

Abstract
Mechanisms inducing neuronal death at defined times during embryogenesis remain enigmatic. We show in explants that a developmental switch occurs between embryonic day 12 (E12) and E13 in rats that is 72-48 hr before programmed cell death. Half the motoneurons isolated from peripheral tissues at E12 escape programmed cell death, whereas 90% of motoneurons isolated at E13 enter a death program. The surrounding somite commits E12 motoneurons to death. This effect requires macrophage cells, is mimicked by tumor necrosis factor α (TNFα), and is inhibited by anti-TNFα antibodies. In vivo, TNFα is detected within somite macrophages, and TNF receptor 1 (TNFR1) is detected within motoneurons precisely between E12 and E13. Although motoneuron cell death occurs normally in TNFα-/- mice, this process is significantly reduced in explants from TNFα-/- and TNFR1 -/- mice. Thus, embryonic motoneurons acquire the competence to die, before the onset of programmed cell death, from extrinsic signals such as macrophage-derived TNFα
Keywords: motoneuron, macrophages, somite, TNFα, apoptosis, developmental death
Introduction
During development of the vertebrate nervous system, more neurons are generated than are required to innervate their target cells (Oppenheim, 1991; Pettmann and Henderson, 1998). Excess neurons are eliminated by programmed cell death at specified times, usually just after the period of target innervation. The spinal motoneuron has been the locus classicus to study this process (Oppenheim, 1996). Developing myotubes are initially innervated by supernumerary motoneurons; however, 48 hr after contact with myotubes, excess neurons are pruned by programmed cell death (Oppenheim and Chu-Wang, 1977). Therefore, the number of motoneurons that escape cell death is matched to the number of myotubes that must receive innervation (McLennan, 1982). Many studies have demonstrated that neurotrophic factors produced by muscle cells are critical for motoneuron survival during this period (Oppenheim, 1991, 1996). Although factors promoting cell death are also involved in achieving the elimination of excess motoneurons (for references, see Davies, 2003), the limiting access to neurotrophic factors is believed to play the central role.
Contrasting with knowledge on the mechanisms of cell death during the developmental time window at which it occurs, upstream processes that instruct newly differentiated neurons to undergo cell death are poorly understood. Previous data suggest that an intrinsic death clock commits neurons to die unless they are saved by neurotrophic factors. For example, spinal commissural neurons isolated at various early developmental stages will die in vitro at the precise time they would have died in vivo if deprived of their targets (Wang and Tessier-Lavigne, 1999). Similarly, motoneurons removed from chick embryos within 2 d before the onset of programmed cell death will undergo cell death just as they would have in vivo (Mettling et al., 1995). The time of death seems to be tailored to match the time required for a particular neuron to reach its target. Sensory neurons from different cranial ganglia were cultured just after their genesis. Neurons, whose cell bodies were close to their targets in vivo, died more rapidly in vitro than neurons with distant targets (Vogel and Davies, 1991). Thus, in the “intrinsic clock model,” the onset of neuronal death appears to be a developmental process timed to coincide with the determination of whether the cell has innervated its target. This model does not exclude that extrinsic signals could tune the clock: transient application of BDNF on cultured cranial ganglionic neurons accelerated their cell death in vitro (Vogel and Davies, 1991).
Developmental cell death may also result from exposition to extrinsic factors. In the zebrafish embryo, the death of an identified primary motoneuron requires cooperative extrinsic signals (Eisen and Melançon, 2001). Another example has been documented in the horn worm Manduca Sexta, in which a defined set of motoneurons do not die unless exposed to a steroid hormone (Hoffman and Weeks, 1998). Experimental paradigms used to study motoneuron developmental death in chicks and rodents have not been designed specifically to discover such extrinsic factors. In chicks or rodent cell cultures, motoneurons were isolated relatively late in development, at a time when the cells have already been exposed to peripheral tissue for several days (Henderson et al., 1995). We also concluded, using rat spinal explants, that embryonic day 13 (E13) motoneurons possessed a death clock (Sedel et al., 1999); however, E13 rat motoneurons have already been exposed to the mesenchyme for 2 d. Spinal cord explants are an alternative to dissociated primary neuronal cultures and can be isolated from peripheral tissues at earlier stages. With this model, we demonstrate that a significant proportion of motoneurons in the spinal cord are not committed to cell death. Rather, these cells receive extrinsic signals that will commit them to die later. One of these signals is tumor necrosis factor α (TNFα), which is secreted from macrophages in the somite. Unlike death executive factors, TNFα acts upstream of programmed cell death as an early developmental signal for motoneuron death.
Materials and Methods
Explant cultures and treatment. Brachial regions of Sprague Dawley E12 (day 0 is the mating day) rat embryos (36-40 somites; Janvier) were dissected. Ectoderm was removed using tungsten needles to expose the dorsal neural tube. After incubation for 10 min with dispase (1 mg/ml; Roche Diagnostics) diluted in L15 medium (Invitrogen, San Diego, CA), somites and developing meninges were removed carefully with the dorsal two-thirds of the neural tube. For asymmetric explants, the dissection procedure was identical, except that a portion of the proximal somite including (explants used in Fig. 2) or excluding (explants used in Fig. 4) the ventral part of the dorsal root ganglia with the corresponding part of the sclerotome (see Figs. 2A, 4A) was kept. Dissection was performed in cold L15 medium supplemented with 10% horse serum (Invitrogen). Ventral explants (two per embryo) were cultured in four-well culture dishes coated with 50 μl of collagen prepared from rat tails. The culture medium contained Neurobasal medium (Invitrogen) supplemented with B27 (Invitrogen), β-mercaptoethanol (25 mm), l-glutamine (200 mm), penicillin-streptomycin (100 U/ml), and 5% horse serum. The same procedure was used for E13 rat embryos, except that dissection was performed without dispase. Dispase treatment did not modify motoneuron survival (data not shown).
Figure 2.
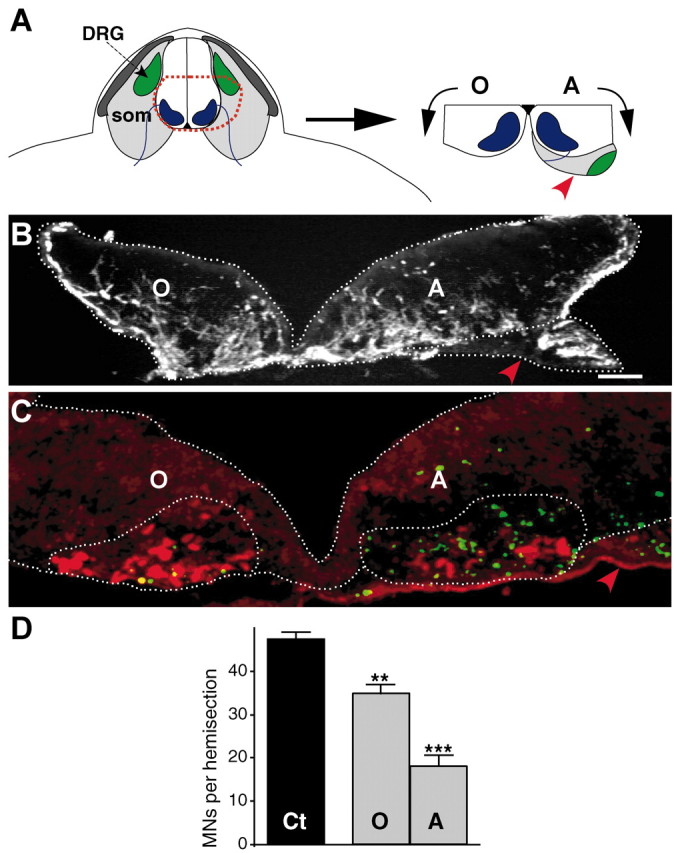
The proximal somite induces motoneuron death in spinal explants. A, Dissection of an E12 asymmetric explant. som, Somite; DRG, dorsal root ganglia; O, ventral horn opposite to the somite; A, ventral horn adjacent to the somite (red arrowhead). B, Transverse section of an E12 asymmetric explant cultured for 2 d in vitro. The explant section is immunolabeled with an anti-neurofilament antibody to show the position of the somite (red arrowhead). C, The section adjacent to that shown in B was double-labeled with TUNEL (green) and islet1/2 immunostaining (red). Note that there are numerous TUNEL- and fewer islet1/2-stained nuclei in the motor area (dotted) close to the somite (red arrowhead). Although some apoptotic nuclei are observed outside the motor area, most of them appear confined to the motor region (dotted). D, Quantification of surviving motoneurons (MNs) opposite (O) and adjacent (A) to the somite compared with control (Ct) E12 explants cultured for 2 d in vitro (8 explants quantified in each condition). **p < 0.01; ***p < 0.0001 (O and A vs Ct), Student's t test. Scale bars, 100 μm.
