

Abstract
Somatostatin receptor 2 (SSTR2) mediates neuromodulatory signals of somatostatin and cortistatin in the cerebral cortex. Recently, SSTR2 has been shown to enhance conserved death ligand- and mitochondria-mediated apoptotic pathways in non-neuronal cells. Whether somatostatin receptors are activated in cerebrocortical neurons and contribute to neurodegeneration after experimental focal ischemia was unknown until now. Here we examined internalization of SSTR2 in a rat model of middle cerebral artery occlusion (MCAO) by confocal microscopy. At 3 and 6 hr after MCAO, SSTR2 was internalized excessively in cerebrocortical neurons adjacent to the infarct, which was prevented by intracerebroventricular application of the SSTR2-selective antagonist BIM-23627. SSTR2 internalization was associated with a transient depletion of somatostatin from axonal terminals and increased expression of SSTR2 mRNA. The initial loss of somatostatin was followed by an increase in somatostatin mRNA levels, whereas cortistatin mRNA expression was decreased. In SSTR2-deficient mice with lacZ under the control of the SSTR2 promoter, MCAO-induced upregulation of SSTR2 gene expression was less pronounced than in wild types. SSTR2-deficient mice exhibited a 40% reduction of infarct size after permanent distal MCAO and a 63% reduction after transient proximal MCAO. In summary, we provide direct evidence for activation of SSTR2 by an endogenous ligand after focal ischemia. Activation of functional SSTR2 receptors contributes to increased SSTR2 gene expression and postischemic neurodegeneration.
Keywords: somatostatin, cortistatin, sst2, internalization, apoptosis, in situ hybridization, immunohistochemistry, neuroprotection
Introduction
All five somatostatin receptors (SSTRs) belong to the G-protein-coupled receptor family and are expressed by numerous neuronal and non-neuronal cells. SSTRs inhibit adenylate cyclase and reduce cAMP levels (Hannon et al., 2002), which are important determinants of neuronal survival in numerous culture models (Rydel and Greene, 1988; Hanson et al., 1998; Putcha et al., 1999; Piiper et al., 2002; Poser et al., 2003). In non-neuronal cells, SSTR2 has been shown to enhance death ligand- and mitochondria-mediated apoptosis (Guillermet et al., 2003), indicating that conserved signaling pathways in programmed cell death can be influenced by SSTRs. Intracerebral applications of SSTR ligands before middle cerebral artery occlusion (MCAO) affect the infarct volume in a dose-dependent manner (Rauca et al., 1999), suggesting that SSTRs may influence neurodegeneration after brain ischemia.
The endogenous peptides somatostatin and cortistatin exhibit similar affinities for the SSTRs. Because cortistatin exerts some functions that are distinct from those of somatostatin, the existence of a cortistatin-specific receptor has been proposed (Spier and de Lecea, 2000). Somatostatin and cortistatin peptides are highly expressed in the cerebral cortex. Somatostatin is localized in cortical GABAergic interneurons, some of which coexpress cortistatin (de Lecea et al., 1997). Both peptides exhibit neuromodulatory roles in the cortex by depressing the activity of pyramidal cells (Moore et al., 1988; de Lecea et al., 1996; Hicks et al., 1998; Schweitzer et al., 2003). Because SSTR2 accounts for most of the somatostatin-binding sites in the cortex (Videau et al., 2003), these neuromodulatory effects are most likely mediated by SSTR2. In addition to the cortex, MCAO also affects the striatum, where somatostatinergic GABAergic interneurons exert neuromodulatory effects on SSTR2-expressing projection neurons (Vilchis et al., 2002; Allen et al., 2003).
Biosynthesis and release of somatostatin is increased after experimental seizures (Binaschi et al., 2003), suggesting that the somatostatin system is activated during hyperexcitation of neuronal circuits. Recently, SSTR2 activation has been shown to cause homologous upregulation of SSTR2 mRNA levels (Boudin et al., 2000; Tannenbaum et al., 2001), a mechanism that may counteract desensitization of SSTR2 during sustained receptor stimulation. To our knowledge, activation of neuropeptide receptors has not been shown in models of neurodegenerative diseases. For SSTR2 in particular, little is known about its activation, regulation, and contribution to brain infarction after focal ischemia.
In the present study, postischemic internalization of SSTR2 immunoreactivity was analyzed in cortical neurons by confocal microscopy, which is an established approach to reveal ligand-induced activation of neuropeptide receptors in the nervous system (Mantyh et al., 1995). By in situ hybridization, upregulation of SSTR2 mRNA expression was detected in areas with SSTR2 internalization and, using SSTR2-deficient mice with a lacZ knock-in allele, functional SSTR2 protein was shown to contribute to SSTR2 upregulation. Finally, reduced infarct volumes in SSTR2-deficient mice in permanent and transient MCAO models provide evidence that SSTR2 enhances neurodegeneration after focal ischemia.
Materials and Methods
For all animal procedures, ethical approval was sought before the experiments according to the requirements of the German National Act on the Use of Experimental Animals.
Animal models of focal cerebral ischemia. Permanent MCAO and sham operation were performed in 6-week-old male Long-Evans rats (Mollegaard) as described previously (Culmsee et al., 1999; Stumm et al., 2001), with the modification that sodium pentobarbital was applied (40 mg/kg, i.p.) for anesthesia. Permanent and transient mouse MCAO models (Stumm et al., 2002; Katchanov et al., 2003) were performed in C57BL/6 SSTR2 knock-out lacZ knock-in mice (Allen et al., 2002) backcrossed for >10 generations to a C57BL/6 background. All experiments were conducted with knock-out and wild-type littermates obtained by heterozygous intercrossing. Infarct volumes 7 d after permanent MCAO were determined by integrating infarct areas, which were measured in 400 μm intervals in cresyl violet-stained cryostat sections using NIH Image 1.62 software. In the transient MCAO model, infarcts were determined after 3 d as described previously (Endres et al., 2000). Differences in infarct size between wild-type and SSTR2-deficient mice were analyzed in both MCAO models by a Mann-Whitney U test. Mice subjected to transient MCAO were tested for neurological deficits before they were killed and were rated on a scale from 0 (no observable deficit) to 3 (severe) (Bederson et al., 1986). Briefly, failure to extend the forepaw when suspended vertically was graded as mild injury (grade 1), circling was graded as moderate (grade 2), and loss of circling or righting reflex was graded as severe (grade 3). Animals were graded by an observer blinded to genotype.
Drug application into the lateral ventricle. Efficacy of the SSTR2 antagonist BIM-23627 (Biomeasure, Milford, MA) in vitro and in vivo has been described previously (Zatelli et al., 2001; Tulipano et al., 2002). In 12 rats (300 gm) anesthetized with sodium pentobarbital (40 mg/kg, i.p.), the skull was exposed and trepanated (1 mm diameter; stereotaxic coordinates, 0.25 mm posterior and 1.6 mm lateral to bregma). One week after surgery, animals were again anesthetized and mounted in a stereotaxic frame. A needle was inserted through the trepanation site to a depth of 4 mm, and 5 μl of saline or 5 μl of saline containing 0.5 nmol/μl BIM-23627 was applied. The needle was left in place for an additional 2 min and then retracted. MCAO and sham operation were performed 30 min after delivery of the drug or saline (n = 3).
In situ hybridization. Generation of [35S]-labeled and digoxigenin-labeled riboprobes, hybridization and washing procedures, as well as detection of hybridization signals were performed according to standard protocols (Stumm et al., 2002). Template rat cDNAs used to transcribe probes for somatostatin, parvalbumin, vesicular glutamate transporter 1 (VGLUT1), and glutamic acid decarboxylase (GAD) (Mr 67,000) have been described recently (Stumm et al., 2004). A rat cortistatin cDNA (GI:6978682; nucleotides 4-286), a rat heat shock protein 70 (HSP70) cDNA (GI:294567; nucleotides 493-696), and a mouse SSTR2 cDNA (GI:54197; nucleotides 611-1510) were isolated from brain RNA by reverse transcription-PCR and cloned into the pGEM-Teasy vector (Promega, Madison, WI) for transcription. Probes for rat SSTRs were generated using previously described cDNAs (Kreienkamp et al., 1997) that correspond to nucleotides 1-2752 of SSTR1 (GI:6981583), nucleotides 368-1477 of SSTR2 (GI:207026), nucleotides 657-1716 of SSTR3 (GI: 34866768), nucleotides 59-1303 of SSTR4 (GI:40254790), and nucleotides 154-1381 of SSTR5 (GI:6981587). Nucleotides 1123-1954 of a lacZ cDNA (GI:41901) were cloned into EcoRV/SacI sites of pBluescript KS(+) (Stratagene, La Jolla, CA) for transcription.
Image analysis. Densitometric analysis of hybridization signals and cell counts were conducted as described previously (Stumm et al., 2001) using NIH Image 1.62. For densitometry, sections of an experimental group, the respective control group, and a [14C] standard (ARC-146; Biotrend Chemicals, Köln, Germany) were exposed to the same x-ray film, and the autoradiograms were digitized. Calibration was performed by plotting the density measurements of the [14C] standard against the given tissue radioactivity equivalents (nanoCuries per gram of tissue). Then, the integrated optical density was determined in selected brain regions by averaging measurements from three sections per individual. Measurements were performed for the SSTRs and somatostatin in rats 6 hr, 1 d, 2 d, and 4 d after MCAO (n = 6) and in stage-matched sham-operated groups (n = 4). Corresponding groups were compared by a non-paired, two-way Student's t test. For cortistatin, naive rats (n = 3) were added to the above experimental groups, and a one-way ANOVA with Newman-Keuls multiple comparison test was performed. To determine the numbers of the neurons expressing SSTR2, cortistatin, and somatostatin mRNA in a given area, the hybridized sections were coated with NTB2 emulsion (Eastman Kodak, Rochester, NY) and photographed under dark-field illumination with an Axioskop microscope (Zeiss, Oberkochen, Germany) equipped with a digital camera (SV MICRO; Electroimage, Great Neck, NY). Cell counts for each individual were averaged from three specimens. MCAO-treated and control groups were compared by Student's t test. In SSTR2-deficient and wild-type mice (n = 8 each), the HSP70 mRNA-positive area, 6 hr after permanent MCAO, was determined in x-ray autoradiograms by setting a threshold level that excluded areas with low-signal intensities. Comparison of the two groups was performed using a Mann-Whitney U test. Levels of SSTR2 mRNA in wild-type mice (n = 11) and lacZ mRNA in SSTR2-deficient mice (n = 14) were determined ipsilaterally and contralaterally 6 hr after MCAO by densitometry. Ipsilateral and contralateral values were compared by a Wilcoxon signed rank test. Then, the fold upregulation was calculated for SSTR2 and lacZ as ipsilateral versus contralateral change, and comparison of both groups was performed by a Mann-Whitney U test.
