

Abstract
Phosphorylation of IκB through IκB kinase (IKK) is the first step in nuclear factor κB (NF-κB) activation and upregulation of NF-κB-responsive genes. Hence, inhibition of IKK activity may be expected to prevent injury-, infection-, or stress-induced upregulation of various proinflammatory genes and may thereby reduce hyperalgesia and inflammation. In the present study, we tested this hypothesis using a specific and potent IKK inhibitor (S1627). In an IKK assay, S1627 inhibited IKK activity with an IC50 value of 10.0 ± 1.2 nm. In cell culture experiments, S1627 inhibited interleukin (IL)-1β-stimulated nuclear translocation and DNA-binding of NF-κB. Plasma concentration time courses after intraperitoneal injection revealed a short half-life of 2.8 hr in rats. Repeated intraperitoneal injections were, therefore, chosen as the dosing regimen. S1627 reversed thermal and mechanical hyperalgesia at 3× 30 mg/kg in the zymosan-induced paw inflammation model and reduced the inflammatory paw edema at 3× 40 mg/kg. S1627 also significantly reduced tactile and cold allodynia in the chronic constriction injury model of neuropathic pain at 30 mg/kg once daily. The drug had no effect on acute inflammatory nociception in the formalin test and did not affect responses to heat and tactile stimuli in naive animals. As hypothesized, S1627 prevented the zymosan-induced nuclear translocation of NF-κB in the spinal cord and the upregulation of NF-κB-responsive genes including cyclooxygenase-2, tumor necrosis factor-α, and IL-1β. Our data indicate that IKK may prove an interesting novel drug target in the treatment of pathological pain and inflammation.
Keywords: behavior, dorsal horn, hyperalgesia, neuropathy, protein kinase, transcription
Introduction
Because glucocorticoids were found to mediate at least part of their anti-inflammatory effects through inhibition of nuclear factor κB (NF-κB) (Auphan et al., 1995), this transcription factor has emerged as a most interesting pharmaceutical target because a dysregulation of NF-κB seems to be involved in various pathological processes, particularly chronic inflammation (Kopp and Ghosh, 1994; Marok et al., 1996; Neurath et al., 1996; Tak et al., 2001; Tegeder et al., 2001), neurodegeneration (Grilli et al., 1996; Aubin et al., 1998; Kaltschmidt et al., 1999), atherosclerosis (Brand et al., 1996), and cancer (Wang et al., 1996; Sovak et al., 1997; Rayet and Gelinas, 1999; Wang et al., 1999; Karin et al., 2002). In most unstimulated cells, NF-κB is retained in a latent form in the cytoplasm by binding to any of several IκB inhibitor proteins. IκBα, which is the best studied IκB protein, is phosphorylated at two N-terminal serine residues on exposure of the cell to various inflammatory or other stress stimuli (DiDonato et al., 1996). This phosphorylation is crucial for the subsequent ubiquitination and proteolytic degradation of IκBα in the 26S proteasome (Gao et al., 2000). The degradation of IκB unmasks the nuclear localization sequence of NF-κB, thus allowing its translocation into the nucleus (Lin et al., 1995) and binding to DNA sites of NF-κB-responsive genes. NF-κB positively regulates the transcription of numerous genes (Pahl, 1999) including cytokines [tumor necrosis factor (TNF)-α, interleukin (IL)-1β, IL-6], chemokines [RANTES (recombinant, human regulated on activation, normal T-cell expressed and secreted), macrophage inflammatory protein, IL-8], adhesion factors (intercellular adhesion molecule, vascular cell adhesion molecule), proinflammatory enzymes [inducible nitric oxide synthase, cyclooxygenase-2 (COX-2)], and proteins involved in the control of cell death (Fas, FasL, inhibitors of apoptosis). The enzymes that catalyze the ubiquitination of phospho-IκBs are constitutively active (Yaron et al., 1998). Hence, the only regulated step that dictates the fate of IκB and NF-κB is, in most cases, the phosphorylation of IκB. This phosphorylation is performed by an IκB kinase (IKK) complex (Zandi et al., 1998), which consists of a dimer of two catalytically active subunits, IKKα (also called IKK1) and IKKβ (IKK2) (DiDonato et al., 1997; Mercurio et al., 1997), and a varying number of regulatory IKKγ subunits (Rothwarf et al., 1998). IKK is activated on inflammatory stimulation through phosphorylation of the β-subunit (Delhase et al., 1999). Known IKK activators are NF-κB-inducing kinase and a MAP kinase kinase kinase (MEKK1) (Nemoto et al., 1998). They are recruited to the IKK complex via the IKKγ subunit (Yamaoka et al., 1998). After stimulation by these upstream kinases, IKK itself controls its activity by autophosphorylation that, at a certain level, results in inhibition of IKK activity and increased sensitivity to phosphatases (Delhase et al., 1999). This autoregulatory loop allows for an attenuation of NF-κB activation. A regulatory failure might cause excessive cytokine production, chronic inflammation, and increased resistance toward apoptotic signals. Interestingly, drugs that have been used for a long time to treat chronic arthritis, such as glucocorticoids, gold salts (Yang et al., 1995; Jeon et al., 2000), and high doses of salicylate, inhibit NF-κB activation (Kopp and Ghosh, 1994; Ray and Prefontaine, 1994; Auphan et al., 1995; Scheinman et al., 1995; Yin et al., 1998). These drugs, however, have potentially serious side effects, thus limiting their clinical usefulness. The toxicity, however, is probably not related to NF-κB inhibition. Hence, given the importance of NF-κB, selective IKK inhibitors might be interesting anti-inflammatory and analgesic drugs. This idea is supported by the finding that intra-articular gene transfer of a dominant-negative IKKβ considerably reduced synovial inflammation in an arthritis model in Lewis rats (Tak et al., 2001). In the present study, we assessed the anti-inflammatory and antinociceptive efficacy of a novel selective and specific IKK inhibitor in various nociceptive models in rats.
Materials and Methods
IKK assay
The in vitro assay for measuring the inhibitory activity of S1627 against the IKK complex uses a biotinylated polypeptide spanning both Ser32 and Ser36 of IκBα and a specific antibody that only binds to the phosphorylated form of this polypeptide (anti-phospho-serine32 IκBα antibody; New England Biolabs, Beverly, MA). The antibody-peptide complex is then detected using ELISA.
The IκBα kinase complex was prepared by diluting 10 ml of HeLa S3 cell extracts S100 fraction with 40 ml of 50 mm HEPES, pH 7.5. Then, 40% ammonium sulfate was added and incubated on ice for 30 min. The precipitated pellet was redissolved with 5 ml of kinase buffer (50 mm HEPES, pH 7.5, 1 mm DTT, 0.5 mm EDTA, and 10 mm 2-glycerophosphate), clarified by centrifugation at 20,000 × g for 15 min and filtration through a 0.22 μm filter unit. The sample was loaded onto a 320 ml Superose-6 FPLC column (Amersham Biosciences, Uppsala, Sweden) equilibrated with kinase buffer operated at a 2 ml/min flow rate at 4°C. Fractions spanning the 670 kDa molecular weight marker were pooled for activation. The kinase-containing pool was then activated by incubation with 100 nm MEKK1Δ, 250 μm MgATP, 10 mm MgCl2, 5 mm DTT, 10 mm 2-glycerophosphate, and 2.5 μm microcystin-LR for 45 min at 37°C. Activated enzyme was stored at -80°C until additional use. Per well of a 96-well plate, S1627 was preincubated at various concentrations in 1 μl of DMSO for 30 min at 25°C with 25 μl of activated enzyme diluted (1:35) with assay buffer (50 mm HEPES, pH 7.5, 5 mm DTT, 10 mm MgCl2, 10 mm 2-glycerophosphate, and 1 μm microcystin-LR). Twenty-five microliters of peptide substrate [biotin-(CH2)6-DRHDSGLDSMKD-CONH2; 40 μm stock solution] was then added to each well and incubated for 45 min before quenching with 50 μl of PBS, 0.1% BSA, and 20 mm EDTA plus (1:250) anti-phospho-IκBα antibody. Quenched kinase reaction samples and phospho-peptide-calibration standards [biotin-(CH2)6- DRHDS[PO3]GLDSMKD-CONH2; serially diluted in PBS/0.1% BSA] at 80 μl per well were transferred to a Protein-G plate (Pierce, Rockford, IL) and incubated for 1-2 hr with shaking. After three washes with PBS plus 0.05% Tween 20 and 100 μl of 0.13 μg/ml streptavidin conjugated with HRP diluted with PBS/0.1% BSA was added for 30 min. After five washes with PBS plus 0.05% Tween 20, 100 μl of TMB substrate (Kirkegaard & Perry Laboratories, Gaithersburg, MD) was added, and color development was stopped by adding 100 μl of 0.18 m H2SO4. Absorbance signals were recorded at 450 nm. Calibration curve standards were fitted by linear regression analysis using a four-parameter dose-response equation.
