

Abstract
Protein movements underlying ligand-gated ion channel activation are poorly understood. The binding of agonist initiates a series of conformational movements that ultimately lead to the opening of the ion channel pore. Although little is known about local movements within the GABA-binding site, a recent structural model of the GABAA receptor (GABAAR) ligand-binding domain predicts that β2Glu155 is a key residue for direct interactions with the neurotransmitter (Cromer et al., 2002). To elucidate the role of the β2Ile154-Asp163 region in GABAAR activation, each residue was individually mutated to cysteine and coexpressed with wild-type α1 subunits in Xenopus laevis oocytes. Seven mutations increased the GABA EC50 value (8- to 3400-fold), whereas three mutations (E155C, S156C, and G158C) also significantly increased the 2-(3-carboxypropyl)-3-amino-6-(4-methoxyphenyl) pyridazinium (SR-95531) KI value. GABA, SR-95531, and pentobarbital slowed N-biotinylaminoethyl methanethiosulfonate modification of T160C and D163C, indicating that β2Thr160 and β2Asp163 are located in or near the GABA-binding site and that this region undergoes structural rearrangements during channel gating. Cysteine substitution of β2Glu155 resulted in spontaneously open GABAARs and differentially decreased the GABA, piperidine-4-sulfonic acid (partial agonist), and SR-95531 sensitivities, indicating that the mutation perturbs ligand binding as well as channel gating. Tethering thiol-reactive groups onto β2E155C closed the spontaneously open channels, suggesting that β2Glu155 is a control element involved in coupling ligand binding to channel gating. Structural modeling suggests that the β2 Ile154-Asp163 region is a protein hinge that forms a network of interconnections that couples binding site movements to the cascade of events leading to channel opening.
Keywords: GABA, SR-95531, gabazine, picrotoxin, piperidine-4-sulfonic acid, mutagenesis, substituted cysteine accessibility method, pentobarbital, Xenopus laevis oocytes, methanethiosulfonate, two-electrode voltage clamp
Introduction
Agonists and antagonists induce different molecular rearrangements in neurotransmitter-binding sites of ligand-gated ion channels (LGICs) (Armstrong and Gouaux, 2000; Boileau et al., 2002; Chang and Weiss, 2002). Agonists, but not antagonists, promote opening of the ion channel pore. It is likely that movements of amino acids near or within the neurotransmitter recognition site trigger the cascade of events leading to channel opening (Boileau et al., 2002; Torres and Weiss, 2002; Unwin et al., 2002; Miyazawa et al., 2003; Chakrapani et al., 2004). Here, we examined the I1e154-Asp163 region of the GABAA receptor (GABAAR) β2 subunit to identify residues that mediate local movements within the binding site that initiate channel gating and residues involved in GABA binding.
GABAARs are heteropentameric LGICs that mediate fast synaptic inhibitory neurotransmission in the brain. The α1β2γ2 GABAAR subtype is the most abundant in vivo, and heterologous expression studies suggest a β-α-β-α-γ stoichiometry and subunit arrangement (Chang et al., 1996; Tretter et al., 1997; Farrar et al., 1999; Baumann et al., 2001, 2002). Expression of α and β subunits also gives rise to functional GABAAR with putative stoichiometries of either 3α:2β (Im et al., 1995) or 3β:2α (Baumann et al., 2001; Horenstein et al., 2001) that lack sensitivity to benzodiazepines (Schofield et al., 1987; Pritchett et al., 1989), are responsive to barbiturates, and have a high apparent affinity for agonists (Boileau et al., 1999, 2002; Wagner and Czajkowski, 2001).
A recent homology model of the GABAAR agonist-binding site predicts that β2Glu155 interacts with the positively charged moiety of GABA (Cromer et al., 2002). In addition, mutagenesis studies have determined that nearby residues, β2Tyr157 and β2Thr160, are important for GABA binding (Amin and Weiss, 1993). Similarly, amino acid residues in aligned regions of the muscle-type nicotinic acetylcholine α1 (Trp148, Tyr151, and Asp152) (Dennis et al., 1988; Galzi et al., 1991; Sugiyama et al., 1996), glycine α1 (Asp148, Gly160, and Tyr161) (Vandenberg et al., 1992, 1993), and serotonin type 3 receptor subunits (Trp160) (Spier and Lummis, 2000) have been determined to be critical for agonist-antagonist binding (see Fig. 1) and define region “B”a of the ligand-binding site. The contributions of these residues in forming their respective agonist binding sites are supported by homology models that place this region (from β-strand 7 and loop 8) within the putative core of LGIC neurotransmitter-binding sites (Cromer et al., 2002; Holden and Czajkowski, 2002; LeNovère et al., 2002; Newell and Czajkowksi, 2003; Reeves et al., 2003).
Figure 1.

The Ile154-Asp163 segment (region B) of the rat GABAAR β2 subunit is aligned with homologous segments of the Torpedo nAChR α, glycine receptor (GlyR) α1, and 5-HT3a subunits. The numbering reflects the position of the residues in the mature GABAAR β2 subunit. Circled residues β2Tyr157 and β2Thr160 are implicated in GABA binding (Amin and Weiss, 1993). Residues important for ligand recognition in other binding sites are boxed. nAChR αTrp149 and possibly Tyr151 and Asp152 have been implicated in forming the acetylcholine-binding site (Dennis et al., 1988; Galzi et al., 1991; Sugiyama et al., 1996). GlyR α1Gly160, Tyr161 (Vandenberg et al., 1992; Schmieden et al., 1993), and Phe159 (Schmieden et al., 1993) are important for antagonist recognition, whereas 5-HT3a Trp160 (Spier and Lummis, 2000) is important for the actions of serotonin. nAChR αG158S has been reported as a naturally occurring mutation in myasthenia gravis patients that gives rise to “slow channels” (Sine et al., 1995; Croxen et al., 1997) GABAAR β2 subunit residues accessible to MTSEA-biotin modification are underlined (E155C, G158C, T160C, and D163C).
Here, we demonstrate that expression of α1β2(E155C) gives rise to spontaneously open GABA channels. Mutation of β2Glu155 alters both channel-gating properties and impairs agonist binding. In addition, we provide evidence that β2Thr160 and β2Asp163 are found on an aqueous surface within or near the GABA-binding site and undergo conformational rearrangements during pentobarbital-mediated gating events. Together, the data suggest that movement in this region of the GABA-binding site is one of the initial triggers for coupling binding to gating.
Materials and Methods
Mutagenesis and expression in oocytes. Rat cDNAs encoding the GABAAR α1 and β2 subunits were used in this study. The β2 cysteine mutants were engineered using a recombinant PCR method, as described previously (Boileau et al., 1999; Kucken et al., 2000). Cysteine substitutions were made in the β2 subunit at Ile154, Glu155, Ser156, Tyr157, Gly158, Tyr159, Thr160, Thr161, Asp162, and Asp163 (see Fig. 1). Cysteine substitutions were verified by restriction endonuclease digestion and double-stranded DNA sequencing.
All wild-type and mutant cDNAs were subcloned into the vector pGH19 (Liman et al., 1992; Robertson et al., 1996) for expression in Xenopus laevis oocytes. Oocytes were isolated as described previously (Boileau et al., 1998). cRNA transcripts were prepared using the T7 mMessage machine (Ambion, Austin, TX). GABAA receptor α1 and β2 or β2 mutant subunits were coexpressed by injection of cRNA (675 pg/subunit) in a 1:1 ratio (α:β). The oocytes were maintained in modified ND96 medium [containing (in mm): 96 NaCl, 2 KCl, 1 MgCl2, 1.8 CaCl2, and 5 HEPES, pH 7.4] that had been supplemented with 100 μg/ml gentamicin and 100 μg/ml bovine serum albumin. Oocytes were used 2-7 d after injection for electrophysiological recordings.
