

Abstract
CNS delivery of Urocortin I (UcnI), a member of the corticotropin-releasing factor family, suppresses feeding behavior and increases plasma glucose. The sites of action necessary and sufficient for these responses remain unclear. The contribution of the caudal brainstem was explored using chronically maintained decerebrate (CD) and neurologically intact control rats given fourth-ventricle injections of UcnI. Ingestive and glycemic responses were evaluated, and Fos immunoreactivity was measured in the paraventricular nucleus of the hypothalamus (PVN), the parabrachical nucleus (PBN), the rostral ventrolateral medulla (RVLM), and the nucleus of the solitary tract (NTS). CD rats, like the neurologically intact controls, decreased intraoral food intake and had elevated plasma glucose in response to Unc1 injections, indicating that forebrain structures are not required for these behavioral and physiological actions of UcnI. Fos immunohistochemistry, however, revealed notable differences in the pattern of UcnI-induced activation between intact and CD rats. UcnI-related activation was observed in each of the four aforementioned brain areas of neurologically intact rats but only in the NTS of CD rats. The intact behavioral and physiological responses to UcnI in the absence of neural activation in the PVN, PBN, and RVLM help limit the list of structures necessary for the stimulation and mediation of these responses to UcnI and suggest that the NTS may serve as a primary site of UcnI action.
Keywords: Urocortin I, CRF, caudal brainstem, hypothalamus, food intake, energy homeostasis
Introduction
Central administration of CRF receptor (CRF-R) agonists [e.g., CRF, Urocortin 1 (UcnI)] evokes responses relevant to energy homeostasis, including an elevation in plasma glucose and a reduction in food intake (Brown et al., 1982; Morley and Levine, 1982; Arase et al., 1988; Krahn et al., 1988; Grill et al., 2000). The anatomical extent of the neural substrates necessary and sufficient for the mediation of these responses, however, is poorly understood. CRF agonist delivered to each of a number of forebrain and caudal brainstem structures produced an increase in plasma glucose (Brown, 1986). Similarly, robust anorexic responses have been observed after UcnI injections into the lateral, third, or fourth ventricles (Morley and Levine, 1982; Arase et al., 1988; Krahn et al., 1988; Grill et al., 2000). Moreover, CRF-R agonists, given at doses below threshold for the response to intracerebroventricular (i.c.v.) administration, decreased food intake when applied directly into the parabrachial nucleus (PBN) (Grill and Kaplan, 2001), dorsal vagal complex (DVC) (Grill et al., 2000), paraventricular hypothalamic nucleus (PVN) (Krahn et al., 1988), or lateral septum (Bakshi et al., 2002).
The neural pathways connecting forebrain and caudal brainstem sites (Ricardo and Koh, 1978; Tribollet and Dreifuss, 1981; Luiten et al., 1985; ter Horst et al., 1989; Larsen and Mikkelsen, 1995) include direct and reciprocal projections between structures sensitive to CRF administration, including the nucleus of the solitary tract (NTS), PVN, and PBN (Luiten et al., 1985; Herbert et al., 1990; Aicher et al., 1995; Yu and Gordon, 1996). Injection of UcnI into the lateral ventricle (Benoit et al., 2000; Bittencourt and Sawchenko, 2000) resulted in the expression of Fos in many structures, including virtually all of those for which local stimulation has been shown to drive behavioral and/or autonomic responses. Interestingly, activation of both forebrain and caudal brainstem structures was also observed after UcnI infusion into either the lateral or fourth ventricle (Markison et al., 2002). The caudal flow of CSF would suggest that the forebrain activation (e.g., of PVN) reported after delivery to the fourth ventricle would be indirect, occurring transsynaptically via ascending pathways such as those from the NTS to PVN. Nevertheless, given the wide distribution of structures activated by UcnI, it is unclear which among them are required for the functional responses observed. It remains an open question whether these pathways drive critical caudal brainstem integrators, or whether the forebrain sites themselves are obligatory contributors to the organized responses triggered by CRF-R stimulation anywhere in the brain. If so, recruitment of these forebrain structures would be required for responses stimulated by CRF-R agonist delivery to sensitive sites within the caudal brainstem.
In the present study, UcnI is delivered to the fourth ventricle of neurologically intact and chronically maintained decerebrate (CD) rats, a well established model for evaluation of caudal brainstem contributions to intake control (Grill and Norgren, 1978a,b; Kaplan et al., 1993; Grill and Kaplan, 2002), to evaluate the relative importance of forebrain and hindbrain structures to the resultant ingestive and glycemic responses. Addressing the same issue, we also evaluate contrasts between intact and CD rats in the pattern of Fos expression after UcnI treatment.
