

Abstract
Activation of the CB1 cannabinoid receptor inhibits neurotransmission at numerous synapses in the brain. Indeed, CB1 is essential for certain types of both short- and long-term synaptic depression. It was demonstrated recently that CB1 is critical for activity-dependent long-term depression (LTD) at glutamatergic corticostriatal synapses in acute brain slice preparations. Here, we show that CB1 activation is necessary, but not solely sufficient, for induction of LTD and that the requisite signaling by endocannabinoids (eCBs) occurs during a time window limited to the first few minutes after high-frequency stimulation delivery. In addition, we have applied intracellularly anandamide membrane transporter inhibitors to provide novel evidence that postsynaptic transport mechanisms are responsible for the release of eCBs from striatal medium spiny neurons. These findings shed new light on the mechanisms by which transient eCB formation participates in the induction of long-lasting changes in synaptic efficacy that could contribute to brain information storage.
Keywords: basal ganglia, cannabinoids, presynaptic, retrograde, transporter, glutamate
Introduction
The principal psychoactive constituent of marijuana is Δ9-tetrohydrocannabinol, a ligand for the CB1 cannabinoid receptor. CB1 couples primarily with Gi/Go G-proteins, and its activation produces presynaptic depression of neurotransmission at a number of different synapses (for review, see Alger, 2002; Freund et al., 2003). Endogenous ligands for these receptors [endocannabinoids (eCBs)], including anandamide (AEA) and 2-arachidonylglycerol (2-AG), are synthesized from phospholipid precursors embedded in the membrane and are released in a nonvesicular manner (Cadas et al., 1996; Stella et al., 1997). There is mounting evidence that eCBs play a major role not only in regulation of basal transmission but synaptic plasticity as well. Indeed, eCB action at the CB1 receptor has been implicated in different forms of synaptic plasticity involving the coupling of postsynaptic activation with decreases in presynaptic release probability (retrograde signaling). These forms of plasticity include depolarization-induced suppression of inhibitory (DSI) and excitatory transmission (Kreitzer and Regehr, 2001; Wilson and Nicoll, 2001) and several types of long-term depression (LTD) at both excitatory and inhibitory synapses (for review, see Gerdeman and Lovinger, 2003).
Convergent glutamatergic inputs from the cortex impinge on the dendrites of striatal medium spiny neurons (MSNs), the principal output neurons of the striatum (Graybiel, 1990; Calabresi et al., 1996). High-frequency activation of MSNs causes a long-lasting decrease in synaptic efficacy, or LTD (Calabresi et al., 1992; Lovinger et al., 1993). Activity-dependent LTD is important not only for striatal-based memory and habit learning (for review, see Jog et al., 1999; Gerdeman et al., 2003) but also for the postnatal development of corticostriatal synapses (Choi and Lovinger, 1997a; Tang et al., 2001). Although LTD induction requires postsynaptic depolarization and an increase in postsynaptic calcium, LTD expression is tightly associated with a decrease in glutamate release probability (Choi and Lovinger, 1997a,b). LTD induction is prevented by blocking both group I metabotropic glutamate receptors (Gubellini et al., 2001; Sung et al., 2001) and D2 dopamine receptors (D2Rs) (Calabresi et al., 1997; Tang et al., 2001). Interestingly, D2R activation in the striatum has been shown to induce de novo synthesis of AEA, a calcium-dependent event (Giuffrida et al., 1999). Moreover, we recently demonstrated that LTD was absent in striatal slices made from CB1 knock-out mice and slices pretreated with a selective CB1 antagonist (Gerdeman et al., 2002). This is a feature shared with LTD at excitatory synapses in the nucleus accumbens (Robbe et al., 2002) and neocortical layer 5 (Sjöström et al., 2003) and LTD at inhibitory synapses in the amygdala (Marsicano et al., 2002) and hippocampus (Chevaleyre and Castillo, 2003). There is strong evidence that these cannabinoid-dependent forms of LTD involve eCBs as retrograde messengers.
