

Abstract
Reactive astrocytes are prominent in the cellular response to spinal cord injury (SCI), but their roles are not well understood. We used a transgenic mouse model to study the consequences of selective and conditional ablation of reactive astrocytes after stab or crush SCI. Mice expressing a glial fibrillary acid protein-herpes simplex virus-thymidine kinase transgene were given mild or moderate SCI and treated with the antiviral agent ganciclovir (GCV) to ablate dividing, reactive, transgene-expressing astrocytes in the immediate vicinity of the SCI. Small stab injuries in control mice caused little tissue disruption, little demyelination, no obvious neuronal death, and mild, reversible functional impairments. Equivalent small stab injuries in transgenic mice given GCV to ablate reactive astrocytes caused failure of blood-brain barrier repair, leukocyte infiltration, local tissue disruption, severe demyelination, neuronal and oligodendrocyte death, and pronounced motor deficits. Moderate crush injuries in control mice caused focal tissue disruption and cellular degeneration, with moderate, primarily reversible motor impairments. Equivalent moderate crush injuries combined with ablation of reactive astrocytes caused widespread tissue disruption, pronounced cellular degeneration, and failure of wound contraction, with severe persisting motor deficits. These findings show that reactive astrocytes provide essential activities that protect tissue and preserve function after mild or moderate SCI. In nontransgenic animals, crush or contusion SCIs routinely exhibit regions of degenerated tissue that are devoid of astrocytes. Our findings suggest that identifying ways to preserve reactive astrocytes, to augment their protective functions, or both, may lead to novel approaches to reducing secondary tissue degeneration and improving functional outcome after SCI.
Keywords: reactive astrocytes, spinal cord injury, inflammation, transgenic mice, neural degeneration, motor function, blood-brain barrier, glial fibrillary acid protein
Introduction
Spinal cord injury (SCI) is characteristically accompanied by a period of secondary cellular degeneration that occurs in injured tissue over a course of hours and days after the initial insult, and that affects both glia and neurons (Young, 1993; Crowe et al., 1997; Liu et al., 1997; Springer et al., 1999). Intervention to reduce inflammation or to block specific membrane receptors during the first hours after SCI can partially protect cells and preserve function in both human patients and experimental animals (Bracken et al., 1990; Behrmann et al., 1994; Constantini and Young, 1994; Agrawal and Fehlings, 1997). Although certain potential triggers of secondary degeneration have been identified, the cell biology of the response to SCI is not well understood, and the roles played by different cell types at different times in the progression of secondary degeneration are not well defined.
Reactive astrocytes are a prominent feature of the cellular response to SCI. Astrocytes exhibit a gradated response to injury that includes changes in gene expression, hypertrophy and process extension, and, in some cases, cell division (Eddleston and Mucke, 1993; Eng and Ghirnikar, 1994). Scar tissue formed in part by reactive astrocytes has long been implicated as a major impediment to axon regeneration (Ramon y Cajal, 1928; Clemente and Windle, 1954; Reier et al., 1983; Liuzzi and Lasek, 1987; Rudge and Silver, 1990), and reactive astrocytes are often regarded as detrimental to functional outcome after SCI and other forms of CNS injury. Nevertheless, the basic phenomena of reactive astrocytosis after CNS injury appear conserved throughout vertebrate evolution (Larner et al., 1995), suggesting that fundamental aspects of the process of reactive astrocytosis convey survival advantage. In this regard, a role for reactive astrocytes has long been postulated in CNS wound healing, but the precise nature and relative importance of that role are unknown. Several lines of evidence suggest that astrocytes might have important tissue-protective functions after CNS injury. Astrocytes clear glutamate and potassium ions from the extracellular space, are a potential energy source, and produce numerous growth factors and cytokines (Kettenmann and Ransom, 1995). The continuation, augmentation, and appropriate regulation of astrocyte functions are likely to be important after SCI, but the activities of reactive astrocytes are not well defined.
Deletion experiments represent powerful means of studying function in biology. To investigate the roles of reactive astrocytes after SCI, we selectively and conditionally targeted their deletion in adult transgenic mice after SCI, using a well characterized transgenic model (Bush et al., 1998, 1999). We show that that the severity of SCI is markedly exacerbated by loss of proliferating reactive astrocytes, and that reactive astrocytes (1) are essential for wound healing, (2) provide essential protective functions for neurons and oligodendrocytes, and (3) preserve motor functions after mild or moderate SCI.
Materials and Methods
Experimental model
To selectively ablate dividing reactive astrocytes, we used transgenic mice in which thymidine kinase (TK) from herpes simplex virus (HSV) was targeted to reactive astrocytes using the mouse glial fibrillary acid protein (GFAP) promoter (Bush et al., 1998). In these GFAP-TK mice, the antiviral agent ganciclovir (GCV) selectively and specifically ablates dividing, reactive GFAP-TK-expressing astrocytes after CNS injury (Bush et al., 1999). These GFAP-TK mice were generated using a fusion gene construct of HSV-TK sequence inserted into a 15 kb GFAP promoter cassette (Bush et al., 1998). To avoid deletion of potential regulatory elements, this GFAP promoter cassette (clone 445) contains the full sequence of the mouse GFAP gene, including all introns and exons, as well as 2 kb of 5′ and 2.5 kb of 3′ flanking regions (Johnson et al., 1995). Two base pairs in exon 1 were mutated to prevent GFAP expression from the transgene. Expression of transgene-derived TK specifically by GFAP-expressing cells has been demonstrated at the single-cell level in the normal and injured forebrain in vivo and in vitro (Bush et al., 1998, 1999; Imura et al., 2003).
Animals
All GFAP-TK transgenic and nontransgenic mice used in this study were obtained by mating heterozygous GFAP-TK females of line 7.1 (Bush et al., 1998, 1999) with wild-type C57Bl6 males. Thus, all experimental and control mice were derived from the same breeding colony backcrossed onto a C57Bl6 background for >10 generations and can be regarded as genetically inbred mice of the C57Bl6 strain. Mice were housed in a 12 hr light/dark cycle in a specific pathogen-free facility with controlled temperature and humidity and allowed ad libitum access to food and water, and experiments were conducted according to protocols approved by the Chancellor's Animal Research Committee of the Office for Protection of Research Subjects at University of California Los Angeles.
Experimental design
Our previous studies indicated that 7 d of continuous subcutaneous GCV delivery was effective in ablating dividing reactive astrocytes immediately adjacent to forebrain stab injury (Bush et al., 1999), and this delivery regimen was used in the present study. All experiments involved comparison of groups of GFAP-TK transgenic and nontransgenic mice that received either a stab or crush SCI (Fig. 1 B,C) followed immediately by a 7 d treatment with either GCV or vehicle (Fig. 1 A). Some mice also received a 4 d Bromodeoxyuridine (BrdU) treatment to label dividing cells, and all mice were perfused for histological evaluation after 14 d (Fig. 1 A). Behavioral evaluations of motor function were conducted before surgery and at various points afterward (Fig. 1 A). Fourteen days was chosen for histological evaluation as a point by which control mice had mostly recovered motor function after mild or moderate SCI.
Figure 1.
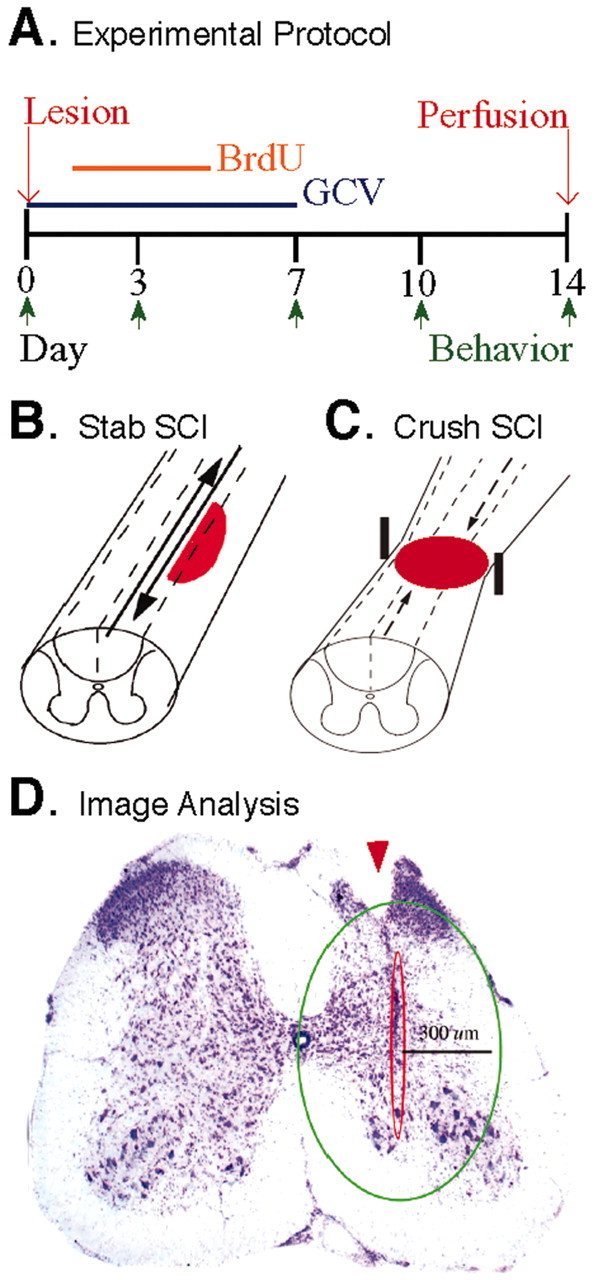
A, Experimental protocol showing time lines of postoperative delivery of GCV and BrdU and time points of behavioral analysis and perfusion fixation. B, C, Schematic illustrations of different SCI models at L1-L2. B, Longitudinal stab SCI was made primarily through gray matter parallel to major ascending and descending long axon tracts. C, Moderate crush SCI was made by compression from both lateral sides using forceps with a 0.5 mm spacer and affected gray matter as well as major ascending and descending long axon tracts. D, Photomicrograph showing tissue area adjacent to stab SCI (arrowhead) used for morphometric analysis as traced using interactive software and a computer-guided microscope stage. Scale bar, 300 μm.
