

Abstract
Nine neurodegenerative diseases including Huntington's disease (HD) and spinocerebellar ataxia type 7 (SCA7) are caused by an expansion of a polyglutamine (polyQ) stretch in the respective proteins. Aggregation of expanded polyQ-containing proteins into the nucleus is a hallmark of these diseases. Recent evidence indicates that transcriptional dysregulation may contribute to the molecular pathogenesis of these diseases. Using SCA7 and HD mouse models in which we recently described a retinal phenotype, we investigated whether altered gene expression underlies photoreceptor dysfunction. In both models, rhodopsin promoter activity was early and dramatically repressed, suggesting that downregulation of photoreceptor-specific genes plays a major role in polyQ-induced retinal dysfunction. Because the rhodopsin promoter drives mutant ataxin-7 expression in our SCA7 mice, we also assessed whether downregulation of mutant SCA7 transgene would reverse retinopathy progression and aggregate formation. Although residual expression of mutant ataxin-7 was found negligible from 9 weeks of age, SCA7 transgenic mice showed a progressive decline of photoreceptor activity leading to a complete loss of electroretinographic responses from 1 year of age. At this age, aggregates were cleared in only half of the photoreceptors, indicating that their formation is not fully reversible in this model. We demonstrate here that abolishing full-length mutant ataxin-7 expression did not reverse retinopathy progression in SCA7 mice, raising the possibility that some polyQ-induced pathological events might be irreversible.
Keywords: neurodegeneration, polyglutamine, retina, transcriptional alteration, reversibility, aggregates
Introduction
Spinocerebellar ataxia type 7 (SCA7) and Huntington's disease (HD) belong to a group of nine inherited neurodegenerative diseases caused by a CAG/polyglutamine (polyQ) expansion and characterized by continuous accumulation of mutant proteins into nuclear inclusions (NIs). Genetic and molecular studies indicate that polyQ tracts confer a novel toxic function to the otherwise unrelated proteins (Zoghbi and Orr, 2000; Ross, 2002). SCA7 is the only polyQ disorder that affects the retina and is characterized by a progressive loss of motor coordination and visual impairment. There is no definitive consensus about the mechanisms leading to cell-specific neuronal dysfunction and degeneration in polyQ diseases (Tobin and Signer, 2000; La Spada and Taylor, 2003).
SCA7 mouse models have been generated and reproduce many aspects of the human pathology such as cleavage and aggregation of an N-terminal fragment of the mutant protein (Yvert et al., 2000, 2001; La Spada et al., 2001; Garden et al., 2002; Yoo et al., 2003). La Spada et al. (2001) proposed that mutant ataxin-7 impairs the normal function of the retina-specific transcription factor Crx, leading to a cone-rod dystrophy. In another SCA7 mouse model, Yoo et al. (2003) reported repression of several photoreceptor-specific genes, whereas Crx levels and activity were not altered. Furthermore, polyQ expansion in another protein can trigger a retinal phenotype, as we described previously in an HD mouse model (Helmlinger et al., 2002). Whether ataxin-7 protein context plays a role in specifying SCA7 retinal involvement needs to be clarified.
Using an inducible HD mouse model (HD94), Yamamoto et al. (2000) showed that HD-like pathology is dependent on continuous expression of a mutant huntingtin fragment. Neuronal dysfunction arrest and aggregate reversal have been observed in the early stages of the disease process in HD94 mice. It remains to be determined at which stage polyQ expansions induce irreversible damages to neurons. This issue is crucial, considering therapeutic strategies interfering with polyQ protein expression. Finally, it is not known whether neurons can recover from the toxicity of full-length mutant proteins and whether these results can be extended to other polyQ disorders.
We previously generated transgenic mice overexpressing normal [Q10] and mutant [Q90] ataxin-7 under rhodopsin promoter control (R7N and R7E mice) (Table 1) (Yvert et al., 2000). We described comparable retinal phenotype in this SCA7 mouse model and in a well characterized HD mouse model overexpressing mutant huntingtin under control of the human HD gene promoter (R6 lines) (Table 1) (Mangiarini et al., 1996; Helmlinger et al., 2002). We report here that polyQ expansions in two different proteins induced an early, progressive, and severe downregulation of rhodopsin expression in both models. This transcriptional alteration was correlated with a progressive thinning of photoreceptor segments, eventually leading to a complete loss of their electrophysiological activity. We also show that mutant ataxin-7 expression, which is controlled by the rhodopsin promoter in R7E mice, was early and dramatically downregulated. Extinction of polyQ protein expression contrasted with a constant worsening of photoreceptor dysfunction. These results suggest that polyQ-induced toxicity might not be reversible beyond a given pathological threshold.
Table 1.
Summary of the different transgenic mouse lines used in this study
Materials and Methods
Animals. R7N and R7E animals were from the R7N.C and R7E.A transgenic lines, respectively, and were maintained on the inbred C57BL/6 background (Yvert et al., 2000). R6/1 and R6/2 lines were obtained from Jackson Laboratories (Bar Harbor, ME), where they were maintained on a mixed C57BL/6:CBA/J genetic background (Table 1). Because the CBA/J inbred strain carries the rd1 mutation, mice were backcrossed on the C57BL/6 background. For genotyping of R7N, R7E, R6, and R7N/R6 mice and of the rd1 mutation, mouse tail genomic DNA was screened by PCR according to procedures described previously (Pittler and Baehr, 1991; Mangiarini et al., 1996; Yvert et al., 2000). For genotyping of R7N/R7E mice, Southern blot of NcoI-digested mouse tail genomic DNA was performed. The experiments were performed in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals.
