

Abstract
Homeodomain proteins play critical roles during development in cell fate determination and proliferation, but few studies have defined gene regulatory networks for this class of transcription factors in differentiated cells. Using a lacZ-knock-in strategy to ablate Nkx6-2, we find that the Nkx6-2 promoter is active embryonically in neuroblasts and postnatally in oligodendrocytes. In addition to neurological deficits, we find widespread ultrastructural abnormalities in CNS white matter and aberrant expression of three genes encoding a paranodal microtubule destabilizing protein, stathmin 1, and the paranodal cell adhesion molecules neurofascin and contactin. The involvement of these downstream proteins in cytoskeletal function and cell adhesion reveals mechanisms whereby Nkx6-2 directly or indirectly regulates axon- glial interactions at myelin paranodes. Nkx6-2 does not appear to be the central regulator of axoglial junction assembly; nonetheless, our data constitute the first evidence of such a regulatory network and provide novel insights into the mechanism and effector molecules that are involved.
Keywords: targeted deletion, homeodomain, Nkx, cytoskeleton, auditory brainstem response, rotarod
Introduction
For more than a century, motor and sensory functions in the CNS have been known to depend on intimate relationships between neurons and oligodendroglia (Ramon y Cajal, 1899). From an electrophysiological standpoint, the most important aspect of this interaction is the assembly and maintenance of heterotypic, adhesive junctions between axons and myelin sheaths (Koles and Rasminsky, 1972). These axoglial junctions, composed of neuronal and oligodendroglial components, serve several essential functions: they flank and define nodes of Ranvier, they impede the movement of interstitial ions and other molecules into the periaxonal space, and they compartmentalize the axolemma into at least three distinct subdomains: nodes of Ranvier, paranodes, and juxtaparanodes (for review, see Arroyo et al., 2001; Bhat, 2003; Salzer, 2003).
Until recently, the composition of axoglial junctions has been essentially unknown. Mutations in model organisms (for review, see Girault and Peles, 2002; Bhat, 2003; Salzer, 2003) have demonstrated structural components of axoglial junctions, including the cell adhesion molecules caspr and contactin expressed by neurons, the cell adhesion molecule neurofascin 155 (NF155; the 155 kDa isoform), and possibly glycolipids, expressed by oligodendrocytes. Contactin is also expressed by oligodendrocytes, and neurofascin (NF186) is also expressed by neurons. In contrast, transcriptional regulation of axoglial junction components is completely unknown. Several transacting protein families are expressed by oligodendrocytes, including Yin Yang 1 (YY1), Olig, Sox, and Nkx homeodomain proteins (Awatramani et al., 1997; Berndt et al., 2001; Lu et al., 2002; Stolt et al., 2004). The downstream targets of these transcription factors are almost completely unknown in oligodendrocytes, and any one or a combination could conceivably regulate the synthesis of axoglial junction components.
Herein, we perform a detailed analysis of postnatal functions of Nkx6-2 in oligodendrocytes, focusing on myelinogenesis, axoglial junctions, and myelin stability in adults. Previous studies in embryos show that Nkx6-2 serves as a transcriptional co-repressor in ventral neural tube patterning and cell fate specification to regulate Dbx and Olig2 expression (Vallstedt et al., 2001). Postnatally, Nkx6-2 is expressed by differentiating and remyelinating oligodendrocytes (Awatramani et al., 1997; Sim et al., 2000); however, targeted deletion of this protein yields mice without a gross CNS phenotype (Cai et al., 2001). Nonetheless, we find in the current study that Nkx6-2 null mice exhibit physiological deficiencies in motor coordination and CNS nerve conduction. Furthermore, we find morphologically abnormal CNS paranodes, vermicular-like processes in adults, and ectopic axoglial junctions in juxtaparanodal myelin. At the level of function, we demonstrate that Nkx6-2 is genetically upstream of cell adhesion and cytoskeleton remodeling at paranodes; oligodendrocyte expression of NF155 and contactin is increased in adult Nkx6-2 null mice, and expression of a microtubule destabilizing protein, stathmin 1, is decreased. Importantly, overexpression of NF155 and contactin may cause inappropriate axoglial junction formation, whereas reduced stathmin 1, a protein that regulates process outgrowth and stability in oligodendrocytes (Liu et al., 2003), may lead to stable vermicular-like processes and abnormal paranode morphology and function. Finally, we find that paranodal widths and lengths are unstable in mutants between 1 and 8 months of age, reflecting ongoing defects at CNS myelin paranodes.
Materials and Methods
Gene targeting construct. The Nkx6-2 gene was cloned from a 129 Sv/Ev genomic library and a 5.8 kb Bam-BglII fragment (supplemental Fig. 1 A, available at www.jneurosci.org as supplemental material) used as a homologous flanking sequence for the targeting construct. A 0.3 kb fragment of this sequence containing the open reading frame in exons 1 and 2 was replaced with a lacZ-PGKneo cassette to generate the mutant allele for transfection into R1 embryonic stem cells. The floxed (arrowheads) PGKneo gene was subsequently removed by injecting circular CMVcre plasmid into male pronuclei of 0.5 d embryos (O'Gorman et al., 1991; Gow et al., 1999). Mutant and wild-type alleles were distinguished by Southern blotting.
Genetic background. Chimeras derived from the homologous recombinant R1 cells were mated with C57BL6/Tac female mice to generate mice that were heterozygous for the targeted Nkx6-2 allele. The mouse colony was expanded and maintained by sibling matings between heterozygous mice, and experiments used mice from multiple litters containing wild-type and null mutant animals.
Southern and Northern blotting. Tail tips and tissues from mice were frozen on dry ice and stored at -80°C. DNA was prepared from proteinase K-digested tail tips, and RNA was purified from tissues over CsCl gradients (Stecca et al., 2000). Mice were genotyped by Southern blotting, and expression was examined by Northern blotting, using a full-length mouse Nkx6-2 cDNA. Expression of mRNAs encoding neurofascin was detected using rat cDNA probes to alternatively spliced exons (Tait et al., 2000). Nf155 was detected with a 515 bp probe encoding the third fibronectin type III repeat domain (primer pair: 5′—GAA ACC CGA GAG ACT TGG AAC-3′ and 5′-TGT CGG ACT CTG AAA CGC CTG-3′), and Nf186 was detected with a 525 bp probe encoding the mucin domain (primer pair: 5′-CCC AAC ACC TCC AAA TGA AG-3′ and 5′-GTG ATG ACG GTA CTG CTG ATG-3′). Before reprobing, Northern blots were stripped for 30 min at 70°C in 0.1× SSPE/0.05% SDS and exposed to film for 24 hr to ensure the absence of residual radiolabeled probe.
Histochemistry, immunocytochemistry, and electron microscopy. Whole-mount 5-bromo-4-chloro-3-indolyl-β-d-galactopyranoside (X-gal) staining and immunocytochemical labeling of fixed tissue have been described previously (Gow et al., 1998, 1999). Primary antibodies were from the following: Dr. J. Ericson (Karolinska Institute, Stockholm, Sweden) (Gp α Nkx6-2, 1:3000); Dr. M. Bhat (Bhat et al., 2001) (University of North Carolina, Chapel Hill, NC) (Gp α caspr, 1:500); Dr. J. Salzer (Rios et al., 2000) (New York University) (Rb α contactin, 1:500, after 10 min acetone and overnight block); Dr. S. Tait (Tait et al., 2000) (University of Edinburgh, UK) (Rb α neurofascin, 1:400, after 1 min Bouin's fixation); Dr. M. Rasband (Rasband et al., 1999), University of Connecticut (Ms α Nav1.2, 1:250 and Rb α Nav1.6, 1:400); Alomone Labs, Jerusalem, Israel (Rb α Kv1.1 and 1.2, 1:60); Calbiochem (La Jolla, CA) (Rb α stathmin 1, 1:3000 for ICC and 1:100,000 for Western blots); and Dr. J. A. Girault (Denisenko-Nehrbass et al., 2003) (Institut National de la Santé et de la Recherche Médicale U114, College de France, Paris, France) (Rb α protein 4.1, 1:1000). Secondary antibodies were from Jackson ImmunoResearch (West Grove, PA), Molecular Probes (Eugene, OR), and Southern Biotechnology (Birmingham, AL). Cryostat sections (10 μm) were permeabilized with 0.1% Triton X-100 and labeled with primary antibodies overnight (Gow et al., 1999). For electron microscopy, mice were perfused intracardially with 4% glutaraldehyde/0.1 m cacodylate buffer, pH 7.2, for 60 min at room temperature and dissected after 3 hr. Optic nerve, cerebellum, and cervical spinal cord were postfixed in osmium tetroxide/ferricyanide and embedded in Epon (Stecca et al., 2000). Whole-mount images were obtained using a Nikon SMZ1500 microscope equipped with a Sony camera. Fluorescence images were obtained using a Leica DMRA2 microscope equipped with a Hamamatsu Orca ER camera and OpenLab software. Electron micrographs were obtained using a Hitachi H7000 TEM.