Materials and Methods
Animals. Adult male Sprague Dawley rats weighing between 300 and 350 gm were obtained from Charles River Laboratories (Wilmington, MA). Animals were maintained in a temperature-controlled vivarium with a reverse light/dark cycle and were provided with ad libitum access to food and water, except as noted below. All experimental protocols described here were approved by the Institutional Animal Care and Use Committee and conform to the regulations described in the National Institutes of Health Guide for the Care and Use of Laboratory Animals.
Surgery. A subset of rats received a complete transection of the neuraxis at the mesodiencephalic juncture according to the procedure detailed previously (Grill and Norgren, 1978b). Briefly, all rats were anesthetized by intramuscular injection of ketamine (90 mg/kg) and xylazine (15 mg/kg), and a subset was subjected to decerebration via blunt hand-held spatula in two hemitransection procedures that were separated by at least 7 d. Both neurologically intact control animals and CD rats were fitted with two intraoral cannulas (PE-100) and with a chronic indwelling fourth-ventricle cannula during the second period of anesthesia. Each intraoral cannula was placed just lateral to the first maxillary molar and led to emerge at the top of the head. The fourth-ventricular cannulas were stereotaxically placed at midline 2.5 mm anterior to the occipital structure and 4.5 mm below the dura. Stainless steel tubing (19 gauge) was press fitted into the distal end of the tube from the intraoral cannulas and secured to the skull along with the fourth-ventricular cannula with screws and dental acrylic. At least 1 week was allowed for recovery before habituation training and testing began.
Maintenance. Beginning 1 week before surgery, nutrition for presumptive control and CD rats was supplied via four daily gastric intubations of 9 ml of liquid diet [1:1 Borden's (Austin, TX) sweetened condensed milk:water, with 1 ml of Polyvisol (with iron) added per 600 ml of diet]. The first feeding was delivered 2 hr before the rat's scheduled testing time, with the remaining feedings separated by intervals of at least 2 hr.
Experimental design. In experiment 1, intraoral intake of 12.5% glucose was measured in neurologically intact (n = 7) and CD (n = 9) rats 30 min after treatment with either vehicle [3 μl of artificial CSF (aCSF); Harvard Apparatus, Holliston, MA] or 3.0 μg of human UcnI (Sigma, St. Louis, MO). All animals received vehicle and UcnI injections according to a counterbalanced repeated-measures design with at least 3 d between testing. In experiment 2, plasma glucose was measured in neurologically intact control (n = 4) or CD (n = 6) rats before, 60, and 120 min after fourth-ventricle injection of either vehicle (3.0 μl of saline) or UcnI (3.0 μg). All rats received both injection conditions as described for experiment 1. In experiment 3, the expression of the early-immediate gene Fos was measured (see below) in intact (n = 10) and CD (n = 11) rats. Two hours before termination by transcardial perfusion, intact and CD rats received either 3.0 μl of aCSF (n = 5 per group) or 3.0 μg of UcnI (intact, n = 5; CD, n = 6).
Intraoral intake. Intake tests (Grill et al., 1987) followed 2 d of habituation training. For both habituation and the experimental days, groups of rats were run, 2-3 hr after being tube fed, in individual hanging cages (18 × 25 × 36 cm). Intraoral infusions of 12.5% glucose were driven by an infusion pump (Pump 44; Harvard Apparatus) at a rate of 0.5 ml/min. Each rat's infusion line was led through a computer-controlled miniature three-way solenoid valve either to a waste dish or to the animal. Animals were allowed to consume the glucose solution until fluid was observed leaving the mouth, at which time the infusion was stopped for 30 sec. The test was terminated if a second rejection event was observed within the 60 sec of reinstated delivery or was allowed to continue until the satiety criterion, two rejections within 90 sec, was met.
Plasma glucose. In experiment 2, blood samples (∼250 ml) were taken from the tail of unrestrained rats. Blood was drawn into heparinized capillary tubes and was centrifuged. Plasma glucose concentration was determined via the glucose oxidase method (Glucose Analyzer II; Beckman Instruments, Fullerton, CA).