Immunohistology and light microscopy. For histology, enucleated eyes were fixed by immersion in Bouin or 4% paraformaldehyde. Fixed eyes were dehydrated and embedded in paraffin, and 5 μm sections were stained with hematoxylin and eosin or processed for immunochemistry. For immunofluorescence, enucleated eyes were dissected to remove the lens and cornea and fixed in 4% paraformaldehyde. Fixed eyecups were cryoprotected in 30% sucrose and frozen in Cryomatrix (Thermo Shandon). Cryostat sections (10 μm) were mounted on SuperFrost/Plus slides (O. Kindler, Freiburg, Germany). Sections were permeabilized and blocked for 1 hr with 10% normal goat serum, 0.5% bovine serum albumin, 0.1% Tween 20, and 1 × PBS. We used anti-ataxin-7 2A10 monoclonal and affinity-purified 1261 polyclonal antibodies and anti-huntingtin 1259 polyclonal antibody, as described previously (Yvert et al., 2000, 2001; Lunkes et al., 2002). For immunofluorescence, the secondary antibodies used were CY3- and Oregon Green-conjugated goat anti-rabbit and anti-mouse IgG (Jackson ImmunoResearch, West Grove, PA), and nuclei were counterstained with 0.5 μg/ml 4,6-diaminido-2-phenylindole (DAPI). For immunochemistry, biotinylated goat anti-rabbit IgG (Vector Laboratories, Burlingame, CA), streptavidin-biotin complex solution (Vector Laboratories), and diaminobenzidine (Vector Laboratories) were used according to the instructions of the manufacturer. Sections were counterstained with methyl green.
Immunoblot analysis. Retinal homogenates were prepared as described previously (Helmlinger et al., 2002). We used mouse anti-ataxin-7 1C1 (Yvert et al., 2000), anti-rhodopsin 4D2 (Hicks and Molday, 1986), and anti-β-actin (Abcam, Cambridge, UK) monoclonal antibodies.
RNA isolation and Northern blot analysis. We prepared total RNA by single-step extraction of guanidium thiocyanate homogenates. Glyoxylated RNA (8 μg) was electrophoresed through agarose gels and transferred onto Hybond N+ membranes (Amersham Biosciences, Little Chalfont, England). cDNA probes were obtained by reverse transcriptase (RT)-PCR from wild-type mouse retinal RNA (Rho) and from cloned human cDNAs (SCA7 and GAPD). Probes were labeled with [α32P]dCTP by random priming. We performed hybridizations using ULTRAhyb solution (Ambion, Austin, TX) according to the instructions of the manufacturer.
Real-time RT-PCR analysis of retinal RNA. Reverse transcription was performed on 1 μg of total retinal RNA using SuperScriptII (Invitrogen, Carlsbad, CA) and random hexamers according to the manufacturer instructions. We performed PCR amplifications of cDNA using LightCycler-FastStart DNA Master SYBR Green I (Roche Products, Basel, Switzerland) on a LightCycler instrument. PCR primers for detection of recombinant SCA7, endogenous Sca7, Rho, Hprt, Ppia, and Arbp mRNA were designed using Primer3 software and are available on request.
Electroretinogram recordings. Electroretinographic analysis was performed as described previously (Helmlinger et al., 2002). Briefly, dark-adapted mice were anesthetized. Pupils were dilated, and the cornea was locally anesthetized. Scotopic rod electroretinograms (ERGs) were recorded by using a gold loop electrode placed on the corneal surface and referenced to a stainless steel reference electrode inserted subcutaneously on the head of the animal. A 150 W xenon lamp in a Ganzfeld stimulator (Multiliner Vision, Jaeger Toennies, Germany) provided the light stimulus, increasing from -4 to 1.4 log cd sec m-2, as measured with a luxmeter at eye level. The duration of the light stimulus was constant. Responses were digitized using a data acquisition labmaster board (Multiliner Vision). Responses were computer averaged at all intensities.
Clinical examination. Funduscopic examinations were performed as described on dark-adapted mice (Helmlinger et al., 2002). Anesthetized mice were clinically evaluated by indirect ophthalmoscopy with a Kowa Genesis (Tokyo, Japan) small animal fundus camera and a condensing Volk (Mentor, OH) 90-diopter lens.
Results
PolyQ expansions downregulate recombinant normal ataxin-7
Several studies showed that nonexpanded polyQ proteins can be recruited into nuclear aggregates, suggesting that polyQ toxicity might lie, in part, in a loss of function of the corresponding normal proteins (Kazantsev et al., 1999; Cattaneo et al., 2001; Uchihara et al., 2001). We asked whether aggregation and cleavage of mutant ataxin-7 could affect wild-type ataxin-7. Endogenous mouse ataxin-7 is expressed at very low levels and cannot be detected by Western blotting or immunostaining in our conditions. To follow normal ataxin-7 fate in photoreceptors expressing mutant ataxin-7, we generated R7N-R7E double transgenic mice in which most rods will express both transgenes (Table 1). In R7N rod photoreceptors, recombinant normal ataxin-7 [Q10] displayed a homogenous nuclear ring-like staining, whereas in R7E mice, an N-terminal fragment of mutant ataxin-7 [Q90] accumulated into NIs (Yvert et al., 2000). Immunofluorescence analysis of retinas from 10-week-old R7N-R7E mice revealed a complete disappearance of recombinant normal ataxin-7 staining. Almost all rods contained NIs immunoreactive for N-terminal but not C-terminal (data not shown) ataxin-7 antibodies, as in R7E retinas (Fig. 1A-C). These observations could be explained by recombinant ataxin-7 cleavage and recruitment into NIs. However, this question cannot be assessed in R7N-R7E mice, because no available antibody discriminates recombinant normal ataxin-7 from the mutant form.
Figure 1.
