

Abstract
Minocycline has been demonstrated to be neuroprotective after spinal cord injury (SCI). However, the cellular consequences of minocycline treatment on the secondary injury response are poorly understood. We examined the ability of minocycline to reduce oligodendrocyte apoptosis, microglial/macrophage activation, corticospinal tract (CST) dieback, and lesion size and to improve functional outcome after SCI. Adult rats were subjected to a C7-C8 dorsal column transection, and the presence of apoptotic oligodendrocytes was assessed within the ascending sensory tract (AST) and descending CST in segments (3-7 mm) both proximal and distal to the injury site. Surprisingly, the numbers of dying oligodendrocytes in the proximal and distal segments were comparable, suggesting more than the lack of axon-cell body contiguity played a role in their demise. Minocycline or vehicle control was injected into the intraperitoneal cavity 30 min and 8 hr after SCI and thereafter twice daily for 2 d. We report a reduction of apoptotic oligodendrocytes and microglia within both proximal and distal segments of the AST after minocycline treatment, using immunostaining for active caspase-3 and Hoechst 33258 staining in combination with cell-specific markers. Activated microglial/macrophage density was reduced remote to the lesion as well as at the lesion site. Both CST dieback and lesion size were diminished after minocycline treatment. Footprint analysis revealed improved functional outcome after minocycline treatment. Thus, minocycline ameliorates multiple secondary events after SCI, rendering this clinically used drug an attractive candidate for SCI treatment trials.
Keywords: oligodendrocyte, apoptosis, spinal cord injury, caspase-3, minocycline, microglia, macrophages, neuroprotection, axonal dieback, footprint analysis
Introduction
The loss of function after spinal cord injury (SCI) results from both the primary mechanical insult and the subsequent, multifaceted secondary degenerative response (Sekhon and Fehlings, 2001; Dumont et al., 2001; McDonald and Sadowsky, 2002). The secondary degenerative response spreads from the site of impact, causing demyelination, axonal injury, and destruction to neuronal tissue that survived the initial insult. Apoptosis of oligodendrocytes is a prominent feature of the secondary degeneration after SCI (Li et al., 1996; Crowe et al., 1997; Liu et al., 1997; Shuman et al., 1997; Abe et al., 1999; Casha et al., 2001; Warden et al., 2001; Beattie et al., 2002; Dong et al., 2003; McBride et al., 2003), a process that may contribute to the chronic demyelination of spared axons often seen after SCI (Blight and Decrescito, 1986; Blight et al., 1991; Bunge et al., 1993; Waxman, 1993). The secondary degenerative response also involves glutamate excitotoxicity, ionic dysregulation, free radical generation, and ischemia (Balentine and Spector, 1977; Young and Flamm, 1982; Panter et al., 1990; Choi, 1992; Wrathall et al., 1994; Agrawal and Fehlings, 1997). A massive inflammatory response results from the release of cytokines and chemokines from resident and blood-derived inflammatory cells, which may further aggravate cellular and axonal damage (Bethea, 2000; Popovich, 2000; Hausmann, 2003). Therefore, treatment strategies designed to target several aspects of the secondary injury will likely be of value in the treatment of human SCI.
Minocycline, a second-generation tetracycline, may be an attractive candidate in the treatment of many neurodegenerative and trauma-induced CNS injuries attributable to both anti-inflammatory and neuroprotective properties. Collectively, the biological effects of minocycline include inhibition of microglial activation, reduction of mRNA of both interleukin 1β (IL-1β) and inducible nitric oxide synthase (Yrjanheikki et al., 1998), cyclooxygenase 2 expression, and prostaglandin E2 production (Yrjanheikki et al., 1999). Minocycline has also been shown to attenuate production of matrix metalloproteinases (MMPs) and to decrease T-lymphocyte transmigration (Brundula et al., 2002; Power et al., 2003). In addition, minocycline has been shown to inhibit caspase expression (Chen et al., 2000), cytochrome c release (Zhu et al., 2002) and caspase-dependent and -independent cell death (Wang et al., 2003).
Recently, minocycline has been shown to improve functional outcome, to reduce lesion size, to reduce cell death, and to alter cytokine expression after SCI (Lee et al., 2003; Wells et al., 2003). In a murine model of SCI, minocycline treatment was superior to methylprednisolone, the only treatment available to SCI patients, in promoting functional improvement (Wells et al., 2003). However, the cellular and molecular basis for the neuroprotective effects of minocycline remains for the most part unknown. We therefore sought to elucidate the cellular consequences of minocycline treatment and how it leads to attenuation of the pathophysiological response after SCI. Specifically, we used a dorsal column transection model at the cervical C7-C8 level of the spinal cord to examine the effects of minocycline treatment on delayed oligodendrocyte death within the distal degenerating and proximal dorsal columns, the microglial/macrophage response, corticospinal tract (CST) dieback, lesion size, and functional outcome after SCI. The results indicate that minocycline influences several aspects of the secondary degenerative response, a characteristic that is likely responsible for its ability to promote improved functional outcome after SCI.
Materials and Methods
Spinal cord dorsal column transection and corticospinal tract tracing. All experiments were conducted in accordance with the University of British Columbia Animal Care Ethics Committee, adhering to guidelines of the Canadian Council on Animal Care. Adult Wistar rats were anesthetized with an intraperitoneal injection of a mixture of ketamine hydrochloride (72 mg/kg; Bimeda-MTC, Cambridge, Ontario, Canada) and xylazine hydrochloride (9 mg/kg; Bayer Inc., Etobicoke, Ontario, Canada). The animals were placed in a stereotaxic frame, and a laminectomy was performed at the seventh cervical vertebra. An adjustable wire knife (model 120; David Kopf Instruments, Tujunga, CA) was used to minimize the variability in depth of the lesion. The knife was lowered to the dura mater 1 mm lateral to midline on the animal's right side. After a prepuncture of the dura mater with a fine needle, the wire knife was lowered 1.1 mm into the dorsal horn between the dorsal roots of C7 and C8. The wire knife blade (3 mm curvature length) was extended to a further depth of 0.5 mm (total, 1.6 mm) and horizontally to a total diameter of 1.9-2 mm. The wire knife was drawn up while gently pushing the dorsal columns down with a cotton swab. This severs most dorsal column axons, but it does not sever the dura except at its point of entry and its extreme distal end that points upward. The wire knife was removed, and a pair of microscissors was introduced into the two small holes produced by the wire knife to cut the dura and dorsal vein and to ensure complete transection of the dorsal columns and central canal. Efficacy of the lesion was confirmed with histology at the end of the study. Two weeks before the dorsal column transection, rats were injected with the anterograde tracer biotin dextran amine (BDA; Molecular Probes, Eugene, OR) to visualize the CST both proximal and distal to the lesion and to assess CST dieback. A burr hole overlying the right side of the cortex was made to access the cell bodies of the CST. To effectively label the CST, 200 nl of (25%) BDA was injected into each of eight sites spanning the rat sensorimotor cortex using a glass micropipette fitted to a Hamilton syringe.
Minocycline administration. Rats were randomly assigned into two treatment groups and were treated twice per day for 2 d with intraperitoneal injections of either saline or 50 mg/kg minocycline (Apotex Inc., Weston, Ontario, Canada) in 1.0 ml of saline (nonbuffered), beginning 30 min after injury. There was a minor grooming response at the site of injection in some animals, lasting a few seconds. Otherwise, these treatments were well tolerated.
Preparation of tissue. At 7 and 14 d after injury, animals were anesthetized with a lethal dose of chloral hydrate (BDH Chemicals, Toronto, Ontario, Canada) and perfused with PBS, followed by perfusion fixation with a solution of 4% paraformaldehyde in PBS. The spinal cords (brainstem to midthoracic region) were removed, postfixed in 4% paraformaldehyde overnight, and subsequently cryoprotected in 30% sucrose. The spinal cords were then cut into three blocks, frozen, and stored at -80°C until sectioned. Specifically, a 6 mm lesion block, centered at the lesion site, was used to assess lesion size, CST dieback, and ED1 density (percent area). A rostral block (+7 to +3 mm) and caudal block (-3 to -7 mm) were used to assess remote oligodendrocyte apoptosis and ED1 density (percent area). Blocks were cryosectioned in the sagittal plane at a thickness of 10 μm (apoptosis studies) or 14 μm (lesion studies), and tissue sections were collected on Superfrost slides (Fisher Scientific, Houston, TX) organized into five adjacent section series.