Two-electrode voltage-clamp analysis. Oocytes under two-electrode voltage clamp were perfused continuously with ND96 recording solution [containing (in mm): 96 NaCl, 2 KCl, 1 MgCl2, 1.8 CaCl2, 5 HEPES, pH 7.4] at a rate of ∼5 ml/min. The holding potential was -80 mV. The volume of the recording chamber was 200 μl. Standard two-electrode voltage-clamp procedures were performed using a GeneClamp500 Amplifier (Axon Instruments, Foster City, CA). Borosilicate electrodes were filled with 3 m KCl and had resistances of 0.5-3.0 MΩ in ND96. Stock solutions of GABA, 2-(3-carboxypropyl)-3-amino-6-(4-methoxyphenyl) pyridazinium (SR-95531) and piperidine-4-sulfonic acid (P4S) (Sigma, St. Louis, MO) were prepared in water, whereas stock solutions of picrotoxin (PTX) (Sigma) and N-biotinylaminoethyl methanethiosulfonate (MTSEA-biotin; 100 mm; Biotium, Hayward, CA) were prepared in dimethylsulfoxide (DMSO). All compounds were diluted appropriately in ND96 such that the final concentration of DMSO was ≤2%. The vehicle did not affect GABA-activated currents.
To measure the sensitivity to GABA or P4S, the agonist (0.0001-100 mm) or partial agonist (0.00001-10 mm) was applied via gravity perfusion or by pipette application (∼5-8 sec) with a 3-15 min washout period between each application to ensure complete recovery from desensitization. Peak GABA- or P4S-activated current (IGABA or IP4S) was recorded. To correct for slow drift in the amplitude of the response as a function of time, concentration-response data were normalized to a low concentration of agonist (EC2-EC5). The apparent affinity of pentobarbital using concentrations between 0.01 and 10 mm was determined via gravity perfusion (∼5-8 sec) with a 3-5 min washout period between each application. Peak pentobarbital-activated current was recorded. Concentration-response data for pentobarbital were normalized to a previous application of pentobarbital (100 μm). Concentration-response curves for GABA, P4S, or pentobarbital were generated for each recombinant receptor, and the data were fitted by nonlinear regression analysis using Prism software (GraphPad, San Diego, CA). Data were fitted to the following equation: I = Imax/(1 + (EC50/[A])n), where I is the peak amplitude of the current for a given concentration of GABA, P4S, or pentobarbital ([A]), Imax is the maximum current, EC50 is the concentration required for half-maximal receptor activation, and n is the Hill coefficient.
To determine SR-95531 or PTX IC50 values, GABA (EC50) was applied via gravity perfusion followed by a brief washout period (20 sec) before application of GABA (EC50) and increasing concentrations of SR-95531 or PTX. The response to the application of SR-95531/PTX and GABA was normalized to the response elicited by the agonist alone. Concentration-inhibition curves were generated by nonlinear regression analysis using GraphPad Prism software. Data were fitted to the following equation: 1 - 1/(1 + (IC50/[Ant])n), where IC50 is the concentration of antagonist ([Ant]) that reduces the amplitude of the GABA-evoked current by 50%, and n is the Hill coefficient. SR-95531 KI values were calculated using the Cheng-Prussof correction: KI = IC50/(1 + ([A]/EC50)), where [A] is the concentration of GABA used in each experiment, and EC50 is the concentration of GABA that elicits a half-maximal response for each receptor (Cheng and Prussof, 1973).
Modification of cysteine residues by MTSEA-biotin. MTSEA-biotin was the cysteine-specific reagent used in this study because it is a relatively impermeant compound (Daniels and Amara, 1998), the dimensions (14.5 Å unreacted moiety; 11.2 Å reacted moiety) of which are similar to SR-95531 (13.5 Å) but longer than GABA (4.5 Å). Therefore, it is reasonable to assume that MTSEA-biotin can occupy the GABA-binding site and that this reagent will principally modify extracellular cysteine residues. We used the following criterion for stability of the response for these studies: ≤10% variation in IGABA (EC50) on two consecutive applications at regular intervals (10 min). Oocytes were then allowed to recover fully, after which a high concentration of MTSEA-biotin (2 mm) was applied (2 min). After MTSEA-biotin application, cells were washed (5 min) with ND96, after which GABA (EC50) was reapplied to determine the effect of MTSEA-biotin application on IGABA. Effects of MTSEA-biotin were calculated as the difference in the amplitude of the GABA-gated current before and after MTSEA-biotin application as follows: (IGABAPRE - IGABAPOST/IGABAPRE) × 100, where “post” refers to the amplitude of the GABA-gated current after MTSEA-biotin application, and “pre” refers to the amplitude of the stabilized GABA-gated current before covalent modification by MTSEA-biotin.
Rate of MTSEA-biotin reaction. The rate at which MTSEA-biotin modified introduced cysteine residues (E155C, T160C, and D163C) was measured using low MTSEA-biotin concentrations, as described previously (Newell and Czajkowski, 2003). The concentrations of MTSEA-biotin used were 50 nm (D163C), 100 nm (T160C), and 2 mm (E155C). The experimental protocol is described as follows: GABA (EC50) application (5 sec), ND96 washout (25 sec), MTSEA-biotin application (10-20 sec), ND96 washout (2.2-2.3 min). The sequence was repeated until IGABA no longer changed after the MTSEA-biotin treatment (i.e., the reaction had proceeded to apparent completion). The individual abilities of GABA, SR-95531, and pentobarbital to alter the rate of cysteine modification by MTSEA-biotin were determined by coapplying GABA (5 × EC50), SR-95531 (6 × KI), or pentobarbital (50 or 500 μm) during the MTSEA-biotin pulse. In all cases, the wash times were adjusted to ensure that currents obtained from test pulses of GABA (EC50) after exposure to high concentrations of GABA, SR-95531, or pentobarbital were stable. This ensured (1) a complete washout of drugs and that (2) reductions in the current amplitude resulted from the application of MTSEA-biotin.
For all rate experiments, the decrease in IGABA was plotted as a function of the cumulative time of MTSEA-biotin exposure and fit to a single-exponential decay function using GraphPad Prism software. A pseudo-first-order rate constant (k1) was determined, and the second-order rate constant (k2) was calculated by dividing k1 by the concentration of MTSEA-biotin used in the assay (Pascual and Karlin, 1998). Second-order rate constants were determined using at least two different concentrations of MTSEA-biotin to ensure accuracy of the protocol.
Statistical analysis. Log (EC50) values, log (KI) values, and log (k2) rates were analyzed using a one-way ANOVA followed by a Dunnett's post hoc test to determine levels of significance.
Structural modeling. The mature protein sequences of the rat α1 and β2 subunits were homology modeled with a subunit of the ACh binding protein (AChBP) (Brejc et al., 2001). The crystal structure of the AChBP was downloaded from RCSB Protein Data Bank (code 1I9B) and loaded into Swiss Protein Bank Viewer (SPDBV). The α1 protein sequence from Thr12-Ile227 and the β2 protein sequence from Ser10-Leu218 were aligned with the AChBP primary amino acid sequence as depicted by Cromer et al. (2002) and threaded onto the AChBP tertiary structure using the “Interactive Magic Fit” function of SPDBV. The threaded subunits were imported into SYBYL (Tripos, St. Louis, MO), in which energy minimization was performed (<0.5 kcal/Å). The first 100 iterations were performed using Simplex minimization (Press et al., 1988) followed by 1000 iterations using the Powell conjugate gradient method (Powell, 1977). A β2/α1 GABA-binding site interface was assembled by overlaying the monomeric subunits on the AChBP scaffold, and the resulting structure was imported into SYBYL and energy minimized. Neither water nor entropy factors were included during the minimizations. After the global energy minimization, several TRIPOS programs were run to evaluate the accuracy of the model. Ramachandran plots, χ plots, side-chain positions, and cis- and trans-bonds were all examined. Problems in the structure that were revealed by these evaluations were fixed manually, and energy minimizations were run again as needed. Our model is quite similar to models published recently for the nicotinic ACh receptor (nAChR) and GABAAR ligand-binding domains (Cromer et al., 2002; LeNovère et al., 2002). Regions with insertions were modeled by fitting structures from a loop database. Because the sequence identity of the AChBP and the GABAAR extracellular ligand-binding domain is only 18%, caution must be used in interpreting the absolute positions of individual side-chain residues in the model.
Results
Expression and functional characterization of cysteine mutants
Cysteine substitutions were engineered at 10 individual positions (Fig. 1) in the GABAAR β2 subunit (Ile154, Glu155, Ser156, Tyr157, Gly158, Tyr159, Thr160, Thr161, Asp162, and Asp163), coexpressed with wild-type α1 subunits in X. laevis oocytes, and analyzed using two-electrode voltage clamp. Expression of most mutant β2 subunits produced functional channels (IGABA = 1-10 μA), with the exceptions Y157C and Y159C. We speculate that introduction of cysteine residues at these positions impaired assembly of mutant receptors.
