

Abstract
Tumor necrosis factor type I receptor (TNFRI), a death receptor, mediates apoptosis and plays a crucial role in the interaction between the nervous and immune systems. A direct link between death receptor activation and signal cascade-mediated neuron death in brains with neurodegenerative disorders remains inconclusive. Here, we show that amyloid-β protein (Aβ), a major component of plaques in the Alzheimer's diseased brain, induces neuronal apoptosis through TNFRI by using primary neurons overexpressing TNFRI by viral infection or neurons from TNFRI knock-out mice. This was mediated via alteration of apoptotic protease-activating factor (Apaf-1) expression that in turn induced activation of nuclear factor κB (NF-κB). Aβ-induced neuronal apoptosis was reduced with lower Apaf-1 expression, and little NF-κB activation was found in the neurons with mutated Apaf-1 or a deletion of TNFRI compared with the cells from wild-type (WT) mice. Our studies suggest a novel neuronal response of Aβ, which occurs through a TNF receptor signaling cascade and a caspase-dependent death pathway.
Keywords: Alzheimer, apoptosis, death, degeneration, neuron, receptor
Introduction
Apoptosis is a normal biological process during brain development. However, it can become a harmful factor for mature brains under injured or diseased circumstances, such as brain neuronal loss in neurodegenerative diseases (Anderson et al., 1996; Estus et al., 1997; Troy et al., 2000; Yuan and Yankner, 2000; Jellinger and Stadelmann, 2001; Morishima et al., 2001; Cregan et al., 2002; Galvan et al., 2002; Kumar-Singh et al., 2002; Wei et al., 2002; Eberhardt and Schulz, 2003; Koriyama et al., 2003; Lopez Salon et al., 2003; Tamagno et al., 2003). Alzheimer disease (AD) is a neurodegenerative disease and is characterized by significant neuronal loss. There are two major neuropathological changes in the AD brain: senile plaques and neurofibrillary tangles. Amyloid-β proteins (Aβ) are major constituents of senile plaques and are produced from cleavage of the amyloid precursor protein (APP) (Selkoe and Kopan, 2003; Sisodia and St. George-Hyslop, 2002). The majority of Aβ fragments include Aβ1-40 and Aβ1-42. Although Aβ1-42 is believed to be the protein initially deposited in diffuse plaques as the seed molecule for amyloid fibril formation, the subsequent deposition of Aβ1-40 is more closely associated with the onset of clinical symptoms in late-onset AD (Jarret and Lansbury, 1993; Ishii et al., 1997). During the past decade, various insults have been proposed as a part of the molecular mechanisms involved in Aβ-induced neurotoxicity. These include receptor mediation, free radicals, imbalances of cellular calcium distribution, and many others (Yan et al., 1996; Sheehan et al., 1997; Geula et al., 1998; Lorenzo et al., 2000; Mattson et al., 2000; Troy et al., 2000; Wang et al., 2000; Butterfield et al., 2002; Heneka et al., 2002; Kaltschmidt et al., 2002; Mettenburg et al., 2002; Xiao et al., 2002; Xie et al., 2002; Chaudhury et al., 2003; Tamagno et al., 2003). Recently, Ferrer et al. (2001) reported that expression of one particular death receptor, Fas, in AD brains is increased. However, Fas expression seemed not to be directly correlated with tangles in AD brains (Ferrer et al., 2001). The other death receptor, tumor necrosis factor type I receptor (TNFRI), has shown increased expression in AD brains and is related to the apoptotic process in AD brains (our unpublished observation). By using double-labeling methods, our unpublished data demonstrated that TNFRI is expressed in most pyramidal glutamatergic neurons in both normal and AD brains. Caspases and transcription factor mechanisms have also been proposed (Nakagawa et al., 2000; Cregan et al., 2002; Galvan et al., 2002; Haas et al., 2002; Kuperstein and Yavin, 2002; Lee et al., 2002; Walsh et al., 2002; Yao et al., 2002; Yu et al., 2002; Zhang et al., 2002; Zau et al., 2002; Raina et al., 2003) as underlying defects in several types of neurodegeneration. However, molecular mechanisms of Aβ-induced neuronal death remain inconclusive.
Recent studies on apoptotic protease-activating factor-1 (Apaf-1) transgenic mice (Li et al., 1997; Cecconi et al., 1998; Yoshida et al., 1998) demonstrate that activation of Apaf-1 induces obvious abnormality in tissues in which cellular development depends on apoptosis (Srinivasula et al., 1998; Cregan et al., 2002; Ferraro et al, 2003). This observation suggests a critical role of Apaf-1 in apoptotic cell death. Apaf-1 is a member of the protein family that contains a caspase recruitment domain and regulates apoptosis. Nod1 and Nod2 contain a caspase recruitment domain (CARD), which can interact with NF-κB and promote NF-κB activation, which requires IKKγ and is inhibited by dominant negative mutants of IκBα, IKKα, IKKβ, and IKKγ (Inohara et al., 2001; Ogura et al., 2001). Furthermore, the activation of NF-κB translocation causes apoptosis in vitro (McCarthy et al., 1998; Thome et al., 1998; Inohara et al., 2001; Ogura et al., 2001). Another NF-κB-related mechanism of apoptosis is TNFRI, which contains a “death domain” (DD). TNFRI and its intracellular mediators may activate common pathways that lead to degradation of IκBα, which relocates NF-κB from the cytoplasm into the nucleus (Miyamoto et al., 1994; Hsu et al., 1995). The translocation of NF-κB might play an anti-apoptotic role in various cells (Guo et al., 1998; Kaltschmidt et al., 1999) and an apoptotic role in neuronal cells (Schneider et al., 1999; Yang et al., 2002). This discrepancy may be caused by different cell types or the involvement of distinct receptors in neurons. In AD, Aβ peptide has been shown to activate NF-κB (Akama et al., 1998; Kuner et al., 1998; Ghribi et al., 2001). A recent report demonstrated that the soluble Aβ1-40-induced toxicity correlates with its association with the cell membrane (Mathews et al., 2002; Morishima-Kawashima and Ihara, 1998). These findings, together with the pronounced toxicity of soluble Aβ at low concentrations, suggests that cell membrane receptor(s) might be involved in Aβ-induced cell death (Kuner et al., 1998; Janciauskiene et al., 1999; Lorenzo et al., 2000; Wang et al., 2000; Mettenburg et al., 2002; Xie et al., 2002).
Although a previous study has demonstrated that knock-out of both TNFRI and TNFRII can simultaneously increase neurodegeneration (Bruce et al., 1996), the specific contributions of each TNF receptor subtype to neuronal cell death has not been clearly identified. We demonstrated recently that TNF-α-induced neuronal cell death is mediated by the NF-κB translocation and is TNFRI dependent (Yang et al., 2002). Here, we explore whether Aβ-induced neuronal apoptosis is related to TNFRI and its unique signal transduction pathway. By using a gene targeting approach, we provide novel evidence that treatment with Aβ1-40 increases Apaf-1 expression and neuronal NF-κB translocation. Our data suggest that, on treatment with Aβ1-40, Apaf-1 activates NF-κB via binding the intracellular death domain of TNFRI with high affinity, eventually leading to neuronal death.
Materials and Methods
All chemicals were purchased from Sigma (St. Louis, MO), unless otherwise stated. Aβ1-40 and reverse Aβ40-1 peptides were purchased from Bachem (Torrance, CA). The peptides were dissolved in DMSO or formic acid to maintain soluble form and then diluted to indicated concentrations with culture media for toxicity or binding experiments. Antibodies for TNF receptors were purchased from R & D Systems (Minneapolis, MN) and the antibody for NF-κB p65 was purchased from Santa Cruz Biotechnology (Santa Cruz, CA). A polyclonal rabbit antibody to Apaf-1 was purchased from Chemicon (Temecula, CA), and monoclonal antibodies to actin and laminin were purchased from Sigma.
Molecular cloning and plasmid construction. For the construction of TNF receptor subtype cDNA plasmids, we isolated poly(A)+ RNA from human brain tissue and synthesized cDNA using reverse transcriptase (RT) methods. PCR was used to amplify TNFRI, complement C1qB chain, complement C9, and the epidermal growth factor receptor (EGFR) coding region. For transient transfection, cDNA was inserted into a pcDNA3.1 expression vector (Invitrogen, Gaithersburg, MD) that directs expression from both cytomegalovirus (CMV) promoter and simian virus 40 (SV40) origin.
Primary hippocampal neuron cultures from TNFRI knock-out mice. TNFRI knock-out mice (C57BL/6J background) were purchased from The Jackson Laboratory (Bar Harbor, ME). Primary hippocampal neuron cultures were prepared as described previously (Kamegai et al., 1990; Yang et al., 2002). Briefly, the hippocampal regions from embryonic 15-d-old mice fetuses were dissected and the neurons were recovered by enzymatic digestion with trypsin and mechanical dissociation. Cells were then plated at a density of ∼6 × 105 cells in 3 ml of media per 35 mm dish (Primaria, Falcon Instruments, Florence, Italy) coated with poly-l-lysine (Sigma). Cultures were maintained at 37°C in an incubator humidified with 95% O2 and 5% CO2. The culture medium was a 1:1 mixture of DMEM and Ham's F12 supplemented with 15 mm HEPES, 100 μg/ml human transferrin, 25 μg/ml bovine insulin, 20 nm progestenne, 20 nm hydrocortisine-21-phosphate, 10 mm l-carnitine, 30 nm 3,3,5-triiodo-l-thyronine, 7 ng/ml α-tocopherol, 7 ng/ml retinol acetate, 1 μm thioctic acid, and 100 μm putrescine (Invitrogen). No serum was used in the dissociation or in the cultures. Neurons were used for experiments 5 d after plating.
Neurotypic cell line culture. Human SH-SY5Y cells, which can be terminally differentiated into cells bearing numerous specific characteristics of neurons (Shen et al., 1997), were cultured using 50% MEM plus 50% F12 medium, 10% heat-inactivated fetal calf serum, and 10 μm retinoic acid. The culture medium was replaced every 3 d and cells were differentiated for 6 d.
RT-PCR for TNFRI-/- and Apaf-1 mRNA detection. Total RNA was extracted from neurons with TNFRI-/- or wild-type mice (Qiagen, Valencia, CA). Pilot experiments were done to determine the linear range of amplification with respect to the amount of starting template and PCR cycles. The cDNA was synthesized from 1.5 ng of total RNA using the SuperScript One-Step RT-PCR kit (Invitrogen) at 62°C for 30 min. The PCR-forward oligonucleotide primer used for TNFRI was 5′TCGATTTGCTGTACCAAGTG3′ and the reverse oligonucleotide primer used for TNFRI was 5′GAAAATGACCAGGGGCAACAG3′. After an initial denaturation step at 94°C for 5 min, the cycle was initiated, which consisted of denaturing for 1 min at 94°C, annealing for 2 min at 53°C, and extending for 2 min at 72°C. The cycle was repeated 35 times. The PCR-forward oligonucleotide primer used for wild-type and knock-out Apaf-1 was 5′AGATAGCCTAGGGGGGTGCAT3′ and the reverse oligonucleotide primer used for Apaf-1 was 5′ATCAGTTTCCAATCDCTGCT3′. After an initial denaturation step at 94°C for 5 min, the cycle was initiated, which consisted of denaturing for 1.5 min at 94°C, annealing for 1 min at 66°C, and extending for 1.5 min at 72°C. The cycle was repeated 35 times, with one final extension cycle of 10 min at 72°C.
Nuclear and cytoplasmic isolation. To prepare nuclear extracts, 4 × 106 treated cells were trypsinized and incubated for 20 min in a hypo-osmotic buffer containing the following: 10 mm HEPES, pH 7.8, 10 mm KCl, 2 mm MgCl2, 0.1 mm EDTA, 10 mg/ml aprotinin, 0.5 mg/ml leupeptin, 3 mg/ml PMSF, and 3 mm DTT with 25 ml of 10% NP-40. The nuclei were pelleted by centrifugation for 5 min in a microcentrifuge. The supernatants containing the cytoplasmic proteins were removed and stored at -70°C. T solubilize the DNA binding proteins, the pelleted nuclei were resuspended in a high salt buffer containing the following: 50 mm HEPES, pH 7.4, 50 mm KCl, 300 mm NaCl, 0.1 mm EDTA, 10% v/v glycerol, 3 mm DTT, and 3 mm PMSF. The resuspended nuclei were gently shaken for 30 min at 4°C. The extracts were spun in a microcentrifuge for 10 min and the clear supernatant, containing nuclear protein, was either aliquoted for determination of the protein concentration or stored at -70°C for future assays.