For cortistatin and parvalbumin mRNAs as well as cortistatin and somatostatin mRNAs, semiquantitative proportional analysis of coexpression was performed in the cingulate and frontal cortical areas by counting single- and double-labeled neurons in four rats 2 d after MCAO and in three naive rats. Coexpression of SSTR2 and GAD mRNAs as well as SSTR2 and VGLUT1 mRNAs was analyzed in the same way in layers 2/3 and 5/6 of the forelimb cortical area in four sham-operated and four MCAO-treated rats after 6 hr.
Immunohistochemistry. Immunohistochemistry for SSTR2a and somatostatin was performed in naive rats (n = 3); 3 hr, 6 hr, 1 d, 2 d, and 4 d after MCAO (n = 5); as well as after sham operation at each time point (n = 3), using the affinity-purified anti-SSTR2a antibody [6291] at 1 μg/ml as described previously (Schulz et al., 1998; Allen et al., 2002) and the monoclonal SOM-14 antibody (Biomeda) at a dilution of 1:50. For immunohistochemical controls, brains of SSTR2-deficient mice were shown to be SSTR2a immunonegative. In addition, primary antibodies were omitted (SOM-14; [6291]), replaced by preimmune sera, or absorbed with several concentrations (ranging from 1 to 10 μg/ml) of homologous or heterologous peptides for 2 hr at room temperature [6291]. Specimens were examined using a TCS-NT laser scanning confocal microscope (Leica, Heidelberg, Germany) equipped with a krypton-argon laser. Cy3 was imaged with a 568 nm excitation and 570-630 nm bandpass emission filter. To analyze internalization of SSTR2a, a 100× objective was used. Neuronal cell bodies in the cerebral cortex that did not contain internalized SSTR2a showed faint SSTR2a-like immunoreactivity (SSTR2a-LIR) on the cell surface and neuropil (Csaba et al., 2001). In neurons with internalized SSTR2, the cytoplasm contained 5-10 bright, immunofluorescent structures (weak internalization) or was filled with confluent SSTR2a-LIR (strong internalization).
Immunocytochemistry. Human embryonic kidney (HEK) 293 cells stably transfected with T7-tagged SSTR2a (Pfeiffer et al., 2002) were grown overnight on poly-l-lysine-treated coverslips. Somatostatin-14 (100 nm), BIM-23627 (1 μm), or both compounds were applied for 30 min. Cells were fixed, washed, stained with 1 μg/ml affinity-purified serum [6291], and detected as described previously (Stumm et al., 2002). For quantitative analysis of internalization, an established ELISA was used (Pfeiffer et al., 2002). The transfected cells were preincubated with 1 μg of affinity-purified rabbit anti-T7 antibody for 2 hr in OPTIMEM (Invitrogen, San Diego, CA) at 4°C. Then, they were treated with 100 nm somatostatin-14, 1 μm BIM-23627, or both compounds for 1 hr, fixed, and incubated with peroxidase-conjugated anti-rabbit antibody (1:1000; Amersham Biosciences, Braunschweig, Germany) for 2 hr at room temperature. After washing, the plates were developed with 250 μl of 2,2′-azino-di-[3-ethylbenzthiazoline sulfonate] solution (Roche, Penzberg, Germany). After 30 min, 200 μl of the substrate solution from each well was transferred to a 96-well plate and analyzed at 405 nm using a microplate reader (Bio-Rad, Munich, Germany).
Results
Internalization of SSTR2a by endogenous ligands after MCAO
To test whether SSTR2a is activated by endogenously released ligands after focal ischemia, we characterized the spatial and temporal patterns of SSTR2a internalization in a rat model of permanent distal MCAO. In controls, which comprised brains of naive and sham-operated rats as well as the contralateral hemisphere of ischemic rats, SSTR2a-LIR diffusely labeled the neuropil in the infragranular layers V and VI of the cingulate, frontal, and parietal cerebrocortical subregions (Fig. 1A,D) (contralateral hemisphere 6 hr after MCAO). High-power analysis showed that the cytoplasm of neuronal perikarya in infragranular layers (Fig. 1F) and dendrites in supragranular layers (Fig. 1H) were either spared from or slightly decorated with SSTR2a-LIR. The observed pattern of SSTR2a-LIR in the forebrain of controls matched exactly previous reports (Csaba et al., 2001), and SSTR2a-LIR in ischemic and control brains was fully absorbed by preincubation with the immunizing peptide (Fig. 1B). At 3 and 6 hr after focal ischemia, profound internalization of SSTR2a-LIR was seen in cerebrocortical tissues adjacent to the primary infarct dorsally and ventrally. For simplicity, the perifocal cortical areas (Fig. 1A, arrowheads) will now be referred to as ventral and dorsal penumbra. In the dorsal penumbra, internalization of SSTR2a was seen frequently in perikarya in infragranular layers (Fig. 1C,E) (6 hr after MCAO) and occasionally in supragranular layers. The strongly internalizing neurons showed confluent SSTR2-LIR in the cytoplasm of the cell bodies. SSTR2a-like immunoreactive dendrites, which were sparsely labeled in controls (Fig. 1D,H), exhibited an accumulation of internalized SSTR2a-LIR after focal ischemia (Fig. 1C,G) and could be easily traced from infragranular to supragranular layers. Also in the ventral penumbra, striking internalization of SSTR2a-LIR was seen both at 3 hr (Fig. 1I,I′--K,K′) and 6 hr after MCAO (see below). Outside the penumbra, an increase in internalized SSTR2a-LIR was seen ipsilaterally in the nonlesioned cingulate and frontal cortical areas at 3 and 6 hr after MCAO. The internalized SSTR2a-LIR typically formed several intracellular clusters but was not confluent (data not shown), suggesting that the amount of internalized SSTR2a-LIR per cell was less in nonlesioned areas than in the penumbra. An increase in internalized SSTR2a-LIR was not observed in the hemisphere contralateral to MCAO compared with the naive specimen. At the late stages (2 and 4 d after MCAO), an increase in internalized SSTR2a-LIR was observed neither ipsilaterally nor contralaterally.
Figure 1.
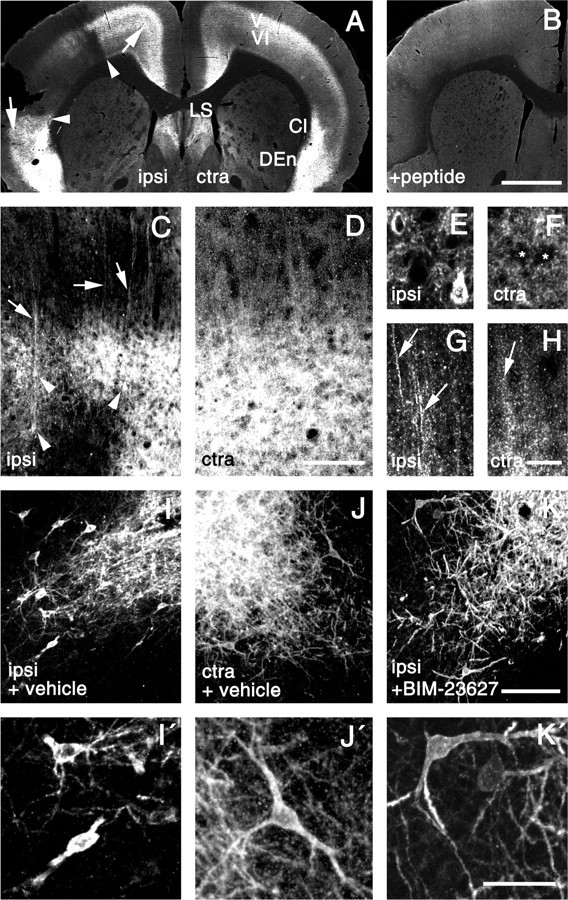
Internalization of SSTR2a after focal ischemia in the rat. A, C-H, Confocal images of SSTR2a-LIR in the ipsilateral (ipsi) and contralateral (ctra) brain hemispheres 6 hr after MCAO. B, Neutralization of SSTR2a-like staining in the ipsilateral hemisphere by preincubation with the immunizing peptide. A, Note the absence of SSTR2a-LIR from the infarct (penumbral margins are marked by arrowheads) and stronger SSTR2a-like staining in noninfarcted ipsilateral cortical regions (arrows) compared with the contralateral side, which shows SSTR2a-LIR in cortical infragranular layers 5/6 (V/VI), the claustrum (Cl), the dorsal endopiriform nucleus (DEn), and the lateral septum (LS). C-H, Details of the perifocal cortex (C, E, G) and the corresponding area on the contralateral side (D, F, H). D, F, H, In the contralateral cortex, SSTR2a-LIR is diffusely distributed over the cortical neuropil (D), with neuronal cell bodies (F; asterisks) and dendrites (H; arrow) exhibiting weak staining. C, E, G, Perifocally, strong SSTR2a-like staining is present in pyramidal-shaped neuronal cells (C; arrowheads) and dendrites (C; arrows), and a neuron in layer 5 (E), and dendrites in layer 4 (G; arrows) exhibit internalized cytoplasmic SSTR2-LIR. I-K, Details of the piriform cortex 3 hr after MCAO with intraventricular application of vehicle (I, J) and SSTR2 antagonist BIM-23627 (K) 30 min before MCAO. I′-K′, Magnifications taken from I-K. J, J′, Contralaterally, SSTR2a-LIR decorates the membrane and cytoplasm of perikarya as well as proximal and distal dendrites. I, I′, Ipsilaterally, high levels of internalized SSTR2a-LIR are present in the cytoplasm of perikarya and proximal dendrites, whereas distal dendrites are spared from any labeling. K, K′, Internalization of SSTR2a-LIR on the ipsilateral side is fully prevented by pretreatment with BIM-23627. Scale bars: A, B, 1 mm; C, D, I-K, 80 μm; E-H, I′-K′, 20 μm.
In addition to internalization, our immunohistochemical analysis revealed an increase in SSTR2a-like labeling in the neuropil in distinct ipsilateral brain regions after 6 hr. In ipsilateral versus contralateral comparison (Fig. 1A), these comprised the cingulate, frontal, insular, and piriform areas of the cortex (arrows), claustrum (Cl), and dorsal endopiriform nucleus (DEn).