Other kinase assays
To assess the specificity of S1627, we determined its effects on 12 other kinases (Table 1) at a concentration of 20 μm, which is 2000-fold higher than the in vitro IC50 value for IKK. For kinases that were inhibited by >50% at that concentration, we additionally assessed the IC50 value. Kinase assays were performed by Cerep (Paris, France). The source of the kinase used in the respective assay, substrate, measured product, and experimental conditions are available at Cerep.
Table 1.
Effects of S1627 on various kinases
|
Kinase |
Percentage inhibition at 20 μm S1627 |
IC50 (μm) of S1627 |
Reference compound |
IC50 (μm) of reference compound |
|---|---|---|---|---|
| Casein kinase II | 25 | Hypericin | 7 | |
| Cdk1 (cyclin-dependent protein kinase) | 23 | Roscovitine | 2.1 | |
| CaMKII (Ca2+-calmodulin-dependent kinase) | 86 | 14.6 | Staurosporine | 0.007 |
| Erk-2 (p42 MAPK) | 0 | Staurosporine | 7.4 | |
| p38β MAPK | 11 | SB-202190 | 0.034 | |
| MEK1 (MAP kinase kinase) | 84 | 18.2 | Staurosporine | 0.03 |
| Protein kinase A (stimulated) | 21 | H89 | 0.23 | |
| PKCα | 0 | Staurosporine | 0.037 | |
| EGF receptor tyrosine kinase | 16 | PD 153035 | 0.005 | |
| Abl (nonreceptor tyrosine kinase) | 5 | Staurosporine | 0.04 | |
| Src (nonreceptor tyrosine kinase) | 103 | 2.18 | Staurosporine | 0.04 |
| ZAP70 (Src family tyrosine kinase) |
93 |
2.43 |
Staurosporine |
0.016 |
Kinase activity was determined by Cerep (Paris, France). MAPK, MAP kinase; EGF, epidermal growth factor.
Nuclear extracts
Human umbilical vein endothelial cells were used to assess the effects of S1627 on NF-κB activation. Cells were cultured in endothelial growth medium supplemented with 1% penicillin-streptomycin, 2% FCS, hydrocortisone, human endothelial growth factor (EGF), human FGF, vascular endothelial growth factor, R3-insulin like growth factor-1, ascorbic acid, GA-1000, and heparin (Cell Systems/Clonetics). Cells were preincubated for 30 min with S1627 or vehicle and then stimulated for 60 min with 1 nm IL-1β in the presence or absence of S1627.
For the preparation of nuclear and cytosolic fractions, cell pellets were resuspended in 1 ml of lysis buffer I (10 mm Tris-HCl, pH 7.4, 10 mm NaCl, 3 mm MgCl2, 1 mm PMSF, and 2 mm DTT) and incubated for 10 min on ice. After the addition of NP-40 (final concentration, 0.5%), the solution was vortexed and centrifuged at 400 × g for 5 min. The supernatant was kept as the cytosolic fraction. The nuclear pellet was washed with lysis buffer I. Pellets were then resuspended in 2 volumes of lysis buffer II (20 mm HEPES-KOH, pH 7.4, 600 mm KCl, 0.2 mm EDTA, 1 mm PMSF, and 2 mm DTT) and incubated for 30 min on ice. After centrifugation (10,000 × g for 10 min) 1 volume of lysis buffer III (20 mm HEPES-KOH, pH 7.4, 0.2 mm EDTA, 0.5 mm PMSF, and 2 mm DTT) and glycerol was added to the supernatant to obtain a final glycerol concentration of 20%. Samples were stored at -80°C.
Preparation of nuclear extracts from lumbar spinal cord was done as described (An et al., 1993). Briefly, spinal cord tissue (L3-L5) samples were homogenized quickly in 3 volumes of buffer A (10 mm HEPES, pH 7.9, 1.5 mm MgCl2, 10 mm KCl, 1 mg/ml leupeptin and aprotinin, and 0.5 mm DTT) and then kept on ice for 10 min. After a 15 min centrifugation at 3300 × g at 4°C, the pellet was resuspended in 3 ml of buffer B (20 mm HEPES, pH 7.9, 20 mm KCl, 1.5 mm MgCl2, 0.2 mm EDTA, 0.2 mm DTT, 0.2 mm PMSF, 1 mg/ml leupeptin and aprotinin, and 25% glycerol), mixed with 1 ml of buffer C (20 mm HEPES, pH 7.9, 1.2 mm KCl, 0.2 mm EDTA, 0.5 mm DTT, 0.2 mm PMSF, and 1 mg/ml leupeptin and aprotinin), and then incubated on ice for 30 min. After another centrifugation step (30 min; 15.000 × g; 4°C), the supernatant was dialyzed against buffer D (20 mm HEPES, pH 7.9, 100 mm KCl, 0.2 mm PMSF, 0.5 mm DTT, and 20% glycerol) overnight and was then used for Western blot analysis.
Transcription factor assay
Transcription factor analysis was performed with an ELISA kit (Active Motif, Rixensart, Belgium) that allowed for the detection and quantification of transcription factor activation by a combination of NF-κB-specific oligonucleotide binding and subsequent detection of the p65 subunit of NF-κB with a specific antibody. The assay was performed as recommended by the manufacturer. Briefly, nuclear protein extracts (10 μg) were added onto the oligonucleotide-coated ELISA plate and then incubated for 1 hr at room temperature. The plate was rinsed with washing buffer and then incubated for 1 hr at room temperature with the primary antibody directed against p65. After incubation with an HRP-conjugated secondary antibody for 1 hr, the HRP substrate was added. The reaction was stopped after 5-10 min, and the absorbance was measured at 450 nm.
Western blot analysis
Animals were finally anesthesized and debleeded and the lumbar spinal cord was quickly dissected, frozen in liquid nitrogen, and kept at -80°C until analysis.
Tissue samples were homogenized in homogenization buffer [10 mm Tris/HCl, pH 7.4, containing 20 mm 3-[(3-cholamidopropyl)dimethylammonio]-1-propanesulfonate, 0.5 mm EDTA, 1 mm DTT, 0.5 mm PMSF, and 1 mm pefabloc (Alexis, Grünberg, Germany)] and centrifugated at 40,000 × g for 1 hr. Extracted proteins (30 μg) were mixed with Laemmli buffer [50 mm Tris, pH 6.8, 2.5% (w/v) SDS, 10% (w/v) glycerol, and 0.001% bromphenol blue], heated for 5 min at 95°C, separated by SDS-PAGE, and transferred onto nitrocellulose membranes (Amersham Biosciences, Freiburg, Germany) by electroblotting (Bio-Rad, Munich, Germany). Membranes were blocked with blocking buffer (5% w/v skimmed milk powder and 0.3% Tween 20 dissolved in PBS, pH 7.6) and incubated overnight with primary antibodies directed against COX-2 (1:100), Erk-2 (1:1000), or p65 (1:100) (Santa Cruz Biotechnology, Heidelberg, Germany) and for 1 hr with the secondary antibody coupled with HRP (dilution 1:20,000). Blots were analyzed with ECL (Amersham Biosciences) according to the manufacturer's protocol.