Surgical procedures
All surgical procedures were performed under full general anesthesia with isoflurane in oxygen-enriched air. Two types of SCI were made: (1) unilateral longitudinal stab injuries that ran parallel to the long ascending and descending tracts and passed primarily through gray matter at the level of L1-L2 (Fig. 1 B) and (2) moderate crush injuries at the level of L1-L2 (Fig. 1C). All surgeries were performed using an operating microscope (Zeiss, Oberkochen, Germany) and a rodent stereotaxic apparatus (David Kopf, Tujunga, CA). Laminectomy of a single vertebra was performed in all cases. The longitudinal stab injuries were 2 mm in length and 1 mm in depth and were made with a retractable wire knife (David Kopf); care was taken not to rupture the dura (except for the small puncture wound created by the retractable wire knife), thereby greatly reducing the variability of lesion. Crush injuries were made with number 5 Dumont forceps (Fine Science Tools, Foster City, CA) ground down to a tip width of 0.5 mm. For the moderately severe injuries reported here, we used forceps modified with a spacer so that at maximal closure a 0.5 mm space remained. This closure distance was selected by comparing results achieved with forceps that closed to 1.0 or 0 mm spaces. Forceps were used to compress the cord laterally from both sides (Fig. 1C) for 10 sec. To deliver GCV, osmotic minipumps (Alzet, Cupertino, CA) were implanted subcutaneously according to the manufacturer's instructions. To evaluate the blood-brain barrier (Broadwell and Sofroniew, 1993), a macromolecular dextran tracer, 70 kDa, labeled with Alexa Fluor 488 (Molecular Probes, Eugene, OR) was infused intravenously via the subclavian vein exposed under general anesthesia and cannulated with a glass micropipette using an operating microscope; 45 min after infusion, mice were fixed and processed for histological evaluation. To selectively kill spinal cord neurons, the excitotoxic glutamate agonist NMDA (25 nmol in 1.0 μl; Sigma, St. Louis, MO), was stereotaxically injected into spinal cord gray matter at L1-L2. To attract infiltration of blood-borne inflammatory cells, lipopolysaccharide (LPS, serotype 011-B4 from Escherichia coli; Sigma; 8 μg in 2 μl), was stereotaxically injected into spinal cord gray matter at L1-L2.
GCV and BrdU treatments
GCV (Hoffmann-La Roche, Nutley, NJ), was administered continuously at a rate of 100 mg · kg -1 · d -1 in sterile physiological saline for the first 7 d (Fig. 1A) after surgery via subcutaneously implanted osmotic minipumps (Alzet) (Bush et al., 1998, 1999). BrdU (Sigma) was administered by intraperitoneal injections at 100 mg/kg dissolved in saline plus 0.007N NaOH given once daily for 4 d on days 1-4 after surgery (Fig. 1A).
Behavioral analysis
Mice were evaluated using three behavioral tasks selected to assess hindlimb function on days 0, 3, 7, 10, and 14 after SCI (Fig. 1 A).
Open-field locomotor task. The objective of this evaluation was to assess gross voluntary use of the hindlimbs and did not attempt to define subtle differences in usage that might be correlated with specific neural mechanisms that might underlie dysfunctions. For this reason we did not use a 21-point Basso, Beattie, and Bresnahan scale designed to evaluate many subtle aspects of hindlimb motor performance in rats to delineate differences in severity of midthoracic SCI (Basso et al., 1995). Instead, we used a simpler six-point scale (Fehlings and Tator, 1995), which focused on gross aspects of hindlimb function and which we found more appropriate for our goals and our model of upper lumbar SCI. Mice were evaluated in an open field by the same one or two observers blind to the experimental condition and received a score for gross voluntary movement of each hindlimb using an operationally defined six-point scale: (0) no voluntary hindlimb movement, (1) little voluntary hindlimb movement, (2) hindlimb movements obvious but did not assist in weight support or stepping, (3) hindlimb assisted in occasional weight support and plantar placement but not in stepping, (4) hindlimb used for weight support and stepping, but obvious disability, and (5) hindlimb function essentially normal (Fehlings and Tator, 1995). Left and right hindlimbs were scored separately. For mice with unilateral stab SCI, scores are presented for the ipsilateral hindlimb. For mice with crush SCI the two hindlimb scores were averaged.
Rotorod. To assess balance and ability to coordinate stepping, animals were placed on a single-lane rotorod (Med Associates, St. Albans, VT) for three trials per session. The rotorod was set for constant acceleration from 3.0 to 30 rpm over 300 sec, and animals were scored on seconds to fall. Each trial was scored individually and averaged for a final score per session.
Footprint analysis. To assess stepping patterns of forelimbs and hindlimbs after injury, animals were required to run along a paper-lined runway (3 feet long, 3 inches wide) to obtain an edible treat in a darkened box at the end. The plantar surfaces of forelimbs and hindlimbs were brushed with red and black nontoxic paint, respectively, and qualitative analysis was performed on plantar stepping, stride length and width, toe or dorsal foot drags, and overall stepping ability (Ma et al., 2001).
Histological procedures
After terminal anesthesia by barbiturate overdose, mice were perfused transcardially with buffered 4% paraformaldehyde. Spinal cords were removed, postfixed for a further 2 hr, and cryoprotected in buffered 30% sucrose overnight. Forty micrometer transverse or 30 μm horizontal frozen sections were prepared using a cryostat microtome (Leica, Nussloch, Germany). Bright-field immunohistochemistry was performed using biotinylated secondary antibodies (Vector Laboratories, Burlingame, CA), biotin-avidin-peroxidase complex (Vector Laboratories), and diaminobenzidine (Sigma) as the developing agent. Fluorescence immunohistochemistry was performed using Alexa Fluor tagged secondary antibodies Alexa 488 (green), Alexa 568 (red), and Alexa 350 (blue) (Molecular Probes). Primary antibodies were rabbit anti-HSV-TK (1:20,000; Bush et al., 1999), rabbit anti-GFAP (1:20,000; Dako, Carpinteria, CA), rabbit anti-mouse IgG (1:200,000; Dako), sheep anti-BrdU (1:6000; Maine Biotechnology Services, Portland, ME), rat anti-mouse CD-45 (1:2000; PharMingen, La Jolla, CA), rat anti-mouse CD-3 (1:1000; PharMingen), rabbit anti-fibronectin (1:5000; Dako), rabbit anti-S100β (1:4000; Sigma), rat anti-myelin basic protein (MBP, 1:10,000; Chemicon, Temecula, CA), and rabbit anti-neurofilament M (NFM, 1:10,000; Chemicon). Sections stained for BrdU were pretreated with 2 m HCl for 30 min and then neutralized with three rinses of PBS before addition of primary antibody. Staining with cresyl violet or Luxol fast blue was conducted according to standard procedures. Stained sections were examined and photographed using bright-field and fluorescence microscopy (Zeiss) and scanning confocal laser microscopy (Leica).
Morphometric and statistical evaluation
Cell counts of nerve cells stained with cresyl violet or of astrocytes or leukocytes stained by immunohistochemistry, as well as lesion volume estimates and three-dimensional lesion reconstruction, were performed using unbiased sampling (Gundersen et al., 1988) and stereological image analysis software (StereoInvestigator or NeuroLucida; MicroBrightField, Williston, VT) operating a computer-driven microscope regulated in the x-, y-, and z-axes (Zeiss). For neuronal or white blood cell counts, areas of specimens to be sampled were traced at low power as an oval region 300 μm around the center of a stab wound (Fig. 1 D) and through the depth of the tissue as determined by the computer software. Counting frames within the traced region were selected at random by the image analysis software (StereoInvestigator) and computer-driven stage. Oligodendrocyte counts were restricted to white matter. The number of positive cells per counting frame and tissue depth was determined according to exclusion criteria of the stereology program, and final counts were expressed as cells per 100 μm3. To estimate severity of demyelination, the area of intact and normally myelinated white matter was as identified by Luxol fast blue staining and traced in dorsal and lateral columns on the left and right sides. The total area of intact white matter on the injured side was expressed as a percentage of that on the uninjured side. Three-dimensional reconstructions of lesions were performed by tracing lesion areas and spinal cord outlines in series of sequentially spaced sections, after which lesions were reconstructed and volumes were estimated using image analysis software (NeuroLucida). Assessment of lesion area was conducted in cresyl violet-stained sections according to commonly accepted qualitative criteria (Zhang et al., 1996; Inman and Steward, 2003); degenerating tissue exhibited staining pallor, pyknotic nuclei, and cellular disorder, and intact parenchyma contained cells with clear nuclei and regular distribution of cells. All statistical evaluations were performed by ANOVA with post hoc independent pair-wise analysis (Prism; GraphPad, San Diego, CA).
Results
The effects of ablating dividing reactive astrocytes after SCI were studied in two models of experimental SCI at L1-L2: (1) a 2-mm-long and 1-mm-deep longitudinal stab injury (Fig. 1B) and (2) a moderate crush injury (Fig. 1C). Both injury models involved neuronal pools in upper lumbar gray matter. The longitudinal stab injury model was used to study the cell biology of penetrating SCI in vivo in a context that caused no grossly detectable behavioral impairments in control mice. Moderate crush injury was used as an SCI model that caused mild and mostly reversible motor impairments, in conjunction with a small lesion surrounded by reactive scar-forming astrocytes in control mice. Dividing reactive astrocytes were ablated by treating GFAP-TK transgenic mice with the antiviral agent GCV for the first 7 d after SCI (Fig. 1A). Previous studies, including quantitative analysis using double-labeling immunocytochemistry at the single-cell level, have demonstrated the precise and selective targeting of HSV-TK expression to GFAP-expressing cells in the forebrain and peripheral nervous system of the GFAP-TK mice of line 7.1 used here (Bush et al., 1998, 1999). Further characterization of the overlap of GFAP and HSV-TK expression specifically in spinal cord astrocytes is described below.
Conditional ablation of reactive, scar-forming astrocytes after SCI
We first validated our experimental model for studying SCI by determining the degree to which GFAP-expressing reactive astrocytes in the spinal cords of GFAP-TK transgenic mice exhibited (1) transgene-derived TK expression, (2) cell division, and (3) ablation after treatment with GCV. To do so, we examined GFAP-TK transgenic as well as nontransgenic mice 14 d after a longitudinal stab SCI. In control mice, this injury caused a robust reactive astrocytosis, including upregulation of GFAP expression, cellular hypertrophy, and division of astrocytes (Fig. 2A-H). Three groups of control mice were examined: (1) GFAP-TK transgenic mice not given GCV, (2) nontransgenic mice not given GCV, and (3) nontransgenic mice given GCV. Control mice were compared with GFAP-TK mice treated with GCV to ablate dividing reactive astrocytes.
Figure 2.