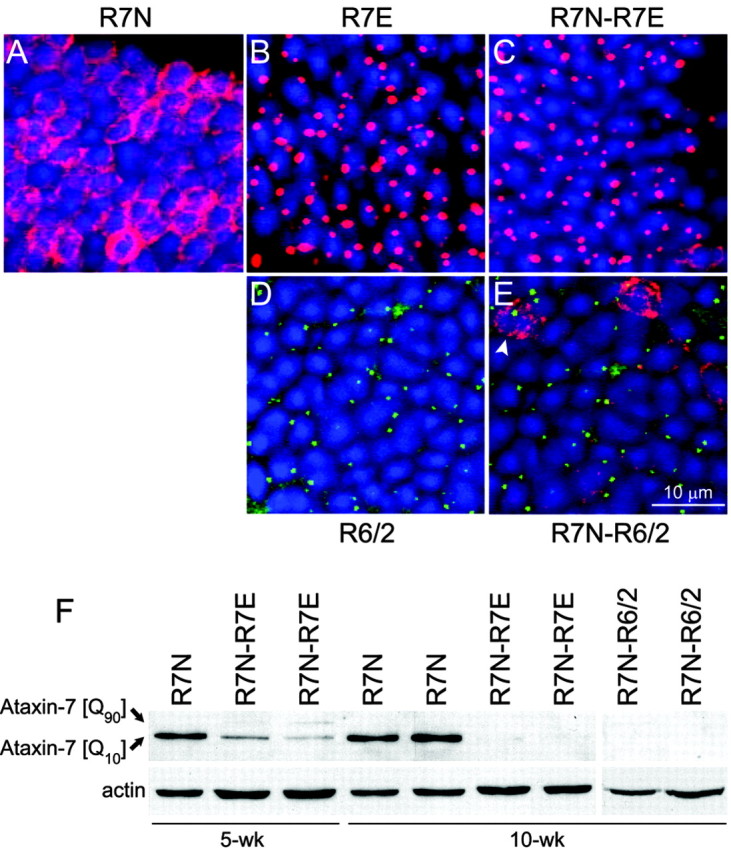
PolyQ expansions downregulate normal recombinant ataxin-7 in R7E and R6 retinas. A-E, Immunofluorescence analysis of 10 μm retinal cryosections from 10-week-old R7N (A), R7E (B), R7N-R7E (C), R6/2 (D), and R7N-R6/2 (E) mice using anti-ataxin-7 (2A10 in red; A-C, E) and anti-Huntingtin (1259 in green; D, E) antibodies. Nuclei were counterstained with DAPI (blue), and merged images are shown. Normal recombinant ataxin-7 displayed a homogenous nuclear ring-like staining (A), whereas mutant ataxin-7 (B) or mutant Huntingtin (D) formed aggregates. Rod photoreceptors from double transgenic animals contained only NIs (C, E). A small number of rods contained Huntingtin aggregates together with ataxin-7 [Q10] (E, arrowhead). Scale bar, (A-F) 10 μm. F, Western blot analysis of retinal extracts from R7N, R7N-R7E, and R7N-R6/2 animals at 5 and 10 weeks of age probed with anti-ataxin-7 (1C1) antibody. Anti-actin antibody was used as an internal loading control. PolyQ expansion in ataxin-7 or Huntingtin induced an early decrease of normal recombinant ataxin-7 that progressively led to undetectable levels in retinal homogenates from 10-week-old R7N-R7E and R7N-R6/2 mice.
We then crossed R7N with R6 transgenic mice, in which we recently described aggregation of mutant huntingtin in rod photoreceptors from R6/1 and R6/2 lines (Davies et al., 1997; Helmlinger et al., 2002). A similar disappearance of ataxin-7 [Q10] nuclear ring-like staining was observed in rods from 10-week-old R7N-R6/2 mice (Fig. 1D,E). NIs were never immunoreactive for ataxin-7, even in the rare cells where both huntingtin and ataxin-7 could be seen, excluding recruitment of ataxin-7 [Q10] into aggregates.
Western blot analysis confirmed an early decrease of recombinant normal ataxin-7 that progressively led to undetectable levels in retinal homogenates from 10-week-old R7N-R7E and R7N-R6/2 mice (Fig. 1F). Together, these results demonstrate that polyQ expansion in two different proteins downregulates recombinant normal ataxin-7.
PolyQ expansions downregulate SCA7 transgene expression
We then assessed whether recombinant normal ataxin-7 downregulation occurred at the transcriptional level by quantifying recombinant normal SCA7 mRNA levels in retinas from R7N-R7E and R7N-R6 animals. Northern blot analysis performed on R7N-R7E mice from different ages (3, 5, and 9 weeks) revealed an early, progressive, and dramatic reduction of SCA7 transgene mRNA levels (Fig. 2A). A similar strong downregulation of recombinant normal SCA7 mRNA was observed in RNA from 32-week-old R7N-R6/1 retinas (Fig. 2B). At these ages, R7E and R6/1 mice do not show significant photoreceptor cell loss that would explain this downregulation (Yvert et al., 2000; Helmlinger et al., 2002). Repression of normal SCA7 transgene was observed in both R7N-R7E and R7N-R6 mice, in which expression of expanded polyQ proteins are driven by two unrelated promoters. This demonstrates that normal SCA7 transgene downregulation is caused by polyQ expansions and not by the presence of multiple copies of rhodopsin promoter in R7N-R7E mice.
Figure 2.