IL-1β and TNF-α ELISA
Animals were treated with S1627 or vehicle, and the spinal cord was dissected 48 hr after zymosan injection (6 mg). Crude protein extracts were prepared from spinal cord tissue as described above (Western blot), and the protein content was assessed with the Bradford method. IL-1β and TNF-α levels were assessed with commercially available ELISA kits according to the instructions of the manufacturer (Amersham Biosciences) and are expressed as picograms per microgram of protein.
Animals and treatments
Male Sprague Dawley rats (150-200 gm; Charles River, Sulzfeld, Germany) were maintained in climate- and light-controlled rooms (21 ± 0.5°C; 12 hr dark/light cycle). They had ad libitum access to food and water before and during the experiments. All experiments were approved by the local Ethics Committee for research in laboratory animals.
Study medication. S1627 was dissolved in a 1:1 mixture of polyethyleneglycol (PEG400) and water at a maximum concentration of 10 mg/ml. Control rats received an equal volume of vehicle. The drug was administered by intraperitoneal injection. Different doses and dosing schedules were used for different models as indicated in the respective figures and figure legends.
Animal models of nociception
Formalin assay. Rats were adapted to the test perspex chamber for at least 30 min. Fifty microliters of a 5% formaldehyde solution was then injected into the subcutaneous space at the dorsal side of the right hind paw. The formalin-induced typical flinching behavior of the injected paw was counted in 1 min intervals for 60 min, starting immediately after formalin injection. Flinches were counted always by the same observer, who was unaware of the actual medication.
Zymosan-evoked inflammation. Unilateral hind paw inflammation was induced by subcutaneous injection of 100 μl of a 10 mg/ml zymosan (Sigma, Germany) suspension in PBS (0.1 m; pH 7.4) into the mid-plantar region of the right hind paw.
The zymosan-induced paw inflammation was assessed by measurement of the paw volume using a plethysmometer (Ugo Basile, Varese, Italy) (Sawynok et al., 1997; Bileviciute et al., 1998). Measurements were obtained at baseline and at 0.5, 1, 2, 4, 6, 8, and 24 hr after zymosan injection. Four rats per group were used. The ratio between the paw volume before and after zymosan injection (fold increase) was used to assess anti-inflammatory effects.
Chronic constriction injury of the sciatic nerve model. During general anesthesia with ketamine and midazolam, the right sciatic nerve was constricted with three loose ligatures as described (George et al., 2000). Dexon 4/0 (Braun, Melsungen, Germany) was used as suture material. The day of the surgery is referred to as day 0.
Thermal hyperalgesia. Nociceptive paw withdrawal latency (PWL) to radiant heat was assessed according to Hargreaves et al. (1988), using a commercially available special device (Ugo Basile). Rats were placed in perspex chambers (18 × 29 × 12.5 cm) on a metal grid (5 × 5 mm). During the week before the experiments, animals were acquainted with the plastic chambers for 1 hr on 2 different days. On the day of the experiments, rats were placed into the chamber 15-20 min before each measurement. The heat source consisting of a high-intensity projector lamp bulb (8 V; 50 W) was placed underneath the mid-plantar surface of the hind paw. Correct positioning was ensured by a mirror system. The bulb and an electronic timer were simultaneously activated at test start, and both were automatically inactivated when a photocell detected a paw withdrawal in response to the heat. PWLs of the right and left paw were recorded before and every hour up to 8 hr after zymosan injection. To prevent tissue damage, the cutoff latency was set at 40 sec (baseline latency, 15-25 sec). The percentage of relative difference in PWL (ΔPWL) between the zymosan-treated right and the untreated left hind paw was calculated as: ΔPWL = (right - left)/left × 100.
Mechanical hyperalgesia. Mechanical hyperalgesia was assessed before zymosan injection and then hourly up to 8 hr after zymosan injection. In chronic constriction injury of the sciatic nerve (CCI) rats, mechanical hyperalgesia/allodynia was assessed at baseline (3, 2, and 1 d before surgery) and after induction of CCI (day 0) on days 4, 5, 6, 7, 8, 11, and 13 after surgery. The threshold to mechanical stimuli was assessed by means of a punctuated stimulation using von Frey hairs of different strength (0.04, 0.07, 0.16, 0.4, 0.6, 1, 1.4, 2, 4, 6, 8, 10, 15, 26, 60, and 100 gm; Stoelting). They were placed perpendicularly onto the plantar surface of the right or left paw and bent slightly to apply punctuated pressure. The stimuli were applied at seven repetitions each and at increasing order until the paw was withdrawn. Stimuli were then applied at decreasing order until paw withdrawal disappeared. This up and down testing was repeated after a short rest. The geometric mean of first reaction (increasing testing) and lowest test result (decreasing testing) was taken as the mechanical paw withdrawal threshold (MPWT). These data were log transformed, and the percentage of decrease of the withdrawal threshold was then calculated in relation to the withdrawal threshold before surgery (baseline) or, in case of the zymosan model, in relation to the withdrawal of the untreated left paw. Hence, the percentage of decrease of the MPWT was ΔMPWT = (CCI paw - baseline)/baseline × 100 or ΔMPWT = (zymosan paw - left paw)/left paw × 100.
Cold allodynia. To assess cold allodynia in the CCI model, a drop of acetone was applied onto the right hindpaw. The time the rat spent lifting, shaking, or licking the acetone-treated paw was recorded with a stopwatch during an observation period of 2 min starting right after acetone application.
Data analysis and statistics
Data are presented as means ± SE. To assess statistical differences, the areas under the effect versus time curves (AUC) were calculated using the linear trapezoidal rule. Effects of the study medications were then assessed by submitting the AUCs to univariate ANOVA (for more than two groups) or Student's t tests (for two groups). For the formalin assay, the total number of flinches was used for statistics. In case of a significant ANOVA result, groups were mutually compared using t tests with a Bonferroni α correction for multiple comparisons. The α level was set to 0.05. Statistical evaluation was done with SPSS 11.0 for Windows (SPSS, Chicago, IL).
Pharmacokinetic parameters for S1627 were calculated according to standard procedures with WinNonlin using a one-compartment model with first-order input and first-order elimination.
Analysis of plasma and CSF concentrations of S1627 and its primary metabolite
Concentrations of S1627 and its metabolite S658 were determined by liquid chromatography coupled with mass spectrometry. The drug was extracted from rat plasma and CSF samples with acetonitrile. To 25 μl of the sample, 25 μl of water, 25 μl of 0.1% formic acid, and 200 μl of acetonitrile were added. After centrifugation (12 × g for 5 min), the supernatant was used for injection onto the HPLC system.
Stock solutions for S1627 and S658 were prepared in methanol (100 μg/ml) and stored at 4°C. Standards were prepared daily. An aliquot of 100 μl of each stock solution was evaporated under a gentle stream of nitrogen in a heating block at a temperature of 60°C. The residue was dissolved in 1 ml of water and contained 10 μg/ml S1627 and 10 μg/ml S658. Additional dilutions were prepared with water to obtain standards with the concentrations 1, 2, 5, 10, 20, 50, 100, 200, 500, 1000, 2000, and 5000 ng/ml. Standards were mixed 1:1 with drug-free plasma or CSF and prepared with formic acid and acetonitrile, as described above.
The HPLC consisted of a pump, three-line degasser, ternary gradient unit, and autosampler (all from Jasco, Gross-Umstadt, Germany). A triple quadrupole mass spectrometer PE Sciex API 3000 was used for detection (Applied Biosystems, Langen, Germany). High-purity nitrogen for the mass spectrometer was produced by a nitrogen generator (Parker, Kaarst, Germany). The interface between HPLC and mass spectrometer was a turbo ion spray.
For the chromatographic separation, a Nucleosil C18 Nautilus column was used (125 × 4 mm inner diameter, 5 μm particle size; and 100 Å pore size; Macherey-Nagel, Düren, Germany). The mobile phase consisted of acetonitrile (75%, v/v), water (25%, v/v), and formic acid (0.025%, v/v). The run time was 8 min at a flow rate of 0.5 ml/min. The injection volume was 10 μl. Retention times of S1627 and S658 were 2.97 min and 5.02 min, respectively.