Figure 4.

Macrophages provide the death signal to E12 but not to E13 motoneurons. A, Scheme of dissection of an asymmetric explant in which the most proximal part of the somite was left in contact with one ventral horn (same abbreviations and representation as in Fig. 2A). B, Macrophages and motoneurons simultaneously labeled with anti-Iba1 (green) and anti-islet1/2 (red) antibodies on a 36-somite (E12) rat embryo section passing through the brachial region. Motoneuron areas (mn), somites (som), and dorsal root ganglia (drg, only visible on one side) are outlined (dotted line). Note that numerous macrophages are within the somite, and some of them (arrows) are also detected in the proximal somite and occasionally within the neural tube (asterisks). C1-D2, Asymmetric explants cultured for 2 d without (C1, C2) or with (D1, D2) the macrophage-specific toxin IB4sap. Triple labeling identifying Iba1 (C1, D1), islet1/2 (C2, D2, red) immunoreactivities and TUNEL (C2, D2, green) are shown. The position of the somite is indicated by a large arrow, and Iba1-positive macrophages are indicated by small arrows. E, F, Quantification of macrophages (E1, F1) and motoneurons (E2, F2) on same sections from E12 (E1, E2) and E13 (F1, F2) explants cultured for 2 d. E12-ct, Explants cultured without somite; ct-asym-O, ct-asym-A, asymmetric explants cultured without IB4sap, ventral horn opposite (ct-asym-O) and adjacent (ct-asym-A) to the somite; IB4sap-O, IB4sap-A, asymmetric explants cultured with IB4sap, ventral horn opposite (IB4sap-O) and adjacent (OB4sap-A) to the somite. Note that in E12 explants, the somite prodeath effect is abolished in IB4sap-treated explants, which contain almost no macrophages. In E13 explants, the elimination of macrophages has no effect on motoneuron survival. ***p < 0.0001, Student's t test. Numbers of explants quantified in each condition are indicated on the histograms. Scale bars: B, 250 μm; C1-D2, 100 μm.
For soluble tissue extracts, embryonic tissues were homogenized in 400 μl of L15 medium and centrifuged at 15,000 × g for 45 min. The supernatant was assayed for protein concentration and used at final concentration of 125 μg/ml. Conditioned medium was prepared from ∼200 whole somites cultured for 2 d in 1 ml of culture medium (see above). The medium was centrifuged for 5 min at 2000 × g. The supernatant was diluted 1:1 in fresh culture medium before use.
Explants were treated with the following exogenous factors or blocking antibodies: recombinant rat TNFα produced in baculovirus-infected insect cells (PharMingen, San Diego, CA), soluble Fas ligand (Alexis Corp.), NGF (Tebu), bone morphogenetic protein 4 (BMP4) (a kind gift from Dr. C. Henderson, Institut Nacional de la Santé et de la Recherche Médicale U382, Marseille, France), CNTF (Tebu), rabbit anti- mouse TNFα (10 μg/ml; Biosource, Camarillo, CA), FasFc (4 μg/ml; Alexis Corp.), anti-TGFβs (monoclonal antibody 1835, 10 μg/ml; R & D Systems, Minneapolis, MN), the laspase inhibitor OEVD-CHO (20 μm; Enzyme Systems Products, Livermore, CA), and anti-p75 low-affinity neurotrophin receptor (1:100; Rex; Weskamp and Reichardt, 1991). To eliminate macrophages, explants were treated with isolectin B4 saporin (1 μg/ml; Advanced Targeting Systems) for the first 24 hr in vitro and incubated another 24 hr with control medium
Double terminal deoxynucleotidyl transferase-mediated biotinylated UTP nick end-labeling staining and immunochemistry. Explants were fixed for 2 hr in 4% paraformaldehyde (PFA) and PBS at 4°C, transferred to 30% sucrose and PBS overnight, and frozen in a Tissue-Tek cryostat (Oxford Laboratories). Transverse cryostat sections, 12 μm thick, were mounted on Superfrost Plus glass slides (Menzel), washed for 5 min in PBS and 0.1% Triton X-100, preincubated for 15 min with terminal deoxynucleotidyl transferase (TdT) buffer (Roche Diagnostics), and then incubated for 4 hr at 37°C in the terminal deoxynucleotidyl transferase-mediated biotinylated UTP nick end-labeling (TUNEL) reaction solution as described (Sedel et al., 1999). Slides were then washed in PBS and incubated for 30 min with PBS, 0.2% gelatin, and 0.1% Triton X-100 (PBGT) and then overnight at 4°C with the 4D5 monoclonal mouse anti-islet1/2 antibody (1:100; Developmental Studies Hybridoma Bank) and streptavidin-FITC (1:500; Amersham Biosciences, Arlington Heights, IL) diluted in PBGT. After three washes in PBS, sections were incubated for 2 hr at room temperature with a goat anti-mouse IgG coupled with cyanin 3 (CY3) (Jackson ImmunoResearch, West Grove, PA) diluted 1:500 in PBGT. Sections were washed three times in PBS and mounted in Vectashield (Vector Laboratories, Burlingame, CA). The same procedure was used for neurofilament immunochemistry, except that the TdT reaction was omitted, and the 4D5 antibody was replaced by the 2H3 monoclonal antibody (Developmental Studies Hybridoma Bank). For double or triple immunolabeling experiments using an anti-Iba1 antibody (dilution, 1:1000; a gift from Dr. Y. Imai, National Institute of Neuroscience, Tokyo, Japan), comparable procedures were used. For TNF receptor 1 (TNFR1) labeling, embryos were fixed for 1 hr and processed as described above, using a polyclonal rabbit anti-TNFR1 antibody (1:100; Santa Cruz Biotechnology, Santa Cruz, CA) and a biotinylated goat anti-rabbit antibody (1:500; Vector Laboratories). The immunoperoxidase reaction was achieved with an ABC kit (Vector Laboratories).
In situ hybridization. Embryos were fixed for 1 hr in 4% PFA and PBS at 4°C and then processed for cryostat sections as described above. Hybridization and washes were performed as described (Schaeren-Wiemers and Gerfin-Moser, 1993). For synthesis of TNFα mRNA probes, the N-terminal portion (100 bp) of human TNFα cDNA (86% homology with rat TNFα cDNA) was subcloned into the pBluescript KS+ plasmid. Digoxigenin-labeled antisense and sense probes were synthesized using T3 and T7 RNA polymerases, respectively. For combined in situ hybridization with immunochemistry, in situ hybridization was followed by incubating sections for 30 min in PBGT and for the next 4 hr at room temperature with biotin-conjugated isolectin-B4 (10 μg/ml; Sigma, St. Louis, MO) and anti-islet1/2 (4D5, 1:50) diluted in PBGT. Sections were washed three times in PBS, incubated for 2 hr at room temperature with streptavidin FITC (1:500) and CY3-conjugated goat anti-mouse secondary antibody (1:500), washed again, and mounted in Vectashield.
TNFα-/- and TNFR1-/- mice. Homozygous TNFR1 null mutation mice generated on a 129 background and backcrossed with C57BL/6 (B6) mice for >10 generations were kindly provided by Dr. Horst Bluethmann (Hoffmann-La Roche, Basel, Switzerland; Rothe et al., 1993). TNFα-/- mice (Marino et al., 1997) backcrossed on a B6 genetic background were obtained from CDTA (Orleans, France). B6 mice obtained from Charles River were used as the wild-type controls.
Quantifications. For in vitro experiments, each fifth section was analyzed with a Leica (Nussloch, Germany) epifluorescence microscope (∼10 sections per explant). Quantification was performed on digitized images focused on one motoneuron area. For each section, islet1/2 and TUNEL images were superimposed using Adobe Photoshop software. Both islet1/2-positive, TUNEL-negative nuclei (i.e., surviving motoneurons) and TUNEL-positive nuclei within the motor area were quantified, and mean numbers per section were calculated for each explant. These means were used for statistical analysis. Each value on histograms represents the mean of means ± SEM from at least three independent experiments.
For in vivo motoneuron quantification, brachial embryonic regions of wild-type and TNFα-/- mice were fixed in 4% PFA and PBS and cut into 10-μm-thick cryostat sections as described above. E12.5 and E14.5 embryonic sections were processed for islet1/2 immunochemistry with a polyclonal rabbit anti-islet1/2 antibody (K5; a kind gift from T. M. Jessell, Howard Hughes Medical Institute, Columbia University, New York, NY) as described above. At postnatal day 0, when most motoneurons are islet1/2-negative, they were identified in Nissl-stained sections. Counts were made in a single lateral motor column for each spinal cord.
