

Abstract
The Akt signaling pathway contributes to regulation of apoptosis after a variety of cell death stimuli. A novel proline-rich Akt substrate (PRAS) was recently detected and found to be involved in apoptosis. In our study, Akt activation was modulated by growth factors, and treatment with nerve growth factor (NGF) reduced apoptotic cell death after ischemic injury. However, the role of the PRAS pathway in apoptotic neuronal cell death after ischemia remains unknown. Phosphorylated PRAS (pPRAS) and the binding of pPRAS/phosphorylated Akt (pPRAS/pAkt) to 14-3-3 (pPRAS/14-3-3) were detected, and their expression transiently decreased in mouse brains after transient focal cerebral ischemia (tFCI). Liposome-mediated pPRAS cDNA transfection induced overexpression of pPRAS, promoted pPRAS/14-3-3, and inhibited apoptotic neuronal cell death after tFCI. The expression of pPRAS, pPRAS/pAkt, and pPRAS/14-3-3 increased in NGF-treated mice but decreased with inhibition of phosphatidylinositol-3 kinase and the NGF receptor after tFCI. These results suggest that PRAS phosphorylation and its interaction with pAkt and 14-3-3 might play an important role in neuroprotection mediated by NGF in apoptotic neuronal cell death after tFCI.
Keywords: cerebral ischemia, apoptosis, proline-rich Akt substrate, nerve growth factor, Akt, gene transfection
Introduction
Cell survival pathways are the focus for clarifying the apoptotic neuronal cell death machinery. Serine-threonine kinases, such as Akt/protein kinase B, are key regulators of neuronal cell death and survival after cerebral ischemia (Noshita et al., 2003). Akt functions as a major downstream target of phosphatidylinositol 3-kinase (PI3-K), and after phosphorylation, it phosphorylates some substrates on the serine or threonine residues, including glycogen synthase kinase-3, Caenorhabditis elegans DAF-16 transcription factor, Bad, phosphodiesterase 3B, a Rab GTPase-activating protein, ATP-citrate lyase, and the tuberous sclerosis complex 2 tumor suppressor gene product tuberin (Lizcano et al., 2000; Berwick et al., 2002; Cantley, 2002; Kane et al., 2002; Manning et al., 2002; McManus and Alessi, 2002). A novel proline-rich Akt substrate (PRAS) has been identified and found to be phosphorylated by activated Akt on the same site that was phosphorylated in insulin-treated cells. PRAS phosphorylation led to its binding to 14-3-3 in vitro (Kovacina et al., 2003). However, the PRAS signaling pathway is unknown in other apoptotic cell death models.
Gene transfer is a means of introducing neurotrophins into the CNS (Karpati et al., 1996; Zou et al., 1999). Cationic liposomes are suitable candidates for gene transfection because of their nonimmunogenicity, nontoxicity, and relative biological safety compared with viral gene transfection (Dokka et al., 2000). They condense DNA and introduce it into cells, even in postmitotic, nondividing cells, including neurons by membrane fusion and endocytosis (Zou et al., 1999; Cao et al., 2002). Recent advances in the development of more efficient liposome formulations have raised the possibility that liposome-mediated neuroprotective gene transfection may prove useful for treating in vivo CNS injury (Yang et al., 1997; Zou et al., 1999; Cao et al., 2002). N,N,N-Trimethyl-2-3-bis ((1-oxo-9-octa-decenyl)oxy)-(Z,Z)-1-propanaminium methyl sulfate, C43H83NO8S CH3SO4- (DOTAP) liposomes have been used as a gene delivery system in a number of in vivo applications (McLachlan et al., 1995; Porteous et al., 1997; Cao et al., 2002). In the present study, the transgene phosphorylated PRAS (pPRAS) complexed with DOTAP liposomes was delivered into neuronal cells by means of a single intrathecal injection.
The PI3-K pathway is involved in nerve growth factor (NGF)-dependent neuronal survival (Crowder and Freeman, 1998). NGF and its tyrosine kinase receptor trkA are induced after in vivo cerebral ischemia. NGF administration ameliorates apoptotic cell death after cerebral ischemia (Buchan et al., 1990; Shigeno et al., 1991; Yamamoto et al., 1992; Lee et al., 1998); however, the cell survival machinery mediated by NGF after ischemic injury in vivo remains unclear. The present study was designed to clarify the role of the novel Akt substrate PRAS in the apoptotic machinery mediated by NGF administration after transient focal cerebral ischemia (tFCI).
Materials and Methods
Focal cerebral ischemia. Adult male mice (3 months old, 35-40 gm) were subjected to tFCI by intraluminal middle cerebral artery (MCA) blockade with a nylon suture as described previously (Yang et al., 1994). The mice were anesthetized with 1.5% isoflurane in 30% oxygen and 70% nitrous oxide using a face mask. The rectal temperature was controlled at 37°C with a homeothermic blanket. Cannulation of a femoral artery allowed the monitoring of blood pressure and arterial blood gases, with samples for analysis taken immediately after cannulation, 10 min after occlusion, and 10 min after reperfusion. Blood gas was analyzed with a pH-blood gas analyzer (Chiron Diagnostics, Ltd., Essex, UK). After a midline skin incision, the left external carotid artery was exposed, and its branches were electrocoagulated. An 11 mm 5-0 surgical monofilament nylon suture, blunted at the end, was introduced into the left internal carotid artery through the external carotid artery stump. After 60 min of MCA occlusion, blood flow was restored by withdrawal of the nylon suture.
pPRAS cloning and generation of an expression construct. An expressed sequence tag containing the entire coding region of human cDNA (GenBank accession number BC007416, IMAGE clone number 2988648) was purchased (Incyte Genomics, Wilmington, DE), amplified via PCR with the addition of a BamHI site (5′) and an EcoRI site (3′), and subcloned into the pcDNA3.1(+) vector (Invitrogen, Carlsbad, CA) containing either an N-terminal Myc tag or a C-terminal hemagglutinin tag for mammalian cell expression. The pPRAS plasmid was grown overnight, and DNA was extracted using a Qiagen (Valencia, CA) kit.
Liposome-mediated Myc-tagged pPRAS cDNA transfection. We used the Myc-tagged plasmid of pPRAS, which was generated by Dr. Richard A. Roth (Stanford University School of Medicine), for liposomal gene transfection (Kovacina et al., 2003). We used the cationic liposome preparation DOTAP (Roche Diagnostics, Mannheim, Germany) for intraventricular cDNA transfection (Porteous et al., 1997; Dokka et al., 2000; Cao et al., 2002). Both the DOTAP liposome and the plasmid or control DNA were allowed to complex at room temperature for 15 min before the injection (Cao et al., 2002). On the basis of previous studies of liposome-mediated intraventricular gene transfection, we initially used four different ratios of pPRAS cDNA to DOTAP liposomes [1:2, 1:4, 1:8, and 1:16, cDNA (micrograms) to liposome (microliters)] for the transfection (Zou et al., 1999; Cao et al., 2002). After the formation of cDNA-liposome complexes, we stereotactically injected these complexes (4 μl, bregma; 1.0 mm lateral, 0.2 mm posterior, 3.1 mm deep) after withdrawal of 2 μl of CSF using a 10 μl Hamilton (Reno, NV) microsyringe. After 0, 12, 24, and 48 hr of incubation, the brains were removed, and Myc expression was immunohistochemically traced for evaluation of gene transfection.
Drug treatment. We administered a PI3-K inhibitor, 2-(4-morpholinyl)-8-phenyl-4H-1-benzopyran-4-one (LY294002), after tFCI to examine the role of PRAS in the PI3-K signaling pathway. LY294002 was purchased from Cell Signaling Technology (Beverly, MA) and dissolved in dimethylsulfoxide and PBS (LY294002, 50 nmol in 25% dimethylsulfoxide in PBS; vehicle, 25% dimethylsulfoxide in PBS) as described previously (Noshita et al., 2001). To clarify the role of PRAS in NGF-mediated neuroprotection, we administered NGF and the tyrosine kinase receptor inhibitor K-252a after tFCI. NGF was purchased from Harlan Bioproducts for Science (Madison, WI), and K-252a was purchased from Calbiochem (La Jolla, CA). NGF was dissolved in PBS to prepare 0.5 μg/μl as described previously (Kent et al., 1999). K-252a was dissolved in 1 μm PBS as described previously (Guilhem et al., 1996). We injected PBS as the vehicle for both NGF and K-252a. The scalp was incised on the midline, and the skull was exposed. These drugs and the vehicle were injected intracerebroventricularly (2 μl, bregma; 1.0 mm lateral, 0.2 mm posterior, 3.1 mm deep). All drugs and the vehicle were injected 1 hr before MCA occlusion.
Immunohistochemistry. Anesthetized animals, as well as normal controls (n = 4 each), were perfused with 10 U/ml heparin and subsequently with 4% paraformaldehyde in 0.1 m PBS, pH 7.4, at 1, 2, 4, 8, and 24 hr of reperfusion. The brains were removed, postfixed for 12 hr, sectioned at 50 μm on a vibratome, and processed for immunohistochemistry. The sections were incubated with blocking solution and reacted with anti-pPRAS antibody (BioSource International, Hopkinton, MA) at a dilution of 1:400 or anti-Myc antibody (Covance Inc., Berkeley, CA) at a dilution of 1:400. Immunohistochemistry was performed using the avidin-biotin technique, and then the nuclei were counterstained with methyl green solution for 2 min. Alternate slices from each brain section were stained with cresyl violet for histological assessment.
Terminal deoxynucleotidyl transferase-mediated biotinylated UTP nick end-labeling staining. To clarify the spatial distribution of DNA fragmentation, we performed terminal deoxynucleotidyl transferase-mediated biotinylated UTP nick end labeling (TUNEL). The sections fixed by 4% paraformaldehyde were prepared as described above and were incubated with NeuroPore (Trevigen, Gaithersburg, MD) for 30 min. They were placed in 1× terminal deoxynucleotidyl transferase (TdT) buffer (Invitrogen) for 30 min, followed by reaction with a TdT enzyme (Invitrogen) and biotinylated 16-dUTP (Roche Diagnostics) at 37°C for 90 min. The sections were washed two times in saline-sodium citrate (150 mmol/l sodium chloride and 15 mmol/l sodium citrate, pH 7.4) for 15 min, followed by washing in PBS two times for 15 min. The avidin-biotin technique was applied, and then the nuclei were counterstained with methyl green solution for 2 min.