Electrophoretic mobility shift assay. Electrophoretic mobility shift assay (EMSA) was performed using a double-stranded 15 bp oligonucleotide (5′CTAGGGGGACTTTCC3′) containing the NF-κB consensus sequence, and radiolabeled with 32P-ATP. For the binding reaction, the nuclear protein extract (10 μg) and 0.25 ng (25,000 cpm) of labeled oligonucleotide was incubated for 15 min at 4°C in a total volume of 30 μl binding buffer containing the following: 10 mm Tris-HCl, pH 7.5, 50 mm NaCl, 50 mm KCl, 1 mm EDTA, 5 mm DTT, 5% glycerol and 2 μg poly dIDC. DNA protein complexes were separated from unbound probe on native 4.5% polyacrylamide gels in 250 mm Tris-boric EDTA at 200-250 V for 2-3 hr. The resultant gel was vacuum-dried and exposed to Kodak film (Eastman Kodak, Rochester, NY) for 8-15 hr at -70°C. The visual inspection of the free probe band at the bottom of the gel confirmed that equivalent amounts of radiolabeled probe were used for each sample. The amount of DNA-protein complex present was analyzed using densitometry or a PhosphorImager (Molecular Dynamics, Sunnyvale, CA).
Lactate dehydrogenase release measurement and statistical analysis. For quantitative assessment of neuron damage, lactate dehydrogenase (LDH) release from degenerating neurons was measured using a CytoTox 96 nonradioactive cytotoxicity assay kit (Promega, Madison, WI). The percentage of LDH release was calculated as the ratio of LDH contained in the supernatant relative to total LDH contained in both the supernatant and cell lysate. Data were analyzed by an ANOVA Student paired t test.
Western blot analysis. The samples of neurons and cells were lysed in buffer containing the following: 10 mm Tris-HCl, pH 7.4, 25 mm NaCl, 50 mm EDTA, 1 mm EGTA plus 0.5% Triton X-100, 10% SDS, and a protease inhibitor mixture [1 mm phenylmethylsulfonyl fluoride, 1 μg/ml pepstatin A, 5 μg/ml leupeptin, and 2 μg/ml aprotinin (Boehringer Mannheim, Indianapolis, IN)]. For each sample, 10 μg protein was separated on a 15% SDS Tricine gel and transferred to a polyvinylidene difluoride (PVDF) membrane electrophoretically for 2 hr with 0.1% Tween 20 in TBS. Either NF-κB p65 or Apaf-1 was detected with antibodies against NF-κB p65 or Apaf-1 at a 1:1000 dilution for 16 hr at 4°C, followed by incubation with an HRP-conjugated secondary antibody and processed using ECL detection (Amersham Biosciences, Piscataway, NJ). Actin and laminin were examined as housekeeping proteins in the cytoplasm and nucleus, respectively.
Expression plasmids and transfection. Plasmid DNA used for transfections was purified using Qiagen Maxiprep kits. Exogenous DNA transfection followed the procedures of Lipofectamine (Invitrogen). For transient transfection, the cDNA of TNFRI, complement C1qB chain, complement C9, or EGFR was inserted into a pcDNA3-histamine (HA) expression vector containing a C-terminal HA tag that directs its expression from both CMV and SV40 promoters. The vector alone was used as a control. These constructs were transiently transfected into neurons. For stable transfection, the cDNA was subcloned into the pcDNA3.1 vector and transfected into neurons using Lipofectamine Plus (Invitrogen). Forty-eight hours later, 96 well plates at 103 cells per well were seeded with cells, selected with G418 (600 μg/ml; Invitrogen) and maintained with 350 μg/ml G418. The selected cells were screened by Western dot-blotting to confirm TNFRI, C1qB, C9, and EGFR expression.
Viral transfection of neurons with IΚΚα, mutant IΚΚα, Apaf-1, and mutant Apaf-1. The adenovirus vector we have constructed was described previously (Choi-Lundberg et al., 1997). In brief, the replication defective adenoviral vector (AdCMV-lacZ), contains the enhancer/early promoter of CMV drives lacZ transcription with an SV40 polyadenylation sequence downstream, but the vector has deleted E1A, E1B, and E3 regions, which impair the ability of the virus to replicate and transform nonpermissive cells. A viral stock solution of 1.0 × 1012 particles/ml was used for AdCMV-lacZ; 10- or 102-fold dilutions of these stocks were prepared in PBS immediately before affection in primary neurons. Control affections were performed with PBS and AdCMV-lacZ without the IKKα, mutant IKKα (mIKKα), Apaf-1, and mutant Apaf-1 plasmids [gifts from Dr. D. V. Goeddel (Tularik Inc., South San Francisco, CA), Dr. P. Baeuerle (Tularik Inc.), and Dr. I. M. Verma (The Salk Institute, San Diego, CA)]. Vector titers were matched for total particles at ∼1-2 × 109 particles/2 μl (Mochizuki and Mizumo, 2003).
Terminal deoxynucleotidyl transferase-mediated biotinylated UTP nick end labeling staining and DNA fragmentation. Neurons were grown on poly-l-lysine-coated chamber slides. Terminal deoxynucleotidyl transferase-mediated biotinylated UTP nick end labeling (TUNEL) was used to visualize neurons with fragmented DNA. Neurons were harvested 16 hr after Aβ1-40 treatment, fixed in 4% paraformaldehyde for 20 min, washed in three changes of PBS, and then incubated for 1 hr at 37°C with 75 μl of a TUNEL mixture (Boehringer Mannheim) containing the following (in μl): 0.5 terminal transferase, 0.95 biotin-deoxyUTP, 6.0 CoCl2, 15.0 TdT buffer, and 52.55 distilled water. The reaction was stopped by incubation in 4× SSC buffer followed by three washes in PBS. Neurons were then labeled with a streptavidin Cy2 secondary antibody (Jackson ImmunoResearch, West Grove, PA) for 45 min at room temperature and counterstained with Hoechst 33258 (0.5 μg/ml) for 5 min. The fraction of TUNEL-positive cells as a percentage of total cell number was determined. To analyze DNA integrity, DNA was extracted from neurons with or without Aβ1-40 treatment from TNFRI-/- mice and its optical density was measured at 260 nm and separated on a 1% agarose gel to confirm DNA loading after staining the gel with ethidium bromide. Equal amounts of DNA were separated by conventional agarose gel electrophoresis and Southern blotted, and apoptotic DNA ladders were visualized by hybridization with digoxigenin-labeled total mouse genomic DNA probe (Boehringer Mannheim).
Transfection and NF-κB activity assay. NF-κB activity was analyzed using a luciferase reporter gene; 3 × 105 cells were transfected with 5× κB-luciferase reporter plasmid and various expression plasmids, including IKKα and mutant IKKα, using Lipofectamine per the manufacturer's instructions. To normalize for transfection efficiency, all transfections included a LacZ-expressing plasmid, and the lysates were assayed for β-galactosidase activity.
Preparation of Apaf-1 antisense oligonucleotide. Both sense and antisense Apaf-1 oligonucleotide phosphorothioates were synthesized on an Applied Biosystems (Foster City, CA) model 392 DNA synthesizer using phosphoramidite chemistry. Oligonucleotides were purified by reverse-phase chromatography using Oligo-Pak oligonucleotide purification columns (Millipore, Billerica, MA) as reported previously (Shen et al., 1997). The Apaf-1 antisense oligonucleotide sequence was 5′CTTCTCCATCGCTCACCT3′, overlapping the code for the initiation site of methionine on the Apaf-1 gene (Zou et al., 1997). The sense oligonucleotides for Apaf-1 were the exact inverse complement of the antisense oligonucleotides. Lyophilized oligonucleotides were dissolved in sterile water to prepare stock solutions.
In vitro 125I-Aβ1-40 binding assay. Cells (2 × 107) were transfected with expression vectors containing TNFRI, complement C1qB, complement component C9, and EGFR. Sixteen hours after transfection, membranes were prepared from transfected cells. For Figure 6B, membrane lysates containing 10 mg of protein were incubated with 600 ng of 125I-Aβ1-40 (Amersham), 5 μg of anti-FLAG M2 antibody (Sigma), 10 μl of protein A-Sepharose, and 10 μl of protein G-Sepharose at 4°C for 2 hr. Proteins bound to the matrix were washed six times with 1 ml of the washing buffer. The bound radioactivity was measured using a Beckman (Fullerton, CA) liquid scintillation counter. For Figure 6C, proteins were immunoprecipitated first from 40 mg of membrane lysate as described above and then incubated with 600 ng of 125I-Aβ1-40 in the presence of 20 mg of bovine serum albumin Fraction V (Sigma) at 4°C for 2 hr. After six washes with 1 μl of washing buffer, the bound radioactivity was measured. To evaluate protein expression, proteins in 25 μg of membrane lysates were detected by immunoblotting with anti-FLAG antibody.
Figure 6.
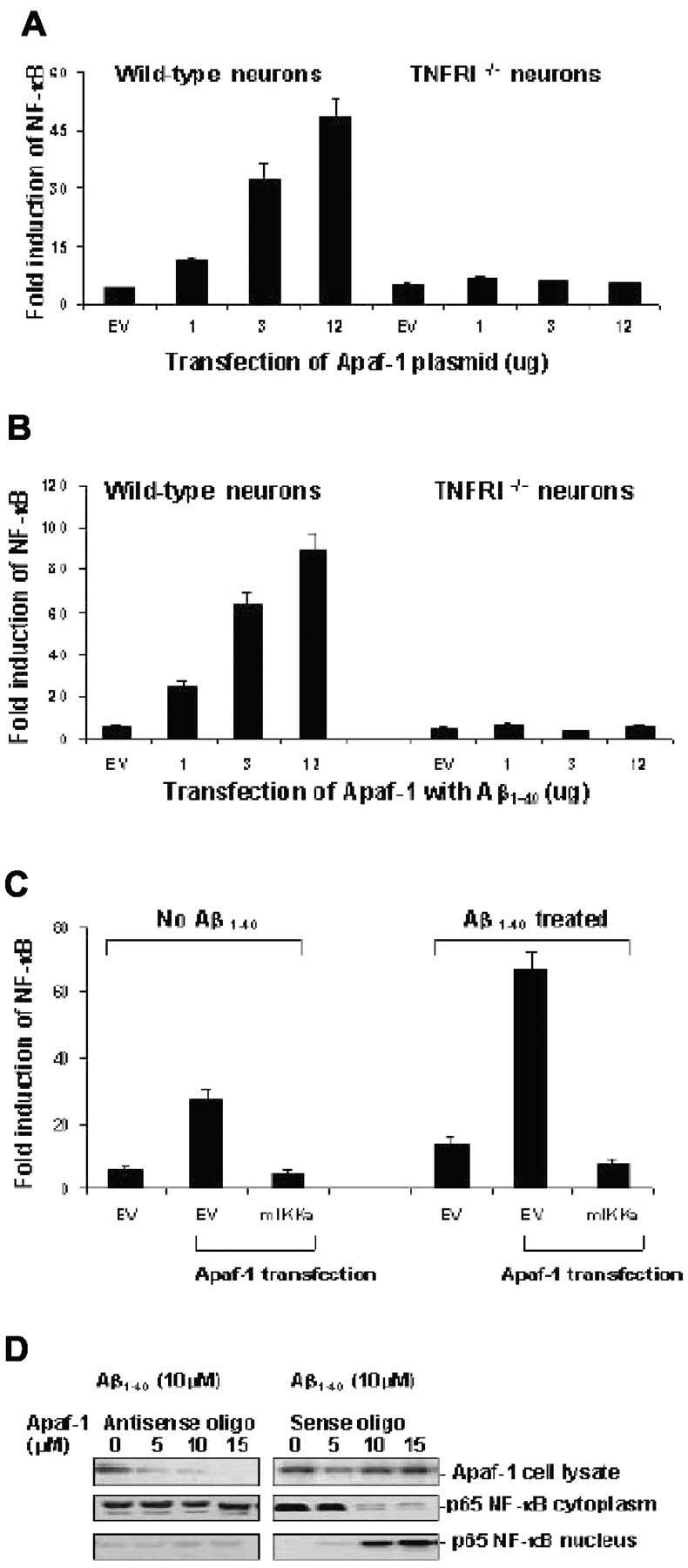
Neuronal NF-κB activation by Apaf-1 expression induced by Aβ. A, Neurons from wild-type and TNFRI-/- mice were transfected with an empty vector (EV) or increasing amounts of FLAG-tagged Apaf-1 construct together with an NF-κB luciferase reporter construct. The cells were harvested 24 hr after transfection and lysed in a luciferase assay buffer. The lysates were assayed for luciferase activity as described in Materials and Methods and subjected to Western blot analysis. The relative amounts of Apaf-1 in the lysates were determined by densitometric scanning of the immunoreactive bands on the Western blots. B, Induction of Apaf-1-induced NF-κB activation by Aβ1-40. Induction of NF-κB activation was determined from triplicate culture of wild-type and TNFRI-/- neurons cotransfected with the same amounts of FLAG-tagged Apaf-1 as (A) with the NF-κB luciferase reporter construct; 23 hr after transfection the neurons were treated with 10 μm Aβ1-40 for 60 min. C, Apaf-1-induced NF-κB activation is inhibited by kinase-inactive IKKα protein. Cells were transfected with 1.5 μg of empty vector, vector plus NF-κB luciferase construct, empty vector plus Apaf-1, or Apaf-1 plus mIKKα. After 23 hr of transfection, one group of neurons was treated with 10 μm Aβ1-40 for 60 min and then cells were harvested and assayed for NF-κB activity by the luciferase assay as described in Materials and Methods. D, Western blot analysis of the abundance of Apaf-1 in the protein extracts isolated neurons by transfection of Apaf-1 and mutant Apaf-1. Pretreatment of neurons using antisense oligonucleotides for Apaf-1 at 5, 10, and 15 μm previous Aβ1-40 treatment (10 μm) resulted in a significant reduction of Apaf-1 expression (top). However, there was no significant change of p65 NF-κB expression level in cytoplasm (middle), and little p65 NF-κB was detected in the nucleus (bottom), suggesting that inhibition of Apaf-1 did not result in the translocation of NF-κB. In the neurons with sense oligonucleotides for Apaf-1 treatment at the same series of concentrations, no Apaf-1 reduction was observed. However, p65 NF-κB was decreased in the cytoplasm and was increased in the nucleus after Aβ1-40 treatment, indicating that Aβ1-40 treatment triggers the activation of NF-κB translocation. One hundred microgram aliquots of cytosolic protein extracts isolated from neurons were subjected to 5% (for Apaf-1) or 10% (for p65 NF-κB) SDS-PAGE and transferred to a nitrocellulose filter. The filters were probed with a polyclonal anti-Apaf-1 antibody (AB16941; Chemicon). The antigen-antibody complexes were visualized by an ECL method, as described in Materials and Methods.