Immunohistochemistry. Fluorescent immunohistochemistry was performed on slides containing sections of 7 or 14 d postinjured spinal cords. In general, slides were rinsed three times in PBS and blocked in 10% normal goat serum in PBS for 30 min at room temperature. To detect active caspase-3-positive apoptotic cells within the remote distal and proximal dorsal ascending sensory tract (AST) and CST, slides were incubated with a rabbit monoclonal antibody for active caspase-3 (1:500; PharMingen, San Diego, CA). After three washes in PBS, slides were incubated for 1 hr at room temperature with the Alexa Fluor 488 goat anti-rabbit secondary antibody (1:200; Molecular Probes) and Cy3-conjugated streptavidin (Jackson ImmunoResearch, West Grove, PA) to visualize active caspase-3 and the BDA-labeled CST, respectively. Slides were then washed three times in PBS and submerged in Hoechst 33258 (1 μg/ml; Sigma, Oakville, Ontario, Canada) to detect condensed nuclei indicative of apoptosis. To assess cell identity of apoptotic profiles, the following mouse primary antibodies were used: adenomatous polyposis coli (CCI clone) (antibody 7, 1:200; Oncogene Research Products, Boston, MA) for oligodendrocyte cell bodies, GFAP (1:1000; Sigma) for astrocytes, OX42 (1:600; Serotec, Oxford, UK) for microglia/macrophages, and ED1 (1:500; Serotec) for activated microglia/macrophages. A Cy3-conjugated donkey anti-mouse secondary antibody (Jackson ImmunoResearch) or Alexa Fluor 488 goat anti-mouse secondary antibody (1:200; Molecular Probes) was used to visualize the mouse primary antibodies listed above.
To determine the number of apoptotic profiles within the distal degenerating and proximal segments remote from injury, active caspase-3-positive cells with a condensed nucleus were counted. Tracing the CST with BDA before the lesion allowed us to discriminate the AST from the CST. Using the central canal as a reference point, 10-μm-thick sagittal sections (three sections 50 μm apart) from either 3-7 mm rostral or 3-7 mm caudal to injury were assessed, and the amount of apoptosis was expressed as the mean number of active caspase-3 positive profiles with condensed nuclei per 10 μm section. Cell counts of active caspase-3 positive oligodendrocytes and microglia were expressed in a similar manner. Counts were performed blind with respect to treatment using a Zeiss (Thornwood, NY) Axioskop microscope. Three sections (spaced 50 μm apart) from four to six animals in each group per time point were analyzed.
Microglial/macrophage activation and recruitment. To assess microglial/macrophage activation and recruitment within the corticospinal or ascending sensory tracts 3-3.5 mm remote from lesion, an overlay of three rectangular boxes (dimensions of each box, 100 μm width × 500 μm length) was placed onto digitally captured sagittal sections containing BDA-labeled axons (red channel) using SigmaScan Pro software (SPSS, Chicago, IL). With regard to the placement of the rectangles, the dorsal most edge of the CST-AST interface was set at 0 μm. The CST rectangle was placed so its top edge was aligned with the dorsal edge of the CST and captured an area (50,000 μm2) within the CST. The AST100 rectangle was positioned 100 μm from the dorsal edge of the labeled CST and captured an area (50,000 μm2) within the adjacent AST. The AST300 rectangle was positioned 100 μm from the dorsal edge of the AST100 rectangle and captured an area (50,000 μm2) 300 μm dorsal to the CST. The overlay of the rectangles was then copied onto thresholded images depicting the ED1-positive cells (green channel), and the percentage of the area occupied by ED1 signal was reported. One or two sections from at least four animals per group were assessed. Similarly, ED1 density within the proximal CST stump was quantified using the same approach with a standardized overlay box (250 μm width × 750 μm length) positioned within the proximal CST stump. At least two sections from five animals per group were assessed.
CST dieback. To quantify CST dieback, 14-μm-thick sections containing the lesion site were subjected to GFAP immunohistochemistry to delineate the lesion area and Cy3-conjugated streptavidin (Jackson ImmunoResearch) to visualize the BDA-traced CST. Digital images were captured, and CST dieback was measured using SigmaScan Pro software (SPSS). At least two sections from each animal (7 d, six or seven animals per group; 14 d, four or five animals per group) were assessed, and the mean distance of leading intact CST fibers from the center of the lesion site per 14 μm section was reported. To ensure equal sampling, we used the central canal as the reference point (midline) and sampled the midline section as well as the 5th and 10th 14 μm section at ∼70 and 140 μm lateral to the midline, respectively. Thus, all sections run through the gracile funiculus containing the axons from the hindlimbs and do not contain unlesioned cervical sensory afferents.
Lesion area. The digital images captured and used for CST dieback analysis (see above) were used to quantify the lesion area (delineated by GFAP immunohistochemistry). Lesion area was measured using SigmaScan Pro software (SPSS). At least two sections from each animal (7 d, six or seven animals per group; 14 d, four or five animals per group) were assessed, and the mean lesion area (square millimeters) per 14 μm section was reported.
Footprint analysis. Walking track footprint analysis was modified from the method of de Medinaceli et al. (1982). The animal's forepaws were dipped in green dye, and the hindpaws were dipped in red dye (nontoxic). The rats were trained to avoid a brightly illuminated starting box by walking across a narrow wooden board (1 m long and 7 cm wide) leading to a darkened box containing their familiar housing mates. Two marks were placed 70 cm apart, centered on the platform, to ensure the rat footprints were not analyzed during the beginning (acceleration) or ending (deceleration) of the movement. The rats were timed as they passed from mark 1 to mark 2. If the animal failed to complete the test within 1-2 sec or paused while traversing the track, the trial was excluded from analysis, and the test was repeated. Each animal (observer was blind to treatment conditions) performed one training session without paws being dipped, followed by two separate baseline sessions taken before injury and one session on 3, 5, 7, and 14 d after injury. Each session consisted of three separate traverses of the track. The footprints were scanned, and digitized images were measured using Photoshop version 6.0 (Adobe Systems, Ottawa, Ontario, Canada). Toe spread was measured as the distance between the first and fifth toe in the hindpaw. Interlimb coordination was measured as the distance between the ipsilateral forepaw (center of pad) and hindpaw (center of pad). Angle of rotation was measured as the angle made by the third toe, center of the footpad, and a line parallel to the direction of travel. To assess toe spread, the maximal toe spread index (TSI) was calculated according to the Brown TSI formula (Brown et al., 1989): TSI = (ETS - NTS)/NTS, where ETS is experimental toe spread, and NTS is normal toe spread.
Similar to toe spread, an index was used to calculate both interlimb coordination and angle of rotation. At least nine footprints per side from three sessions were measured per animal per group (n = 8, 7 d; n = 4, 14 d) to determine the mean values of each parameter assessed.
Statistics. Statistical analysis was performed using SigmaStat software (SPSS). Data were compared between groups using a Student's t test. In the case of unequal variances, the Mann-Whitney rank sum test was used. Differences with p < 0.05 were considered significant.
Results
Active caspase-3-mediated apoptosis of oligodendrocytes and microglia within both proximal and degenerating white matter after C7-C8 dorsal column transection
To assess the efficacy of minocycline treatment in the prevention of delayed white matter oligodendrocyte cell death after SCI, we chose a dorsal column transection model to compare cell death within both proximal and distal regions of two well defined systems, the descending dorsal CST and AST, namely the cuneate and gracile funiculi (see supplemental Fig. 1A,B, available at www.jneurosci.org).