For the cysteine mutants that did express, seven of eight significantly increased GABA EC50 values, demonstrating that this region is particularly sensitive to structural perturbation. Expression of receptors containing I154C, E155C, S156C, G158C, T160C, D162C, and D163C increased GABA EC50 values 8-, 3375-, 22-, 260-, 23-, 18-, and 9-fold relative to wild type (1.6 μm) (Fig. 2A, Table 1). The Hill coefficients for GABA activation of G158C- and D163C-containing receptors were significantly lower than wild type. The KI values for the competitive antagonist SR-95531 were significantly different from wild type (KI = 163 nm) for E155C, S156C, and G158C by 11-, 3-, and 18-fold, respectively (Table 1, Fig. 2B). Small currents (Imax < 90 nA) of receptors containing G158C precluded additional analysis.
Figure 2.

A, Concentration-response curves of GABA-activated current for wild type (▪), E155C (▴), G158C (○), T160C (♦), and D163C (□) expressed in Xenopus oocytes. Data were normalized to peak IGABA for each experiment. Data points represent the mean ± SE of three to five independent experiments. B, Concentration dependence of SR-95531 inhibition of IGABA (EC50) current for wild type (▪), G158C (○), and E155C (▴). Data points represent the mean ± SE of three independent experiments. The GABA EC50 values, SR-95531 KI values, and calculated Hill coefficients are reported in Table 1.
Table 1.
Concentration-response data for GABA activation and SR-95531 inhibition of wild-type and mutant receptors expressed in Xenopus oocytes
|
|
GABA |
SR-95531 |
||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Receptor |
EC50 (μm) |
nH
|
n
|
mut/wt |
KI (nm) |
nH
|
n
|
mut/wt |
||||||
| α1β2 | 1.6 ± 0.2 | 1.35 ± 0.15 | 5 | 1 | 163 ± 83 | 1.13 ± 0.10 | 3 | 1 | ||||||
| α1β2(I154C) | 12.1 ± 4.7** | 1.13 ± 0.22 | 3 | 7.6 | 143 ± 63 | 0.87 ± 0.10 | 3 | 0.9 | ||||||
| α1β2(E155C) | 5400 ± 1500** | 1.21 ± 0.07 | 3 | 3375 | 1810 ± 401* | 0.66 ± 0.10 | 3 | 11.1 | ||||||
| α1β2(S156C) | 34.5 ± 8.8** | 1.32 ± 0.15 | 3 | 22 | 561 ± 178* | 0.76 ± 0.15 | 3 | 3.4 | ||||||
| α1β2(Y157C) | No current | |||||||||||||
| α1β2(G158C) | 416 ± 184** | 0.65 ± 0.09** | 3 | 260 | 2920 ± 1100* | 1.35 ± 0.40 | 3 | 17.9 | ||||||
| α1β2(Y159C) | No current | |||||||||||||
| α1β2(T160C) | 37.4 ± 11.7** | 1.40 ± 0.10 | 6 | 23 | 250 ± 88 | 1.03 ± 0.09 | 3 | 1.5 | ||||||
| α1β2(T161C) | 1.8 ± 0.4 | 1.10 ± 0.14 | 3 | 1.1 | 132 ± 38 | 1.03 ± 0.06 | 3 | 0.8 | ||||||
| α1β2(D162C) | 28.5 ± 5.6** | 1.48 ± 0.13 | 4 | 18 | 187 ± 115 | 1.49 ± 0.7 | 3 | 1.1 | ||||||
| α1β2(D163C) |
13.8 ± 3.8*
|
0.82 ± 0.05*
|
3 |
8.6 |
120 ± 32 |
0.84 ± 0.12 |
3 |
0.7 |
||||||
Data represent the mean ± SE for three to six experiments (n). EC50 values, KI values, and Hill slopes (nH) were determined from concentration-response data using nonlinear regression analysis with GraphPad Prism software. Hill slopes, log(EC50), and log(KI values were analyzed using a one-way ANOVA followed by a Dunnett's test to determine the levels of significance (*p < 0.05; **p < 0.01; from wild type). mut, Mutant; wt, wild type.
Pentobarbital is a barbiturate that exerts its pharmacological effects (allosteric modulation and channel opening) via interactions with the GABAA receptor at a binding site distinct from the GABA site (Akk and Steinbach, 2000). Pentobarbital is therefore useful for assessing the consequences of mutating residues located near the GABA-binding site on overall receptor structure-function. Pentobarbital activated wild-type receptors with an EC50 of 1.1 ± 0.3 mm (n = 4) (Table 2) but failed to elicit current in receptors containing Y157C or Y159C, again suggesting that these mutant β2 subunits did not assemble into functional channels. Sensitivity to pentobarbital was increased approximately twofold for E155C- and T160C-containing receptors [EC50 values = 0.53 ± 0.1 mm (n = 4) and 0.32 ± 0.03 mm (n = 3), respectively], whereas expression of D163C (EC50 = 2.0 ± 0.3 mm; n = 3) had no significant effect on pentobarbital EC50. These data suggest that the rightward GABA EC50 shifts (Table 1) measured for E155C-, T160C-, and D163C-containing receptors are attributable to local effects at the GABA-binding site.
Table 2.
Concentration-response data for GABA, P4S, SR-95531, PTX, and pentobarbital for α1β2 and α1β2(E155C) receptors expressed in Xenopus oocytes
| α1β2
|
α1β2(E155C) |
|||||||
|---|---|---|---|---|---|---|---|---|
| Ligand |
EC50/IC50(μm) |
nH
|
EC50/IC50 (μm) |
nH
|
mut/wt |
|||
| GABAa | 1.6 ± 0.2 | 1.35 ± 0.15 | 5400 ± 1500* | 1.21 ± 0.07 | 3375 | |||
| P4S | 4.7 ± 0.4 | 1.59 ± 0.22 | 730.7 ± 52.1* | 1.30 ± 0.11 | 152 | |||
| SR-95531a | 0.2 ± 0.08 | 1.13 ± 0.10 | 1.8 ± 0.4* | 0.66 ± 0.1 | 11.1 | |||
| PTX | 4.7 ± 1.2 | 0.84 ± 0.02 | 3.1 ± 0.1 | 0.81 ± 0.10 | 0.6 | |||
| Pentobarbital |
1100 ± 300 |
1.56 ± 0.07 |
530 ± 100*
|
1.08 ± 0.02 |
0.5 |
|||
Data represent the mean ± SE for more than three experiments. EC50 values, IC50 values, KI values, and Hill slopes (nH) were determined from concentration-response data using nonlinear regression analysis with GraphPad Prism software. Hill slopes, log(EC50), log(IC50), and log (KI) values were analyzed using a two-tailed unpaired t test to determine the levels of significance (*p < 0.01; from α1β2). mut, Mutant, wt, wild type.
Values are from Table 1.
Although it is impossible to know whether the introduced cysteine residues occupy positions equivalent to wild-type residues, because SR-95531 and pentobarbital apparent affinities were similar or, in certain cases, more potent for some mutant receptors in which there were large rightward shifts in GABA EC50, we believe gross structural reorganizations of the GABAA receptor did not occur as a result of these mutations.
Spontaneous openings at α1β2(E155C)
Expression of α1β2(E155C) receptors gave rise to higher than normal resting conductances (Ileak), the magnitudes of which (-609 ± 76 nA; n = 9) were ∼12-fold greater than injection-matched wild-type receptors (-51 ± 9 nA; n = 9). To determine the nature of this high resting conductance, we applied the GABAAR channel blocker PTX. PTX (100 μm) reduced Ileak by 72.1 ± 4.2% (n = 3) (Fig. 3A), demonstrating that spontaneously open GABAAR channels accounted for the high resting conductance. PTX inhibited GABA-activated currents elicited from α1β2(E155C) receptors with an IC50 value of 3.1 μm, which was not significantly different from wild-type receptor values (4.7 μm) (Fig. 3B-D, Table 2).
Figure 3.

A, Representative current trace demonstrating that the high resting leak conductance for α1β2(E155C) receptors expressed in Xenopus oocytes is sensitive to blockade by PTX (100 μm). PTX (100 μm) reduced the resting conductance (-609 ± 76 nA; n = 9) by 72.1 ± 4.2%. Representative current traces obtained from PTX-mediated inhibition of GABA-evoked currents (EC50) for wild-type (WT; B) and α1β2(E155C) (C) receptors are shown. Note that the PTX IC50 value for the E155C mutant was determined by using the baseline leak current as the zero. D, Concentration dependence of PTX-mediated reduction of IGABA (EC50) current for wild type (▪) and E155C (○) expressed in Xenopus oocytes. Data points represent the mean ± SE of three independent experiments. Data were normalized to IGABA in the absence of PTX. IC50 values are summarized in Table 2.