TNF receptor binding in whole cells. TNFRI transfected SH-SY5Y cells (1 × 106 per assay) were incubated with increasing concentrations of 125I-Aβ1-40 alone or with a 100-fold excess of unlabeled 125I-Aβ1-40 for 2 hr at 4°C. 125I-Aβ1-40 receptor complexes were precipitated at 4°C by the addition of 500 μl of 25% polyethylene glycol and 500 μl of 0.1% rabbit γ-globulin. Cells were incubated with increasing concentrations of 125I-Aβ1-40 alone or with a 200-fold excess of unlabeled Aβ1-40 for 2 hr at 4°C and then washed three times. Specific binding (the difference between binding of 125I-Aβ1-40 in the absence or presence of cold 125IAβ1-40) was usually >80% of the total 125I-Aβ1-40 binding activity. After centrifugation, 3 ml of ice-cold buffer was added to the tubes, followed by filtration through a Brandel (Gaithersburg, MD) cell harvester, using GF/C glass fiber filters previously soaked in 0.05% polyethylenimine. Radioactivity on the filters was determined in a liquid scintillation counter with a counting efficiency of ∼40% (Wallac, Gaithersburg, MD). The data preprocessing was performed using a Prism program and the actual nonlinear curve-fitting was performed using Ligands software.
Results
TNFRI contributes to Aβ1-40-induced neuronal apoptosis
Because TNFRI contains a DD, we thus assume that neurons with high levels of TNFRI might be more vulnerable to insults such as Aβ. To test this hypothesis, we overexpressed TNFRI in neurons by adenoviral affection (Fig. 1A) and used LDH release as a cell death marker to evaluate Aβ-induced neurotoxicity in the infected neurons. We found that Aβ-induced LDH release from the TNFRI-infected neurons is significantly increased in a dose-dependent manner after 8 hr infection. The dose dependency of Aβ1-40 treatment was also observed at low doses (100-10,000 nm), at which these concentrations are generally not toxic to noninfected control neurons (Fig. 1B). Our data indicate that TNFRI-infected neurons are more vulnerable to Aβ-induced toxicity compared with noninfected control neurons (Fig. 1B), which usually need at least 10-20 times higher concentrations of Aβ1-40 to cause neuron death (Estus et al., 1997; Morishima et al., 2001). This is consistent with our recent findings as well as with studies from other groups that TNFRI-affected neurons are more vulnerable to insults (Bruce et al., 1996; Estus et al., 1997; Morishima et al., 2001; Yang et al., 2002). Moreover, to determine whether Aβ-induced cell death is associated with apoptosis, we performed DNA fragmentation experiments and found that DNA fragments were greatly increased in Aβ-treated TNFRI-affected neurons compared with the noninfected control or control vector-infected neurons (Fig. 1C). Moreover, the appearance of DNA fragments was observed at the much lower dose (100 nm) of Aβ treatment (Fig. 1C) in the infected cells; similarly, DNA fragments were found in nontransfected neurons at ∼100-fold greater doses of Aβ treatment (Fig. 1C).
Figure 1.
Overexpressing TNFRI reveals vulnerability to Aβ1-40. Neurons were infected with defective adenoviral vector containing TNFRI, TNFRII, empty-defective adenoviral vector, or noninfection. A, The protein expression of TNFRI after infection was confirmed by Western blot. B, Overexpression of TNFRI resulted in neuronal vulnerability to Aβ1-40-induced cell death even at low doses (100 or 10,000 nm), whereas no such effects were observed in nonaffected neurons or neurons transfected with an empty-defective viral vector in the presence of Aβ1-40 at the same doses. All results were repeated four times from independent experiments, 8 hr after infection. **p < 0.01. C, DNA fragmentation was observed in neurons with TNFRI overexpression at 100, 1000, and 10,000 nm of Aβ1-40, but noninfected or control vector-infected neurons and TNFRII-infected neurons exhibited DNA fragmentation only at the high dose of Aβ1-40 (10,000 nm) after 8 hr of infection.
To further examine how Aβ causes neuronal apoptosis, and further verify whether TNFRI is linked to Aβ-induced neuronal apoptosis, we examined Aβ-induced toxicity in the cultured hippocampal neurons from TNFRI knock-out or WT mice. Studies from our Southern blot and Western blot hybridizations confirmed no expression of TNFRI in TNFRI-/- mice (data not shown). Morphologically, the hippocampal neurons from TNFRI-/- mice treated with vehicle grew rather healthily with multiple and long neurites (Fig. 2A). To further confirm our morphological observation, LDH release was used as a cell death marker to quantitatively measure cell death in the cultured neurons under each treatment condition. Interestingly, we found that treatment with Aβ1-40 at various doses in the TNFRI-/- neurons produced no changes in LDH release compared with neurons from WT mice, even at high doses of Aβ1-40 (0.1-10 μm) for 48 hr (Fig. 2B). These data suggest that TNFRI gene deletion might have shut down a cellular death-related signal pathway and neurons without an IDD became “insensitive” to insults. Likewise, TNFRI-/- neurons treated with vehicle grew well, suggesting that mature neurons are able to survive in the absence of TNFRI (Fig. 2A). Furthermore, to determine whether this neuronal degeneration is associated with apoptosis, we used TUNEL staining and DNA fragmentation techniques to evaluate neuronal apoptosis and found that most TNFRI-/- neurons were negative for TUNEL staining after Aβ1-40 treatment, compared with very positive TUNEL staining in the Aβ-treated WT neurons (Fig. 2C,D).
Figure 2.

Hippocampal neurons from knock-out TNFRI exhibit clear neurodegeneration after exposures of Aβ1-40 peptide. A, Morphological changes of hippocampal neurons from TNFRI knock-out (TNFRI-/-) or wild-type mice after exposure to Aβ1-40 for 48 hr. Nikon (Tokyo, Japan) microscope at 200×. TNFRI-/- neurons show resistance to Aβ1-40 insults, but wild-type neurons demonstrated degeneration after Aβ1-40 treatment. B, The primary cultured hippocampal neurons from six TNFRI-/- or wild-type mice were incubated for 48 hr with Aβ1-40. The dead or detached neurons were removed with the supernatant, and the remaining neurons were lysed. LDH levels were measured in both supernatant and cell lysates. The data in B are from four independent experiments and represent the means ± SD, which is the percentage of supernatant LDH values relative to total LDH contained in both the supernatant and cell lysate as described in Materials and Methods. Hippocampal neuronal apoptosis induced by Aβ1-40 treatment is TNFRI dependent. Aβ1-40-induced apoptosis in hippocampal neurons in the absence of TNFRI is demonstrated in TUNEL staining (C) and DNA fragmentation (D). The hippocampal neurons from TNFRI-/- or wild-type mice were incubated with Aβ1-40 at 10, 000 nm for 18 hr. The cells were fixed and neurons were analyzed for apoptosis using TUNEL for fragmented nuclear DNA (C). These images are typical of apoptotic single neurons cultured with Aβ1-40 at 10,000 nm in wild-type mice. For DNA fragmentation, Southern blots of DNA isolated from TNFRI-/- or wild-type mice were analyzed. TNFRI-/- and wild-type neurons were treated with Aβ1-40 (100 nm to 20 μm) for 48 hr (D) and cycloheximide was used as a positive control (data not shown). The results from TUNEL, Hoechst staining, and DNA fragmentation indicate that neurons with TNFRI deletion were resistant to Aβ1-40-induced apoptosis.