Immunofluorescent double-labeling staining. To evaluate colocalization of pPRAS and neuron-specific nuclear protein (NeuN) and activated caspase-3, we performed double immunofluorescent staining for these proteins. The sections fixed by 4% paraformaldehyde were immunostained with anti-pPRAS antibody (BioSource International) or anti-Myc antibody (Covance), with biotinylated goat anti-rabbit IgG (Vector Laboratories, Burlingame, CA) followed by fluorescein-avidin DCS (a cell sorter grade of fluorescein avidin D) (Vector Laboratories). The sections were then incubated with a blocking solution and reacted with anti-NeuN antibody (Chemicon, Temecula, CA) or the anticleaved caspase-3 antibody (Cell Signaling Technology), followed by Texas Red-conjugated donkey anti-mouse IgG antibody (Jackson ImmunoResearch, West Grove, PA) at a dilution of 1:400. The sections were placed on slides, which were then covered with Vectashield mounting medium with 4′,6-diamidino-2-phenylindole (DAPI; Vector Laboratories). Fluorescence of fluorescein was observed at excitation (Ex) of 495 nm and emission (Em) of >515 nm, and fluorescence of Texas Red was observed at Ex of 510 nm and Em of >580 nm. Fluorescence of DAPI was observed at Ex of 360 nm and Em of >460 nm.
Immunofluorescent double labeling with pPRAS immunohistochemistry and TUNEL. We performed double staining for pPRAS and TUNEL using a fluorescent method to clarify the spatial relationship between pPRAS expression and DNA fragmentation. The sections fixed by 4% paraformaldehyde were immunostained with the pPRAS antibody as described above, with biotinylated goat anti-rabbit IgG (Vector Laboratories) followed by fluorescein-avidin DCS (Vector Laboratories). Then the sections were incubated with NeuroPore (Trevigen) for 30 min and placed in 1× TdT buffer (Invitrogen) for 30 min, followed by reaction with a TdT enzyme (Invitrogen) and biotinylated 16-dUTP (Roche Diagnostics) at 37°C for 90 min. The sections were washed two times in saline-sodium citrate (150 mmol/l sodium chloride and 15 mmol/l sodium citrate, pH 7.4) for 15 min, followed by washing in PBS two times for 15 min. Texas Red-avidin DCS (Vector Laboratories) was applied to the sections for 30 min. Subsequently, the slides were covered with Vectashield mounting medium with DAPI (Vector Laboratories). Fluorescence was assessed as described previously.
Western blot analysis. Protein extraction of the cytosolic fraction was performed as described previously with some modification (Fujimura et al., 1999). Samples were obtained from the MCA territory brain tissue on the ischemic sides and from nonischemic controls (n = 4 each). Fresh brain tissue was removed after 1, 2, 4, 8, and 24 hr (n = 4 each) of reperfusion and homogenized by gently douncing 35 times in a glass tissue grinder (Wheaton, Millville, NJ) in 7 volumes of cold suspension buffer [in mm: 20 HEPES-KOH, pH 7.5, 250 sucrose, 10 KCl, 1.5 MgCl2, 1 EDTA, and 1 EGTA plus 0.7% protease and phosphatase inhibitor mixtures (Sigma, St. Louis, MO)]. The homogenate was centrifuged at 750 × g for 10 min at 4°C and then at 8000 × g for 20 min at 4°C. The 8000 × g pellets were used to obtain the mitochondrial fraction. The supernatant was further centrifuged at 100,000 × g for 60 min at 4°C. This supernatant was used for the cytosolic analysis. After adding the same volume of Tris-glycine SDS sample buffer (Invitrogen) to the supernatant, we loaded equal amounts of the samples per lane. The primary antibodies were 1:2000 dilution of rabbit polyclonal antibody against pPRAS (BioSource International), 1:1000 dilution of rabbit polyclonal antibody against Akt (Cell Signaling Technology), 1:800 dilution of rabbit polyclonal antibody against phosphorylated Akt (pAkt; Cell Signaling Technology), 1:1000 dilution of mouse polyclonal antibody against 14-3-3 (Cell Signaling Technology), and 1:10000 dilution of anti-β-actin monoclonal antibody (Sigma). Western blots were performed with horseradish peroxidase-conjugated anti-rabbit IgG (Cell Signaling Technology) or anti-mouse IgG (Chemicon) using enhanced chemiluminescence Western blot detection reagents (Amersham Biosciences, Buckinghamshire, UK). The film was scanned with a GS-700 imaging densitometer (Bio-Rad, Hercules, CA), and the results were quantified using Multi-Analyst software (Bio-Rad).
Coimmunoprecipitation. Protein extraction of both the cytosolic and mitochondrial fractions was performed as described previously with some modification (Fujimura et al., 1999). The procedure for precipitation was performed as described previously (Saito et al., 2003). Samples were obtained from the MCA territory brain tissue on the ischemic side and from nonischemic controls (n = 4 each). Fresh brain tissue was removed after 1, 2, 4, 8, and 24 hr (n = 4 each) of reperfusion and was prepared for collection of the cytosolic and mitochondrial fractions in the same manner as for Western blotting. Protein concentrations were determined by the Bradford method (Bio-Rad). Three hundred micrograms of protein from the cytosolic fraction were used for coimmunoprecipitation. Whole-brain extract was included as a positive control. The protein sample was incubated with a 50% slurry of protein G-Sepharose (Amersham Biosciences, Uppsala, Sweden) for 1 hr at 4°C, and this mixed sample was centrifuged at 12,000 × g for 1 min. The supernatant was incubated with 2 μg of polyclonal mouse anti-14-3-3 antibody (Cell Signaling Technology) and 15 μl of protein G-Sepharose (50% slurry) for 1 hr at 4°C. The negative control was prepared with protein G-Sepharose without an antibody. The 14,000 × g pellets were washed three times and used as the samples bound to each antibody. After adding the same volume of Tris-glycine SDS sample buffer (Invitrogen) to the samples, we boiled these samples to remove the Sepharose beads. After centrifugation at 14,000 × g for 1 min, the supernatant was immunoblotted with a 1:600 dilution of anti-Bad antibody (Cell Signaling Technology) or a 1:600 dilution of the anti-pPRAS antibody (BioSource International) as described for the Western blot method.
Cell death assay. For quantification of apoptosis-related DNA fragmentation, we used a commercial enzyme immunoassay to determine cytoplasmic histone-associated DNA fragments (Roche Diagnostics), which detect apoptotic but not necrotic cell death (Bonfoco et al., 1995). Samples were obtained from the entire MCA territory on the ischemic side and from nonischemic controls (n = 4 each). Fresh brain tissue was cut into pieces after 1, 2, 4, 8, and 24 hr of reperfusion and homogenized with a Teflon homogenizer in 5 volumes of ice-cold buffer (50 mm KH2PO4 and 0.1 mm EDTA, pH 7.8) and spun for 10 min at 750 × g. The supernatant was collected and spun for 20 min at 10,000 × g and was centrifuged further at 100,000 × g for 60 min at 4°C. The resulting supernatant was collected, and the protein concentration was determined. A cytosolic volume containing 20 μg of protein was used for the ELISA, following the manufacturer's protocol.
Caspase-3 activity assay. For quantification of caspase-3 activation, we used a commercial enzyme immunoassay (Chemicon) to specifically determine by active caspase-3, the cleaved bioluminescent substrate containing the cleavage site DEVD (Gill et al., 2002). The cytosolic samples were prepared as described for the Western blot method (n = 4 each). Cytosolic samples (20 μg of protein) were used for the ELISA, following the manufacturer's protocol.
Quantification and statistical analysis. The data are expressed as mean ± SD. Comparisons among multiple groups were performed with a one-way ANOVA with appropriate post hoc tests (SigmaStat software; Jandel Corp., San Rafael, CA). Comparisons between two groups were achieved with Student's t test. Significance was accepted at p < 0.05.
Results
Physiological data and cerebral infarction
Physiological data demonstrated no significant differences in body temperature, mean arterial blood pressure, or arterial blood gas analysis between the groups. The preischemic physiological values were as follows (in mmHg): 36.4 ± 0.3°C body temperature, 83 ± 3.1 mmHg mean arterial blood pressure, 7.3 ± 0.1 pH, 164.9 ± 19 mmHg PaO2, and 30 ± 10 mmHg PaCO2 (values are mean ± SD; n = 4). There was no deviation from these values over the period of assessment. An ischemic lesion of the core of the caudate putamen was visible as a pale, slightly stained area in the ischemic hemisphere as early as 1 hr after reperfusion and extended to the entire MCA territory at 4 hr by cresyl violet staining (data not shown). The time-dependent increase in infarction in the mouse brains with the intraluminal suture blockade is consistent with previous reports that used the same FCI model in mice (Yang et al., 1994).
Specificity of anti-pPRAS and transient decrease in neuronal pPRAS expression after tFCI
To examine the phosphospecificity of the reaction of the anti-pPRAS antibody, we observed this reaction with the pPRAS peptide or a non-pPRAS peptide (BioSource International). Western blot analysis demonstrated that a single band of 40 kDa was shown in the lane of the phospho-specific PRAS peptide but not in that of the non-pPRAS peptide (n = 3; Fig. 1A). We confirmed that pPRAS expression occurred in the cytosolic samples of mouse brains and was observed as a 40 kDa band (n = 3; Fig. 1A). Double immunofluorescent staining for pPRAS and NeuN demonstrated that pPRAS protein expression colocalized with neurons in the ischemic cortex 24 hr after reperfusion (n = 4; Fig. 1B). pPRAS was transiently reduced at 2 hr and began to recover to the control level 4 hr after tFCI (n = 4; p < 0.05; Fig. 1C). These results suggest that pPRAS was in neurons after tFCI and that pPRAS expression was transiently reduced during the early period of reperfusion injury.