A previous study using a similar injury model at the thoracic level reported numerous terminal deoxynucleotidyl transferase-mediated biotinylated UTP nick end-labeling (TUNEL)-positive glia within degenerating and proximal (to a lesser extent) segments of the AST and CST and identified a subpopulation of the dying cells as oligodendrocytes (Warden et al., 2001). We examined death of oligodendodrocytes within the proximal and distal regions of the AST and CST remote from a cervical lesion, a region not previously studied using this model in mature rodents. Prelabeling of the right motor cortex with the anterograde tracer BDA allowed us to discriminate the proximal and distal segments of the CST from the proximal and distal segments of the AST after injury (see supplemental Fig. 1B, available at www.jneurosci.org). A role for caspase-3-mediated oligodendrocyte apoptosis after SCI has been shown in rat, mouse, chick, and human (Emery et al., 1998; Springer et al., 1999; Beattie et al., 2002; Dong et al., 2003; McBride et al., 2003). We therefore assessed active cell death using immunohistochemistry for active caspase-3 and Hoechst 33258 stain to visualize nuclear condensation, a hallmark of apoptosis. Figure 1 shows apoptotic cells within both rostral (Fig. 1A-E) and caudal (Fig. 1F-J) segments of the AST and CST 14 d after injury. Using cell-specific markers CC1 and OX42, we confirmed that most these profiles were oligodendrocytes (Fig. 1K-N,S) and microglia/macrophages (Fig. 1O-R,T), respectively. No apoptotic astrocytes (GFAP) were observed (data not shown). To ensure that microglia and oligodendrocytes were truly apoptotic and did not represent apoptotic bodies contained within a phagocyte, which would possess a normal nucleus as well, we used confocal microscopy to assess the nucleus within the entire boundaries of the cell. Examples of apoptotic nuclei (Fig. 1S′) within the boundaries of an oligodendrocyte are shown in Figure 1S and within a microglial/macrophage in Figure 1, T and T′, confirming their apoptotic phenotype. Within the AST, active caspase-3 positive-profiles were abundant in both the proximal segment, i.e., caudal to the lesion (Figs. 1U, 2B), and distal segment, i.e., rostral to the lesion (Figs. 1V, 2B). Approximately 10-15% of apoptotic profiles were oligodendrocytes (CC1-positive), whereas most apoptotic profiles (∼65-70%) were of microglial/macrophage origin (OX42-positive; Fig. 2A).
Figure 1.

Cell death of oligodendrocytes and microglia/macrophages within both proximal and distal segments of the CST and AST after SCI. The dorsal funiculus 5 mm rostral (A-E) or caudal (F-J) to the injury site, 14 d after the lesion, is shown. The BDA-labeled CST (red) is easily distinguished from the adjacent ascending fibers, allowing for analysis of apoptotic profiles within the CST or AST. B, Most CST fibers are intact, but some degenerate (arrowheads). C-E, Boxed area in B, an active caspase-3-positive profile (C, arrow) with a condensed nucleus (D, arrow). E, Merged image of C, D, and BDA (red). Caudal to the injury site, the CST completely degenerates (F). Apoptotic profiles are found within both the degenerating CST (arrow) and within the proximal segment of the ascending fibers (boxed area in G). H-J, Boxed area in G showing an active caspase-3-positive profile (H) with a condensed nucleus (I, arrow). J, Merged image of H, I, and BDA (red). K-N, Colocalization of CC1 (K), active caspase-3 (L), and a condensed nucleus (M) revealing an apoptotic oligodendrocyte within the proximal AST 7 d after injury (N, merged image of K-M). O-R, OX42 immunoreactivity (O) revealing that apoptotic microglia/macrophages were also present within the proximal AST (P, active caspase-3; Q, Hoechst 33528; R, merged image of O-Q). S, T, Representative merged confocal images of CC1-positive (S, arrow) and OX42-positive (T, arrow), active caspase-3-positive profiles with (S′, T′) or without a condensed nucleus (S, arrowhead). U-W, Numerous active caspase-3-positive profiles (green) are present within the proximal (U) and distal (V) AST at 7 d after the lesion. Minocycline treatment greatly reduces the number of active caspase-3-positive profiles within both the proximal and distal AST (W, distal AST). Scale bars: A, F, U-W, 100 μm; B, G, 25 μm; C-E, H-T, 10 μm.
Figure 2.
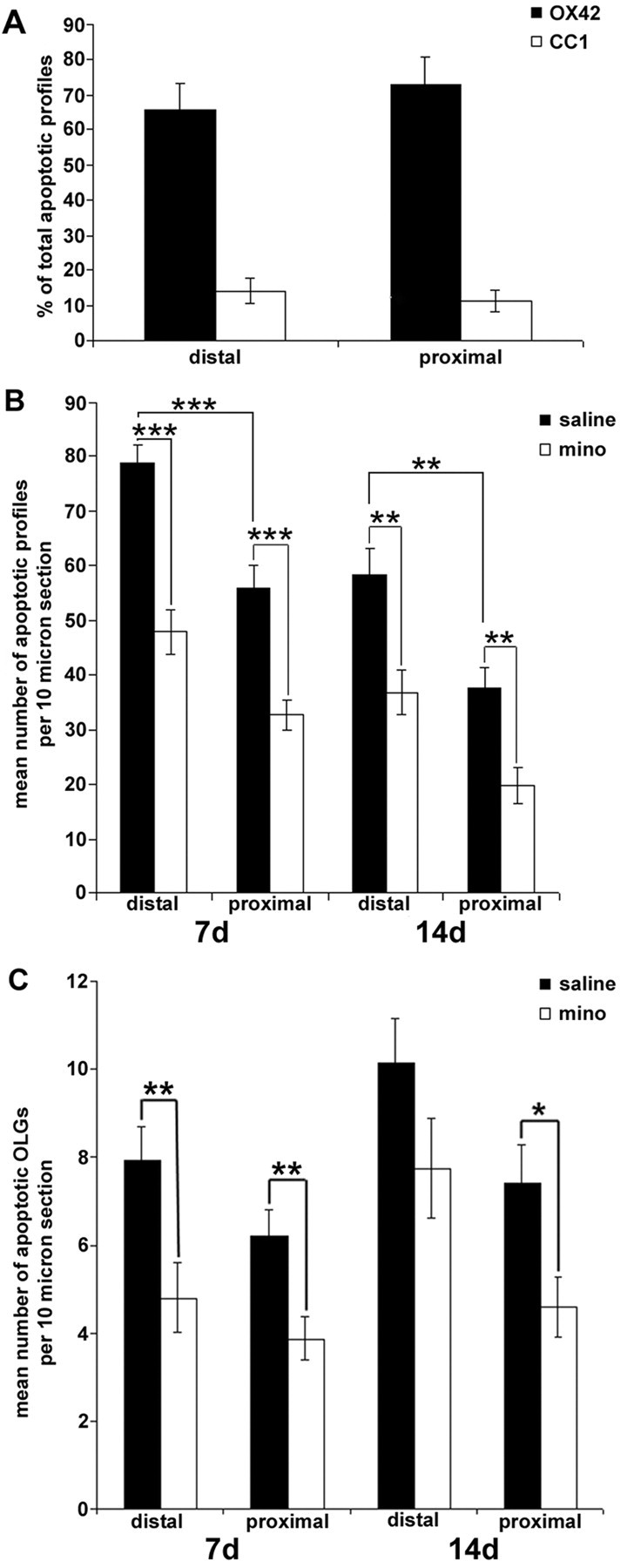
Minocycline treatment inhibited dorsal column transection-induced glial cell death within the distal and proximal AST. A, Percentage of apoptotic microglia/macrophages (OX42-positive) or oligodendrocytes (CC1-positive) within the proximal and distal AST 7 d after the lesion (n = 6 per group). Microglia/macrophages are the main cell type to undergo apoptosis in this model. B, Quantification of the mean number of apoptotic profiles within the distal and proximal segments of the AST. Significantly fewer apoptotic profiles are located within the proximal versus distal segments of the AST. Minocycline (mino) treatment significantly reduced the mean number of apoptotic profiles per 10 μm section ± SEM within distal and proximal segments of the AST at both 7 and 14 d after injury compared with saline-treated controls. (*p < 0.05; **p < 0.01; ***p < 0.001; 7 d, n = 6 per group; 14 d, n = 4 or 5 per group). C, Apoptotic oligodendrocytes (CC1-positive), active caspase-3-positive profiles with a condensed nucleus, were significantly decreased in the minocycline-treated group compared with saline controls within the proximal AST at both 7 and 14 d after injury. However, only the proximal segment of the AST from minocycline-treated animals contained significantly fewer apoptotic oligodendrocytes at 14 d after injury. There was no difference between the mean number of apoptotic oligodendrocytes within the proximal versus distal segments of the AST at both 7 and 14 d after injury (data represent mean ± SEM; *p < 0.05; 7 d, n = 5 per group; 14 d, n = 4 or 5 per group).