Spontaneously active LGICs often arise as a consequence of mutations in the M2 channel-lining segment, and a characteristic of these constitutively open channels is a leftward shift in agonist concentration responses (Bertrand et al., 1992; Filatov and White, 1995; Labarca et al., 1995; Tierney et al., 1996; Chang and Weiss, 1998, 1999; Thompson et al., 1999; Findlay et al., 2000). However, this was not the case on expression of β2E155C, wherein we observed a 3375-fold decrease in GABA sensitivity. The sensitivity of the partial agonist (P4S) was also reduced (with no apparent reduction in efficacy) (Fig. 4A,B), albeit to a lesser extent than that of GABA (152-fold) (Table 2, Fig. 4C). The mutation also decreased the sensitivity of the competitive antagonist SR-95531 (11.1-fold rightward shift) (Table 2). Again, it should be noted that for this mutation, pentobarbital sensitivity was increased twofold relative to wild type (Table 2, Fig. 4D). Mutation of ρ1Y102 located in the GABAC receptor D region of the agonist binding site (i.e., β-strand 2) has also been reported to result in spontaneously open channels with similar properties (Torres and Weiss, 2002).
Figure 4.

Representative current traces obtained from GABA- and P4S-mediated activation of wild-type (WT; A) and α1β2(E155C) (B) receptors are shown. P4S efficacy at wild-type (0.41 ± 0.05; n = 7) and α1β2(E155C) (0.47 ± 0.04; n = 6) receptors was not significantly different, where efficacy is reported as Imax P4S/Imax GABA. Values are given as mean ± SE. C, Concentration-response curves for P4S-activated current for wild-type (▪) and α1β2(E155C) (○) receptors expressed in Xenopus oocytes. D, Concentration-response curves for pentobarbital-activated current for wild-type (▪) and α1β2(E155C) (○) receptors. Data points represent the mean ± SE of three to four independent experiments. The EC50 values for P4S and pentobarbital are reported in Table 2.
Alterations in EC50 values are difficult to evaluate because changes in either ligand binding and/or channel gating can alter this macroscopic constant (Colquhoun, 1998). The increase in open probability for the E155C mutant indicates that the mutation altered GABAA receptor channel gating. If the mutation altered gating exclusively, similar-fold changes in the apparent affinities of a series of ligands as well as leftward shifts in their concentration responses would be expected (Zhang et al., 1994). The apparent affinities for GABA, P4S, and SR-95531 were altered by different factors (Table 2), and rightward shifts in their concentration responses were observed. Thus, these data indicate that, besides altering gating, E155C decreased the binding of orthosteric ligands to the GABA-binding site. We can exclude the possibility that these differential effects arise from a mixed population of receptors [i.e.,α1β2(E155C) and β2(E155C)] because expression of β2E155C alone produced no currents.
In addition to PTX, covalent modification of E155C by thiol-specific reagents closed the spontaneously open channels. The leak current was reduced by MTSEA-biotin (2 mm; 57.2 ± 0.7%; n = 3), MTSEA-biotin-CAP (N-biotinylcaproylaminoethyl methanethiosulfonate) (2 mm; 63.0 ± 1.8%; n = 3), 2-aminoethyl methanethiosulfonate (MTSEA) (2 mm; 39.4 ± 1.4%; n = 3), MTSET (2-(trimethylammonium)ethyl methanethiosulfonate) (2 mm; 30.5 ± 0.9%; n = 3), and MTSES (2-sulfonatoethyl methanethiosulfonate) (2 mm; 26.8 ± 7.5%; n = 3) (Fig. 5) The observation that Ileak is reduced by tethering different chemical groups directly onto E155C suggests that this region of the binding site may play a key role in the triggering of allosteric transitions from the closed to open state.
Figure 5.

Representative traces demonstrating the effects of PTX (100 μm; A) and MTSEA (2 mm; B) on Ileak for α1β2(E155C) receptors are shown. Reagents were applied in the absence of GABA and resulted in a reduction in the leak current. Note that the PTX trace is the same as that presented in Figure 3A but is included here for comparison purposes. C, Bar graph summarizing the effects of all methanethiosulfonate reagents on Ileak. Note that the maximum effect of each MTS reagent is normalized to the maximum effect obtained with 100 μm PTX for comparison purposes. The mean values for blockade of the resting conductance by MTS reagents are reported in Results. Error bars represent SE.
MTSEA-biotin modification of cysteine residues
To further examine the β2 subunit Ile154-Asp163 region, we assessed the accessibility of cysteine residues introduced into this region. Wild-type and mutant receptors were exposed to MTSEA-biotin (2 mm; 2 min), a thiol-specific reagent that modifies water-accessible cysteine residues (Karlin and Akabas, 1998). MTSEA-biotin significantly reduced IGABA for receptors containing E155C (91.8 ± 1.5%; n = 6), G158C (34.6 ± 4.5%; n = 4), T160C (60.9 ± 3.0%; n = 10), and D163C (98.9 ± 3.7%; n = 8) (Fig. 6). MTSEA-biotin had no effect on wild-type receptors or those containing I154C, S156C, T161C, and D162C. Lack of effect indicates that the thiol group was not accessible to modification or that modification produced no detectable functional effect. The accessibility pattern of the residues is consistent with the predicted side-chain positions observed in homology models of the GABAA receptor, which envision this region of the GABAA receptor forming a loop structure (Cromer et al., 2002).
Figure 6.

Summary of the effects of MTSEA-biotin (2 mm) on wild-type (WT) and mutant receptors. A, Representative current traces demonstrating the effects of MTSEA-biotin application on GABA-mediated current (EC50) at wild-type and T160C- and D163C-containing receptors. The arrows in the current traces represent MTSEA-biotin application (2 min), and the breaks represent the subsequent wash (5 min). B, Summary of the effect of MTSEA-biotin at all receptors. The effect (percentage of inhibition or potentiation) is calculated using the following: ([IGABA-POST MTSEA-biotin/IGABA-PRE MTSEA-biotin] - 1) × 100. Bars represent the mean ± SE for 3-10 experiments. The filled bars indicate values that were statistically different from wild-type values (*p < 0.001). Note that expression of Y157C or Y159C failed to yield functional receptors.
MTSEA-biotin rates of reaction
The rate of reaction of MTSEA-biotin with an introduced cysteine mainly depends on the ionization of the thiol group and the access route of the methanethiosulfonate reagent (Karlin and Akabas, 1998). Cysteine residues with ionized sulfhydryls react 108 to 109 times faster than nonionized sulfhydryls (Roberts et al., 1986). Second-order rate constants therefore provide information about the local environment of a substituted cysteine. The fast second-order rate constants (in the absence of other ligands) for MTSEA-biotin modification of D163C (604,771 m-1sec-1) and T160C (286,100 m-1sec-1) indicate that both residues are found in an open, aqueous environment. The second-order rate constant for E155C is significantly slower (27.9 m-1sec-1), suggesting that the thiol group is not well ionized and/or that the introduced cysteine residue is in a restricted-buried environment (Table 3, Fig. 7).
Table 3.
Second-order rate constants for MTSEA-biotin-mediated modification of accessible engineered cysteine residues in the absence (control) and presence of SR-95531, GABA, and pentobarbital
|
|
Control |
SR-95531 |
GABA |
Pentobarbital(500) |
Pentobarbital(50) |
||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Receptor |
k2 (m−1sec−1) |
n
|
k2 (m−1sec−1) |
n
|
k2 (m−1sec−1) |
n
|
k2 (m−1sec−1) |
n
|
k2 (m−1sec−1) |
n
|
|||||
| α1β2(E155C) | 27.9 ± 11 | 3 | 24.8 ± 3.8 | 3 | 23.7 ± 3.7 | 3 | 27.5 ± 9.8 | 3 | N.D. | ||||||
| α1β2(T160C) | 288, 664 ± 24, 911 | 11 | 132, 200 ± 3, 524* | 4 | 122, 828 ± 37, 753* | 4 | 131, 768 ± 16, 610* | 4 | 107, 493 ± 17, 751* | 3 | |||||
| α1β2(D163C) |
604, 771 ± 22, 681 |
7 |
293, 200 ± 43, 267*
|
3 |
280, 018 ± 43, 416*
|
3 |
281, 760 ± 56, 625*
|
3 |
479, 100 ± 48, 932 |
4 |
|||||
Data represent the mean ± SE of 3-11 independent experiments (n). k2 values were calculated by dividing the pseudo-first-order rate constant by the concentration of MTSEA-biotin used in the experiments. The concentrations of MTSEA-biotin used were 2 mm (E155C), 100 nm (T160C), and 50 nm (D163C). GABA (5 × EC50), SR-95531 (6 × KI), and pentobarbital (500 and 50 μm) were coapplied as described in Materials and Methods to determine their ability to alter the rate of covalent cysteine modification. N.D., Not determined. *p < 0.01; from control.