Figure 1.
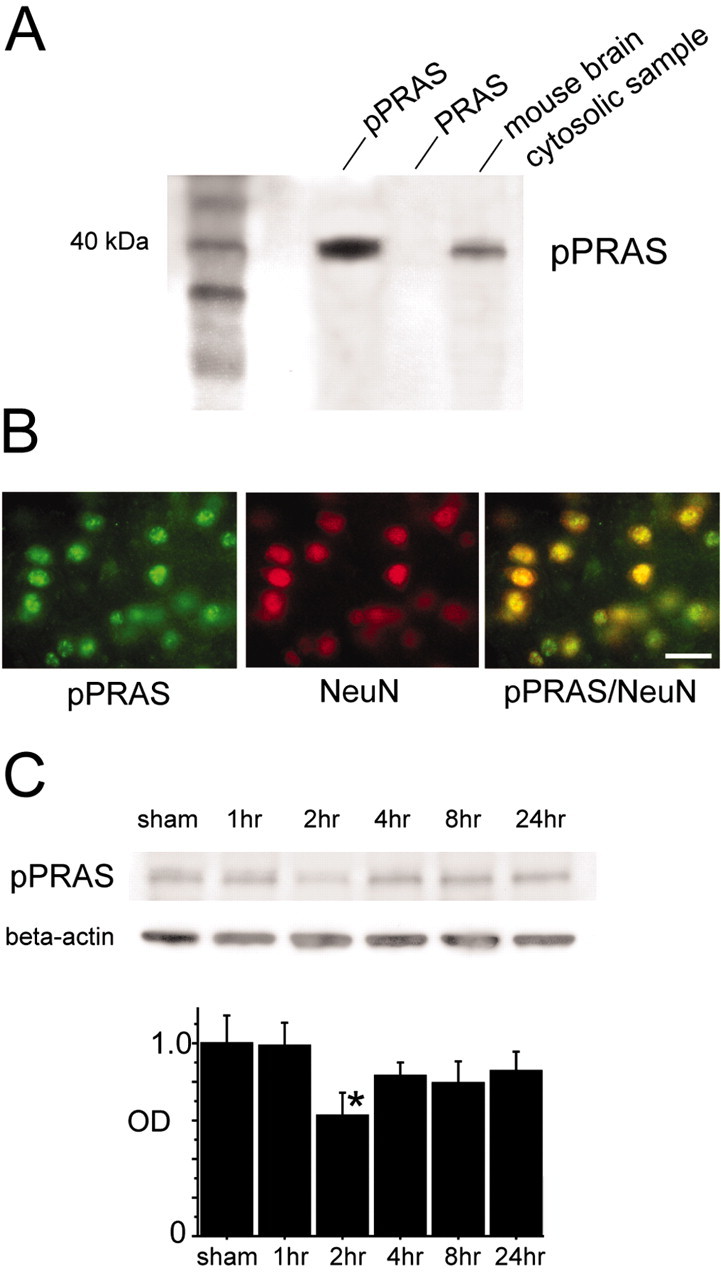
A, Western blot analysis demonstrates that the anti-pPRAS antibody reacted with the phospho-specific PRAS peptide, and it showed a single band of 40 kDa (n = 3; pPRAS, left lane). No band was shown in the lane of the nonphosphorylated PRAS peptide (n = 3; PRAS, middle lane). The single 40 kDa band was observed in the cytosolic samples of the mouse brains (n = 3; mouse brain cytosolic sample, right lane). B, Representative photomicrographs show double immunofluorescent staining for pPRAS and NeuN 24 hr after tFCI. Expression of pPRAS (green, left panel) was observed in the ischemic cortical penumbra. NeuN immunoreactivity (red, middle panel) showed the distribution of neurons in the same view. An overlapped image of pPRAS and NeuN immunoreactivity (yellow, right panel) demonstrated that pPRAS expression colocalized with neurons in the same view. Scale bar, 20 μm. C, pPRAS (40 kDa, antibody pPRAS) was expressed constitutively in the sham-operated brains (sham, top) and showed a significant transient decrease after ischemia by Western blot analysis (2 hr; n = 4; *p < 0 0.05; top). The results of the β-actin analysis are shown as an internal control (bottom). OD, Optical density.
Direct binding of pPRAS to phosphorylated Akt and the 14-3-3 protein after tFCI
The 14-3-3 protein is known as an anchor protein for some Akt substrates in inactivated states. In our model, pAkt transiently increased 2 and 4 hr after tFCI, and 14-3-3 expression was not different among any of the time points after tFCI (Noshita et al., 2001; Saito et al., 2003). 14-3-3 protein-immunopositive cells were seen in neurons of the cortex and the caudate putamen in both the nonischemic and ischemic hemispheres (data not shown). Coimmunoprecipitation showed the direct binding of pPRAS to pAkt (pPRAS/pAkt) and to 14-3-3 (pPRAS/14-3-3) after tFCI (n = 4; Fig. 2A,B). Interestingly, expression of both pPRAS/pAkt and pPRAS/14-3-3 was transiently reduced 2 hr after reperfusion (n = 4; p < 0.05; Fig. 2A,B). These results suggest that both pPRAS expression and pPRAS/14-3-3 were transiently reduced during the early period of reperfusion injury. In an in vitro study, phosphorylation of PRAS and formation of the pPRAS/14-3-3 complex required a direct reaction with pAkt (Kovacina et al., 2003). Our results did not conflict with these previous findings.
Figure 2.
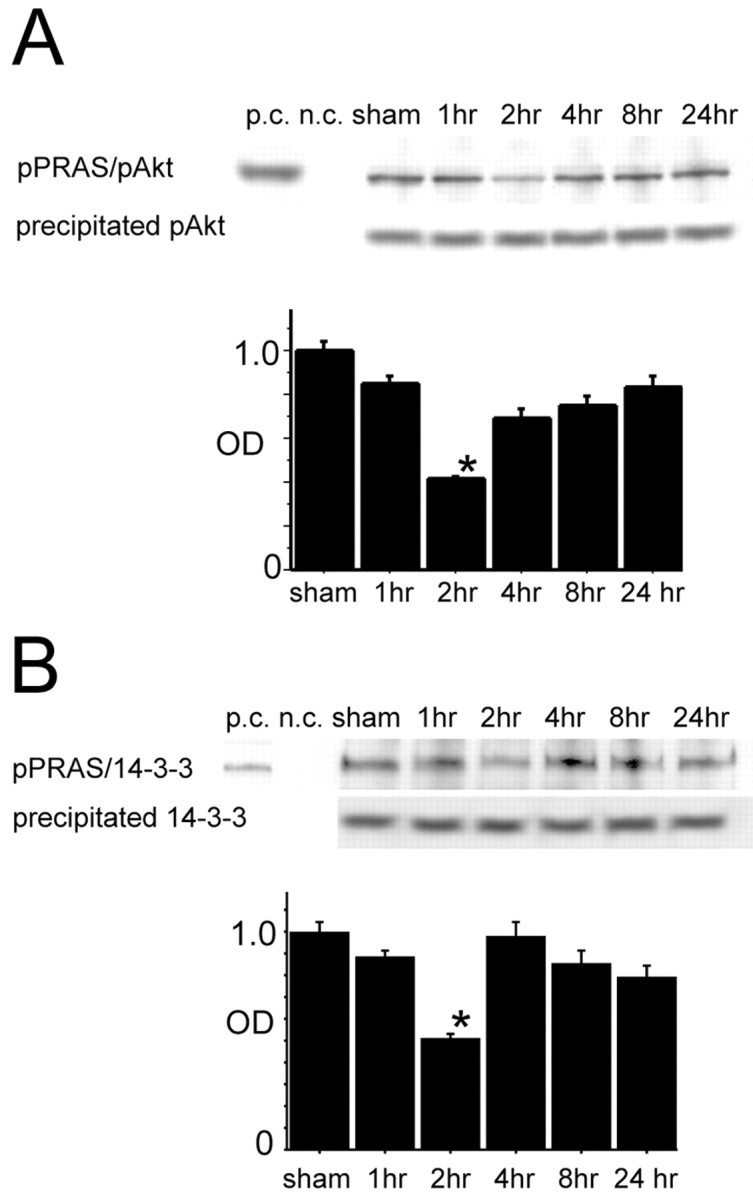
A, Coimmunoprecipitation analysis for pPRAS immunoreactivity precipitated by pAkt in the cytosolic fraction (pPRAS/pAkt) transiently decreased 2 hr after tFCI (top row, graph; n = 4; *p < 0.05). Expression of precipitated pAkt was constant at any time point (bottom row). B, Coimmunoprecipitation analysis for pPRAS immunoreactivity precipitated by 14-3-3 in the cytosolic fraction (pPRAS/14-3-3) transiently decreased 2 hr after tFCI (top row, graph; n = 4; *p < 0 0.05). Expression of precipitated 14-3-3 protein was constant at any time point (bottom row). p.c., Positive control; n.c., negative control; sham, sham-operated sample; OD, optical density.
PI3-K inhibition reduced pPRAS and pPRAS/14-3-3 expression after tFCI
Western blot analysis revealed that expression of pPRAS, pPRAS/pAkt, and pPRAS/14-3-3 was reduced in the LY294002-treated mice compared with the vehicle-treated mice 2 hr after tFCI (n = 4; p < 0.05; Fig. 3A,B). In our model, pAkt was reduced by LY294002 treatment, and phosphorylation of Akt was affected by a reaction downstream of PI3-K (Noshita et al., 2001). 14-3-3 expression did not change in the LY294002-treated or the vehicle-treated mice (data not shown). These results suggest that a pPRAS reaction may be involved in the PI3-K pathway as a downstream target of PI3-K as well as Akt after tFCI.