Physical interaction between Aβ1-40 and TNFRI in neurons
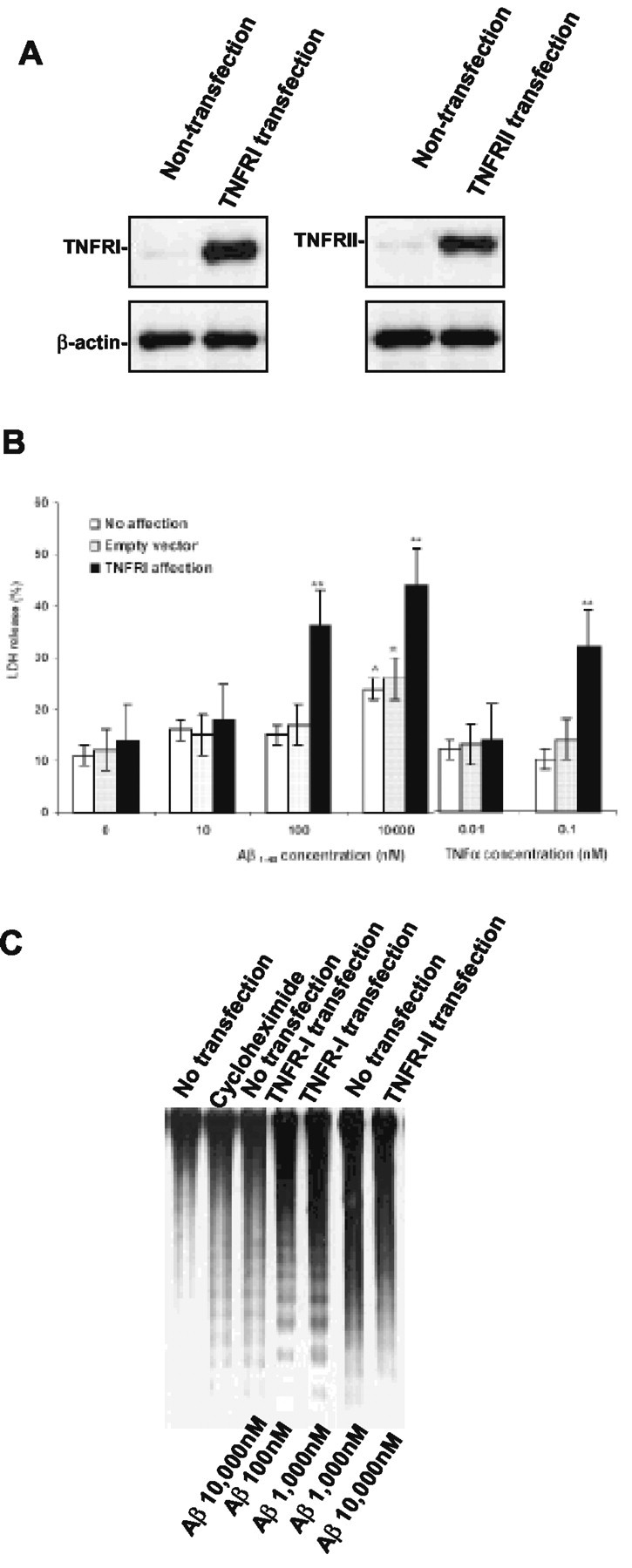
Having shown that Aβ exposure resulted in less neuron degeneration and apoptosis in neurons from target-deleted TNFRI mice, we further addressed the issue of how Aβ1-40 interacts with TNFRI on the neuronal surface to transduce receptor activation to the death signal cascade. A high level of expression of TNFRI in the infected neurons was confirmed by mRNA and protein expression using RT-PCR and Western blots (Fig. 3B). To examine whether Aβ1-40 binds TNFRI directly, cell membranes were prepared from neurons expressing TNFRI as described in Materials and Methods. The ability of TNFRI to bind radiolabeled 125I-Aβ1-40 was tested by a modified immunoprecipitation assay. To further rule out nonspecific binding, we included cold reverse Aβ40-1 peptide to compete for our specific binding by using radiolabeled Aβ1-40. We did not find any specific inhibitory binding in our assays (data not shown). Aβ1-40 was coimmunoprecipitated with FLAG-tagged TNFRI only, not with other FLAG-tagged control proteins (i.e., EGFR, C1qB, IL receptor, and C9) (Fig. 3B). Furthermore, to rule out nonspecific immunoprecipitation, we also used reverse Aβ40-1 to coimmunoprecipitate with FLAG-tagged TNFRI, as well as EGFR, C1qB, IL-1R, and C9, but no significant immunoprecipitation was observed (data not shown). Thus, TNFRI is associated with an Aβ-binding activity present in the membrane of neurons. However, there is a possibility that TNFRI does not bind directly to Aβ1-40 and that the association requires other membrane factors, such as fatty lipids (Avdulov et al., 1997; Corsico et al., 1998; Waschuk et al., 2001). To test this possibility, we first immunoprecipitated TNFRI and a complement component protein, C9, as a control protein, with anti-FLAG antibody, and the ability of the immunoprecipitated proteins to bind Aβ1-40 was tested in lipid-free buffer. Immunoprecipitated TNFRI exhibited Aβ1-40 binding activity, but control protein, C9, did not (Fig. 3C). These results suggest that TNFRI binds directly to Aβ1-40.
Figure 3.
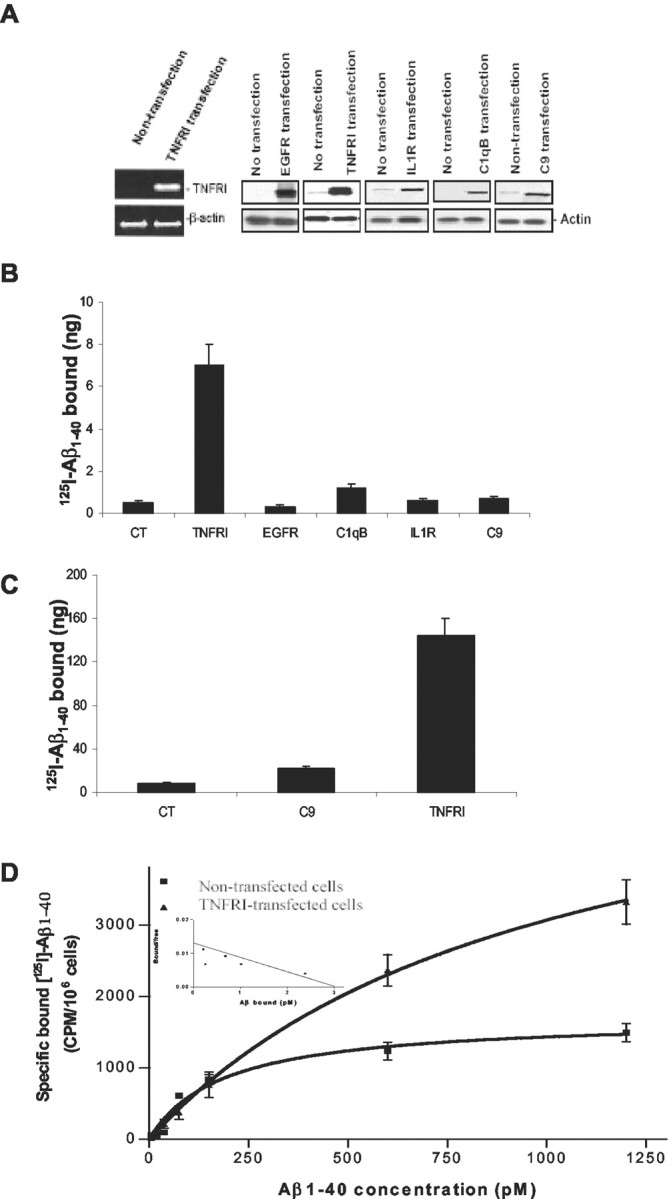
Interaction between TNFRI and Aβ1-40. A, Detection of mRNA and protein expression of TNFRI by using RT-PCR and Western blot techniques. For RT-PCR, the negative control reaction in the absence of RT with RNA template yielded no detectable product, whereas the positive control reaction in the TNFRI transfection with RNA template, yielded a highly abundant band, as expected. For Western blot analysis, the same SH-SY5Y cells transfected with TNFRI, EGFR, complement component C1qB, complement component C9, and IL-1R were used for confirmation of the expression of this receptor. The results demonstrated little expression of the endogenous TNFRI in SH-SY5Y cells (A). B, Physical interaction between TNFRI and Aβ1-40. 5 × 107 SH-SY5Y cells were transfected with 20 μg of pcDNA3.1-FLAG-TNFRI, pcDNA3.1-FLAG EGFR, pcDNA3.1-FLAG-C1qB, pcDNA3.1-FLAG-C9, and pcDNA3.1-FLAG-IL1R. Twenty-four hours after transfection, cell membranes were prepared from transfected cells as described in Materials and Methods. The radioactivity of 125I-Aβ1-40 coimmunoprecipitated with anti-FLAG antibody was determined as described in Materials and Methods. C, Proteins were immunoprecipitated first from 40 mg of membrane lysate and incubated with 125I-Aβ1-40 in the presence of 20 mg bovine serum albumin. The coimmunoprecipitated radioactivity was determined as described in Materials and Methods. The expression of each protein in 40 μg of membrane lysates was immunoprecipitated with anti-FLAG antibody. D, Pharmacological binding analysis of the 125I-Aβ1-40 binding in TNFRI transfected SH-SY5Y cells. The assays with transfected, and control cells contained 1 × 106 cells per assay. Five independent binding assays were performed, and the Kd average for TNFRI is 0.42 nm.
To further characterize the pharmacological binding affinity of Aβ to TNFRI, we established an in vitro model by subcloning TNFRI cDNA into the expression vector pcDNA3.1 that contains the human CMV promoter and SV40 ori. This construct was transfected into human SH-SY5Y neurotypic cells, which have low levels of endogenous TNFRI (Fig. 3D) and exhibit neuronal properties (Shen et al., 1997), followed by a modified calcium phosphate transfection technique. Protein expression in whole cells was measured by 125I-specific binding (Yang et al., 2002). To ensure the specific binding was from soluble Aβ1-40 instead of nonspecific binding caused by “stickiness” of the physical property of the protein, specific 125I-Aβ1-40 binding was analyzed in the absence and presence of a 100-fold excess of cold-soluble Aβ1-40. To prevent peptide aggregation that can occur at higher temperatures (Estus et al., 1997; Morishima et al., 2001), all binding assays were performed on ice at all times to maintain the soluble form of Aβ1-40 peptide, as described in Materials and Methods. We found that 125I-Aβ1-40 binds specifically to TNFRI in the transfected SH-SY5Y cells, but little specific binding was seen in nontransfected cells (Fig. 3D). After analyses of 125I-Aβ1-40 saturation binding and Scatchard properties in TNFRI transfected cells, we found that 125I-Aβ1-40 exhibits receptor binding dissociate constant (Kd) for TNFRI (Kd = 0.42 nm), as illustrated in Figure 3D. This was determined using the SH-SY5Y cell transfectants and is comparable with the Kd of the native TNFRI expressed constitutively on Chinese hamster ovary cells and endogenous TNF receptors in HeLa cells (data not shown). We have also examined whether 125I-Aβ1-40 binds to other cytokine receptors such as the p75 NGF or EGF receptor in transfected SH-SY5Y cells and no specific or high-affinity binding was observed (data not shown). Together, these results indicate that Aβ1-40 binds with high affinity and specificity to the TNFRI receptor.
Activated NF-κB translocation by Aβ is diminished in neurons from TNFRI-/- mice
To further study the possible mechanisms by which Aβ1-40 enhances apoptosis in neurons with TNFRI overexpression, we examined TNF-receptor-related signal transduction pathways. Moreover, several studies have shown that Aβ induces the activity of NF-κB DNA binding (Akama et al., 1998; Ghribi et al., 2001) and that NF-κB is also reported to be TNFRI dependent not only in the peripheral immune system (Tartaglia et al., 1993; Hsu et al., 1995; Sipe et al., 1996), but also in the brain (Yang et al., 2002). To determine whether TNFRI is involved in the Aβ1-40-induced NF-κB neuronal activity, an experiment for examining translocation of p65, a subunit of NF-κB, was performed using Western blotting. Nuclear and cytoplasmic extracts were isolated from the neurons of WT and TNFRI-/- mice after a 30 min incubation with Aβ1-40 at 0, 0.1, 1, and 10 μm. We found that Aβ1-40 induced increases in nuclear NF-κB p65 in a dose-dependent manner, whereas decreased cytoplasmic NF-κB p65 was observed in hippocampal neurons of WT mice (Fig. 4A). This suggests that Aβ1-40 induces NF-κB translocation into the nucleus. However, no significant NF-κB p65 translocation was observed in either the cytoplasm or the nucleus in neurons of TNFRI-/- mice after Aβ treatment (Fig. 4B). This result indicates that translocation of NF-κB p65 in neurons induced by Aβ1-40 is at least partially TNFRI dependent. To examine changes of the binding activity of NF-κB, we conducted NF-κB binding studies using EMSA and found low NF-κB binding activity in TNFRI-/- neurons, whereas WT neurons still contained high levels of Aβ1-40-induced NF-κB binding activity (Fig. 4C). Although we found that the translocation of NF-κB by Aβ is TNFRI dependent, whether translocation of NF-κB causes neuronal death or enhances cell survival or plays a compensatory role was not clear. To examine this possibility, we used adenovirus to deliver IΚΚα, which on phosphorylation, releases p65 NF-κB into the nucleus. Adenovirus was also used to deliver mutant IΚΚα, which is unable to be phosphorylated. We found that Aβ1-40 treatment in neurons with viral transduction of IΚΚα resulted in DNA fragmentation, whereas little DNA fragmentation was observed in mutant IΚΚα-affected neurons even at a relatively high dose of Aβ1-40 treatment (Fig. 4D), suggesting that NF-κB translocation activated by IΚΚα leads to apoptosis in neurons.
Figure 4.