However, a number of issues concerning retrograde signaling by eCBs in striatal LTD remain unresolved. The timing of eCB release and CB1 activation relative to LTD induction and expression is unknown. In addition, the mechanism of postsynaptic eCB release is unknown. The existence of an AEA membrane transporter (AMT) has been postulated to explain cellular uptake of both AEA (Beltramo et al., 1997) and 2-AG (Bisogno et al., 2001). The AMT seems to act via facilitated diffusion and, therefore, should mediate bidirectional transport (Hillard et al., 1997). We have used AMT inhibitors (apparent competitive substrates for the AMT) as a first test of the possibility that the AMT is involved in postsynaptic release of eCBs during retrograde signaling involved in striatal LTD.
Materials and Methods
Brain slice preparation. Brain slices containing both striatum and cortex were prepared, as described previously (Gerdeman et al., 2002), from postnatal day (P) 15-21 Sprague Dawley rats (Charles River Laboratories, Wilmington, MA). For eCB loading experiments, animals ranged in age from P13 to P17 to ensure comparable paired-pulse ratios (PPRs) in vehicle-loaded cells (Choi and Lovinger, 1997a). For DSI experiments, hippocampal slices were made from P16-P21 rats. Animals were killed by decapitation, and the brains were transferred rapidly to ice-cold modified artificial CSF (aCSF) containing (in mm): 194 sucrose, 30 NaCl, 4.5 KCl, 1 MgCl2, 26 NaHCO3, 1.2 NaH2PO4, and 10 d-glucose. Modified aCSF was brought to pH 7.4 by aeration with 95% O2/5% CO2. Coronal sections (350 μm thick) were cut in ice-cold modified aCSF using a manual vibroslice. Slices were transferred immediately to a nylon net submerged in normal aCSF containing (in mm): 124 NaCl, 4.5 KCl, 2 CaCl2, 1 MgCl2, 26 NaHCO3, 1.2 NaH2PO4, and 10 d-glucose. Normal aCSF was maintained at pH 7.4 by bubbling with 95% O2/5% CO2 at room temperature (19-21°C), and osmolarity was adjusted to 330 mOsm. After at least a 30 min incubation at room temperature, hemislices were transferred to a recording chamber, submerged in normal aCSF containing 25 μm picrotoxin (to prevent EPSC contamination with GABAA receptor-mediated responses), and maintained at a temperature between 29 and 32°C stable within ±1°C during any given experiment.
Whole-cell voltage-clamp recording. Whole-cell recordings from MSNs were performed as described previously (Gerdeman et al., 2002). We performed all of the striatal recordings in this study in the dorsolateral striatum, where the predominant form of high-frequency stimulation (HFS)-induced synaptic plasticity is LTD (Partridge et al., 2000). Pipettes were made from borosilicate glass capillaries pulled on a micropipette puller (Flaming-Brown, Novato, CA). Test stimuli were delivered via a Master-8 stimulator (A.M.P.I., Jerusalem, Israel) at a frequency of 0.05 Hz through a bipolar twisted tungsten wire that was placed in the white matter dorsal to the lateral striatum. Pipette resistance ranged from 2.5 to 5 MΩ, when filled with an internal solution (in mm: 120 cesium methane sulfonate, 5 NaCl, 10 tetraethylammonium chloride, 10 HEPES, 4 lidocaine N-ethyl bromide, 1.1 EGTA, 4 Mg-ATP, and 0.3 Na-GTP, pH adjusted to 7.2 with CsOH), and osmolarity was set to 298 mOsm with sucrose. Internal solution was filtered using a nylon (0.45 μm pore size) 4 mm syringe filter. Recordings were made from medium-sized cells with the aid of differential interference contrast-enhanced visual guidance from neurons three to four layers below the slice surface. Cells were voltage clamped at -70 mV during the test period before and subsequent to HFS. The stimulus intensity was set to the level at which EPSC amplitude was 200-400 pA, unless noted otherwise. The HFS protocol used to induce LTD was paired with depolarization of the postsynaptic cell to 0 mV and consisted of four 1 sec 100 Hz trains delivered every 10 sec. Series resistance was <10 MΩ and was uncompensated. Both series and input resistances were monitored throughout the experiment by measuring the capacitive transient amplitude after a 5 mV voltage step. If series resistance changed by >20% during the course of an experiment, the cell was discarded.