Rotarod and auditory brainstem response measurements. For rotarod analyses, mice were trained and tested using a 2 d protocol (Gow et al., 1999). At least five mice from each genotype were analyzed at each time point. Auditory brainstem responses (ABRs) were evoked in the left and right ears of avertin-anesthetized mice (0.375 mg/gm) with 2048 pure tone stimuli at 8, 16, and 32 kHz between 80 and 50 dB sound pressure level (SPL) at a rate of 29.1/sec and analyzed using Intelligent Hearing Systems equipment (Miami, FL) with minor modifications (Gow et al., 2004) from published methods (Zheng et al., 1999). Peak V latencies were normalized by subtracting peak I latencies (i.e., peak V - I) to correct for age- and stimulation frequency-dependent changes that otherwise increase variability in the EEG data. For each animal, these data are averages of 80, 70, 60, and 50 dB SPL latencies at 32, 16, and 8 kHz from both cochleas (i.e., 24 latency estimates per mouse).
Analysis of paranodal width and length. Longitudinal cryostat sections (10 μm) of optic nerve were labeled with anti-caspr antibodies (Bhat et al., 2001) and visualized using a secondary antibody directly conjugated to fluorescein (Invitrogen-Molecular Probes, Carlsbad, CA). Five sections per animal were photographed using a 63× Plan-Apo lens (numerical aperture = 1.4) on a Leica DMRA2 microscope. Paranode widths and lengths were measured from these images after calibration, using the OpenLab advanced measurements software module (Improvision, Lexington, MA).
Results
To define the function of Nkx6-2 and track its developmental expression in mice, we replaced the coding region for this protein with lacZ by homologous recombination (supplemental Fig. 1A, available at www.jneurosci.org as supplemental material). Southern blots from embryonic stem cells (supplemental Fig. 1B, available at www.jneurosci.org as supplemental material) identify two clones, 254 and 377, suitable for blastocyst injections. Clone 321 failed to proliferate well in culture. Northern blots of adult brain (from line 254) reveal two mRNAs for Nkx6-2 in wild-type (+/+) and heterozygous (+/-) mice at 1.6 kb (major) and 2 kb (minor) using an exon 3 probe that likely reflect utilization of polyadenylation signals in exons 3 or 4, respectively (supplemental Fig. 1A, available at www.jneurosci.org as supplemental material). Neither one of these mRNAs is detected in null (-/-) mice (supplemental Fig. 1C, available at www.jneurosci.org as supplemental material); however, two hybridization signals are observed near 5 kb that also hybridize to a lacZ probe (data not shown) and likely encode β-galactosidase derived from the mutant Nkx6-2 allele. Null mutants from embryonic stem cell clone 377 are indistinguishable from clone 254-derived animals.
Nkx6-2 is expressed in cranial nerves and motoneurons in embryos and oligodendrocytes postnatally
Previous studies have revealed Nkx6-2 expression in ventral regions of neural tube from mouse and chick embryos (Cai et al., 1999, 2001; Vallstedt et al., 2001). In this study, whole-mount X-gal staining revealed the extent of Nkx6-2 expression in the neural tube of embryonic day 12.5 (E12.5) null embryos and, in particular, in cranial nerves and motoneurons extending axons into the periphery. Cranial nerves V, VII, and X are β-galactosidase+ (Fig. 1a), and cranial nerves III, IX, and XI are stained between E10.5 and 11.5 (data not shown). Transverse sections of neural tube from Nkx6-2 null embryos show β-galactosidase staining (Fig. 1c) in a pattern similar to Nkx6-2 in situ hybridization in controls (Fig. 1b). V0/V1 interneuron precursors (asterisk) and motoneurons (black arrowheads) are labeled. Control embryos are β-galactosidase- (Fig. 1d).
Figure 1.
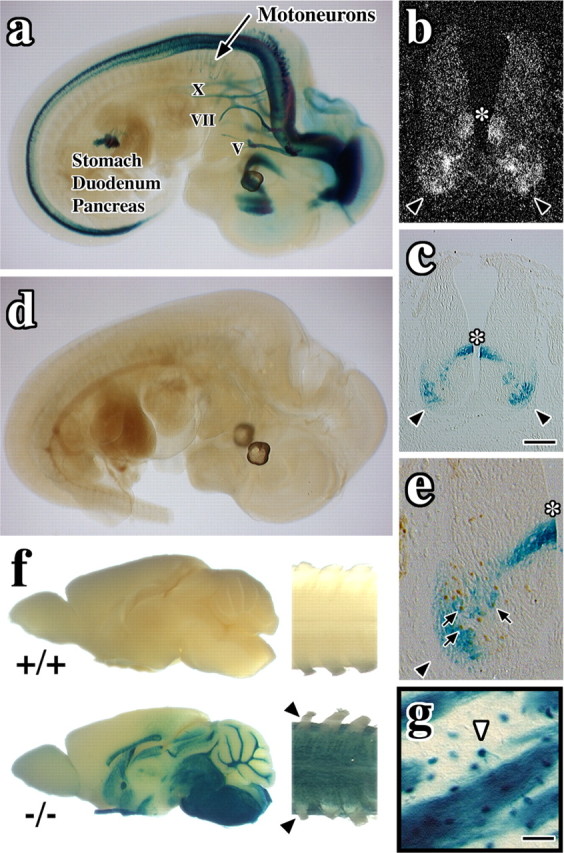
Nkx6-2 is expressed prenatally in neurons and postnatally in oligodendrocytes. a, Whole-mount X-gal staining in E12.5 null embryos. Cranial nerves V, VII, and X, cervical motoneurons, thalamus, pylorus, duodenum, and pancreatic primordium are stained. b, In situ hybridization for Nkx6-2 in neural tube of wild-type E12.5 embryo. The asterisk indicates V0/V1 precursors; arrowheads indicate motoneurons. c, X-gal staining in neural tube of E12.5 null embryo. d, Whole-mount X-gal staining of E12.5 wild-type embryo. e, Colocalization of X-gal and Islet 1 staining (arrows) in E12.5 null embryo. f, X-gal staining of P28 brains (left) and cervical spinal cords (right) from wild-type (+/+) and null (-/-) mice. Arrowheads indicate PNS nerve roots that are unstained. g, Striatum from a null mutant showing an X-gal-labeled oligodendrocyte (arrowhead) extending two processes into an axon fascicle. Scale bars: (in c) b, c, 100 μm; e, 43 μm; g, 40 μm.
β-Galactosidase+ cells in Nkx6-2 null embryos are not labeled by early oligodendrocyte lineage markers such as olig2 (data not shown); however, colocalization of X-gal staining with Islet 1 (Fig. 1e, arrows), En1, and Dbx2 (data not shown) indicates that Nkx6-2-expressing cells are fated to motoneurons and V0 and V1 interneurons, similar to Nkx6-2+ cells in controls. Recent studies by others suggest that there may be subtle changes in fate specification of V1 and V0 interneurons in Nkx6-2 null embryos (Vallstedt et al., 2001), but the postnatal consequences are currently unclear. Null mutants also strongly express β-galactosidase in the pyloric region of the stomach, primordia of the pancreas, and gall bladder between E9.5 and 14.5; however, we detect no gross abnormalities in these animals stemming from the absence of Nkx6-2 in the gut, perhaps because of redundant expression of other Nkx family members (Sander et al., 2000).
Whole-mount X-gal histochemistry of postnatal day 28 (P28) brain and spinal cord from null mutants (Fig. 1f, -/-) reveals strong β-galactosidase expression in all major CNS white matter tracts as shown previously for endogenous Nkx6-2 expression (Komuro et al., 1993; Awatramani et al., 1997). Figure 1g shows β-galactosidase-positive cells exhibiting the canonical morphology of myelinating oligodendrocytes (Gow et al., 1992) in and around striatal axons (white arrowhead), which demonstrates that the presence of the lacZ sequence does not appreciably alter cell-type specificity. On the other hand, β-galactosidase activity in wild-type CNS tissue is negligible (Fig. 1f, +/+).