Perfusions and tissue preparation for examination of Fos. Two hours after vehicle or UcnI was injected into the fourth ventricle of neurologically intact or CD rats, the animals were anesthetized with ketamine (90 mg/kg) and xylazine (15 mg/kg) and transcardially perfused with normal saline followed by 4% paraformaldehyde in 0.1 m phosphate buffer (PB). Brains were removed from the crania and postfixed for no more than 24 hr. After being submerged in 20% sucrose in 0.1 m PB for at least 48 hr, brains were frozen and cut by microtome into four sets of 30 μm coronal sections. Sections were stored in cryoprotectant (Watson et al., 1986) at -20° until processing by immunohistochemistry as described below.
Fos immunohistochemistry. Immunohistochemistry for the immediate-early gene Fos was performed on one set of free-floating sections from each animal. Sections were removed from cryoprotectant, washed in Tris-buffered saline (TBS), pH = 7.4, and incubated for 15 min in TBS containing 0.3% H2O2 before being incubated overnight at room temperature in rabbit anti-Fos (1:20,000; Santa Cruz Biotechnology, Santa Cruz, CA) diluted in TBS containing 0.2% Triton X-100 and 3% normal donkey serum. Sections then were washed in TBS and incubated for 2 hr with biotinylated donkey anti-rabbit IgG (1:1000; Jackson ImmunoResearch, West Grove, PA). After a brief wash with TBS, sections were incubated for 1 hr in an avidin-biotin-peroxidase complex (1:333; Elite kit; Vector Laboratories, Burlingame, CA). Sections were washed again with TBS and then with 50 mm Tris, pH = 7.4, before immunoreactivity was visualized by reacting the sections with 3,3′-diaminobenzidine (0.2 mg/ml) and 0.025% H2O2 in 50 mm Tris for 5 min. The reaction was stopped with TBS washes, after which the sections were floated onto gelatinized slides, dehydrated with increasing concentrations of alcohol followed by Hemo-De (Fisher Scientific, Pittsburgh, PA), and cover-slipped with Permount (Fisher Scientific).
Data analysis. Fos immunoreactivity was quantified by counting labeled nuclei on printed digital micrographs that had been coded such that the treatment condition was unknown. A total of three hemisections per brain area per animal were quantified in duplicate with an error rate lower than 5%. For the PBN, PVN, and RVLM, the data were expressed as the mean ± SEM number of cells per hemisection, whereas the data for the NTS were quantified on a per section basis. Statistical comparisons were made using Systat (Systat Inc., Point Richmond, CA).
Results
UcnI decreases food intake and increases plasma glucose in both intact and CD rats
Previous studies demonstrated virtually identical suppression of food intake or elevation of plasma glucose when UcnI was injected into either the lateral or the fourth ventricle (Grill et al., 2000). To determine whether caudal brainstem activation by UcnI requires concomitant forebrain activity, presumably recruited by brainstem neurons, we measured intraoral glucose intake in intact and CD rats after fourth ventricle injection of vehicle or UcnI. Fourth ventricle administration of UcnI produced a hypophagic response in both neurologically intact and CD rats, despite a reduced baseline intake observed in the CD rats (Fig. 1) (mixed design two-way ANOVA; intact vs CD, F(1,14) = 19.11, p < 0.001; vehicle vs UcnI, F(1,14) = 42.16, p = 0.0001; interaction, F(1,14) = 11.25, p < 0.01). Similarly, injection of UcnI resulted in a statistically significant elevation of plasma glucose in both neurologically intact and decerebrate animals (Fig. 2) (mixed design three-way ANOVA; intact vs CD, F(1,8) = 7.04, p = 0.03; vehicle vs UcnI, F(1,8) = 17.11, p < 0.01; time, F(2,16) = 11.34, p < 0.001). Examination of the interactions between group (intact vs CD), drug (vehicle vs UcnI), and time (0, 60, and 120 min) revealed a significant interaction only between drug and time (F(2,16) = 13.30; p < 0.001), indicating that decerebration did not affect the changes in plasma glucose occurring over time after UcnI injection. Thus, despite any differences in baseline measures, UcnI elicited a clear and statistically significant decrease in food intake and increase in plasma glucose in both intact and CD rats.
Figure 1.

The hypophagia induced by UcnI does not require a forebrain contribution. Neurologically intact and CD rats were treated with vehicle (aCSF; open bars) or UcnI (filled bars) before intraoral glucose intake was measured. Asterisks are used to indicate statistical differences from the corresponding control group (p < 0.01). Error bars represent SEM.
Figure 2.