Results
Motoneurons become committed to cell death at day E13
In the rat brachial spinal cord, neuronal precursors differentiate into ventral horn motoneurons between embryonic day 10 (E10) and E12 (Altman and Bayer, 1984). Motor axons start to leave the spinal cord at E11, contact muscle cells at E13, and form end plates at E14. From E15 to E17, ∼50% of the motoneurons die (Harris and McCaig; 1984; Oppenheim, 1986). If motoneuron death is triggered exclusively by an intrinsic clock, then one can make the following predictions: (1) motoneurons isolated at E13 should activate programmed cell death after 2 d in vitro, at the equivalent of E15; and (2) motoneurons isolated at E12 should activate programmed cell death after 3 d in vitro. We tested these predictions by quantifying motoneuron survival and death in cultured ventral spinal cord explants. Motoneuron nuclei were labeled with islet1/2 immunostaining (Tsuchida et al., 1994). Dying cells were identified in the same explant sections by TUNEL labeling of apoptotic nuclei (Gavrieli et al., 1992). As shown in Figure 1, TUNEL-stained nuclei in explant motor areas were weakly labeled or negative for islet1/2 immunostaining, suggesting that islet1/2 expression is downregulated in apoptotic motoneurons. Therefore, surviving motoneurons in each explant section were defined as intact nuclei stained positively with anti-islet1/2 antibodies but negatively with TUNEL in superimposed digitized images (Fig. 1C). Both surviving motoneurons (Fig. 1F) and TUNEL-stained nuclei (Fig. 1G) were counted. As expected, almost all motoneurons died in E13 explants deprived of exogenous neurotrophic factors (Fig. 1B,C,F,G). After 2 d in vitro (equivalent to E15), 70% of motoneurons were lost, and by 3 d in vitro (equivalent to E16), 90% had died (Fig. 1B,C,F). Similarly, 90% of motoneurons die by E16 in vivo if deprived of muscle containing survival factors (Kablar and Rudnicki, 1999). Losses of islet1/2-positive nuclei in E13 explants were mirrored by a proportional increase in the number of TUNEL-stained nuclei in the motor area (Fig. 1B,C,G). Note that 55% of motoneurons were lost at the equivalent of E14 (after only 1 d in vitro). This initial wave of death is likely to be caused by both the dissection and programmed cell death.
Figure 1.
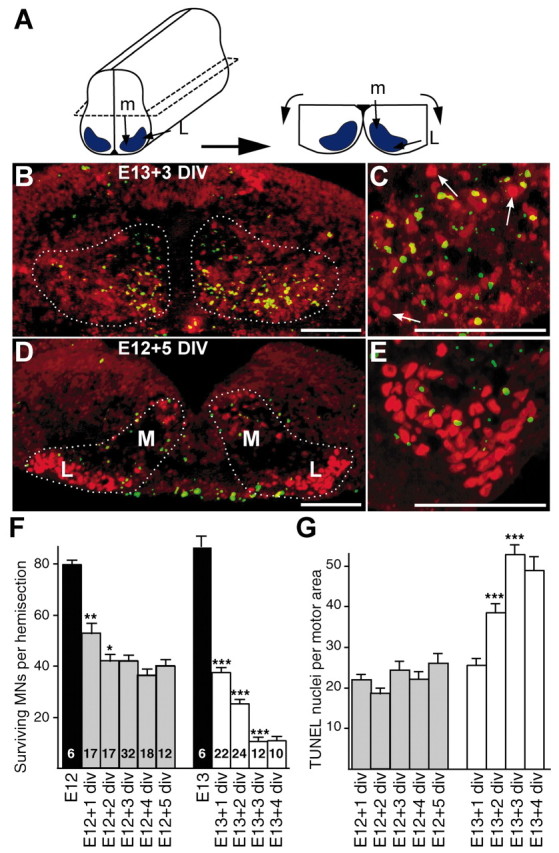
Motoneuron cell death in E12 and E13 ventral horn explants cultured in vitro. A, Dissection of an E12 ventral horn explant. Motoneuron areas are in blue. The relative positions of medial (m) and lateral (L) motoneurons are indicated. B-E, Explant sections were colabeled with TUNEL staining of apoptotic nuclei (green) and islet1/2 immunolabeling of motoneuron nuclei (red). B, C, Transverse sections of an E13 explant cultured for 3 d in vitro (DIV) at low (10×; B) and high (25×; C) magnifications. Arrows indicate some surviving motoneurons, e.g., intact nuclei positive for islet1/2 but negative for TUNEL. D, E, E12 explant cultured for 5 DIV at low (D) and high (E) magnification. Note that more surviving motoneurons are observed in E12+5 DIV explants compared with E13+3 div explants. Note also that in E12 explants the localization of apoptotic nuclei and of motoneuron loss corresponds to the medial part of the motor area. The observed number of apoptotic nuclei in explants is higher than in vivo at similar embryonic stages (Kablar and Rudnicki, 1999), suggesting that, in contrast to the in vivo situation, TUNEL-positive cells are not cleared by phagocytes in explant cultures. F, Absolute numbers of surviving motoneurons (MNs) in E12 and E13 explants cultured for the indicated numbers of days (+n) in vitro. The number of explants in each condition is marked inside the bars of histograms. G, Quantification of TUNEL-positive nuclei in same explants as in F. For both E12 and E13 explants, each value obtained for a given time of culture was compared with the value from the previous day. *p < 0.05; **p < 0.01; ***p < 0.0001, Student's t test. Scale bars, 100 μm.
In E12 explants, ∼35% of the motoneurons were lost during the first day after explantation (Fig. 1F). Motoneuron loss and apoptotic nuclei were in the most medial portion of the motor area (data not shown), where motoneurons are the first to die in vivo (Gould et al., 1999; Yamamoto et al., 1999). These motoneurons could be more vulnerable to the dissection procedure, programmed cell death, or both. However, no additional wave of death was observed in the E12 explants at the equivalent of E15 (Fig. 1D). Even after 5 d in vitro (equivalent to E17), motoneuron survival remained at ∼50% of the starting population (Fig. 1D-F). The large number of living motoneurons in E12 explants compared with E13 explants was not attributable to different starting population sizes. First, in accordance with Kablar and Rudnicki (1999), motoneuron numbers in E12 and E13 explants were the same at the time of dissection based on islet1/2 expression (Fig. 1F). Second, the number of dying cells determined by TUNEL labeling was decreased in the E12 explants, directly demonstrating that cell death is reduced (Fig. 1G). Thus, at the equivalent of E17, only 50% of motoneurons have died in E12 explants despite the absence of muscle-derived neurotrophic factors, whereas in E13 explants, 90% of motoneurons have died. Together, these results indicate that (1) the program that commits rat motoneurons to cell death is fully activated not at E12 but at E13; and (2) a large proportion of motoneurons do not autoactivate this program if they are isolated from peripheral tissues at E12. These data already suggested that developmental cell death is not an entirely intrinsic differentiation process but also depends on extrinsic signals from peripheral tissues.
Cell death of motoneurons is induced by a mesenchyme-derived factor
The somitic mesenchyme lies adjacent to motoneuron somata between E12 and E13 (Fig. 2A). To test whether a signal from this tissue could initiate a death program in E12 motoneurons, we examined motoneuron survival and death in E12 spinal cord-somite explants (Fig. 2). In these asymmetric explants, one ventral horn remained in contact with the proximal somite. Contralateral motoneurons, separated from peripheral tissue, served as internal controls (Fig. 2A,B). After 2 d in vitro, many apoptotic nuclei were observed in the motor area adjacent to the mesenchyme (Fig. 2C). Only 37.5% of neurons survived, compared with explants without the attached mesenchyme tissue (p < 0.0001; Fig. 2D). Although some cell death was observed in the ventral horn opposite to the attached mesenchyme, most motoneurons survived; specifically, 73% of the cells survived compared with simple explants of the ventral horn (p < 0.01; Fig. 2D). These results indicate that the proximal mesenchyme is able to drive a death program in E12 motoneurons. Furthermore, the signal from the mesenchyme appears to be diffusible because motoneuron survival in the opposite ventral horn was also decreased.