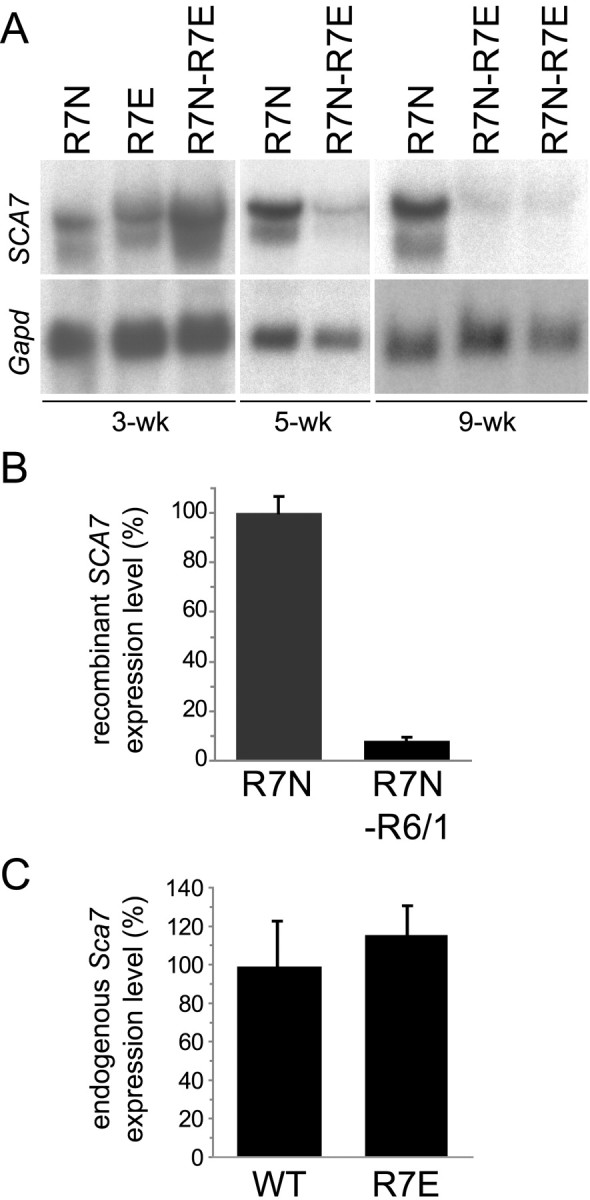
PolyQ expansions downregulate recombinant ataxin-7 but not endogenous ataxin-7 expression. A, Northern blots of total RNA isolated from R7N, R7E, and R7N-R7E retinas at 3, 5, and 9 weeks of age were hybridized with SCA7 and Gapd probes. At 3 weeks of age, high levels of transgene mRNA were detected in single and double transgenic animals. Later, SCA7 transgene expression was sustained in R7N mice but severely reduced in R7N-R7E mice. B, Real-time RT-PCR analysis of total RNA extracted from 32 week R7N and R7N-R6/1 retinas, using primers specific for recombinant human SCA7 and Hprt as an internal control. Accordingly, we observed a reduced expression of normal SCA7 transgene in R7N-R6/1 mice. SCA7 mRNA level is quantified as a percentage of R7N mice, after normalization to Hprt levels. Each bar represents the mean value ± SEM (n = 2). C, Quantification of endogenous mouse Sca7 expression in wild-type and R7E mice at 9 weeks of age by real-time RT-PCR using primers specific for endogenous Sca7 and Arbp as an internal control. No difference could be detected between wild-type and R7E animals at 10 weeks of age. Sca7 levels are represented as a percentage of the mean of wild-type littermate mice after normalization to Arbp levels. Each bar represents the mean value ± SEM (n = 2). Each panel is representative of at least two independent experiments performed on different mice. WT, Wild type.
We then checked whether there is also a downregulation of endogenous mouse ataxin-7 that would lead to a partial loss of its function, contributing to polyQ-induced retinopathy. This led us to examine the expression level of endogenous Sca7 by quantitative RT-PCR. Sca7 mRNA levels were not modified in R7E retinas (Fig. 2C). Steady-state levels of other proteins with glutamine repeats, like endogenous huntingtin, ataxin-3, CBP, and TBP, were not modified in R7E or R6 retinas (data not shown).
PolyQ expansions repress rhodopsin promoter activity
We then hypothesized that downregulation of the SCA7 transgene could be mediated by repression of the rhodopsin promoter driving its expression in our transgenic lines. Therefore, we examined expression levels of gene-encoding rhodopsin. Northern blot revealed a strong decrease of Rho mRNA levels in R7E mice (Fig. 3A). Already at 3.5 weeks of age, real-time RT-PCR showed a threefold decrease of Rho mRNA levels (Fig. 3B). A time course analysis revealed that this downregulation was progressive, leading to a fivefold reduction of Rho mRNA levels from 6 weeks of age. This effect appeared dose dependent, because homozygous 9-week-old R7E mice had 4 ± 0.3% residual Rho expression, whereas age-matched heterozygous showed 13 ± 7% residual Rho mRNA. Interestingly, we found a similar downregulation of Rho expression in 10-week-old R6/2 mice (Fig. 3B). Western blotting of retinal homogenates from 10-week-old R7E and R6/2 mice confirmed this dramatic reduction of Rho expression at the protein level (Fig. 3C). Together, these results indicated that polyQ expansions repress rhodopsin promoter activity, because both endogenous rhodopsin and Rho promoter driven ataxin-7 expression are affected. These transcriptional alterations are induced by two unrelated polyQ proteins and are therefore not specific to mutant ataxin-7.
Figure 3.

Rhodopsin mRNA (Rho) and protein levels are severely reduced in SCA7 and HD mice. A, Northern blot of total RNA extracted from R7N and R7E mice at 3, 6, and 9 weeks of age was hybridized with Rho and Gapd probes. Constant expression of Rho in aging R7N mice contrasted with a progressive diminution of Rho mRNA in R7E mice from 3 to 9 weeks of age. B, Quantification of Rho expression in wild-type, R7E (at 3.5, 6, and 9 weeks of age), homozygous R7E (at 9 weeks of age), and R6/2 mice (at 10 weeks of age) (n = 3 for each group) by real-time RT-PCR using primers specific for Rho and Ppia as an internal control. This time-course analysis revealed that Rho downregulation was progressive, leading to a fivefold reduction of Rho mRNA levels from 6 weeks of age. Downregulation of Rho mRNA in homozygous 9-week-old R7E mice is approximately twice that in age-matched heterozygous R7E mice. A similar downregulation of Rho expression was detected in 10-week-old R6/2 mice. Rho levels are represented as a percentage of the mean of wild-type littermate mice (n = 12) after normalization to Ppia levels. Each bar represents the mean value ± SEM (n = 3). C, Western blot analysis of retinal homogenates from R7N, R7E, and R6/2 animals at 10 weeks of age, using anti-rhodopsin and anti-actin antibodies, confirmed this dramatic reduction of Rho expression at the protein level. Each panel is representative of at least two independent experiments performed on different mice. WT, Wild type.
Early suppression of mutant ataxin-7 expression contrasts with continuous worsening of retinal phenotype in R7E mice
Because mutant SCA7 transgene expression is controlled by the human rhodopsin promoter in R7E mice, we wondered at which extent mutant ataxin-7 [Q90] expression would also be affected. Northern blot analysis of R7N and R7E retinal extracts at 3 weeks of age showed similar levels of normal and mutant SCA7 mRNA, respectively (Fig. 2A). In contrast, almost undetectable levels of SCA7 mRNA were found in 9-week-old R7N-R7E mice, whereas SCA7 transgene expression is not modified over time in R7N mice (Fig. 2A). We then used real-time RT-PCR to precisely quantify SCA7 residual expression in R7E retinas at different ages. We observed an early and rapidly progressing downregulation of mutant SCA7 mRNA with more than a 20-fold reduction in 6-week-old R7E animals. Mutant ataxin-7 expression was almost completely shut off, because <2% of residual expression was detected from 9 weeks of age up to late stages of R7E mice life-span (Fig. 4A). Thus, expression of mutant ataxin-7 in R7E rod photoreceptors is restricted to a narrow time window of ∼3-4 weeks.