Nkx6-2 is expressed long term by oligodendrocytes in vivo
To determine the expression profile of Nkx6-2 with age in wild-type mice, we performed a Northern blotting analysis with spinal cord RNA from P16 to 13 months (Fig. 2A). Full-length Nkx6-2 cDNA probe hybridization to the 1.6 kb major band (identical to supplemental Fig. 1C, available at www.jneurosci.org as supplemental material) is shown, and expression is maximal around P29. The probe does not label null mutant samples. Immunofluorescence labeling of brain sections from wild-type P33 mice (Fig. 2Ba) show that myelin basic protein (MBP)+ myelinating oligodendrocytes (green) express Nkx6-2 (arrowheads, red), and that this transcription factor is localized to the nucleus (inset). Furthermore, oligodendrocytes continue to express Nkx6-2 at 4 months of age (Fig. 2 Bc). As expected, sections from the null mutants are not stained with anti-Nkx6-2 antibodies (Fig. 2 Bb, P33).
Figure 2.

Nkx6-2 is expressed continuously in most oligodendrocytes from an early age. A, Northern blot of spinal cord RNA shows continuous Nkx6-2 expression up to 13 months. Loading control, 18S rRNA. B, Immunocytochemistry showing Nkx6-2 (red) localized to nuclei (blue) in MBP+ oligodendrocytes (green) from thalamus of P33 (a) and 4 month (c; P120) wild-type (+/+) mice and in P33 Nkx6-2 null (-/-) mice (b). Insets, MBP+ oligodendrocytes. Scale bars: low magnification, 20 μm; insets, 8.4 μm. C, Nkx6-2+ cells as a proportion of MBP+ oligodendrocytes in brains of P33 and 4 month wild-type (+/+) and Nkx6-2 null (-/-) mice.
Morphometry in brains from P33 wild-type mice (n = 3 animals; >200 oligodendrocytes per animal) indicates that 92% of MBP+ oligodendrocytes throughout brain are also Nkx6-2+ and localize this protein to the nucleus (Fig. 2C). This proportion of double-positive cells is undiminished at 4 months of age. In contrast, we do not find any MBP+-Nkx6-2+ cells in the null mutants, although the number of oligodendrocytes in these mutants is comparable with controls. Thus, from these data we conclude that most of the Nkx6-2+ cells are myelinating oligodendrocytes that express this protein from an early age and maintain expression into adulthood. Importantly, we observe no changes in expression of other neural cell-specific Nkx family genes such as Nkx2-2 and Nkx6-2 (data not shown) that could potentially effect compensation in our mutants (Qi et al., 2001; Pattyn et al., 2003).
Nkx6-2 null mice exhibit motor coordination and CNS-evoked potential defects
Nkx6-2 null mice reproduce normally and in the cage are indistinguishable from littermate controls as reported previously (Cai et al., 2001). Nevertheless, persistent behavioral defects are revealed in a rotarod analysis (Fig. 3A). Although the performance of null mice in this test is similar to wild-type (+/+) and heterozygous (+/-) littermates at 3 weeks of age, by 6 weeks the mutants exhibit a pronounced deficiency that persists into adulthood. In view of the fact that myelinogenesis in mouse CNS is maximal between 3 and 6 weeks of age and Nkx6-2 is expressed by myelinating oligodendrocytes, Nkx6-2 transcriptional activity appears to play a critical role in latter stages of myelinogenesis.
Figure 3.
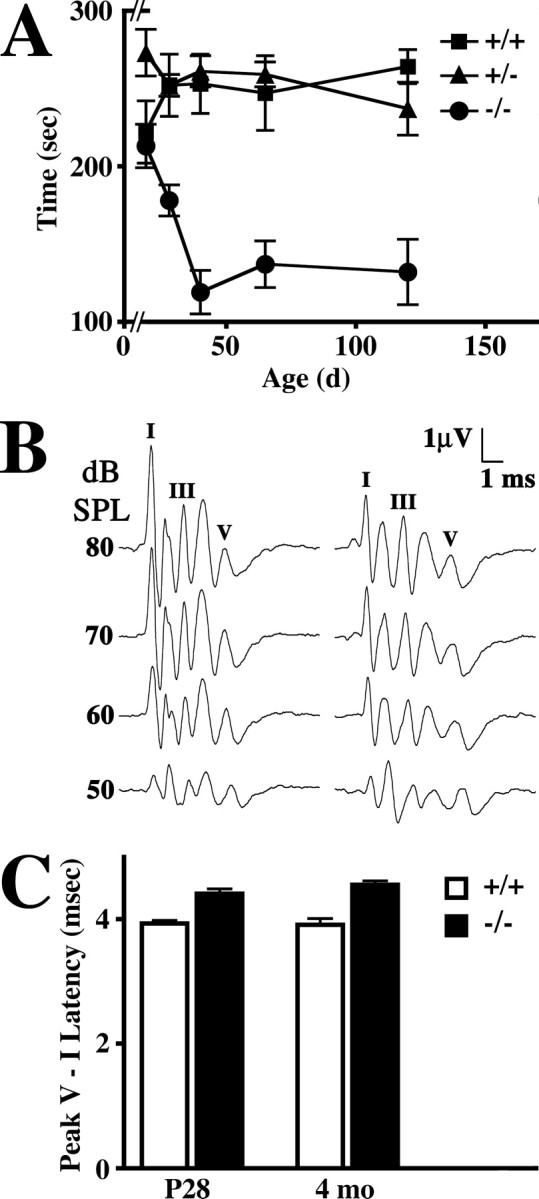
Progressive rotarod phenotype and conduction abnormalities in Nkx6-2 null mice. A, Rotarod analysis of wild-type (+/+), heterozygous (+/-), and null mutant (-/-) mice from P21 to 4 months of age. Data are expressed as mean ± SEM; 5 ≥ n ≥ 10. B, EEGs elicited from 4-month-old wild-type (+/+) and Nkx6-2 null (-/-) mice using 2048 tone pips at 32 kHz between 80 and 50 dB SPL. Peaks I, III, and V are labeled. C, Averaged peak V - I latencies (mean ± SEM; n ≥ 5) derived from auditory brainstem responses at 8, 16, and 32 kHz in wild-type (+/+) and mutant (-/-) mice at 1 and 4 months of age.
To determine whether the rotarod phenotype might be associated with changes in conduction velocity in the CNS of Nkx6-2 null mice, we examined evoked potentials derived from ABRs in a manner similar to that of previous studies (Fujiyoshi et al., 1994; Gow et al., 2004). Presentation of 102 μsec pure tone pips into the auditory canals of anesthetized mice at different sound pressure levels elicits a series of EEGs comprising five or six well resolved peaks with latencies ∼1 msec apart (Fig. 3B). The latency of the fifth peak is proportional to central nerve conduction velocity. Amplitudes (microvolts) and latencies (milliseconds) of the initial peaks in EEGs obtained from Nkx6-2 null mice are similar to littermate controls, indicating that the cochlea from the mutants functions relatively normally; however, the peak V latencies are longer in the mutants. In Figure 3C, we show average peak V - I latencies as a function of age in Nkx6-2 null mice and littermate controls. These latencies are invariant in controls between 1 and 4 months of age, indicating that CNS conduction in the auditory tract is stable shortly after the onset of hearing. Measurements before P25 are less reliable. In Nkx6-2 null mice, peak V - I latencies are also stable with age but are increased by 15%, which is a small but statistically significant change (p < 0.001). Thus, the ABR data indicate that motor impairment revealed by the rotarod analysis is likely caused, at least in part, by slowed conduction velocity in the CNS of Nkx6-2 null mice.
Nkx6-2 null mice exhibit widespread paranodal abnormalities
In view of the physiological defects revealed in Figure 3, we examined myelin ultrastructure in adult Nkx6-2 null mice at 3, 7, and 12 months of age from cervical spinal cord, brainstem, and optic nerve. This latter CNS white matter tract is of particular interest for several reasons: it is composed primarily of small-to-medium myelinated fibers (Fig. 4a), it is well defined and amenable to morphometric studies, and it is sensitive to damage in myelin diseases such as leukodystrophy and multiple sclerosis.
Figure 4.