Figure 7.

Rate of MTSEA-biotin modification of T160C and D163C. A, Representative GABA-evoked (EC50) current traces after successive application (10-20 sec) of MTSEA-biotin on α1β2(D163C) receptors in the absence or presence of SR-95531 (6 × KI) and GABA (5 × EC50). Sequential application of MTSEA-biotin reduced the amplitude of subsequent GABA-mediated (EC50) currents at receptors containing T160C (B) and D163C (C). Data were normalized to the current measured at t = 0 for each experiment and plotted as a function of cumulative MTSEA-biotin exposure. Data were fitted to a single exponential function to get k1. k2 values were calculated by dividing the pseudo-first-order rate constant by the concentration of MTSEA-biotin used. Data points represent the mean ± SE for control (▪), GABA (□), SR-95531 (○), and pentobarbital (500 μm; •) for at least three independent experiments. Data are summarized in Table 3.
To determine whether a given residue lies near the neurotransmitter binding site, MTSEA-biotin reaction rates were measured in the presence of GABA and the competitive antagonist SR-95531. These ligands promote different conformational changes in the binding site (Boileau et al., 2002), and thus, if the rate at which MTSEA-biotin reacts with an introduced cysteine is slowed by both ligands, then it is likely that the ligands are sterically blocking the reaction and that the sulfhydryl side chain is facing into or near the GABA-binding site. Both GABA (at EC90 concentration) and SR-95531 (at IC90 concentration) significantly slowed modification of T160C and D163C approximately twofold (Fig. 8), suggesting that these residues are found within or near the GABA-binding site (Fig. 9). Although the data are consistent with GABA and SR-95531 causing a steric block, it is feasible that the binding of either ligand induces local allosteric changes in the receptor that leads to the slowing of MTSEA-biotin reaction rates. Neither ligand slowed modification of E155C (Table 3). Because α1E155C receptors are spontaneously open, the control rate of MTSEA-biotin modification of E155C likely reflects reaction to a “ligand-bound, active” conformation of the binding site. Thus, the result that GABA and SR-95531 had no effect on modification rate is not surprising.
Figure 8.

Summary of the effects of GABA, SR-95531, and pentobarbital on MTSEA-biotin second-order rate constants. Data were normalized to the control k2 (rate measured when no other compound was present; denoted as a dashed line). Coapplication of GABA (5 x EC50; black), SR-95531 (6 x KI; gray), and pentobarbital [500 μm (white) or 50 μm (lined)] slows reaction of MTSEA-biotin at receptors carrying T160C and D163C (*p < 0.001; from control). Note that 50 μm pentobarbital significantly slows the rate of reaction at T160C but not at D163C. The ability of 50 μm pentobarbital to alter the rate of reaction at E155C was not determined (N.D.). Error bars represent SE.
Figure 9.
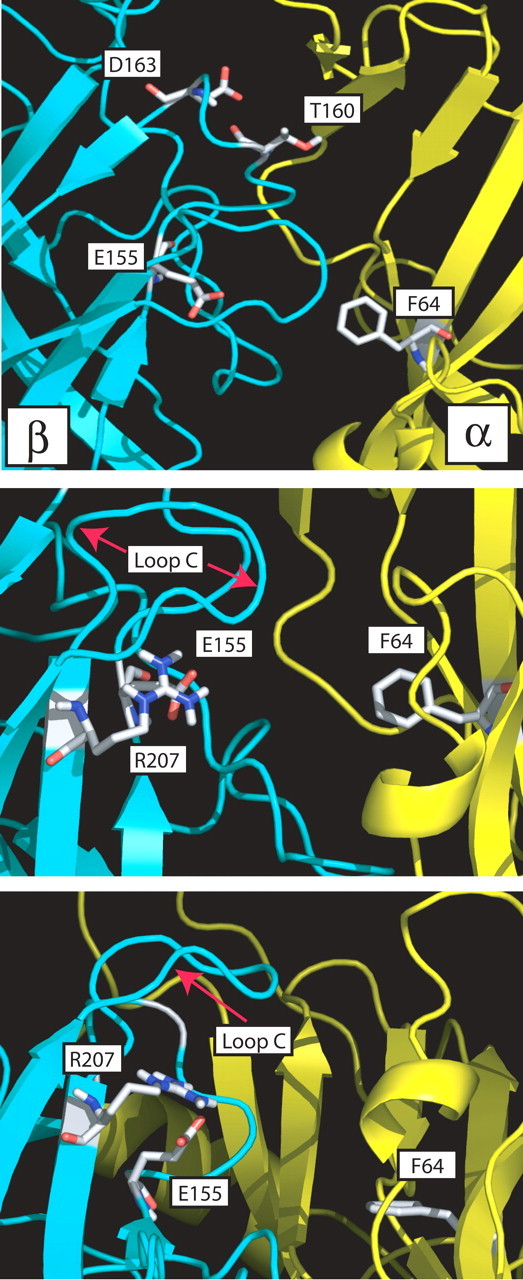
Top, Side view of a homology model of the GABA-binding site β (cyan)-α (yellow) subunit interface with residues β2Glu155, β2Thr160, β2Asp163 in region B and residue α1Phe64 in region D displayed as sticks. Middle, Position of β2Glu155 (region B) relative to β2Arg207 (region C) is highlighted. β2Arg207 has been identified previously as a GABA-binding site residue (Wagner and Czajkowski, 2001; Wagner et al., 2004). Bottom, View of the binding site from below, which shows the close apposition of β2Glu155 and β2Arg207 (2.5 Å), suggesting an electrostatic interaction between the two residues.
Effect of pentobarbital on MTSEA-biotin second-order rate constants
To identify whether movements occur in and near the Ile154-Asp163 region of the GABA-binding site during channel gating and modulation, we measured the rates of reaction of MTSEA-biotin with T160C, D163C, and E155C in the presence pentobarbital. The ability of pentobarbital to alter the rates of modification provides an indirect measure of changes that occur within this region of the binding cleft in the transition from the resting to the active-desensitized states. As a result of the slowness of drug application using the oocyte expression system, we cannot easily determine whether the movement is associated with open or desensitized states. Nevertheless, coapplication of pentobarbital and MTSEA-biotin should capture receptor states that differ from that captured by application of MTSEA-biotin alone. Concentrations of pentobarbital (500 μm) that activate the receptor slowed the rate of MTSEA-biotin modification of T160C and D163C approximately twofold but had no effect on the second-order rate constant for E155C (Table 3). Thus, T160C and D163C act as reporters of barbiturate-mediated channel gating. Concentrations of pentobarbital that do not open the channel but potentiate GABA current (e.g., 50 μm) (Table 3) slowed modification of T160C but not D163C (Fig. 7B,C; Table 3). These data suggest that movements within the binding site associated with channel gating versus allosteric modulation are distinct.
Discussion
Binding-site movements that initiate ligand-gated ion channel activation are not well established. Here, we provide evidence that movement in the β2 Ile154-Asp163 region of the GABAAR is involved in coupling GABA binding to channel gating, and we describe a role for β2Glu155 as an initial trigger for ion channel opening.