If such a diffusible factor exists, mesenchyme conditioned medium should be able to initiate cell death. When E12 ventral explants were treated for 2 d with conditioned medium prepared from cultured E12 somites, motoneuron numbers were decreased by 39% compared with controls (Fig. 3B,D). Because cultured somites may release nonspecific toxic factors in the culture medium, we also tested the effect of soluble extracts from noncultured E12 somites. Again, motoneuron numbers were decreased by 35% in E12 explants cultured for 2 d with these extracts (Fig. 3C,D). This was accompanied by an increase in TUNEL-stained nuclei in the motoneuron area (Fig. 3C). Interestingly, the effect of somite extracts was totally abolished after heat denaturation, suggesting that the signal is a diffusible protein. The motoneuron death induced by this protein involves caspase activation because it was inhibited by the caspase inhibitor DEVD-CHO (20 μm; Li et al., 1998) (Fig. 3D). By contrast, soluble extracts prepared from limb bud, heart, or E13 body wall did not enhance cell death (Fig. 3D). Our data therefore suggest that a diffusible protein present in the somite mesenchyme at E12 commits motoneurons to cell death.
Figure 3.

Induction of motoneuron death by a somite-derived soluble protein. A-C, Double islet1/2-TUNEL labeling of E12 explants cultured for 2 d with culture medium alone (A), somite conditioned medium (B), or soluble somite extracts (C). Numerous apoptotic nuclei (green) and few surviving motoneurons (red) are present in the motor area of somite-treated explants. D, Numbers of surviving motoneurons (MNs) in E12 explants cultured for 2 d with control medium or medium supplemented with various soluble extracts as indicated. The number of explants quantified is indicated inside the bars of the histogram. **p < 0.01; ***p < 0.0001, Student's t test. Scale bar, 100 μm.
Macrophage cells produce the death signal from the somite
Using asymmetric explant experiments (see above), we observed that the most proximal part of the somite could induce motoneuron cell death in the adjacent ventral horn but not in the opposite one (Fig. 4). In these asymmetric explants, the somite prodeath effect was weaker than that observed when larger portions of the proximal somite were included (compare Figs. 2D, 4E2). This most proximal part of the somite corresponds partly to the developing meninges, which start to differentiate at ∼E12 in rats (Kamiryo et al., 1990). Developing meninges contain macrophages (Cuadros et al., 1992) known to induce cell death in cultured cortical neurons (Thery et al., 1991) or in the retina (Frade and Barde, 1998). Therefore, we postulated that embryonic macrophages could be the source of the somite-derived motoneuronal prodeath signal. In our experiments, macrophages were identified with an anti-Iba1 antibody (Ito et al., 1998). In 36-somite old rat embryos (corresponding to E12), numerous Iba-1-positive macrophages were found in the somite mesenchyme (Fig. 4B). These cells were dispersed throughout the somite, including its proximal portion and occasionally within the neural tube (Fig. 4B). In E12 asymmetric explants with the most medial somite cultured for 2 d, there were 2.5 times more macrophages in the side containing the proximal somite than in the opposite one (Fig. 4C1, quantified in E1). To evaluate whether these macrophages were responsible for the induction of motoneuron cell death, explants were cultured for 2 d with the toxin isolectin B4-saporin (IB4sap), which specifically binds macrophages (Maddox et al., 1982) and kills them already after 12 hr (data not shown). Explants were analyzed using double-labeling experiments for simultaneous identification of motoneurons (islet1/2) and macrophages (Iba1). When these explants were cultured for 2 d without IB4sap, the number of motoneurons in the somite side was 60% of that in the opposite side (p < 0.0001; Fig. 4C2,E2). In contrast, when explants were cultured for the first 24 hr with IB4sap and then 24 hr without IB4sap, the prodeath effect of the proximal somite was totally abolished (Fig. 4D2,E2). Despite a dramatic reduction, rare macrophages were occasionally observed in IB4sap-treated explants (Fig. 4E1) but did not induce significant motoneuron death. Altogether, these experiments underline the involvement of macrophages in the prodeath activity of the proximal mesenchyme on E12 motoneurons.
Interestingly, when the macrophages were killed in E13 explants cultured for 2 d, motoneuron numbers remained unaffected (Fig. 4F1,F2). Therefore, macrophages are instrumental at E12 but not at E13 for motoneuron developmental cell death.
This suggests that macrophages are involved in the developmental switch that commits motoneurons to cell death between E12 and E13 rather than in the active killing of motoneurons after E13.
TNFα is a diffusible activator of motoneuron cell death
To identify the mesenchyme-derived death-inducing factor, we screened known proapoptotic diffusible signaling proteins using the induction of cell death in E12 explants as our bioassay. We found that TNFα added at 10 ng/ml for 2 d induced 25% of the motoneurons to die compared with control E12 explants cultured for 2 d with medium alone (p < 0.0001; Fig. 5A). When TNFα was added at 100 ng/ml, 35% of the motoneurons died compared with controls (Fig. 5A). This decrease in motoneuron survival was in the same range as that obtained with somite extracts (Fig. 3D). The decrease in survival was mirrored by an increasing number of TUNEL-stained nuclei in the motor area of TNFα treated explants (data not shown). Cell death was not induced by BMP4 (Graham et al., 1994), NGF (Sedel et al., 1999), soluble Fas ligand (Raoul et al., 1999), and CNTF (Kessler et al., 1993), tested at concentrations reported to induce neuronal apoptosis. Thus, exogenous TNFα specifically mimics the prodeath effect of somite extracts on E12 motoneurons.
Figure 5.
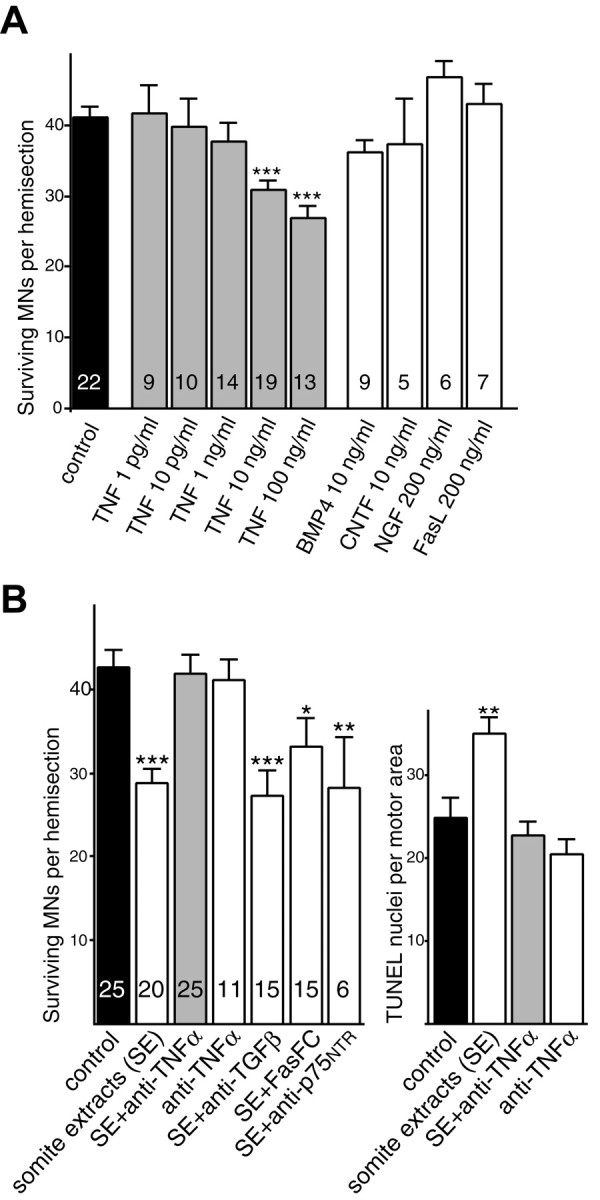
TNFα activates a death program in E12 motoneurons, and the somite prodeath effect is specifically inhibited by anti-TNFα blocking antibodies. A, E12 explants were treated for 2 d with increasing concentrations of TNFα or other proteic factors known to trigger neuronal death. B, E12 explants were treated for 2 d with somite extracts alone, somite extracts preincubated with anti-TNFα antibodies, or other blocking antibodies as indicated. An analysis of double islet1/2-TUNEL labeling experiments is shown. Note that anti-TNFα reverses the somite prodeath effect. Values corresponding to each test condition are compared with the control condition (black bar). **p < 0.01; ***p < 0.0001, Student's t test.