The mass spectrometer operated in positive ion mode with a voltage of 4500 V at 400°C and was supplied by an auxiliary gas flow of 6 l/min. The nebulizer gas flow was set at 1.23 l/min and the curtain gas flow at 1.25 l/min. Every gas was high-purity nitrogen.
Multiple reaction monitoring was used for quantification. The mass transitions used were m/z 547.2 → m/z 182.2 for S1627 (collision energy, 35 eV) and m/z 507.2 → m/z 294.2 for the metabolite S658 (collision energy, 49 eV), with a dwell time of 300 msec for each substance. All quadruples were working at unit resolution. Quantitation was performed with Analyst software version 1.1 (Applied Biosystems) using peak areas and 1/x weighting. The coefficient of variation (CV) over the calibration range of 1-5000 ng/ml was <5%.
Results
Inhibition of IKK activity and NF-κB nuclear translocation
S1627 inhibited the activity of purified IKK with an IC50 value of 10 ± 1.2 nm (Fig. 1). It also prevented IL-1β-stimulated nuclear translocation and DNA binding of NF-κB in human cells in a concentration-dependent manner with an IC50 value of ∼2 μm (Fig. 2). The different potency is most likely caused by high protein binding of the drug, which is 99.9%.
Figure 1.

Concentration-dependent inhibition of IKK activity with S1627 assessed in an in vitro kinase assay. The IC50 value (10.0 ± 1.2 nm; mean ± SE) was calculated with a standard sigmoidal Emax model.
Figure 2.
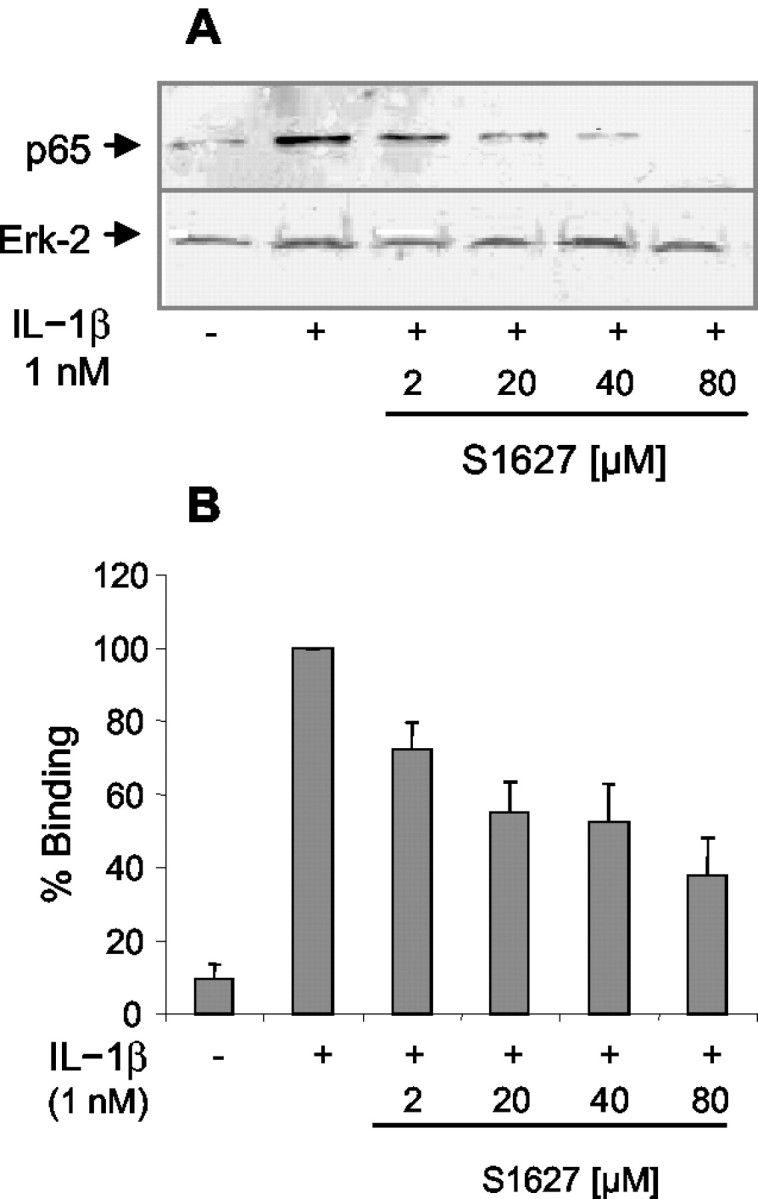
A, Western blot analysis showing the concentration-dependent inhibition of IL-1β-stimulated nuclear translocation of NF-κB (p65 subunit) in human umbilical vein endothelial cells. Cells were preincubated with S1627 or vehicle for 30 min, stimulated for 60 min with 1 nm IL-1β in the presence or absence of S1627, and then harvested for preparation of nuclear and cytosolic extracts. Nuclear p65 was detected with a specific antibody. Erk-2 was used as loading control. B, Concentration-dependent inhibition of IL-1β-stimulated nuclear translocation and DNA binding of NF-κB in human umbilical vein endothelial cells (mean ± SE) assessed using an ELISA that uses an oligonucleotide-coated plate and detection of protein binding with a p65-specific antibody. Results of four repeated experiments are shown.
Effect of S1627 on other kinases
At a concentration of 20 μm (∼2000-fold higher than the IC50 value for IKK), S1627 had no or only a marginal effect on protein kinase A, PKC, Erk, p38, casein kinase II, EGF kinase, Abl tyrosine kinase, and Cdk-1. CamK-II, MEK1, Src, and Zap70 were inhibited by >80% at this high concentration (Table 1). The in vitro IC50 values for the most sensitive of these kinases (Src and Zap70), however, are still ∼200-fold higher than the IC50 value for IKK. Because of the high protein binding of the drug, total plasma concentrations of S1627 would have to be in the millimolar range to inhibit these kinases in vivo. Such concentrations are not reached after treatment with antinociceptive doses of the drug.
Plasma and CSF concentrations
Plasma concentration time courses of the mother substance (Fig. 3A) and its main metabolite, S658 (Fig. 3B), which has minor IKK inhibitory activity in vitro (data not shown), were assessed after intraperitoneal administration of one, two, or three doses of 10 or 30 mg/kg. The absorption rate constant K01 was 2.963 hr-1 (CV%, 14.81), and the elimination rate constant K10 was 0.245 hr-1 (CV%, 8.53), resulting in an elimination half-life of 2.8 hr. The plasma Tmax was 0.91 ± 0.09 hr (CV%, 8.53), and Cmax in plasma was 1533 ± 38.5 ng/ml (2.63 ± 0.07 μm) and 538 ± 22.0 ng/ml (0.92 ± 0.04 μm) after 30 and 10 mg/kg, respectively. Because of the short half-life, the drug was injected repeatedly in the behavioral models.
Figure 3.