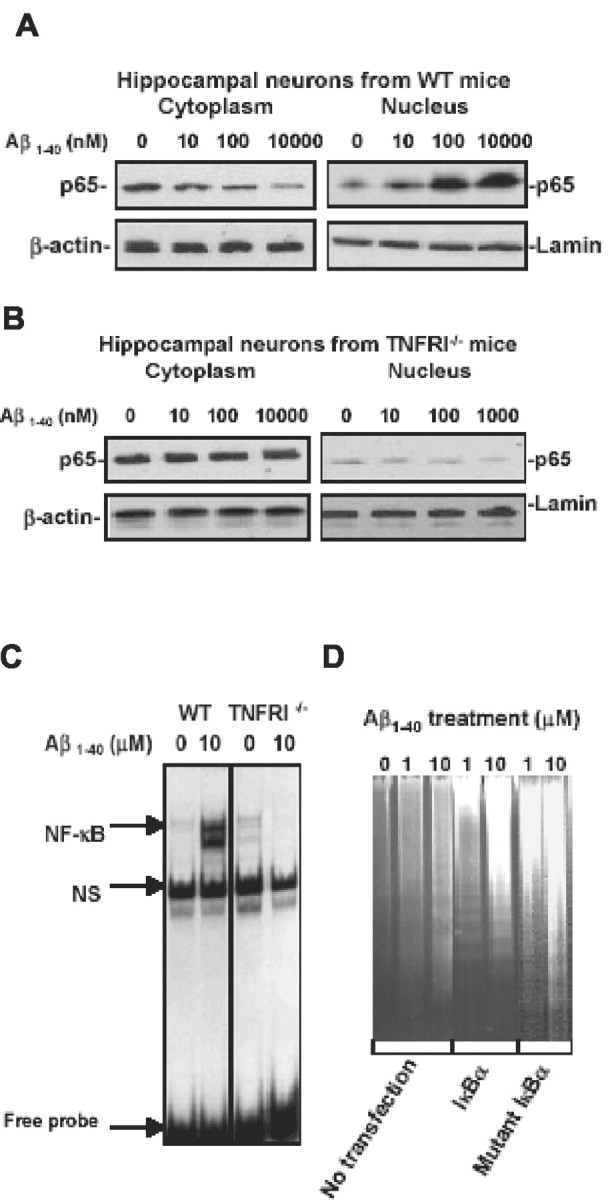
Aβ-induced neuronal NF-κB p65 translocation and activity are TNFRI dependent. A, Western blots of cytoplasmic and nuclear NF-κB p65 in hippocampal neurons of wild-type mice. Neurons were treated with soluble Aβ1-40 at 0, 0.1, 1, and 10 μm for 30 min. At the various indicated doses of Aβ1-40 treatment, cell cytoplasmic or nuclear samples were subjected to SDS-PAGE and the blots were probed with an antibody to NF-κB p65 or actin and laminin as the cytoplasmic and nucleic housekeeping proteins, respectively. Visualization of the proteins was performed with ECL. This result demonstrates that cytoplasmic NF-κB p65 was at high levels, whereas nuclear NF-κB p65 without Aβ1-40 treatment was low. However, Aβ1-40 treatment resulted in low levels of cytoplasmic NF-κB p65, and high levels were seen in the nucleus, suggesting that NF-κB p65 was translocated from the cytoplasm to the nucleus in hippocampal neurons after Aβ1-40 treatment. B, Western blot of nuclear NF-κB p65 in hippocampal neurons from TNFRI knock-out mice (TNFRI-/-). The neurons from TNFRI knock-out brains were treated with Aβ1-40 at the same doses (0.1-10 μm) for 30 min. The cytoplasmic and nuclear samples were subjected to SDS-PAGE, and the blots were probed with an antibody to NF-κB p65 or actin and laminin. We found that little NF-κB p65 was induced by Aβ1-40 at 100 nm compared with that in wild-type neurons (A). C, EMSA analysis of NF-κB binding activity in hippocampal neurons from TNFRI-/- and wild-type mice. EMSA assay with nuclear extracts prepared from neurons of TNFRI-/- and wild-type mice with Aβ1-40 treatment (10 μm) for 30 min. The 32P-labeled oligonucleotide probe that was used contained the NF-κB p65 subunit and a 50-fold molar excess of an unlabeled AP2 (nonspecific competitor, NS) probe. The NF-κB activity was abolished in TNFRI-deleted neurons but not in neurons from wild-type mice. D, DNA fragmentation was examined in neurons affected with IKKα and mIKKα adenoviral affection after Aβ1-40 treatment (0, 1, or 10 μm). Interestingly, Aβ-induced DNA fragmentation was observed only in IKKα-transfected neurons but not in neurons with mIKKα transfection.
Apaf-1 is essential for Aβ-induced neuronal NF-κB translocation in a TNFRI-dependent manner
Apoptotic protease activating factor-1 (Apaf-1) has been reported to be essential in apoptosis during development (Cecconi et al., 1998; Yoshida et al., 1998; Fortin et al., 2001). Recent studies have also shown that insults, including ischemic injury, may enhance Apaf-1 expression only in certain types of cells, such as neurons in the brain. The neuronal-cell-specific increases in Apaf-1 are involved in an apoptotic process mediated by caspases (Li et al., 1997, 1998; Yakovlev et al., 1997), suggesting a critical role of Apaf-1 in neuronal degeneration (Yuan and Yankner, 2000). To examine the relationship between the Aβ-induced NF-κB activation and Apaf-1 expression in neurons, we tested whether Aβ treatment can alter Apaf-1 expression levels. We examined Apaf-1 mRNA levels using semiquantitative RT-PCR. As shown in Figure 5A, TNFRI-infected neurons treated with Aβ1-40 exhibited a significant increase in Apaf-1 mRNA levels in comparison with Aβ1-40-treated non-TNFRI-transduced neurons, and the Apaf-1 mRNA increase was in an Aβ1-40 dose-dependent manner. This increase in Apaf-1 mRNA levels was observed after 8 hr of Aβ1-40 (10 μm) treatment and remained elevated after 24 hr of treatment. To confirm that the induction of Apaf-1 expression was caused by Aβ1-40 administration, the levels of Apaf-1 mRNA were examined in neurons treated with control peptide, a reversed Aβ1-40 peptide. The Apaf-1 mRNA levels did not increase in the control peptide treated neurons (Fig. 5A). Our data confirm the requirement of Aβ1-40 for Apaf-1 induction. To examine whether Aβ-induced Apaf-1 upregulation is associated with TNFRI, we treated primary neurons from TNFRI knock-out mice with Aβ1-40 for 8 and 24 hr and examined whether the Apaf-1 mRNA level was changed. Apaf-1 mRNA levels were not increased in TNFRI knock-out mice, but instead were decreased after Aβ1-40 treatment at 8 and 24 hr (Fig. 5A).
Figure 5.
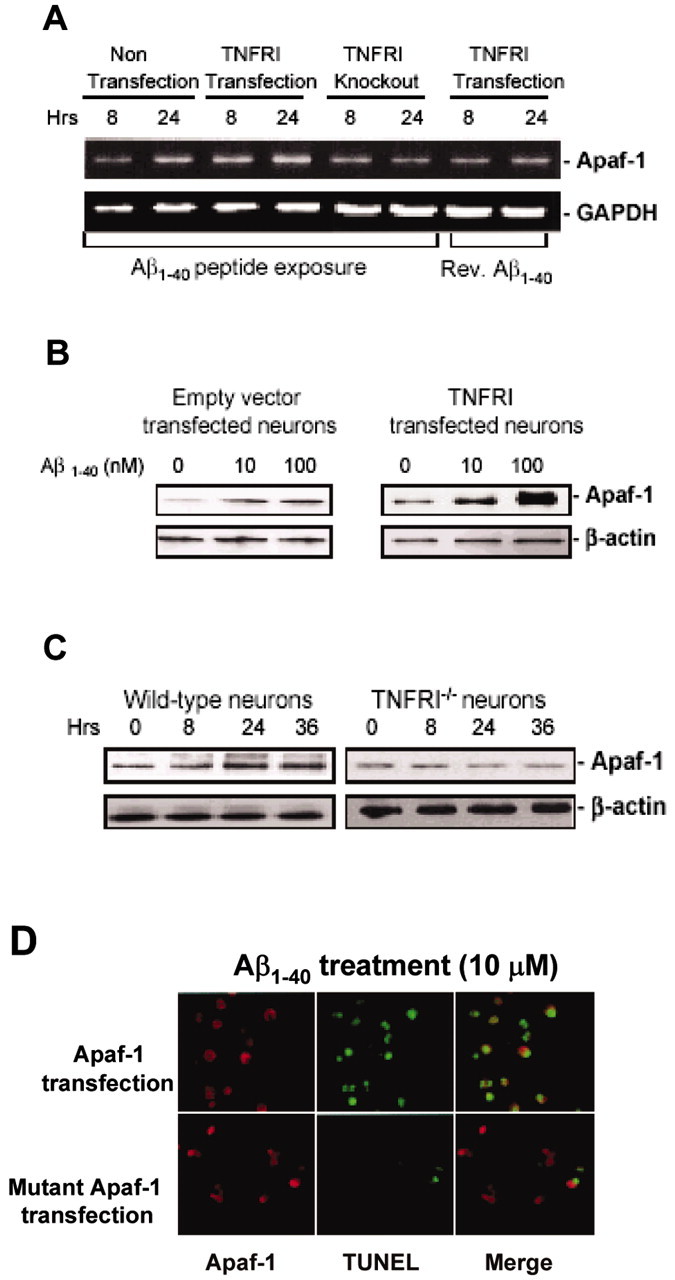
Apaf-1-induced expression by Aβ in hippocampal neurons is TNFRI dependent. A, RNA was extracted from neurons of wild-type and TNFRI-/- mice 8 and 24 hr after Aβ1-40 treatment (1 μm) and analyzed for Apaf-1 or β-actin expression using semiquantitative RT-PCR. B, Hippocampal neurons from TNFRI-/- mouse brains were treated with Αβ1-40 (0, 10, 100 μm) for 24 hr. Extracts of cell lysates were fractionated by SDS-PAGE on 12% Tris-glycine gels and transferred to PVDF membranes; 10 μg of cell lysates was loaded per lane. The Apaf-1 expression was detected with affinity-purified antibody using a horseradish peroxidase-conjugated secondary antibody and processed using ECL detection. C, We found that Aβ1-40 increased Apaf-1 activity in hippocampal neurons from wild-type mice. These effects are dose dependent. However, no alteration of Apaf-1 expression was observed in these TNFRI-/- neurons by Aβ1-40 peptide treatment, suggesting that Apaf-1 expression is induced by Aβ peptide. D, TUNEL staining was performed, and representative photomicrographs are shown in the Apaf-1-transfected neurons with Aβ treatment. Primary neurons were treated with Aβ1-40 and then stained with anti-Apaf-1 (red, left) and TUNEL (green, middle). The rightmost panels are merged images of the Apaf-1 stain and TUNEL (red and green). Most TUNEL-positive neurons with Apaf-1 transfection after Aβ1-40 treatment show condensed nuclei, suggestive of apoptosis.
To determine whether the alteration in mRNA levels of Apaf-1 was accompanied by a change in its protein expression, we first conducted Western blot analysis of Apaf-1 protein expression from TNFRI-infected and control (empty vector-infected wild-type) neurons, undergoing Aβ1-40-induced apoptosis. Cell lysates were obtained from Aβ1-40 treated neurons at various concentrations and Apaf-1 protein expression was examined. A significant increase in Apaf-1 protein expression in TNFRI-transfected neurons was observed at 100 nm of Aβ1-40 treatment compared with that of control neurons, although both groups of neurons were treated with Aβ1-40 (Fig. 5B), but such phenomena were not observed by using reverse Aβ40-1 peptide (data not shown). A time-dependent increase in Aβ1-40-induced Apaf-1 protein levels was found only in control neurons. When we compared the time course of Aβ treatment, the Aβ1-40-induced Apaf-1, mRNA upregulation appeared earlier than protein expression. To verify whether Aβ-upregulated Apaf-1 protein expression occurred through TNFRI, we examined Apaf-1 protein levels in neurons from TNFRI-/- mice after Aβ1-40 treatment and found no significant change in Apaf-1 protein expression in neurons from TNFRI-/- mice after Aβ1-40 treatment at 10 μm for 24 and 36 hr (Fig. 5C).