Figure 3.
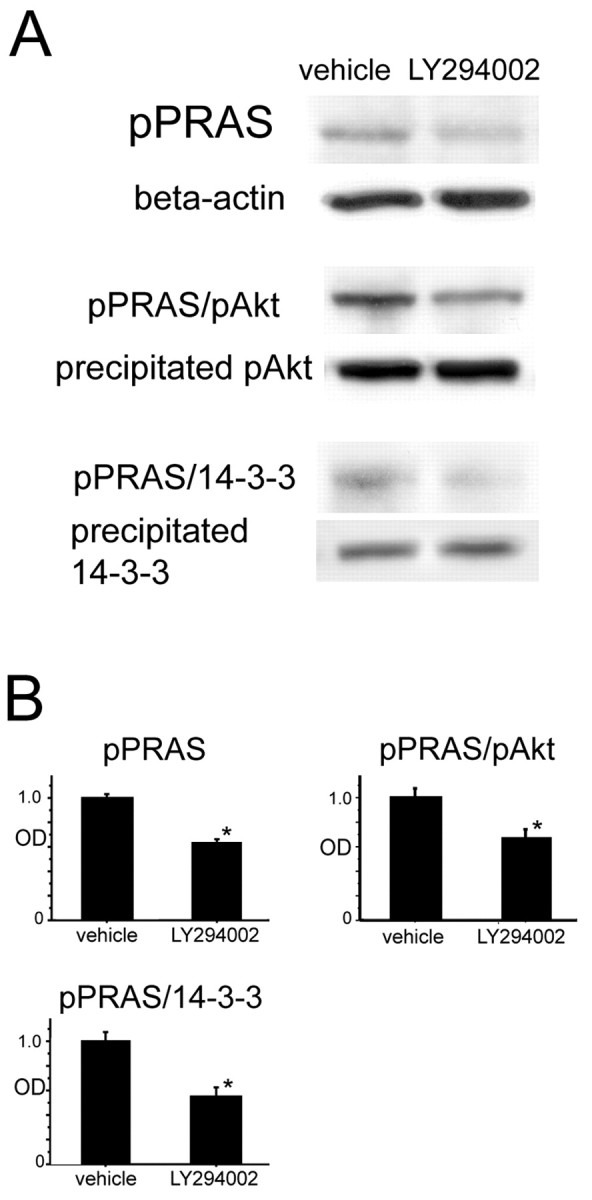
A, Western blot analysis for pPRAS and β-actin in the cytosolic fraction in the LY294002- and vehicle-treated mice 2 hr after tFCI. pPRAS expression was significantly stronger in the vehicle-treated mice compared with the LY294002-treated mice at the same time point (n = 4; *p < 0.05; top row). β-Actin was used as an internal control for the cytosolic fraction and no difference was seen between the samples (second row). Coimmunoprecipitation showed that both pPRAS/pAkt expression (n = 4; *p < 0.05; third row) and pPRAS/14-3-3 expression (n = 4; *p < 0.05; fifth row) were significantly stronger in the vehicle-treated mice compared with the LY294002-treated mice at the same time point. The expression of both precipitated pAkt (fourth row) and precipitated 14-3-3 protein (bottom row) was constant between the vehicle- and LY294002-treated mice. B, Optical density (OD) of these expressions was quantified.
Liposome-mediated pPRAS gene transfer led to induction of pPRAS in mouse brains after tFCI
We intraventricularly injected the complex of Myc-tagged pPRAS cDNA and DOTAP liposomes or the DOTAP liposome only as a control and examined Myc expression as a trace marker in the ischemic lesion after tFCI. Immunohistochemical analysis revealed that only a 1:2 [pPRAS cDNA (micrograms) to DOTAP liposome (microliters)] transfer ratio led to remarkable Myc expression in neuronal cells located in the cortical surface of the MCA territory and around the ventricular regions, and other transfer ratios did little to induce Myc expression (n = 6; Fig. 4A). Myc expression was not observed in the control samples, and the double immunofluorescent study demonstrated that Myc-immunopositive cells colocalized with NeuN immunoreactivity (n = 6; Fig. 4A). Western blot analysis showed that 24 hr of incubation after injection of a 1:2 cDNA-to-liposome ratio led to a prominent increase in pPRAS expression in the cytosolic fraction of the mouse brains after tFCI (n = 4; p < 0.05; Fig. 4B). The Myc-positive cells showed stronger expression of pPRAS compared with the Myc-negative cells in the same cortical ischemic lesion (n = 4; Fig. 4C). The prominent increase in pPRAS expression was induced by the gene transfection and was observed until 48 hr after injection (data not shown). These results suggest that liposome-mediated pPRAS gene transfer led to induction of pPRAS in neurons in the ischemic lesion after tFCI.
Figure 4.
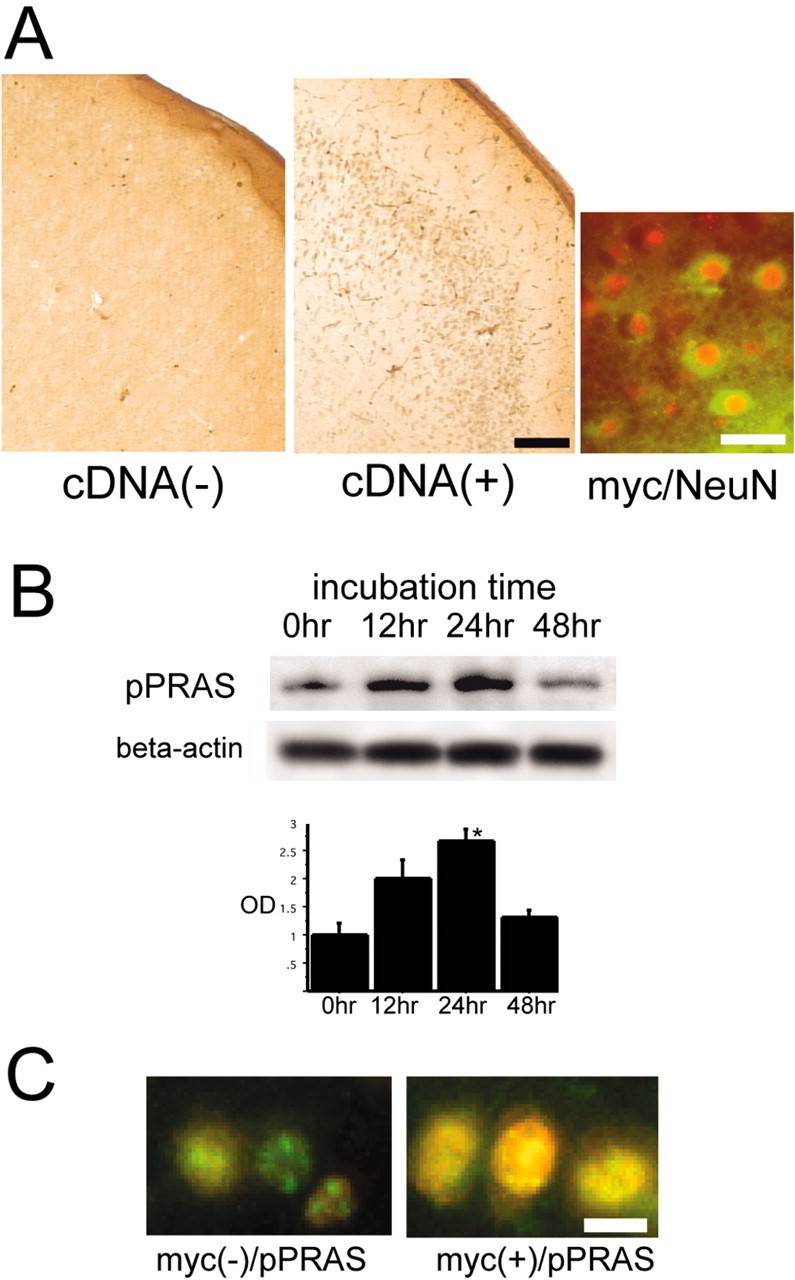
A, Representative photomicrographs show immunohistochemical staining for Myc 24 hr after injection of the complex Myc-tagged pPRAS cDNA and DOTAP liposomes at a 1:2 transfer ratio [pPRAS cDNA (micrograms) to DOTAP liposome (microliters)] or DOTAP liposome only as a control and 2 hr after tFCI. Myc immunopositivity was shown in the cortical penumbra of the MCA territory in the pPRAS cDNA-injected mice (n = 6; middle panel). No clear staining was observed in the control (n = 6; left panel). Scale bar, 100 μm. A representative photomicrograph shows double immunofluorescent staining for Myc (green) and NeuN (red) and demonstrates that colocalization of Myc and NeuN (yellow) was observed in the cortical penumbra of the MCA territory in the pPRAS cDNA-injected mice (n = 6; right panel). Scale bar, 15 μm. B, Western blot analysis shows that pPRAS expression began to increase 12 hr after injection of the complex Myc-tagged pPRAS cDNA and DOTAP liposomes, and it significantly increased 24 hr after injection (n = 4; *p < 0.05; top). β-Actin was used as an internal control for the cytosolic fraction, and no difference was seen between the samples (bottom). C, Representative photomicrographs show double immunofluorescent staining for Myc (red) and pPRAS (green) and demonstrates that pPRAS expression was stronger in the colocalized cells of Myc and pPRAS (right panel) compared with the Myc-negative cells (left panel) in the same view of the cortical penumbra in the pPRAS cDNA-injected mice (n = 4). Scale bar, 10 μm. OD, Optical density.
pPRAS gene transfer overexpressed pPRAS and promoted pPRAS/14-3-3 after tFCI
We used the 1:2 transfer ratio [pPRAS cDNA (micrograms) to DOTAP liposome (microliters)] and 24 hr of incubation for gene transfection and used the liposome-injected sample only as a cDNA-negative control. Reperfusion injury occurred after 24 hr of incubation with the cDNA-liposome complex. Immunohistochemistry demonstrated that pPRAS expression was stronger in the cortex of the pPRAS cDNA-transfected mice compared with the control mice 2 hr after tFCI (n = 4; Fig. 5A). Western blot analysis demonstrated that pPRAS expression increased in the pPRAS cDNA-transfected mice compared with the control mice 2 hr after tFCI (n = 4; p < 0.05; Fig. 5B). Coimmunoprecipitation demonstrated that pPRAS/pAkt expression did not significantly differ between the pPRAS cDNA-transfected mice and the control mice (data not shown). The expression of pPRAS/14-3-3 significantly increased in the pPRAS cDNA-transfected mice compared with the control mice 2 hr after tFCI (n = 4; p < 0.05; Fig. 5C). These results suggest that liposome-mediated pPRAS cDNA transfer overexpresses pPRAS and promotes pPRAS/14-3-3 after tFCI.