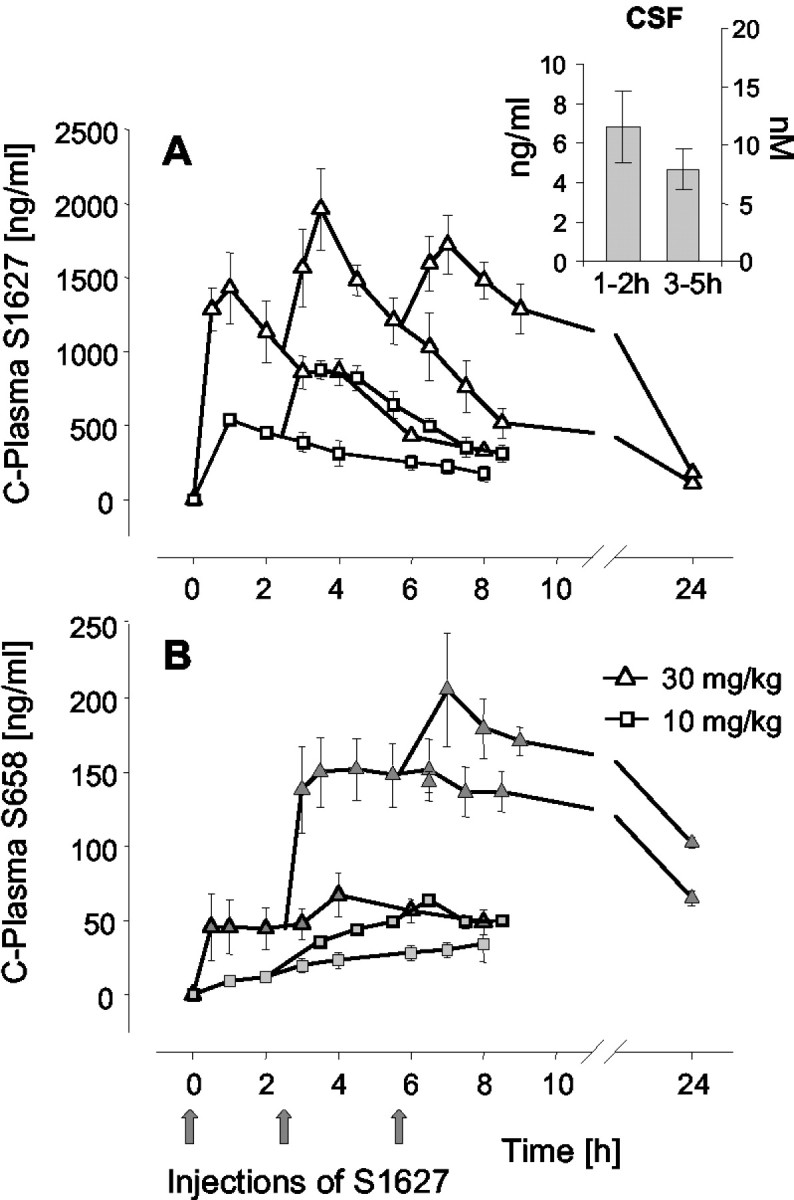
Plasma concentration time course of S1627 (A) and its primary metabolite S658 (B) after intraperitoneal injection of one, two, or three doses of S1627 at the indicated times (4 rats in each group). The inset in A shows CSF concentrations after a single dose of 30 mg/kg (6 rats per time). Plasma and CSF concentrations were determined with liquid chromatography coupled with tandem mass spectrometry. The data represent the mean ± SE.
CSF concentrations are shown in the inset of Figure 3A. Because only one CSF sample can be obtained per rat, we have summarized the data as mean concentrations between 1 and 2 hr and 3-5 hr (six rats in each group). The CSF concentrations after a single dose of 30 mg/kg range from 2.1 to 11.9 ng/ml (3.6-20.4 nm) and from 1.2 to 7.9 ng/ml (2.1-13.7 nm) at 1-2 hr and 3-5 hr after drug injection, respectively. Because CSF contains almost no protein, these are free (i.e., protein unbound) drug levels. Differences between CSF and plasma concentrations (∼100-fold lower CSF levels at 3-5 hr) are most likely caused by the high plasma protein binding of the drug (99.9%).
Antinociceptive and anti-inflammatory effects
Response to heat and tactile stimuli in naive rats
Heat and von Frey stimuli were applied to noninflamed paws to evaluate effects of the drug on motor and sensory function and alertness. Three doses of 30 mg/kg S1627 or vehicle were injected at the indicated times. Drug injection did neither affect the withdrawal latency to radiant heat (Fig. 4A) nor the mechanical threshold to von Frey stimuli (Fig. 4B).
Figure 4.

Effects of S1627 in naive animals on thermal PWL (A) and paw withdrawal thresholds (B) to von Frey stimulation. Three doses of 30 mg/kg S1627 (n = 6; ▵) or vehicle (n = 6; •) were injected intraperitoneally at the indicated times. Data represent the mean ± SE.
Formalin assay (Fig. 5A)
Figure 5.

A, Effects of S1627 in the formalin assay. Two doses of 30 mg/kg were injected intraperitoneally 90 and 30 min before injection of formalin into the hind paw. The total number of flinches did not differ between groups. The data represent the mean ± SE of five rats in each group. B, Dose-dependent reduction of thermal hyperalgesia (Hargreaves test) in the zymosan-induced paw inflammation model. Either 10 mg/kg S1627 (n = 6; □) or 30 mg/kg (n = 6; ▵) was injected intraperitoneally at the indicated times. Controls (n = 12; •) received the appropriate volume of vehicle (1:1, vol/vol; polyethyleneglycol:water). The data represent the mean ± SE. ANOVA comparing the AUCs revealed significant effects for both doses (p < 0.05). C, Dose-dependent reduction of tactile hyperalgesia assessed with von Frey hairs in the zymosan-induced paw inflammation model. Either 10 mg/kg S1627 (n = 6; □) or 30 mg/kg (n = 6; ▵) was injected intraperitoneally at the indicated times (controls, n = 12; •). The data represent the mean ± SE. Antinociceptive effects of 3× 30 mg/kg (comparison of AUCs) were statistically significant with p < 0.05. 5. D, Reduction of pressure hyperalgesia (Randall Selitto test) in the zymosan-induced paw inflammation model. Three doses of 40 mg/kg (n = 4) were injected intraperitoneally 0.5 hr before and 2.25 and 5 hr after zymosan injection. The data represent the mean ± SE. The pressure pain threshold significantly differed between groups with p < 0.05 (indicated with the asterisk). E, Reduction of the zymosan-induced inflammatory paw edema with S1627. Two doses of 30 mg/kg or three doses of 40 mg/kg were injected intraperitoneally at the indicated times (n = 4 in each group). Controls received an equal volume of vehicle (n = 8). The data represent the mean ± SE. The reduction of paw swelling was statistically significant (comparison of AUCs) with the higher dose of 3× 40 mg/kg at p < 0.05.
Two doses of 30 mg/kg were injected intraperitoneally 90 and 30 min before injection of formalin. S1627 caused a prolongation of the latency period and, therefore, a reduction of flinches in phase 2a (10-30 min after formalin injection). However, the total number of flinches did not differ from that of control rats (583.8 ± 83.5 vs 672 ± 73.4 flinches in S1627 and control rats, respectively).
Thermal hyperalgesia caused by zymosan injection into the hind paw (Fig. 5B)
Three doses of 10 or 30 mg/kg were administered at the indicated times before and after zymosan injection. During the first 2 hr after zymosan injection, the thermal nociceptive threshold was similar in control and S1627-treated rats. In the next hours, the thermal nociceptive threshold further declined in control animals but returned to baseline levels in rats treated with 3× 30 mg/kg S1627. Hence, thermal hyperalgesia was completely reversed with the high dose of S1627. The lower dose also provided a considerable reduction of thermal hyperalgesia. ANOVA comparing the AUCs revealed statistically significant differences between groups (F = 43.54; df 2,20; p < 0.001). Mutual comparison showed significant differences between control and S1627-treated groups (p < 0.001 for both doses) and between the 3× 30 mg/kg versus the 3× 10 mg/kg group (p = 0.041).
Tactile hind paw hyperalgesia caused by zymosan injection (Fig. 5C)
Tactile hyperalgesia after zymosan injection was also reduced at the dose of 3× 30 mg/kg S1627. The onset of antinociceptive effects was again 1.5-2 hr after zymosan injection (2-2.5 hr after injection of the first dose). ANOVA comparing the AUCs revealed statistically significant differences between groups (F = 4.95; df 2,20; p = 0.018). Mutual comparison showed significant differences between control and the 3× 30 mg/kg S1627-treated group (p = 0.027). There was no significant difference between control and the 3× 10 mg/kg group (p = 0.146).
Pressure hyperalgesia caused by zymosan injection (Fig. 5D)
The Randall Selitto test performed 8 hr after zymosan injection revealed significantly higher-pressure nociceptive thresholds in S1627-treated rats (3× 40 mg/kg) than in controls (p < 0.001; t test).