Figure 4.

Early loss of mutant SCA 7 expression contrast with continuous worsening of retinal dysfunction. A, Real-time RT-PCR analysis of total RNA extracted from R7E retinas at 3, 6, 9, 20, 45, and 100 weeks of age using primers specific for recombinant human SCA7 and Arbp as an internal control. Maximal levels of mutant transgene expression were detected at 3 weeks of age when photoreceptor differentiation was complete. Early and dramatic downregulation of mutant SCA7 mRNA was detected from 6 weeks of age and led to negligible levels of expression. SCA7 mRNA levels are quantified as a percentage of 3-week-old R7E mice after normalization to Arbp. Each bar represents the mean value ± SEM (n = 4-6). B, Scotopic ERG response of wild-type and R7E mice at 3.5, 6, 9, 14, 29, 53, 79, and 100 weeks of age. A complete ERG time-course analysis of R7E mice revealed a progressive and continuous worsening of rod photoreceptor function. Rod activity is quantified as the mean amplitude of the a-wave recorded at the four maximal light stimulus intensities (from 0 to 1.4 log cd sec m-2). Each bar represents the mean value ± SEM (n = 4-7).
Such an early and long-lasting shut-off of mutant ataxin-7 expression prompted us to assess whether SCA7 pathology is reversible in R7E mice. Indeed, Yamamoto et al. (2000) showed that abolishing mutant huntingtin exon 1 expression reversed neurological phenotype in an inducible HD mouse model (HD94). R7E transgenic mice present the unique opportunity to assess the long-term progression of the neurological phenotype, because these animals have a normal life span resulting from restricted expression of mutant ataxin-7 in the retina. Rod photoreceptor function can be reproducibly quantified on living animals by measuring the a-wave from ERG recordings performed on dark-adapted mice (scotopic ERG). Rod response was reduced by twofold in 5-week-old R7E mice and progressively worsened up to flat recordings from 1.5 years of age (Fig. 4B). At 6 weeks of age, residual expression of mutant SCA7 transgene was only 4% and contrasted with a twofold reduction of the a-wave amplitude. Continuous decrease of ERG response was concomitant to progressive thinning of the segment layer (Fig. 5A,B). Progression of the retinopathy was also evidenced at the histological level by the appearance of numerous waves disrupting the photoreceptor (outer) nuclear layer (ONL). Later, flattening of the ONL was concordant with the disappearance of white spots by funduscopy performed in 100-week-old R7E mice (Fig. 5B,C). Late-onset but restricted retinal degeneration was evidenced by the reduction of the ONL thickness from 1 year of age (Fig. 5B), suggesting that retinopathy is still progressing in aging R7E mice.
Figure 5.

Long-term rod photoreceptor dysfunction eventually leads to a late onset and limited neurodegeneration. A, Time-course analysis of scotopic ERG response from wild-type and R7E mice at 5, 9, 26, 52, and 100 weeks of age. Each recording corresponds to the average response of one animal at maximal light intensities (from 0 to 1.4 log cd sec m-2). Single recordings revealed a progressive decrease of rod electrophysiological activity up to flat responses. B, C, Progression of retinopathy in aging R7E mice assessed by histological (B) and clinical (C) examination. Decrease of ERG response was concomitant to progressive thinning of the segment layer. Histologically, the photoreceptor layer showed first abnormalities at 5 weeks of age and was then completely disrupted by numerous whorls. Later, flattening of the ONL was concordant with the disappearance of white spots by funduscopy performed in 100-week-old R7E mice that showed areas of abnormal pigmentation. Each panel is representative of observations performed on at least three other mice. WT, Wild type; RPE, retinal pigment epithelium; OS, outer segment; IS, inner segment; INL, inner nuclear layer. Scale bar, (A-C) 40 μm.
Partial and slow clearance of aggregates
Finally, we investigated whether loss of mutant SCA7 expression could reverse NI formation. Using HD94 mice, Martin-Aparicio et al. (2001) showed that aggregate reversal is a rapid process in vivo, because 3 weeks of transgene expression shut off were sufficient for complete disappearance of huntingtin staining. Immunohistochemistry performed on 26- and 52-week-old R7E retinal paraffin sections showed that the proportion of rods bearing NIs was >95% in 26-week-old R7E mice and was reduced to approximately one-half in 52-week-old R7E mice (Fig. 6). These results were confirmed in two different 26-week-old and three different 52-week-old R7E mice. Ataxin-7 aggregate clearance is a long-lasting process, because NI abundance is similar at 10 and 26 weeks of age, whereas expression of mutant ataxin-7 became negligible from 9 weeks of age (Figs. 1, 4, 6). Finally, 2-year-old R7E mice still showed a significant proportion of rods bearing NIs, suggesting that this event was never complete (data not shown).
Figure 6.