Figure 5.
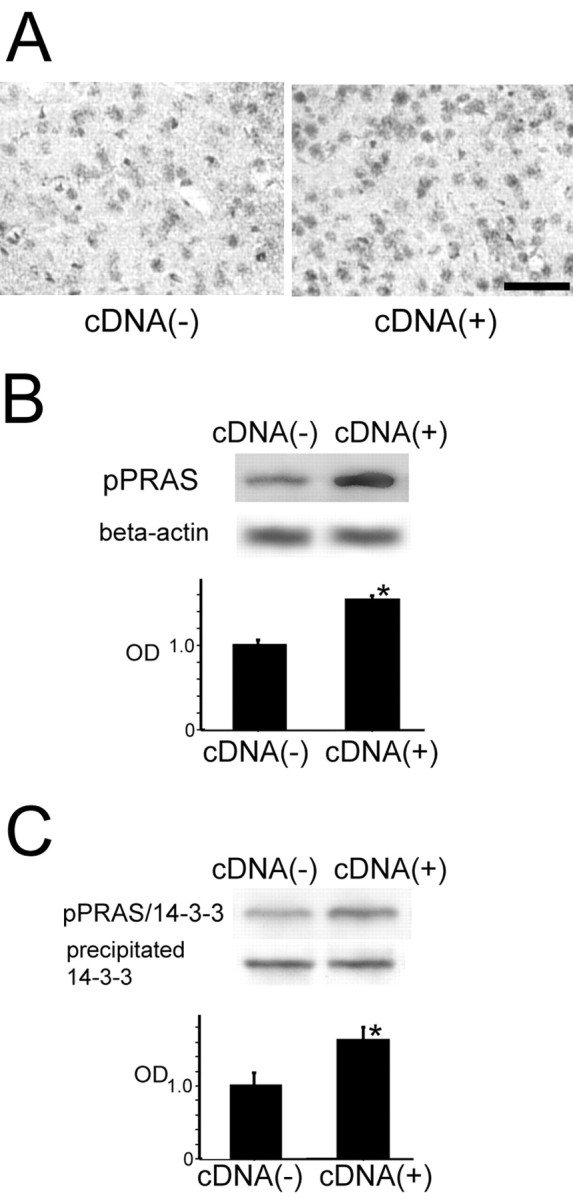
A, Representative photomicrographs show immunohistochemical staining for pPRAS in the cortical penumbra of the MCA territory in the pPRAS cDNA-transfected mice and the control mice 2 hr after tFCI (n = 4). pPRAS immunoreactivity was stronger in the pPRAS cDNA-transfected mice (right panel) than in the control mice (left panel). Scale bar, 50 μm. B, Western blot analysis for pPRAS and β-actin in the cytosolic fraction in the pPRAS cDNA-transfected and control mice 2 hr after tFCI. pPRAS expression was significantly stronger in the pPRAS cDNA-transfected mice compared with the control mice 2 hr after tFCI (n = 4; *p < 0.05; top). β-Actin was used as an internal control for the cytosolic fraction, and no difference was seen between the samples (bottom). C, Coimmunoprecipitation showed that pPRAS/14-3-3 expression was significantly stronger in the pPRAS cDNA-transfected mice compared with the control mice at the same time point (n = 4; *p < 0.05; top). Expression of the precipitated 14-3-3 protein was constant between the pPRAS cDNA-transfected mice and the control mice (bottom). OD, Optical density.
Overexpression of pPRAS attenuated apoptotic neuronal cell death after tFCI
DNA fragmentation was significantly increased 24 hr after reperfusion in the same model (data not shown) (Saito et al., 2003). We used the cytosolic fraction sample of the cortical surface of the MCA territory in the ischemic hemisphere as an ischemic penumbra lesion. DNA fragmentation was significantly reduced in the pPRAS cDNA-transfected mice compared with the control mice 24 hr after tFCI (n = 4; p < 0.05; Fig. 6A). TUNEL-positive cells were seen in the ischemic penumbra of the MCA territory and were observed most obviously 24 hr after reperfusion (Saito et al., 2003). TUNEL-positive cells were reduced in the pPRAS cDNA-transfected mice compared with the control mice 24 hr after tFCI (n = 4; Fig. 6B). We examined the infarction size and looked for hemiparesis, hemihypesthesia, and change in pupils after tFCI. Quantitative and histochemical assessment showed that overexpression of pPRAS improved the DNA damage after tFCI, but there was no significant difference in the infarction size or neurological deficits between the samples (data not shown). These results suggest that overexpression of pPRAS attenuates apoptotic cell death after tFCI.
Figure 6.
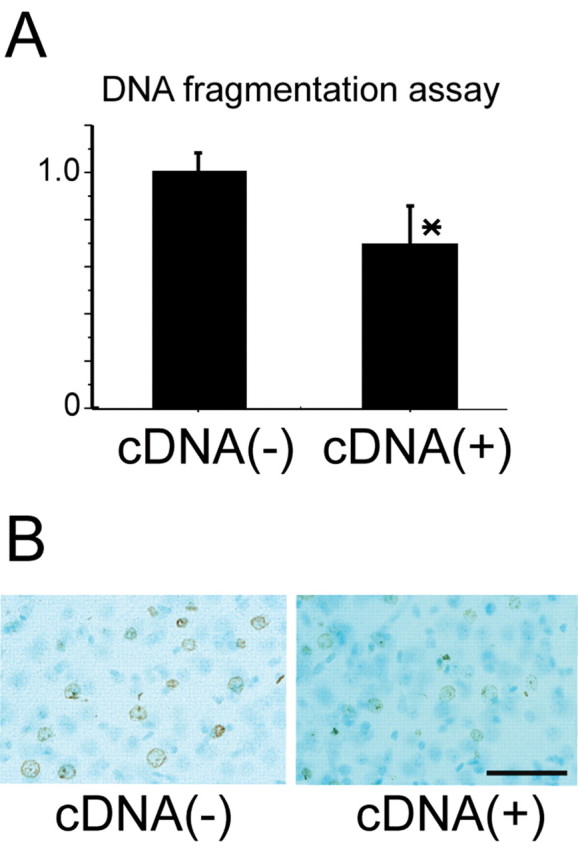
A, Apoptotic-related DNA fragmentation significantly decreased in the pPRAS cDNA-transfected mice compared with the control mice 24 hr after tFCI. B, TUNEL reactivity decreased in the pPRAS cDNA-transfected mice compared with the control mice 24 hr after tFCI (n = 4). Scale bar, 50 μm.
pPRAS positivity did not colocalize with activated caspase-3 reactivity or TUNEL reactivity
We examined the immunoreactive localization of pPRAS and activated caspase-3 and TUNEL reactivity to ascertain the relationship between PRAS phosphorylation and apoptotic cell death after tFCI. Caspase-3 is one of the well known executors of the apoptotic cascade. When caspase-3 is activated by apoptotic stimuli, it is cleaved at aspartate 175 to the large fragment (17 and 19 kDa), resulting in the caspase chain reaction in apoptotic cell death (Namura et al., 1998). First, we examined cleaved caspase-3 activity using the specific substrate from a commercial assay kit (Chemicon). Quantitative assay revealed that caspase-3 activity began to increase 4 hr after tFCI and significantly increased 8 and 24 hr after tFCI (n = 4; p < 0.05; Fig. 7A). Double immunofluorescent staining for pPRAS and cleaved caspase-3 demonstrated that the most pPRAS-reactive cells did not colocalize with cleaved caspase-3 reactivity in the ischemic penumbra 24 hr after reperfusion (n = 4; Fig. 7B). Double immunofluorescent staining for pPRAS and TUNEL reactivity revealed that the strong pPRAS immunoreactivity was barely observed in TUNEL-positive cells 24 hr after tFCI (n = 4; Fig. 7C). These results suggest that pPRAS expression was reduced in cells that were progressing toward apoptosis or had progressed to apoptosis after reperfusion.
Figure 7.