Synaptic currents were recorded with an Axopatch 1D amplifier (Axon Instruments, Foster City, CA), filtered at 5 kHz, digitized at 10 kHz, and stored on a microcomputer (Dell, Round Rock, TX). EPSC amplitudes were examined using peak detection software in pCLAMP8 (Axon Instruments). The PPR was determined by calculating the ratio of the amplitude of the second EPSC peak to that of the first EPSC. For experiments involving HFS, the amplitude of the response over the 10 min before HFS was averaged and compared with the average amplitude of the response 20-30 min after HFS using a paired t test, in which p < 0.05 was considered statistically significant. For eCB loading experiments, stimulus intensity was set to yield an EPSC amplitude of ∼200 pA. The mean PPR from 10 traces for each cell was compiled with the means of other cells for each condition, and the mean of that group was compared with the vehicle-loaded cell group using an unpaired t test, in which p < 0.05 was considered statistically significant.
For DSI experiments, CA1 pyramidal neurons were voltage clamped at -60 mV with an internal solution containing (in mm): 150 CsCl, 10 HEPES, 1 MgCl2, 0.2 BAPTA, 5 lidocaine N-ethyl bromide, 3.0 Mg-ATP, and 0.3 Na-GTP. The NMDA receptor antagonist AP-5 (25 μm) and the AMPA receptor antagonist NBQX (5 μm) were included in the external solution. Test stimuli were given every 3 sec via a twisted tungsten bipolar electrode placed in the stratum radiatum. DSI was induced with a 4 sec voltage step to 0 mV. The average amplitude of the two IPSCs immediately after the step was compared with the average amplitude of the seven IPSCs preceding the step.
Field potential recording. Extracellular recordings were obtained with pipettes (1 MΩ) filled with a 0.9% NaCl solution. Stimulus intensity was set to yield an evoked population spike (PS) amplitude that was approximately one-half the amplitude of the maximal evoked response. Maximum responses were 1.0-2.5 mV and were typically evoked with stimulus intensities ranging from 0.5 to 1 mA at a 0.04-0.15 msec duration.
Pharmacology. SR14716A (National Institute of Mental Health Chemical Synthesis and Drug Supply Program, Bethesda, MD) and WIN22,515-2 (Tocris, St. Louis, MO) were diluted from DMSO (10 mm) stock solutions in aCSF with 25 μm picrotoxin (Sigma, St. Louis, MO) and 0.05% BSA as carrier. For extracellular application, VDM11 (10 μm; Tocris) was similarly diluted from a Tocrisolve100 (9 mg/ml) stock solution. In slices pretreated with drug, no carrier was used, and the slices were preincubated with drug in the recording chamber for at least 30 min. For internal AMT inhibitor experiments, VDM11 (10 μm; provided by V. Di Marzo, Endocannabinoid Research Group, Institute of Biomolecular Chemistry, Pozzuoli, Italy) and AM404 (2 μm for LTD and 10 μm for eCB loading experiments; Tocris) were diluted from DMSO stock solutions (50 and 10 mm, respectively). For plasticity experiments, the internal solution contained no more than 0.02% DMSO, because previous experiments indicated that intracellular postsynaptic application of 0.1% DMSO blocked striatal LTD. For eCB loading experiments, AEA (50 μm; Tocris) and 2-AG (50 μm; Tocris) were also diluted from DMSO stock solutions (25 mm). For DSI experiments, dl-AP-5 (Tocris) and NBQX disodium salt (Tocris) were diluted from water stock solutions in aCSF.