Inflammatory paw edema caused by zymosan injection (Fig. 5E)
The inflammatory paw edema was reduced at 3× 40 mg/kg S1627 (p = 0.048) (i.e., reduction of the edema required a higher dose than reduction of thermal hyperalgesia). A lower dose of 2× 30 mg/kg had no effect (p = 1).
Tactile allodynia induced by CCI model (Fig. 6A)
Figure 6.

A, Tactile allodynia after chronic constriction of the sciatic nerve (CCI model). Rats (n = 12 in each group) received 30 mg/kg S1627 or vehicle once daily for 5 d starting on day 4 after surgery. The nociceptive threshold was assessed daily with von Frey hairs before injection of the daily dose. The data represent the mean ± SE. The differences between the AUCs of control and S1627-treated rats was statistically significant at p < 0.05. B, Cold allodynia after chronic constriction of the sciatic nerve (CCI model). Rats (n = 12 in each group) received 30 mg/kg S1627 or vehicle once daily for 5 d starting at day 4 after surgery. Cold allodynia was assessed daily before drug injection by application of a drop of acetone onto the plantar surface of the hind paw. The time the rats spent licking, shaking, or lifting the paw after acetone application was measured with a stopwatch during an observation time of 2 min. The data represent the mean ± SE. The differences between the AUCs of control and S1627-treated rats was statistically significant at p < 0.05.
Treatment with S1627 was started when allodynia had established at the fourth day after surgery. Measurements were always performed before injection of the daily dose of 30 mg/kg. Injection of S1627 caused a reduction of tactile allodynia compared with allodynia before starting drug treatment (day 4), whereas allodynia in control animals increased with a maximum at day 7. After stopping daily injections, nociceptive thresholds remained higher in S1627-treated animals than those in controls up to the end of the observation period (day 15), indicating an ongoing antiallodynic effect. The t test comparing AUCs revealed a significant difference between S1627-treated and control rats with p < 0.001.
Cold allodynia induced by CCI model (Fig. 6B)
The effects of daily S1627 injections on cold allodynia were comparable with the effects on tactile allodynia (i.e., S1627 caused a reduction of cold allodynia during the treatment period, and the effect lasted for several days after stopping treatment). The t test comparing AUCs revealed a significant difference between S1627-treated and control rats with p = 0.013.
Inhibition of COX-2, TNF-α, and IL-1β upregulation
To evaluate the effects of S1627 on the upregulation of NF-κB-dependent genes in the spinal cord, protein levels of COX-2, TNF-α, and IL-1β were determined using Western blots or ELISAs, respectively. COX-2, TNF-α, and IL-1β are known to be positively regulated by NF-κB.
Western blots of nuclear protein extracts from lumbar spinal cord tissue revealed that S1627 inhibited the nuclear translocation of NF-κB (Fig. 7A). S1627 inhibited the zymosan-induced upregulation of COX-2 at the dose of 3× 30 mg/kg (Fig. 7B). There was no effect with 3× 10 mg/kg. Similarly, 3× 30 mg/kg S1627 reduced the zymosan-induced upregulation of TNF-α (Fig. 7C) and IL-1β (Fig. 7D) in the spinal cord. There was no significant effect at 3× 10 mg/kg.
Figure 7.

A, Western blot analysis of nuclear p65 (NF-κB subunit) using nuclear extracts from lumbar spinal cord tissue (L3-L5). Rats received a single dose of 100 mg/kg or vehicle 30 min before injection of 6 mg of zymosan into the hindpaw. The spinal cord was dissected 1 hr after zymosan injection. B, Western blot analysis of COX-2 in lumbar spinal cord tissue (L3-L5). Rats were treated as indicated, and the spinal cord was dissected 24 hr after zymosan injection. The times of drug or vehicle injection were 0.5 hr before zymosan and 2.25 and 5.5 hr after zymosan injection. C, D, TNF-α and IL-1β levels in lumbar spinal cord homogenates (L3-L5) assessed with commercially available ELISA assays. Rats were treated as indicated, and the spinal cord was dissected 48 hr after zymosan injection (6 mg). The times of drug or vehicle injection were 0.5 hr before zymosan and 2.25 and 5.5 hr after zymosan injection. Data are the mean ± SE. Zymosan-induced upregulation of TNF-α and IL-1β was significantly inhibited with 3× 30 mg/kg S1627, indicated with the asterisk (p < 0.05).
Discussion
The present study shows that specific inhibition of IKK results in a reduction of zymosan-induced paw inflammation, complete reversal of inflammatory hyperalgesia, and reduction of neuropathic pain without affecting “pain” thresholds in naive animals. The latency between first-dose injection and the earliest manifestation of antinociception strongly suggests that the effect is mediated through inhibition of stimulated gene transcription. The drug has no acute analgesic effect. In most experiments, the first dose was administered before induction of inflammation. However, results with the CCI model and the supplementary data show that a postinjury start of treatment works as well, suggesting that it does not matter at what phase of the inflammatory of neuropathic process NF-κB-stimulated gene transcription is stopped. Reduction of inflammation was expected on the basis of known anti-inflammatory effects of glucocorticoids and was also suggested by results with p50 (NF-κB) knock-out mice that are refractory to induction of both chronic and acute arthritis (Campbell et al., 2000). The strong antihyperalgesic effects were more surprising. They were not caused by unspecific inhibition of other kinases. The effects were stronger than those of nonsteroidal anti-inflammatory drugs in equivalent models and were more pronounced than could be expected from the rather moderate anti-inflammatory effects in the model used. Hence, the antihyperalgesic effects are probably not exclusively attributable to peripheral anti-inflammatory effects, although reduction of inflammation surely is associated with a reduction of nociceptor activation at the site of inflammation. The evaluation of NF-κB-dependent gene expression in the spinal cord revealed that only the higher dose of 3× 30 mg/kg inhibited the upregulation of COX-2, TNF-α, and IL-1β. This dose reversed zymosan-induced hyperalgesia completely, whereas 3× 10 mg/kg only partly reduced it. This comparison again suggests that the effects at the level of the spinal cord are required for full antinociceptive efficacy and require free (i.e., protein unbound) central drug levels in the range of the in vitro IC50 values. Free CSF concentrations of S1627 were surely not sufficient to inhibit any of the other kinases tested because the in vitro IC50 for the most sensitive one of these kinases was 200-fold higher than that for IKK.
NF-κB is activated in neurons on NMDA receptor stimulation (Ko et al., 1998), and NF-κB-stimulated gene transcription contributes to the development of hyperexcitability of nociceptive neurons. Hence, IKK inhibition in the spinal cord is probably important for the strong antihyperalgesic effects of S1627. Because of the poor solubility of the drug, however, we did not use intrathecal drug delivery to directly assess the relative contribution of peripheral versus central effects.
Compared with the well studied function of NF-κB in inflammation, its function in neuropathic pain is still elusive, although various cytokines are known to be involved in the development of allodynia after nerve injury. The CCI model used in the present study has an inflammatory component (Wagner et al., 1998; Levy et al., 1999), and inhibition of perineural inflammation and TNF-α release reduces nociception in this model (George et al., 2000). Hence, S1627 may act at the site of the peripheral nerve. Chronic constriction of the sciatic nerve additionally causes a microglia activation in the spinal cord (Colburn et al., 1999; Stuesse et al., 2000; Raghavendra et al., 2002) that may depend in part on NF-κB activation (Wilms et al., 2003) and probably is involved in nociceptive responses (Watkins et al., 1997). Inhibition of IKK may, therefore, protect from an excessive glial reaction and may thereby reduce neuropathic pain. The central effects of the drug may broaden the potential clinical usefulness of a specific IKK inhibitor because neurodegenerative diseases also involve inflammatory reactions that are possibly mediated by activated glia cells (Kaltschmidt et al., 1997; Bales et al., 1998).