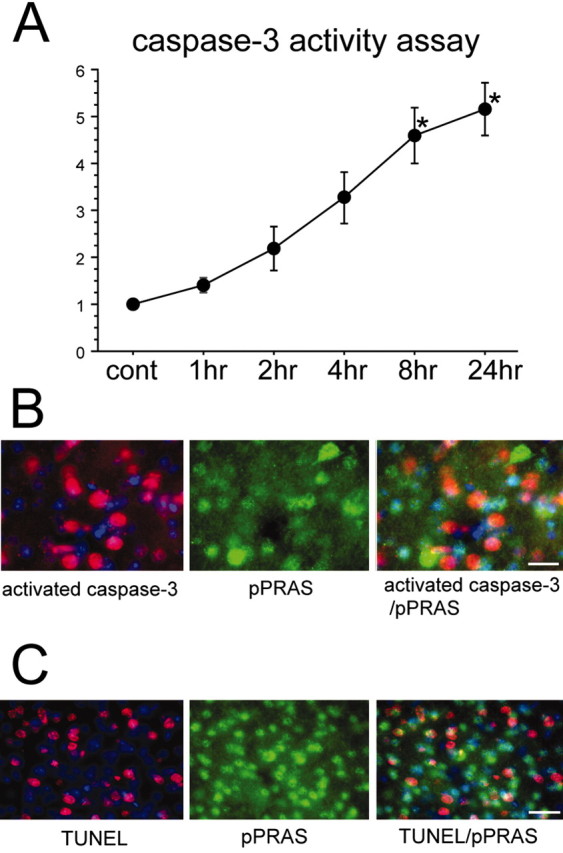
A, C aspase-3 activity assay demonstrated that activation gradually increased from 4 hr after tFCI, and it significantly increased 8 and 24 hr after tFCI (n = 4; *p < 0.05). B, Representative photomicrographs show double immunofluorescent staining for pPRAS and cleaved caspase-3 24 hr after tFCI. Expression of cleaved caspase-3 immunoreactivity (red, left panel) was observed in the ischemic cortical penumbra. pPRAS (green, middle panel) showed the distribution of neurons in the same view. An overlapped image of pPRAS and cleaved caspase-3 immunoreactivity (red, green, right panel) demonstrated that pPRAS expression did not colocalize with cleaved caspase-3-positive cells. Scale bar, 15 μm. C, Representative photomicrographs show double immunofluorescent staining for pPRAS and TUNEL reactivity 24 hr after tFCI. Expression of TUNEL immunoreactivity (red, left panel) was observed in the ischemic cortical penumbra. pPRAS (green, middle panel) showed the distribution of neurons in the same view. Overlapped image of pPRAS and TUNEL immunoreactivity (red, green, right panel) demonstrated that pPRAS expression was hardly seen in the TUNEL-positive cells. Scale bar, 25 μm. cont, Control.
NGF administration attenuated and NGF/trkA inhibition promoted apoptotic neuronal cell death after tFCI
DNA fragmentation was significantly reduced in the NGF-treated mice compared with the vehicle-treated mice 24 hr after tFCI but was significantly increased in the K-252a-treated mice compared with the vehicle-treated mice at the same time point (n = 4; p < 0.05; Fig. 8A). We performed intraventricular injection of K-252a as previously reported (Guilhem et al., 1996) and confirmed that it effectively reduced NGF receptor activity. TUNEL-positive cells were reduced in the NGF-treated mice compared with the vehicle-treated mice 24 hr after tFCI, but they were increased in the K-252a-treated mice compared with the vehicle-treated animals at the same time point (n = 4; Fig. 8B). Quantitative and histochemical assessment showed that NGF treatment improved the DNA damage after tFCI, but there was no significant difference in the infarction size or neurological deficits between the samples (data not shown). These results suggest that NGF administration attenuates but inhibition of NGF/trkA activity aggravates apoptotic cell death after tFCI.
Figure 8.

A, Apoptotic-related DNA fragmentation significantly decreased in the NGF-treated mice (n = 4; *p < 0.05) and significantly increased in the K-252a-treated mice (n = 4; #p < 0.05), as a tyrosine kinase inhibitor sample, compared with the vehicle-treated mice 24 hr after tFCI. B, TUNEL reactivity decreased in the NGF-treated mice (middle) and increased in the K-252a-treated mice (right) compared with the vehicle-treated mice (left) 24 hr after tFCI (n = 4). Scale bar, 50 μm.
NGF administration promoted but tyrosine kinase receptor inhibition reduced pAkt and pPRAS expression and binding of pPRAS to 14-3-3 after tFCI
Western blot analysis revealed that Akt expression was constantly present in the NGF-treated mice, the K-252a-treated mice, and the vehicle-treated mice 2 hr after reperfusion (n = 4; Fig. 9A,B). pAkt expression increased in the NGF-treated mice and decreased in the K-252a-treated mice compared with the vehicle-treated mice 2 hr after reperfusion (n = 4; p < 0.05; Fig. 9A,B). pPRAS, pPRAS/pAkt, and pPRAS/14-3-3 also increased in the NGF-treated mice and decreased in the K-252a-treated mice compared with the vehicle-treated mice 2 hr after reperfusion (n = 4; p < 0.05; Fig. 9A,B). These results suggest that NGF administration promoted but NGF/trkA inhibition reduced Akt activation, pPRAS, pPRAS/pAkt, and pPRAS/14-3-3 after tFCI.
Figure 9.
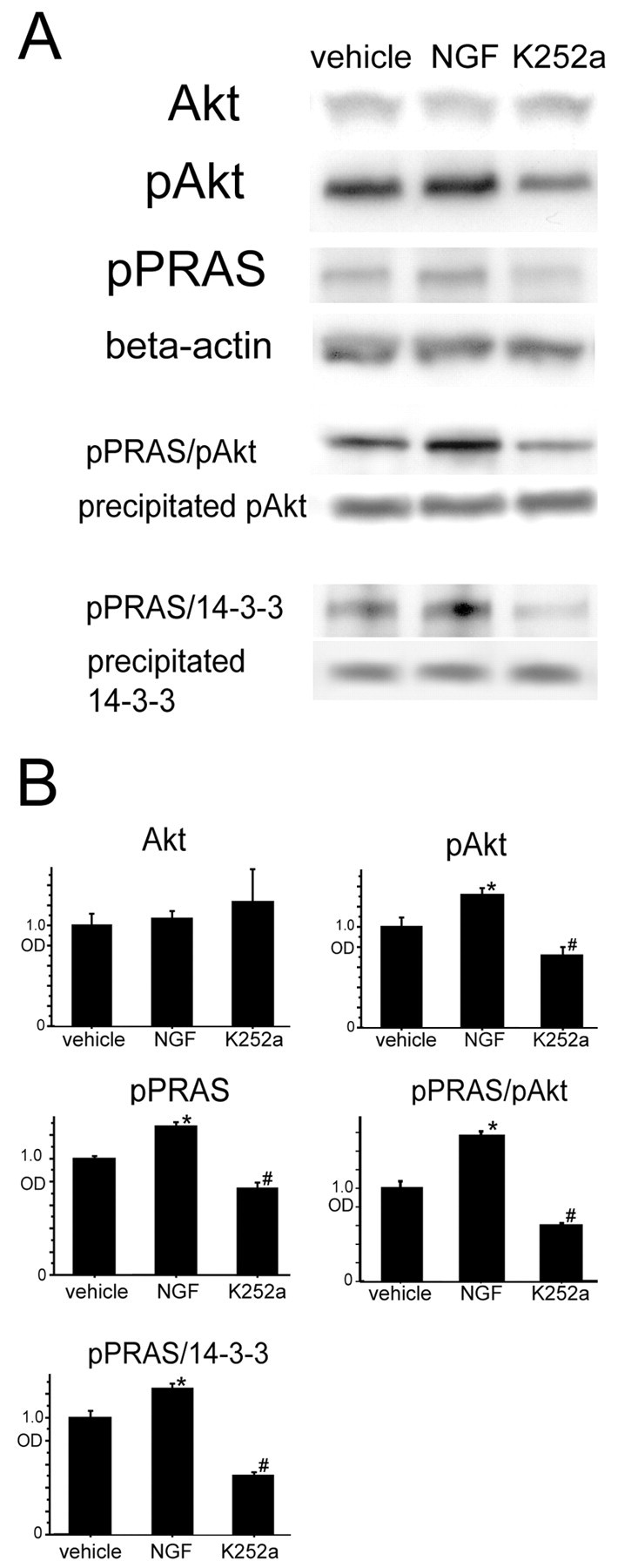
A, There was no significant difference in Akt expression among the NGF-, K-252a-, and vehicle-treated mice 2 hr after tFCI (n = 4; top row). pAkt expression was significantly increased in the NGF-treated mice (*p < 0.05) and was significantly reduced in the K-252a-treated mice compared with the vehicle-treated mice (#p < 0.05) at the same time point (n = 4; second row). pPRAS expression was significantly increased in the NGF-treated mice (*p < 0.05) and was significantly reduced in the K-252a-treated mice (#p < 0.05) compared with the vehicle-treated mice at the same time point (n = 4; third row). β-Actin was used as an internal control for the cytosolic fraction, and no difference was seen between the samples (n = 4; fourth row). Coimmunoprecipitation revealed that both pPRAS/pAkt (n = 4; fifth row) and pPRAS/14-3-3 (n = 4; seventh row) expression was significantly increased in the NGF-treated mice (*p < 0.05) and was significantly reduced in the K-252a-treated mice (#p < 0.05) compared with the vehicle-treated mice 2 hr after tFCI. The expression of precipitated pAkt (n = 4; sixth row) and precipitated 14-3-3 (n = 4; bottom row) was constant between the samples. B, Optical density (OD) of these expressions was quantified.
Discussion
The serine-threonine protein kinase Akt is a multifunctional regulator of cell survival, growth, and glucose metabolism (Datta et al., 1999). Akt contains an N-terminal pleckstrin homology domain, which binds phosphorylated lipids at the membrane in response to PI3-K activation (Franke et al., 1995). Akt is activated by phospholipid binding and activation loop phosphorylation at threonine 308 by phosphatidylinositol-triphosphate-dinase 1 and also within the C terminus at serine 473 by integrin-linked kinase (Alessi et al., 1996). Several substrates of Akt, including Bad, DAF-16 homologs, and the tuberous sclerosis complex 2 tumor suppressor gene tuberin, bind to 14-3-3 after their phosphorylation (Cahill et al., 2001; Masters et al., 2002; Nellist et al., 2002). In apoptotic neuronal cell death after in vivo cerebral ischemia, Akt is regulated downstream of the PI3-K pathway and regulates substrates, such as phosphorylated Bad at serine 136, and a Forkhead transcription factor, FKHR (Friguls et al., 2001; Noshita et al., 2001, 2003; Kawano et al., 2002).