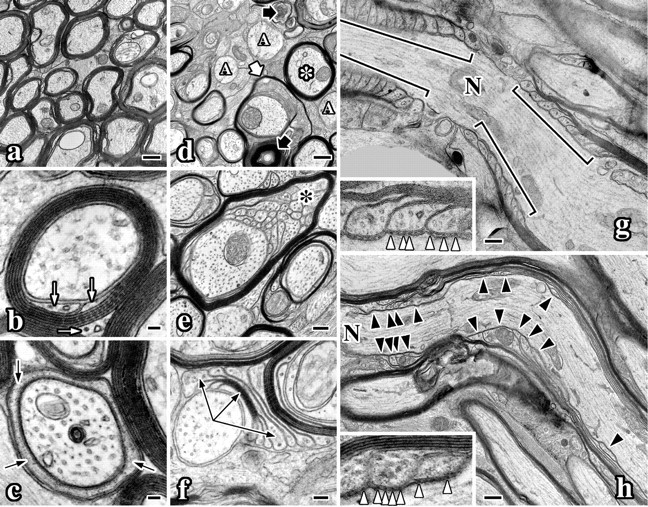
CNS myelin ultrastructural abnormalities in Nkx6-2 null mice. Electron micrographs of optic nerves from 7-month-old wild-type (a-c, g) and Nkx6-2 null (d-f, h) mice. a, Transverse section of wild-type optic nerve. b, Wild-type myelinated axon sectioned through the internode. The inner tongue process is visible (white arrows). c, Wild-type myelinated axon sectioned through the paranode. Cytoplasm in a paranodal loop is visible (black arrows). d, Transverse section of mutant optic nerve shows redundant loops (thick white arrow), thin myelin (asterisk), unmyelinated axons (“A”), and axonal degeneration (thick black arrows). e, Stable vermicular-like processes (black asterisk) underneath a myelin sheath in mutant optic nerve. f, Compact myelin flap flanked by paranodal loops (arrows) in optic nerve from null mice. g, Longitudinal section of a wild-type node of Ranvier (N). h, Longitudinal section of a node of Ranvier from a null mouse. Paranodal loops are disorganized (black arrowheads). g, h, Insets show axoglial junctions (white arrowheads). Scale bars: a, d, g, h, 500 nm; b, c, 50 nm; e, 200 nm; f, 100 nm; insets, g, h, 150 nm.
Transverse sections through compact regions of myelin sheaths reveal the high degree of organization of this membrane (Fig. 4b). Compaction at cytoplasmic surfaces of the lipid bilayer is evident from the dark spiral, which is interposed by a lightly stained spiral representing juxtaposed extracellular surfaces of the bilayer. Cytoplasm has been excluded from this internodal region but persists at the inner and outer loops (white arrows). At the paranode (Fig. 4c), only a few membrane wraps are observed, and the inner wrap contains cytoplasm around the axon (black arrows). Importantly, the closer a transverse section is to the node of Ranvier, the fewer compact myelin lamellas there are around the axon (for review, see Raine, 1984). This feature is apparent by comparing the axons in Figure 4, b and c.
All white matter tracts that we examined in Nkx6-2 null mice exhibit qualitatively similar myelin defects. At low magnification, unmyelinated fibers (Fig. 4d, A) are readily apparent, whereas most axons are ensheathed in thin myelin. The white asterisk identifies such an axon sectioned through the internode. Redundant sheaths around axons (thick white arrow) are common in null mutants, and Wallerian degeneration is infrequent (thick black arrow). These abnormalities are rarely observed in normal adult animals but are common in many myelin mutants, including md rats, quaking mice, and more recently in Mag-null and Cgt-null mice (Friedrich, 1974; Rosenbluth, 1987; Dupree et al., 1998; Trapp et al., 1998).
Several abnormalities observed in Nkx6-2 null mice are more pronounced than in other mutants. In Figure 4e, >30 cytoplasmic processes underlie an inter-nodal myelin sheath (black asterisk). These processes contain one or more microtubules and are reminiscent of vermicular processes that are observed during normal myelinogenesis (Knobler et al., 1974). Indeed, the similar composition and cytoplasmic density in these processes, and the continuity of several with the overlying sheath, reveal their derivation from myelin. Notably, the absence of intermediate filament-containing processes rules out a contribution to this pathology by astrocytes that can invade nodes of Ranvier and extend processes along the periaxonal space (Rosenbluth, 1987; Bhat et al., 2001).
We do not observe vermicular-like processes underneath compact myelin sheaths or myelin flaps in optic nerve or spinal cords from P16 mutants. Perhaps the relatively late appearance of this pathology coincides with the formation of myelin sheaths in the latter stages of myelinogenesis, and in this regard we note that, at P16, a portion of axons destined to be ensheathed are unmyelinated. Another common abnormality found in Nkx6-2 null mice is shown in Figure 4f. The axon to the left is associated with a myelin “flap,” which is flanked by cytoplasmic loops that contact the axon and several adjacent cell processes (long black arrows). Cytoplasmic components have been excluded from the three innermost membrane layers of this flap, indicating that myelin compaction proceeds independently of circumferential wrapping (Peters et al., 1991).
In longitudinal sections of optic nerve (Fig. 4g), lateral myelin loops are normally tightly clustered at paranodes (brackets) and are symmetrical across the axon. Three distinct homotypic and heterotypic junctions contribute to the structure and function of the paranode: claudin 11 tight junctions, which minimize electrical capacitance of the sheath (Gow et al., 1999; Southwood and Gow, 2001); adherens junctions, which maintain the apposition of lateral myelin loops (Schnapp and Mugnaini, 1978); and axoglial junctions, which are dominant features of the paranode composed of NF155, caspr, contactin, and probably other proteins (Salzer, 2003). These junctions appear as electron-dense particles connecting the axolemma and myelin loops (Fig. 4g, white arrowheads).
A longitudinal section of optic nerve from a null mutant is shown in Figure 4h, with a node of Ranvier to the left. In contrast to the serried appearance of lateral myelin loops in controls, paranodes in Nkx6-2 null mice are often disorganized, extend for large distances along the axon (black arrowheads), and are non-symmetrical. Nevertheless, axoglial junctions underlie paranodal loops (inset, white arrowheads), which likely accounts for the mild phenotype in Nkx6-2 null mice compared with other mutants that lack these adhesive junctions (Coetzee et al., 1996; Bosio et al., 1998; Bhat et al., 2001; Boyle et al., 2001).
A morphometric analysis in optic nerve from 50 random fields (60,000× overall magnification) in thin sections of P120 wild-type, heterozygote, and two Nkx6-2 null mice (Table 1) shows that 27% of axons are unmyelinated in the mutants. Of myelinated axons, >11% exhibit disorganized paranodes, and these data accord with counts from transverse optic nerve sections, in which 13% of axons from two null mice exhibit abnormalities. Such pathology is observed in <1% of control axons, despite similar proportions of paranodes found in controls (associated with 2.2% of axons) and mutants (associated with 2.4%).
Table 1.
Morphometric analysis of myelinated fiber ultrastructure in optic nerves from P120 Nkx6-2 null mice
|
|
-/- |
+/+ and +/- |
||||
|---|---|---|---|---|---|---|
| Morphological feature |
% |
Number of axons |
% |
Number of axons |
||
| Paranodesa | 2.4 | 990 | 2.2 | 190 | ||
| Internodal loopsa | 11 | 990 | 0.5 | 190 | ||
| Unmyelinated axonsb | 27 | 1064 | 1 | 903 | ||
| Internodal loopsb
|
13 |
1064 |
0.7 |
903 |
||
Longitudinal sections of optic nerve.
Transverse sections of optic nerve.
Paranode and juxtaparanode domains are abnormal in Nkx6-2 null mice
Poor ultrastructural organization of paranodal loops in Nkx6-2 null mice prompted us to examine the localization of several paranodal proteins in brain, optic nerve, the CNS region of the trigeminal nerve, and cervical ventral white matter tracts of P120 mice using immunocytochemistry.
Longitudinal sections of ventral cervical spinal cord from adult wild-type and Nkx6-2 null mice at low magnification reveal abundant caspr+ myelin paranodes (Fig. 5Aa,Ab, black arrows). Caspr labeling (green) flanks nodes of Ranvier on both sides and in turn is flanked on both sides by Kv channel labeling at juxtaparanodes (red). In wild-type mice, paranodes and juxtaparanodes are nonoverlapping domains except for one or two caspr+ spirals coursing through the internode (Fig. 5Ac, black arrowheads). Furthermore, the width of the juxtaparanode across the axon revealed by Kv channel staining (short white arrowheads) is significantly greater than that of the paranode (white arrows), particularly for large-diameter axons. In this regard, large axons in Nkx6-2 null mice are abnormal (Fig. 5Ad). Numerous caspr+ spirals and deposits (black arrowhead) are found in juxtaparanodes and appear to segregate this internodal region (Bhat, 2003) of the axon into multiple subdomains. These data are consistent with the observed disorganization of paranodal loops in electron micrographs (Fig. 4f). In addition, Kv 1.1/1.2 channel labeling suggests that paranodes and juxtaparanodes in the mutants are of similar widths, which suggests that mechanisms regulating axonal width at these sites are abnormal.
Figure 5.