UcnI causes an elevation in plasma glucose in neurologically intact (control) as well as CD rats. UcnI treatment resulted in an elevation of plasma glucose 60 and 120 min after administration (mixed design, three-way ANOVA; main effect of vehicle vs UcnI, p < 0.01; main effect of time, p < 0.001; drug vs time interaction, p < 0.001; other interactions, NS). The data are shown here as the percentage of the mean response of the vehicle-treated, neurologically intact group of animals. The mean level of plasma glucose for the neurologically intact and decerebrate rats at the 0 time point were 127.0 ± 4.3 and 105.7 ± 4.8 mg/dl, respectively (F(1,8) = 9.58; p = 0.015). Error bars represent SEM.
Fourth ventricle UcnI injections increase Fos expression in the PVN, PBN, RVLM, and NTS in the intact rat but only in the NTS of the CD rat
To test the possibility that UcnI-induced Fos expression in the PVN requires connectivity with caudal brainstem sites of primary UcnI action, we quantified vehicle- or UcnI-induced Fos immunoreactivity in the PVN, PBN, RVLM, and NTS of intact and CD rats. Fos immunoreactivity was relatively absent in the PVN, PBN, RVLM, and NTS of neurologically intact rats after fourth ventricle injection of vehicle. Fourth ventricle injection of UcnI, however, was associated with robust increases in the number of Fos-immunoreactive cells in all four brain areas examined. Specifically, the number of Fos-immunoreactive neurons increased by nearly threefold in the PVN, 25-fold in the PBN, eightfold in the RVLM, and sixfold in the NTS compared with counts from vehicle-injected rats (Figs. 3, 4). The UcnI-induced increase in Fos expression in the PBN of intact rats was noteworthy in terms of both the magnitude and the finding that Fos in this brain area was generally confined to the portion of the PBN termed by both Paxinos and Watson (1988) and Swanson (1999) as the external part of the lateral parabrachial nucleus (Fig. 3C).
Figure 3.
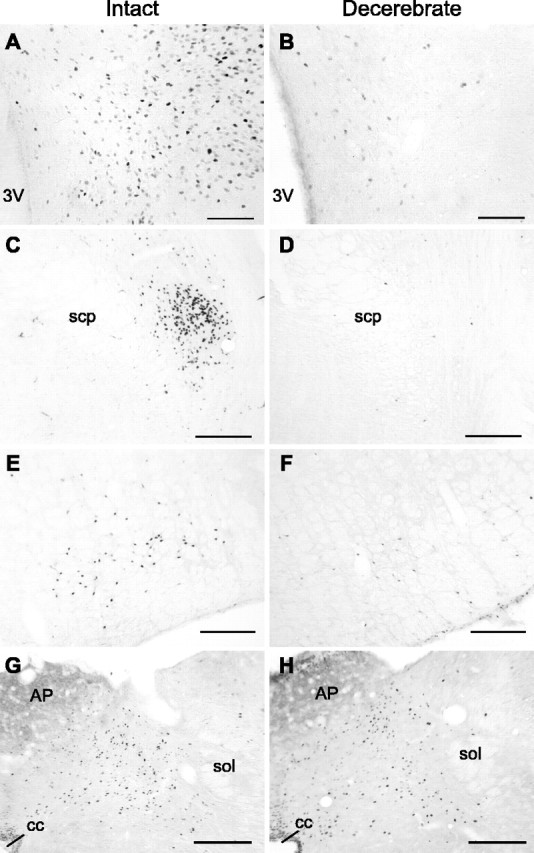
UcnI-induced Fos expression in neurologically intact and CD rats. Representative coronal sections from neurologically intact (A, C, E, G) and CD (B, D, F, H) rats from the PVN (A, B), PBN (C, D), RVLM (E, F), and NTS (G, H) are shown. 3V, Third ventricle; AP, area postrema; cc, central canal; scp, superior cerebellar peduncle; sol, solitary tract. Scale bars, 200 μm.
Figure 4.