PRAS is a novel Akt substrate that was purified, sequenced, and identified by Kovacina et al. (2003) as a proline-rich molecule. They demonstrated that PRAS can be phosphorylated by purified Akt, that the activation of an inducible Akt/mer-Akt is alone sufficient to induce PRAS phosphorylation, that PRAS phosphorylation decreases in cells lacking Akt1 and Akt2, and that phosphorylation of this protein leads to its binding to 14-3-3 (Kovacina et al., 2003). In the present study, we have demonstrated the following for the first time: (1) pPRAS was detected in neurons in the ischemic penumbra after tFCI, and its expression was transiently reduced during the early period of reperfusion injury; (2) direct binding of pPRAS to 14-3-3 was detected after tFCI and was transiently reduced concomitant with the decrease in pPRAS expression; (3) PI3-K inhibition reduced pPRAS and pPRAS/14-3-3 expression after tFCI; (4) liposome-mediated pPRAS cDNA transfection led to overexpression of pPRAS in neurons in the cortical ischemic penumbral lesion after tFCI; (5) overexpression of pPRAS also promoted pPRAS/14-3-3 expression after tFCI; (6) overexpression of pPRAS attenuated apoptotic neuronal cell death after tFCI; (7) pPRAS did not colocalize with activated caspase-3 or TUNEL immunoreactivity in the ischemic penumbral lesion after tFCI; (8) intraventricular NGF administration attenuated apoptotic neuronal cell death, and inhibition of its receptor aggravated apoptotic cell death after tFCI; and (9) NGF treatment promoted pPRAS and pPRAS/14-3-3 expression, and inhibition of its receptor reduced it after tFCI. Akt activation transiently increased during the early period of cerebral ischemia as observed in previous studies (Friguls et al., 2001; Noshita et al., 2001). However, both phosphorylated Bad at serine 136 and phosphorylated FKHR at serine 256 were remarkably reduced during the early ischemic period (Friguls et al., 2001; Kawano et al., 2002). pAkt transiently increased 4 hr after tFCI in the model used by Noshita et al. (2001). This discrepancy in time-dependent expression between pAkt and pPRAS was also observed after tFCI in our study. There is a possibility that the decrease in pPRAS might be involved in irreversible proapoptotic progression, which cannot be inhibited by survival factors, including activated Akt, after cerebral ischemia. PI3-K inhibition reduced not only pAkt expression but also pPRAS expression after tFCI. These results suggest that pPRAS might be regulated by the PI3-K/Akt pathway after tFCI in the same manner as pPRAS regulation in the in vitro study by Kovacina et al. (2003). Neuronal cell damage after tFCI includes processes involving both necrotic neuronal cell death in the ischemic core and apoptotic neuronal cell death in the peri-infarct area. We cannot completely exclude the Akt or PRAS pathways from playing roles in other cell death mechanisms after tFCI. TUNEL-positive neuronal cells, which showed postischemic apoptotic cell death, occupied <10% of the cortex in the MCA territory, and these apoptotic cells may not be enough to affect the total ischemic injury volume and neurological deficits after tFCI. Activation of the Akt cell survival pathway by pPRAS overexpression and NGF treatment did not extensively affect brain damage to a degree that improves neurological deficits. There is a possibility that these regulators may partially affect the cell survival pathway, but the neuroprotective role of these interventions remains unclear. In our study, we could not demonstrate the precise proapoptotic reaction after the transient decrease in pPRAS or overexpression of PRAS; however, the phosphorylation of PRAS might induce the anchoring to 14-3-3 to prevent progression to apoptosis, and the decrease in its phosphorylation might indicate the shift to apoptosis. There is a possibility that PRAS is phosphorylated by pAkt to inhibit proapoptotic progression after reperfusion injury following cerebral ischemia, in a manner similar to other Akt substrates, such as Bad and FKHR.
Cationic liposome-mediated gene transfection, which can condense DNA and increase transfection efficiency both in vitro and in vivo, is a potential therapeutic strategy for CNS disorders (Karpati et al., 1996; Zou et al., 1999; Cao et al., 2002). One of the most attractive features of this method for the treatment of CNS disorders is that DNA carriers can effectively transfect postmitotic, nondividing cells, such as neurons, by intrathecal injection (Zou et al., 1999; Cao et al., 2002). Another is that cationic liposomes can be used to deliver multiple genes of any type, such as linear or supercoiled nucleic acids, and a simply prepared liposomal complex can be administered in multiple ways (Yang et al., 1997; Zou et al., 1999).
Intraventricular liposomal transfection of the NGF gene was reported to be effective in traumatic brain injury, and hepatocyte growth factor, vascular endothelial growth factor, and bcl-2 genes have also been effective in neuronal cell damage after cerebral ischemia by liposome-mediated gene transfection (Zou et al., 1999; Wang et al., 2000; Cao et al., 2002; Yoshimura et al., 2002). There is a possibility that many other genes of neurotrophins or neuroprotective factors may effectively be induced in neurons to prevent neuronal cell damage after cerebral ischemia by liposome-mediated gene transfection. In this study, we found that a ratio of 1 μg of pPRAS cDNA to 2 μl of liposomes, with 24 hr of incubation after injection, produced high transfection efficiency in neurons by intrathecal injection. Overexpression of pPRAS in the cortical penumbral lesion of the MCA territory effectively inhibited apoptotic neuronal cell death after tFCI. However, a prominent increase in the target proteins was observed in localized areas, such as the periventricular regions and cortical surface, but this expression was not seen in the deep cortical region or the ischemic core. Further studies are required to improve the efficiency of the transfection into brains and to find the therapeutic possibility against neuronal cell death after cerebral ischemia.
Akt positively mediates survival in skeletal muscles and in neurons subjected to NGF withdrawal (Eves et al., 1998; Fujio et al., 1999). Akt is activated by several growth factors, including NGF, platelet-derived growth factor, basic fibroblast growth factor, insulin, and insulin-like growth factor-1, which results in the phosphorylation of the tyrosine kinase receptor (Franke et al., 1997). The cell survival role of NGF is thought to be involved in the Akt signaling pathway via the tyrosine kinase receptor (Eves et al., 1998; Fujio et al., 1999). However, the relationship between tyrosine kinase activation downstream and the complicated Akt signaling pathway remains unknown. Furthermore, the mechanism of NGF-mediated neuroprotection in apoptotic cell death models in vivo is also little understood. In our study, we quantitatively demonstrated for the first time the neuroprotective effect of NGF administration against apoptotic neuronal cell death and the proapoptotic effect of tyrosine receptor inhibition in mouse brains after tFCI. NGF treatment and inhibition of its receptor were much involved in Akt activation, pPRAS expression, and the interaction of pPRAS with pAkt and the 14-3-3 protein after tFCI. These results suggest that the neuroprotective machinery of pPRAS might play a critical role in neuronal cell survival pathways mediated by NGF after cerebral ischemia. However, little is known about the mechanisms of this link after apoptosis, and further studies are needed for clarification.
In summary, our results imply that overexpression of pPRAS has a neuroprotective role in apoptotic neuronal cell death after tFCI and that the pPRAS reaction with pAkt and 14-3-3 might be involved in its neuroprotective pathway downstream of PI3-K. Neuronal cell protection mediated by NGF may affect pPRAS reactions after cerebral ischemia. We suggest that pPRAS may play a critical role in the NGF cell survival pathway and should be considered as a therapeutic target for stroke.
Footnotes
This work was supported by National Institutes of Health Grants P50NS14543, R01NS25372, R01NS36147, and R01NS38653 and an American Heart Association Bugher Foundation award. P.H.C. is a recipient of the Jacob Javits Neuroscience Investigator Award. We thank Dr. Richard A. Roth (Department of Molecular Pharmacology, Stanford University School of Medicine) for preparation of the plasmid of the phosphorylated proline-rich Akt substrate, Cheryl Christensen for editorial assistance, Liza Reola and Bernard Calagui for technical assistance, and Elizabeth Hoyte for figure preparation.
Correspondence should be addressed to Dr. Pak H. Chan, Neurosurgical Laboratories, Stanford University, 1201 Welch Road, MSLS P314, Stanford, CA 94305-5487. E-mail: phchan@stanford.edu.