Effect of minocycline treatment on active caspase-3-mediated oligodendrocyte death
Apoptotic profiles were counted within both proximal and distal segments of the AST and CST remote from a C7-C8 dorsal column transection site and compared between saline- versus minocycline-treated groups (Fig. 2B). In the untreated controls, there were significantly fewer apoptotic profiles within the proximal segment compared with the distal segment within the AST at both 7 and 14 d after injury (7 d, p < 0.001; 14 d, p < 0.01; Fig. 2B). Minocycline treatment significantly reduced the number of apoptotic profiles within the proximal AST (32.7 ± 2.7, mean ± SEM) versus saline-treated animals (56.1 ± 3.9; p < 0.001) at 7 d after injury. Within the degenerating (distal) AST, minocycline treatment also reduced the mean number of apoptotic profiles (47.9 ± 4.0) when compared with saline treatment (78.7 ± 3.5; p < 0.001; Figs. 1W, 2B). Minocycline treatment continued to prevent cell death at 14 d after the lesion within both proximal (minocycline, 19.8 ± 3.3; vs saline, 37.6 ± 3.8; p < 0.01) and distal (minocycline, 36.8 ± 4.1; vs saline, 58.2 ± 5.0; p < 0.01) segments. These results suggest that minocycline treatment continued for 48 hr after injury is sufficient to reduce secondary cell death by ∼39% (distal) and 42% (proximal) at 7 d and by 37% (distal) and 47% (proximal) at 14 d after injury.
Using triple labeling with CC1 as a marker for oligodendrocytes combined with the two apoptotic markers used above, we counted the number of apoptotic oligodendrocytes from minocycline- or saline-treated rats. Minocycline significantly reduced the mean number of apoptotic oligodendrocytes within both the proximal (minocycline, 3.9 ± 0.5; vs saline, 6.2 ± 0.6; p < 0.01) and distal (minocycline, 4.8 ± 0.8; vs saline, 7.9 ± 0.8; p < 0.01) segments of the AST at 7 d after injury compared with saline-treated animals (Fig. 2C). At 14 d after the lesion, only the proximal segment from minocycline-treated rats (4.6 ± 0.7) had significantly fewer apoptotic oligodendrocytes compared with saline-treated animals (7.4 ± 0.9; p < 0.05). Interestingly, in the untreated group, the number of oligodendrocytes in the distal segments at both time points was only marginally higher than their proximal counterparts but did not reach significance (7 and 14 d, p > 0.05).
Effect of minocycline treatment on ED1 density 7 d after SCI
Previously, it has been shown that the microglial/macrophage response peaks at 7 d after SCI and is associated with demyelination and axonal degeneration (Blight, 1985; Giulian and Robertson, 1990; Blight, 1992; Popovich et al., 1997, 1999). In addition, Shuman et al. (1997) previously hypothesized that activated microglia may induce oligodendrocyte apoptosis within degenerating tracts after SCI. We therefore assessed whether minocycline treatment would inhibit microglial/macrophage activation in areas of oligodendrocyte apoptosis, i.e., the segments several millimeters distant to the lesion site as well as within the lesion site itself. Untreated injured animals had large numbers of ED1-positive microglia/macrophages located within the degenerating distal segments of the AST and CST (Fig. 3A,B,E,F). Treatment with minocycline significantly reduced ED1 density within the distal (rostral to lesion site) degenerating segments of the AST measured from 100 μm (AST100, minocycline, 1.6 ± 0.5%, mean ± SEM; vs saline, 6.0 ± 1.1%; p < 0.01) and 300 μm dorsal to the CST (AST300, minocycline, 2.0 ± 0.3%; vs saline, 4.7 ± 0.7%; p < 0.01; Fig. 3C,G,M). Within the distal (caudal to lesion site) degenerating segment of the CST, minocycline treatment also significantly reduced ED1 density (minocycline, 5.1 ± 2.1%; vs saline, 11.8 ± 1.4%; p < 0.05; Fig. 3D,H,M). Although there was a trend toward a reduction of ED1 density within the proximal segment of the AST (caudal to the lesion) closest to the CST (Fig. 3, compare B,F, D,H) and within the proximal CST (rostral to the lesion) in minocycline-treated rats, this failed to reach significance (Fig. 3M).
Figure 3.
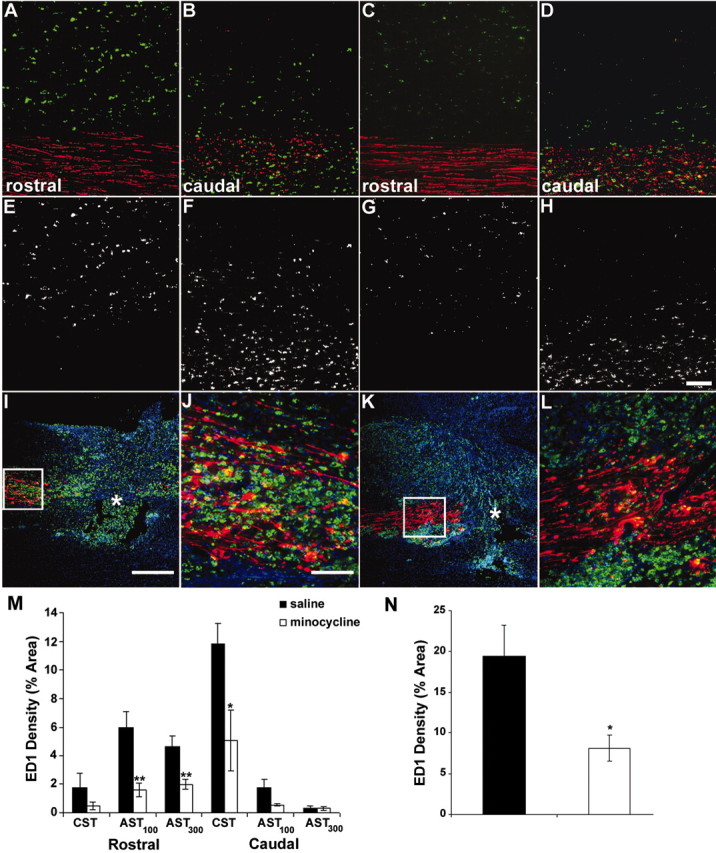
Minocycline treatment reduced ED1-positive (microglial/macrophage) density 7 d after injury. A-H, ED1-positive profiles (green) 3 mm rostral (A, C, E, G) and caudal (B, D, F, H) to injury. Minocycline significantly reduced ED1 density within both rostral AST (100 and 300 μm dorsal to CST; C, G, M) and within the caudal CST (D, H, M) compared with saline-treated animals (A, B, E, F, M). Triple immunofluorescence images of the lesion site from saline-treated (I, J) and minocycline-treated (K, L) animals are shown. The proximal CST is red; ED1-positive microglia/macrophages are green; and Hoechst 33258 is blue. Note the reduced ED1-positive signal in K compared with I. J, L, Higher magnification of the boxed area in I and K. Less ED1-positive signal is evident within the proximal CST of minocycline-treated animals compared with saline-treated animals. N, Quantification of the density of the ED1-positive signal within the proximal CST. Data represent mean percentage ± SEM (n = 4 or 5 per group). *p < 0.05; **p < 0.01. Scale bars: A-H, J, L, 100 μm; I, K, 500 μm.
At the injury site, large numbers of microglia/macrophages were found within the proximal stump of the anterogradely traced CST, i.e., immediately adjacent to the lesion, as well as in areas of cavitation in saline-treated rats (Fig. 3I,J). This ED1 density was reduced by minocycline treatment at 7 d after injury (Fig. 3K,L). Quantification of ED1 density within the proximal CST stump confirmed the visual impression given in Figure 3, revealing a significant reduction in ED1 density in minocycline-treated animals (8.2 ± 1.6%, mean ± SEM) compared with saline-treated controls (19.4 ± 3.8%; p < 0.01; Fig. 3N).