Moreover, TNFα is necessary for activation of the death program by the somite. In E12 explants treated for 2 d with somite extracts, islet1/2-positive nuclei counts decreased by 35%, and TUNEL profiles within the motor area increased by 40% compared with controls (Fig. 4B). This effect was totally abolished when somite extracts were pretreated with anti-TNFα blocking antibodies (10 μg/ml; Fig. 5B). Importantly, anti-TNFα alone did not modify motoneuron survival, indicating that the protective effect of these antibodies does not result from a direct action on motoneurons (Fig. 5B). Furthermore, blocking agents against TGFβs (Krieglstein et al., 2001), Fas ligand (Raoul et al., 1999), and NGF signaling (Sedel et al., 1999), which have been reported to inhibit motoneuron apoptosis at later embryonic stages, did not suppress the death effect of E12 somite extracts (Fig. 5B). These results indicate that TNFα is specifically required for the somite-dependent activation of a death program in E12 motoneurons.
TNFα is expressed within macrophages and TNFR1 is expressed within motoneurons during spinal cord development in rats
In the simplest model, TNFα secreted by somite macrophages commits motoneurons to cell death between E12 and E13 in rats. Thus, TNFα should be expressed in macrophages, and it should be expressed in these cells at E12. TNFα-producing cells were identified using in situ hybridization. Digoxigenin-labeled antisense and sense RNA probes were synthesized from the N-terminal region of TNFα cDNA. Before the 36-somite stage (corresponding to E12), no TNFα mRNA labeling was observed in the environment of the developing neural tube (Fig. 6B; data not shown). After the 36-somite stage, some TNFα-expressing cells were observed within the proximal somite close to, but outside, the neural tube (Fig. 6A). The TNFα-positive cells were round in shape and were located both surrounding the ventral horn near the motor area and also near the intermediate and dorsal parts of the cord. Labeled cells were also found rarely within the neural tube (Fig. 6A; data not shown). Expression of TNFα close to the neural tube was observed at all stages examined between E12 and E12.5 (36-41 somites). However, by E13, TNFα expression was no longer detected in the vicinity of the neural tube (Fig. 6C). This transient expression of TNFα mRNA was previously reported by Ohsawa and Natori (1989) using Northern analysis in mouse embryos.
Figure 6.
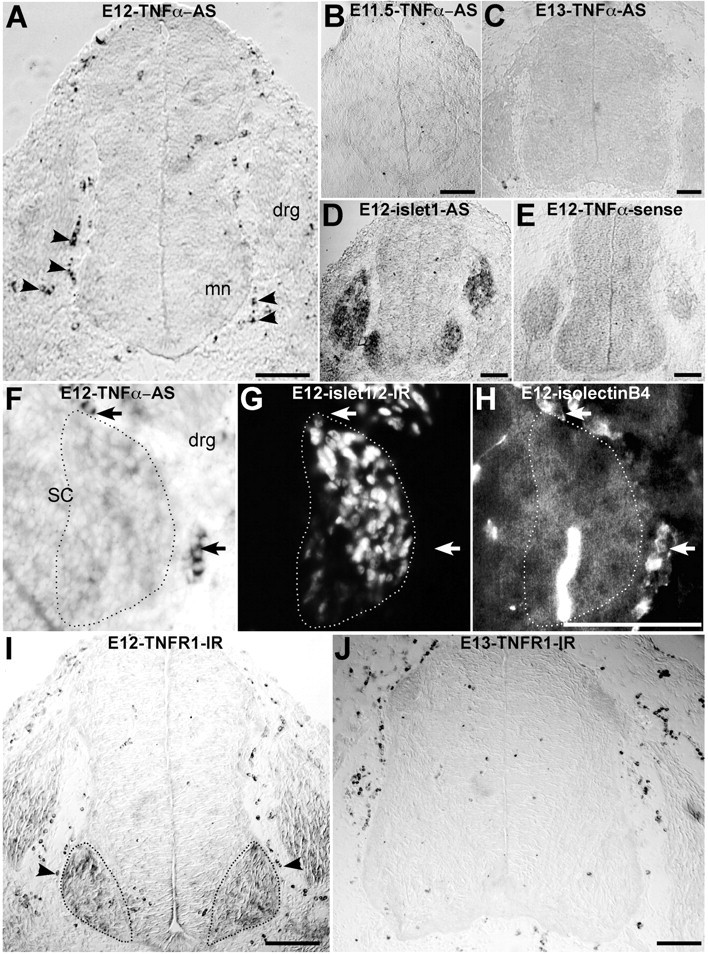
Transient TNFα mRNA and TNFR1 protein expression in spinal cord region during embryonic development in rats. A-C, In situ hybridization to detect TNFα mRNA with digoxigenin-labeled TNFα antisense (AS) probe. A, E12 (38 somites); B, E11.5 (31 somites); C, E13 spinal cord sections at the brachial level. Note that at E12, numerous round-labeled cells (arrowheads) are located close to the neural tube in the somite. Labeling is absent at E11.5 and E13. D, E, In situ hybridization of an E12 section labeled with islet-1 antisense (AS) probe, which labels motoneurons and dorsal root ganglia (D) and TNFα sense probe (E) as a negative control. F-H, E12 section at the brachial level triple-stained with TNFα antisense probe (F), Islet1/2 immunolabeling (G), and isolectin B4 (H). TNFα-expressing cells (arrows) are located in the somite close to motoneurons (G, dotted) and are stained with the macrophage marker isolectin B4 (H). SC, Spinal cord; DRG, dorsal root ganglia. I, J, TNFR1 immunoperoxidase of E12 (I) and E13 (J) spinal cord sections at the brachial level. On E12 (I), immunoreactivity (IR) is detected in the motor area (dotted), in the dorsal root ganglia, and in macrophage cells (arrowheads), whereas on E13 (J), immunoreactivity is only detected within macrophages. Scale bars, 100 μm.
To determine whether TNFα-expressing cells are macrophages, we labeled E12 embryo sections simultaneously with isolectin B4 to identify macrophages, a TNFα antisense RNA probe to identify TNFα-expressing cells, and islet1/2 antibodies to identify motoneurons. All cells that expressed TNFα mRNAs were also colabeled with isolectin B4 (Fig. 6F-H; data not shown). However, the converse was not true; not all isolectin B4-labeled cells expressed TNFα, suggesting that TNFα expression is restricted to a subpopulation of macrophages or that, because of the transient expression of TNFα within macrophages, this expression is only observed in a subset of macrophages at a given time. The arrest of TNFα expression around the neural tube at E13 was not attributable to migration of macrophages because isolectin B4-labeled cells (but TNFα-negative) were still detected at this stage (data not shown; Fig. 6J). Therefore, macrophages located in the proximal somite around the motor area only transiently express TNFα in vivo between E12 and E13.
TNFα mediates its intracellular effects by binding to the cognate receptors TNFR1 and TNFR2, of which TNFR1 is primarily responsible for its death-inducing activity (Vandenabeele et al., 1995). TNFR1 expression was studied in the rat spinal cord by immunochemistry. At E12, TNFR1 immunoreactivity was observed within the motor area and appeared not to be restricted to any specific motoneuron pool (Fig. 6I). The TNFR1 immunoreactivity was weak and restricted to somata. Our results therefore favor an action of macrophage-derived TNFα via somatic TNFR1, although one cannot exclude the possibility that TNFR1 was also expressed in axons but below the immunodetection threshold.
Expression of TNFR1 was also detected within macrophages in the proximal somite and in dorsal root ganglia (Fig. 6I). At E12, TNFR2 expression was only detected within macrophages, as revealed by in situ hybridization (data not shown). Interestingly, TNFR1 expression was no longer detected in the motor area from E13 onward (Fig. 6J), indicating that, as for TNFα, TNFR1 expression is transient. Therefore, motoneurons specifically express TNFR1 between E12 and E13, at the same time that TNFα is expressed within macrophages in the proximal somite and motoneurons become committed to cell death.
Studies on TNFα-/- and TNFR1 -/- mice reveal a role for TNFα in the early commitment of motoneurons to cell death
To further analyze the role of TNFα, we examined both the motoneuron commitment to cell death using explant cultures and the period of motoneuron developmental death in TNFα-/- and TNFR1 -/- mice. Developmental stages in rat embryos are extended by 1.5 d compared with mouse embryos (Butler and Juurlink, 1987). Thus, E12 rat embryos are at the equivalent stage of E10.5 mouse embryos (36 somites). This stage just precedes the in vivo exposure to TNFα and the commitment to cell death (see above). Similarly, E13 rat embryos are at the equivalent stage of E11.5 mouse embryos, i.e., potentially after exposure to TNFα and the commitment to cell death have occurred. Motoneuron death was quantified by counting TUNEL-stained nuclei in the motor area of explant sections. When explants were isolated from E11 wild-type mouse embryos and cultured for 2 d in vitro, two times more TUNEL profiles were detected compared with E10.5 explants cultured for 2.5 d in vitro (Fig. 7; p < 0.0001). These results suggest that, as in rats, mouse motoneurons become committed to cell death between E10.5 and E11. By contrast, in TNFα-/- or TNFR1 -/- mice, numbers of apoptotic nuclei were the same in both E10.5 explants cultured for 2.5 d and in E11 explants cultured for 2 d in vitro (Fig. 7). Thus, motoneurons do not become committed to cell death at E11 in TNFα-/- and TNFR1 -/- mice, demonstrating that both TNFα and TNFR1 are required in this early process.