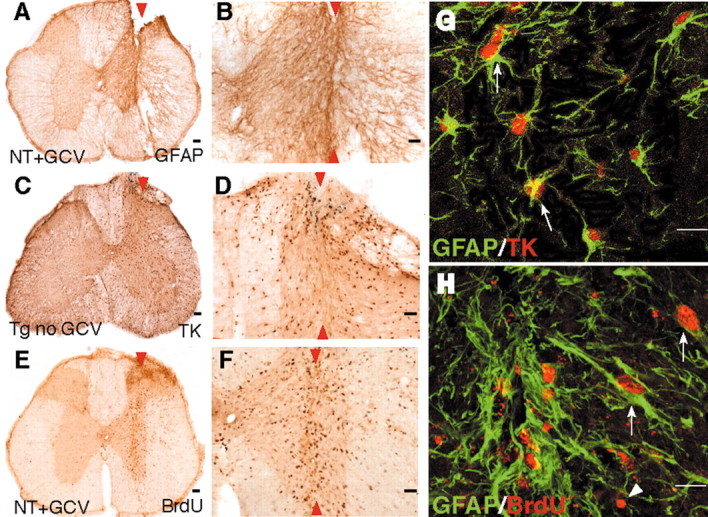
Expression of GFAP and transgene-derived TK by dividing reactive astrocytes adjacent to stab SCI in control mice. A-H, Transverse sections of upper lumbar spinal cord after longitudinal stab SCI (arrowheads) in either nontransgenic mice given GCV (A, B, E, F, H; NT+GCV) or GFAP-TK transgenic mice not given GCV (C, D, G; Tg no GCV). B, D, F, Details, respectively, of A, C, and E. A-F, Single immunohistochemical staining for GFAP (A, B), TK (C, D), or BrdU (E, F) viewed by bright-field microscopy. G, H, Double immunohistochemical staining for GFAP and TK (G) or GFAP and BrdU (H) viewed by laser-scanning confocal microscopy. Staining for GFAP is prominent in astrocyte processes and rare in cell bodies (A, B, G), whereas staining for TK is prominent in astrocyte cell bodies and rare in cell processes (C, D, G). After stab SCI, astrocytes along the wound margin hypertrophy and upregulate their expression of both GFAP (A, B, G) and TK (C, D, G). GCV had no effect on astrocyte scar formation in nontransgenic mice (A, B, E, F). Double staining and analysis using laser-scanning confocal microscopy reveal that TK staining is found only in GFAP-positive astrocytes (G, arrows). Many cells along the wound margin divide and take up BrdU (E, F). Many of these dividing BrdU-labeled cells are GFAP-positive reactive astrocytes (H, arrows); others are not (G, arrowheads). Scale bars: A, C, E, 100 μm; B, D, F, 50 μm; G, H, 15 μm.
In the stab-injured spinal cord of GFAP-TK mice not given GCV, we found that all TK-expressing cells also expressed GFAP, and that >98%, but not all, of GFAP-expressing reactive astrocytes also expressed detectable levels of transgene-derived TK (Fig. 2A-D,G). The inability to detect TK in all GFAP-expressing cells may be attributable to the discontinuous activity of the GFAP gene and differences in the intracellular half-life of the GFAP and TK proteins, as discussed elsewhere (Bush et al., 1999; Imura et al., 2003). We found no evidence for TK expression by cells that did not express GFAP.
To evaluate the degree of astrocyte cell division induced by SCI, we administered BrdU during the first 4 d after injury (Fig. 1A). At 14 d after injury, control mice exhibited many BrdU-labeled cells in tissue immediately adjacent to the stab wound (Fig. 2E,F); most of these BrdU-labeled cells expressed GFAP (Fig. 2H) and TK.
There was no qualitatively detectable difference in the response to SCI in all three control groups with regard to upregulation of GFAP expression, cellular hypertrophy, and numbers of dividing astrocytes (Fig. 2). In contrast, GCV treatment of GFAP-TK mice ablated essentially all GFAP-immunoreactive astrocytes immediately adjacent to the wound such that at 14 d, only a few viable astrocytes and scattered cell fragments remained (Fig. 3B). As previously observed in the forebrain (Bush et al., 1999), the pattern of immunohistochemical staining at 14 d after SCI suggested that the death of astrocytes that occurred during the 7 d of GCV treatment served as a trigger for astrocytosis in the immediately surrounding tissue during the next 7 d after GCV delivery had stopped, resulting in the formation of a surrounding rim of GFAP-positive scar-forming astrocytes at some distance away from the original wound (Fig. 3B). Cell counts showed a statistically significant >95% decline in the number of TK-immunoreactive astrocytes in the immediate vicinity of the SCI (Fig. 3D,H). Our qualitative and quantitative evaluations of immunohistochemically stained cells did not distinguish between dividing and nondividing astrocytes. Thus, a >95% reduction in the number of counted cells indicated a massive reduction in the total number of reactive astrocytes within the tissue area evaluated immediately next to the wound. This observation suggests that the process of transgene-induced astrocyte death stimulated adjacent quiescent astrocytes to divide, which in turn would cause these cells to be ablated, leading to the observed almost complete depletion of reactive astrocytes in the immediate wound vicinity (Fig. 3B,D,H). A small number of BrdU-positive cells that were GFAP- or TK-negative remained (Fig. 3G), and most of these cells were found to express CD45 and appeared to be proliferating microglia/macrophages or other inflammatory cells (Fig. 3I,J). The area immediately adjacent to the wound, from which astrocytes were almost entirely ablated (Fig. 3B,D), was studied further.
Figure 3.

Transgenically targeted ablation of reactive scar-forming astrocytes adjacent to stab SCI. A-G, J, I, Transverse sections of upper lumbar spinal cord after longitudinal stab SCI (arrowheads) in nontransgenic mice given GCV (A, E; NT+GCV), a GFAP-TK transgenic mouse not given GCV (C; Tg no GCV), or GFAP-TK transgenic mice given GCV (B, D, F, G, J, I; Tg+GCV). A-F, Single immunohistochemical staining for GFAP (A, B), TK (C, D), or BrdU (E, F) viewed by bright-field microscopy. G, I, J, Double immunohistochemical staining for GFAP and BrdU (G) or CD45 and BrdU (I, J) viewed by laser-scanning confocal microscopy. After stab SCI in control mice (A, C, E), astrocytes along the wound margin hypertrophy, upregulate their expression of GFAP (A) and TK (C), and take up BrdU. After stab SCI in transgenic mice given GCV for 7 d (B, D, F), GFAP- and TK-expressing astrocytes (B, D) and BrdU-labeled dividing cells (F) are substantially depleted from a large area around the center of the stab wound. Within this area, only GFAP-positive fragments of astrocytes remain (B, G, arrows). Astrocytes at the borders of this area have upregulated GFAP or TK during the 7 d after GCV delivery (B, D). Some dividing cells are present in the area depleted of astrocytes (F); all of these cells were GFAP-negative (G, arrowheads), and most were positive for the inflammatory cell marker CD45 (I, J). H, Graph showing mean ± SEM number of TK-positive cells within the 300 μm wound margin adjacent to the stab injury (see Fig. 1 D) in GFAP-TK transgenic mice not given GCV (Tg-GCV) or given GCV (Tg+GCV); n = 4 per group. **Significantly different from control, p < 0.01 (t test). Scale bars: A-F, 100 μm; G, 15 μm; J, 10 μm; I, 5 μm.
Together, these findings indicate that transgene-derived HSV-TK is regulated similarly to endogenous GFAP after SCI in these GFAP-TK mice, and essentially all reactive astrocytes, but not other cell types, express transgene-derived TK after SCI. In addition, these findings demonstrate the presence of dividing GFAP-positive astrocytes after stab SCI and show that GCV treatment in GFAP-TK transgenic mice kills essentially all reactive astrocytes in the immediate vicinity of stab SCI.
Failure of blood-brain barrier repair after loss of reactive astrocytes
After penetrating injury to the CNS, the BBB is damaged and becomes leaky to endogenous and exogenous blood-borne macromolecules. In rodents, this leakiness is repaired within 10-14 d (Bush et al., 1999). Various lines of evidence suggest that astrocytes contribute to establishing and maintaining the BBB. To evaluate BBB function after SCI, we examined the entry into spinal cord parenchyma of serum IgG and an intravenously injected tagged macromolecule (70 kDa dextran-Alexa 488) as examples of endogenous and exogenous macromolecules normally excluded from brain parenchyma except at sites that do not exhibit a BBB (Broadwell and Sofroniew, 1993). At 14 d after longitudinal stab SCI in control mice, there was little or no entry of IgG or 70 kDa dextran at the wound site, indicating that the BBB had resealed in the presence of reactive astrocytes (Figs. 4A,C). In contrast, after similar SCI in GCV-treated GFAP-TK mice, there was pronounced entry of IgG or 70 kDa dextran into parenchyma adjacent to wound margins (Fig. 4B,D), indicating that the BBB had failed to repair in the absence of reactive astrocytes.
Figure 4.

Failure of blood-brain barrier repair after ablation of reactive astrocytes adjacent to stab SCI. A-D, Transverse sections of upper lumbar spinal cord after longitudinal stab SCI (arrowheads) in GFAP-TK transgenic mice not given GCV (A, C; Tg no GCV) or GFAP-TK transgenic mice given GCV (B, D; Tg+GCV). A, B, Immunohistochemical staining for IgG viewed by bright-field microscopy. C, D, Fluorescence microscopy of Alexa 488-tagged dextran (molecular weight, 70,000). At 14 d after stab SCI in control mice, IgG and dextran are excluded from parenchyma adjacent to the injury, indicating that the blood-brain barrier has repaired as expected (A, C); similar results were obtained in n = 5 mice. At 14 d after stab SCI in GFAP-TK mice given GCV, IgG and dextran continue to enter neural parenchyma, indicating that the blood-brain barrier has failed to repair in the absence of reactive astrocytes (B,D); similar results were obtained in n = 5 mice. Scale bars, 100 μm.
Increased inflammation after loss of reactive astrocytes
Astrocytes produce various pro- or anti-inflammatory chemokines, cytokines, and growth factors that are thought to participate in the regulation of inflammation. Nevertheless, the roles of astrocytes in attracting or restricting the entry into the CNS of inflammatory cells at different times after CNS injury are not understood. To determine the effects of astrocyte ablation on inflammation after SCI, we counted the number of CD45-positive inflammatory cells. At 14 d after longitudinal stab injury in the three groups of control mice, the immediate wound area contained many CD45-positive activated microglia, but there were few phagocytic macrophages (Figs. 5A,B,E). In contrast, GFAP-TK mice given GCV exhibited a sevenfold greater number of large, rounded CD45-postive inflammatory cells in tissue adjacent to the SCI after ablation of astrocytes (Fig. 5C,D,F,G,L). Most of these cells had the appearance of activated, phagocytic macrophages (Fig. 5F,L), and some stained positively for the lymphocyte marker CD3 (data not shown). To examine whether this pronounced increase in inflammatory cells might merely be attributable to the presence of cellular debris after ablation of astroglia, we injected low doses of NMDA into the spinal cord gray matter at L1-L2 in nontransgenic mice. As expected, NMDA injections killed many neurons but also triggered pronounced local reactive astrocytosis. In the presence of large numbers of reactive astrocytes, there was only a moderate increase in the number of large, round CD45-positive inflammatory cells despite pronounced local cell death (Fig. 5H-K). We conclude that the presence of dead cells and cellular debris alone (Fig. 5I-K) cannot account for the massive increase in inflammatory cells observed after astrocyte ablation (Figs. 5C,D,L). Taken together, these findings show that loss of reactive astrocytes after SCI leads to markedly increased and prolonged infiltration of inflammatory cells, in particular macrophages.
Figure 5.