β2 Ile154-Asp163 mutations affect ligand binding and channel gating
If the β2 Ile154-Asp163 region plays a pivotal role for coupling binding to gating, one would expect that mutations within this domain would affect both processes. Seven cysteine substitutions significantly increase GABA EC50 values, which reflect changes in either microscopic binding affinity and/or channel gating (Colquhoun, 1998). Three of the seven mutations that shift the GABA EC50 (E155C, S156C, and G158C) also significantly reduce SR-95531 KI, suggesting that at least one effect of these mutations is to alter ligand binding, because SR-95531 does not gate the channel (but see Ueno et al., 1997). α1β2(G158C) and α1β2(D163C) receptors have significantly reduced Hill coefficients for GABA activation, consistent with a reduction in gating efficacy (Colquhoun, 1998). β2E155C results in spontaneously open GABAARs, clearly demonstrating that this mutation alters channel gating. In addition, expression of β2E155C differentially shifts the concentration dependencies rightward for GABA, SR-95531, and P4S, indicating that the mutation also perturbs ligand binding (Zhang et al., 1994). Furthermore, tethering thiol-reactive groups onto β2E155C closes the spontaneously open channels. Together, the data suggest that β2Glu155 occupies a key position in the activation pathway involved in coupling ligand binding site to channel gating. Although detailed kinetic analyses of these mutations are required to quantitatively tease apart the effects of each of these mutations on microscopic binding affinity and channel gating properties, the above results indicate that mutations in β2 Ile154-Asp163 region of the GABA-binding site disrupt both affinity and efficacy.
Structural rearrangements during gating transitions
The β2 subunit forms the principal side of the GABA-binding site. We conclude that β2Asp163 and β2Thr160 are found within or near the GABA-binding site, based on a slowing of the rate of MTSEA-biotin modification of T160C and D163C by both GABA and SR-95531. Protection by both ligands suggests steric hindrance of MTSEA-biotin modification, because agonists and antagonists promote different conformational changes within the binding site (Armstrong and Gouaux, 2000). In addition, mutagenesis and homology modeling studies suggest that the β2 Ile154- Asp163 region lines part of the GABA-binding site (Amin and Weiss, 1993).
Tierney et al. (1996) predict that movements within the β2 subunit are critical for channel gating. To test the hypothesis that the β2 Ile154-Asp163 region of the binding site undergoes structural rearrangements during channel activation, we measured the rate of MTSEA-biotin modification of introduced cysteines in the presence of pentobarbital (500 μm). Although the binding sites for GABA and pentobarbital differ, the final structure of the activated GABAA receptor channel is likely similar because both drugs produce similar single channel conductances (Jackson et al., 1982; Akk and Steinbach, 2000). We can therefore monitor pentobarbital-induced movements in the GABA-binding site during state transitions from resting to open-desensitized states. Coapplication of pentobarbital and MTSEA-biotin should capture this region of the receptor in a conformation that differs from that captured by application of MTSEA-biotin alone. The observation that pentobarbital significantly slows modification of T160C and D163C (approximately twofold) indicates that the environment surrounding these residues changes and that they are “conformationally sensitive” to channel activation, supporting our hypothesis that movements within this region are critical for channel gating. T160C also appears to act as a reporter for movements that occur during allosteric modulation, because the rate of MTSEA-biotin modification of T160C was slowed almost twofold in the presence of a low concentration of pentobarbital (50 μm) that potentiates but does not gate the channel.
β2 Ile154-Asp163 is a protein hinge
Local agonist-induced movements within LGIC binding sites precede a conformational wave that leads to channel gating (Chakrapani et al., 2004). Structural studies suggest that the binding of ACh induces a 15° clockwise rotation of the inner β-sheets of the N-terminal ligand binding domain of the nAChR α1 subunits. This, in turn, brings the β1-β2 loop (loop 2) into contact with the extracellular M2-M3 loop, and movement of the M2-M3 loop then causes the M2 region to rotate, which leads to opening of the channel gate (Miyazawa et al., 2003). Linear free energy analysis of the nAChR suggest that a conformational wave begins at the binding site and region A of the ACh-binding site (β-strand 4 and loop 5), followed by movements of loops 2, 7 (Cys-Cys loop) and the M2-M3 linker at the extracellular juxtapore region, and finally movement of the transmembrane domains (Grosman et al., 2000; Chakrapani et al., 2004). Studies examining the structural mechanisms of GABAAR activation have identified pairs of interacting residues within these regions that are necessary for coupling GABA binding to channel gating-desensitization. These include electrostatic interactions between negatively charged residues in loops 2 (α1Asp57) and 7 (α1Asp149) of the GABAAR and a positively charged lysine (α1Lys279) in the M2-M3 loop (Kash et al., 2003). Recently, a study using a chimeric receptor comprised of AChBP fused to the transmembrane pore domain of the 5-HT3A receptor demonstrated that only when loops 2, 7, and 9 (region F of the binding site) from AChBP were replaced with 5-HT3A receptor sequences did ACh binding trigger channel opening (Bouzat et al., 2004). This indicates that loops 2, 7, and 9 are critical elements involved in coupling the extracellular binding site domain to the transmembrane channel gating domain. During examination of homology models of the GABAAR, we noticed that the β2 Ile154-Asp163 region of the binding site (β-strand 7 and loop 8) physically links loops 2 and 9. Thus, we speculate that the β2 Ile154-Asp163 region may act as a protein hinge and that structural rearrangements within this region of the binding site could therefore be an efficient means for simultaneously propagating movements to both loops 2 and 9.
Movements in region B are also likely to be transmitted to the region C of the GABA-binding site (i.e., end of β-strand 9, loop 10 and beginning of β-strand 10). Based on homology models, possible interactions within regions B and C include the following amino acid pairs: β2Glu153 and β2Lys196, β2Glu155 and β2Arg207, as well as β2Glu165 and β2Lys197. Previously, we demonstrated that β2Arg207 stabilizes GABA binding (Wagner and Czajkowski, 2001; Wagner et al., 2004). We predict that the carboxylate side chain of Glu155 is within 2.5 Å from the guanido group of β2Arg207 (Fig. 9) and may play a role in positioning the β2Arg207 side chain. This may explain why mutation of β2Glu155 disrupts orthosteric ligand binding. However, mutation of β2Glu155 also produces spontaneously open channels and indicates that perturbation of this residue has additional long-range allosteric effects that are likely propagated to the channel gate by changes in the positions of regions B and C located in the binding site as well as loops 2 and 9 near the juxtapore region. Additional experiments are needed to test these hypotheses. Support for interactions between regions B and C of LGIC binding sites comes from studies of the nAChR. It has been reported that a hydrogen bond between a residue in region B (G152K) and in region C (P193I) of the nAChR α7 subunit is important for nAChR activation (Grutter et al., 2003) and may serve to explain how the α1G153S human polymorphism gives rise to a slow channel myasthenic syndrome (Sine et al., 1995).
Finally, the β2 Ile154-Asp163 region may also be involved in intersubunit interactions. Models of the N-terminal domains of GABAAR predict that β2Asp163 forms a salt bridge with α1Arg119 and β2Arg28. The roles of salt bridges in GABAAR function are not presently known, but it is believed that salt bridges may limit the number of conformations of a protein complex, be key participants in determining ligand binding geometry, or be important for the association of subunits in multiprotein complexes (Hendsch and Tidor, 1994). The disruption or formation of bonds among these charged residues at subunit interfaces may be important for conformational changes that occur during activation and/or desensitization. Mutation of β2Asp163 significantly decreased the Hill coefficient for GABA activation of the receptor and is consistent with this hypothesis.
Conclusions
The β2 Ile154-Asp163 region of the GABAAR-binding site appears to be a protein hinge that is uniquely positioned to transduce binding site movements to the cascade of events that lead to opening of the ion channel. We demonstrate that the region undergoes conformational rearrangements during pentobarbital-mediated gating events, and mutation of β2Glu155 gives rise to spontaneously open channels, suggesting that movements in this region of the GABA-binding site are one of the initial triggers for coupling binding to gating. Ultimately, precise mapping of inter-residue contacts will be required to test this activation mechanism and to define the pathway leading from the binding site to the channel gate.
Footnotes
This work was supported by National Institute of Neurological Disorders and Stroke Grant 34727 to C.C. and a Postdoctoral Fellowship from the Natural Sciences and Engineering Research Council of Canada to J.G.N.
Correspondence should be addressed to Cynthia Czajkowski, Department of Physiology, University of Wisconsin-Madison, 601 Science Drive, Madison, WI 53711. E-mail: czajkowski@physiology.wisc.edu.
J. G. Newell's present address: Department of Physiology, University of Toronto, Room 3318, Medical Sciences Building, 1 King's College Circle, Toronto, Ontario, Canada M5S 1A8.