Localization of juxtaparanodal, paranodal, and nodal proteins in Nkx6-2 null mice. A, Longitudinal section of ventral white matter from P120 wild-type (a, c) and Nkx6-2 null (b, d) mice stained for caspr (green) and Kv1.2 (red) reveal paranodes and juxtaparanodes (black arrows). c, Black arrowheads show a juxtaparanodal caspr+ domain. White arrows and white arrowheads delineate paranodal and juxtaparanodal widths, respectively. Scale bars: a, b, 50 μm; c, d, 5 μm. B, Localization of caspr (green) and six other proteins (red) to paranodes and nodes of Ranvier in ventral white matter from P120 wild-type (+/+) and null (-/-) mice. Short black arrowheads indicate stathmin 1 localization in oligodendrocyte paranodal loops. Scale bars, 5 μm. C, Immunofluorescence labeling of optic nerve for caspr (green) and stathmin 1 (red) from P18 wild-type (a) and Nkx6-2 null (b) mice. Insets, Paranode staining. Scale bars: a, b, 5 μm; insets, 2.2 μm. D, Morphometric analysis of stathmin 1+ paranodes as a proportion of total paranodes from P18 wild-type (+/+) and null mutant (-/-) mice.
Despite these abnormalities, a number of proteins (red in each panel) are normally targeted to the paranodal region in null mutant mice (Fig. 5B, caspr labeling in green). Thus, NF186 and Nav1.6 channels are localized to nodes of Ranvier, and the age-dependent sodium α-subunit switch from Nav1.2 to Nav1.6 occurs normally (data not shown). Furthermore, NF155, contactin, and protein 4.1B colocalize with caspr at paranodes. Nonetheless, paranode morphology revealed in Figure 5B appears disorganized and accords with the data in Figure 4. Similar to others (Rios et al., 2000), we find that contactin localizes to a subpopulation of nodes in optic nerve and spinal cord (data not shown).
Reduced levels of stathmin 1 in paranodes of Nkx6-2 null mice
In light of the vermicular-like processes underneath myelin sheaths of Nkx6-2 null mice, we postulated that aberrant regulation of microtubule dynamics might be involved with this pathology. Stathmin 1 is a microtubule-destabilizing protein expressed by neurons and oligodendrocytes that is thought to be instrumental in neural cell process formation (Ozon et al., 1999; Liu et al., 2003), and we find that this protein is targeted to paranodes (Fig. 5B, +/+). Short black arrowheads show the outer edges of stathmin 1 staining at paranodes; this labeling is wider than the axonal diameter delineated by caspr. Thus, stathmin 1 is targeted to myelin paranodes by oligodendrocytes. Hitherto, stathmin 1 has not been known to be localized to paranodes, and its presence illustrates the importance of cytoskeletal dynamics and remodeling at these remote outposts of oligodendrocyte cytoplasm. Stathmin 1 labeling of paranodes is more difficult to detect in Nkx6-2 null mice and suggests lower levels of protein that may perturb paranode function.
To further explore stathmin 1 localization to paranodes, we labeled optic nerve sections from P18 mice with anti-stathmin 1 antibodies (Fig. 5C). In controls (Fig. 5Ca, black arrows), stathmin 1 (red) colocalizes with caspr (green) in most paranodes and is also present in oligodendrocyte cell bodies (white arrows). The inset shows a node of Ranvier at high magnification, and stathmin 1 is clearly evident in the myelin paranodal loops (white arrowheads). In contrast, paranodes from Nkx6-2 null mice are poorly labeled by stathmin 1 antibodies (Fig. 5Cb, inset), and levels of stathmin 1 in cell bodies also appear to be lower than in controls (white arrows). Indeed, a morphometric analysis of stathmin 1 labeling in optic nerves (Fig. 5D) from three wild-type mice reveals that 66 ± 4% of 209 caspr+ paranodes are stathmin 1+; however, this protein is detectable in only 13 ± 2% of 291 paranodes from three null mutants.
Paranodal widths and lengths are abnormal in Nkx6-2 null mice
In view of the abnormalities apparent in Figure 5Ad, we performed a morphometric analysis of paranodal widths and lengths as functions of age in Nkx6-2 null mice. Frequency histograms in Figure 6 show the distributions of paranodal widths and lengths measured from caspr-stained cryostat sections of optic nerve at P18 and at 4 and 8 months of age. The distributions of paranodal width for myelinated axons in wild-type (+/+) and null mutants (-/-) at P18 overlap almost completely (Fig. 6a), and supplemental Table 1 (available at www.jneurosci.org as supplemental material) highlights the similarity of these distributions with arithmetic means (0.72 ± 0.16 vs 0.74 ± 0.16 μm, respectively). Regarding CNS hypomyelination in the mutants (Table 1), the normal distribution of myelinated fiber diameters shown in Figure 6a indicates that the absence of myelin is probably stochastic because there are no specific subpopulations of axon sizes that are disproportionately affected.
Figure 6.

Abnormal paranodal widths and lengths in Nkx6-2 null mice. Relative frequency distributions of caspr+ paranodal widths (a-c) and lengths (d-f) from wild-type (+/+) and null (-/-) mice at P18 (a, b), 4 months (P115; b, d), and 8 months (P230; c, f). n, Number of mice examined.
At 4 months of age, the distribution for null mutants is shifted to the right ∼10% (0.80 ± 0.23 μm), whereas the wild-type distribution is unchanged from P18 (0.72 ± 0.16 μm). The right-shift of the distribution for the mutants at 8 months appears markedly increased (Fig. 6c); however, in fact, this distribution is virtually unchanged from 4 months (0.79 ± 0.21 μm). Rather, the wild-type distribution is left-shifted (0.61 ± 0.16 μm). These surprising data may reflect a previously unrecognized process of paranodal contraction between 4 and 8 months, during a period after the completion of myelinogenesis in mice. The mechanism and purpose of this remodeling process are unknown, and we posit the involvement of the cytoskeleton at myelin paranodes that may normally serve to compress the axon in this region. If so, the absence of regulation of paranode diameter in null mutants may indicate cytoskeletal abnormalities in oligodendrocytes.
In a manner similar to paranodal width, the distributions of paranodal length in 3-week-old Nkx6-2 null mice and littermates are comparable (Fig. 6d) (supplemental Table 1, available at www.jneurosci.org as supplemental material). At 4 months of age, paranodal length is increased in both wild-type and mutant mice to the same extent, and the distributions again are similar (Fig. 6e). At 8 months of age, paranodal length in wild-type mice is essentially unchanged from that at 4 months; however, length distribution for the mutants is substantially right-shifted (Fig. 6f). These data indicate that paranodal length normally increases during the most active phase of myelination and becomes invariant in adults over an extended period. In contrast, paranodal length in the null mutant mice continues to expand with age and may reflect an ongoing process of maturation or remodeling. Currently, we do not know whether the abnormalities in paranodal width and length in the mutants stem from a common pathology or are manifested as a result of the dysregulation of multiple independent pathways, but these data are consistent with Figures 5 and 6.
Dysregulated expression of paranodal genes in Nkx6-2 null mice
Defects in Nkx6-2 null mice are not limited to structural abnormalities at axoglial junctions. Densitometric analysis of Northern blots from spinal cord (Fig. 7A) and brain (data not shown) reveal that steady-state Nf155 mRNA is elevated in adult mutants. At early ages, hybridization signals are similar to controls; however, Nf155 mRNA levels are fourfold higher in older mutants, which likely reflects a derepression of transcriptional activity on the Neurofascin gene in oligodendrocytes. In contrast, the Neurofascin and Caspr genes are normally regulated in neurons from Nkx6-2 null mice at all ages, as are mRNAs encoding other myelin-specific genes, including the following: Tag-1, Mag, Mbp, and PlpI. Two other homeodomain transcription factors, Nkx6-1 and Nkx2-2, that could conceivably compensate for Nkx6-2 loss of function are not detectable in spinal cord of the mutants or controls at any age (data not shown). Finally, changes in Claudin 11 expression are observed at 4 and 13 months but are less than twofold and not consistent with age.
Figure 7.

Dysregulation of genes encoding paranodal proteins in Nkx6-2 null mice. A, Northern blots of spinal cord RNA from wild-type (+/+), heterozygous (+/-), and null (-/-) mice from P21 to 13 months. Loading control, 28S rRNA. B, Northern blots of retina and optic nerve (Opt. n.) RNAs from wild-type (+/+) and null (-/-) mice at P16 and 4 months. Loading control, 28S rRNA. C, Western blots of optic nerve proteins from wild-type (+/+) and null (-/-) mice at P16 and 4 months.
Steady-state mRNA levels of Contactin and Stathmin 1 in spinal cord are consistently altered in null mutants (increased and decreased, respectively); however, both genes are expressed by oligodendrocytes and neurons, and we sought to determine whether dysregulation occurs in one or both cell types. One of the only regions of CNS in which neuronal and myelinating oligodendrocyte cell bodies are easily separated is optic tract, and in Figure 7B we show Northern blots for gene expression in retina (neurons) and optic nerve (oligodendrocytes). As expected, Contactin and Stathmin 1 mRNAs are present in both retina and optic nerve, but only in mutant optic nerves are these genes dysregulated. Thus, Nkx6-2 regulates expression of Contactin and Stathmin 1 in oligodendrocytes, but neuronal expression is normal.