Increased leukocyte infiltration after ablation of reactive astrocytes adjacent to stab SCI. A-F, L, Transverse sections of upper lumbar spinal cord 14 d after longitudinal stab SCI (arrowheads) in a nontransgenic mouse not given GCV (A, B, E; NT no GCV) or in a GFAP-TK transgenic mouse given GCV (C, D, F, L; Tg+GCV). A-F, Immunohistochemical staining for the leukocyte marker CD45 viewed by bright-field microscopy. B, D, Details, respectively, of boxed areas in A and C. E, F, Details, respectively, of B and D showing CD45-positive cells with the typical appearance of microglia (E) and activated macrophages (F). In the control mouse, there is moderate microglial activation (arrow) but few intensely stained CD45-positive cells with round cell bodies typical of leukocytes in the neural parenchyma adjacent to the injury (A, B, E). In the GFAP-TK transgenic mouse given GCV, the region of astrocyte ablation is densely packed with many intensely stained CD45-positive cells with rounded cell bodies typical of leukocytes, in particular, activated macrophages (C, D, F). G, Mean cell number ± SEM of CD45-immunoreactive cells within the wound margin adjacent to the stab injury (see Fig. 1D); GFAP-TK mice given GCV exhibit a sevenfold greater number compared with control mice; n = 5 per group. ***Significantly different from control, p < 0.001 (ANOVA). H-K, Transverse sections of upper lumbar ventral horn 14 d after injection of vehicle (H) or NMDA (I-K). Cresyl violet staining (H, I) reveals pronounced loss of ventral horn neurons induced by NMDA (I). Immunohistochemical staining for CD45 in an adjacent tissue section reveals modest inflammatory response in ventral horn after NMDA (J). K, L, Laser-scanning confocal microscopy of triple staining for astrocytes (GFAP), inflammatory cells (CD45), and neurons (NeuN) to compare the inflammatory response after injection of NMDA (K) or ablation of astrocytes in GFAP-TK transgenic mice treated with GCV (L). Both experimental conditions cause severe loss of NeuN-positive neurons (K, L). CD45-positive inflammatory cells exhibit a mild inflammatory response after NMDA, which induces pronounced GFAP-positive astrogliosis (K). In contrast, CD45-positive inflammatory cells exhibit a pronounced inflammatory response in the absence of reactive astrocytes (L). Scale bars: A, C, 100 μm; B, D, 50 μm; E, F, 5 μm; H-J, 50 μm; K, L, 15 μm.
Pronounced neuronal degeneration after loss of reactive astrocytes
Astrocytes perform a number of functions that may be important for neuronal health and survival, such as uptake of glutamate and potassium, or production of growth factors (Kettenmann and Ransom, 1995). We therefore examined the effects of astrocyte ablation after longitudinal stab SCI on local neurons in the L1-L2 gray matter. In all three groups of control mice, tissue adjacent to longitudinal SCI exhibited many healthy neurons surrounded by reactive astrocyte processes immediately adjacent to the lesion, and there was no qualitatively appreciable loss of neurons compared with uninjured tissue (Fig. 6A,B,F). In contrast, GFAP-TK mice treated with GCV exhibited pronounced loss of neurons after ablation of astrocytes adjacent to the SCI (Fig. 6C,D), which extended over several upper lumbar segments. Morphometric analysis showed a statistically significant 80% decline in the number of cresyl violet-stained neurons, including both motor neurons and interneurons, adjacent to SCI in tissue depleted of reactive astrocytes compared with tissue containing reactive astrocytes (Fig. 6E). To determine whether this substantial neuronal death might simply be attributable to increased inflammation after ablation of reactive astrocytes, we injected bacterial LPS into the spinal cord at L1-L2 to induce an inflammatory response (Andersson et al., 1992). As expected, LPS induced pronounced infiltration of CD45-positive inflammatory cells but also triggered pronounced reactive astrocytosis. In the presence of large numbers of reactive astrocytes, there was no obvious loss of spinal cord neurons despite the pronounced inflammation (Fig. 6G-I), and these mice exhibited no obvious functional deficits. We conclude that the loss of neurons observed after ablation of reactive astrocytes was not merely attributable to increased infiltration of inflammatory cells but was attributable to loss of astrocyte-related functions. Together these findings show that astrocyte loss after SCI is associated with the death of most local neurons and suggest that reactive astrocytes provide essential protection or support for neurons after SCI.
Figure 6.
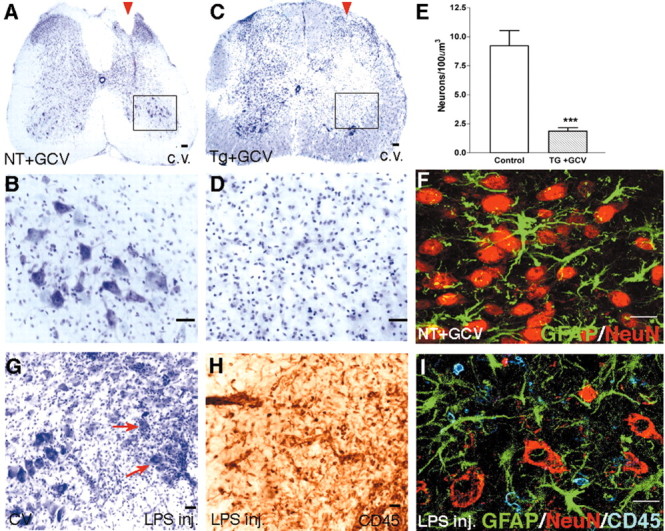
Degeneration of neurons after ablation of reactive astrocytes adjacent to stab SCI. A-D, F, Transverse sections of upper lumbar spinal cord after longitudinal stab SCI (arrowheads) in a nontransgenic mouse given GCV (A, B, F; NT+GCV) or in a GFAP-TK transgenic mouse given GCV (C, D; Tg+GCV). A-D, Staining with cresyl violet (c.v.) reveals no detectable loss of neurons adjacent to stab SCI in the nontransgenic mouse (A, B), in contrast to pronounced neuronal loss in the GFAP-TK transgenic mouse given GCV (C, D). B, D, Details, respectively, of boxed areas in A and C, showing that neurons of all sizes appear healthy in the neural parenchyma adjacent to the SCI in the control mouse, and there is little or no evidence of neuronal degeneration (B); in contrast, there is pronounced neuronal degeneration in the region from which astrocytes have been ablated in the GFAP-TK transgenic mouse given GCV (D). E, Mean cell number ± SEM of cresyl violet-stained neurons within the wound margin adjacent to the stab injury (see Fig. 1D); GFAP-TK mice given GCV exhibited fourfold fewer neurons compared with control mice; n = 5 per group. ***Significantly different from control, p < 0.001 (ANOVA). F, Laser-scanning confocal microscopy of double staining for astrocytes (GFAP) and neurons (NeuN) showing that reactive astrocytes and their processes intermingle extensively among NeuN-positive neurons in the ventral horn after SCI in a control mouse. H-I, Transverse sections of upper lumbar ventral horn 14 d after injection of LPS. Cresyl violet staining reveals no detectable loss of ventral horn neurons induced by LPS (G). Immunohistochemical staining for CD45 in an adjacent tissue section reveals a pronounced inflammatory response and infiltration of leukocytes into the ventral horn induced by LPS (H). I, Laser-scanning confocal microscopy of triple staining for astrocytes (GFAP), neurons (NeuN), and inflammatory cells (CD45) showing that, in the presence of astrocytes, NeuN-positive neurons survive well despite the presence of many CD45-positive inflammatory cells in the ventral horn after LPS injection. Scale bars: A, C, G,H, 100 μm; B, D ,50 μm; F, I, 15 μm.
Demyelination and oligodendrocyte degeneration after loss of reactive astrocytes
We next examined the effects of astrocyte loss on Luxol fast blue staining for myelin and the number of oligodendrocytes stained for MBP in L1-L2 white matter immediately adjacent to longitudinal stab SCI. All three groups of control mice exhibited a similar ∼18% decrease in the area of normally appearing white matter stained for Luxol fast blue in the dorsal and lateral columns immediately adjacent to longitudinal SCI compared with the uninjured side (Fig. 7A,F), as well as a 24% decline in the number of oligodendrocytes stained for MBP compared with uninjured tissue (Fig. 7C,D,G). In contrast, GFAP-TK mice treated with GCV exhibited a severe and significantly greater demyelination of >70% of the white matter and a loss of >90% of the oligodendrocytes in white matter tissue that was depleted of reactive astrocytes adjacent to SCI (Fig. 7A-G). To determine whether demyelination might simply be attributable to increased inflammation after ablation of reactive astrocytes, we examined L1-L2 spinal cord tissue injected with LPS as described above. As expected, LPS induced pronounced infiltration of CD45-positive inflammatory cells but also triggered pronounced reactive astrocytosis. These mice exhibited no obvious demyelination in Luxol fast blue-stained sections. These findings show that astrocyte loss after SCI markedly exacerbates demyelination and degeneration of oligodendrocytes and suggest that reactive astrocytes provide essential protection or support for oligodendrocytes and myelinated white matter after SCI.
Figure 7.