Figure 7.
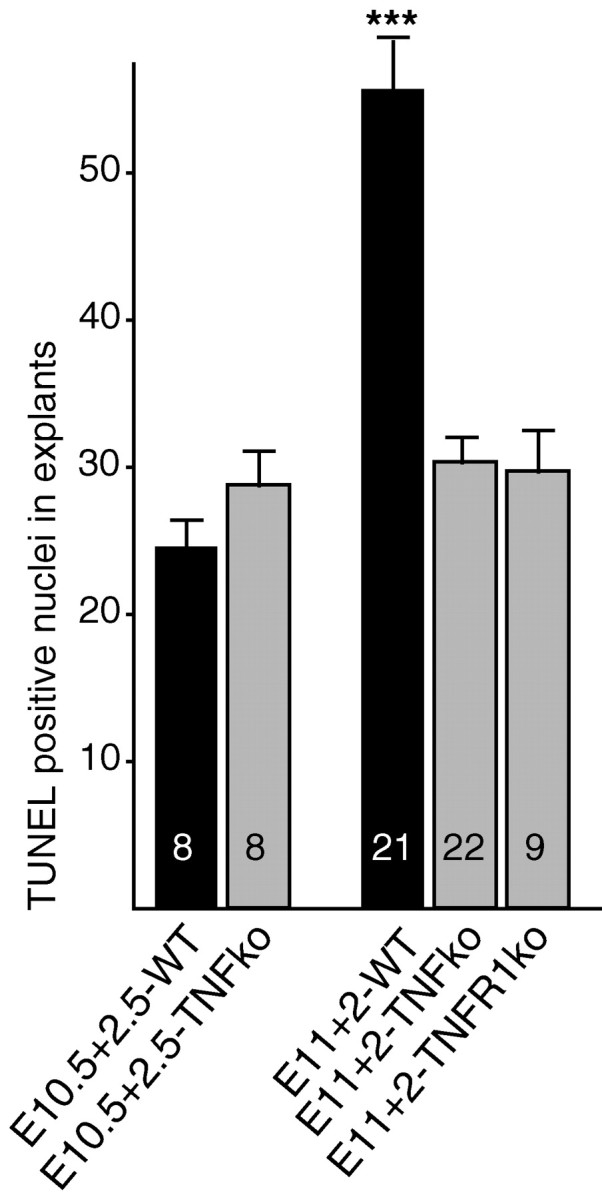
Motoneuron death in TNFα-/- and TNFR1 -/- mouse explants. A comparison of apoptotic nuclei numbers per motor area in E10.5 explants cultured for 2.5 d in vitro (E10.5+2.5) or in E11 explants cultured for 2 d (E11+2) is shown. Note that in explants from wild-type mice (WT), there are twice as many apoptotic nuclei in E11+2 explants than in E10.5+2.5 explants. In contrast, no difference in apoptotic nuclei numbers is observed between E11+2 and E10.5+2.5 explants from both TNFα-/- and TNFR1 -/- mice. ***p < 0.0001, Student's t test. ko, Knock-out.
Previous in vivo studies have shown that motoneuron death occurs principally between E13 and E15 in the brachial region of the mouse spinal cord (Lance-Jones, 1982; Yamamoto and Henderson, 1999). Brachial motoneurons were identified and quantified in both wild-type and TNFα-/- mouse embryos by using islet1/2 immunostaining on E12.5 and E14.5 and Nissl staining on postnatal day 0. Unfortunately, the analysis revealed no difference in motoneuron numbers between wild-type and TNFα-/- mouse embryos (data not shown). The same negative results were observed with TNFR1-/- and double TNFR1/TNFR2-/- mice (data not shown). As discussed below, these results suggest that compensatory mechanisms can trigger motoneuron death in vivo when TNFα or TNF receptor genes are lacking.
Discussion
Our results indicate that the process that causes motoneurons to undergo programmed cell death at a specific time in development is not exclusively an intrinsic differentiation process. The surrounding macrophages constitute an early source of death-inducing signals. TNFα secreted from macrophages in the somite mesenchyme is one of the signaling molecules, and its effects on motoneurons are mediated through the TNFR1 receptor.
The motoneuron commitment to cell death requires extrinsic signals
Very little is known about the process that commits motoneurons to undergo programmed cell death. Evidence from previous in vivo and in vitro work indicates that motoneurons die in an orderly manner depending on when they are born, earliest born motoneurons of the medial motor column dying ∼3 d before latest born motoneurons of the lateral motor column (Mettling et al., 1995; Gould et al., 1999; Yamamoto et al., 1999). This fixed interval between neuron birth and death might depend on an intrinsic clock set within the neuroepithelium. However, in in vivo experiments, motoneurons were still in association with adjacent tissues. Similarly, in cell cultures, motoneurons are isolated relatively late in development. We have used explant cultures to determine the time after which motoneurons will die in vitro if deprived of target-derived neurotrophic factors. Our experiments indicate that a developmental switch occurs in vivo between E12 and E13 in rats: whereas only 10% of E13 motoneurons in explants survive after 3 d, 50% of E12 motoneurons escape cell death even after 5 d in vitro. Furthermore, part of the motoneuron loss in explants probably results from the explantation procedure, suggesting that >50% of E12 motoneurons are unable to autonomously activate a death program. This indicates for the first time that the countdown for death of motoneurons is not activated from neurogenesis, which occurs at E10-E12, but at ∼E13, which is 2 d before the onset of programmed cell death (Altman and Bayer, 1984; Oppenheim, 1986). Almost all motoneurons activate a death program if they are isolated after this critical point in development; if they are isolated 24 hr before, most of them remain uncommitted to cell death, unless they are exposed to extrinsic factors such as TNFα. Interestingly, TNFα induces the death of only 35% of motoneurons in E12 explants, indicating that other extrinsic factors are required to commit all motoneurons to a cell death fate.
Somite macrophages provide an early death signal to motoneurons
The appearance of macrophages within the developing brain often coincides with cell death phenomena (Cuadros and Navascues, 1998). In many cases, brain macrophages migrate to regions of cell death where they phagocyte cellular debris. Macrophages can themselves induce cell death by secreting molecules such as reactive oxygen intermediates, glutamate, and NGF (Thery et al., 1991; Giulian et al., 1993, Frade and Barde, 1998). Similarly, in the nematode Caenorhabditis elegans, extrinsic signals from ungulfing cells are implicated in the execution of motoneuron programmed cell death (Reddien et al., 2001). Here we show that the proximal somite provides a death signal to E12 motoneurons, and this prodeath effect is mediated by macrophages. These results are in apparent contradiction with those of Phelan and Holliday (1991), who found that the in vivo extirpation of the somite in E2 chick embryos induced additional death in the operated side, indicating that the somite has a trophic rather than a killing effect on motoneurons. However, in the chick, macrophages originating in the yolk sac are first seen near the neural tube from E3 onward (Cuadros et al., 1992). Therefore, the somite removal at E2 is not likely to prevent the subsequent migration of macrophages through remaining operated tissues. As a consequence, the additional motoneuron death seen in vivo on the operated side is likely to result from death factors issued from macrophages combined with the absence of neurotrophic factors derived from muscle cells, which normally differentiate in the external portion of the somite.
Macrophage-derived TNFα acts before motoneuron programmed cell death
Our results identify a new function for macrophages to commit a neuronal population to a cell death fate 2 d before this process begins in vivo. This function is mediated by TNFα between E12 and E13 in rats, and TNFα is not involved in the execution of the cell death program after E13. Actually, at E12 in rats, TNFα is present in somite macrophages surrounding motoneurons, which express the corresponding receptor (TNFR1). However, both TNFα and TNFR1 were no longer detected from E13 onward. This observation is consistent with a physiological effect of TNFα on E12 motoneurons but not on older ones. This notion was further reinforced by the use of knock-out animals: explants from TNFα-/- and TNFR1 -/- mice displayed reduced motoneuron cell death when isolated at a stage equivalent to rat E13, suggesting that the in vivo exposure to TNFα at E12 provides a death signal for motoneurons. However, TNFα or anti-TNFα antibodies had no effect on E13 rat motoneurons in explants or in E14 dissociated cultures of purified motoneurons (data not shown), indicating that TNFα has no role after E13.