Effect of minocycline treatment on CST dieback after SCI
Previously, it has been shown that after transection of the adult rat CST, the proximal axon stumps degenerate during the first few weeks after injury (Pallini et al., 1988). This extensive axonal dieback may have implications for regeneration and sprouting after injury. We measured the distance between the closest, intact, anterogradely traced CST fibers and the center of the lesion site to test whether minocycline treatment would prevent proximal CST degeneration after SCI. In agreement with previous findings (Pallini et al., 1988), the proximal stump of the lesioned CST axons showed pronounced axonal dieback (Fig. 4A,C,E). After treatment with minocycline, CST dieback was significantly attenuated at both 7 and 14 d after injury compared with saline-treated animals (7d, p < 0.001; 14 d, p < 0.001; Fig. 4B,D,E). At 7 d after the lesion, the mean distance between the proximal CST axons to the center of the lesion site was 598.4 ± 48.6 μm (mean ± SEM) in minocycline-treated animals versus 877.1 ± 47.7 μm in saline-treated animals. At 14 d after injury, CST dieback reached 751.2 ± 58.0 μm in minocycline-treated animals compared with 1120.2 ± 80.5 μm in saline-treated controls. On average, minocycline treatment reduced the distance of the leading CST axons from the injury site by ∼32 and 33% at 7 and 14 d, respectively.
Figure 4.
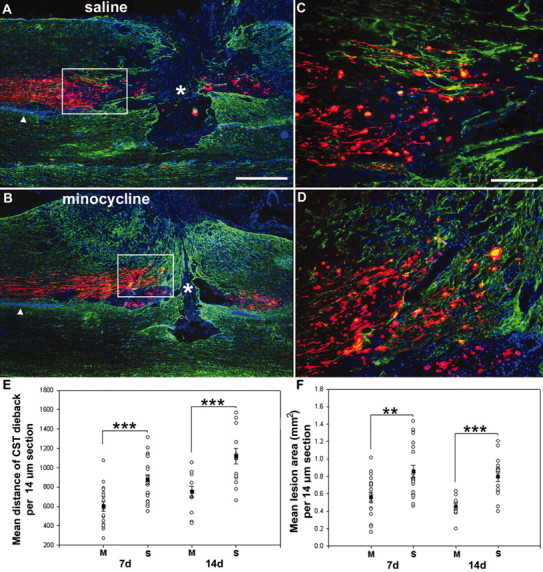
Minocycline treatment reduced CST dieback and lesion size both 7 and 14 d after the lesion. A, B, Images of the dorsal columns revealing the lesion site at 7 d after injury from saline-treated (A) versus minocycline-treated (B) rats. Note that the lesion area (delineated by GFAP staining, green) and CST dieback (red) is clearly reduced in minocycline- versus saline-treated animals. C, Boxed area in A; D, boxed area in B showing a higher magnification of the proximal BDA-traced CST axons (red). E, Quantification of CST dieback. Overall minocycline (M) treatment significantly reduced CST axonal dieback by ∼32 and ∼33% at 7 and 14 d, respectively. S, Saline. F, Quantification of lesion area. Minocycline treatment significantly reduced the lesion area by ∼34 and ∼43% at 7 and 14 d. Data represent mean ± SEM (n = 4-7 per group). **p < 0.01; **p < 0.001. Scale bars: A, B, 500 μm; C, D, 100 μm.
Effect of minocycline treatment on lesion size after SCI
Recently, Wells et al. (2003) reported that minocycline treatment significantly reduced lesion area after SCI in mice. However, the sequelae of secondary damage after SCI in the mouse vary from those in humans (Guth et al., 1999; Inman et al., 2002). Therefore, we assessed whether minocycline treatment would reduce lesion size in our rat model of SCI. The region demarcated by activated astrocytes (determined by GFAP immunofluorescence) was used to delineate the boundaries of the lesion. Lesion area was greatly enhanced in saline-treated animals (Fig. 4A,C,F) versus minocycline-treated animals (Fig. 4B,D,F). Quantification of these results showed that minocycline treatment significantly reduced lesion area at both 7 and 14 d after injury compared with saline-treated animals (7 d, p < 0.01; 14 d, p < 0.001; Fig. 4F).
Improved functional outcome after minocycline treatment
To assess functional outcome after SCI, rat footprints were collected and assessed for perturbations in limb coordination, angle of rotation, and toe spread. Footprints from animals during presurgical training revealed highly coordinated forelimb and hindlimb foot placements (Fig. 5A). After a cervical dorsal column transection, forelimb-hindlimb coordination was compromised (saline-treated animals), whereas minocycline treatment significantly improved forelimb-hindlimb coordination at both 7 and 14 d after injury (p < 0.05; Fig. 5B). Minocycline treatment also reduced the injury-induced hindlimb angle of rotation compared with saline-treated animals (p < 0.05; Fig. 5C). In addition, minocycline treatment significantly attenuated the SCI-induced increase in hindlimb toe spread compared with saline-treated animals at 7 d after injury (toe spread index, mean ± SEM, minocycline, 0.07 ± 0.02; vs saline, 0.13 ± 0.01; p < 0.01). However, at 14 d, there was no significant difference between treatment groups, suggesting that minocycline reduced the time of the recovery process in this paradigm.
Figure 5.
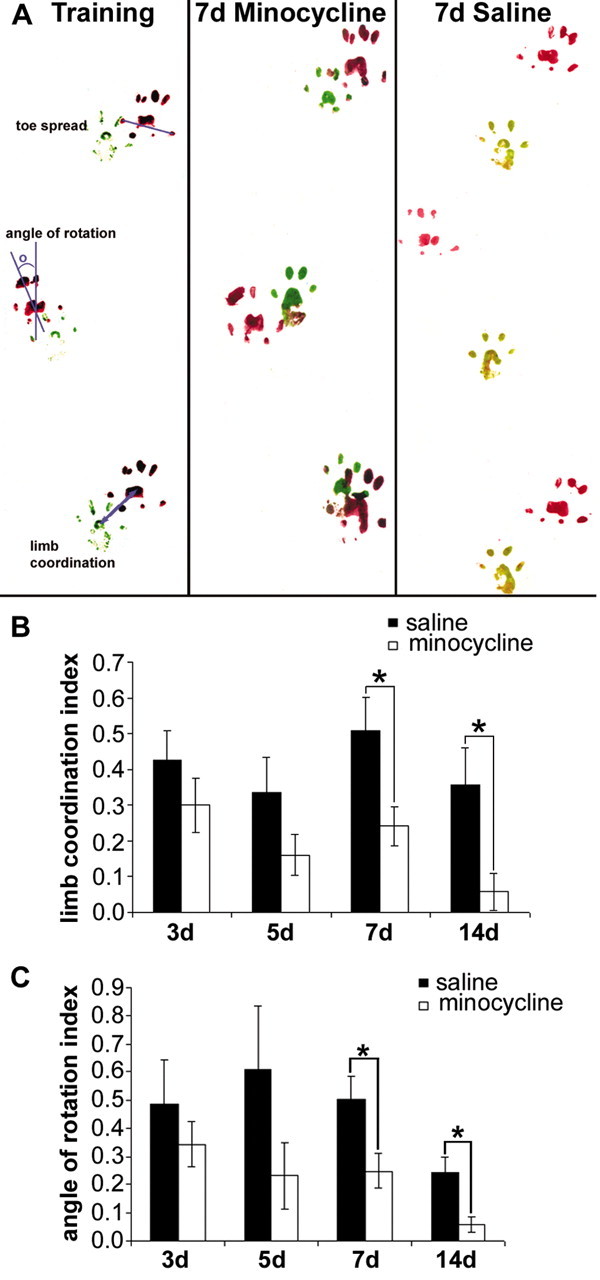
Minocycline treatment improved interlimb coordination and reduced hindlimb angle of rotation after SCI. A, Representative footprints collected from saline- and minocycline-treated animals. Minocycline treatment improved limb coordination (B) and reduced the hindlimb angle of rotation (C) at both 7 and 14 d after injury. Indexes were calculated as experimental value - baseline value/baseline value. Data represent mean ± SEM (7 d, n = 8 per group; 14 d, n = 4 per group). *p < 0.05.
Discussion
Here, we report that after a cervical dorsal column transection, minocycline treatment reduced delayed white matter oligodendrocyte apoptosis, attenuated microglial/macrophage activation and recruitment, prevented corticospinal axonal dieback, and reduced lesion size. In addition, minocycline treatment improved interlimb coordination and normalized the hindpaw angle of rotation and toe spread after SCI.