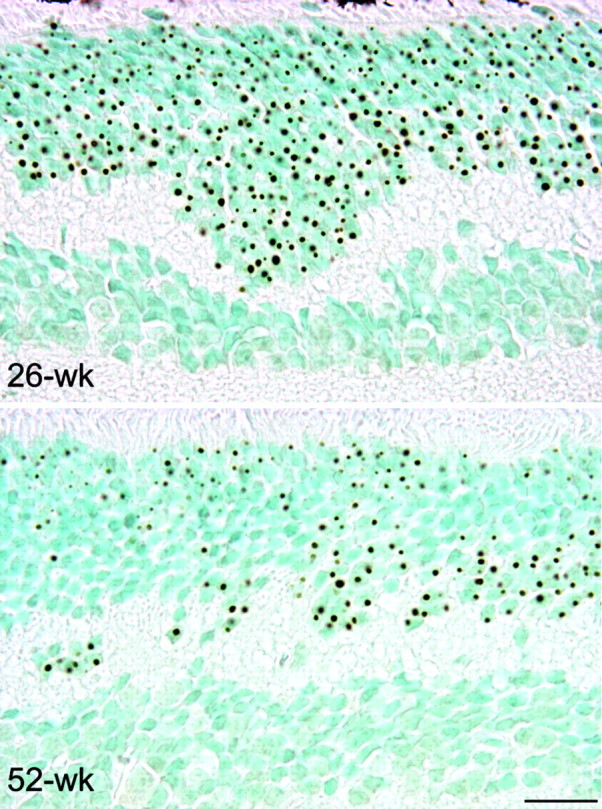
Partial disappearance of mutant ataxin-7 NIs in the photoreceptor nuclear layer of 52-week-old R7E mice. DAB immunostaining was performed on 5 μm paraffin-embedded retinal sections from 26-week-old (top panel) and 52-week-old (bottom panel) R7E mice using anti-ataxin-7 (1261) antibody. Although aggregates were present in most rods at 26 weeks of age, the proportion of rods bearing NIs was reduced to approximately one-half in 52-week-old R7E mice. Counterstaining with methyl green revealed a significant number of rod nuclei without any ataxin-7 immunoreactivity. Thus, partial aggregate clearance is not attributable to photoreceptor cell loss. Scale bar, 20 μm.
Discussion
R7E mice present the unique opportunity to assess the long-term effect of polyQ toxicity in vivo, because these animals have a normal life span resulting from restricted expression of mutant ataxin-7 in retina (Fig. 7). This allowed us to show that mutant protein expression was restricted to a narrow time window of ∼3-4 weeks in R7E mice. This was sufficient to trigger an early transcriptional dysregulation leading to a long-lasting photoreceptor dysfunction without massive neuronal loss until late ages. Altered rhodopsin expression, concomitant with decreased rod ERG response, was the earliest anomaly and appeared at 3.5 weeks of age. These defects steadily worsened to very low levels of rhodopsin expression, which correlated with flat rod ERG response and complete loss of segments from 1.5 years of age.
Figure 7.

Time-course assessment of phenotypic and molecular events occurring during R7E mice lifespan. High expression of mutant ataxin-7 in rod photoreceptors is restricted to a narrow time window of ∼3-4 weeks. Photoreceptor differentiation is normal in R7E mice, because we did not detect any abnormalities until 3 weeks of age. Altered rhodopsin expression, concomitant with decreased rod ERG response, is the earliest anomaly and occurred at 3-4 weeks of age. At 8 weeks of age, most rods contain NIs, and the photoreceptor layer is disrupted by numerous whorls. From this time point, drastic reduction of mutant SCA7 expression leads to partial aggregate reversion. Despite negligible influx of mutant protein, we observed a progressive and complete loss of rod normal function that strikingly does not immediately lead to their degeneration.
Transcriptional abnormalities in SCA7 and HD mice
Although retinal degeneration is only observed in SCA7, we described a retinopathy in an HD mouse model. Expression of a mutant huntingtin fragment in R6/2 mice leads to a widespread pathology that may not be representative of what is occurring in patients (Sathasivam et al., 1999). Regardless, comparison of retinopathy in HD and SCA7 mice offers the opportunity to assess whether the molecular pathways underlying rod dysfunction are identical. We showed that HD and SCA7 mouse models develop comparable photoreceptor abnormalities as revealed by progressive thinning of their segments, decreased rod ERG response, and concomitant reduction of Rho mRNA levels.
Crx has a major role in the transcriptional regulation of photoreceptor-specific genes such as Rho (Chen et al., 1997; Furukawa et al., 1997). La Spada et al. (2001) reported an interaction between ataxin-7 and Crx and proposed that expanded ataxin-7 impairs Crx normal function leading to SCA7-specific retinopathy. Their SCA7 transgenic mice presented mild transcriptional abnormalities, whereas Crx protein levels and activity were reduced. Surprisingly, Yoo et al. (2003) reported early and robust transcriptional alterations without modification of Crx levels and activity in a SCA7 model, in which 266 CAG repeats were knocked in the Sca7 mouse locus. We demonstrate here that repression of rhodopsin promoter activity is induced by polyQ expansion in two unrelated proteins. Together, these observations indicate that the specific retinal dysfunction in SCA7 patients cannot be exclusively explained by a specific interference of mutant ataxin-7 with Crx-dependent transcriptional regulation.
SCA7 transgenic mice (R7E), which highly overexpress ataxin-7 with 90 glutamines and SCA7 knock-in mice that express endogenous levels of ataxin-7 with a very large polyQ expansion (266 glutamines), each present their own strengths and limitations. Early and dramatic downregulation of Rho was observed in both SCA7 transgenic and knock-in models, identifying common pathological mechanisms that might underlie photoreceptor dysfunction in SCA7 patients. These results support the hypothesis that polyQ-induced retinal dysfunction observed in mice might be caused by reduced expression of photoreceptor-specific genes such as Rho. Accordingly, mice heterozygous for Rho null allele present late onset and progressive photoreceptor degeneration (Humphries et al., 1997; Lem et al., 1999). PolyQ expansions also downregulated recombinant ataxin-7, in which expression is controlled by the rhodopsin promoter. In R7N-R7E and R7N-R6/2 double transgenic mice, the normal SCA7 transgene can be considered a reporter gene, indicating that a 2.2 kb fragment of the human rhodopsin promoter is abnormally regulated in SCA7 and HD mice.
Reversibility of polyQ-induced toxicity in different models
In R7E mice, early suppression of full-length mutant ataxin-7 expression was neither sufficient to reverse nor halt retinal phenotype progression. However, high and stable expression levels of mutant ataxin-7 may have more deleterious effects, suggesting that transgene silencing might have slowed retinopathy progression in R7E mice. Regardless, our observations contrast with previous studies showing that phenotypic abnormalities were reversed after suppression of mutant huntingtin expression in a conditional HD mouse model (HD94) (Yamamoto et al., 2000) and in rats injected with tetracycline-regulated lentiviral vectors (Regulier et al., 2003). This apparent contradiction is not attributable to differences in residual transgene expression, because only highly sensitive RT-PCR, a technique not reported in HD94 and lentiviral-based models, allowed us to detect mutant ataxin-7 residual expression in R7E mice from 9 weeks of age. HD94 mice express an interrupted polyQ expansion (Q41RQ52) that might be less deleterious to neurons than the pure Q90 expansion from R7E animals, in which high expression levels of mutant ataxin-7 led to an early and severe neuronal dysfunction.