The SSTR2 antagonist BIM-23627 blocks SSTR2a internalization in vitro and in vivo
To substantiate that SSTR2 was internalized by endogenously released ligands, we next attempted to block this process pharmacologically. The SSTR2-selective antagonist BIM-23627 proved to be a suitable compound to block ligand-induced SSTR2 internalization in HEK293 cells stably expressing SSTR2a (Fig. 2A-D). Quantitatively, internalization of SSTR2a-LIR by 0.1 μm somatostatin-14 was reduced from 58% to 15% by coapplication of 1 μm BIM-23627 (Fig. 2E) (p < 0.05). When BIM-23627 was applied at 1 μm without somatostatin-14, it did not affect the subcellular distribution of SSTR2a (Fig. 2D,E). Next, 2.5 nmol of the compound or vehicle was injected into the ventricle 30 min before MCAO and sham operation. Analysis was performed in the ventral cortical penumbra (piriform cortex) 3 hr after the operation. In this area, SSTR2a-LIR strongly decorated the plasma membrane and, to a weaker extent, the cytoplasm of neurons in naive rats, sham-operated rats receiving BIM-23627 or vehicle, and contralaterally in ischemic rats receiving BIM-23627 or vehicle (Fig. 1J,J′) (shown for the latter group). Thus, BIM-23627 had no effect on the subcellular distribution of SSTR2a under normal conditions. After ischemia, however, internalization of SSTR2a in the ipsilateral hemisphere, as seen after vehicle treatment (Fig. 1I,I′), was absent after BIM-23627 treatment (Fig. 1K,K′).
Figure 2.

Somatostatin-14 (SS14)-induced internalization of SSTR2a is blocked by the SSTR2 antagonist BIM-23627 in vitro. A-D, Confocal images of SSTR2a-LIR in HEK293 cells stably expressing SSTR2a. A, In untreated (control) cells, SSTR2a-LIR is present at the plasma membrane. B, After stimulation with 0.1 μm SS14, most SSTR2a-LIR is internalized. C, Compared with B, SS14-induced internalization is reduced by coapplication of 1 μm BIM-23627. D, Compared with A, 1 μm BIM-23627 does not alter the subcellular distribution of SSTR2a-LIR. E, Quantitative analysis of SSTR2a internalization by ELISA as described in Materials and Methods. Differences in membrane-associated SSTR2a-LIR between control cells and ligand-exposed cells (internalization) were expressed as percentages of membrane-associated SSTR2a-LIR in control cells. Note the strong reduction of somatostatin-14 (SS-14)-induced internalization after coapplication of BIM-23627 and no internalization after BIM-23627.
Depletion of somatostatin from axonal terminals
Given the evidence for activation of SSTR2a by endogenous ligands after focal ischemia, we assessed postischemic changes of the somatostatin content in terminals by immunohistochemistry (Fig. 3). Somatostatin-LIR in terminals was strongly decreased ipsilaterally in the entire cortex, claustrum, and dorsal endopirifom nucleus at 3 hr, 6 hr, and 1 d after MCAO (shown in detail for the cortex at 6 hr as ipsilateral versus contralateral comparison) (Fig. 3B,C). During this time, somatostatin-LIR was unchanged in the ipsilateral striatum and septal area (Fig. 3A), as well as contralaterally (compared with naive animals; data not shown). Thus, the spatial pattern of the depletion of somatostatin from axonal terminals during the first 24 hr after MCAO mimicked that of SSTR2a internalization. After 2 and 4 d, somatostatin-LIR was similar to naive rats in all noninfarcted areas, and a slight increase was observed ipsilaterally in the cortex (data not shown), suggesting that absence of somatostatin-LIR from terminals in this area during the first 24 hr after MCAO was not attributable to degeneration of axons.
Figure 3.
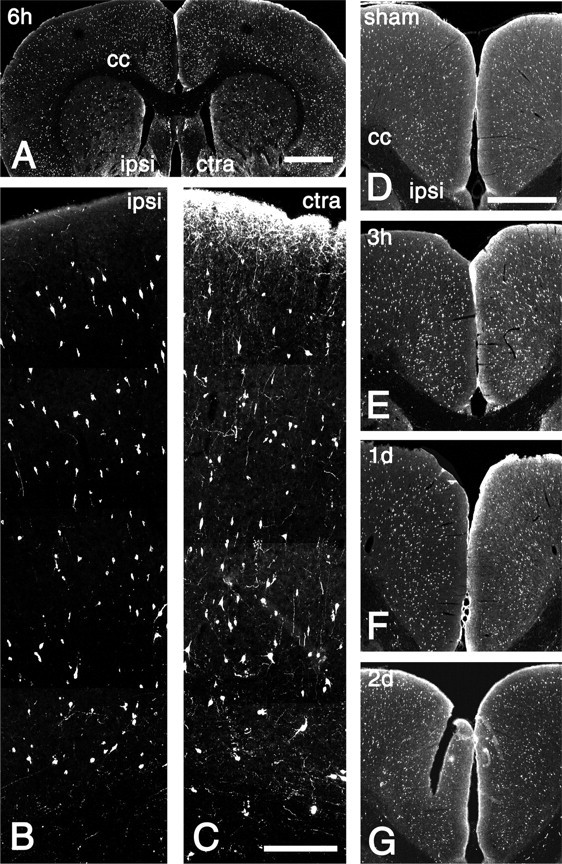
Somatostatin-LIR in the cerebral cortex after MCAO. A, Confocal image of a coronal section through the rat forebrain 6 hr after MCAO. The ipsilateral (ipsi) and contralateral (ctra) hemispheres as well as the corpus callosum (cc) are indicated. Note the decreased intensity of somatostatin-LIR in the ipsilateral cerebral cortex but not in the striatal and septal areas compared with the contralateral side. B, C, Layers 1-6 of the ipsilateral frontal cortex (B) and the corresponding contralateral area (C) 6 hr after MCAO. C, Contralaterally, somatostatin-immunoreactive fibers form a dense network in layer 1 and a loose network in the other layers. B, Ipsilaterally, only few somatostatin-immunoreactive fibers are detectable. B, C, There are no ipsilateral versus contralateral differences in the number of somatostatin-immunoreactive perikarya. D-G, Somatostatin-LIR in the ipsilateral and contralateral areas of the cingulate and frontal cortex 1 d after sham operation (sham) and 3 hr, 1 d, and 2 d after MCAO. Note the reduced levels of somatostatin-LIR in the ipsilateral cortex 3 hr and 1 d, but not 2 d, after MCAO. Scale bars: A, 2 mm; B, C, 300 μm; D-G, 1 mm.
Differential regulation of somatostatin and cortistatin mRNA expression after focal ischemia
To determine whether ischemia-induced changes of somatostatin-LIR were related to alterations in somatostatin gene expression, levels of somatostatin mRNA were quantified at 6 hr, 1 d, 2 d, and 4 d after MCAO and sham operation by in situ hybridization ipsilaterally and contralaterally in the noninfarcted brain regions identified in Figure 4P, as well as in the dorsal and ventral penumbra (Fig. 4G, arrows). Areas exhibiting significant changes compared with the stage-matched, sham-operated groups are marked by asterisks in Figure 4. Significant increases (p < 0.05) in somatostatin mRNA expression were found ipsilaterally in the cortex after 2 d (+71 ± 32%) and 4 d (+61 ± 33%) as well as in the dorsal striatum after 2 d (+50 ± 32%) and 4 d (+59 ± 38%). Expression of somatostatin mRNA was unchanged in all noninfarcted areas after 6 hr and at 1 d. Thus, the loss of somatostatin from cortical axonal terminals 3 hr to 1 d after MCAO was associated with increased somatostatin mRNA levels after 2 and 4 d, possibly reflecting a reactive increase in the synthesis of the peptide.
Figure 4.
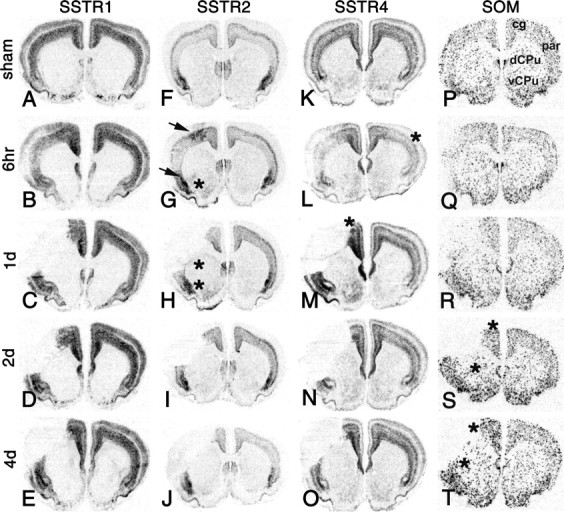
Changes in SSTR1 (A-E), SSTR2 (F-J), SSTR4 (K-O), and pre-prosomatostatin (SOM; P-T) mRNA expression in the rat forebrain after MCAO. X-ray autoradiograms of hybridized coronal sections 6 hr, 1 d, 2 d, and 4 d after focal ischemia and selected time points after sham operation [time points with the most pronounced changes after MCAO: 1 d (A, K); 6 hr (F); 4 d (P)]. In the infarcted parietal cortex, mRNA expression of the four genes is reduced at 6 hr and undetectable at later time points. A-E, Outside the infarct, expression of SSTR1 mRNA is unaltered after MCAO. F-J, SSTR2 mRNA expression is strongly increased in perifocal cortical areas 6 hr after MCAO (G; arrows). H-J, SSTR2 expression in the cortex is unchanged after 1, 2, and 4 d. In the striatum, SSTR2 mRNA is expressed at very low constitutive levels (F) and increased ipsilaterally in the ventral part after 6 hr (G; asterisk) as well as in the ventral and dorsal parts after 1 d (H; asterisks). K-O, SSTR4 mRNA levels are decreased contralaterally in the parietal cortex after 6 hr (L; asterisk) and increased in noninfarcted cortical areas after 1 d (M; asterisk). SSTR4 mRNA levels at 2 and 4 d are unchanged. M, The depicted animal exhibits a strong upregulation of SSTR4 mRNA levels in the ipsilateral striatum (changes in SSTR4 expression in the striatum were observed only in a subpopulation of animals 1 d after MCAO). P-T, Somatostatin mRNA levels are slightly increased ipsilaterally in the cortex and striatum after 2 and 4 d (S, T; asterisks). cg, Cingulate cortex; par, parietal cortex; dCPu, dorsal caudate-putamen; vCPu, ventral caudate-putamen.
Cortistatin is the second endogenous ligand at the SSTRs and is expressed selectively in the cerebral cortex. Surprisingly, cortistatin mRNA levels exhibited a strong and sustained decrease in both cortical hemispheres after MCAO (Fig. 5, asterisks), with the maximal change in nonlesioned areas occurring in the cingulate/frontal cortex after 2 d. Because sham operation provoked changes in cortistatin expression, a group of naive rats was used as reference to quantify changes in the sham-operated and ischemic groups (Table 1). The downregulation of cortistatin expression was further confirmed by cell counts in the ipsilateral cingulate/frontal cortical area, which revealed a decrease in the number of cortistatin mRNA-positive neurons per millimeters squared by 73% (p < 0.01) (Fig. 6A,D).