Our experimental data demonstrate that Aβ can induce the upregulation of Apaf-1 mRNA and protein expression in a TNFRI-dependent manner. To determine whether Apaf-1 plays an essential role in Aβ-induced apoptosis mediated by TNFRI activation (Yang et al., 2002), we examined the incidence of TUNEL-labeled neuron death after Aβ1-40 treatment in Apaf-1 mutant and control neurons. As shown in Figure 5D, Aβ induced widespread cell death in control neurons, whereas sparsely distributed apoptotic neurons are evident in the Apaf-1 mutant neuron culture. This may be caused by a caspase-independent form of neuron death or a delayed cell death program as reported for some cells in the Apaf-1 knock-out mouse (Cecconi et al., 1998; Yoshida et al., 1998). Thus, in addition to physiological apoptosis, Apaf-1 plays a central role in pathological apoptosis engaged by Aβ.
To determine whether Aβ-induced Apaf-1 protein could activate NF-κB translocation in neurons containing high levels of TNFRI, we transfected neurons with NF-κB luciferase reporter plasmid in combination with increasing amounts of an expression vector encoding the Apaf-1 protein. We used these transfected neurons as a system to examine whether Apaf-1 is able to regulate NF-κB translocation in neurons. The neurons were lysed 16 hr after the transfection and assayed for NF-κB activity and protein expression. As shown in Figure 6A, transfection of Apaf-1 caused NF-κB activation in a transfection dose-dependent manner. Interestingly, Aβ1-40 treatment induced approximately fourfold to fivefold more activation of NF-κB than that in vehicle treatment in the Apaf-1 transfected neurons (Fig. 6B). As many other research groups have indicated, IKKα can cause degradation of IκBα (an NF-κB inhibitor) by phosphorylation, and result in the release of cytoplasmic NF-κB, which is then able to translocate into the nucleus (Miyamoto et al., 1994; Verma et al., 1995; Baeuerle and Baltimore, 1996). However, a mutated IKKα fails to phosphorylate IκBα and blocks NF-κB activation (Malinin et al., 1997; Regnier et al., 1997). Thus, to further verify that Aβ does indeed induce NF-κB activation through Apaf-1, we transiently cotransfected neurons with NF-κB control luciferase reporter plasmid with Apaf-1 and mutated IKKα. As shown in Figure 6C, the kinase-inactive mutant IKKα completely blocked the Apaf-1-induced NF-κB translocation regardless of the presence or absence of Aβ1-40 treatment. This result shows that the kinase activity of IKKα is essential for Apaf-1-induced NF-κB activity and suggests that Apaf-1 may contribute to Aβ-induced transduction of NF-κB activation signals from TNFRI in neurons.
To further verify the above result that Aβ-induced Apaf-1 activates NF-κB, we first used antisense oligonucleotides (ASO) to knock down Apaf-1 expression. As Figure 6D demonstrates, when Apaf-1 expression is reduced by ASO, Aβ1-40 treatment did not result in translocation of p65 NF-κB whereas, in our control group, Aβ-induced p65 NF-κB translocation is clearly observed. Furthermore, we examined Apaf-1 and NF-κB in neurons from TNFRI-/- mice. Our results demonstrated that either Apaf-1 or NF-κB failed to be induced and transduce the Aβ signal in TNFRI-/- neurons (Fig. 6). This supports our hypothesis that Aβ-induced Apaf-1 expression is related to neuronal death in a TNFRI-dependent manner. Together, these data suggest that TNFRI plays a critical role in the degeneration of neurons responding to insults such as Aβ.
Discussion
Although plaques containing Aβ are considered an early event in the development of AD, and Aβ is believed to cause neuronal loss, the mechanisms of Aβ-induced neuronal death remain unclear. The results presented in this paper are the first to demonstrate that Aβ induces Apaf-1 elevation, in turn activating NF-κB translocation in a TNFRI-dependent manner, which may contribute to neuronal apoptosis in the AD brain.
Activation of Apaf-1 appears to be a critical event in the execution of Aβ-induced neuronal apoptosis in the brain, whereas caspases may also be involved in Aβ-induced Apaf-1 and NF-κB signal transduction. In the intrinsic pathway of this elevation, Apaf-1, NF-κB, and caspases (caspases 3, 9, and 12) may form a functional complex contributing to apoptosis during development as well as in diseased brains (Li et al., 1997; Zou et al., 1999; Yakovlev et al., 2001). In the present study, we examined the intrinsic pathway of Apaf-1-activated NF-κB in mouse hippocampal neurons after an Aβ-induced lesion. We also investigated the expression of the Apaf-1 gene at mRNA and protein levels, given the role of this protein in injury-induced neuronal apoptosis. Our data show that Aβ induces a relatively small but steady increase of Apaf-1. Also, a robust decrease of p65 NF-κB in was seen in the cytoplasm, whereas there was a significant increase in the nucleus. This again supports the critical role of Apaf-1 in the apoptosis of neurons, because depletion of Apaf-1 in mice reduces tissue damage (Li et al., 1997; Yoshida et al., 1998). We also demonstrated that the ability of Aβ to induce Apaf-1 elevation and NF-κB translocation is partially abolished in TNFRI knock-out neurons, suggesting TNFRI dependence and involvement. Thus, induction of Apaf-1-induced NF-κB translocation may be necessary for the induction of apoptosis by death receptors such as TNFRI after Aβ stimulation in neurons. This is consistent with studies in other types of cells with different apoptotic stimuli. We have shown recently that high expression of TNFRI-induced by TNF-α is sufficient to trigger the apoptotic cascade in mature neurons and that depletion or loss of functional TNFRI reduces neuronal cell death (Shen et al., 1997; Yang et al., 2002). This suggests that Apaf-1 acts as a key regulator of the death cascades in injured neurons.
Our results also demonstrate a physical and pharmacological interaction between Aβ1-40 and the extracellular domains of TNF receptors, indicating novel Aβ involvement with TNFRI. Our unpublished data from ligand dot-blot binding experiments show no interaction between the last 100 aa of APP or reverse Aβ40-1 peptide (data not shown) and TNFRI, or between other growth factors or immune molecules such as EGF, p75 NGF receptor, and Aβ1-40 peptide. Radiolabeled Aβ binding assay data also support this finding (Fig. 6), indicating the selectivity of Aβ1-40 for TNFRI. The interaction between purified TNFRI (extracellular fractions) and Aβ1-40 was confirmed on SDS-PAGE using labeled Ab (data not shown). The interaction between Aβ and other amyloid-binding proteins was evaluated by an 125I-Aβ1-40 binding assay (data not shown). The results from this binding assay indicated that the affinity of interaction between Aβ and TNFRI was comparable with or higher than the affinity of interaction between Aβ and insulin, α2-macroglobulin, receptor for advanced glycation end product, apolipoprotein (apo) E3, and apoE4 (Yamauchi et al., 1999; Mettenburg et al., 2002; Xie et al., 2002).
A recent report (Kayed et al., 2003) on the conformation of Aβ suggests that soluble amyloid oligomers may be a common mechanism of pathogenesis of AD. To understand the conformation of Aβ1-40 we used for neuronal toxicity and binding activity, we identified our Aβ1-40 at the conditions for neurotoxicity and 125I-Aβ1-40 for binding experiments by using PAGE gel and Western blot and found that most of Aβ1-40 were in a soluble conformation, not fibrils or protofibrils (data not shown), suggesting that Aβ1-40 seems to be soluble when binding TNFRI as a ligand. However, to further verify the absolute conformation, in future experiments, we will need to use oligomers to conduct TNFRI radioligand binding to determine whether binding affinity would be same or different and whether such binding would affect neuronal degeneration.
TNFRI is expressed not only in many non-neuronal cells such as lymphocytes, but also in other types of cells in the CNS, including microglia, astrocytes, and neurons of diseased brains (Y. Konishi and Y. Shen, unpublished observation). This suggests that these TNF receptor subtypes might not be mediated exclusively by the native TNF-α ligand. Evidence from this study shows that TNFRI binds Aβ, and other studies show that Fas ligand binds TNF receptors, supporting this notion. We believe that different types of neurons have different expression ratios of the two TNF receptors. For example, the expression level of the two types of TNF receptors in a single neuron in a specific brain area is different from other regions of the brain. Moreover, in AD, this ratio could be changed such that more TNFRI is upregulated, leading to increased neuronal loss. Additional studies are aimed at whether more rigorous techniques or double-labeling in situ hybridization techniques will reveal single neuron expression. The TNF receptor type I family contains several members with homologous cytoplasmic domains known as a death domain (DD). The DD is critical in initiating apoptosis and other signaling pathways after ligand binding by the receptors (Tartaglia et al., 1993; Troy et al., 2002; Yang et al., 2002). In the absence of a ligand, DD-containing receptors are maintained in an inactive state. However, under pathological conditions, in which glial cells are activated in the brain and Aβ or other secreted molecules behave like TNF-α, TNFRI may then be activated. TNFRI contains a cytoplasmic DD required for signaling pathways that is responsible for NF-κB activation and apoptosis (Tartaglia et al., 1993; Hsu et al., 1995; Yang et al., 2002). Now, a newly discovered role of Apaf-1 in neurons provides a bridge for Aβ protein and TNF receptor binding in NF-κB translocation and neuronal death.
Our present studies suggest that Apaf-1 and NF-κB may be direct regulatory target candidates for Aβ in neuronal cell death and that the upregulation of Apaf-1 by Aβ and activated NF-κB translocation plays an important role in the apoptosis-signaling cascade after brain injury. Furthermore, neuronal death mediated by the Apaf-NF-κB signal cascade seems to be involved in the activation of TNFRI, which may interact through Aβ binding. The binding complex of Aβ to TNFRI may be involved in more than one distinct signal transductions, such as JNK (c-Jun N-terminal protein kinase; Morishima et al., 2001), Src protein kinase (Williamson et al., 2002), and Cdk5 (cyclin-dependent kinase 5) activity (Patrick et al., 1999; Lee et al., 2002). On the other hand, neuronal death mediated by the Apaf-NF-κB signal cascade might interact with more than one surface protein of neurons, such as APP itself (Lorenzo et al., 2000), a receptor for advanced glycation end products (Yan et al., 1996), or both. Additional studies on structural features specific to these binding proteins, including the TNF receptor, by site-directed mutagenesis and their differential expression in the AD brain using in situ hybridization and immunohistochemistry, need to be conducted. Activation of the Apaf-1 and NF-κB signal cascades provides a mechanism by which Aβ may be coupled to signal transduction pathways exerting a major influence on neuron fate. Elucidation of the multiple pathways and their components is an important step in understanding Aβ-induced neuronal loss in AD brains and may lead to alternative therapeutic targets in AD and better strategies for treatments.
Footnotes
This work was supported by grants from the Alzheimer's Disease Association, Neuroscience Education Research Foundation, Arizona Disease Control Research Center Program Project Category II (Y.S., R.L.), and the National Nature and Science Foundation of China (D.Z.). We thank Dr. D. V. Goeddel and Dr. P. Baeuerle from Tularik, Inc., for their constructive and helpful discussion and encouragement; Dr. I. M. Verma from The Salk Institute for providing NF-κB p65, IKKα, and mutant IKKα plasmids; Dr. Ted Dawson from Johns Hopkins University Medical School for his critical reading of this manuscript and correction; and G. Arnold for her technical support and careful manuscript reading during the preparation of this manuscript.
Correspondence should be addressed to either of the following: Dr. Yong Shen, Haldeman Laboratory of Molecular and Cellular Neurobiology, Sun Health Research Institute, 10515 West Santa Fe Drive, Sun City, AZ 85351, E-mail: yong.shen@sunhealth.org; or Dr. Rena Li, Alzheimer's Research Laboratory, Sun Health Research Institute, 10515 West Santa Fe Drive, Sun City, AZ 85351, E-mail: rena.li@sunhealth.org.
L. Yang's present address: Earle Chiles Institute of Molecular Biology, Oregon Health Sciences University, Portland, OR 97213.