Optic tract Northern blots show gene expression changes for several other genes. Nf155 is not expressed in retina but is present in optic nerve and, consistent with the spinal cord data in Figure 7A, is derepressed in adult null mutants. Nf186 mRNA is present in retina but not optic nerve and is expressed normally in mutants. Finally, Tag-1 expression in oligodendrocytes is reduced to a small (twofold), probably insignificant extent at P16 and is expressed normally in adult mice.
To determine whether changes in steady-state mRNA levels are reflected at the protein level, we performed Western blots on single optic nerves from P16 and adult mice (Fig. 7C). In young mice, levels of NF155 and contactin are comparable with controls, but the levels of these proteins are elevated in null mice compared with controls and are consistent with mRNA levels in Figure 7, A and B. On the other hand, stathmin 1 levels in the mutants are similar to controls at both ages, which likely reflects a large background contribution to the total from neurons. Levels of the other proteins examined, including NF186, caspr, MAG, MBP, PLPI, and DM-20 are comparable with wild-type controls. Thus, in general, Western blot data can be accounted for or are consistent with the Northern blotting data.
Discussion
Our analysis of Nkx6-2 null mice reveals widespread abnormalities in myelinated axons in lieu of a strong behavioral phenotype. Importantly, many of these abnormalities are common to other myelin mutants harboring divergent genetic defects that perturb myelin or axoglial junctions. Such commonality suggests convergence of pathology on a cellular pathway that is critical for myelinogenesis and about which we know little. So, what are the major players in this cellular pathway and how are they regulated? The Nkx6-2 null phenotype affords a partial answer to this question but, moreover, provides novel insight into the transcriptional regulation of this pathway, identifies a network of effector molecules regulated by this pathway, and sheds sufficient light on this pathway to generate an integrated model of pathology.
The schematics shown in supplemental Figure 2 (available at www.jneurosci.org as supplemental material) depict salient ultrastructural features of a normal myelinated axon (Raine, 1984) as well as abnormalities that we observe in Nkx6-2 null mice. Although we cannot formally exclude the possibilities that pathology stems from abnormal myelin maintenance or demyelination, our data are most easily explained by current views of paranode formation (Pedraza et al., 2001) rather than by myelin destruction (supplemental Fig. 2, available at www.jneurosci.org as supplemental material). Accordingly, we integrate our observations in Nkx6-2 null mice and formulate a coherent model of pathogenesis stemming from the absence of a transcription factor by considering four issues.
First, the paranodal abnormalities highlighted in supplemental Figure 2B (available at www.jneurosci.org as supplemental material) may stem from a single structural defect that arises during myelinogenesis, that of obstructed myelin membrane expansion. In wild-type mice, circumferential myelin sheath expansion occurs at the inner tongue process, and the direction of expansion (clockwise or counterclockwise) is determined to a large extent by local factors (Knobler et al., 1974). Thus, adjacent membrane segments in the same sheath often expand in opposite directions. Adding to this turmoil, transient microtubule-based protrusions arising locally from the lateral edges of the myelin membrane, termed vermicular processes, extend along axons and frequently interfere with membrane expansion. Nonetheless, competing membrane segments eventually merge and capitulate, presumably by the process of cytoskeletal remodeling, to synchronized lamellipodial expansion and elongation along the axon to generate a myelin trapezoid (supplemental Fig. 2A, available at www.jneurosci.org as supplemental material). In Nkx6-2 null mice, this elongation phase is incomplete. The presence of stable vermicular-like processes subjacent to compact myelin (supplemental Fig. 2B, “e”; available at www.jneurosci.org as supplemental material) may interfere with or block longitudinal membrane expansion toward the paranodes and give rise to myelin flaps seen in adult Nkx6-2 null mice (Fig. 4f). The mechanism by which these vermicular-like processes persist is not clear, but one possibility is defective cytoskeletal remodeling. In this regard, it is tempting to speculate about the consequences of reduced stathmin 1 in myelin sheaths from the mutants (Fig. 7).
Stathmin 1 is a ubiquitous α-tubulin-binding protein that functions to destabilize microtubules, thereby facilitating cytoskeletal dynamics (Andersen, 2000). Drosophila mutants that are hypomorphic for stathmin 1 exhibit strong cell migration and axonal pathfinding phenotypes (Ozon et al., 2002), and aging Stathmin 1-null mice exhibit axonal degeneration (Liedtke et al., 2002). Oligodendrocyte process outgrowth is regulated by stathmin 1 in vitro, whereby lowering the activity of this protein promotes microtubule stability as well as process extension and branching (Liu et al., 2003). In the same vein, lower stathmin 1 levels in Nkx6-2 null oligodendrocytes would be expected to increase the stability of vermicular processes and lead to their persistence in myelin sheaths. If so, our data demonstrate an important regulatory function of Nkx6-2 during development, that of cytoskeletal remodeling. In a broader context, regulation of the cytoskeleton by homeodomain proteins is not without precedence. The engrailed protein has been found to regulate expression of two cytoskeletal proteins, β3-tubulin in Drosophila (Serrano et al., 1997) and MAP 1B in chick (Montesinos et al., 2001).
A second aspect of pathology in Nkx6-2 null mice to be considered is altered expression of genes encoding myelin proteins. Sustained expression of Nf155 mRNA (Fig. 7A,B), a message that is expressed only transiently in oligodendrocytes during normal development (Collinson et al., 1998), indicates that the Neurofascin gene is genetically downstream of Nkx6-2 in this cell type. NF155 is a member of the L1 family of neural cell adhesion molecules, which bind to the cytoskeleton through cytoplasmic tail ankyrin-binding domains, mediate intercellular signaling, and coordinate cell growth (Hortsch, 1996; Zhang et al., 1998; Bennett and Baines, 2001). At least a portion of the NF155 pool in oligodendrocytes is phosphorylated early in myelinogenesis, a posttranslational modification that inhibits ankyrin-binding activity (Tuvia et al., 1997; Tait et al., 2000). Nonetheless, NF155 is efficiently confined to mature paranodal loops, likely interacts with the cytoskeleton through unknown intermediaries, and likely recruits adhesion proteins in the axolemma, such as caspr and contactin, to form axoglial junctions (Rasband et al., 1999; Charles et al., 2002). Overexpression of Contactin in null mutant mice indicates that Nkx6-2 regulates multiple cell adhesion proteins in oligodendrocytes during myelinogenesis. Contactin is a member of the immunoglobulin superfamily and likely mediates homophilic interactions with neuronal contactin at paranodes (Rios et al., 2000). It is presently unclear how overexpression of the Neurofascin and Contactin genes contributes to pathology; however, excessive accumulation of NF155 and contactin at the lateral edges of myelin sheaths may trigger premature assembly of axoglial junctions before the expanding membrane reaches paranodes.
Currently, we find no evidence from Northern and Western blotting that Nkx6-2 regulates the transcription of previously proposed myelin genes (Awatramani et al., 1997; Farhadi et al., 2003). Previous studies had identified cis-elements in the proximal promoters of the MBP and PLP1 genes, but these sites do not appear to be functional in vivo (Cai et al., 2001). Of course, given the limitations of quantitation techniques used in the current study, we cannot rule out the possibility that small expression changes (approximately less than twofold) in these genes occur in Nkx6-2 null mice or that functional redundancy in regulatory networks controlling these genes maintains normal expression. Bearing these caveats in mind, the simplest conclusion from our data is that Nkx6-2 regulates expression of a limited set of genes involved in cell adhesion and cytoskeletal remodeling.
The third consideration is the absence of a strong behavioral phenotype in Nkx6-2 null mice in view of the ultrastructural abnormalities that we observe. Although internodal loops, flaps, thin myelin, and naked axons are widespread in these animals, our data indicate that outer lateral loops in most sheaths reach the paranode and form axoglial junctions sufficiently well to enable appropriate targeting of NF155, caspr, contactin, K+ channels, NF186, and Na+ channels. Previous studies showed that only a few wraps of compact myelin membrane are necessary for reliable nerve conduction in CNS (Koles and Rasminsky, 1972); together, these data account for the relatively minor physiological consequences for null mutants without the need to invoke compensatory mechanisms from other neural Nkx family members. Nevertheless, the subtlety of this phenotype does not diminish the importance of the gene regulatory network uncovered in the current study. Indeed, we find that Nkx6-2 is genetically upstream of genes encoding a cytoskeleton-remodeling protein and two cell adhesion molecules that interact with the cytoskeleton. Regardless of whether these genes are direct or indirect targets of Nkx6-2, our data demonstrate novel aspects of the processes involved in generating the nervous system and provide an important animal model in which to further explore myelinogenesis.