Figure 5.
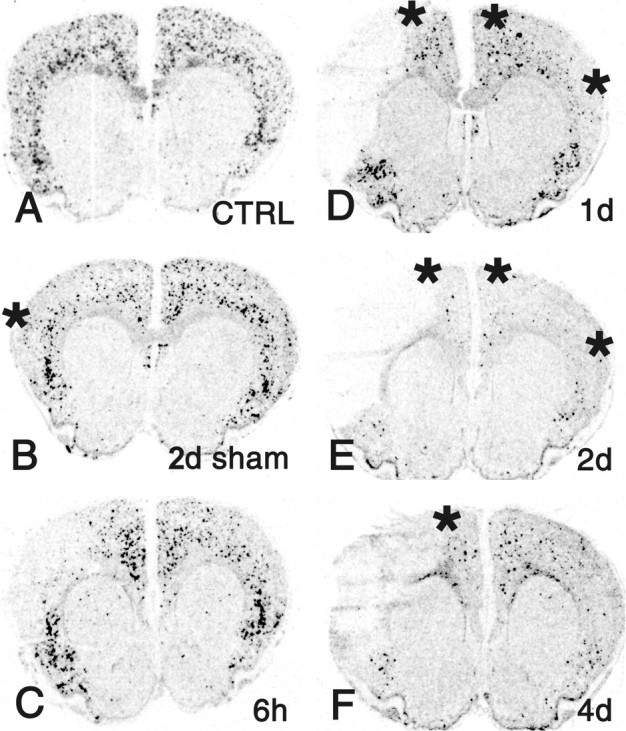
Changes in cortistatin mRNA expression in the rat forebrain after MCAO. X-ray autoradiograms of hybridized coronal sections from an untreated rat (CTRL; A), 2 d after sham operation (B), and 6 hr, 1 d, 2 d, and 4 d after focal ischemia (C-F). Cortistatin expression levels are strongly decreased in ipsilateral and contralateral cerebrocortical areas 1, 2, and 4 d after MCAO (asterisks). Note also the decreased cortistatin mRNA expression ipsilaterally in the cortex in response to sham operation (B; asterisk).
Table 1.
Influence of MCAO on cortistatin mRNA levels in the cerebral cortex
|
|
Sham |
MCAO |
||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Areas of interest |
6 hr |
1 d |
2 d |
4 d |
6 hr |
1 d |
2 d |
4 d |
||||||
| Cg/Fr (i) | 60 ± 24 | 63 ± 33 | 53 ± 36* | 57 ± 21 | 62 ± 14 | 34 ± 10** | 12 ± 5**† | 16 ± 4**† | ||||||
| Cg/Fr (co) | 52 ± 26 | 72 ± 23 | 78 ± 41 | 76 ± 27 | 49 ± 20 | 26 ± 12** | 12 ± 5**†† | 57 ± 21 | ||||||
| Par (i) | 52 ± 18 | 66 ± 48 | 87 ± 47 | 94 ± 24 | n.d. | n.d. | n.d. | n.d. | ||||||
| Par (co) |
51 ± 19 |
54 ± 18 |
85 ± 35 |
93 ± 37 |
42 ± 16 |
32 ± 14 |
23 ± 14*†
|
78 ± 48 |
||||||
Values express mean mRNA levels ± SD at 6 hr, 1 d, 2 d, and 4 d after sham operation (n = 4) and MCAO (n = 6) as a percentage of naive animals (n = 3). Expression in the infarcted parietal cortex was not determined (n.d.). *p < 0.05, **p < 0.01 versus naive group; †p < 0.05, ††p < 0.01 versus stage-matched, sham-operated group; one-way ANOVA followed by Newman-Keuls multiple comparison test. Cg/Fr, Cingulate and frontal regions of the cortex; Par, parietal cortex; i, ipsilateral side; co, contralateral side.
Figure 6.
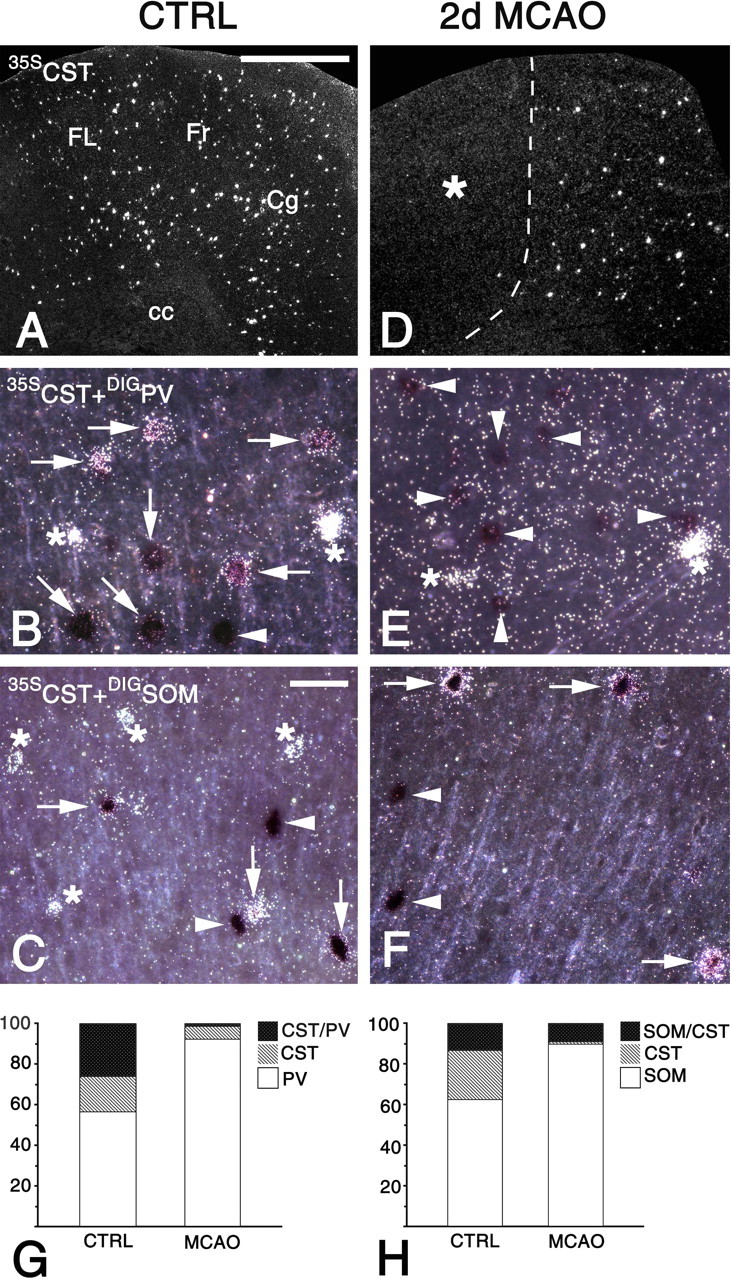
Phenotypes of neurons expressing cortistatin (CST) mRNA 2 d after MCAO. A-F, In situ hybridizations with a 35S-labeled probe for CST mRNA (35SCST). A, D, Dark-field micrographs of the cingulate (Cg), frontal (Fr), and forelimb (FL) areas of the cortex of an untreated rat (CTRL; A) and the corresponding region ipsilateral to MCAO (D). A, CST mRNA is expressed by numerous neurons throughout all cortical layers. D, The infarcted (asterisk) and noninfarcted areas are segregated by a dashed line. Note that in the noninfarcted cortical area, the number of CST-expressing cells is strongly reduced. B, C, E, F, For phenotype identification of CST-expressing neurons, simultaneous hybridization of a 35S-labeled probe for CST mRNA (seen as grains) and digoxigenin-labeled probes for parvalbumin (DIGPV; B, E) and somatostatin (DIGSOM; C, F) mRNAs was performed (DIGPV and DIGSOM are detected by colored enzymatic reaction products). Shown are high-power dark-field micrographs from layer 6 of the frontal cortex of a CTRL (B, C) and 2 d after MCAO (E, F). B, The coexpression analysis reveals a PV-positive neuron (arrowhead), CST/PV-positive neurons (arrows), and CST-positive neurons (asterisks) in the untreated cortex. E, After focal ischemia, CST-positive (asterisks) and parvalbumin-positive (arrowheads) neurons but no CST/parvalbumin-positive neurons are observed. C, Detection of an SOM-positive neuron (arrowhead), CST-positive neurons (asterisks), and CST/SOM-positive neurons (arrows) in the untreated cortex. F, Presence of SOM-positive (arrowheads) as well as CST/SOM-positive (arrows) neurons and absence of CST-positive neurons 2 d after focal ischemia. G, H, Semiquantitative analysis of the coexpression in layers 2-6 of the cingulate and frontal cortex 2 d after MCAO and the corresponding area of naive rats (CTRL). G, Proportions (percentages) of PV/CST-, CST-, and PV-expressing neurons in CTRL versus MCAO were 25.9 ± 5.1 versus 1 ± 0.7, 17.5 ± 5.1 versus 5.6 ± 2.2, and 56.6 ± 5.7 versus 93.3 ± 2.4, respectively. H, Proportions (percentages) of SOM/CST-, CST-, and SOM-expressing neurons in CTRL versus MCAO were 13.2 ± 4.6 versus 8.7 ± 1.7, 24.5 ± 1.0 versus 1.7 ± 0.8, and 62.3 ± 11.9 versus 89.6 ± 2.1, respectively. cc, Corpus callosum. Scale bars: A, D, 1.25 mm; B, C, E, F, 100 μm.
Cortistatin is known to be synthesized by somatostatin-expressing and parvalbumin-expressing cortical GABAergic neurons (de Lecea et al., 1997). Because these types comprise two nonoverlapping populations with distinct intracortical connectivity (for review, see Kawaguchi and Kondo, 2002), the effects of cortistatin derived from parvalbumin-positive and somatostatin-positive circuits are likely to affect different targets. To test whether cortistatin mRNA levels were differentially regulated in the two neuronal types, digoxigenin-labeled probes for parvalbumin and somatostatin were each cohybridized with a 35S-labeled probe for cortistatin (Fig. 6B,C,E,F), and coexpression analysis was performed ipsilaterally in the cingulate/frontal cortex 2 d after MCAO compared with naive animals. In controls, cortistatin mRNA was frequently detected in the two neuronal types (Fig. 6B,C, arrows). After MCAO, cortistatin expression was virtually absent from the parvalbumin-expressing population (Fig. 6E, arrowheads) and restricted to the somatostatin-expressing population (Fig. 6F, arrows), whereas numbers of parvalbumin- and somatostatin-expressing neurons per area were unaltered. The observation that cortistatin was downregulated in parvalbumin-expressing neurons was further substantiated by semiquantitative analyses, in which marker-positive cells, cortistatin-positive cells, and marker/cortistatin-positive cells were counted and the relative proportions were calculated (Fig. 6G,H). Together, the analyses of somatostatin and cortistatin mRNA expression showed that the levels of the two mRNAs were oppositely regulated during the late stages after focal ischemia (1-4 d), which was attributable to massive downregulation of cortistatin in nonsomatostatinergic neurons.