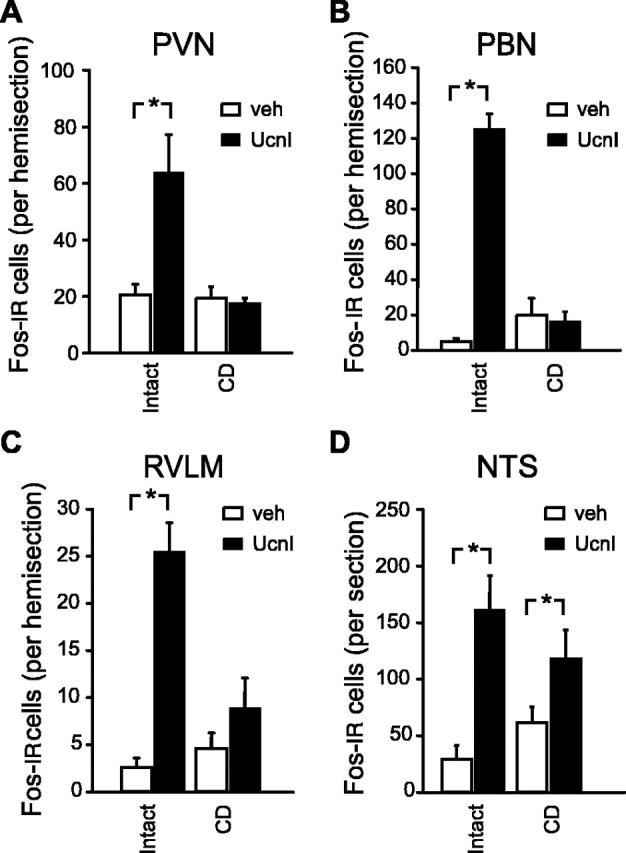
Quantification of the Fos immunoreactivity observed after neurologically intact or CD rats were treated with vehicle (veh) or UcnI. Data from the PVN (A), PBN (B), RVLM (C), and NTS (D) are shown. Asterisks are used to indicate statistical differences from the corresponding control groups (p < 0.05). Error bars represent SEM. IR, Immunoreactive.
The pattern of Fos expression in CD rats, however, was markedly different from that observed in control animals. As with the neurologically intact rats, the number of Fos-immunoreactive cells was greater in the NTS of CD rats that received fourth ventricle injections of UcnI compared with vehicle (main effect of drug, F(1,17) = 18.03, p = 0.001; group, F = 0.056, NS; interaction, F(1,17) = 2.82, NS). In contrast, however, UcnI-induced Fos expression in the PVN, PBN, and RVLM was absent in the CD rats [significant main effects of group (p values <0.01) and drug (p values <0.02) and significant interaction (p values <0.02) for each of the three ANOVAs].
Discussion
UcnI injected into the fourth ventricle elicited significant hyperglycemic and anorexic responses in the chronically maintained decerebrate rat. There were quantitative differences between CD and intact controls, including lower baseline intake values and a smaller magnitude of intake inhibition to UcnI in the CD. It is important to note that even identical CD and control response profiles would not allow us to rule out the possibility of a forebrain contribution to the responses observed in the neurologically intact rat. The clear and significant results observed in CD rats, however, establish that circuits contained within the caudal brainstem are sufficient for mediating responses to UcnI that are reminiscent in direction, if not magnitude, to those observed in intact controls.
The indication that the caudal brainstem is sufficient for the behavioral and physiological responses to UcnI rests on an assumed central, rather than peripheral, site of action. The direct application of UcnI to the brain suggests a central site of action, but the possibility that UcnI was able to leave the brain to act peripherally cannot be ruled out. The use of UcnI in the present study reduces this concern, however, because unlike CRF, a saturable transport system does not appear to mediate the efflux of UcnI from the brain (Kastin et al., 2000). It is unlikely as well that access to the anterior pituitary mediated any of these effects, because previous reports showed that CRF-R-induced hyperglycemia and anorexia were not attenuated by hypophysectomy (Brown et al., 1982; Morley and Levine, 1982).
In contrast to the behavioral and physiological results, there were prominent differences between intact and CD rats in the pattern of Fos expression associated with fourth ventricle UcnI delivery. For the intact rat, UcnI-induced activation was observed in each of the four structures evaluated (PVN, PBN, RVLM, and NTS), a pattern consistent with that attending forebrain i.c.v. administration of the same peptide (Benoit et al., 2000; Bittencourt and Sawchenko, 2000). For the decerebrate, however, a much more restricted distribution of Fos expression was observed. One prominent difference was the absence of UcnI-related Fos expression in the PVN. This finding is consistent with the expectation that the PVN activation observed in neurologically intact rats after fourth-ventricle administration of UcnI is driven via projections ascending from the caudal brainstem. Perhaps more interesting in relationship to the present functional observations was the absence of UcnI-induced activation in two (PBN and RVLM) of the three regions evaluated within the caudal brainstem of CD rats. Because these structures are situated at appreciable distances from the transection plane and do not appear abnormal on gross inspection of Nissl-stained sections, it is not likely that the lack of Fos expression in these areas was attributable to generalized damage after decerebration. Furthermore, Fos expression in these regions under vehicle-baseline conditions was no lower in CD than in control rats, suggesting that the absence of a treatment effect was not secondary to a global depression of activity. It remains possible, however, that there was a loss or metabolic disruption specifically affecting neurons responsive to UcnI.