Our findings revealed large numbers of active caspase-3-positive apoptotic profiles located within both degenerating and proximal segments of the AST remote to the lesion. Systemic minocycline treatment significantly attenuated the number of apoptotic profiles, including oligodendrocytes within both degenerating and proximal segments. These results are in agreement with a recent study that assessed the effects of minocycline treatment on cell death at the lesion site after SCI (Lee et al., 2003).
Surprisingly, microglia/macrophages were found to be the principle cell type to undergo apoptosis after dorsal column transection. At 7 d after injury, ∼70% of apoptotic profiles within the proximal and degenerating AST were positive for OX42, a cell-specific marker of microglia/macrophages, whereas only ∼11% were identified as oligodendrocytes (CC1-immunopositive). Our results are in agreement with previous studies that found that large numbers of apoptotic microglia are present after contusion SCI in rats (Shuman et al., 1997; Yong et al., 1998). However, other studies have reported that microglia/macrophages were not among the apoptotic profiles present after partial or complete transection injuries in rat (Abe et al., 1999; Warden et al., 2001). Differences in apoptotic detection methods (TUNEL vs active caspase-3), microglial/macrophage markers (OX42 vs Bandeiraea simplicifolia agglutinin or OX6), or injury paradigms may partially account for the discrepancy between studies. We cannot entirely rule out the possibility that phagocytes engulfing apoptotic cells may be included in the counts of apoptotic microglia/macrophages, thereby inflating their number. In an attempt to limit this problem, confocal microscopy was used to visualize the entire boundary of the cell. Of the 20 randomly selected cells analyzed in this manner, all displayed an apoptotic nucleus within an active caspase-3-positive profile, confirming their apoptotic phenotype.
Previously, it has been shown that activation and proliferation of microglia within the ventral gray matter after sciatic nerve injury are followed by the elimination of these cells via apoptosis (Gehrmann and Banati, 1995). Because minocycline treatment reduces both microglial/macrophage activation and proliferation (Yrjanheikki et al., 1998; Tikka and Koistinaho, 2001; Tikka et al., 2001), a resultant decrease in microglial/macrophage apoptosis would be expected as well. In addition, minocycline has been shown to inhibit caspase-dependant cell death (Chen et al., 2000; Lee et al., 2003; Wang et al., 2003). Therefore, the reduction in microglial/macrophage cell death after minocycline treatment in this study is probably attributable to a direct effect on microglial/macrophage activation as well as their death.
Interestingly, apoptotic oligodendrocytes were present at the same levels in both proximal (beyond the level of axonal dieback) and degenerating segments within the transected dorsal columns, suggesting that lack of axonal-derived trophic support may not be the only mechanism to induce oligodendrocyte apoptosis after SCI. These results are in contrast to previous studies that showed that apoptotic profiles are predominantly localized within white matter tracts undergoing Wallerian degeneration (Crowe et al., 1997; Shuman et al., 1997; Abe et al., 1999; Casha et al., 2001; Warden et al., 2001). However, these studies did not directly assess oligodendrocyte apoptosis within distinguished boundaries of degenerating and intact tracts within the injured dorsal columns. In the present study, we applied the anterograde tracer BDA to label the CST previous to injury. This approach facilitates a clear distinction between the boundaries of the AST and CST and allowed the precise localization of apoptotic profiles, including oligodendrocytes, to these two tracts. We cannot rule out the possibility that some of the proximal fibers degenerate after injury and, therefore, may induce oligodendrocyte apoptosis. However, large numbers of ascending and CST fibers are present within their proximal stumps, suggesting that oligodendrocyte apoptosis within the proximal segments remote from injury is not attributable to pronounced axonal degeneration. If this were the case, we would not expect the same number of apoptotic oligodendrocytes within both segments because the distal segment completely degenerates. Regardless of the precise stimulus that induces oligodendrocyte apoptosis, its prevention may prevent the chronic demyelination observed in areas where axons are left intact after SCI (Blight and Decrescito, 1986; Blight et al., 1991; Bunge et al., 1993; Waxman, 1993; Werner et al., 2000).
Several studies provide strong evidence that activated mononuclear phagocytes (CNS resident microglia, blood-derived macrophages, or both) and products associated with their activation contribute to the secondary damage after SCI, including demyelination or axonal degeneration of previously spared axons, or both (Blight, 1985, 1992, 1994; Giulian and Robertson, 1990; Popovich et al., 1999, 2002). After SCI in cats, demyelination of intact axons within the outer rim of the spinal cord is temporally correlated with an invasion of the CNS parenchyma by phagocytes (Blight, 1985). Treatments designed to inhibit the macrophage response after SCI have promoted survival of motoneurons, reduced the loss of myelinated axons, reduced cavitation, and improved function after SCI (Giulian and Robertson, 1990; Blight, 1994; Popovich et al., 1999). Here, we show that minocycline treatment effectively reduced microglial/macrophage activation at the lesion site within the proximal CST stump and remote from injury within the distal segments of the dorsal column tracts. In addition, inhibition of the microglial/macrophage response was associated with reduced oligodendrocyte death, lesion size, and axonal dieback and improved functional outcome.
In agreement with previous studies, minocycline treatment reduced lesion size after SCI (Lee et al., 2003; Wells et al., 2003). Although the mechanism is unknown, lesion size is often decreased after attenuation of the inflammatory response (Bethea et al., 1999; Oudega et al., 1999; Popovich et al., 1999). Indeed, treatment with the anti-inflammatory cytokine IL-10 decreases spinal cord injury-induced and monocyte/macrophage production of the proinflammatory cytokine tumor necrosis factor α (TNF-α), with a concurrent decrease in lesion size (Bethea et al., 1999; Takami et al., 2002). Interestingly, minocycline treatment increased mRNA levels of the anti-inflammatory cytokine IL-10, with a concomitant decrease in the proinflammatory cytokine TNF-α, mirroring the neuroprotective effects of IL-10 treatment (Lee et al., 2003). However, despite a large reduction in macrophages after methylprednisolone treatment, the loss of spinal tissue and macrophage number was not correlated (Oudega et al., 1999). Further studies are needed to elucidate the mechanisms underlying spinal tissue loss after SCI.
A possible limitation to successful regeneration and plasticity of severed axons after SCI is the progressive dieback of the proximal stump that may cause additional loss of function. The mechanisms underlying axonal dieback are currently unknown. Our CST dieback data from saline-treated animals are consistent with those of Pallini et al. (1988), who showed that CST dieback occurs at a rate of ∼40-90 μm/d over the first 2 weeks after midthoracic spinal cord transection. We demonstrate here that minocycline treatment prevents CST axonal dieback compared with saline-treated animals at both 7 and 14 d after injury. Interestingly, methylprednisolone treatment reduced vestibulospinal tract dieback but failed to reduce corticospinal tract dieback (Oudega et al., 1999), suggesting that minocycline may be more effective in preventing proximal corticospinal tract axonal degeneration after SCI. We also report reduced ED1 density within the proximal CST stump in minocycline-treated animals. Inhibition of the microglial/macrophage response may have prevented axonal dieback by preventing phagocytosis of axonal ends or reducing the liberation of cytotoxic or axonal repellant molecules associated with their activation. Furthermore, minocycline has been shown to reduce macrophage production of nitric oxide (Amin et al., 1997), a molecule that may be involved in axonal dieback (He et al., 2002). However, we cannot rule out a direct effect of the drug on axonal dieback. Minocycline has been shown to be a strong chelator and may sequester excess Ca2+ released after injury (Chopra and Roberts, 2001). Reduced axonal Ca2+ levels may prevent activation of calpains and may preserve axonal integrity (Balentine and Spector, 1977; George et al., 1995). Future mechanistic studies are needed to elucidate the role of minocycline treatment in preventing axonal degeneration.
Previously, it has been shown that minocycline treatment improved functional outcome after SCI (Lee et al., 2003; Wells et al., 2003). Furthermore, the murine study showed that minocycline was more effective in improving functional outcome when compared with methylprednisolone, the current treatment option to patients with SCI (Wells et al., 2003). We extend these results to show that minocycline improved interlimb coordination and reduced both hindpaw angle of rotation and toe spread after SCI.