Upregulation of SSTR2 mRNA expression in the cortical penumbra
Because activation and internalization of SSTR2 has been reported to increase SSTR2 mRNA expression in cortical slices (Boudin et al., 2000), we next determined changes in SSTR2 mRNA levels (regions of interest corresponded to those for somatostatin) (Fig. 4P). All changes in SSTR2 mRNA expression were essentially confined to the ipsilateral side (Fig. 4G-J, highlighted area). After 6 hr, SSTR2 mRNA levels in the cortex were increased in the dorsal penumbra (+106 ± 42%; p < 0.01), ventral penumbra (+178 ± 22%; p < 0.01), and cingulate/frontal area (+37 ± 23%; p < 0.05). In addition, SSTR2 mRNA expression was increased in the ventral striatum after 6 hr (+263 ± 158%; p < 0.05) and 1 d (+395 ± 97%; p < 0.01) as well as in the dorsal striatum after 1 d (+199 ± 116%; p < 0.05). Thus, cortical regions with internalized SSTR2a-LIR exhibited increased SSTR2 mRNA levels 6 hr after MCAO.
Unchanged mRNA expression of SSTR1, SSTR4, and SSTR5 in the penumbra
To test whether the observed regulation was selective for SSTR2, changes in mRNA levels of SSTR1, SSTR4, and SSTR5 were analyzed. SSTR1 mRNA was abundantly expressed in the cortex, but the levels were unchanged in nonlesioned areas (Fig. 4A-E). Expression levels of SSTR4 mRNA were decreased contralaterally in the parietal cortex after 6 hr (-29 ± 18%; p < 0.05) and increased ipsilaterally in the cingulate/frontal cortex after 1 d (+73 ± 49%; p < 0.05) (Fig. 4L,M, asterisks). Two animals showed a strong increase in SSTR4 mRNA levels in the ipsilateral striatum after 1 d (Fig. 4M). Striatal SSTR4 mRNA levels of the entire group, however, were not statistically different when compared with the respective sham-operated group. Levels of SSTR5 mRNA were below the detection limit in the cortex and striatum (data not shown). Thus, upregulated mRNA expression in the cortical penumbra during the first 6 hr after focal ischemia was selective for the SSTR2 gene and not observed for the SSTR1, SSTR4, and SSTR5 genes.
Induction of SSTR2 in penumbral pyramidal cells
The induction of SSTR2 mRNA expression in the dorsal cortical penumbra (dorsomedial forelimb area) after 6 hr was then analyzed in detail (Fig. 7). Cell counts in this area showed that the number of SSTR2 mRNA-positive cells per millimeters squared were increased by 420% in supragranular layers (p < 0.01) and by 190% in infragranular layers (p < 0.05) compared with the corresponding area of sham-operated rats (Fig. 7A-D), indicating that expression of the SSTR2 gene was induced in a substantial proportion of penumbral cells. To further characterize the transmitter types of SSTR2-expressing neurons in the penumbra and normal cortex, coexpression analyses were performed. Digoxigenin-labeled probes for GAD67 mRNA and VGLUT1 mRNA, which identify GABAergic neurons and glutamatergic pyramidal cells, respectively, were used. Together, these two neuronal populations comprise the preponderant part of cortical neurons. The probes for the marker mRNAs were each cohybridized with the probe for SSTR2 mRNA in sections from rats 6 hr after sham operation and MCAO (Fig. 7E-H). For both combinations of probes, marker-positive cells, SSTR2-positive cells, and marker/SSTR2-positive cells were counted, and the proportions of the three populations from all counted cells were calculated (Fig. 7I,J). Because SSTR2 expression differs strongly between infragranular and supragranular layers (Fig. 7C), segregated analyses were performed for layers 2/3 and 5/6. After sham treatment, SSTR2/GAD-expressing neurons formed a small population, both in layers 2/3 and 5/6 (Fig. 7I). SSTR2/VGLUT1-positive neurons constituted only a very small population in layers 2/3 but the predominant population in layers 5/6 (Fig. 7J). These patterns suggest that in the cortex of the controls, SSTR2 mRNA was expressed at high levels by the majority of pyramidal cells in layers 5/6, sparsely by layer 2/3 pyramidal cells, and at high levels by few GABAergic neurons in layers 2-6. After ischemia, the proportion of SSTR2/VGLUT1-positive neurons increased strongly both in layers 2/3 and 5/6 (Fig. 7J). Correspondingly, the proportion of SSTR2-positive/GAD-negative neurons increased, and the proportion of SSTR2-positive/GAD-positive neurons decreased (Fig. 7I). This proportional shift showed that SSTR2 mRNA expression was upregulated in almost all pyramidal cells in the penumbra. In contrast, SSTR2 mRNA was detected only in a small population of GABAergic inhibitory neurons, both after sham treatment and ischemia.
Figure 7.
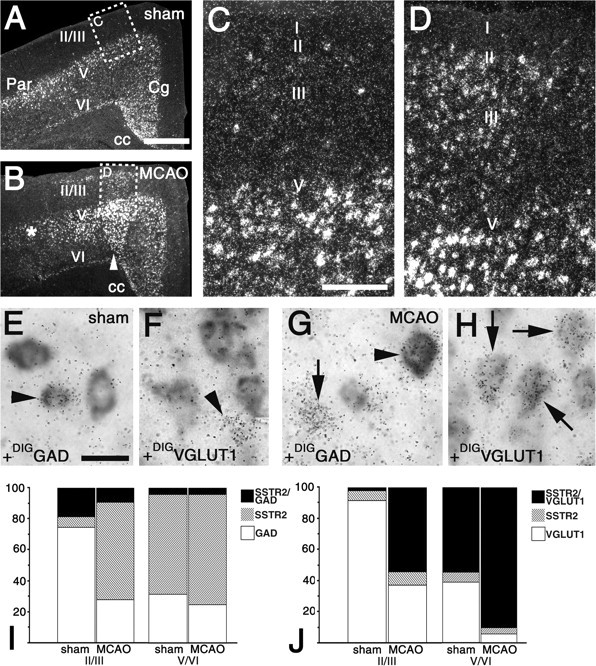
Upregulation of SSTR2 expression in glutamatergic neurons in the perifocal cerebral cortex. A-D, Dark-field micrographs of the cingulate (Cg), frontal, forelimb, and parietal (Par) areas after hybridization for SSTR2 mRNA. Pictures were taken from the ipsilateral side of a rat 6 hr after sham operation (A, C) and 6 hr after MCAO (B, D). B, The infarcted parietal area and the noninfarcted perifocal area are marked by an asterisk and an arrowhead, respectively. A, C, SSTR2 mRNA is constitutively expressed at high levels by numerous cells in cortical layers 5/6 (V/VI) and at moderate levels by very few cells in layers 2/3 (II/III). B, D, In the perifocal area of the ischemic cortex, SSTR2 expression is induced in numerous layer 2/3 cells and increased in layers 5/6 compared with A and C. E-H, For phenotype identification of SSTR2-expressing neurons, simultaneous hybridization of a 35S-labeled probe for SSTR2 mRNA and digoxigenin-labeled probes for GAD (DIGGAD; E, G) and VGLUT 1 (DIGVGLUT1; F, H) mRNAs was performed. The SSTR2 probe is seen as grains in E-H, and DIGGAD and DIGVGLUT1 are detected as gray enzymatic reaction products. Pictures are high-power micrographs from layers 2/3 of the perifocal cortex of a rat 6 hr after MCAO and the corresponding region of a rat 6 hr after sham operation. E, F, Arrowheads point to a GAD-positive neuron (E) and a VGLUT1-negative cell (F) that are SSTR2 mRNA positive. F, Note, SSTR2 expression is not observed in VGLUT1-positive neurons in layers 2/3. G, H, In the perifocal area, SSTR2 mRNA is detected in a GAD-negative cell (G; arrow) and three VGLUT1-positive neurons (H; arrows). G, Arrowhead points to a GAD-positive SSTR2-expressing neuron. I, J, Semiquantitative analysis of the coexpression in the perifocal cortex 6 hr after MCAO and the corresponding area of sham-operated rats. I, SSTR2/GAD-, SSTR2-, and GAD-expressing neurons. Proportions (percentages) in layers 2/3 of sham versus MCAO were 18.7 ± 1.8 versus 9.2 ± 1.8 (SSTR2/GAD), 6.8 ± 2.1 versus 62.9 ± 2.1 (SSTR2), and 74.5 ± 3.1 versus 27.9 ± 5.8 (GAD). Proportions (percentages) in layers 5/6 of sham versus MCAO were 4.2 ± 1.2 versus 4.4 ± 1.2 (SSTR2/GAD), 64.2 ± 2.5 versus 70.9 ± 5.0 (SSTR2), and 31.6 ± 3.0 versus 24.7 ± 3.8 (GAD). J, SSTR2/VGLUT1-, SSTR2-, and VGLUT1-expressing neurons. Proportions (percentages) in layers 2/3 of sham versus MCAO were 2.4 ± 0.23 versus 54.1 ± 8.4 (SSTR2/VGLUT1), 5.9 ± 1.0 versus 8.9 ± 3.3 (SSTR2), and 91.6 ± 1.0 versus 36.9 ± 11.7 (VGLUT1). Proportions (percentages) in layers 5/6 of sham versus MCAO were 54.6 ± 5.6 versus 90.1 ± 2.7 (SSTR2/VGLUT1), 6.4 ± 0.8 versus 3.9 ± 1.0 (SSTR2), and 39.0 ± 6.1 versus 6.0 ± 1.9 (VGLUT1). Scale bars: A, B, 1 mm; C, D, 200 μm; E-H, 25 μm.