Copyright © 2004 Society for Neuroscience 0270-6474/04/231584-10$15.00/0
References
- Alessi DR, Andjelkovic M, Caudwell B, Cron P, Morrice N, Cohen P, Hemmings BA (1996) Mechanism of activation of protein kinase B by insulin and IGF-1. EMBO J 15: 6541-6551. [PMC free article] [PubMed] [Google Scholar]
- Berwick DC, Hers I, Heesom KJ, Moule SK, Tavaré JM (2002) The identification of ATP-citrate lyase as a protein kinase B (Akt) substrate in primary adipocytes. J Biol Chem 277: 33895-33900. [DOI] [PubMed] [Google Scholar]
- Bonfoco E, Krainc D, Ankarcrona M, Nicotera P, Lipton SA (1995) Apoptosis and necrosis: two distinct events induced, respectively, by mild and intense insults with N-methyl-d-aspartate or nitric oxide/superoxide in cortical cell curtures. Proc Natl Acad Sci USA 92: 7162-7166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buchan AM, Williams L, Bruederlin B (1990) Nerve growth factor: pretreatment ameliorates ischemic hippocampal neuronal injury. Stroke 21: 177. [Google Scholar]
- Cahill CM, Tzivion G, Nasrin N, Ogg S, Dore J, Ruvkun G, Alexander-Bridges M (2001) Phosphatidylinositol 3-kinase signaling inhibits DAF-16 DNA binding and function via 14-3-3-dependent and 14-3-3-independent pathways. J Biol Chem 276: 13402-13410. [DOI] [PubMed] [Google Scholar]
- Cantley LC (2002) The phosphoinositide 3-kinase pathway. Science 296: 1655-1657. [DOI] [PubMed] [Google Scholar]
- Cao Y-J, Shibata T, Rainov NG (2002) Liposome-mediated transfer of the bcl-2 gene results in neuroprotection after in vivo transient focal cerebral ischemia in an animal model. Gene Ther 9: 415-419. [DOI] [PubMed] [Google Scholar]
- Crowder RJ, Freeman RS (1998) Phosphatidylinositol 3-kinase and Akt protein kinase are necessary and sufficient for the survival of nerve growth factor-dependent sympathetic neurons. J Neurosci 18: 2933-2943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Datta SR, Brunet A, Greenberg ME (1999) Cellular survival: a play in three Akts. Genes Dev 13: 2905-2927. [DOI] [PubMed] [Google Scholar]
- Dokka S, Malanga CJ, Shi X, Chen F, Castranova V, Rojanasakul Y (2000) Inhibition of endotoxin-induced lung inflammation by interleukin-10 gene transfer in mice. Am J Physiol 279: L872-L877. [DOI] [PubMed] [Google Scholar]
- Eves EM, Xiong W, Bellacosa A, Kennedy SG, Tsichlis PN, Rosner MR, Hay N (1998) Akt, a target of phosphatidylinositol 3-kinase, inhibits apoptosis in a differentiating neuronal cell line. Mol Cell Biol 18: 2143-2152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franke TF, Yang SI, Chan TO, Datta K, Kazlauskas A, Morrison DK, Kaplan DR, Tsichlis PN (1995) The protein kinase encoded by the Akt proto-oncogene is a target of the PDGF-activated phosphatidylinositol 3-kinase. Cell 81: 727-736. [DOI] [PubMed] [Google Scholar]
- Franke TF, Kaplan DR, Cantley LC (1997) PI3K: downstream AKTion blocks apoptosis. Cell 88: 435-437. [DOI] [PubMed] [Google Scholar]
- Friguls B, Justicia C, Pallas M, Planas AM (2001) Focal cerebral ischemia causes two temporal waves of Akt activation. NeuroReport 12: 3381-3384. [DOI] [PubMed] [Google Scholar]
- Fujimura M, Morita-Fujimura Y, Kawase M, Copin J-C, Calagui B, Epstein CJ, Chan PH (1999) Manganese superoxide dismutase mediates the early release of mitochondrial cytochrome c and subsequent DNA fragmentation after permanent focal cerebral ischemia in mice. J Neurosci 19: 3414-3422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujio Y, Guo K, Mano T, Mitsuuchi Y, Testa JR, Walsh K (1999) Cell cycle withdrawal promotes myogenic induction of Akt, a positive modulator of myocyte survival. Mol Cell Biol 19: 5073-5082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gill R, Soriano M, Blomgren K, Hagberg H, Wybrecht R, Miss M-T, Hoefer S, Adam G, Niederhauser O, Kemp JA, Loetscher H (2002) Role of caspase-3 activation in cerebral ischemia-induced neurodegeneration in adult and neonatal brain. J Cereb Blood Flow Metab 22: 420-430. [DOI] [PubMed] [Google Scholar]
- Guilhem D, Dreyfus PA, Makiura Y, Suzuki F, Onteniente B (1996) Short increase of BDNF messenger RNA triggers kainic acid-induced neuronal hypertrophy in adult mice. Neuroscience 72: 923-931. [DOI] [PubMed] [Google Scholar]
- Kane S, Sano H, Liu SCH, Asara JM, Lane WS, Garner CC, Lienhard GE (2002) A method to identify serine kinase substrates: Akt phosphorylates a novel adipocyte protein with a Rab GTPase-activating protein (GAP) domain. J Biol Chem 277: 22115-22118. [DOI] [PubMed] [Google Scholar]
- Karpati G, Lochmüller H, Nalbantoglu J, Durham H (1996) The principles of gene therapy for the nervous system. Trends Neurosci 19: 49-54. [DOI] [PubMed] [Google Scholar]
- Kawano T, Morioka M, Yano S, Hamada J-i, Ushio Y, Miyamoto E, Fukunaga K (2002) Decreased Akt activity is associated with activation of Forkhead transcription factor after transient forebrain ischemia in gerbil hippocampus. J Cereb Blood Flow Metab 22: 926-934. [DOI] [PubMed] [Google Scholar]
- Kent TA, Quast M, Taglialatela G, Rea C, Wei J, Tao Z, Perez-Polo JR (1999) Effect of NGF treatment on outcome measures in a rat model of middle cerebral artery occlusion. J Neurosci Res 55: 357-369. [DOI] [PubMed] [Google Scholar]
- Kovacina KS, Park GY, Bae SS, Guzzetta AW, Schaefer E, Birnbaum MJ, Roth RA (2003) Identification of a proline-rich Akt substrate as a 14-3-3 binding partner. J Biol Chem 278: 10189-10194. [DOI] [PubMed] [Google Scholar]
- Lee TH, Kato H, Chen ST, Kogure K, Itoyama Y (1998) Expression of nerve growth factor and trkA after transient focal cerebral ischemia in rats. Stroke 29: 1687-1697. [DOI] [PubMed] [Google Scholar]
- Lizcano JM, Morrice N, Cohen P (2000) Regulation of BAD by cAMP-dependent protein kinase is mediated via phosphorylation of a novel site, Ser155 Biochem J 349: 547-557. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Manning BD, Tee AR, Logsdon MN, Blenis J, Cantley LC (2002) Identification of the tuberous sclerosis complex-2 tumor suppressor gene product tuberin as a target of the phosphoinositide 3-kinase/Akt pathway. Mol Cell 10: 151-162. [DOI] [PubMed] [Google Scholar]
- Masters SC, Subramanian RR, Truong A, Yang H, Fujii K, Zhang H, Fu H (2002) Survival-promoting functions of 14-3-3 proteins. Biochem Soc Trans 30: 360-365. [DOI] [PubMed] [Google Scholar]
- McLachlan G, Davidson DJ, Stevenson BJ, Dickinson P, Davidson-Smith H, Dorin JR, Porteous DJ (1995) Evaluation in vitro and in vivo of cationic liposome-expression construct complexes for cystic fibrosis gene therapy. Gene Ther 2: 614-622. [PubMed] [Google Scholar]
- McManus EJ, Alessi DR (2002) TSC1-TSC2: a complex tale of PKB-mediated S6K regulation. Nat Cell Biol 4: E214-E216. [DOI] [PubMed] [Google Scholar]
- Namura S, Zhu J, Fink K, Endres M, Srinivasan A, Tomaselli KJ, Yuan J, Moskowitz MA (1998) Activation and cleavage of caspase-3 in apoptosis induced by experimental cerebral ischemia. J Neurosci 18: 3659-3668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nellist M, Goedbloed MA, de Winter C, Verhaaf B, Jankie A, Reuser AJJ, van den Ouweland AMW, van der Sluijs P, Halley DJJ (2002) Identification and characterization of the interaction between tuberin and 14-3-3ζ. J Biol Chem 277: 39417-39424. [DOI] [PubMed] [Google Scholar]
- Noshita N, Lewén A, Sugawara T, Chan PH (2001) Evidence of phosphorylation of Akt and neuronal survival after transient focal cerebral ischemia in mice. J Cereb Blood Flow Metab 21: 1442-1450. [DOI] [PubMed] [Google Scholar]
- Noshita N, Sugawara T, Lewén A, Hayashi T, Chan PH (2003) Copper-zinc superoxide dismutase affects Akt activation after transient focal cerebral ischemia in mice. Stroke 34: 1513-1518. [DOI] [PubMed] [Google Scholar]
- Porteous DJ, Dorin JR, McLachlan G, Davidson-Smith H, Davidson H, Stevenson BJ, Carothers AD, Wallace WAH, Moralee S, Hoenes C, Kallmeyer G, Michaelis U, Naujoks K, Ho L-P, Samways JM, Imrie M, Greening AP, Innes JA (1997) Evidence for safety and efficacy of DOTAP cationic liposome mediated CFTR gene transfer to the nasal epithelium of patients with cystic fibrosis. Gene Ther 4: 210-218. [DOI] [PubMed] [Google Scholar]
- Saito A, Hayashi T, Okuno S, Ferrand-Drake M, Chan PH (2003) Overexpression of copper/zinc superoxide dismutase in transgenic mice protects against neuronal cell death after transient focal ischemia by blocking activation of the Bad cell death signaling pathway. J Neurosci 23: 1710-1718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shigeno T, Mima T, Takakura K, Graham DI, Kato G, Hashimoto Y, Furukawa S (1991) Amelioration of delayed neuronal death in the hippocampus by nerve growth factor. J Neurosci 11: 2914-2919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang R, Zhang X, Zhang J, Xiu R (2000) Gene transfer of vascular endothelial growth factor plasmid/liposome complexes in glioma cells in vitro: the implication to the treatment of cerebral ischemic diseases. Clin Hemorheol Microcirc 23: 303-306. [PubMed] [Google Scholar]
- Yamamoto S, Yoshimine T, Fujita T, Kuroda R, Irie T, Fujioka K, Hayakawa T (1992) Protective effect of NGF atelocollagen mini-pellet on the hippocampal delayed neuronal death in gerbils. Neurosci Lett 141: 161-165. [DOI] [PubMed] [Google Scholar]
- Yang G, Chan PH, Chen J, Carlson E, Chen SF, Weinstein P, Epstein CJ, Kamii H (1994) Human copper-zinc superoxide dismutase transgenic mice are highly resistant to reperfusion injury after focal cerebral ischemia. Stroke 25: 165-170. [DOI] [PubMed] [Google Scholar]
- Yang K, Clifton GL, Hayes RL (1997) Gene therapy for central nervous system injury: the use of cationic liposomes: an invited review. J Neurotrauma 14: 281-297. [DOI] [PubMed] [Google Scholar]
- Yoshimura S, Morishita R, Hayashi K, Kokuzawa J, Aoki M, Matsumoto K, Nakamura T, Ogihara T, Sakai N, Kaneda Y (2002) Gene transfer of hepatocyte growth factor to subarachnoid space in cerebral hypoperfusion model. Hypertension 39: 1028-1034. [DOI] [PubMed] [Google Scholar]
- Zou LL, Huang L, Hayes RL, Black C, Qiu YH, Perez-Polo JR, Le W, Clifton GL, Yang K (1999) Liposome-mediated NGF gene transfection following neuronal injury: potential therapeutic applications. Gene Ther 6: 994-1005. [DOI] [PubMed] [Google Scholar]