Finally, it is presently unclear whether the CNS phenotype in Nkx6-2 null mice stems entirely from a primary defect in oligodendrocytes. Null embryos express high levels of lacZ in brain nuclei contributing to cranial nerves and neurons throughout the ventral neural tube (Fig. 1a) from E8.5 to E14.5. Committed oligodendrocyte precursors arise only at the end of this developmental window; thus, abnormal differentiation of neurons in null embryos could conceivably perturb interactions between these cells and oligodendrocytes in subsequent developmental stages. In support of this notion, altered expression of the transcription factors Dbx1 and Dbx2 in p0 and p1 domain progenitors from Nkx6-2 null mice (Vallstedt et al., 2001) may portend cell fate changes that impact postnatal axon- glial interactions. On the other hand, we do not observe Nkx6-2 expression in neural retina either in embryos during the proliferation and differentiation phases of this tissue (E9.5-12.5) or postnatally during myelination of the optic tract (2-12 weeks of age). Thus, retinal ganglion cell defects seem an unlikely source of ultrastructural abnormalities in optic nerve myelin sheaths from null mutants.
Footnotes
This work was supported by grants to A.G. from the National Institute on Deafness and Other Communication Disorders, National Institutes of Health (DC006262), and the National Multiple Sclerosis Society (PP0755, RG2891) and by a grant to J.K. and A.G. from the National Multiple Sclerosis Society (RG2769). We are grateful for guidance, discussion, and insights from Dr. Victor Friedrich Jr, Mount Sinai School of Medicine (MSSM) (New York, NY). We thank Dr. Kevin Kelley, director of the transgenic core at MSSM, who performed ES cell injections into blastocysts, Dr. Scott Henderson and Valerie Williams in the electron microscopy core at MSSM, and Dr. James Hatfield in the confocal microscopy core at the Veterans Administration Medical Center (Detroit, MI). We also thank Dr. R. Awatramani, who identified the mouse Nkx6-2 gene from a 129 Sv/Ev genomic library; Dr. M. Bhat (University of North Carolina, Chapel Hill, NC), Dr. M. Rasband (University of Connecticut Health Center), Dr. E. Peles (Weisman Institute, Jerusalem, Israel), Dr. S. Tait (University of Edinburgh, UK), Dr. J. Salzer (New York University), Dr. J. A. Girault (Institut National de la Santé et de la Recherche Médicale U114, College de France) for cDNA probes and antibodies, and Dr. D. Neusteadter.
Correspondence should be addressed to Dr. Alexander Gow, Center for Molecular Medicine and Genetics, 3216 Scott Hall, 540 East Canfield Avenue, Wayne State University School of Medicine, Detroit, MI 48201. E-mail: agow@med.wayne.edu.
Copyright © 2004 Society for Neuroscience 0270-6474/04/2411215-11$15.00/0
References
- Andersen SS (2000) Spindle assembly and the art of regulating microtubule dynamics by MAPs and Stathmin/Op18. Trends Cell Biol 10: 261-267. [DOI] [PubMed] [Google Scholar]
- Arroyo EJ, Xu T, Poliak S, Watson M, Peles E, Scherer SS (2001) Internodal specializations of myelinated axons in the central nervous system. Cell Tissue Res 305: 53-66. [DOI] [PubMed] [Google Scholar]
- Awatramani R, Scherer S, Grinspan J, Collarini E, Skoff R, O'Hagan D, Garbern J, Kamholz J (1997) Evidence that the homeodomain protein Gtx is involved in the regulation of oligodendrocyte myelination. J Neurosci 17: 6657-6668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennett V, Baines AJ (2001) Spectrin and ankyrin-based pathways: metazoan inventions for integrating cells into tissues. Physiol Rev 81: 1353-1392. [DOI] [PubMed] [Google Scholar]
- Berndt JA, Kim JG, Tosic M, Kim C, Hudson LD (2001) The transcriptional regulator Yin Yang 1 activates the myelin PLP gene. J Neurochem 77: 935-942. [DOI] [PubMed] [Google Scholar]
- Bhat MA (2003) Molecular organization of axo-glial junctions. Curr Opin Neurobiol 13: 552-559. [DOI] [PubMed] [Google Scholar]
- Bhat MA, Rios JC, Lu Y, Garcia-Fresco GP, Ching W, Martin MS, Li J, Einheber S, Chesler M, Rosenbluth J, Salzer JL, Bellen HJ (2001) Axon-glia interactions and the domain organization of myelinated axons requires neurexin iv/caspr/paranodin. Neuron 30: 369-383. [DOI] [PubMed] [Google Scholar]
- Bosio A, Bussow H, Adam J, Stoffel W (1998) Galactosphingolipids and axono-glial interaction in myelin of the central nervous system. Cell Tissue Res 292: 199-210. [DOI] [PubMed] [Google Scholar]
- Boyle ME, Berglund EO, Murai KK, Weber L, Peles E, Ranscht B (2001) Contactin orchestrates assembly of the septate-like junctions at the paranode in myelinated peripheral nerve. Neuron 30: 385-397. [DOI] [PubMed] [Google Scholar]
- Cai J, St. Amand T, Yin H, Guo H, Li G, Zhang Y, Chen Y, Qiu M (1999) Expression and regulation of the chicken Nkx-6.2 homeobox gene suggest its possible involvement in the ventral neural patterning and cell fate specification. Dev Dyn 216: 459-468. [DOI] [PubMed] [Google Scholar]
- Cai J, Qi Y, Wu R, Modderman G, Fu H, Liu R, Qiu M (2001) Mice lacking the Nkx6.2 (Gtx) homeodomain transcription factor develop and reproduce normally. Mol Cell Biol 21: 4399-4403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charles P, Tait S, Faivre-Sarrailh C, Barbin G, Gunn-Moore F, Denisenko-Nehrbass N, Guennoc AM, Girault JA, Brophy PJ, Lubetzki C (2002) Neurofascin is a glial receptor for the paranodin/Caspr-contactin axonal complex at the axoglial junction. Curr Biol 12: 217-220. [DOI] [PubMed] [Google Scholar]
- Coetzee T, Fujita N, Dupree J, Shi R, Blight A, Suzuki K, Popko B (1996) Myelination in the absence of galactocerebroside and sulfatide: normal structure with abnormal function and regional instability. Cell 86: 209-219. [DOI] [PubMed] [Google Scholar]
- Collinson JM, Marshall D, Gillespie CS, Brophy PJ (1998) Transient expression of neurofascin by oligodendrocytes at the onset of myelinogenesis: implications for mechanisms of axon-glial interaction. Glia 23: 11-23. [DOI] [PubMed] [Google Scholar]
- Denisenko-Nehrbass N, Oguievetskaia K, Goutebroze L, Galvez T, Yamakawa H, Ohara O, Carnaud M, Girault JA (2003) Protein 4.1B associates with both Caspr/paranodin and Caspr2 at paranodes and juxtaparanodes of myelinated fibres. Eur J Neurosci 17: 411-416. [DOI] [PubMed] [Google Scholar]
- Dupree JL, Coetzee T, Suzuki K, Popko B (1998) Myelin abnormalities in mice deficient in galactocerebroside and sulfatide. J Neurocytol 27: 649-659. [DOI] [PubMed] [Google Scholar]
- Farhadi HF, Lepage P, Forghani R, Friedman HC, Orfali W, Jasmin L, Miller W, Hudson TJ, Peterson AC (2003) A combinatorial network of evolutionarily conserved myelin basic protein regulatory sequences confers distinct glial-specific phenotypes. J Neurosci 23: 10214-10223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedrich Jr VL (1974) The myelin deficit in quacking mice. Brain Res 82: 168-172. [DOI] [PubMed] [Google Scholar]
- Fujiyoshi T, Hood L, Yoo TJ (1994) Restoration of brain stem auditory-evoked potentials by gene transfer in shiverer mice. Ann Otol Rhinol Laryngol 103: 449-456. [DOI] [PubMed] [Google Scholar]
- Girault J, Peles E (2002) Development of nodes of Ranvier. Curr Opin Neurobiol 12: 476-485. [DOI] [PubMed] [Google Scholar]
- Gow A, Friedrich VL, Lazzarini RA (1992) Myelin basic protein gene contains separate enhancers for oligodendrocytes and Schwann cell expression. J Cell Biol 119: 605-616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gow A, Southwood CM, Lazzarini RA (1998) Disrupted proteolipid protein trafficking results in oligodendrocyte apoptosis in an animal model of Pelizaeus-Merzbacher disease. J Cell Biol 140: 925-934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gow A, Southwood CM, Li JS, Pariali M, Riordan GP, Brodie SE, Danias J, Bronstein JM, Kachar B, Lazzarini RA (1999) CNS myelin and Sertoli cell tight junction strands are absent in Osp/Claudin 11-null mice. Cell 99: 649-659. [DOI] [PubMed] [Google Scholar]