R7E mice present a long period of neuronal dysfunction during which most rods are not committed to early death and are even able to clear up their aggregates. One could expect a reversion of the retinal phenotype by suppressing mutant ataxin-7 expression at the beginning of this period. In contrast, we never observed a recovery of rhodopsin expression or an improvement of ERG responses, suggesting that mutant transgene shut-off might have taken place when neuronal dysfunction reached an irreversible pathological threshold. Transcriptional repression of photoreceptor-specific genes might constitute such an irreversible event. Alternatively, loss of mutant ataxin-7 expression only led to a partial clearance of aggregates in approximately one-half of the photoreceptors, suggesting that aggregates themselves may also contribute to maintain the long-term neuronal dysfunction in R7E mice. In these mice, disease progression despite long-term extinction of mutant ataxin-7 expression could be attributable to a combination of irreversible transcriptional abnormalities and partial persistence of aggregates.
Furthermore, aggregate clearance was observed in 1-year-old R7E mice in which mutant ataxin-7 expression was lost since 2 months of age. Using HD94 mice, Martin-Aparicio et al. (2001) showed that aggregate formation and clearance is a rapid process that results from a balance between polyQ protein synthesis and degradation by the proteasome. Despite long-term loss of mutant ataxin-7 expression and normal proteasomal activity (D.H., unpublished results), a significant number of rods retained aggregates, even in 2-year-old R7E mice. Our results suggest that in this model, aggregate formation is rapid, whereas their clearance is partial and much slower than reported previously in the HD94 model.
RNA interference (RNAi) appeared as an attractive approach to silence the expression of genes harboring gain of function mutations (Xia et al., 2002). Several studies recently demonstrated the feasibility of targeting transcripts with expanded CAG repeats (Caplen et al., 2002; Miller et al., 2003). RNAi achieve 5-10% residual expression, which could slow or even prevent disease progression. Whether RNAi-mediated downregulation of polyQ proteins is sufficient to rescue neurological phenotype has never been tested in mammalian models. In R7E mice, 1% residual expression of mutant ataxin-7 lasts more than 2 years and contrasts with a progressive worsening of neuronal dysfunction. Our findings suggest that some pathological processes might be irreversibly triggered by expression of polyQ proteins and that therapeutic strategies aimed at reducing expression of the mutant genes should be initiated early in pathogenesis.
Footnotes
This study was supported by a grant from the Hereditary Disease Foundation and by funds from the Institut National de la Santé et de la Recherche Médicale, the Centre National de la Recherche Scientifique, and the Hôpital Universitaire de Strasbourg. D.H. is a recipient of the Ministère de l'Education Nationale et de la Recherche. We thank S. Picaud, K. Lindenberg, K. Merienne, and L. Ben-Haïem for fruitful discussions and E. Metzger and the staff of the Mouse Clinical Institute.
Correspondence should be addressed to Didier Devys, Department of Molecular Pathology, Institut de Génétique et de Biologie Moléculaire et Cellulaire, BP 10142, 67404 Illkirch Cedex, France. E-mail: devys@igbmc.u-strasbg.fr.
G. Yvert's present address: Centre G. Durand, Centre National de la Recherche Scientifique, 31077 Toulouse, France.
Copyright © 2004 Society for Neuroscience 0270-6474/04/241881-07$15.00/0
References
- Caplen NJ, Taylor JP, Statham VS, Tanaka F, Fire A, Morgan RA (2002) Rescue of polyglutamine-mediated cytotoxicity by double-stranded RNA-mediated RNA interference. Hum Mol Genet 11: 175-184. [DOI] [PubMed] [Google Scholar]
- Cattaneo E, Rigamonti D, Goffredo D, Zuccato C, Squitieri F, Sipione S (2001) Loss of normal huntingtin function: new developments in Huntington's disease research. Trends Neurosci 24: 182-188. [DOI] [PubMed] [Google Scholar]
- Chen S, Wang QL, Nie Z, Sun H, Lennon G, Copeland NG, Gilbert DJ, Jenkins NA, Zack DJ (1997) Crx, a novel Otx-like paired-homeodomain protein, binds to and transactivates photoreceptor cell-specific genes. Neuron 19: 1017-1030. [DOI] [PubMed] [Google Scholar]
- Davies SW, Turmaine M, Cozens BA, Difiglia M, Sharp AH, Ross CA, Scherzinger E, Wanker EE, Mangiarini L, Bates GP (1997) Formation of neuronal intranuclear inclusions underlies the neurological dysfunction in mice transgenic for the HD mutation. Cell 90: 537-548. [DOI] [PubMed] [Google Scholar]
- Furukawa T, Morrow EM, Cepko CL (1997) Crx, a novel otx-like homeobox gene, shows photoreceptor-specific expression and regulates photoreceptor differentiation. Cell 91: 531-541. [DOI] [PubMed] [Google Scholar]
- Garden GA, Libby RT, Fu YH, Kinoshita Y, Huang J, Possin DE, Smith AC, Martinez RA, Fine GC, Grote SK, Ware CB, Einum DD, Morrison RS, Ptacek LJ, Sopher BL, La Spada AR (2002) Polyglutamine-expanded ataxin-7 promotes non-cell-autonomous Purkinje cell degeneration and displays proteolytic cleavage in ataxic transgenic mice. J Neurosci 22: 4897-4905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Helmlinger D, Yvert G, Picaud S, Merienne K, Sahel J, Mandel JL, Devys D (2002) Progressive retinal degeneration and dysfunction in R6 Huntington's disease mice. Hum Mol Genet 11: 3351-3359. [DOI] [PubMed] [Google Scholar]