S1627 primarily interacts with the β-subunit of IKK, which is essential for IκB phosphorylation. Interestingly, the α-subunit of IKK is translocated to the nucleus and binds to DNA (Anest et al., 2003), suggesting that IKKα may directly modulate gene transcription. This is probably not affected by S1627. Treatment with S1627 also still allows for IKK-independent NF-κB activation mediated by an IKK-independent breakdown of IκB (Pianetti et al., 2001; Romieu-Mourez et al., 2002), for example, by the protease calpain (Pianetti et al., 2001) or caspases (Chaudhary et al., 2000; Hu et al., 2000). These alternative pathways may preserve some NF-κB activation even in the presence of an IKK inhibitor and may explain why we did not achieve complete inhibition of NF-κB nuclear translocation and DNA binding in IL-1β-stimulated cells despite high concentrations of S1627. Pharmacologically, it may be advantageous to target the upstream kinase of the NF-κB pathway to prevent side effects that might be caused by complete inhibition of NF-κB.
In summary, our results suggest that specific inhibitors of IKK may be promising novel drugs for treatment of pain and inflammation.
Footnotes
This work was supported in part by the Deutsche Forschungsgemeinschaft (DFG TE322_2-1), Bundesministerium für Bildung und Forschung (BMBF 01 EM 0103), and an unrestricted grant from Aventis Pharma, Germany. We are grateful to Christine Schäfer and Ovidiu Coste for excellent assistance.
Correspondence should be addressed to Dr. Irmgard Tegeder, Neural Plasticity Research Group, Department of Anesthesia, Massachusetts General Hospital and Harvard Medical School, Room 4309, 149 13th Street, Charlestown, MA 02129. E-mail: itegeder@partners.org.
Copyright © 2004 Society for Neuroscience 0270-6474/04/241637-09$15.00/0
I.T. and E.N. contributed equally to this work.
References
- An G, Lin TN, Liu JS, Xue JJ, He YY, Hsu CY (1993) Expression of c-fos and c-jun family genes after focal cerebral ischemia. Ann Neurol 33: 457-464. [DOI] [PubMed] [Google Scholar]
- Anest V, Hanson JL, Cogswell PC, Steinbrecher KA, Strahl BD, Baldwin AS (2003) A nucleosomal function for IkappaB kinase-alpha in NF-kappaB-dependent gene expression. Nature 423: 659-663. [DOI] [PubMed] [Google Scholar]
- Aubin N, Curet O, Deffois A, Carter C (1998) Aspirin and salicylate protect against MPTP-induced dopamine depletion in mice. J Neurochem 71: 1635-1642. [DOI] [PubMed] [Google Scholar]
- Auphan N, DiDonato JA, Rosette C, Helmberg A, Karin M (1995) Immunosuppression by glucocorticoids: inhibition of NF-kappa B activity through induction of I kappa B synthesis. Science 270: 286-290. [DOI] [PubMed] [Google Scholar]
- Bales KR, Du Y, Dodel RC, Yan GM, Hamilton-Byrd E, Paul SM (1998) The NF-kappaB/Rel family of proteins mediates Amyloid-beta-induced neurotoxicity and glial activation. Brain Res Mol Brain Res 57: 63-72. [DOI] [PubMed] [Google Scholar]
- Bileviciute I, Stenfors C, Theodorsson E, Lundeberg T (1998) Unilateral injection of calcitonin gene-related peptide (CGRP) induces bilateral oedema formation and release of CGRP-like immunoreactivity in the rat hindpaw. Br J Pharmacol 125: 1304-1312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brand K, Page S, Rogler G, Bartsch A, Brandl R, Knuechel R, Page M, Kaltschmidt C, Baeuerle PA, Neumeier D (1996) Activated transcription factor nuclear factor-kappa B is present in the atherosclerotic lesion. J Clin Invest 97: 1715-1722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell IK, Gerondakis S, O'Donnell K, Wicks IP (2000) Distinct roles for the NF-kappaB1 (p50) and c-Rel transcription factors in inflammatory arthritis. J Clin Invest 105: 1799-1806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaudhary PM, Eby MT, Jasmin A, Kumar A, Liu L, Hood L (2000) Activation of the NF-kappaB pathway by caspase 8 and its homologs. Oncogene 19: 4451-4460. [DOI] [PubMed] [Google Scholar]
- Colburn RW, Rickman AJ, DeLeo JA (1999) The effect of site and type of nerve injury on spinal glial activation and neuropathic pain behavior. Exp Neurol 157: 289-304. [DOI] [PubMed] [Google Scholar]
- Delhase M, Hayakawa M, Chen Y, Karin M (1999) Positive and negative regulation of IkappaB kinase activity through IKKbeta subunit phosphorylation. Science 284: 309-313. [DOI] [PubMed] [Google Scholar]
- DiDonato J, Mercurio F, Rosette C, Wu-Li J, Suyang H, Ghosh S, Karin M (1996) Mapping of the inducible IkappaB phosphorylation sites that signal its ubiquitination and degradation. Mol Cell Biol 16: 1295-1304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DiDonato JA, Hayakawa M, Rothwarf DM, Zandi E, Karin M (1997) A cytokine-responsive IkappaB kinase that activates the transcription factor NF-kappaB. Nature 388: 548-554. [DOI] [PubMed] [Google Scholar]
- Gao Y, Lecker S, Post MJ, Hietaranta AJ, Li J, Volk R, Li M, Sato K, Saluja AK, Steer ML, Goldberg AL, Simons M (2000) Inhibition of ubiquitin-proteasome pathway-mediated I kappa B alpha degradation by a naturally occurring antibacterial peptide. J Clin Invest 106: 439-448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- George A, Marziniak M, Schafers M, Toyka KV, Sommer C (2000) Thalidomide treatment in chronic constrictive neuropathy decreases endoneurial tumor necrosis factor-alpha, increases interleukin-10 and has long-term effects on spinal cord dorsal horn met-enkephalin. Pain 88: 267-275. [DOI] [PubMed] [Google Scholar]
- Grilli M, Pizzi M, Memo M, Spano P (1996) Neuroprotection by aspirin and sodium salicylate through blockade of NF-kappaB activation. Science 274: 1383-1385. [DOI] [PubMed] [Google Scholar]
- Hargreaves K, Dubner R, Brown F, Flores C, Joris J (1988) A new and sensitive method for measuring thermal nociception in cutaneous hyperalgesia. Pain 32: 77-88. [DOI] [PubMed] [Google Scholar]
- Hu WH, Johnson H, Shu HB (2000) Activation of NF-kappaB by FADD, Casper, and caspase-8. J Biol Chem 275: 10838-10844. [DOI] [PubMed] [Google Scholar]
- Jeon KI, Jeong JY, Jue DM (2000) Thiol-reactive metal compounds inhibit NF-kappa B activation by blocking I kappa B kinase. J Immunol 164: 5981-5989. [DOI] [PubMed] [Google Scholar]
- Kaltschmidt B, Uherek M, Volk B, Baeuerle PA, Kaltschmidt C (1997) Transcription factor NF-kappaB is activated in primary neurons by amyloid beta peptides and in neurons surrounding early plaques from patients with Alzheimer disease. Proc Natl Acad Sci USA 94: 2642-2647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaltschmidt B, Uherek M, Wellmann H, Volk B, Kaltschmidt C (1999) Inhibition of NF-kappaB potentiates amyloid beta-mediated neuronal apoptosis. Proc Natl Acad Sci USA 96: 9409-9414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karin M, Cao Y, Greten FR, Li ZW (2002) NF-kappaB in cancer: from innocent bystander to major culprit. Nat Rev Cancer 2: 301-310. [DOI] [PubMed] [Google Scholar]