The expression of CRF-R1 and/or CRF-R2 in the PVN, PBN, and RVLM and NTS (Bittencourt and Sawchenko, 2000) is in agreement with the expression of Fos in these brain areas after ventricular UcnI application to neurologically intact rats. From the present and previous data (Bittencourt and Sawchenko, 2000), one might conclude that UcnI accesses these brain areas directly to stimulate Fos expression. The lack of Fos expression in the PVN, PBN, and RVLM of CD rats, however, would otherwise suggest that after fourth-ventricle delivery of UcnI, these brain areas are normally activated through a transsynaptic mechanism that is disrupted in the decerebrate animal. The persistence of UcnI-induced Fos expression in the NTS of the CD rat, together with the known expression of both CRF-R1 and CRF-R2 in this structure, would suggest direct stimulation by UcnI of at least a subset of the Fos-positive cells in the NTS. It is therefore tempting to speculate that at least in the CD rat, the NTS serves as a primary site of action for UcnI. Similar to the PBN and RVLM, the NTS gives rise to long ascending pathways and receives direct projections from forebrain (Ricardo and Koh, 1978; Tribollet and Dreifuss, 1981; Luiten et al., 1985; ter Horst et al., 1989; Larsen and Mikkelsen, 1995), all of which, of course, are eliminated by the transection. Given that projections within the caudal brainstem (e.g., from the NTS to PBN) are likely to remain intact in the CD rat, it is the loss of ascending projections from the NTS and descending projections from the forebrain [e.g., from PVN (Luiten et al., 1985)] to the PBN and RVLM that may be responsible for the lack of activation by UcnI in the CD. The projections from the PVN and/or other forebrain structures, then, perhaps constitute the descending arm of a long-loop mechanism stimulated by the delivery of UcnI to the caudal brainstem of intact animals.
The Fos-immunohistochemical results for the CD rat appear to limit the set of structures, even within the caudal brainstem, that might be considered part of the circuitry sufficient for behavioral and glycemic responses to UcnI. Thus, to the extent that absence of UcnI-related Fos expression indeed indicates lost or disrupted function, the contribution of PBN and RVLM would appear to be of diminished significance. RVLM is known to contribute to a variety of sympathetic reflexes through interactions with other sympathetic “command” centers (Jansen et al., 1995) and via direct projections to intermediolateral column, including those terminating on preganglionic neurons innervating the adrenal medulla (Morrison et al., 1988; Cao and Morrison, 2000; Morrison and Cao, 2000). Yet, a robust hyperglycemic response to UcnI in CD rats was observed without attendant activation of this structure. Similarly, for the PBN, a contribution of which might have been supposed given its role as a visceral afferent relay does not appear to be part of the necessary control circuitry for the sympathoadrenal response in CD rats. The lack of activation within the PBN, in fact, is consistent with two previous studies with decerebrate preparations in which Fos expression in response to 2,5-anhydro-d-mannitol or taste stimulation were evaluated (Horn et al., 1998; Travers et al., 1999). The quiescence of this structure, then, is not unique to the response to UcnI and may represent a stable feature of the CD rat. Nevertheless, the expression of intact-like behavioral and physiological responses to UcnI despite the lack of activation in PBN suggests that this brain area is not among the list of required structures for UcnI-induced anorexia and hyperglycemia. Our interpretation of the caudal brainstem circuits underlying the physiological results, with emphasis on NTS and a discounting of PBN and RVLM contributions, is based on the Fos results, which provide suggestive support for, but not proof of, structure-function relationships. Additional work is required to strengthen or qualify this interpretation. If correct, we would expect, for example, that UcnI delivered to the PBN in the CD rat would be without effect and that PBN lesions would not disrupt responses to fourth ventricle UcnI in the decerebrate.
In summary, the present results are consistent with the hypothesis that an anatomically limited circuit within the caudal brainstem is sufficient for mediation of ingestive and sympathetic responses to UcnI. The observations in the decerebrate rat are consistent with a mechanism involving direct activation of CRF-R-expressing neurons in the NTS, which then engage, directly or indirectly, patterning mechanisms for the production of ingestive behavior in the brainstem core and sympathetic preganglionic neurons within the spinal cord.