Functional SSTR2 receptors contribute to the upregulation of SSTR2 mRNA
Given the close spatial and temporal correlation between SSTR2 activation and SSTR2 mRNA upregulation, we hypothesized that an SSTR2-mediated signal may contribute to the enhanced SSTR2 mRNA expression after focal ischemia. A similar mechanism has been proposed previously to counteract ligand-induced downregulation of SSTR2 after application of agonist to brain slices (Boudin et al., 2000). To test this hypothesis, wild-type mice and mutant mice with a sstr2 knock-out/lacZ knock-in were subjected to 6 hr MCAO. SSTR2 gene expression was then visualized by hybridization with a probe for SSTR2 mRNA in the wild types and a probe for lacZ mRNA in the mutants. Ipsilateral versus contralateral comparison of mRNA levels in wild types and mutants revealed strongly increased SSTR2 and moderately increased lacZ mRNA expression in the dorsal and ventral cortical penumbra (Fig. 8D,E, arrows). Quantitative analysis in the ventral penumbra and comparison with the corresponding contralateral area (Fig. 8A,B) showed a 2.7-fold upregulation of SSTR2 mRNA in the wild types (p < 0.01) and a 1.5-fold upregulation of lacZ mRNA in the mutants (p < 0.01). Thus, an SSTR2-derived signal was not required for enhanced SSTR2 gene expression. However, SSTR2 mRNA upregulation was significantly larger than lacZ mRNA upregulation (p < 0.01), suggesting that an SSTR2-derived signal might contribute to enhanced SSTR2 gene expression after MCAO.
Figure 8.

SSTR2 gene expression in SSTR2 knock-out/lacZ knock-in mice after focal ischemia. SSTR2 gene expression in wild-type mice (sstr2+/+; A, D) and SSTR2 knock-out/lacZ knock-in mice (sstr2-/-; B, E) subjected to 6 hr permanent MCAO was detected by isotopic in situ hybridization with probes for SSTR2 mRNA (A, D) and lacZ mRNA (B, E), respectively. D, E, Comparison of the ipsilateral and contralateral brain hemispheres reveals increased SSTR2 gene expression levels in dorsal and ventral penumbral regions (arrows) both in sstr2+/+ (D) and sstr2-/- (E) mice. A, B, Scatter plots showing quantification of hybridization signal intensities (nanoCuries per gram) performed in the area boxed in D and the corresponding contralateral area. A, In 11 sstr2+/+ mice, median values were significantly larger (p < 0.01) on the ipsilateral side (ipsi; 0.90 nCi/mg) than in the contralateral side (contra; 0.33 nCi/mg). B, A significant ipsilateral versus contralateral increase (p < 0.01) was seen also in 14 sstr2-/- mice (ipsi, 1.39 nCi/mg; contra, 0.91 nCi/mg). C, Scatter plot showing relative SSTR2 gene upregulation in the sstr2+/+ and sstr2-/- mice. The fold upregulation was calculated for each animal dividing ipsilateral by contralateral SSTR2 gene expression levels. Note the stronger upregulation in sstr2+/+ mice (median value, 2.7-fold) than in sstr2-/- mice (median value, 1.5-fold). A-C, Median values are indicated by dashed lines.
Reduced infarct size in SSTR2-deficient mice in models of transient and permanent focal ischemia
To address whether the endogenous somatostatin system might protect from or contribute to the infarction process via SSTR2, infarct volumes in SSTR2-deficient mutants and wild types were compared in models of transient and permanent MCAO (Fig. 9A,B). In both models, the infarcts were significantly smaller in the mutant mice than in the wild types. At 7 d after permanent MCAO, the infarct volume was reduced by 40% (p < 0.01). After transient MCAO, a 63% reduction of the infarct volume was found after 3 d (p < 0.05). The animals exhibited a neurological deficit score of ≥2 (i.e., moderate or severe deficit) 30 min after the onset of ischemia. At 3 d, the deficits were improved in the SSTR2-deficient group compared with wild type (Table 2).
Figure 9.

Reduced infarct size in SSTR2 knock-out/lacZ knock-in mice after transient and permanent MCAO. A, B, Scatter plots showing infarct volume in wild-type mice (sstr2+/+) and SSTR2 knock-out/lacZ knock-in mice (sstr2-/-) 3 d after transient MCAO and 7 d after permanent MCAO, respectively. In both models, median values in sstr2-/- mice are smaller than in sstr2+/+ mice: transient MCAO, 52.2 mm3 (sstr2+/+) versus 19.5 mm3 (sstr2-/-); permanent MCAO, 24.2 mm3 (sstr2+/+) versus 14.5 mm3 (sstr2-/-). C-E, Area expressing HSP70 mRNA 6 hr after permanent MCAO. D, E, X-ray autoradiograms of hybridized coronal sections showing a smaller HSP70 mRNA-positive area in sstr2-/- mice (E) than in sstr2+/+ mice (D). C, Scatter plots showing quantification of the HSP70 mRNA-positive area in eight sstr2+/+ mice and eight sstr2-/- mice. Note the smaller HSP70-positive area in sstr2-/- mice (median value, 1.26 mm2) than in sstr2+/+ mice (median value, 2.28 mm2). A-C, Median values are indicated by dashed lines.
Table 2.
Reduced neurological deficit in SSTR2 knock-out/lacZ knock-in mice
|
Score |
0 |
1 |
2 |
3 |
Mean ± SD |
|---|---|---|---|---|---|
| sstr2+/+ (n = 10) | 0 | 4 | 6 | 0 | 1.6 ± 0.5 |
| sstr2-/- (n = 10) |
3 |
5 |
2 |
0 |
0.9 ± 0.7 |
Neurological scoring was determined in sstr2+/+ versus sstr2-/- mice using a previously published method (Bederson et al., 1986) performed 72 hr after MCAO/reperfusion. The mean neurological deficit in sstr2-/- mice is reduced compared with sstr2+/+ mice. Scoring: 0, normal function; 1, flexion of contralateral torso and forelimb on lifting the animal by the tail; 2, circling; 3, loss of walking or righting reflex.
If excessive activation of SSTR2 in perifocal regions hours after MCAO contributes to infarct expansion, early markers of neuronal damage are expected to be differentially activated in SSTR2-deficient mice and wild-type mice. Previously, perifocal regions expressing HSP70 mRNA during the first hours after MCAO have been shown to correspond to the penumbral region undergoing degeneration until the lesion gained its full size (Hata et al., 2000). As determined by in situ hybridization 6 hr after permanent MCAO, the HSP70 mRNA-positive area was reduced by 45% (p < 0.01) in SSTR2-deficient mice compared with wild-type mice. Thus, SSTR2-deficient mice exhibited smaller mature infarcts and smaller HSP70-positive penumbral regions after MCAO.
Discussion
Here we provide the first evidence for a role of the endogenous somatostatin system in focal cerebral ischemia. We show internalization and upregulation of SSTR2 hours after MCAO in perifocal pyramidal neurons. Using SSTR2-deficient mice with lacZ expression under the control of the SSTR2 promoter, functional SSTR2 signaling was shown to contribute to ischemia-induced upregulation of SSTR2. In two distinct models of cerebral ischemia, SSTR2-deficient mice exhibited smaller infarct volumes than wild-type mice, strongly suggesting the involvement of SSTR2 in neurodegeneration under ischemic conditions. In addition, we show strong downregulation of cortistatin expression in exofocal cortical GABAergic neurons days after MCAO.
Activation and upregulation of SSTR2 in the cerebral cortex after focal ischemia
Previous studies established SSTR2 internalization in cortical neurons in vivo as an indicator for SSTR2 activation after intracortical application of SSTR2 ligand (Csaba et al., 2001). This study provides the first evidence for in vivo internalization of SSTR2 by endogenous ligands and shows that SSTR2 internalization can be blocked pharmacologically by intracerebral application of an SSTR2 antagonist. The close association of SSTR2 internalization and SSTR2 mRNA upregulation in cortical regions suggests an involvement of an SSTR2-dependent signal in SSTR2 upregulation. SSTR2 activation is known to trigger upregulation of SSTR2 mRNA expression in tumor cells in vitro (Bruno et al., 1994; Hukovic et al., 1996) and in vivo (Froidevaux et al., 1999). Recently, intravenous application of SSTR2-preferring ligands has been shown to be associated with increased SSTR2 mRNA levels in the arcuate nucleus, which is accessible for systemically administered drugs (Tannenbaum et al., 2001). In brain slices, SSTR2 activation induces SSTR2 mRNA upregulation by a mechanism that involves SSTR2 internalization (Boudin et al., 2000). The assumption that an SSTR2-dependent mechanism contributes to ischemia-induced upregulation of SSTR2 gene expression is further supported by our finding that, in mice expressing lacZ instead of SSTR2, upregulation of SSTR2 gene expression was reduced. Notably, the observation that lacZ mRNA levels were increased in SSTR2-deficient mice after ischemia provides evidence for additional SSTR2-independent mechanisms. Thus, SSTR2-dependent and SSTR2-independent signals contribute to postischemic upregulation of SSTR2 expression that may represent a regulatory mechanism to compensate for SSTR2 desensitization. Although activation of SSTR2 is apparently linked to enhanced SSTR2 expression, sustained downregulation of cortistatin was not associated with changes in SSTR expression. Our comparative expression analysis of the five SSTRs showed further that upregulation in perilesional areas during the first hours after MCAO was selective for SSTR2 and points to a particular relevance of this SSTR subtype in the pathobiology of cerebral ischemia.
Involvement of the endogenous somatostatin system in post-stroke neurodegeneration
Internalization of SSTR2 in ipsilateral perifocal and exofocal cortical regions hours after MCAO was paralleled by depletion of somatostatin from axonal terminals, suggesting excessive release of the peptide. In perifocal regions, reduced protein synthesis (Christensen et al., 1996) may contribute to the observed depletion of axonal somatostatin. SSTR2 expression was detected in the vast majority of perifocal neurons that are at risk to die. Smaller mature infarcts in SSTR2-deficient mice suggest that activation of SSTR2 in perifocal neurons contributes to neurodegeneration under ischemic conditions. The reduced final infarct size corresponded to a smaller perifocal HSP70-positive area in the mutants 6 hr after MCAO. Because HSP70 is an established marker of neuronal stress, this finding supports the assumption that rapid activation of SSTR2 in the penumbra contributes to infarct growth. Along with the reduction in infarct size, we observed a significant improvement in neurological sensory-motor scores, which supports the notion that lack of SSTR2 had functional consequences in post-stroke neurodegeneration.
The MCAO models we applied are different with respect to the infarcted brain regions. Although permanent occlusion of the distal middle cerebral artery causes neurodegeneration in the cerebral cortex, transient occlusion of the proximal middle cerebral artery affects the striatum and cortex (Welsh et al., 1987; Katchanov et al., 2003). In both brain regions, somatostatin is contained in local circuits and acts on SSTR2-expressing projection neurons (Hicks et al., 1998; Vilchis et al., 2002; Allen et al., 2003).