Y. Konishi's present address: Department of Physiology, Ehime University School of Medicine, Ehime 791-0296, Japan.
Copyright © 2004 Society for Neuroscience 0270-6474/04/241760-12$15.00/0
References
- Akama KT, Albanese C, Pestell RG, Van Eldik LJ (1998) Amyloid β-peptide stimulates nitric oxide production in astrocytes through an NFκB-dependent mechanism. Proc Natl Acad Sci USA 95: 5795-5800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson AJ, Su JH, Cotman CW (1996) DNA damage and apoptosis in Alzheimer's disease: colocalization with c-Jun immunoreactivity, relationship to brain area, and effect of postmortem delay. J Neurosci 16: 1710-1719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Avdulov NA, Chochina SV, Igbavboa U, Warden CS, Vassiliev AV, Wood WG (1997) Lipid binding to amyloid β-peptide aggregates: preferential binding of cholesterol as compared with phosphatidylcholine and fatty acids. J Neurochem 69: 1746-1752. [DOI] [PubMed] [Google Scholar]
- Baeuerle PA, Baltimore D (1996) NF-κB: ten years after. Cell 87: 13-20. [DOI] [PubMed] [Google Scholar]
- Bruce AJ, Boling W, Kindy MS, Peschon J, Kraemer PJ, Carpenter MK, Holtsberg FW, Mattson MP (1996) Altered neuronal and microglial responses to excitotoxic and ischemic brain injury in mice lacking TNF receptors. Nat Med 2: 788-794. [DOI] [PubMed] [Google Scholar]
- Butterfield DA, Castegna A, Lauderback CM, Drake J (2002) Evidence that amyloid β-peptide-induced lipid peroxidation and its sequelae in Alzheimer's disease brain contribute to neuronal death. Neurobiol Aging 23: 655-664. [DOI] [PubMed] [Google Scholar]
- Cecconi F, Alvarez-Bolado G, Meyer BI, Roth KA, Gruss P (1998) Apaf1 (CED-4 homolog) regulates programmed cell death in mammalian development. Cell 94: 727-737. [DOI] [PubMed] [Google Scholar]
- Chaudhury AR, Gerecke KM, Wyss JM, Morgan DG, Gordon MN, Carroll SL (2003) Neuregulin-1 and erbB4 immunoreactivity is associated with neuritic plaques in Alzheimer disease brain and in a transgenic model of Alzheimer disease. J Neuropathol Exp Neurol 62: 42-54. [DOI] [PubMed] [Google Scholar]
- Choi-Lundberg DL, Lin Q, Chang YN, Chiang YL, Hay CM, Mohajeri H, Davidson BL, Bohn MC (1999) Dopaminergic neurons protected from degeneration by GDNF gene therapy. Science 275: 838-841. [DOI] [PubMed] [Google Scholar]
- Corsico B, Cistola DP, Frieden C, Storch J (1998) The helical domain of intestinal fatty acid binding protein is critical for collisional transfer of fatty acids to phospholipid membranes. Proc Natl Acad Sci USA 95: 12174-12178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cregan SP, Fortin A, MacLaurin JG, Callaghan SM, Cecconi F, Yu SW, Dawson TM, Dawson VL, Park DS, Kroemer G, Slack RS (2002) Apoptosis-inducing factor is involved in the regulation of caspase-independent neuronal cell death. J Cell Biol 158: 507-517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eberhardt O, Schulz JB (2003) Apoptotic mechanisms and antiapoptotic therapy in the MPTP model of Parkinson's disease. Toxicol Lett 139: 135-151. [DOI] [PubMed] [Google Scholar]
- Estus S, Tucker HM, van Rooyen C, Wright S, Brigham EF, Wogulis M, Rydel RE (1997) Aggregated amyloid-β protein induces cortical neuronal apoptosis and concomitant “apoptotic” pattern of gene induction. J Neurosci 17: 7736-7745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferraro E, Corvaro M, Cecconi F (2003) Physiological and pathological roles of Apaf1 and the apoptosome. J Cell Mol Med 7: 21-34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrer I, Blanco R, Carmona M, Puig B (2001) Fas and Fas ligand expression in Alzheimer's disease. Acta Neuropathol (Berl) 27: 343-351. [DOI] [PubMed] [Google Scholar]
- Fortin A, Cregan SP, MacLaurin JG, Kushwaha N, Hickman ES, Thompson CS, Hakim A, Albert PR, Cecconi F, Helin K, Park DS, Slack RS (2001) Apaf-1 is a key transcriptional target for p53 in the regulation of neuronal cell death. J Cell Biol 155: 207-216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galvan V, Chen S, Lu D, Logvinova A, Goldsmith P, Koo EH, Bredesen DE (2002) Caspase cleavage of members of the amyloid precursor family of proteins. J Neurochem 82: 283-294. [DOI] [PubMed] [Google Scholar]
- Geula C, Wu CK, Saroff D, Lorenzo A, Yuan M, Yankner BA (1998) Aging renders the brain vulnerable to amyloid β-protein neurotoxicity. Nat Med 4: 394-395. [DOI] [PubMed] [Google Scholar]
- Ghribi O, Herman MM, DeWitt DA, Forbes MS, Savory J (2001) Aβ(1-42) and aluminum induce stress in the endoplasmic reticulum in rabbit hippocampus, involving nuclear translocation of gadd 153 and NF-κB. Brain Res Mol Brain Res 96: 30-38. [DOI] [PubMed] [Google Scholar]
- Guo Q, Robinson N, Mattson MP (1998) Secreted β-amyloid precursor protein counteracts the proapoptotic action of mutant presenilin-1 by activation of NF-κB and stabilization of calcium homeostasis. J Biol Chem 273: 12341-12351. [DOI] [PubMed] [Google Scholar]
- Haas J, Storch-Hagenlocher B, Biessmann A, Wildemann B (2002) Inducible nitric oxide synthase and argininosuccinate synthetase: co-induction in brain tissue of patients with Alzheimer's dementia and following stimulation with β-amyloid 1-42 in vitro. Neurosci Lett 322: 121-125. [DOI] [PubMed] [Google Scholar]
- Heneka MT, Galea E, Gavriluyk V, Dumitrescu-Ozimek L, Daeschner J, O'Banion MK, Weinberg G, Klockgether T, Feinstein DL (2002) Noradrenergic depletion potentiates β-amyloid-induced cortical inflammation: implications for Alzheimer's disease. J Neurosci 22: 2434-2442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsu H, Xiong J, Goeddel DV (1995) The TNF receptor 1-associated protein TRADD signals cell death and NF-κB activation. Cell 81: 495-504. [DOI] [PubMed] [Google Scholar]
- Inohara N, Ogura Y, Chen FF, Muto A, Nunez G (2001) Human Nod1 confers responsiveness to bacterial lipopolysaccharides. J Biol Chem 276: 2551-2554. [DOI] [PubMed] [Google Scholar]
- Ishii K, Tamaoka A, Mizusawa H, Shoji S, Ohtake T, Fraser PE, Takahashi H, Tsuji S, Gearing M, Mizutani T, Yamada S, Kato M, St George-Hyslop PH, Mirra SS, Mori H (1997) Aβ1-40 but not Aβ1-42 levels in cortex correlate with apolipoprotein E ϵ4 allele dosage in sporadic Alzheimer's disease. Brain Res 748: 250-252. [DOI] [PubMed] [Google Scholar]
- Janciauskiene S, Wright HT, Lindgren S (1999) Fibrillar Alzheimer's amyloid peptide Aβ(1-42) stimulates low density lipoprotein binding and cell association, free radical production and cell cytotoxicity in PC12 cells. Neuropeptides 33: 510-516. [DOI] [PubMed] [Google Scholar]
- Jarret JT, Lansbury PT (1993) Seeding “one-dimensional crystallization” of amyloid: a pathogenetic mechanism in Alzheimer's disease and scrapie? Cell 73: 1055-1058. [DOI] [PubMed] [Google Scholar]
- Jellinger KA, Stadelmann C (2001) Problems of cell death in neurodegeneration and Alzheimer's disease. J Alzheimers Dis 3: 31-40. [DOI] [PubMed] [Google Scholar]
- Kaltschmidt B, Uherek M, Wellmann H, Volk B, Kaltschmidt C (1999) Inhibition of NF-κB potentiates amyloid β-mediated neuronal apoptosis. Proc Natl Acad Sci USA 96: 9409-9414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaltschmidt B, Heinrich M, Kaltschmidt C (2002) Stimulus-dependent activation of NF-κB specifies apoptosis or neuroprotection in cerebellar granule cells. Neuromol Med 2: 299-309. [DOI] [PubMed] [Google Scholar]
- Kamegai M, Niijima K, Kunishita T, Nishizawa M, Ogawa M, Araki M, Ueki A, Konishi Y, Tabira T (1990) Interleukin 3 as a trophic factor for central cholinergic neurons in vitro and in vivo Neuron 4: 429-436. [DOI] [PubMed] [Google Scholar]
- Kayed R, Head E, Thompson JL, McIntire TM, Milton SC, Cotman CW, Glabe CG (2003) Common structure of soluble amyloid oligomers implies common mechanism of pathogenesis. Science 300: 486-489. [DOI] [PubMed] [Google Scholar]
- Koriyama Y, Chiba K, Mohri T (2003) Propentofylline protects β-amyloid protein-induced apoptosis in cultured rat hippocampal neurons. Eur J Pharmacol 458: 235-241. [DOI] [PubMed] [Google Scholar]
- Kumar-Singh S, Julliams A, Nuydens R, Ceuterick C, Labeur C, Serneels S, Vennekens K, Van Osta P, Geerts H, De Strooper B, Van Broeckhoven C (2002) In vitro studies of Flemish, Dutch, and wild-type β-amyloid provide evidence for two-staged neurotoxicity. Neurobiol Dis 11: 330-340. [DOI] [PubMed] [Google Scholar]
- Kuner P, Schubenel R, Hertel C (1998) β-amyloid binds to p57NTR and activates NFκB in human neuroblastoma cells. J Neurosci Res 54: 798-804. [DOI] [PubMed] [Google Scholar]
- Kuperstein F, Yavin E (2002) ERK activation and nuclear translocation in amyloid-β peptide- and iron-stressed neuronal cell cultures. Eur J Neurosci 16: 44-54. [DOI] [PubMed] [Google Scholar]
- Lee MS, Kwon YT, Li M, Peng J, Friedlander RM, Tsai LH (2002) Neurotoxicity induces cleavage of p35 to p25 by calpain. Nature 405: 360-364. [DOI] [PubMed] [Google Scholar]
- Li H, Zhu H, Xu CJ, Yuan J (1998) Cleavage of BID by caspase 8 mediates the mitochondrial damage in the Fas pathway of apoptosis. Cell 94: 491-501. [DOI] [PubMed] [Google Scholar]
- Li P, Nijhawan D, Budihardjo I, Srinivasula SM, Ahmad M, Alnemri ES, Wang X (1997) Cytochrome c and dATP-dependent formation of Apaf-1/caspase-9 complex initiates an apoptotic protease cascade. Cell 91: 479-489. [DOI] [PubMed] [Google Scholar]
- Lopez Salon M, Pasquini L, Besio Moreno M, Pasquini JM, Soto E (2003) Relationship between β-amyloid degradation and the 26S proteasome in neural cells. Exp Neurol 180: 131-143. [DOI] [PubMed] [Google Scholar]
- Lorenzo A, Yuan M, Zhang Z, Paganetti PA, Sturchler-Pierrat C, Staufenbiel M, Mautino J, Vigo FS, Sommer B, Yankner BA (2000) Amyloid β interacts with the amyloid precursor protein: a potential toxic mechanism in Alzheimer's disease. Nat Neurosci 3: 460-464. [DOI] [PubMed] [Google Scholar]
- Miyamoto S, Maki M, Schmitt MJ, Hatanaka M, Verma IM (1994) Tumor necrosis factor α-induced phosphorylation of IκBα is a signal for its degradation but not dissociation from NF-κB. Proc Natl Acad Sci USA 91: 12740-12744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malinin NL, Boldin MP, Kovalenko AV, Wallach D (1997) MAP3K-related kinase involved in NF-κB induction by TNF, CD95 and IL-1. Nature 385: 540-544. [DOI] [PubMed] [Google Scholar]
