOI] [PubMed] [Google Scholar]
- Selkoe DJ (2002) Alzheimer's disease is a synaptic failure. Science 298: 789-791. [DOI] [PubMed] [Google Scholar]
- Smalla KH, Matthies H, Langnase K, Shabir S, Bockers TM, Wyneken U, Staak S, Krug M, Beesley PW, Gundelfinger ED (2000) The synaptic glycoprotein neuroplastin is involved in long-term potentiation at hippocampal CA1 synapses. Proc Natl Acad Sci USA 97: 4327-4332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Syken J, Shatz CJ (2003) Expression of T cell receptor beta locus in central nervous system neurons. Proc Natl Acad Sci USA 100: 13048-13053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terry RD, Katzman R (2001) Life span and synapses: will there be a primary senile dementia? Neurobiol Aging 22: 347-348. [DOI] [PubMed] [Google Scholar]
- Tomasello E, Desmoulins PO, Chemin K, Guia S, Cremer H, Ortaldo J, Love P, Kaiserlian D, Vivier E (2000) Combined natural killer cell and dendritic cell functional deficiency in KARAP/DAP12 loss-of-function mutant mice. Immunity 13: 355-364. [DOI] [PubMed] [Google Scholar]
- Vedeler C, Ulvestad E, Grundt I, Conti G, Nyland H, Matre R, Pleasure D (1994) Fc receptor for IgG (FcR) on rat microglia. J Neuroimmunol 49: 19-24. [DOI] [PubMed] [Google Scholar]
- Verdoorn TA, Burnashev N, Monyer H, Seeburg PH, Sakmann B (1991) Structural determinants of ion flow through recombinant glutamate receptor channels. Science 252: 1715-1718. [DOI] [PubMed] [Google Scholar]
- Vitry S, Bertrand JY, Cumano A, Dubois-Dalcq M (2003) Primordial hematopoietic stem cells generate microglia but not myelin-forming cells in a neural environment. J Neurosci 23: 10724-10731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams K, Russell SL, Shen YM, Molinoff PB (1993) Developmental switch in the expression of NMDA receptors occurs in vivo and in vitro. Neuron 10: 267-278. [DOI] [PubMed] [Google Scholar]
- Wu K, Xu JL, Suen PC, Levine E, Huang YY, Mount HT, Lin SY, Black IB (1996) Functional trkB neurotrophin receptors are intrinsic components of the adult brain postsynaptic density. Brain Res Mol Brain Res 43: 286-290. [DOI] [PubMed] [Google Scholar]
- Wyneken U, Smalla KH, Marengo JJ, Soto D, de la Cerda A, Tischmeyer W, Grimm R, Boeckers TM, Wolf G, Orrego F, Gundelfinger ED (2001) Kainate-induced seizures alter protein composition and N-methyl-d-aspartate receptor function of rat forebrain postsynaptic densities. Neuroscience 102: 65-74. [DOI] [PubMed] [Google Scholar]
- Wyss-Coray T, Mucke L (2002) Inflammation in neurodegenerative disease-a double-edged sword. Neuron 35: 419-432. [DOI] [PubMed] [Google Scholar]
- Zheng F, Gingrich MB, Traynelis SF, Conn PJ (1998) Tyrosine kinase potentiates NMDA receptor currents by reducing tonic zinc inhibition. Nat Neurosci 1998 1: 185-191. [DOI] [PubMed] [Google Scholar]