This role of TNFα in developing motoneurons differs from its known action on embryonic sympathetic and trigeminal sensory neurons (Barker et al., 2001). In the latter case, TNFα triggers neuronal death during the period of programmed cell death and is expressed in neurons together with TNFR1, thus functioning as an autocrine executive signal. Analogous autocrine death-executive functions have been described for Fas ligand (FasL) and TGFβs on motoneurons (Raoul et al., 1999; Krieglstein et al., 2001). These factors were shown to trigger motoneuron death during the period of cell death and were expressed together with their cognate receptors within motoneurons. Although FasL triggered the death of motoneurons at E14 in rats (Raoul et al., 1999), we did not find a proapoptotic effect on E12 motoneurons in explants. This suggests that different prodeath molecules act in an orderly manner: whereas TNFα is involved before motoneuron cell death (i.e., between E12 and E13 in rat), FasL and TGFβs act during the period of motoneuron elimination.
The notion that an external factor is an early signal for motoneuron cell death has been demonstrated in the hawk moth Manduca sexta (Hoffman and Weeks, 1998). In this model, some identified accessory planta retractor (APR) motoneurons die within 24 to 48 hr after pupal ecdysis. Exposure to the steroid hormone 20-hydroxyecdysone 24 hr before pupal ecdysis triggers the death of these APR motoneurons. If motoneurons are isolated before this 24 hr period, they escape cell death, and if they are isolated just after (at the pupal stage or later), they autonomously activate a death program on schedule. Our results now suggest that a similar mechanism exists in vertebrates.
Motoneuron cell death occurred normally in vivo in TNFα-/- mice. Similarly, although caspase 3 and Fas are known to be required for the induction of in vitro motoneuron death (Li et al., 1998; Raoul et al., 1999), no change in motoneuron number after the cell death period has been reported in vivo in caspase 3 or Fas knock-out mice (Oppenheim et al., 2001; Ugolini et al., 2003). In our experiments, the discrepancy between in vitro and in vivo data can indeed result from the explant isolation procedure. However, our results favor the notion that TNFα plays a physiological role. First, mRNAs for TNFα are transiently detected in vivo at E12-E13 in the vicinity of motoneurons, whereas the TNFR1 protein is detected at the same stage within these cells. Second, when spinal cord explants from TNF-/- and TNFR1-/- mice are isolated just after this stage, motoneurons are partially protected from in vitro cell death compared with wild-type mouse explants. Therefore, the in vivo exposure to TNFα at E12-E13 has played a role in sensitizing motoneurons to cell death as revealed in vitro. Thus, in vivo, between the TNFα exposure at E12-E13 and the onset of the in vivo motoneuron death period at E15, other factors are likely to compensate for the lack of TNFα. The absence of compensation in isolated spinal cords from TNFα-/- and TNFR1 -/- mice indicates that redundant factor(s) may originate from tissues surrounding the spinal cord.
TNFα, a link between physiological and degenerative neuronal death?
TNFα is overexpressed in several human neurodegenerative diseases, including amyotrophic lateral sclerosis (Poloni et al., 2000), Alzheimer's disease (Fillit et al., 1991), and Parkinson's disease (Boka et al., 1994). In the latter study, TNFα was found in activated brain macrophages surrounding degenerating neurons. In an experimental mouse model of amyotrophic lateral sclerosis, TNFα was shown to mediate neuronal death (Robertson et al., 2001), whereas data from Parkinsonian patients suggest that TNFα signaling is involved in death of dopaminergic neurons (Hartmann et al., 2002). Moreover, polymorphism in the regulatory region of the human TNFα gene constitutes a risk factor for Alzheimer's disease (McCusker et al., 2001). An intriguing link therefore exists between a role for TNFα in developmentally regulated and pathological neuronal death. The TNFα-signaling system may constitute a potent target for therapeutics designed to impede neurodegenerative diseases.
Footnotes
This work was supported by the Association Francaise contre les Myopathies and by the Institut pour la Recherche sur la Moelle Epinière. We thank Prof. E. Jorgensen, R. Miles, A. Bessis, and J. L. Bessereau for helpful suggestions, other members of Unité 497 for helpful discussions and encouraging support, Dr. C. Vesque for technical advice on explant cultures, Dr. L. F. Reichardt for the REX antibody, Dr. D. Wallach for the TNFα cDNA, and Dr. H. Bluethmann and Dr. M. Marino for knock-out mice.
Correspondence should be addressed to Antoine Triller, Laboratoire de Biologie Cellulaire de la Synapse Normale et Pathologique, Institut National de la Santé et de la Recherche Médicale Unité 497, Ecole Normale Supérieure, 46 rue d'Ulm, 75005 Paris, France. E-mail: triller@wotan.ens.fr.
Copyright © 2004 Society for Neuroscience 0270-6474/04/242236-11$15.00/0
References
- Altman J, Bayer SA (1984) The development of the rat spinal cord. Adv Anat Embryol Cell Biol 85: 1-164. [DOI] [PubMed] [Google Scholar]
- Barker V, Middleton G, Davey F, Davies AM (2001) TNFalpha contributes to the death of NGF-dependent neurons during development. Nat Neurosci 4: 1194-1198. [DOI] [PubMed] [Google Scholar]
- Boka G, Anglade P, Wallach D, Javoy-Agid F, Agid Y, Hirsch EC (1994) Immunocytochemical analysis of tumor necrosis factor and its receptors in Parkinson's disease. Neurosci Lett 172: 151-154. [DOI] [PubMed] [Google Scholar]
- Butler H, Juurlink BHJ (1987) An atlas for staging mammalian and chick embryos. Boca Raton, FL: CRC.
- Cuadros MA, Navascues J (1998) The origin and differentiation of microglial cells during development. Prog Neurobiol 56: 173-189. [DOI] [PubMed] [Google Scholar]
- Cuadros MA, Coltey P, Carmen Nieto M, Martin C (1992) Demonstration of a phagocytic cell system belonging to the hematopoietic lineage and originating from the yolk sac in the early avian embryo. Development 115: 157-168. [DOI] [PubMed] [Google Scholar]
- Davies AM (2003) Regulation of neuronal survival and death by extracellular signals during development. EMBO J 22: 2537-2545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eisen JS, Melançon E (2001) Interactions with identified muscle cells break motoneuron equivalence in embryonic zebrafish. Nat Neurosci 4: 1065-1070. [DOI] [PubMed] [Google Scholar]
- Fillit H, Buee WH, Kalman J, Altstiel L, Lawlor B, Wolf-Klein G (1991) Elevated circulating tumor necrosis factor levels in Alzheimer's disease. Neurosci Lett 129: 318-320. [DOI] [PubMed] [Google Scholar]
- Frade JM, Barde Y-A (1998) Microglia-derived nerve growth factor causes cell death in the developing retina. Neuron 20: 35-41. [DOI] [PubMed] [Google Scholar]
- Gavrieli Y, Sherman Y, Ben-Sasson B (1992) Identification of programmed cell death in situ via specific labeling of nuclear DNA fragmentation. J Cell Biol 119: 493-501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giulian D, Vaca K, Corpuz M (1993) Brain glia release factors with opposing actions upon neuronal survival. J Neurosci 13: 29-37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gould TW, Burek MJ, Sosnowski JM, Prevette D, Oppenheim RW (1999) The spatial-temporal gradient of naturally occurring MN death reflects the time of prior exit from the cell cycle and position within the lateral motor column. Dev Biol 216: 611-621. [DOI] [PubMed] [Google Scholar]
- Graham A, Francis-West P, Brickell P, Lumsden A (1994) The signaling molecule BMP4 mediates apoptosis in the rhombencephalic neural crest. Nature 372: 684-686. [DOI] [PubMed] [Google Scholar]
- Harris AJ, McCaig CD (1984) Motor neuron death and motor unit size during embryonic development of the rat. J Neurosci 4: 13-24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartmann A, Mouatt-Prigent A, Faucheux BA, Agid Y, Hirsch EC (2002) FADD: a link between TNF family receptors and caspases in Parkinson's disease. Neurology 22: 08-310. [DOI] [PubMed] [Google Scholar]
- Henderson CE, Bloch-Gallego E, Camu W (1995) Purified embryonic motoneurons. In: nerve cell culture: a practical approach (Cohen J, Wilkin G, eds), pp 69-81. London: Oxford UP.