- Mathews PM, Jiang Y, Schmidt SD, Grbovic OM, Mercken M, Nixon RA (2002) Calpain activity regulates the cell surface distribution of amyloid precursor protein: inhibition of clapains enhances endosomal generation of β-cleaved C-terminal APP fragments. J Biol Chem 277: 36415-36424. [DOI] [PubMed] [Google Scholar]
- Mattson MP, LaFerla FM, Chan SL, Leissring MA, Shepel PN, Geiger JD (2000) Calcium signaling in the ER: its role in neuronal plasticity and neurodegenerative disorders. Trends Neurosci 23: 222-229. [DOI] [PubMed] [Google Scholar]
- McCarthy JV, Ni J, Dixit VM (1998) RIP2 is a novel NF-κB-activating and cell death-inducing kinase. J Biol Chem 273: 16968-16975. [DOI] [PubMed] [Google Scholar]
- Mettenburg JM, Webb DJ, Gonias SL (2002) Distinct binding sites in the structure of α2-macroglobulin mediate the interaction with β-amyloid peptide and growth factors. J Biol Chem 277: 13338-13345. [DOI] [PubMed] [Google Scholar]
- Mochizuki H, Mizuno Y (2003) Gene therapy for Parkinson's disease. J Neural Transm Suppl 65: 205-213. [DOI] [PubMed] [Google Scholar]
- Morishima Y, Gotoh Y, Zieg J, Barrett T, Takano H, Flavell R, Davis RJ, Shirasaki Y, Greenberg ME (2001) β-amyloid induces neuronal apoptosis via a mechanism that involves the c-Jun N-terminal kinase pathway and the induction of Fas ligand. J Neurosci 21: 7551-7560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morishima-Kawashima M, Ihara Y (1998) The presence of amyloid β-protein in the detergent-insoluble membrane compartment of human neuroblastoma cells. Biochemistry 37: 15247-15253. [DOI] [PubMed] [Google Scholar]
- Nakagawa T, Zhu H, Morishima N, Li E, Xu J, Yankner BA, Yuan J (2000) Caspase-12 mediates endoplasmic-reticulum-specific apoptosis and cytotoxicity by amyloid-β. Nature 403: 98-103. [DOI] [PubMed] [Google Scholar]
- Ogura Y, Inohara N, Benito A, Chen FF, Yamaoka S, Nunez G (2001) Nod2, a Nod1/Apaf-1 family member that is restricted to monocytes and activates NF-κB. J Biol Chem 276: 4812-4818. [DOI] [PubMed] [Google Scholar]
- Patrick GN, Zukerberg L, Nikolic M, de la Monte S, Dikkes P, Tsai LH (1999) Conversion of p35 to p25 deregulate Cdk5 activity and promotes neurodegeneration. Nature 402: 615-622. [DOI] [PubMed] [Google Scholar]
- Raina AK, Hochman A, Ickes H, Zhu X, Ogawa O, Cash AD, Shimohama S, Perry G, Smith MA (2003) Apoptotic promoters and inhibitors in Alzheimer's disease: who wins out? Prog Neuropsychopharmacol Biol Psychiatry 27: 251-254. [DOI] [PubMed] [Google Scholar]
- Regnier CH, Song HY, Gao X, Goeddel DV, Cao Z, Rothe M (1997) Identification and characterization of an IκB kinase. Cell 90: 373-383. [DOI] [PubMed] [Google Scholar]
- Schneider A, Martin-Villalba A, Weih F, Vogel J, Wirth T, Schwaninger M (1999) NF-κB is activated and promotes cell death in focal cerebral ischemia. Nat Med 5: 554-559. [DOI] [PubMed] [Google Scholar]
- Selkoe D, Kopan R (2003) Notch and Presenilin: regulated intramembrane proteolysis links development and degeneration. Annu Rev Neurosci 26: 565-597. [DOI] [PubMed] [Google Scholar]
- Sheehan J, Swerdlow RH, Miller SW, Davis RE, Parks JK, Parker WD, Tuttle JB (1997) Calcium homeostasis and reactive oxygen species production in cells transformed by mitochondria from individuals with sporadic Alzheimer's disease. J Neurosci 17: 4612-4622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen Y, Li R, Shiosaki K (1997) Inhibition of p75 tumor necrosis factor receptor by antisense oligonucleotides increases hypoxic injury and β-amyloid toxicity in human neuronal cell line. J Biol Chem 272: 77-81. [PubMed] [Google Scholar]
- Siman R, Reaume AG, Savage MJ, Trusko S, Lin YG, Scott RW, Flood DG (2000) Presenilin-1 P264L knock-in mutation: differential effects on Aβ production, amyloid deposition, and neuronal vulnerability. J Neurosci 20: 8717-8726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sipe KJ, Srisawasdi D, Dantzer R, Kelley KW, Weyhenmeyer JA (1996) An endogenous 55 kDa TNF receptor mediates cell death in a neural cell line. Brain Res Mol Brain Res 38: 222-232. [DOI] [PubMed] [Google Scholar]
- Sisodia SS, St George-Hyslop PH (2002) Gamma-Secretase, Notch, Aβ and Alzheimer's disease: where do the presenilins fit in? Nat Rev Neurosci 3: 281-290. [DOI] [PubMed] [Google Scholar]
- Srinivasula SM, Ahmad M, Fernandes-Alnemri T, Alnemri ES (1998) Autoactivation of procaspase-9 by Apaf-1-mediated oligomerization. Mol Cell 1: 949-957. [DOI] [PubMed] [Google Scholar]
- Tamagno E, Robino G, Obbili A, Bardini P, Aragno M, Parola M, Danni O (2003) H2O2 and 4-hydroxynonenal mediate amyloid β-induced neuronal apoptosis by activating JNKs and p38MAPK. Exp Neurol 180: 144-155. [DOI] [PubMed] [Google Scholar]
- Tartaglia LA, Merrill A, Wong GW, Goedell DV (1993) A novel domain within the 55 kd TNF receptor signals cell death. Cell 74: 845-854. [DOI] [PubMed] [Google Scholar]
- Thome M, Hofmann K, Burns K, Martinon F, Bodmer JL, Mattmann C, Tschopp J (1998) Identification of CARDIAK, a RIP-like kinase that associates with caspase-1. Curr Biol 8: 885-888. [DOI] [PubMed] [Google Scholar]
- Troy CM, Rabacchi SA, Friedman WJ, Frappier TF, Brown K, Shelanski ML (2000) Caspase-2 mediates neuronal cell death induced by β-amyloid. J Neurosci 20: 1386-1392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Troy CM, Friedman JE, Friedman WJ (2002) Mechanisms of p75-mediated death of hippocampal neurons: role of caspases. J Biol Chem 277: 34295-34302. [DOI] [PubMed] [Google Scholar]
- Verma IM, Stevenson JK, Schwartz EM, Van Antwerp D, Miyamoto S (1995) Rel/NF-κB/IκB family: intimate tales of association and dissociation. Genes Dev 9: 2723-2735. [DOI] [PubMed] [Google Scholar]
- Walsh DT, Montero RM, Bresciani LG, Jen AY, Leclercq PD, Saunders D, EL-Amir AN, Gbadamoshi L, Gentleman SM, Jen LS (2002) Amyloid-β peptide is toxic to neurons in vivo via indirect mechanisms. Neurobiol Dis 10: 20-27. [DOI] [PubMed] [Google Scholar]
- Wang HY, Lee DH, D'Andrea MR, Peterson PA, Shank RP, Reitz AB (2000) β-Amyloid(1-42) binds to α7 nicotinic acetylcholine receptor with high affinity: implications for Alzheimer's disease pathology. J Biol Chem 275: 5626-5632. [DOI] [PubMed] [Google Scholar]
- Waschuk SA, Elton EA, Darabie AA, Fraser PE, McLaurin JA (2001) Cellular membrane composition defines Aβ-lipid interactions. J Biol Chem 276: 33561-33568. [DOI] [PubMed] [Google Scholar]
- Wei W, Norton DD, Wang X, Kusiak JW (2002) Aβ 17-42 in Alzheimer's disease activates JNK and caspase-8 leading to neuronal apoptosis. Brain 125: 2036-2043. [DOI] [PubMed] [Google Scholar]
- Williamson R, Scales T, Clark BR, Gibb G, Reynolds CH, Kellie S, Bird IN, Varndell IM, Sheppard PW, Everall I, Anderton BH (2002) Rapid tyrosine phosphorylation of neuronal proteins including tau and focal adhesion kinase in response to amyloid-β peptide exposure: involvement of Src family protein kinases. J Neurosci 22: 10-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao AY, Wang XQ, Yang A, Yu SP (2002) Slight impairment of Na+, K+-ATPase synergistically aggravates ceramide- and β-amyloid-induced apoptosis in cortical neurons. Brain Res 955: 253-259. [DOI] [PubMed] [Google Scholar]
- Xie L, Helmerhorst E, Taddei K, Plewright B, Van Bronswijk W, Martins R (2002) Alzheimer's β-amyloid peptides compete for insulin binding to the insulin receptor. J Neurosci 22: RC221(1-5). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yakovlev AG, Knoblach SM, Fan L, Fox GB, Goodnight R, Faden AI (1997) Activation of CPP32-like caspases contributes to neuronal apoptosis and neurological dysfunction after traumatic brain injury. J Neurosci 17: 7415-7424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yakovlev AG, Ota K, Wang G, Movsesyan V, Bao WL, Yoshihara K, Faden AI (2001) Differential expression of apoptotic protease-activating factor-1 and caspase-3 genes and susceptibility to apoptosis during brain development and after traumatic brain injury. J Neurosci 21: 7439-7446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamauchi K, Tozuka M, Nakabayashi T, Sugano M, Hidaka H, Kondo Y, Katsuyama T (1999) Higher avidity binding of apolipoprotein (E-AII) complex than of apolipoprotein E monomer to β-amyloid. J Neurosci Res 58: 301-307. [PubMed] [Google Scholar]
- Yan SD, Chen X, Fu J, Chen M, Zhu H, Roher A, Slattery T, Zhao L, Nagashima M, Morser J, Migheli A, Nawroth P, Stern D, Schmidt AM (1996) RAGE and amyloid-β peptide neurotoxicity in Alzheimer's disease. Nature 382: 685-691. [DOI] [PubMed] [Google Scholar]
- Yang L, Lindholm K, Konishi Y, Li R, Shen Y (2002) Target depletion of distinct tumor necrosis factor receptor subtypes reveals hippocampal neuron death and survival through different signal transduction pathways. J Neurosci 22: 3025-3032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao ZX, Brown RC, Teper G, Greeson J, Papadopoulos V (2002) 22R-Hydroxycholesterol protects neuronal cells from β-amyloid-induced cytotoxicity by binding to β-amyloid peptide. J Neurochem 83: 1110-1119. [DOI] [PubMed] [Google Scholar]
- Yoshida H, Kong YY, Yoshida R, Elia AJ, Hakem A, Hakem R, Penninger JM, Mak TW (1998) Apaf1 is required for mitochondrial pathways of apoptosis and brain development. Cell 94: 739-750. [DOI] [PubMed] [Google Scholar]
- Yu J, Bakhos L, Chang L, Holterman MJ, Klein WL, Venton DL (2002) Per-6-substituted β-cyclodextrin libraries inhibit formation of β-amyloid-peptide (Aβ)-derived, soluble oligomers. J Mol Neurosci 19: 51-55. [DOI] [PubMed] [Google Scholar]
- Yuan J, Yankner BA (2000) Apoptosis in the nervous system. Nature 407: 802-809. [DOI] [PubMed] [Google Scholar]
- Zhang Y, McLaughlin R, Goodyer C, LeBlanc A (2002) Selective cytotoxicity of intracellular amyloid β peptide1-42 through p53 and Bax in cultured primary human neurons. J Cell Biol 156: 519-529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zou H, Henzel WJ, Liu X, Lutschg A, Wang X (1997) Apaf-1, a human protein homologous to C. elegans CED-4, participates in cytochrome c-dependent activation of caspase-3. Cell 90: 405-413. [DOI] [PubMed] [Google Scholar]
- Zou H, Li Y, Liu X, Wang X (1999) An APAF-1. cytochrome c multimeric complex is a functional apoptosome that activates procaspase-9. J Biol Chem 274: 11549-11556. [DOI] [PubMed] [Google Scholar]
- Zou K, Gong JS, Yanagisawa K, Michikawa M (2002) A novel function of monomeric amyloid β-protein serving as an antioxidant molecule against metal-induced oxidative damage. J Neurosci 22: 4833-4841. [DOI] [PMC free article] [PubMed] [Google Scholar]