Which SSTR2-activated pathways may contribute to neurodegeneration under ischemic conditions? Elevated cAMP levels are important survival and differentiation signals in cultured neurons (Rydel and Greene, 1988; Tanaka et al., 1997; Hanson et al., 1998; Putcha et al., 1999; Piiper et al., 2002; Vaudry et al., 2002). The Gs-coupled EP2 E-prostanoid receptor reduces neuronal death in cultured neurons subjected to oxygen glucose deprivation in a cAMP-dependent manner and is protective in focal cerebral ischemia (McCullough et al., 2004). Conversely, Gi-coupled SSTR2 may interfere with neuronal survival in the penumbra by reducing neuronal cAMP levels. In addition, SSTR2 may induce apoptosis by activating SHP-1 (Guillermet et al., 2003), a tyrosine phosphatase that blocks neurotrophin- and nicotine-induced neuroprotective signals and induces neuronal death (Marsh et al., 2003; Shaw et al., 2003). Furthermore, somatostatin has been shown to activate a large variety of potassium channels (Patel et al., 1995). Potassium efflux from mitochondria and the cytoplasm through activated potassium channels has been identified in neurons subjected to chemical hypoxia, excitotoxicity, and staurosporine treatment (Liu et al., 2003). In a variety of in vitro and in vivo models, activated potassium channels contribute to neuronal death (Yu et al., 1998, 1999; McLaughlin et al., 2001; Liu et al., 2003; Pal et al., 2003; Wei et al., 2003).
Regulation of somatostatin and cortistatin in GABAeric cortical neurons
The initial depletion of somatostatin from axonal terminals was followed by a slight increase in somatostatin mRNA and a strong decrease in cortistatin mRNA levels 2 and 4 d after MCAO. These changes occurred in nonlesioned brain areas at stages when the infarction process was complete and reflect most likely adaptive changes of the cortical neuronal network. Somatostatin and cortistatin are known to depress neuronal activity in the cerebral cortex (Spier and de Lecea, 2000). In our studies, SSTR2 was detected only in a very small fraction of the inhibitory cortical GABAergic neurons. In contrast, strong expression of SSTR2 was found in 58% of pyramidal cells in infragranular layers under normal conditions and in >90% of pyramidal cells in all perifocal layers 6 hr after MCAO. This suggests that cortistatin and somatostatin exert their SSTR2-mediated effects directly on excitatory neurons, both under normal and postischemic conditions. However, additional SSTR-independent effects of cortistatin have to be considered (Spier and de Lecea, 2000).
As reported previously for the somatosensory cortex (de Lecea et al., 1997), we found cortistatin in somatostatinergic and parvalbumin-positive neurons in frontal and cingulate regions. Parvalbumin-positive and somatostatin-positive neurons are two distinct populations of GABAergic local circuit neurons in the frontal cerebral cortex (Kawaguchi and Kondo, 2002). Although somatostatinergic neurons have ascending axons that tend to target the distal dendrites of pyramidal cells, parvalbumin-positive neurons tend to target the proximal dendrites and perisomatic regions (Freund, 2003). Dendritic inhibition is considered to control the excitatory input and plasticity of pyramidal cells, whereas perisomatic inhibition is thought to control output and synchronization of pyramidal cells (Freund, 2003). Thus, although cortistatin and somatostatin share similar pharmacological characteristics at the known SSTRs, the localization of cortistatin in neurons with a connectivity different from that of somatostatinergic neurons may enable distinct SSTR-mediated functions of both peptides in the cortical circuitry. The regulation pattern of cortistatin and somatostatin we observed after MCAO was completely different from the regulation of these genes in a kainic acid seizure model, in which somatostatin expression was strongly increased in pyramidal cells and cortistatin expression was unaltered (Calbet et al., 1999). To our knowledge, we provide the first report of sustained downregulation of cortistatin expression after cortical lesion. Given the neuromodulatory role of cortistatin (Spier and de Lecea, 2000), loss of cortistatin as an inhibitory cotransmitter in GABAergic circuits may contribute to known alterations in cortical network activity after focal ischemia (Carmichael and Chesselet, 2002).
Conclusion
Together, this study provides the first evidence for activation and upregulation of SSTR2 in perifocal pyramidal neurons after focal ischemia. Upregulation of SSTR2 is most likely the result of both SSTR2-derived and heterologous signals and may counteract SSTR2 desensitization. Excessive activation of SSTR2 in perifocal neurons by endogenous ligands is likely to account for the larger infarcts in wild-type mice than in SSTR2-deficient mice. Sustained loss of cortistatin expression in inhibitory circuits regulating output and synchronization of pyramidal cells may contribute to altered excitability of nondegenerating cortical areas during late stages after ischemia.
Footnotes
This work was supported by grants from the Deutsche Forschungsgemeinschaft (Ra 698/4 to C. Rauca and V.H.; En343/7-1 to M.E.); the Bundesministerium für Bildung, Wissenschaft, Forschung und Technologie (NBL3 to R.K.S.); and Volkswagen-Stiftung (M.E.). We thank Dr. Michael Culler (Biomeasure, Milford, MA) for the generous gift of BIM-23627 and Dr. Hans-Jürgen Kreienkamp (Institut für Zellbiochemie und Klinische Neurobiologie, Universität Hamburg, Hamburg, Germany) for kindly providing rat SSTR cDNAs. Excellent technical assistance by Anke Reichenauer and Karina Schäfer is gratefully acknowledged.
Correspondence should be addressed to Dr. Ralf K. Stumm, Institut für Pharmakologie und Toxikologie, Otto-von-Guericke-Universität Magdeburg, Leipziger Strasse 44, 39120 Magdeburg, Germany. E-mail: ralf.stumm@medizin.uni-magdeburg.de.
G. Tulipano's present address: Department of Biomedical Sciences and Biotechnology, University of Brescia, 25123 Brescia, Italy.
Copyright © 2004 Society for Neuroscience 0270-6474/04/2411404-12$15.00/0
R.K.S. and C.Z. contributed equally to this work.
References
- Allen JP, Canty AJ, Schulz S, Humphrey PP, Emson PC, Young HM (2002) Identification of cells expressing somatostatin receptor 2 in the gastrointestinal tract of Sstr2 knockout/lacZ knockin mice. J Comp Neurol 454: 329-340. [DOI] [PubMed] [Google Scholar]
- Allen JP, Hathway GJ, Clarke NJ, Jowett MI, Topps S, Kendrick KM, Humphrey PP, Wilkinson LS, Emson PC (2003) Somatostatin receptor 2 knockout/lacZ knockin mice show impaired motor coordination and reveal sites of somatostatin action within the striatum. Eur J Neurosci 17: 1881-1895. [DOI] [PubMed] [Google Scholar]
- Bederson JB, Pitts LH, Tsuji M, Nishimura MC, Davis RL, Bartkowski H (1986) Rat middle cerebral artery occlusion: evaluation of the model and development of a neurologic examination. Stroke 17: 472-476. [DOI] [PubMed] [Google Scholar]
- Binaschi A, Bregola G, Simonato M (2003) On the role of somatostatin in seizure control: clues from the hippocampus. Rev Neurosci 14: 285-301. [DOI] [PubMed] [Google Scholar]
- Boudin H, Sarret P, Mazella J, Schonbrunn A, Beaudet A (2000) Somatostatin-induced regulation of SST(2A) receptor expression and cell surface availability in central neurons: role of receptor internalization. J Neurosci 20: 5932-5939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruno JF, Xu Y, Berelowitz M (1994) Somatostatin regulates somatostatin receptor subtype mRNA expression in GH3 cells. Biochem Biophys Res Commun 202: 1738-1743. [DOI] [PubMed] [Google Scholar]
- Calbet M, Guadano-Ferraz A, Spier AD, Maj M, Sutcliffe JG, Przewlocki R, de Lecea L (1999) Cortistatin and somatostatin mRNAs are differentially regulated in response to kainate. Brain Res Mol Brain Res 72: 55-64. [DOI] [PubMed] [Google Scholar]
- Carmichael ST, Chesselet MF (2002) Synchronous neuronal activity is a signal for axonal sprouting after cortical lesions in the adult. J Neurosci 22: 6062-6070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christensen T, Bruhn T, Frank L, Diemer NH (1996) Differential effect of NMDA and AMPA receptor blockade on protein synthesis in the rat infarct borderzone. Acta Neurol Scand 93: 160-167. [DOI] [PubMed] [Google Scholar]
- Csaba Z, Bernard V, Helboe L, Bluet-Pajot MT, Bloch B, Epelbaum J, Dournaud P (2001) In vivo internalization of the somatostatin sst2A receptor in rat brain: evidence for translocation of cell-surface receptors into the endosomal recycling pathway. Mol Cell Neurosci 17: 646-661. [DOI] [PubMed] [Google Scholar]
- Culmsee C, Stumm RK, Schafer MK, Weihe E, Krieglstein J (1999) Clenbuterol induces growth factor mRNA, activates astrocytes, and protects rat brain tissue against ischemic damage. Eur J Pharmacol 379: 33-45. [DOI] [PubMed] [Google Scholar]
- de Lecea L, Criado JR, Prospero-Garcia O, Gautvik KM, Schweitzer P, Danielson PE, Dunlop CL, Siggins GR, Henriksen SJ, Sutcliffe JG (1996) A cortical neuropeptide with neuronal depressant and sleep-modulating properties. Nature 381: 242-245. [DOI] [PubMed] [Google Scholar]
- de Lecea L, del Rio JA, Criado JR, Alcantara S, Morales M, Danielson PE, Henriksen SJ, Soriano E, Sutcliffe JG (1997) Cortistatin is expressed in a distinct subset of cortical interneurons. J Neurosci 17: 5868-5880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Endres M, Meisel A, Biniszkiewicz D, Namura S, Prass K, Ruscher K, Lipski A, Jaenisch R, Moskowitz MA, Dirnagl U (2000) DNA methyltransferase contributes to delayed ischemic brain injury. J Neurosci 20: 3175-3181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freund TF (2003) Interneuron diversity series: rhythm and mood in perisomatic inhibition. Trends Neurosci 26: 489-495. [DOI] [PubMed] [Google Scholar]
- Froidevaux S, Hintermann E, Torok M, Macke HR, Beglinger C, Eberle AN (1999) Differential regulation of somatostatin receptor type 2 (sst 2) expression in AR4-2J tumor cells implanted into mice during octreotide treatment. Cancer Res 59: 3652-3657. [PubMed] [Google Scholar]