R. A. McDevitt's present address: Graduate Program in Neurobiology and Behavior, University of Washington, Box 357270, Room T-471, Seattle, WA 98195.
Copyright © 2004 Society for Neuroscience 0270-6474/04/2411226-10$15.00/0
Throughout this paper, the six previously identified binding site regions are named by the letters A-F (Galzi and Changeux, 1994; Lester et al., 2004). Based on the structure of AChBP (Brejc et al., 2001), these regions can be defined as follows: region A, β strand 4 and loop 5; region B, β strand 7 and loop 8; region C, β strand 9, loop 10 and β strand 10; region D, β strand 2; region E, β strands 5′ and 6; region F, loop 9. When we refer to regions that are not part of the binding site, these are named by numbers based on AChBP structure.
References
- Akk G, Steinbach JH (2000) Activation and block of recombinant GABAA receptors by pentobarbitone: a single-channel study. Br J Pharmacol 130: 249-258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amin J, Weiss DS (1993) GABAA receptor needs two homologous domains of the β-subunit for activation by GABA but not by pentobarbital. Nature 366: 565-569. [DOI] [PubMed] [Google Scholar]
- Armstrong N, Gouaux E (2000) Mechanisms of activation and antagonism of an AMPA-sensitive glutamate receptor: crystal structures of the GluR2 ligand binding core. Neuron 28: 165-181. [DOI] [PubMed] [Google Scholar]
- Baumann SW, Baur R, Sigel E (2001) Subunit arrangement of γ-aminobutyric acid type A receptors. J Biol Chem 276: 36275-36280. [DOI] [PubMed] [Google Scholar]
- Baumann SW, Baur R, Sigel E (2002) Forced subunit assembly in α1β2γ2 GABAA receptors: insight into the absolute arrangement. J Biol Chem 277: 46020-46025. [DOI] [PubMed] [Google Scholar]
- Bertrand D, Devillers-Thiéry A, Revah F, Galzi JL, Hussy N, Mulle C, Berstrand S, Ballivet M, Changeux JP (1992) Unconventional pharmacology of a neuronal nicotinic receptor mutated in the channel domain. Proc Natl Acad Sci USA 89: 1261-1265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boileau AJ, Kucken AM, Evers AM, Czajkowski C (1998) Molecular dissection of benzodiazepine binding and allosteric coupling using chimeric γ-aminobutyric acidA receptor subunits. Mol Pharmacol 53: 295-303. [DOI] [PubMed] [Google Scholar]
- Boileau AJ, Evers AM, Davis AF, Czajkowski C (1999) Mapping the agonist binding site of the GABAA receptor: evidence for a β-strand. J Neurosci 19: 4847-4854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boileau AJ, Newell JG, Czajkowksi C (2002) GABAA receptor β2Tyr97 and Leu99 line the GABA-binding site: insights into the mechanisms of agonist and antagonist actions. J Biol Chem 277: 2931-2937. [DOI] [PubMed] [Google Scholar]
- Bouzat C, Gumilar F, Spitzmaul G, Wang HL, Rayes D, Hansen SB, Taylor P, Sine SM (2004) Coupling agonist binding to channel opening in an ACh-binding protein linked to an ion channel. Nature 430: 896-900. [DOI] [PubMed] [Google Scholar]
- Brejc K, van Dijk W, Klaassen RV, Schuurmans M, van der Gost J, Smit AB, Sixma T (2001) Crystal structure of an ACh-binding protein reveals the ligand-binding domain of nicotinic receptors. Nature 17: 269-276. [DOI] [PubMed] [Google Scholar]
- Chakrapani S, Bailey TD, Auerbach A (2004) Gating dynamics of the acetylcholine receptor extracellular domain. J Gen Physiol 123: 341-356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang Y, Weiss DS (1998) Substitution of the highly conserved M2 leucine create spontaneously opening ρ1 γ-aminobutyric acid receptors. Mol Pharmacol 53: 511-523. [DOI] [PubMed] [Google Scholar]
- Chang Y, Weiss DS (1999) Allosteric activation mechanism of the α1β2γ2 γ-aminobutyric acid type A receptor revealed by mutation of the conserved M2 leucine. Biophys J 77: 2543-2551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang Y, Weiss DS (2002) Site-specific fluorescence reveals distinct structural changes with GABA receptor activation and antagonism. Nat Neurosci 5: 1163-1168. [DOI] [PubMed] [Google Scholar]
- Chang Y, Wang R, Barot S, Weiss DS (1996) Stoichiometry of a recombinant GABAA receptor. J Neurosci 16: 5415-5424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng Y, Prussof WH (1973) Relationship between the inhibition constant (KI) and the concentration of inhibitor which causes 50 percent inhibition (I50) of an enzymatic reaction. Biochem Pharmacol 22: 3099-3108. [DOI] [PubMed] [Google Scholar]
- Colquhoun D (1998) Binding, gating, affinity and efficacy: the interpretation of structure-activity relationships for agonists and the effects of mutating receptors. Br J Pharmacol 125: 924-927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cromer BA, Morton CJ, Parker MW (2002) Anxiety over GABAA receptor structure relieved by AChBP. Trends Biochem Sci 27: 280-287. [DOI] [PubMed] [Google Scholar]
- Croxen R, Newland C, Beeson D, Oosterhuis H, Chauplannaz G, Vincent A, Newsom-Davis J (1997) Mutations in different functional domains of the human muscle acetylcholine receptor α subunit in patients with slow-channel myasthenia gravis. Hum Mol Genet 6: 767-774. [DOI] [PubMed] [Google Scholar]
- Daniels GM, Amara S (1998) Selective labeling of neurotransmitter transporters at the cell surface. Methods Enzymol 296: 307-318. [DOI] [PubMed] [Google Scholar]
- Dennis M, Giraudat J, Kotzyba-Hibert F, Goeldner M, Hirth C, Chang JY, Lazure C, Chrétien M, Changeux JP (1988) Amino acids of the Torpedo marmorata acetylcholine receptor α subunit labeled by a photoaffinity ligand for the acetylcholine binding site. Biochemistry 27: 2346-2357. [DOI] [PubMed] [Google Scholar]
- Farrar SJ, Whiting PJ, Bonnert TP, McKernan RM (1999) Stoichiometry of a ligand-gated ion channel determined by fluorescence energy transfer. J Biol Chem 274: 10100-10104. [DOI] [PubMed] [Google Scholar]
- Filatov GN, White MW (1995) The role of leucine residues in the M2 domain of the acetylcholine receptor in channel gating. Mol Pharmacol 48: 379-384. [PubMed] [Google Scholar]
- Findlay G, Ueno S, Harrison NL, Harris RA (2000) Allosteric modulation in spontaneously active mutant γ-aminobutryic acidA receptors in frogs. Neurosci Lett 293: 155-158. [DOI] [PubMed] [Google Scholar]
- Galzi JL, Changeux JP (1994) Neurotransmitter-gated ion channels as unconventional allosteric proteins. Curr Opin Struct Biol 4: 554-565. [Google Scholar]
- Galzi JL, Revah F, Black D, Goeldner M, Hirth C, Changeux JL (1991) Identification of a novel amino acid α-tyrosine 93 within the cholinergic ligands-binding sites of the acetylcholine receptor by photoaffinity labeling: additional evidence for a three-loop model of the cholinergic ligands-binding sites J Biol Chem 265: 10430-10437. [PubMed] [Google Scholar]
- Grosman C, Zhou M, Auerbach A (2000) Mapping the conformational wave of acetylcholine receptor gating. Nature 403: 773-776. [DOI] [PubMed] [Google Scholar]
- Grutter T, de Carvalho LP, Le Novère N, Corringer PJ, Edelstein S, Changeux JP (2003) An H-bond between two residues from different loops of the acetylcholine binding site contributes to the activation mechanism of nicotinic receptors. EMBO J 22: 1990-2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hendsch SB, Tidor B (1994) So salt bridges stabilize proteins? A continuum electrostatic analysis. Protein Sci 3: 211-226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holden JH, Czajkowski C (2002) Different residues in the GABAA receptor α1T60-α1K70 region mediate GABA and SR-95531 actions. J Biol Chem 277: 18785-18792. [DOI] [PubMed] [Google Scholar]