- Hicks D, Molday RS (1986) Differential immunogold-dextran labeling of bovine and frog rod and cone cells using monoclonal antibodies against bovine rhodopsin. Exp Eye Res 42: 55-71. [DOI] [PubMed] [Google Scholar]
- Humphries MM, Rancourt D, Farrar GJ, Kenna P, Hazel M, Bush RA, Sieving PA, Sheils DM, McNally N, Creighton P, Erven A, Boros A, Gulya K, Capecchi MR, Humphries P (1997) Retinopathy induced in mice by targeted disruption of the rhodopsin gene. Nat Genet 15: 216-219. [DOI] [PubMed] [Google Scholar]
- Kazantsev A, Preisinger E, Dranovsky A, Goldgaber D, Housman D (1999) Insoluble detergent-resistent aggregates form between pathological and non-pathological lengths of polyglutamine in mammalian cells. Proc Natl Acad Sci USA 96: 11404-11409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- La Spada AR, Taylor JP (2003) Polyglutamines placed into context. Neuron 38: 681-684. [DOI] [PubMed] [Google Scholar]
- La Spada AR, Fu YH, Sopher BL, Libby RT, Wang X, Li LY, Einum DD, Huang J, Possin DE, Smith AC, Martinez RA, Koszdin KL, Treuting PM, Ware CB, Hurley JB, Ptacek LJ, Chen S (2001) Polyglutamine-expanded ataxin-7 antagonizes CRX function and induces cone-rod dystrophy in a mouse model of SCA7. Neuron 31: 913-927. [DOI] [PubMed] [Google Scholar]
- Lem J, Krasnoperova NV, Calvert PD, Kosaras B, Cameron DA, Nicolo M, Makino CL, Sidman RL (1999) Morphological, physiological, and biochemical changes in rhodopsin knockout mice. Proc Natl Acad Sci USA 96: 736-741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lunkes A, Lindenberg KS, Ben-Haiem L, Weber C, Devys D, Landwehrmeyer GB, Mandel JL, Trottier Y (2002) Proteases acting on mutant huntingtin generate cleaved products that differentially build up cytoplasmic and nuclear inclusions. Mol Cell 10: 259-269. [DOI] [PubMed] [Google Scholar]
- Mangiarini L, Sathasivam K, Seller M, Cozens B, Harper A, Hetherington C, Lawton M, Trottier Y, Lehrach H, Davies SW, Bates GP (1996) Exon 1 of the HD gene with an expanded CAG repeat is sufficient to cause a progressive neurological phenotype in transgenic mice. Cell 87: 493-506. [DOI] [PubMed] [Google Scholar]
- Martin-Aparicio E, Yamamoto A, Hernandez F, Hen R, Avila J, Lucas JJ (2001) Proteasomal-dependent aggregate reversal and absence of cell death in a conditional mouse model of Huntington's disease. J Neurosci 21: 8772-8781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller VM, Xia H, Marrs GL, Gouvion CM, Lee G, Davidson BL, Paulson HL (2003) Allele-specific silencing of dominant disease genes. Proc Natl Acad Sci USA 100: 7195-7200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pittler SJ, Baehr W (1991) Identification of a nonsense mutation in the rod photoreceptor cGMP phosphodiesterase beta-subunit gene of the rd mouse. Proc Natl Acad Sci USA 88: 8322-8326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Regulier E, Trottier Y, Perrin V, Aebischer P, Deglon N (2003) Early and reversible neuropathology induced by tetracycline-regulated lentiviral over-expression of mutant Huntingtin in rat striatum. Hum Mol Genet 12: 2827-2836. [DOI] [PubMed] [Google Scholar]
- Ross CA (2002) Polyglutamine pathogenesis: emergence of unifying mechanisms for Huntington's disease and related disorders. Neuron 35: 819-822. [DOI] [PubMed] [Google Scholar]
- Sathasivam K, Hobbs C, Turmaine M, Mangiarini L, Mahal A, Bertaux F, Wanker EE, Doherty P, Davies SW, Bates GP (1999) Formation of polyglutamine inclusions in non-CNS tissue. Hum Mol Genet 8: 813-822. [DOI] [PubMed] [Google Scholar]
- Tobin AJ, Signer ER (2000) Huntington's disease: the challenge for cell biologists. Trends Cell Biol 10: 531-536. [DOI] [PubMed] [Google Scholar]
- Uchihara T, Fujigasaki H, Koyano S, Nakamura A, Yagishita S, Iwabuchi K (2001) Non-expanded polyglutamine proteins in intranuclear inclusions of hereditary ataxias-triple-labeling immunofluorescence study. Acta Neuropathol (Berl) 102: 149-152. [DOI] [PubMed] [Google Scholar]
- Xia H, Mao Q, Paulson HL, Davidson BL (2002) siRNA-mediated gene silencing in vitro and in vivo Nat Biotechnol 20: 1006-1010. [DOI] [PubMed] [Google Scholar]
- Yamamoto A, Lucas JJ, Hen R (2000) Reversal of neuropathology and motor dysfunction in a conditional model of Huntington's disease. Cell 101: 57-66. [DOI] [PubMed] [Google Scholar]
- Yoo SY, Pennesi ME, Weeber EJ, Xu B, Atkinson R, Chen S, Armstrong DL, Wu SM, Sweatt JD, Zoghbi HY (2003) SCA7 knockin mice model human SCA7 and reveal gradual accumulation of mutant ataxin-7 in neurons and abnormalities in short-term plasticity. Neuron 37: 383-401. [DOI] [PubMed] [Google Scholar]
- Yvert G, Lindenberg KS, Picaud S, Landwehrmeyer GB, Sahel JA, Mandel JL (2000) Expanded polyglutamines induce neurodegeneration and trans-neuronal alterations in cerebellum and retina of SCA7 transgenic mice. Hum Mol Genet 9: 2491-2506. [DOI] [PubMed] [Google Scholar]
- Yvert G, Lindenberg KS, Devys D, Helmlinger D, Landwehrmeyer GB, Mandel JL (2001) SCA7 mouse models show selective stabilization of mutant ataxin-7 and similar cellular responses in different neuronal cell types. Hum Mol Genet 10: 1679-1692. [DOI] [PubMed] [Google Scholar]
- Zoghbi HY, Orr HT (2000) Glutamine repeats and neurodegeneration. Annu Rev Neurosci 23: 217-247. [DOI] [PubMed] [Google Scholar]