- Hoffman KL, Weeks JC (1998) Programmed cell death of an identified motoneuron in vitro: temporal requirements for steroid exposure and protein synthesis J Neurobiol 35: 300-322. [PubMed] [Google Scholar]
- Ito D, Imai Y, Ohsawa K, Nakajima K, Fukuuchi Y, Kohsaka S (1998) Microglia-specific localisation of a novel calcium binding protein, Iba1. Brain Res Mol Brain Res 57: 1-9. [DOI] [PubMed] [Google Scholar]
- Kablar B, Rudnicki MA (1999) Development in the absence of skeletal muscle results in the sequential ablation of motor neurons from the spinal cord to the brain. Dev Biol 208: 93-109. [DOI] [PubMed] [Google Scholar]
- Kamiryo T, Orita T, Nishizaki T, Aoki H (1990) Development of the rat meninx: experimental study using bromodeoxyuridine. Anat Rec 227: 207-210. [DOI] [PubMed] [Google Scholar]
- Kessler JA, Ludlam WH, Freidin MM, Hall DH, Michaelson MD, Spray DC, Dougherty M, Batter D (1993) Cytokine-induced programmed death of cultured sympathetic neurons. Neuron 11: 1123-1132. [DOI] [PubMed] [Google Scholar]
- Krieglstein K, Richter S, Farkas L, Schuster N, Dunker N, Oppenheim RW, Unsicker K (2000) Reduction of endogenous transforming growth factors prevents ontogenetic neuron death. Nat Neurosci 3: 1085-1090. [DOI] [PubMed] [Google Scholar]
- Lance-Jones C (1982) Motor neuron cell death in the developing lumbar spinal cord of the mouse. Dev Brain Res 4: 473-479. [DOI] [PubMed] [Google Scholar]
- Li L, Prevette D, Oppenheim RW, Milligan CE (1998) Involvement of specific caspases in motor neuron cell death in vivo and in vitro following trophic factor deprivation. Mol Cell Neurosci 12: 157-167. [DOI] [PubMed] [Google Scholar]
- Maddox DE, Shibata S, Goldstein IJ (1982) Stimulated macrophages express a new glycoprotein receptor reactive with Griffonia simplicifolia I-B4 isolectin. Proc Natl Acad Sci USA 79: 166-170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marino MW, Dunn A, Grail D, Inglese M, Noguchi Y, Richards E, Jungbluth A, Wada H, Moore M, Williamson B, Basu S, Old LJ (1997) Characterization of tumor necrosis factor-deficient mice. Proc Natl Acad Sci USA 94: 8093-8098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCusker SM, Curran MD, Dynan KB, McCullagh CD, Urquhart DD, Middleton D, Patterson CC, McLlroy SP, Passmore AP (2001) Association between polymorphism in the regulatory region of gene encoding tumor necrosis factor α and risk of Alzheimer's disease and vascular dementia: a case-control study. Lancet 357: 436-439. [DOI] [PubMed] [Google Scholar]
- McLennan I (1982) Size of motor neuron pool may be related to number of myotubes in developing muscle. Dev Biol 92: 263-265. [DOI] [PubMed] [Google Scholar]
- Mettling C, Gouin A, Robinson M, El M'Hamdi H, Camu W, Bloch-Gallego E, Buisson B, Tanaka H, Davies AM, Henderson CE (1995) Survival of newly postmitotic motor neurons is transiently independent of exogenous trophic support. J Neurosci 15: 3128-3137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohsawa T, Natori S (1989) Expression of tumor necrosis factor at a specific developmental stage of mouse embryos. Dev Biol 135: 459-461. [DOI] [PubMed] [Google Scholar]
- Oppenheim RW (1986) The absence of significant postnatal motor neuron death in the brachial and lumbar spinal cord of the rat. J Comp Neurol 246: 281-286. [DOI] [PubMed] [Google Scholar]
- Oppenheim RW (1991) Cell death during development of the nervous system. Annu Rev Neurosci 14: 453-501. [DOI] [PubMed] [Google Scholar]
- Oppenheim RW (1996) Neurotrophic survival molecules for motor neurons: an embarrassment of riches. Neuron 17: 195-197. [DOI] [PubMed] [Google Scholar]
- Oppenheim RW, Chu-Wang I-W (1977) Spontaneous cell death of spinal motor neurons following peripheral innervation in the chick embryo. Brain Res 125: 154-160. [DOI] [PubMed] [Google Scholar]
- Oppenheim RW, Flavell RA, Vinsant S, Prevette D, Kuan CY, Rakic P (2001) Programmed cell death of developing mammalian neurons after genetic deletion of caspases. J Neurosci 21: 4752-4760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pettmann B, Henderson CE (1998) Neuronal cell death. Neuron 20: 633-647. [DOI] [PubMed] [Google Scholar]
- Phelan KA, Holliday M (1991) Embryonic development and survival of brachial motoneurons projecting to muscleless chick wings. J Comp Neurol 311: 313-320. [DOI] [PubMed] [Google Scholar]
- Poloni M, Facchetti D, Mai R, Micheli A, Agnoletti L, Mora G, Camana C, Mazzini L, Bachetti T (2000) Circulating levels of tumor-necrosis factor-alpha and its soluble receptors are increased in the blood of patients with amyotrophic lateral sclerosis. Neurosci Lett 287: 211-214. [DOI] [PubMed] [Google Scholar]
- Raoul C, Pettmann B, Henderson CE (1999) Programmed cell death of embryonic motor neurons triggered through the Fas death receptor. J Cell Biol 147: 1049-1062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reddien PW, Cameron S, Horvitz HR (2001) Phagocytosis promotes programmed cell death in C. elegans Nature 412: 198-202. [DOI] [PubMed] [Google Scholar]
- Robertson J, Beaulieu JM, Doroudchi MM, Durham HD, Julien JP, Mushynski WE (2001) Apoptotic death of neurons exhibiting peripherin aggregates is mediated by the proinflammatory cytokine tumor necrosis factor-α. J Cell Biol 155: 217-226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rothe J, Lesslauer W, Lotscher H, Lang Y, Koebel P, Kontgen F, Althage A, Zinkernagel R, Steinmetz M, Bluethmann H (1993) Mice lacking the tumour necrosis factor receptor 1 are resistant to TNF-mediated toxicity but highly susceptible to infection by Listeria monocytogenes. Nature 364: 798-802. [DOI] [PubMed] [Google Scholar]
- Schaeren-Wiemers N, Gerfin-Moser (1993) A single protocol to detect transcripts of various types and expression levels in neural tissue and cultured cells: in situ hybridization using digoxigenin-labeled cRNA probes. Histochemistry 100: 431-440. [DOI] [PubMed] [Google Scholar]
- Sedel F, Béchade C, Triller A (1999) Nerve growth factor induces motor neuron apoptosis in rat embryonic spinal cord in vitro. Eur J Neurosci 11: 3904-3912. [DOI] [PubMed] [Google Scholar]
- Thery C, Chamak B, Mallat M (1991) Cytotoxic effect of brain macrophages on developing neurons. Eur J Neurosci 3: 1155-1164. [DOI] [PubMed] [Google Scholar]
- Tsuchida T, Ensini M, Morton SB, Baldassare M, Edlund T, Jessell TM, Pfaff SL (1994) Topographic organization of embryonic motor neurons defined by expression of LIM homeobox genes. Cell 79: 957-970. [DOI] [PubMed] [Google Scholar]
- Ugolini G, Raoul C, Ferri A, Haenggeli C, Yamamoto Y, Salaun D, Henderson CE, Kato A, Pettmann B, Hueber AO (2003) Fas/tumor necrosis receptor death signaling is required for axotomy-induced death of motoneurons in vivo. J Neurosci 23: 8526-8531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vandenabeele P, Declercq W, Beyaert R, Fiers W (1995) Two tumor necrosis factor receptors: structure and function. Trends Cell Biol 5: 392-399. [DOI] [PubMed] [Google Scholar]
- Vogel KS, Davies AM (1991) The duration of neurotrophic factor independence in early sensory neurons is matched to the time course of target field innervation. Neuron 7: 819-830. [DOI] [PubMed] [Google Scholar]
- Wang H, Tessier-Lavigne M (1999) En passant neurotrophic action of an intermediate axonal target in the developing mammalian CNS. Nature 401: 765-769. [DOI] [PubMed] [Google Scholar]
- Weskamp G, Reichardt LF (1991) Evidence that the biological activity of NGF is mediated through a novel subclass of high affinity receptors. Neuron 6: 649-663. [DOI] [PubMed] [Google Scholar]
- Yamamoto Y, Henderson CE (1999) Patterns of programmed cell death in populations of developing spinal motor neurons in chicken, mouse, and rat. Dev Biol 214: 60-71. [DOI] [PubMed] [Google Scholar]