- Horenstein J, Wagner DA, Czajkowksi C, Akabas MH (2001) Protein mobility and GABA-induced conformational changes in the GABAA receptor pore-lining M2 segment. Nat Neurosci 4: 477-485. [DOI] [PubMed] [Google Scholar]
- Im WB, Pregenzer JF, Binder JA, Dillon GH, Alberts GL (1995) Chloride channel expression with the tandem construct of α6-β2 GABAA receptor subunit requires a monomeric subunit of α6 or γ2. J Biol Chem 270: 26063-26066. [DOI] [PubMed] [Google Scholar]
- Jackson MB, Lecar H, Matthers D, Barker JL (1982) Single channel currents activated by gamma-aminobutyric acid, muscimol and (-)pentobarbital in cultured mouse spinal neurons. J Neurosci 2: 889-894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karlin A, Akabas M (1998) Substituted-cysteine accessibility method. Methods Enzymol 293: 123-145. [DOI] [PubMed] [Google Scholar]
- Kash TL, Jenkins A, Kelley JC, Trudell JR, Harrison NL (2003) Coupling of agonist binding to channel gating in the GABAA receptor. Nature 42: 272-275. [DOI] [PubMed] [Google Scholar]
- Kucken AM, Wagner DA, Ward PH, Teissére JA, Boileau AJ, Czajkowski C (2000) Identification of benzodiazepine binding site residues in the γ2 subunit of the γ-aminobutyric acidA receptor. Mol Pharmacol 57: 932-939. [PubMed] [Google Scholar]
- Labarca C, Nowak MW, Zhang H, Tang L, Despande P, Lester HA (1995) Channel gating governed symmetrically by conserved leucine residues in the M2 domain of nicotinic receptors. Nature 376: 514-516. [DOI] [PubMed] [Google Scholar]
- LeNovère N, Grutter T, Changeux JP (2002) Models of the extracellular domain of the nicotinic receptors and of agonist- and Ca2+-binding sites. Proc Natl Acad Sci USA 99: 3210-3215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lester HA, Dibas MI, Dahan DS, Leite JF, Dougherty DA (2004) Cys-loop receptors: new twists and turns. Trends Neurosci 27: 329-336. [DOI] [PubMed] [Google Scholar]
- Liman ER, Tygat J, Hess P (1992) Subunit stoichiometry of a mammalian K+ channel determined by construction of multimeric cDNAs. Neuron 9: 861-871. [DOI] [PubMed] [Google Scholar]
- Miyazawa A, Fujiyoshi Y, Unwin N (2003) Structure and gating mechanism of the acetylcholine receptor pore. Nature 423: 949-955. [DOI] [PubMed] [Google Scholar]
- Newell JG, Czajkowski C (2003) The GABAA receptor α1 subunit Pro174-Asp191 segment is involved in GABA binding and channel gating. J Biol Chem 278: 13166-13172. [DOI] [PubMed] [Google Scholar]
- Pascual JM, Karlin A (1998) State-dependent accessibility and electrostatic potential in the channel of the acetylcholine receptor: inferences from rates of reaction of thiosulfonates with substituted cysteines in the M2 segment of the α subunit. J Gen Physiol 111: 717-739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Powell JMD (1977) Restart procedures for the conjugate gradient method. Math Program 12: 241-251. [Google Scholar]
- Press WH, Flannery BP, Teukolsky SA, Vetterling WT (1988) Numerical recipes in C: the art of scientific computing. In: Minimization and maximization of functions, Ed 1, Chap 10, pp 301-327. Cambridge, UK: Cambridge UP.
- Pritchett DB, Sontheimer H, Shivers DB, Ymer S, Kettenham H, Schofield PR, Seeburg PH (1989) Importance of a novel GABAA receptor subunit for benzodiazepine pharmacology. Nature 338: 582-585. [DOI] [PubMed] [Google Scholar]
- Reeves DC, Sayed MF, Chau PL, Price KL, Lummis SC (2003) Prediction of 5-HT3 receptor agonist-binding residues using homology modeling. Biophys J 84: 2338-2344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts DD, Lewis SD, Ballou DP, Olson ST, Shafer JA (1986) Reactivity of small thiolate anions and cysteine-25 in papain toward methyl methanethiosulfonate. Biochemistry 25: 5595-5601. [DOI] [PubMed] [Google Scholar]
- Robertson GA, Warmke JM, Ganetzky B (1996) Potassium currents expressed from Drosophila and mouse eag cDNAs in Xenopus oocytes. Neuropharmacology 35: 841-850. [DOI] [PubMed] [Google Scholar]
- Schmieden V, Kuhse J, Betz H (1993) Mutation of glycine receptor subunit creates β-alanine receptor responsive to GABA. Science 262: 256-258. [DOI] [PubMed] [Google Scholar]
- Schofield PR, Darlison MG, Fujita N, Burt DR, Stephenson FA, Rodriguez H, Rhee LM, Ramachandran J, Reale V, Glencorse TA (1987) Sequence identity and functional expression of the GABA-A receptor shows a ligand-gated receptor super-family. Nature 328: 221-227. [DOI] [PubMed] [Google Scholar]
- Sine S, Ohno K, Bouzat C, Auerbach A., Milone M, Pruit JN, Engel G (1995) Mutation of the acetylcholine receptor subunit causes a slow-channel myasthenic syndrome by enhancing agonist binding affinity. Neuron 15: 229-239. [DOI] [PubMed] [Google Scholar]
- Spier AD, Lummis SC (2000) The role of tryptophan residues in the 5-hydroxytryptamine3 receptor ligand binding domain. J Biol Chem 275: 5620-5625. [DOI] [PubMed] [Google Scholar]
- Sugiyama N, Boyd AE, Taylor P (1996) Anionic residue in the α-subunit of the nicotinic acetylcholine receptor contributing to subunit assembly and ligand binding. J Biol Chem 271: 26575-26581. [DOI] [PubMed] [Google Scholar]
- Thompson SA, Smith MZ, Wingrove PB, Whiting PJ, Wafford KA (1999) Mutation at the putative GABAA ion-channel gate reveals changes in allosteric modulation. Br J Pharmacol 127: 1349-1358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tierney ML, Birnir B, Pillai NP, Clements JD, Howitt SM, Cox GB, Gage PW (1996) Effects of mutating leucine to threonine in the M2 segment of the α1 and β1 subunits of the GABAA α1β1 receptors. J Membr Biol 154: 11-21. [DOI] [PubMed] [Google Scholar]
- Torres VI, Weiss DS (2002) Identification of a tyrosine in the agonist binding site of the homomeric ρ1 γ-aminobutyric acid (GABA) receptor that, when mutated, produces spontaneous opening. J Biol Chem 277: 43741-43748. [DOI] [PubMed] [Google Scholar]
- Tretter V, Ehya N, Fuchs K, Sieghart W (1997) Stoichiometry and assembly of a recombinant GABAA receptor subtype. J Neurosci 17: 2728-2737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ueno S, Bracamontes J, Zorumski C, Weiss DS, Steinbach JH (1997) Bicuculline and gabazine are allosteric inhibitors of channel opening of the GABAA receptor. J Neurosci 17: 625-634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Unwin N, Miyazawa A, Li J, Fujiyoshi Y (2002) Activation of the nicotinic acetylcholine receptors involves a switch in conformation of α subunits. J Mol Biol 319: 1165-1176. [DOI] [PubMed] [Google Scholar]
- Vandenberg RJ, French CR, Barry PH, Shine J, Schofield PR (1992) Antagonism of ligand-gated ion channel receptors: two domains of the glycine receptor α subunit form the strychnine-binding site. Proc Natl Acad Sci USA 89: 1765-1769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vandenberg RJ, Rajendra S, French CR, Barry PH, Schofield SC (1993) The extracellular disulfide loop motif of the inhibitory glycine receptor α subunit forms the strychnine-binding site. Mol Pharmacol 44: 198-203. [PubMed] [Google Scholar]
- Wagner DA, Czajkowski C (2001) Structure and dynamics of the GABA binding pocket: a narrowing cleft that constricts during channel activation. J Neurosci 21: 67-74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagner DA, Czajkowski C, Jones MV (2004) An arginine involved in GABA binding and unbinding but not gating of the GABAA receptor. J Neurosci 24: 2733-2741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang HG, ffrench-Constant RH, Jackson M (1994) A unique amino acid of the Drosophila GABA receptor with influence on drug sensitivity by two mechanisms. J Physiol (Lond) 479: 65-75. [DOI] [PMC free article] [PubMed] [Google Scholar]