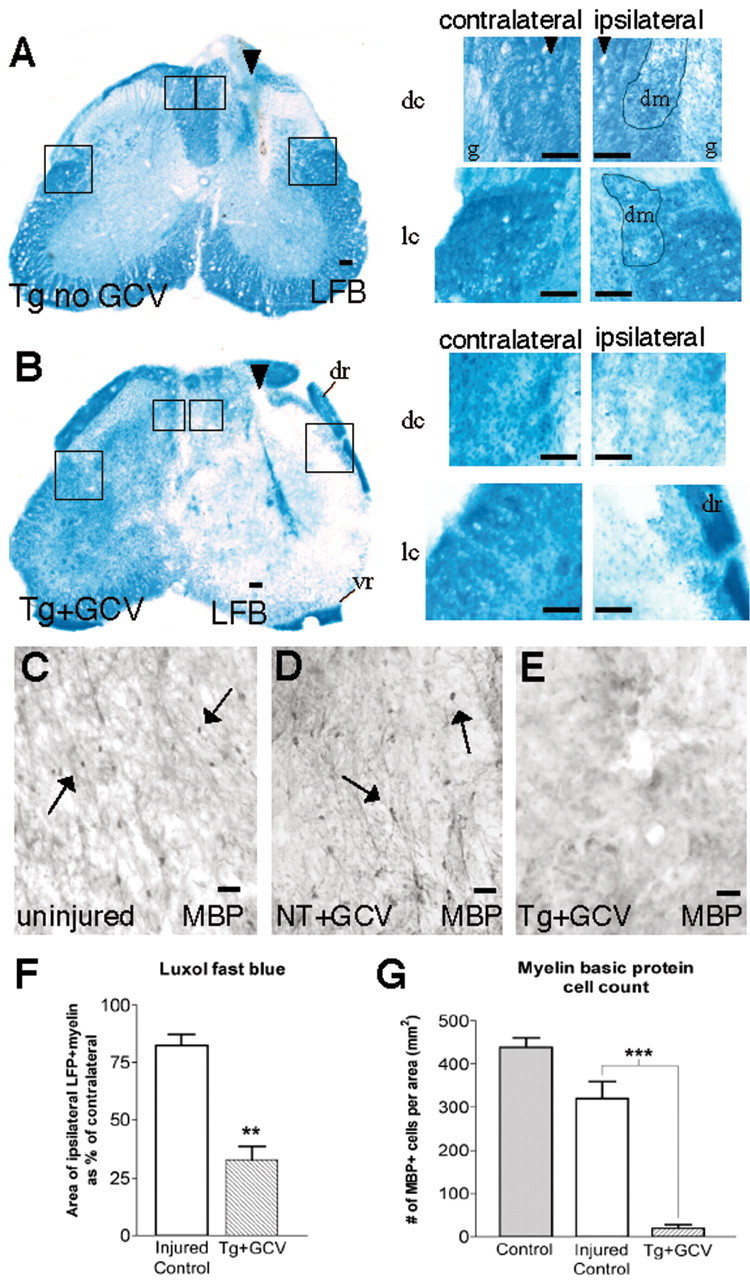
Loss of white matter and degeneration of oligodendrocytes after ablation of reactive astrocytes adjacent to stab SCI. A, B, Transverse sections of upper lumbar spinal cord after longitudinal stab SCI (arrowheads) in control GFAP-TK transgenic mouse not given GCV (A; Tg no GCV) and in a GFAP-TK transgenic mouse given GCV (B; Tg+GCV). Staining with Luxol fast blue (LFB) reveals minimal loss of myelin adjacent to stab SCI in the control mouse (A), in contrast to severe myelin loss in the GFAP-TK transgenic mouse given GCV (B). Boxed areas in survey images are shown as details of dorsal columns (dc) next to gray matter (g) and lateral columns (lc), both contralateral and ipsilateral to the stab SCI. Dotted lines demarcate tissue areas of partially degenerated myelin (dm). Note that dorsal roots (dr) and ventral roots (vr) retain myelin immediately adjacent to severely demyelinated CNS white matter in the GFAP-TK mouse (B), indicating that LFB staining has worked successfully. C-E, Transverse sections of upper lumbar spinal cord showing uninjured tissue (C) or tissue adjacent to longitudinal stab SCI in a nontransgenic mouse (D; NT+GCV) or in a GFAP-TK transgenic mouse given GCV (E; Tg+GCV). Immunohistochemistry for MBP shows no detectable difference in the number of MBP-stained oligodendrocytes (arrows) in uninjured tissue (C) and tissue adjacent to stab SCI in the nontransgenic mouse (D), in contrast to pronounced loss of oligodendrocytes in the GFAP-TK transgenic mouse given GCV (E). F, Mean area ± SEM of Luxol fast blue-stained myelin in dorsal and lateral columns adjacent to stab SCI. The area of white matter exhibiting LFB-stained intact myelin ipsilateral and adjacent to the SCI is expressed as a percentage of the area in the equivalent region on the uninjured contralateral side. GFAP-TK mice given GCV exhibited a significantly greater decline in area of myelinated white matter next to the SCI compared with injured control mice; n = 6 per group. **Significantly different from control, p < 0.001 (ANOVA). G, Mean number ± SEM of MBP-stained oligodendrocyte cell bodies in white matter ipsilateral and adjacent to stab SCI or in uninjured tissue. GFAP-TK mice given GCV exhibited a significantly greater decline in the number of oligodendrocytes next to the SCI compared with injured control mice; n = 5 per group. ***Significantly different from control, p < 0.0005 (ANOVA). Scale bars: A, B, surveys, 100 μm; A, B, details, 50 μm; C-E, = 25 μm.
Impaired limb function after loss of reactive astrocytes in stab SCI
During the course of initial experiments designed to study cell biology after longitudinal SCI, we noted that, in contrast to control mice, GFAP-TK mice treated with GCV to ablate reactive astrocytes consistently exhibited deficits in ipsilateral hindlimb function. We therefore conducted a prospective quantitative analysis of hindlimb function using three behavioral tests (Fig. 8A-C). The aim of this analysis was to determine whether there was reproducible and statistically significant loss of gross motor function in mice that received stab SCI in combination with ablation of reactive astrocytes and did not attempt to delineate the precise nature of any deficit or to identify the neural mechanism that might underlie any deficit. Accordingly, we used (1) open-field evaluation of motor performance scored on a simple six-point scale, (2) performance on the rotorod, and (3) footprint analysis. As in other experiments, GFAP-TK mice treated with GCV, as well as three groups of control mice, were given longitudinal stab SCIs in the lumbar cord gray matter at L1-L2. In all three groups of control mice, these lesions caused no detectable deficits in motor function, as assessed by open-field evaluation and footprint analysis (Fig. 8A,C), and caused only a minor and temporary deficit in rotorod performance that was fully reversible by 14 d after SCI (Fig. 8B). In contrast, GFAP-TK mice given GCV exhibited a gradual but substantial decline in performance on all three tests over the period of ablation of reactive astrocytes, and by 14 d after SCI, performance on all three tests indicated a pronounced, statistically significant deterioration of ipsilateral hindlimb function (Fig. 8A-C). Nevertheless, these mice retained bladder function throughout the time evaluated. These findings show that loss of reactive astrocytes can lead to pronounced functional deterioration after a small stab SCI that would otherwise cause little or no functional deficit.
Figure 8.

Loss of hindlimb function after ablation of reactive astrocytes adjacent to stab SCI. A, Time course of left hindlimb (HL) locomotor performance in an open field over 14 d after ipsilateral stab SCI in control mice and GFAP-TK transgenic mice given GCV (Tg+GCV). Three groups of control mice were evaluated: nontransgenic mice not given GCV, nontransgenic mice given GCV, and transgenic mice not given GCV. No significant difference was detected among these control groups (ANOVA), and their values were pooled to a single control value. Control mice (n = 14) exhibited no visibly detectable impairment of ipsilateral hindlimb performance after stab SCI, whereas transgenic mice given GCV (n = 7) exhibited a gradual but ultimately substantial and significant impairment during and after ablation of reactive astrocytes. ***Significantly different from control, p < 0.001 (ANOVA plus post hoc pair-wise analysis). B, Time course of rotorod performance after SCI of control mice and GFAP-TK transgenic mice given GCV. Control mice (n = 9; pooled values of three groups not significantly different by ANOVA) exhibited an initial mild impairment of rotorod performance after stab SCI that was fully reversible by 14 d. In contrast, transgenic mice given GCV (n = 5) failed to recover rotorod performance during and after ablation of reactive astrocytes and by 14 d exhibited a substantial and significant impairment. ***Significantly different from control, p < 0.001 (ANOVA plus post hoc pair-wise analysis). C, Representative footprint analysis at 14 d after stab SCI of a control mouse and a GFAP-TK transgenic mouse given GCV. Footprints of a control mouse illustrate normal plantar placement of forepaws and hindpaws. In contrast, footprints of a transgenic mouse given GCV show no plantar placement of the left (L, ipsilateral) hindpaw (circles show placement of only the left forepaw), which is indicative of foot dragging on the dorsal surface. In addition, the right (R, contralateral) hindpaw shows smears (arrow) indicative of toe dragging.
Loss of reactive astrocytes exacerbates tissue degeneration and prevents recovery of motor functions after moderate crush SCI
Although longitudinal stab SCI is a useful and appropriate model to study aspects of SCI cell biology in vivo, other experimental models such as contusion or crush injuries are thought to be more relevant to the types of tissue damage seen after SCI in human patients (Behrmann et al., 1994; Ma et al., 2001; Inman et al., 2002). Crush SCI generated with forceps in mice is a well characterized and consistently reproducible model of SCI that allows evaluation of both cell biology and behavior (Inman et al., 2002). We therefore compared the behavioral and cell biological effects of a moderately severe crush SCI in control mice and in mice having transgenically targeted ablation of reactive astrocytes. To generate a moderately severe crush SCI at L1-L2, we used forceps that closed to a 0.5 mm space. Using this crush severity, control mice exhibited initial deficits in motor behavior from which they fully recovered over a 14 d period. Again, the aim of the behavioral analysis was to determine whether there was reproducible and statistically significant loss of gross motor function in mice that received crush SCI in combination with ablation of reactive astrocytes and did not attempt to delineate the precise nature of any deficit or identify the neural mechanism that might underlie any deficit. Accordingly, we again used (1) open-field evaluation of motor performance scored on a simple six-point scale, (2) performance on the rotorod, and (3) footprint analysis.
In all three types of control mice, crush SCI using these forceps reproducibly caused moderately severe motor impairments that were mostly reversible, and over a 14 d period, mice recovered to essentially normal performance levels on all three behavioral tests (Fig. 9A-C). These mice also initially showed loss of bladder function, which most mice recovered by 14 d. In all three types of control mice, this moderate crush SCI reproducibly caused a small focal lesion of similar size. On histological examination at 14 d after injury, tissue degeneration in control mice consisted of a clearly restricted, compact, central lesion surrounded by and clearly delineated from well preserved tissue (Fig. 10A). The central lesion, described in detail below, was surrounded by a rim of reactive dividing astrocytes, which together with fibroblasts formed a scar that walled off the area of degenerated tissue (Figs. 11A,C,G, 12A-F). At the perimeters of the cord, the central discoid lesion was generally surrounded by a rim of well preserved tissue consisting mostly of white matter (Figs. 10A, 11I).
Figure 9.