- Ko HW, Park KY, Kim H, Han PL, Kim YU, Gwag BJ, Choi EJ (1998) Ca2+-mediated activation of c-Jun N-terminal kinase and nuclear factor kappa B by NMDA in cortical cell cultures. J Neurochem 71: 1390-1395. [DOI] [PubMed] [Google Scholar]
- Kopp E, Ghosh S (1994) Inhibition of NF-kappa B by sodium salicylate and aspirin. Science 265: 956-959. [DOI] [PubMed] [Google Scholar]
- Levy D, Hoke A, Zochodne DW (1999) Local expression of inducible nitric oxide synthase in an animal model of neuropathic pain. Neurosci Lett 260: 207-209. [DOI] [PubMed] [Google Scholar]
- Lin YZ, Yao SY, Veach RA, Torgerson TR, Hawiger J (1995) Inhibition of nuclear translocation of transcription factor NF-kappa B by a synthetic peptide containing a cell membrane-permeable motif and nuclear localization sequence. J Biol Chem 270: 14255-14258. [DOI] [PubMed] [Google Scholar]
- Marok R, Winyard PG, Coumbe A, Kus ML, Gaffney K, Blades S, Mapp PI, Morris CJ, Blake DR, Kaltschmidt C, Baeuerle PA (1996) Activation of the transcription factor nuclear factor-kappaB in human inflamed synovial tissue. Arthritis Rheum 39: 583-591. [DOI] [PubMed] [Google Scholar]
- Mercurio F, Zhu H, Murray BW, Shevchenko A, Bennett BL, Li J, Young DB, Barbosa M, Mann M, Manning A, Rao A (1997) IKK-1 and IKK-2: cytokine-activated IkappaB kinases essential for NF-kappaB activation. Science 278: 860-866. [DOI] [PubMed] [Google Scholar]
- Nemoto S, DiDonato JA, Lin A (1998) Coordinate regulation of IkappaB kinases by mitogen-activated protein kinase kinase kinase 1 and NF-kappaB-inducing kinase. Mol Cell Biol 18: 7336-7343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neurath MF, Pettersson S, Meyer zum Buschenfelde KH, Strober W (1996) Local administration of antisense phosphorothioate oligonucleotides to the p65 subunit of NF-kappa B abrogates established experimental colitis in mice. Nat Med 2: 998-1004. [DOI] [PubMed] [Google Scholar]
- Pahl HL (1999) Activators and target genes of Rel/NF-kappaB transcription factors. Oncogene 18: 6853-6866. [DOI] [PubMed] [Google Scholar]
- Pianetti S, Arsura M, Romieu-Mourez R, Coffey RJ, Sonenshein GE (2001) Her-2/neu overexpression induces NF-kappaB via a PI3-kinase/Akt pathway involving calpain-mediated degradation of IkappaB-alpha that can be inhibited by the tumor suppressor PTEN. Oncogene 20: 1287-1299. [DOI] [PubMed] [Google Scholar]
- Raghavendra V, Rutkowski MD, DeLeo JA (2002) The role of spinal neuroimmune activation in morphine tolerance/hyperalgesia in neuropathic and sham-operated rats. J Neurosci 22: 9980-9989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ray A, Prefontaine KE (1994) Physical association and functional antagonism between the p65 subunit of transcription factor NF-kappa B and the glucocorticoid receptor. Proc Natl Acad Sci USA 91: 752-756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rayet B, Gelinas C (1999) Aberrant rel/nfkb genes and activity in human cancer. Oncogene 18: 6938-6947. [DOI] [PubMed] [Google Scholar]
- Romieu-Mourez R, Landesman-Bollag E, Seldin DC, Sonenshein GE (2002) Protein kinase CK2 promotes aberrant activation of nuclear factor-kappaB, transformed phenotype, and survival of breast cancer cells. Cancer Res 62: 6770-6778. [PubMed] [Google Scholar]
- Rothwarf DM, Zandi E, Natoli G, Karin M (1998) IKK-gamma is an essential regulatory subunit of the IkappaB kinase complex. Nature 395: 297-300. [DOI] [PubMed] [Google Scholar]
- Sawynok J, Zarrindast MR, Reid AR, Doak GJ (1997) Adenosine A3 receptor activation produces nociceptive behaviour and edema by release of histamine and 5-hydroxytryptamine. Eur J Pharmacol 333: 1-7. [DOI] [PubMed] [Google Scholar]
- Scheinman RI, Cogswell PC, Lofquist AK, Baldwin Jr AS (1995) Role of transcriptional activation of I kappa B alpha in mediation of immunosuppression by glucocorticoids. Science 270: 283-286. [DOI] [PubMed] [Google Scholar]
- Sovak MA, Bellas RE, Kim DW, Zanieski GJ, Rogers AE, Traish AM, Sonenshein GE (1997) Aberrant nuclear factor-kappaB/Rel expression and the pathogenesis of breast cancer. J Clin Invest 100: 2952-2960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stuesse SL, Cruce WL, Lovell JA, McBurney DL, Crisp T (2000) Microglial proliferation in the spinal cord of aged rats with a sciatic nerve injury. Neurosci Lett 287: 121-124. [DOI] [PubMed] [Google Scholar]
- Tak PP, Gerlag DM, Aupperle KR, van de Geest DA, Overbeek M, Bennett BL, Boyle DL, Manning AM, Firestein GS (2001) Inhibitor of nuclear factor kappaB kinase beta is a key regulator of synovial inflammation. Arthritis Rheum 44: 1897-1907. [DOI] [PubMed] [Google Scholar]
- Tegeder I, Niederberger E, Israr E, Guhring H, Brune K, Euchenhofer C, Grosch S, Geisslinger G (2001) Inhibition of NF-{kappa}B and AP-1 activation by R- and S-flurbiprofen. FASEB J 15: 2-4. [DOI] [PubMed] [Google Scholar]
- Wagner R, Janjigian M, Myers RR (1998) Anti-inflammatory interleukin-10 therapy in CCI neuropathy decreases thermal hyperalgesia, macrophage recruitment, and endoneurial TNF-alpha expression. Pain 74: 35-42. [DOI] [PubMed] [Google Scholar]
- Wang CY, Mayo MW, Baldwin Jr AS (1996) TNF- and cancer therapy-induced apoptosis: potentiation by inhibition of NF-kappaB. Science 274: 784-787. [DOI] [PubMed] [Google Scholar]
- Wang CY, Cusack Jr JC, Liu R, Baldwin Jr AS (1999) Control of inducible chemoresistance: enhanced anti-tumor therapy through increased apoptosis by inhibition of NF-kappaB. Nat Med 5: 412-417. [DOI] [PubMed] [Google Scholar]
- Watkins LR, Martin D, Ulrich P, Tracey KJ, Maier SF (1997) Evidence for the involvement of spinal cord glia in subcutaneous formalin induced hyperalgesia in the rat. Pain 71: 225-235. [DOI] [PubMed] [Google Scholar]
- Wilms H, Rosenstiel P, Sievers J, Deuschl G, Zecca L, Lucius R (2003) Activation of microglia by human neuromelanin is NF-kappaB dependent and involves p38 mitogen-activated protein kinase: implications for Parkinson's disease. FASEB J 17: 500-502. [DOI] [PubMed] [Google Scholar]
- Yamaoka S, Courtois G, Bessia C, Whiteside ST, Weil R, Agou F, Kirk HE, Kay RJ, Israel A (1998) Complementation cloning of NEMO, a component of the IkappaB kinase complex essential for NF-kappaB activation. Cell 93: 1231-1240. [DOI] [PubMed] [Google Scholar]
- Yang JP, Merin JP, Nakano T, Kato T, Kitade Y, Okamoto T (1995) Inhibition of the DNA-binding activity of NF-kappa B by gold compounds in vitro. FEBS Lett 361: 89-96. [DOI] [PubMed] [Google Scholar]
- Yaron A, Hatzubai A, Davis M, Lavon I, Amit S, Manning AM, Andersen JS, Mann M, Mercurio F, Ben-Neriah Y (1998) Identification of the receptor component of the IkappaBalpha-ubiquitin ligase. Nature 396: 590-594. [DOI] [PubMed] [Google Scholar]
- Yin MJ, Yamamoto Y, Gaynor RB (1998) The anti-inflammatory agents aspirin and salicylate inhibit the activity of I(kappa)B kinase-beta. Nature 396: 77-80. [DOI] [PubMed] [Google Scholar]
- Zandi E, Chen Y, Karin M (1998) Direct phosphorylation of IkappaB by IKKalpha and IKKbeta: discrimination between free and NF-kappaB-bound substrate. Science 281: 1360-1363. [DOI] [PubMed] [Google Scholar]