From the results of the present study, we conclude that the positive effects of minocycline on the improved functional outcome after SCI result from its multifaceted impact on the secondary degenerative response. The neuroprotective properties of minocycline combined with its proven safety record in humans and animals suggest its efficacy as an effective treatment option after human SCI and justifies further examination.
Footnotes
This research was funded by a grant from the British Columbia Neurotrauma Fund (BCNTF). D.P.S. and K.K. were funded by studentships provided by the BCNTF.
Correspondence should be addressed to Dr. Wolfram Tetzlaff, International Collaboration On Repair Discoveries, 6270 University Boulevard, Vancouver, British Columbia, Canada V6T 1Z4. E-mail: tetzlaff@icord.org.
Copyright © 2004 Society for Neuroscience 0270-6474/04/242182-09$15.00/0
References
- Abe Y, Yamamoto T, Sugiyama Y, Watanabe T, Saito N, Kayama H, Kumagai T (1999) Apoptotic cells associated with Wallerian degeneration after experimental spinal cord injury: a possible mechanism of oligodendroglial death. J Neurotrauma 16: 945-952. [DOI] [PubMed] [Google Scholar]
- Agrawal SK, Fehlings MG (1997) Role of NMDA and non-NMDA ionotropic glutamate receptors in traumatic spinal cord axonal injury. J Neurosci 17: 1055-1063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amin AR, Patel RN, Thakker GD, Lowenstein CJ, Attur MG, Abramson SB (1997) Post-transcriptional regulation of inducible nitric oxide synthase mRNA in murine macrophages by doxycycline and chemically modified tetracyclines. FEBS Lett 410: 259-264. [DOI] [PubMed] [Google Scholar]
- Balentine JD, Spector M (1977) Calcification of axons in experimental spinal cord trauma. Ann Neurol 2: 520-523. [DOI] [PubMed] [Google Scholar]
- Beattie MS, Harrington AW, Lee R, Kim JY, Boyce SL, Longo FM, Bresnahan JC, Hempstead BL, Yoon SO (2002) ProNGF induces p75-mediated death of oligodendrocytes following spinal cord injury. Neuron 36: 375-386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bethea JR (2000) Spinal cord injury-induced inflammation: a dual-edged sword. Prog Brain Res 128: 33-42. [DOI] [PubMed] [Google Scholar]
- Bethea JR, Nagashima H, Acosta MC, Briceno C, Gomez F, Marcillo AE, Loor K, Green J, Dietrich WD (1999) Systemically administered interleukin-10 reduces tumor necrosis factor-alpha production and significantly improves functional recovery following traumatic spinal cord injury in rats. J Neurotrauma 16: 851-863. [DOI] [PubMed] [Google Scholar]
- Blight AR (1985) Delayed demyelination and macrophage invasion: a candidate for secondary cell damage in spinal cord injury. Cent Nerv Syst Trauma 2: 299-315. [DOI] [PubMed] [Google Scholar]
- Blight AR (1992) Macrophages and inflammatory damage in spinal cord injury. J Neurotrauma 9 [Suppl 1]: S83-S91. [PubMed] [Google Scholar]
- Blight AR (1994) Effects of silica on the outcome from experimental spinal cord injury: implication of macrophages in secondary tissue damage. Neuroscience 60: 263-273. [DOI] [PubMed] [Google Scholar]
- Blight AR, Decrescito V (1986) Morphometric analysis of experimental spinal cord injury in the cat: the relation of injury intensity to survival of myelinated axons. Neuroscience 19: 321-341. [DOI] [PubMed] [Google Scholar]
- Blight AR, Toombs JP, Bauer MS, Widmer WR (1991) The effects of 4-aminopyridine on neurological deficits in chronic cases of traumatic spinal cord injury in dogs: a phase I clinical trial. J Neurotrauma 8: 103-119. [DOI] [PubMed] [Google Scholar]
- Brown CJ, Mackinnon SE, Evans PJ, Bain JR, Makino AP, Hunter DA, Hare GM (1989) Self-evaluation of walking-track measurement using a sciatic function index. Microsurgery 10: 226-235. [DOI] [PubMed] [Google Scholar]
- Brundula V, Rewcastle NB, Metz LM, Bernard CC, Yong VW (2002) Targeting leukocyte MMPs and transmigration: minocycline as a potential therapy for multiple sclerosis. Brain 125: 1297-1308. [DOI] [PubMed] [Google Scholar]
- Bunge RP, Puckett WR, Becerra JL, Marcillo A, Quencer RM (1993) Observations on the pathology of human spinal cord injury: a review and classification of 22 new cases with details from a case of chronic cord compression with extensive focal demyelination. Adv Neurol 59: 75-89. [PubMed] [Google Scholar]
- Casha S, Yu WR, Fehlings MG (2001) Oligodendroglial apoptosis occurs along degenerating axons and is associated with FAS and p75 expression following spinal cord injury in the rat. Neuroscience 103: 203-218. [DOI] [PubMed] [Google Scholar]
- Chen M, Ona VO, Li M, Ferrante RJ, Fink KB, Zhu S, Bian J, Guo L, Farrell LA, Hersch SM, Hobbs W, Vonsattel JP, Cha JH, Friedlander RM (2000) Minocycline inhibits caspase-1 and caspase-3 expression and delays mortality in a transgenic mouse model of Huntington disease. Nat Med 6: 797-801. [DOI] [PubMed] [Google Scholar]
- Choi DW (1992) Excitotoxic cell death. J Neurobiol 23: 1261-1276. [DOI] [PubMed] [Google Scholar]
- Chopra I, Roberts M (2001) Tetracycline antibiotics: mode of action, applications, molecular biology, and epidemiology of bacterial resistance. Microbiol Mol Biol Rev 65: 232-260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crowe MJ, Bresnahan JC, Shuman SL, Masters JN, Beattie MS (1997) Apoptosis and delayed degeneration after spinal cord injury in rats and monkeys. Nat Med 3: 73-76. [DOI] [PubMed] [Google Scholar]
- de Medinaceli L, Freed WJ, Wyatt RJ (1982) An index of the functional condition of rat sciatic nerve based on measurements made from walking tracks. Exp Neurol 77: 634-643. [DOI] [PubMed] [Google Scholar]
- Dong H, Fazzaro A, Xiang C, Korsmeyer SJ, Jacquin MF, McDonald JW (2003) Enhanced oligodendrocyte survival after spinal cord injury in bax-deficient mice and mice with delayed Wallerian degeneration. J Neurosci 23: 8682-8691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dumont RJ, Okonkwo DO, Verma S, Hurlbert RJ, Boulos PT, Ellegala DB, Dumont AS (2001) Acute spinal cord injury, part I: pathophysiologic mechanisms. Clin Neuropharmacol 24: 254-264. [DOI] [PubMed] [Google Scholar]
- Emery E, Aldana P, Bunge MB, Puckett W, Srinivasan A, Keane RW, Bethea J, Levi AD (1998) Apoptosis after traumatic human spinal cord injury. J Neurosurg 89: 911-920. [DOI] [PubMed] [Google Scholar]
- Gehrmann J, Banati RB (1995) Microglial turnover in the injured CNS: activated microglia undergo delayed DNA fragmentation following peripheral nerve injury. J Neuropathol Exp Neurol 54: 680-688. [DOI] [PubMed] [Google Scholar]
- George EB, Glass JD, Griffin JW (1995) Axotomy-induced axonal degeneration is mediated by calcium influx through ion-specific channels. J Neurosci 15: 6445-6452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giulian D, Robertson C (1990) Inhibition of mononuclear phagocytes reduces ischemic injury in the spinal cord. Ann Neurol 27: 33-42. [DOI] [PubMed] [Google Scholar]
- Guth L, Zhang Z, Steward O (1999) The unique histopathological responses of the injured spinal cord: implications for neuroprotective therapy. Ann NY Acad Sci 890: 366-384. [DOI] [PubMed] [Google Scholar]
- Hausmann ON (2003) Post-traumatic inflammation following spinal cord injury. Spinal Cord 41: 369-378. [DOI] [PubMed] [Google Scholar]