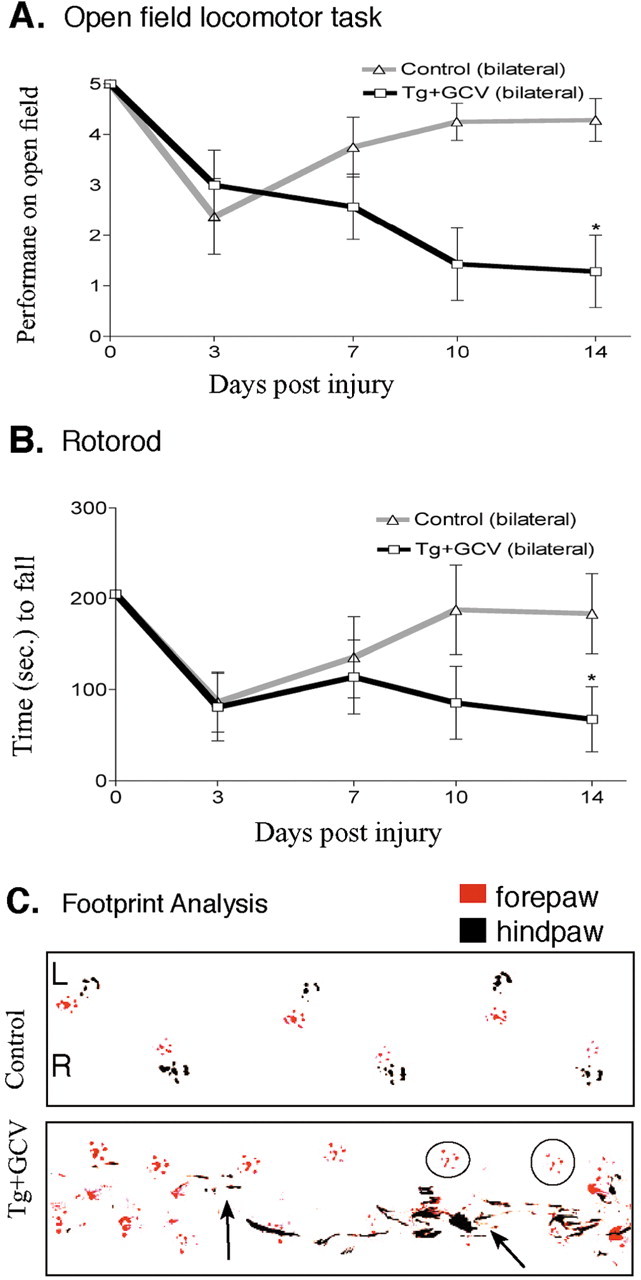
Persisting loss of motor function after ablation of reactive astrocytes in crush SCI. A, Time course of left hindlimb locomotor performance in an open field over 14 d after moderately severe forceps crush SCI in control mice and GFAP-TK transgenic mice given GCV (Tg+GCV). Control mice (n = 8) exhibited an initial impairment of bilateral (both sides scored individually and averaged) hindlimb performance after crush SCI that was fully reversible by 14 d. In contrast, transgenic mice (n = 7) given GCV failed to recover bilateral hindlimb performance during and after ablation of reactive astrocytes and by 14 d exhibited a substantial and significant impairment. *Significantly different from control, p < 0.05 (ANOVA plus post hoc pair-wise analysis). B, Time course of rotorod performance after SCI in control mice and GFAP-TK transgenic mice given GCV. Control mice (n = 8) exhibited an initial mild impairment of rotorod performance after crush SCI that was fully reversible by 14 d. In contrast, transgenic mice (n = 7) given GCV failed to recover rotorod performance during and after ablation of reactive astrocytes and by 14 d exhibited a substantial and significant impairment. *Significantly different from control, p < 0.05 (ANOVA plus post hoc pair-wise analysis). C, Representative footprint analysis at 14 d after crush SCI of a control mouse and a GFAP-TK transgenic mouse given GCV. Footprints of a control mouse illustrate normal plantar placement of forepaws and hindpaws. In contrast, footprints of a transgenic mouse given GCV exhibit smears on the left side (L, left arrow) indicative of toe dragging as well as frequent failure of plantar placement of the left hindpaw (circles show placement of only the left forepaw) indicative of dorsal foot dragging, and on the right (R) side, the hindpaw shows substantial smears (right arrow) indicative of toe dragging and uncoordinated hindpaw placements.
Figure 10.

Increased lesion volume after ablation of reactive astrocytes in crush SCI. A, B, Horizontal sections of the lower thoracic and upper lumber spinal cord (rostral is to the right) 14 d after forceps crush SCI (arrowheads) of similar severity in a nontransgenic mouse given GCV (A; NT+GCV) or in a GFAP-TK transgenic mouse given GCV (B; Tg+GCV). Staining with cresyl violet (c.v.) reveals a compact centrally placed lesion with clearly delineated boundaries in the nontransgenic mouse, and there are many normally appearing neurons immediately next to the lesion (A). In contrast, the lesion is large, diffuse, and without clear boundaries in the GFAP-TK transgenic mouse given GCV, and there is clear evidence of neuronal loss at considerable distances away from the crush center (B). C, D, Dorsal view of three-dimensional reconstructions of the outer walls (white) of the lower thoracic and upper lumber spinal cord (rostral is to the right) generated from horizontal cresyl violet-stained tissue sections using NeuroLucida. Visible inside the cord are the reconstructed lesions (red) after forceps crush stab SCI of similar severity in a nontransgenic mouse given GCV (C) or in a GFAP-TK transgenic mouse given GCV (D). E, Mean lesion volume ± SEM determined from the three-dimensional reconstructions. GFAP-TK mice given GCV exhibit a fivefold greater lesion volume compared with control mice; n = 5 per group. ***Significantly different from control, p < 0.001 (ANOVA). Scale bars, 400 μm.
Figure 11.

Increased inflammation, demyelination, and failure of wound contraction after ablation of reactive astrocytes in crush SCI. A-J, Horizontal sections of the lower thoracic and upper lumber spinal cord 14 d after forceps crush SCI (arrowheads) of similar severity in nontransgenic mice given GCV (A, C, E, G, I; NT+GCV) or in GFAP-TK transgenic mice given GCV (B, D, F, H, J; Tg+GCV). Nontransgenic mice exhibit compact central lesions lined by a clearly demarcated scar comprising GFAP-positive, reactive astrocytes as well as fibronectin-positive fibroblasts (C, G), and the lateral columns immediately adjacent to the central lesion remain well myelinated, as demonstrated by Luxol fast blue (LFB) staining (I, arrow). Many of the scar-forming astrocytes that line the lesion divided after SCI and exhibit staining for both BrdU and GFAP, as demonstrated by scanning confocal laser microscopy (A, C, insets). The lesions are filled with CD45-positive inflammatory cells (E). In contrast, GFAP-TK transgenic mice given GCV exhibit large diffuse lesions that have no obvious boundaries and are not delineated by a detectable scar (B, D, F, H), and the lateral columns adjacent to the lesion are severely demyelinated, as demonstrated by Luxol fast blue staining (J, arrow). Lesions in these mice contain almost no reactive astrocytes that are positive for BrdU or GFAP (B, D) and exhibit widespread and dispersed infiltration of CD45-positive inflammatory cells, many of which have the appearance of large, ovoid, and active macrophages (F). These lesions also exhibit a widespread and diffuse distribution of fibronectin-positive fibroblasts that do not cluster along a detectable border of the lesion (H). Scale bars: A-H, 200 μm; I, J, 100 μm.
Figure 12.
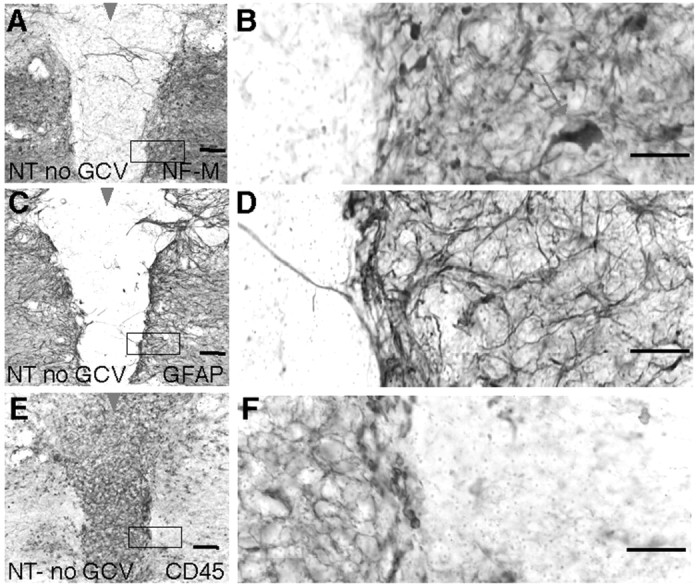
Central lesion exhibiting loss of neurons and nerve fibers, pronounced inflammation, and absence of reactive astrocytes after crush SCI in nontransgenic mice. A-F, Closely neighboring immunohistochemically stained horizontal sections of the lower thoracic and upper lumber spinal cord 14 d after moderately severe forceps crush SCI (arrowheads) in a nontransgenic mouse not given GCV (NT-GCV). The compact central lesion at the crush site contains no viable neurons and only a few scattered nerve fibers, as detected by staining for NFM (A, B), is clearly demarcated by a scar of reactive astrocytes (C, D), and is filled with CD45-positive inflammatory cells (E, F). B, D, F, Details, respectively, of boxed areas in A, C, and E and show the sharply defined border between the lesion and viable tissue, which is defined by densely packed processes of GFAP-expressing reactive astrocytes (C, D). There is a strict and essentially absolute correlation between the presence of neural elements, both nerve fibers and neuronal cell bodies (arrow), and the presence of reactive astrocytes in viable tissue right up to the border of the lesion, whereas the central lesion contains essentially no neural elements or astrocytes (A-D). Conversely, activated inflammatory cells are numerous and densely packed within the central lesion in the absence of astrocytes, whereas they are few and scattered in tissue-containing reactive astrocytes (C-F). Scale bars: A, C, E, 100 μm; B, D, F, 50 μm.
In GFAP-TK mice given GCV, crush SCI using forceps with a 0.5 mm spacer also initially caused moderately severe motor impairments. However, in contrast with control mice, these mice with ablation of reactive astrocytes did not recover but deteriorated further over a 14 d period and exhibited severe, persisting motor impairment on three behavioral tests (Fig. 9A-C). These mice also all failed to recover bladder function over the 14 d evaluated. On histological examination at 14 d after injury, moderate crush SCI combined with reactive astrocyte ablation consistently led to pronounced and widespread tissue degeneration that significantly increased the lesion volume by greater than fivefold (Fig. 10A-E) and that extended over several upper lumbar segments. In these mice, there was no detectable border of scar-forming, dividing astrocytes or fibroblasts that separated damaged from viable tissue, and large ovoid fibronectin-positive fibroblasts were scattered diffusely throughout the lesion area (Fig. 11B,D,H). Astrocyte ablation after moderate crush injury led to markedly increased neuronal degeneration (Fig. 10A,B), severely aggravated demyelination (Fig. 11I,J), and widespread infiltration of CD45-positive inflammatory cells (Fig. 11E,F), many of which had the appearance of activated macrophages (Fig. 11F). Together, these findings show that loss of reactive astrocytes markedly exacerbates tissue degeneration and prevents functional recovery after moderate crush SCI.
Crush SCI in nontransgenic mice routinely exhibits central necrotic regions devoid of reactive astrocytes
During the course of examining sections from nontransgenic control mice after moderate or severe SCI, we noted that all otherwise normal, nontransgenic mice with moderate or severe SCI without exception exhibited central necrotic lesions that did not contain reactive astrocytes. On systematic examination, we noted that these central lesions were consistently devoid of GFAP- and S100β-positive (data not shown) astrocytes and contained no neuronal cell bodies that stained with either cresyl violet or neurofilament M (Fig. 12A-D). These central lesions also contained little or no Luxol fast blue-stained myelin (Fig. 11I) or MBP-stained oligodendrocytes. In addition, these central lesions were always filled with many large CD45-positive inflammatory cells with the appearance of phagocytic macrophages (Fig. 12E,F). The central lesions were always surrounded by a rim of reactive dividing astrocytes that formed a sharp border between areas that contained viable neural elements and areas that did not (Fig. 12C,D). These findings show that in the immediate vicinity of the lesion after forceps crush SCI in otherwise normal, nontransgenic mice, the presence of viable neurons, oligodendrocytes, and myelinated axons correlates with the presence of reactive astrocytes, and areas devoid of reactive astrocytes exhibit degeneration of neurons, oligodendrocytes, and myelin.