Footnotes
This work was supported in part by National Institutes of Health Grants DK21397 (H.J.G.), DK64012 (D.D.), and DK42284 (J.M.K.). These data were reported in preliminary form at the meeting of the Society for the Study of Ingestive Behavior, 2002 (Santa Cruz, CA). We thank Dr. Tracy L. Bale for her thoughtful comments on this manuscript. We are also grateful for the technical assistance provided by L. Faulconbridge, J. Foxhall, and T. Yemini.
Correspondence should be addressed to Derek Daniels, Department of Animal Biology, School of Veterinary Medicine, University of Pennsylvania, 3800 Spruce Street, 254E, Philadelphia, PA 19104. E-mail: derekd@vet.upenn.edu.
S. Markison's present address: Neurocrine Biosciences, San Diego, CA 92130.
Copyright © 2004 Society for Neuroscience 0270-6474/04/2411457-06$15.00/0
References
- Aicher SA, Kurucz OS, Reis DJ, Milner TA (1995) Nucleus tractus solitarius efferent terminals synapse on neurons in the caudal ventrolateral medulla that project to the rostral ventrolateral medulla. Brain Res 693: 51-63. [DOI] [PubMed] [Google Scholar]
- Arase K, York DA, Shimizu H, Shargill N, Bray GA (1988) Effects of corticotropin-releasing factor on food intake and brown adipose tissue thermogenesis in rats. Am J Physiol 255: E255-E259. [DOI] [PubMed] [Google Scholar]
- Bakshi VP, Newman SM, Weinberg LE, Kalin NH (2002) Anatomical substrates and CRH receptor subtype mediation of urocortin-induced anorexia injection of threonine (THR) into the anterior. Society for the Study of Ingestive Behavior Abstracts, Appetite 39: 64. [Google Scholar]
- Benoit SC, Thiele TE, Heinrichs SC, Rushing PA, Blake KA, Steeley RJ (2000) Comparison of central administration of corticotropin-releasing hormone and urocortin on food intake, conditioned taste aversion, and c-Fos expression. Peptides 21: 345-351. [DOI] [PubMed] [Google Scholar]
- Bittencourt JC, Sawchenko PE (2000) Do centrally administered neuropeptides access cognate receptors?: an analysis in the central corticotropin-releasing factor system. J Neurosci 20: 1142-1156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown M (1986) Corticotropin releasing factor: central nervous system sites of action. Brain Res 399: 10-14. [DOI] [PubMed] [Google Scholar]
- Brown MR, Fisher LA, Spiess J, Rivier C, Rivier J, Vale W (1982) Corticotropin-releasing factor: actions on the sympathetic nervous system and metabolism. Endocrinology 111: 928-931. [DOI] [PubMed] [Google Scholar]
- Cao WH, Morrison SF (2000) Responses of adrenal sympathetic preganglionic neurons to stimulation of cardiopulmonary receptors. Brain Res 887: 46-52. [DOI] [PubMed] [Google Scholar]
- Grill HJ, Kaplan JM (2001) Effects of urocortin, leptin and melanocortin receptor ligands delivered to the parabrachial nucleus. Society for the Study of Ingestive Behavior Abstracts, Appetite 37: 140. [Google Scholar]
- Grill HJ, Kaplan JM (2002) The neuroanatomical axis for control of energy balance. Front Neuroendocrinol 23: 2-40. [DOI] [PubMed] [Google Scholar]
- Grill HJ, Norgren R (1978a) The taste reactivity test. II. Mimetic responses to gustatory stimuli in chronic thalamic and chronic decerebrate rats. Brain Res 143: 281-297. [DOI] [PubMed] [Google Scholar]
- Grill HJ, Norgren R (1978b) Chronically decerebrate rats demonstrate satiation but not bait shyness. Science 201: 267-269. [DOI] [PubMed] [Google Scholar]
- Grill HJ, Spector AC, Schwartz GJ, Kaplan JM, Flynn FW (1987) Evaluating taste effects on ingestive behavior. In: Feeding and drinking (Toates FM, Rowland NE, eds), pp 151-188. New York: Elsevier.