- He Y, Yu W, Baas PW (2002) Microtubule reconfiguration during axonal retraction induced by nitric oxide. J Neurosci 22: 5982-5991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inman D, Guth L, Steward O (2002) Genetic influences on the secondary degeneration and wound healing following spinal cord injury in various strains of mice. J Comp Neurol 451: 225-235. [DOI] [PubMed] [Google Scholar]
- Lee SM, Yune TY, Kim SJ, Park do W, Lee YK, Kim YC, Oh YJ, Markelonis GJ, Oh TH (2003) Minocycline reduces cell death and improves functional recovery after traumatic spinal cord injury in the rat. J Neurotrauma 10: 1017-1027. [DOI] [PubMed] [Google Scholar]
- Li G, Brodin G, Farooque M, Funa K, Holtz A, Wang W, Olsson Y (1996) Apoptosis and expression of Bcl-2 after compression trauma to rat spinal cord. J Exp Neuropathol Exp Neurol 55: 280-289. [DOI] [PubMed] [Google Scholar]
- Liu XZ, Xu XM, Hu R, Du C, Zhang SX, McDonald JW, Dong HX, Wu YJ, Fan GS, Jacquin MF, Hsu CY, Choi DW (1997) Neuronal and glial apoptosis after traumatic spinal cord injury. J Neurosci 17: 5395-5406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McBride CB, McPhail LT, Vanderluit JL, Tetzlaff W, Steeves JD (2003) Caspase inhibition attenuates transection-induced oligodendrocyte apoptosis in the developing chick spinal cord. Mol Cell Neurosci 23: 383-397. [DOI] [PubMed] [Google Scholar]
- McDonald JW, Sadowsky C (2002) Spinal-cord injury. Lancet 359: 417-425. [DOI] [PubMed] [Google Scholar]
- Oudega M, Vargas CG, Weber AB, Kleitman N, Bunge MB (1999) Long-term effects of methylprednisolone following transection of adult rat spinal cord. Eur J Neurosci 11: 2453-2464. [DOI] [PubMed] [Google Scholar]
- Pallini R, Fernandez E, Sbriccoli A (1988) Retrograde degeneration of corticospinal axons following transection of the spinal cord in rats: a quantitative study with anterogradely transported horseradish peroxidase. J Neurosurg 68: 124-128. [DOI] [PubMed] [Google Scholar]
- Panter SS, Yum SW, Faden AI (1990) Alteration in extracellular amino acids after traumatic spinal cord injury. Ann Neurol 27: 96-99. [DOI] [PubMed] [Google Scholar]
- Popovich PG (2000) Immunological regulation of neuronal degeneration and regeneration in the injured spinal cord. Prog Brain Res 128: 43-58. [DOI] [PubMed] [Google Scholar]
- Popovich PG, Wei P, Stokes BT (1997) Cellular inflammatory response after spinal cord injury in Sprague-Dawley and Lewis rats. J Comp Neurol 377: 443-464. [DOI] [PubMed] [Google Scholar]
- Popovich PG, Guan Z, Wei P, Huitinga I, van Rooijen N, Stokes BT (1999) Depletion of hematogenous macrophages promotes partial hindlimb recovery and neuroanatomical repair after experimental spinal cord injury. Exp Neurol 158: 351-365. [DOI] [PubMed] [Google Scholar]
- Popovich PG, Guan Z, McGaughy V, Fisher L, Hickey WF, Basso DM (2002) The neuropathological and behavioral consequences of intraspinal microglial/macrophage activation. J Neuropathol Exp Neurol 61: 623-633. [DOI] [PubMed] [Google Scholar]
- Power C, Henry S, Del Bigio MR, Larsen PH, Corbett D, Imai Y, Yong VW, Peeling J (2003) Intracerebral hemorrhage induces macrophage activation and matrix metalloproteinases. Ann Neurol 53: 731-742. [DOI] [PubMed] [Google Scholar]
- Sekhon LH, Fehlings MG (2001) Epidemiology, demographics, and pathophysiology of acute spinal cord injury. Spine 26: S2-12. [DOI] [PubMed] [Google Scholar]
- Shuman SL, Bresnahan JC, Beattie MS (1997) Apoptosis of microglia and oligodendrocytes after spinal cord contusion in rats. J Neurosci Res 50: 798-808. [DOI] [PubMed] [Google Scholar]
- Springer JE, Azbill RD, Knapp PE (1999) Activation of the caspase-3 apoptotic cascade in traumatic spinal cord injury. Nat Med 5: 943-946. [DOI] [PubMed] [Google Scholar]
- Takami T, Oudega M, Bethea JR, Wood PM, Kleitman N, Bunge MB (2002) Methylprednisolone and interleukin-10 reduce gray matter damage in the contused Fischer rat thoracic spinal cord but do not improve functional outcome. J Neurotrauma 19: 653-666. [DOI] [PubMed] [Google Scholar]
- Tikka TM, Koistinaho JE (2001) Minocycline provides neuroprotection against N-methyl-d-aspartate neurotoxicity by inhibiting microglia. J Immunol 166: 7527-7533. [DOI] [PubMed] [Google Scholar]
- Tikka T, Fiebich BL, Goldsteins G, Keinanen R, Koistinaho J (2001) Minocycline, a tetracycline derivative, is neuroprotective against excitotoxicity by inhibiting activation and proliferation of microglia. J Neurosci 21: 2580-2588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Zhu S, Drozda M, Zhang W, Stavrovskaya IG, Cattaneo E, Ferrante RJ, Kristal BS, Friedlander RM (2003) Minocycline inhibits caspase-independent and -dependent mitochondrial cell death pathways in models of Huntington's disease. Proc Natl Acad Sci USA 100: 10483-10487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warden P, Bamber NI, Li H, Esposito A, Ahmad KA, Hsu CY, Xu XM (2001) Delayed glial cell death following wallerian degeneration in white matter tracts after spinal cord dorsal column cordotomy in adult rats. Exp Neurol 168: 213-224. [DOI] [PubMed] [Google Scholar]
- Waxman SG (1993) Aminopyridines and the treatment of spinal cord injury. J Neurotrauma 10: 19-24. [DOI] [PubMed] [Google Scholar]
- Wells JE, Hurlbert RJ, Fehlings MG, Yong VW (2003) Neuroprotection by minocycline facilitates significant recovery from spinal cord injury in mice. Brain 126: 1628-1637. [DOI] [PubMed] [Google Scholar]
- Werner P, Pitt D, Raine CS (2000) Glutamate excitotoxicity: a mechanism for axonal damage and oligodendrocyte death in multiple sclerosis? J Neural Transm [Suppl] 60: 375-385. [DOI] [PubMed] [Google Scholar]
- Wrathall JR, Choiniere D, Teng YD (1994) Dose-dependent reduction of tissue loss and functional impairment after spinal cord trauma with the AMPA/kainate antagonist NBQX. J Neurosci 14: 6598-6607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yong C, Arnold PM, Zoubine MN, Citron BA, Watanabe I, Berman NE, Festoff BW (1998) Apoptosis in cellular compartments of rat spinal cord after severe contusion injury. J Neurotrauma 15: 459-472. [DOI] [PubMed] [Google Scholar]
- Young W, Flamm ES (1982) Effect of high-dose corticosteroid therapy on blood flow, evoked potentials, and extracellular calcium in experimental spinal injury. J Neurosurg 57: 667-673. [DOI] [PubMed] [Google Scholar]
- Yrjanheikki J, Keinanen R, Pellikka M, Hokfelt T, Koistinaho J (1998) Tetracyclines inhibit microglial activation and are neuroprotective in global brain ischemia. Proc Natl Acad Sci USA 95: 15769-15774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yrjanheikki J, Tikka T, Keinanen R., Goldsteins G, Chan PH, Koistinaho J (1999) A tetracycline derivative, minocycline, reduces inflammation and protects against focal cerebral ischemia with a wide therapeutic window. Proc Natl Acad Sci USA 96: 13496-13500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu S, Stavrovskaya IG, Drozda M, Kim BYS, Ona V, Li M, Sarang S, Liu AS, Hartley DM, Wu DC, Gullans S, Ferrante RJ, Przedborski S, Kristal BS, Friedlander RM (2002) Minocycline inhibits cytochrome c release and delays progression of amyotrophic lateral sclerosis in mice. Nature 417: 74-78. [DOI] [PubMed] [Google Scholar]