Discussion
In this study, we used a transgenically targeted cell ablation strategy and two models of experimental SCI to investigate the roles of reactive astrocytes after SCI. In both injury models, the severity of SCI was markedly exacerbated by the loss of reactive astrocytes, and this loss led to failure of both the tissue repair and the functional recovery normally associated with mild or moderate SCI. Our findings show that reactive astrocytes (1) are essential for wound healing and blood-brain barrier repair, (2) restrict inflammation, (3) protect neurons and oligodendrocytes, and (4) preserve motor functions after mild or moderate SCI. These findings are important for understanding the process of secondary tissue degeneration and the regulation of inflammation after SCI and begin to define specific roles of astrocytes during the response to SCI.
Specificity of the transgenic model
Transgenes insert randomly into the genome, and transgene expression cannot be assumed automatically to mimic endogenous promoter activity. Nevertheless, transgene expression is stable within transgenic breeding lines and, once characterized, can be a useful tool (Feng et al., 2000). Transgene expression should be characterized at the single-cell level in every tissue region studied. Our previous studies showed that in the GFAP-TK mice used here, transgene-derived TK was detectable only in GFAP-expressing cells in the forebrain and peripheral nervous system in vivo and in vitro (Bush et al., 1998, 1999; Imura et al., 2003). Here we showed at the single-cell level that all cells expressing transgene-derived TK also expressed GFAP in uninjured and injured spinal cord.
Various observations indicate that the degeneration of other cells after astrocyte ablation is attributable to loss of functions performed by astrocytes and not to nonspecific toxic effects of the transgenic model. In tissue cultures prepared from GFAP-TK mice, GCV killed only cells that expressed the GFAP-TK transgene, whereas neighboring GFAP-TK-negative cells survived well, multiplied, and exhibited no evidence of toxicity (Bush et al., 1998; Imura et al., 2003). In addition, numerous other studies using transgenically targeted TK ablation models reported that cells surrounding ablated TK-expressing cells in vivo do not show evidence of toxicity (Borrelli et al., 1989; Wallace et al., 1991; Canfield et al., 1996; Mathis et al., 2000).
Reactive astrocytes are required for wound and BBB repair after SCI
After SCI in mice, the lesion site rarely forms cavities and instead fills with connective tissue that remodels and contracts to <40% the original size during the first 3 weeks (Inman and Steward, 2003). The fivefold greater lesion size that we observed 2 weeks after ablation of reactive astrocytes and SCI is greater than might be expected solely on the basis of induced and secondary cell loss and suggests that loss of reactive astrocytes contributed to failure of wound contraction. This finding supports evidence that astrocytes have receptor-regulated contractile properties likely to be involved in wound healing (Manning et al., 1998).
The adult CNS exhibits a BBB to blood-borne macromolecules (Broadwell and Sofroniew, 1993; Rubbin and Staddon, 1999) that becomes leaky after CNS injury and is restored by local cellular interactions. Here we show that astrocytes are required for BBB repair after SCI. We have previously shown that grafts of nontransgenic astrocytes can restore blood-brain barrier function after astrocyte ablation in the forebrain (Bush et al., 1999). Our findings support the idea that astrocytes play fundamental roles in maintenance and repair of the BBB throughout the CNS.
Reactive astrocytes restrict inflammation after SCI
Inflammation is a variable and inadequately understood component of the tissue response to SCI. Different studies report that inflammatory cells may either improve or exacerbate outcome after SCI (Rapalino et al., 1998; Popovich et al., 1999). Cellular interactions that regulate the entry and subsequent exit of different types of inflammatory cells after SCI are uncertain. In tissue culture, astrocytes produce both pro- and anti-inflammatory molecules (Ridet et al., 1997). Our findings here and in the forebrain (Bush et al., 1999) show that ablation of reactive astrocytes leads to a pronounced increase in the number of activated inflammatory cells in CNS tissue 14 d after injury. It is noteworthy that tissue from which astrocytes were intentionally ablated in our transgenic mice was similar in appearance to the central necrotic lesion observed in nontransgenic mice after crush SCI here and as reported by others after crush or contusion SCI (Fujiki et al., 1996; Ma et al., 2001). In both transgenic and nontransgenic mice, tissue lacking reactive astrocytes contains activated macrophages, whereas areas that contain viable reactive astrocytes have few phagocytic inflammatory cells. These findings agree with studies showing that astrocytes suppress the phagocytic capacity of microglia in vitro (DeWitt et al., 1998). Taken together, these findings suggest that after SCI, scar-forming reactive astrocytes surround and wall off regions that lack astrocytes but contain activated inflammatory cells and act to restrict infiltration of activated inflammatory cells into surrounding tissue. Nevertheless, our findings need not be interpreted that reactive astrocytes are exclusively anti-inflammatory, particularly immediately after SCI, when they may produce pro-inflammatory cytokines. Available evidence suggests that astrocyte roles after SCI are likely to be dynamic and context-dependent, such that astrocytes may exert either pro- or anti-inflammatory functions at different locations or at different times in the response to injury and during repair.
Reactive astrocytes protect neurons and oligodendrocytes after SCI
Tissue loss after SCI is exacerbated by secondary degeneration that takes place over hours or days after the initial insult. Although various molecules have been implicated in contributing to secondary cell death and tissue disruption, such as glutamate, reactive oxygen species, and certain cytokines (Liu et al., 1997; McDonald et al., 1998), the roles played by specific cell types in the progression of secondary degeneration are not understood. Our findings show that loss of reactive astrocytes after SCI markedly exacerbates cell degeneration in both gray and white matter, perhaps by leading to the accumulation of extracellular glutamate with subsequent excitotoxicity toward both neurons and oligodendrocytes (McDonald et al., 1998; Bush et al., 1999). Blockade of glutamate receptor preserves hippocampal neurons after astrocyte ablation (Bush et al., 1999).
It is interesting that after crush or contusion SCI in normal, nontransgenic mice, the central lesion that is devoid of neurons and axons also lacks reactive astrocytes, as reported by others (Ma et al., 2001; Inman et al., 2002) and noted here. Temporal correlative analysis indicates that astrocyte loss precedes signs of neurodegeneration after traumatic brain injury (Zhao et al., 2003). Normal glia protect spinal cord motor neurons from degeneration in a mutant mouse model of amyotrophic lateral sclerosis (Clement et al., 2003). In vitro models suggest that astrocytes are induced to migrate away from and abandon the centers of injury sites (Fitch et al., 1999). Extending these studies, our astrocyte ablation studies provide direct in vivo evidence that astrocyte loss or dysfunction can contribute causally to posttraumatic neurodegeneration. Together these findings suggest that astrocytes exert essential protective activities for neurons, oligodendrocytes, and myelin after SCI, and identifying the molecular basis of this protection may point toward means of reducing secondary degeneration after SCI.
Reactive astrocytes preserve function after SCI
Our findings also show that ablation of reactive astrocytes exacerbated motor deficits or prevented motor recovery after stab or crush SCI. Ablation of reactive astrocytes transformed mild or moderate injuries that caused little tissue disruption and no or only mild motor deficits into injuries with substantial tissue degeneration and severe loss of motor function. Additional studies will be needed to determine the precise neural basis for the motor deficits observed after astrocyte ablation. Astrocyte ablation leads to substantial loss of neurons among upper lumbar neuron pools important for hindlimb function, and neuronal loss of this kind is reported to result in significant dysfunction (Magnuson et al., 1999). In addition, loss of oligodendrocytes and severe demyelination in descending tracts in lateral and dorsal columns after astrocyte ablation are also likely to have contributed to motor deficits. Taken together, our findings demonstrate that loss of astrocyte activities during the cellular response to SCI can lead to gross functional deficits and failure of functional recovery and suggest that reactive astrocytes not only protect tissue but also preserve function after SCI.
Astrocyte roles in the response to SCI
The roles of specific cell types in the process of wound healing and tissue repair after SCI are only beginning to be understood. Considerable evidence implicates scar-forming astrocytes as inhibitors of axon regeneration (Reier et al., 1983; Bush et al., 1999; Davies et al., 1999; Menet et al., 2003), and in this context, reactive astrocytes are widely regarded as detrimental to overall outcome after SCI. Nevertheless, our findings show that scar-forming reactive astrocytes also exert a number of positive roles after SCI, including repairing the blood-brain barrier, restricting leukocyte infiltration, protecting various neural elements, and preserving motor function. Crush or contusion SCIs in animal models and human patients are associated with central necrotic lesions or cavities that are devoid of reactive astrocytes. It is noteworthy that these normally occurring lesions exhibit a number of similarities in appearance to tissue from which astrocytes were intentionally ablated in our transgenic mice. We speculate that regions of pronounced tissue degeneration after SCI may represent areas where astrocytes have become dysfunctional, died (Zhao et al., 2003), or migrated away from (Fitch et al., 1999) early in response to the initial insult, and the early loss or dysfunction of astrocytes may be a major contributing factor to the process of secondary degeneration after SCI. Identifying means to promote astrocyte survival and to augment specific astrocyte protective activities may lead to novel approaches to reducing secondary degeneration and preserving function after SCI.
Footnotes
This work was supported by National Institutes of Health National Institute of Neurological Disorders and Stroke Grants NS42039 and NS016333, Christopher Reeve Paralysis Foundation Grant SAC1-00005-2, and the Roman Reed Spinal Cord Injury Research Fund of California. J.R.F. and J.E.H. were supported by Roman Reed predoctoral fellowships. We thank N. B. Doan and M. E. Sislak for technical assistance. We are grateful to P. Borgese and Hoffman-La Roche for ganciclovir.
Correspondence should be addressed to Michael V. Sofroniew, Department of Neurobiology, University of California Los Angeles School of Medicine, 10833 Le Conte Avenue, Los Angeles, CA 90095-1763. E-mail: sofroniew@mednet.ucla.edu.