- Guillermet J, Saint-Laurent N, Rochaix P, Cuvillier O, Levade T, Schally AV, Pradayrol L, Buscail L, Susini C, Bousquet C (2003) Somatostatin receptor subtype 2 sensitizes human pancreatic cancer cells to death ligand-induced apoptosis. Proc Natl Acad Sci USA 100: 155-160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hannon JP, Nunn C, Stolz B, Bruns C, Weckbecker G, Lewis I, Troxler T, Hurth K, Hoyer D (2002) Drug design at peptide receptors: somatostatin receptor ligands. J Mol Neurosci 18: 15-27. [DOI] [PubMed] [Google Scholar]
- Hanson Jr MG, Shen S, Wiemelt AP, McMorris FA, Barres BA (1998) Cyclic AMP elevation is sufficient to promote the survival of spinal motor neurons in vitro J Neurosci 18: 7361-7371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hata R, Maeda K, Hermann D, Mies G, Hossmann KA (2000) Dynamics of regional brain metabolism and gene expression after middle cerebral artery occlusion in mice. J Cereb Blood Flow Metab 20: 306-315. [DOI] [PubMed] [Google Scholar]
- Hicks GA, Feniuk W, Humphrey PP (1998) Outward current produced by somatostatin (SRIF) in rat anterior cingulate pyramidal cells in vitro. Br J Pharmacol 124: 252-258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hukovic N, Panetta R, Kumar U, Patel YC (1996) Agonist-dependent regulation of cloned human somatostatin receptor types 1-5 (hSSTR1-5): subtype selective internalization or upregulation. Endocrinology 137: 4046-4049. [DOI] [PubMed] [Google Scholar]
- Katchanov J, Waeber C, Gertz K, Gietz A, Winter B, Bruck W, Dirnagl U, Veh RW, Endres M (2003) Selective neuronal vulnerability following mild focal brain ischemia in the mouse. Brain Pathol 13: 452-464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawaguchi Y, Kondo S (2002) Parvalbumin, somatostatin and cholecystokinin as chemical markers for specific GABAergic interneuron types in the rat frontal cortex. J Neurocytol 31: 277-287. [DOI] [PubMed] [Google Scholar]
- Kreienkamp HJ, Honck HH, Richter D (1997) Coupling of rat somatostatin receptor subtypes to a G-protein gated inwardly rectifying potassium channel (GIRK1). FEBS Lett 419: 92-94. [DOI] [PubMed] [Google Scholar]
- Liu D, Slevin JR, Lu C, Chan SL, Hansson M, Elmer E, Mattson MP (2003) Involvement of mitochondrial K+ release and cellular efflux in ischemic and apoptotic neuronal death. J Neurochem 86: 966-979. [DOI] [PubMed] [Google Scholar]
- Mantyh PW, DeMaster E, Malhotra A, Ghilardi JR, Rogers SD, Mantyh CR, Liu H, Basbaum AI, Vigna SR, Maggio JE (1995) Receptor endocytosis and dendrite reshaping in spinal neurons after somatosensory stimulation. Science 268: 1629-1632. [DOI] [PubMed] [Google Scholar]
- Marsh HN, Dubreuil CI, Quevedo C, Lee A, Majdan M, Walsh GS, Hausdorff S, Said FA, Zoueva O, Kozlowski M, Siminovitch K, Neel BG, Miller FD, Kaplan DR (2003) SHP-1 negatively regulates neuronal survival by functioning as a TrkA phosphatase. J Cell Biol 163: 999-1010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCullough L, Wu L, Haughey N, Liang X, Hand T, Wang Q, Breyer RM, Andreasson K (2004) Neuroprotective function of the PGE2 EP2 receptor in cerebral ischemia. J Neurosci 24: 257-268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLaughlin B, Pal S, Tran MP, Parsons AA, Barone FC, Erhardt JA, Aizenman E (2001) p38 activation is required upstream of potassium current enhancement and caspase cleavage in thiol oxidant-induced neuronal apoptosis. J Neurosci 21: 3303-3311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore SD, Madamba SG, Joels M, Siggins GR (1988) Somatostatin augments the M-current in hippocampal neurons. Science 239: 278-280. [DOI] [PubMed] [Google Scholar]
- Pal S, Hartnett KA, Nerbonne JM, Levitan ES, Aizenman E (2003) Mediation of neuronal apoptosis by Kv2.1-encoded potassium channels. J Neurosci 23: 4798-4802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel YC, Greenwood MT, Panetta R, Demchyshyn L, Niznik H, Srikant CB (1995) The somatostatin receptor family. Life Sci 57: 1249-1265. [DOI] [PubMed] [Google Scholar]
- Pfeiffer M, Koch T, Schroder H, Laugsch M, Hollt V, Schulz S (2002) Heterodimerization of somatostatin and opioid receptors cross-modulates phosphorylation, internalization, and desensitization. J Biol Chem 277: 19762-19772. [DOI] [PubMed] [Google Scholar]
- Piiper A, Dikic I, Lutz MP, Leser J, Kronenberger B, Elez R, Cramer H, Muller-Esterl W, Zeuzem S (2002) Cyclic AMP induces transactivation of the receptors for epidermal growth factor and nerve growth factor, thereby modulating activation of MAP kinase, Akt, and neurite outgrowth in PC12 cells. J Biol Chem 277: 43623-43630. [DOI] [PubMed] [Google Scholar]
- Poser S, Impey S, Xia Z, Storm DR (2003) Brain-derived neurotrophic factor protection of cortical neurons from serum withdrawal-induced apoptosis is inhibited by cAMP. J Neurosci 23: 4420-4427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Putcha GV, Deshmukh M, Johnson Jr EM (1999) BAX translocation is a critical event in neuronal apoptosis: regulation by neuroprotectants, BCL-2, and caspases. J Neurosci 19: 7476-7485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rauca C, Schafer K, Hollt V (1999) Effects of somatostatin, octreotide and cortistatin on ischaemic neuronal damage following permanent middle cerebral artery occlusion in the rat. Naunyn Schmiedebergs Arch Pharmacol 360: 633-638. [DOI] [PubMed] [Google Scholar]
- Rydel R, Greene L (1988) cAMP analogs promote survival and neurite outgrowth in cultures of rat sympathetic and sensory neurons independently of nerve growth factor. Proc Natl Acad Sci USA 85: 1257-1261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schulz S, Schreff M, Schmidt H, Handel M, Przewlocki R, Hollt V (1998) Immunocytochemical localization of somatostatin receptor sst2A in the rat spinal cord and dorsal root ganglia. Eur J Neurosci 10: 3700-3708. [DOI] [PubMed] [Google Scholar]
- Schweitzer P, Madamba SG, Siggins GR (2003) The sleep-modulating peptide cortistatin augments the h-current in hippocampal neurons. J Neurosci 23: 10884-10891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaw S, Bencherif M, Marrero MB (2003) Angiotensin II blocks nicotine-mediated neuroprotection against β-amyloid (1-42) via activation of the tyrosine phosphatase SHP-1. J Neurosci 23: 11224-11228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spier AD, de Lecea L (2000) Cortistatin: a member of the somatostatin neuropeptide family with distinct physiological functions. Brain Res Brain Res Rev 33: 228-241. [DOI] [PubMed] [Google Scholar]
- Stumm R, Culmsee C, Schafer MK, Krieglstein J, Weihe E (2001) Adaptive plasticity in tachykinin and tachykinin receptor expression after focal cerebral ischemia is differentially linked to GABAergic and glutamatergic cerebrocortical circuits and cerebrovenular endothelium. J Neurosci 21: 798-811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stumm RK, Rummel J, Junker V, Culmsee C, Pfeiffer M, Krieglstein J, Hollt V, Schulz S (2002) A dual role for the SDF-1/CXCR4 chemokine receptor system in adult brain: isoform-selective regulation of SDF-1 expression modulates CXCR4-dependent neuronal plasticity and cerebral leukocyte recruitment after focal ischemia. J Neurosci 22: 5865-5878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stumm RK, Zhou C, Schulz S, Hollt V (2004) Neuronal types expressing mu- and delta-opioid receptor mRNA in the rat hippocampal formation. J Comp Neurol 469: 107-118. [DOI] [PubMed] [Google Scholar]
- Tanaka J, Koshimura K, Murakami Y, Sohmiya M, Yanaihara N, Kato Y (1997) Neuronal protection from apoptosis by pituitary adenylate cyclase-activating polypeptide. Regul Pept 72: 1-8. [DOI] [PubMed] [Google Scholar]
- Tannenbaum GS, Turner J, Guo F, Videau C, Epelbaum J, Beaudet A (2001) Homologous upregulation of sst2 somatostatin receptor expression in the rat arcuate nucleus in vivo. Neuroendocrinology 74: 33-42. [DOI] [PubMed] [Google Scholar]
- Tulipano G, Soldi D, Bagnasco M, Culler MD, Taylor JE, Cocchi D, Giustina A (2002) Characterization of new selective somatostatin receptor subtype-2 (sst2) antagonists, BIM-23627 and BIM-23454. Effects of BIM-23627 on GH release in anesthetized male rats after short-term high-dose dexamethasone treatment. Endocrinology 143: 1218-1224. [DOI] [PubMed] [Google Scholar]
- Vaudry D, Stork PJ, Lazarovici P, Eiden LE (2002) Signaling pathways for PC12 cell differentiation: making the right connections. Science 296: 1648-1649. [DOI] [PubMed] [Google Scholar]
- Videau C, Hochgeschwender U, Kreienkamp HJ, Brennan MB, Viollet C, Richter D, Epelbaum J (2003) Characterisation of [125I]-Tyr0DTrp8-somatostatin binding in sst1- to sst4- and SRIF-gene-invalidated mouse brain. Naunyn Schmiedebergs Arch Pharmacol 367: 562-571. [DOI] [PubMed] [Google Scholar]
- Vilchis C, Bargas J, Perez-Rosello T, Salgado H, Galarraga E (2002) Somatostatin modulates Ca2+ currents in neostriatal neurons. Neuroscience 109: 555-567. [DOI] [PubMed] [Google Scholar]
- Wei L, Yu SP, Gottron F, Snider BJ, Zipfel GJ, Choi DW (2003) Potassium channel blockers attenuate hypoxia- and ischemia-induced neuronal death in vitro and in vivo. Stroke 34: 1281-1286. [DOI] [PubMed] [Google Scholar]
- Welsh FA, Sakamoto T, McKee AE, Sims RE (1987) Effect of lactacidosis on pyridine nucleotide stability during ischemia in mouse brain. J Neurochem 49: 846-851. [DOI] [PubMed] [Google Scholar]
- Yu SP, Farhangrazi ZS, Ying HS, Yeh CH, Choi DW (1998) Enhancement of outward potassium current may participate in beta-amyloid peptide-induced cortical neuronal death. Neurobiol Dis 5: 81-88. [DOI] [PubMed] [Google Scholar]
- Yu SP, Yeh CH, Gottron F, Wang X, Grabb MC, Choi DW (1999) Role of the outward delayed rectifier K+ current in ceramide-induced caspase activation and apoptosis in cultured cortical neurons. J Neurochem 73: 933-941. [DOI] [PubMed] [Google Scholar]
- Zatelli MC, Tagliati F, Taylor JE, Rossi R, Culler MD, degli Uberti EC (2001) Somatostatin receptor subtypes 2 and 5 differentially affect proliferation in vitro of the human medullary thyroid carcinoma cell line tt. J Clin Endocrinol Metab 86: 2161-2169. [DOI] [PubMed] [Google Scholar]