- Grill HJ, Markison S, Ginsberg A, Kaplan JM (2000) Long-term effects on feeding and body weight after stimulation of forebrain or hindbrain CRH receptors with urocortin. Brain Res 867: 19-28. [DOI] [PubMed] [Google Scholar]
- Herbert H, Moga MM, Saper CB (1990) Connections of the parabrachial nucleus with the nucleus of the solitary tract and the medullary reticular formation in the rat. J Comp Neurol 293: 540-580. [DOI] [PubMed] [Google Scholar]
- Horn CC, Kaplan JM, Grill HJ, Friedman MI (1998) Brain fos-like immunoreactivity in chronic decerebrate and neurologically intact rats given 2,5-anhydro-d-mannitol. Brain Res 801: 107-115. [DOI] [PubMed] [Google Scholar]
- Jansen AS, Nguyen XV, Karpitskiy V, Mettenleiter TC, Loewy AD (1995) Central command neurons of the sympathetic nervous system: basis of the fight-or-flight response. Science 270: 644-646. [DOI] [PubMed] [Google Scholar]
- Kaplan JM, Seeley RJ, Grill HJ (1993) Daily caloric intake in intact and chronic decerebrate rats. Behav Neurosci 107: 876-881. [PubMed] [Google Scholar]
- Kastin AJ, Akerstrom V, Pan W (2000) Activation of urocortin transport into brain by leptin. Peptides 21: 1811-1817. [DOI] [PubMed] [Google Scholar]
- Krahn DD, Gosnell BA, Levine AS, Morley JE (1988) Behavioral effects of corticotropin-releasing factor: localization and characterization of central effects. Brain Res 443: 63-69. [DOI] [PubMed] [Google Scholar]
- Larsen PJ, Mikkelsen JD (1995) Functional identification of central afferent projections conveying information of acute “stress” to the hypothalamic paraventricular nucleus. J Neurosci 15: 2609-2627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luiten PG, ter Horst GJ, Karst H, Steffens AB (1985) The course of paraventricular hypothalamic efferents to autonomic structures in medulla and spinal cord. Brain Res 329: 374-378. [DOI] [PubMed] [Google Scholar]
- Markison S, Daniels D, Kaplan JM, Grill HJ (2002) Feeding and c-Fos activation in response to urocortin administration in intact and chronic decerebrate rats. Society for the Study of Ingestive Behav Abstracts, Appetite 39: 92. [Google Scholar]
- Morley JE, Levine AS (1982) Corticotrophin releasing factor, grooming and ingestive behavior. Life Sci 31: 1459-1464. [DOI] [PubMed] [Google Scholar]
- Morrison SF, Cao WH (2000) Different adrenal sympathetic preganglionic neurons regulate epinephrine and norepinephrine secretion. Am J Physiol Regul Integr Comp Physiol 279: R1763-R1775. [DOI] [PubMed] [Google Scholar]
- Morrison SF, Milner TA, Reis DJ (1988) Reticulospinal vasomotor neurons of the rat rostral ventrolateral medulla: relationship to sympathetic nerve activity and the C1 adrenergic cell group. J Neurosci 8: 1286-1301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paxinos G, Watson C (1988) The rat brain in stereotaxic coordinates, Ed 4. Sydney: Academic.
- Ricardo JA, Koh ET (1978) Anatomical evidence of direct projections from the nucleus of the solitary tract to the hypothalamus, amygdala, and other forebrain structures in the rat. Brain Res 153: 1-26. [DOI] [PubMed] [Google Scholar]
- Swanson LW (1999) Brain maps: structure of the rat brain, Ed 2. Amsterdam: Elsevier.
- ter Horst GJ, de Boer P, Luiten PG, van Willigen JD (1989) Ascending projections from the solitary tract nucleus to the hypothalamus. A Phaseolus vulgaris lectin tracing study in the rat. Neuroscience 31: 785-797. [DOI] [PubMed] [Google Scholar]
- Travers JB, Urbanek K, Grill HJ (1999) Fos-like immunoreactivity in the brain stem following oral quinine stimulation in decerebrate rats. Am J Physiol 277: R384-R394. [DOI] [PubMed] [Google Scholar]
- Tribollet E, Dreifuss JJ (1981) Localization of neurones projecting to the hypothalamic paraventricular nucleus area of the rat: a horseradish peroxidase study. Neuroscience 6: 1315-1328. [DOI] [PubMed] [Google Scholar]
- Watson RE, Wigand SJ, Clough RW, Hoffman GE (1986) Use of cryoprotectant to maintain long-term peptide immunoreactivity and tissue morphology. Peptides 7: 155-159. [DOI] [PubMed] [Google Scholar]
- Yu D, Gordon FJ (1996) Anatomical evidence for a bi-neuronal pathway connecting the nucleus tractus solitarius to caudal ventrolateral medulla to rostral ventrolateral medulla in the rat. Neurosci Lett 205: 21-24. [DOI] [PubMed] [Google Scholar]