Copyright © 2004 Society for Neuroscience 0270-6474/04/242143-13$15.00/0
References
- Agrawal SK, Fehlings MG (1997) Role of NMDA and non-NMDA ionotropic glutamate receptors in traumatic spinal cord axonal injury. J Neurosci 17: 1055-1063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andersson P-B, Perry VH, Gordon S (1992) The acute inflammatory response to lipopolysaccharide in CNS parenchyma differs from that in other body tissues. Neuroscience 48: 169-186. [DOI] [PubMed] [Google Scholar]
- Basso DM, Beattie MS, Bresnahan JC (1995) A sensitive and reliable locomotor rating scale for open field testing in rats. J Neurotrauma 12: 1-21. [DOI] [PubMed] [Google Scholar]
- Behrmann DL, Bresnahan JC, Beattie MS (1994) Modeling of acute spinal cord injury in the rat: neuroprotection and enhanced recovery with methylprednisolone, U-74006F and YM-14673. Exp Neurol 126: 61-75. [DOI] [PubMed] [Google Scholar]
- Borrelli E, Heyman RA, Arias C, Sawchenko PE, Evans RM (1989) Transgenic mice with inducible dwarfism. Nature 339: 538-541. [DOI] [PubMed] [Google Scholar]
- Bracken MB, Shepard MJ, Collins WF, Holford TR, Young W, Baskin DS, Eisenberg HM, Flamm E, Leo-Summers L, Maroon J, Marshall L, Perot Jr P, Piepmeier J, Sonntag V, Wagner F, Wilberger J, Winn H (1990) A randomized, controlled trial of methylprednisolone in the treatment of acute spinal injury. N Engl J Med 322: 1405-1411. [DOI] [PubMed] [Google Scholar]
- Broadwell RD, Sofroniew MV (1993) Serum proteins by-pass the blood brain barrier for extracellular entry to the CNS. Exp Neurol 120: 246-263. [DOI] [PubMed] [Google Scholar]
- Bush TG, Savidge TC, Freeman TC, Cox HJ, Campbell EA, Mucke L, Johnson MH, Sofroniew MV (1998) Fulminant jejuno-ileitis following ablation of enteric glia in adult transgenic mice. Cell 93: 189-201. [DOI] [PubMed] [Google Scholar]
- Bush TG, Puvanachandra N, Horner CH, Polito A, Ostenfeld T, Svendsen CN, Mucke L, Johnson MH, Sofroniew MV (1999) Leukocyte infiltration, neuronal degeneration and neurite outgrowth after ablation of scar-forming, reactive astrocytes in adult transgenic mice. Neuron 23: 297-308. [DOI] [PubMed] [Google Scholar]
- Canfield V, West AB, Goldenring JR, Levenson R (1996) Genetic ablation of parietal cells in transgenic mice: a new model for analyzing cell lineage relationships in the gastric mucosa. Proc Natl Acad Sci USA 93: 2431-2435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clement AM, Nguyen MD, Roberts EA, Garcia ML, Boillee S, Rule M, McMahon AP, Doucette W, Siwek D, Ferrante RJ, Brown Jr RH, Julien J-P, Goldstein LSB, Cleveland DW (2003) Wild-type nonenuronal cells extend survival of SOD1 mutant motor neurons in ALS mice. Science 302: 113-117. [DOI] [PubMed] [Google Scholar]
- Clemente CD, Windle WF (1954) Regeneration of severed nerve fibers in the spinal cord of the adult cat. J Comp Neurol 101: 691-731. [DOI] [PubMed] [Google Scholar]
- Constantini S, Young W (1994) The effects of methylprednisolone and ganglioside GM1 on acute spinal cord injury in rats. J Neurosurg 80: 97-111. [DOI] [PubMed] [Google Scholar]
- Crowe MJ, Bresnahan JC, Shuman SL, Masters JN, Beattie MS (1997) Apoptosis and delayed degeneration after spinal cord injury in rats and monkeys. Nat Med 3: 73-76. [DOI] [PubMed] [Google Scholar]
- Davies SJA, Goucher DR, Doller C, Silver J (1999) Robust regeneration of adult sensory axons in degenerating white matter of the adult rat spinal cord. J Neurosci 19: 5810-5822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeWitt DA, Perry G, Cohen M, Doller C, Silver J (1998) Astrocytes regulate microglial phagocytosis of senile plaque cores of Alzheimer's disease. Exp Brain Res 149: 329-340. [DOI] [PubMed] [Google Scholar]
- Eddleston M, Mucke L (1993) Molecular profile of reactive astrocytes: implications for their role in neurological disease. Neuroscience 54: 15-36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eng LF, Ghirnikar RS (1994) GFAP and astrogliosis. Brain Pathol 4: 229-237. [DOI] [PubMed] [Google Scholar]
- Fehlings MG, Tator CH (1995) The relationships among the severity of spinal cord injury, residual neurological function, axon counts, and counts of retrogradely labeled neurons after experimental spinal cord injury. Exp Neurol 132: 220-228. [DOI] [PubMed] [Google Scholar]
- Feng G, Mellor R, Bernstein M, Keller-Peck C, Nguyen Q, Wallace M, Nerbonne J, Lichtman J, Sanes J (2000) Imaging neuronal subsets in transgenic mice expressing multiple spectral variants of GFP. Neuron 28: 41-51. [DOI] [PubMed] [Google Scholar]
- Fitch MT, Doller C, Combs CK, Landreth GE, Silver J (1999) Cellular and molecular mechanisms of glial scarring and progressive cavitation: in vivo and in vitro analysis of inflammation-induced secondary injury after CNS trauma. J Neurosci 19: 8182-8198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujiki M, Zhang Z, Guth L, Steward O (1996) Genetic influences on cellular reactions to spinal cord injury: activation of macrophages/microglia and astrocytes is delayed in mice carrying a mutation (WldS) that causes delayed Wallerian degeneration. J Comp Neurol 371: 469-484. [DOI] [PubMed] [Google Scholar]
- Gundersen HJG, Bendtsen TF, Korbo L, Marcussen N, Moller A, Nielsen K, Nyengaard JR, Pakkenberg B, Sorensen FB, Vesterby A, West MJ (1988) Some new, simple and efficient steriological methods and their use in pathological research and diagnosis. Acta Pathol Microbiol Immunol Scand 96: 379-394. [DOI] [PubMed] [Google Scholar]
- Imura T, Kornblum HI, Sofroniew MV (2003) The predominant neural stem cell isolated from postnatal and adult forebrain but not from early embryonic forebrain expresses GFAP. J Neurosci 23: 2824-2832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inman DM, Steward O (2003) Physical size does not determine the unique histopathological response seen in the injured mouse spinal cord. J Neurotrauma 20: 33-42. [DOI] [PubMed] [Google Scholar]
- Inman DM, Guth L, Steward O (2002) Genetic influences on secondary degeneration and wound healing following spinal cord injury in various strains of mice. J Comp Neurol 451: 225-235. [DOI] [PubMed] [Google Scholar]
- Johnson WB, Ruppe MD, Rockenstein EM, Price J, Sarthy VP, Vederber LC, Mucke L (1995) Indicator expression directed by regulatory sequences of the glial fibrillary acidic protein (GFAP) gene: in vitro comparison of distinct GFAP-lacZ transgenes. Glia 13: 174-184. [DOI] [PubMed] [Google Scholar]
- Kettenmann H, Ransom BR, eds (1995) Neuroglia. New York: Oxford UP.
- Larner AJ, Johnson AR, Keynes RJ (1995) Regeneration in the vertebrate central nervous system: phylogeny, ontogeny, and mechanisms. Biol Rev 70: 597-619. [DOI] [PubMed] [Google Scholar]
- Liu XZ, Xu XM, Hu R, Du C, Zhang SX, McDonald JW, Dong HX, Wu YJ, Fan GS, Jacquin MF, Hsu CY, Choi DW (1997) Neuronal and glial apoptosis after traumatic spinal cord injury. J Neurosci 17: 5395-5406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liuzzi FJ, Lasek RJ (1987) Astrocytes block axonal regeneration in mammals by activating the physiological stop pathway. Science 237: 642-645. [DOI] [PubMed] [Google Scholar]
- Ma M, Walters P, Stokes BT, Jakeman LB (2001) Behavioral and histological outcomes following graded spinal cord contusion injury in C57/Bl/6 mouse. Exp Neurol 169: 239-254. [DOI] [PubMed] [Google Scholar]
- Magnuson DS, Trinder TC, Zhang YP, Burke D, Morassutti DJ, Shields CB (1999) Comparing deficits following excitotoxic and contusion injuries in the thoracic and lumbar spinal cord of the adult rat. Exp Neurol 156: 191-204. [DOI] [PubMed] [Google Scholar]
- Manning Jr TJ, Rosenfeld SS, Sontheimer H (1998) Lysophsphatidic acid stimulates actomyosin contraction in astrocytes. J Neurosci Res 53: 343-352. [DOI] [PubMed] [Google Scholar]
- Mathis C, Hindelang C, LeMeur M, Borrelli E (2000) A transgenic mouse model for inducible and reversible dysmyelination. J Neurosci 20: 7698-7705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDonald JW, Althomsons SP, Hyrc KL, Choi DW, Goldberg MP (1998) Oligodendrocytes from forebrain are highly vulnerable to AMPA/kainate receptor-mediated excitotoxicity. Nat Med 4: 291-297. [DOI] [PubMed] [Google Scholar]
- Menet V, Prieto M, Privat A, Gimenez y Ribotta M (2003) Axonal plasticity and functional recovery after spinal cord injury in mice deficient in both glial fibrillary acidic protein and vimentin. Proc Natl Acad Sci USA 100: 8999-9004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Popovich PG, Guan Z, Wei P, Huitinga I, van Rooijen N, Stokes BT (1999) Depletion of hematogenous macrophages promotes partial hindlimb recovery and neuroanatomical repair after experimental spinal cord injury. Exp Neurol 158: 351-365. [DOI] [PubMed] [Google Scholar]
- Ramon y Cajal S (1928) Degeneration and regeneration of the nervous system. London: Oxford UP.
- Rapalino O, Lazarov-Spiegler O, Agranov E, Velan GJ, Yoles E, Fraidakis M, Solomon A, Gepstein R, Katz A, Belkin M, Hadani M, Schwartz M (1998) Implantation of stimulated homologous macrophages results in partial recovery of paraplegic rats. Nat Med 7: 814-821. [DOI] [PubMed] [Google Scholar]
- Reier PJ, Stensaas LJ, Guth J (1983) The astrocytic scar as an impediment to regeneration in the central nervous system. In: Spinal cord reconstruction (Kao CC, Bunge RP, Reier PJ, eds), pp 163-195. New York: Raven.
- Ridet JL, Malhotra SK, Privat A, Gage FH (1997) Reactive astrocytes: cellular and molecular cues to biological function. Trends Neurosci 20: 570-577. [DOI] [PubMed] [Google Scholar]
- Rubbin LL, Staddon JM (1999) The cell biology of the blood-brain barrier. Annu Rev Neurosci 22: 11-28. [DOI] [PubMed] [Google Scholar]
- Rudge JS, Silver J (1990) Inhibition of neurite growth on astroglial scars in vitro. J Neurosci 10: 3594-3603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Springer JE, Azbill RD, Knapp PE (1999) Activation of the caspase-3 apoptotic cascade in traumatic spinal cord injury. Nat Med 5: 943-946. [DOI] [PubMed] [Google Scholar]
- Wallace H, Ledent C, Vassart G, Bishop JO, Al-Shawi R (1991) Specific ablation of thyroid follicle cells in adult transgenic mice. Endocrinology 129: 3217-3226. [DOI] [PubMed] [Google Scholar]
- Young W (1993) Secondary injury mechanisms in acute spinal cord injury. J Emerg Med 11: 13-22. [PubMed] [Google Scholar]
- Zhang Z, Fujiki M, Guth L, Steward O (1996) Genetic influences on cellular reactions to spinal cord injury: a wound-healing response present in normal mice is impaired in mice carrying a mutation (Wlds) that causes delayed Wallerian degeneration. J Comp Neurol 371: 485-495. [DOI] [PubMed] [Google Scholar]
- Zhao X, Ahram A, Berman RF, Muizellar JP, Lyeth BC (2003) Early loss of astrocytes after experimental traumatic brain injury. Glia 44: 140-152. [DOI] [PubMed] [Google Scholar]