- Gow A, Davies C, Southwood CM, Frolenkov G, Chrustowski M, Ng L, Yamauchi D, Marcus DM, Kachar B (2004) Deafness in Claudin 11-null mice reveals the critical contribution of basal cell tight junctions to stria vascularis function. J Neurosci 24: 7051-7062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hortsch M (1996) The L1 family of neural cell adhesion molecules: old proteins performing new tricks. Neuron 17: 587-593. [DOI] [PubMed] [Google Scholar]
- Knobler RL, Stempak JG, Laurencin M (1974) Oligodendroglial ensheathment of axons during myelination in the developing rat central nervous system. A serial section electron microscopical study. J Ultrastruct Res 49: 34-49. [DOI] [PubMed] [Google Scholar]
- Koles ZJ, Rasminsky M (1972) A computer simulation of conduction in demyelinated nerve fibres. J Physiol (Lond) 227: 351-364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Komuro I, Schalling M, Jahn L, Bodmer R, Jenkins NA, Copeland NG, Izumo S (1993) Gtx: a novel murine homeobox-containing gene, expressed specifically in glial cells of the brain and germ cells of testis, has a transcriptional repressor activity in vitro for a serum-inducible promoter. EMBO J 12: 1387-1401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liedtke W, Leman EE, Fyffe RE, Raine CS, Schubart UK (2002) Stathmin-deficient mice develop an age-dependent axonopathy of the central and peripheral nervous systems. Am J Pathol 160: 469-480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu A, Muggironi M, Marin-Husstege M, Casaccia-Bonnefil P (2003) Oligodendrocyte process outgrowth in vitro is modulated by epigenetic regulation of cytoskeletal severing proteins. Glia 44: 264-274. [DOI] [PubMed] [Google Scholar]
- Lu QR, Sun T, Zhu Z, Ma N, Garcia M, Stiles CD, Rowitch DH (2002) Common developmental requirement for Olig function indicates a motor neuron/oligodendrocyte connection. Cell 109: 75-86. [DOI] [PubMed] [Google Scholar]
- Montesinos ML, Foucher I, Conradt M, Mainguy G, Robel L, Prochiantz A, Volovitch M (2001) The neuronal microtubule-associated protein 1B is under homeoprotein transcriptional control. J Neurosci 21: 3350-3359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Gorman S, Fox DT, Wahl GM (1991) Recombinase-mediated gene activation and site-specific integration in mammalian cells. Science 251: 1351-1355. [DOI] [PubMed] [Google Scholar]
- Ozon S, El Mestikawy S, Sobel A (1999) Differential, regional, and cellular expression of the stathmin family transcripts in the adult rat brain. J Neurosci Res 56: 553-564. [DOI] [PubMed] [Google Scholar]
- Ozon S, Guichet A, Gavet O, Roth S, Sobel A (2002) Drosophila stathmin: a microtubule-destabilizing factor involved in nervous system formation. Mol Biol Cell 13: 698-710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pattyn A, Vallstedt A, Dias JM, Sander M, Ericson J (2003) Complementary roles for Nkx6 and Nkx2 class proteins in the establishment of motoneuron identity in the hindbrain. Development 130: 4149-4159. [DOI] [PubMed] [Google Scholar]
- Pedraza L, Huang JK, Colman DR (2001) Organizing principles of the axoglial apparatus. Neuron 30: 335-344. [DOI] [PubMed] [Google Scholar]
- Peters A, Palay SL, Webster H deF (1991) The fine structure of the nervous system: neurons and their supporting cells, Ed 3. New York: Oxford.
- Qi Y, Cai J, Wu Y, Wu R, Lee J, Fu H, Rao M, Sussel L, Rubenstein J, Qiu M (2001) Control of oligodendrocyte differentiation by the Nkx2.2 homeodomain transcription factor. Development 128: 2723-2733. [DOI] [PubMed] [Google Scholar]
- Raine CS (1984) Morphology of myelin and myelination. In: Myelin, Ed 2 (Morell P, ed), pp 1-50. New York: Plenum.
- Ramon y Cajal S (1899) Textura del sistema nervioso del hombre y de los vertebrados. Madrid: Nicolas Moya.
- Rasband MN, Peles E, Trimmer JS, Levinson SR, Lux SE, Shrager P (1999) Dependence of nodal sodium channel clustering on paranodal axoglial contact in the developing CNS. J Neurosci 19: 7516-7528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rios JC, Melendez-Vasquez CV, Einheber S, Lustig M, Grumet M, Hemperly J, Peles E, Salzer JL (2000) Contactin-associated protein (Caspr) and contactin form a complex that is targeted to the paranodal junctions during myelination. J Neurosci 20: 8354-8364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenbluth J (1987) Abnormal axoglial junctions in the myelin-deficient rat mutant. J Neurocytol 16: 497-509. [DOI] [PubMed] [Google Scholar]
- Salzer JL (2003) Polarized domains of myelinated axons. Neuron 40: 297-318. [DOI] [PubMed] [Google Scholar]
- Sander M, Sussel L, Conners J, Scheel D, Kalamaras J, Dela Cruz F, Schwitzgebel V, Hayes-Jordan A, German M (2000) Homeobox gene Nkx6.1 lies downstream of Nkx2.2 in the major pathway of beta-cell formation in the pancreas. Development 127: 5533-5540. [DOI] [PubMed] [Google Scholar]
- Schnapp B, Mugnaini E (1978) Membrane architecture of myelinated fibers as seen by freeze-fracture. In: Physiology and pathobiology of axons (Waxman SG, ed), pp 83-123. New York: Raven.
- Serrano N, Brock HW, Maschat F (1997) beta3-tubulin is directly repressed by the engrailed protein in Drosophila Development 124: 2527-2536. [DOI] [PubMed] [Google Scholar]
- Sim FJ, Hinks GL, Franklin RJ (2000) The re-expression of the homeodomain transcription factor Gtx during remyelination of experimentally induced demyelinating lesions in young and old rat brain. Neuroscience 100: 131-139. [DOI] [PubMed] [Google Scholar]
- Southwood CM, Gow A (2001) Functions of OSP/claudin 11-containing parallel tight junctions: implications from the knockout mouse. In: Tight junctions, Ed 2 (Anderson JM, Cereijido M, eds), pp 719-741. New York: CRC.
- Stecca B, Southwood CM, Gragerov A, Kelley KA, Friedrich VLJ, Gow A (2000) The evolution of lipophilin genes from invertebrates to tetrapods: DM-20 cannot replace PLP in CNS myelin. J Neurosci 20: 4002-4010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stolt CC, Lommes P, Friedrich RP, Wegner M (2004) Transcription factors Sox8 and Sox10 perform non-equivalent roles during oligodendrocyte development despite functional redundancy. Development 131: 2349-2358. [DOI] [PubMed] [Google Scholar]
- Tait S, Gunn-Moore F, Collinson JM, Huang J, Lubetzki C, Pedraza L, Sherman DL, Colman DR, Brophy PJ (2000) An oligodendrocyte cell adhesion molecule at the site of assembly of the paranodal axo-glial junction. J Cell Biol 150: 657-666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trapp BD, Peterson J, Ransohoff RM, Rudick R, Mork S, Bo L (1998) Axonal transection in the lesions of multiple sclerosis. N Engl J Med 338: 278-285. [DOI] [PubMed] [Google Scholar]
- Tuvia S, Garver TD, Bennett V (1997) The phosphorylation state of the FIGQY tyrosine of neurofascin determines ankyrin-binding activity and patterns of cell segregation. Proc Natl Acad Sci USA 94: 12957-12962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vallstedt A, Muhr J, Pattyn A, Pierani A, Mendelsohn M, Sander M, Jessell TM, Ericson J (2001) Different levels of repressor activity assign redundant and specific roles to nkx6 genes in motor neuron and interneuron specification. Neuron 31: 743-755. [DOI] [PubMed] [Google Scholar]
- Zhang X, Davis JQ, Carpenter S, Bennett V (1998) Structural requirements for association of neurofascin with ankyrin. J Biol Chem 273: 30785-30794. [DOI] [PubMed] [Google Scholar]
- Zheng QY, Johnson KR, Erway LC (1999) Assessment of hearing in 80 in-bred strains of mice by ABR threshold analyses. Hearing Res 130: 94-107. [DOI] [PMC free article] [PubMed] [Google Scholar]