Results
CB1 activation is necessary for induction, but not maintenance, of striatal LTD
As demonstrated previously, HFS delivered to the white matter dorsal to the striatum, coupled with postsynaptic depolarization, results in a long-lasting decrease in EPSC amplitude (striatal LTD) (Lovinger et al., 1993) and an increase in the PPR (Choi and Lovinger, 1997a). We had previously shown that activation of CB1 is essential for this form of striatal LTD (Gerdeman et al., 2002). However, it was not known whether continued activation of CB1 is necessary for maintaining striatal LTD. Furthermore, the timing of eCB signaling necessary for induction or for expression of striatal LTD had not been characterized. To determine the period during which CB1 must be activated to achieve LTD expression, we applied the CB1 antagonist SR141716A (3 μm) at different time points relative to HFS. Previously, we had shown that application of the antagonist beginning 40-120 min before HFS, and continuing until 5-10 min after HFS, prevented LTD induction (Gerdeman et al., 2002). If SR141716A was applied beginning 1 min after HFS, only short-lasting depression of transmission was observed, and no decrease in EPSC amplitude was seen at 20-30 min after HFS (EPSC amplitude, 102.4 ± 7.7% of baseline; n = 6) (Fig. 1). Application of SR14176A beginning ∼3 min after HFS impaired LTD measured 20-30 min after HFS (Fig. 1) (EPSC amplitude, 82.4 ± 14.9% of baseline; n = 8) (Fig. 1). However, the effects of antagonist application at this time point were variable, with five slices showing LTD averaging 59.2 ± 3.7% of baseline, one cell showing no change relative to baseline, and two cells showing apparent LTP (161.4% of baseline). When SR141716A was applied 10 min after HFS, the depression of EPSC amplitude was maintained (EPSC amplitude, 57.7 ± 4.5% of baseline; n = 5) (Fig. 1). These findings suggest that release of eCBs and subsequent CB1 activation necessary for LTD induction occurs in a time window during and just after HFS and postsynaptic depolarization. Furthermore, maintenance of LTD tens of minutes after HFS does not require continued CB1 activation.
Figure 1.

CB1 activation is required for induction, but not maintenance, of striatal LTD. After obtaining a stable EPSC for at least 10 min, HFS was paired with a postsynaptic depolarization (black arrow). This stimulation protocol resulted in a long-lasting decrease in EPSC amplitude that was unchanged when the CB1 antagonist SR141716A (3 μm) was bath applied 10 min after HFS (♦). LTD was blocked when SR141716A was applied 1 min after HFS (•), whereas variable results were obtained when SR141716A was applied ∼3 min after HFS (□). Points are the averaged values over 1 min. Bottom, Thirty trace averages of EPSCs recorded 10 min before (left) and 20-30 min after (right) HFS. Left, SR141716A applied 1 min after HFS. Right, SR141716A applied 10 min after HFS. Calibration: 100 pA, 25 msec.
Absence of striatal LTD when an AMT inhibitor is applied intracellularly but not extracellularly: eCB release via the AMT
The mechanism whereby eCBs are released from the postsynaptic cell to act on CB1 receptors found presynaptically is presently unknown. Uptake of eCBs across cellular membranes seems to occur via a process of facilitated diffusion (Beltramo et al., 1997; Hillard et al., 1997), mediated by an AMT that is blocked pharmacologically by compounds such as VDM11 (De Petrocellis et al., 2000) and AM404 (Beltramo et al., 1997). Because these AMT inhibitors have been characterized as competitive substrates for the AMT (Piomelli et al., 1999), we reasoned that synaptic efflux of eCBs might be sensitive to intracellular blockade by AMT inhibitors. We, therefore, examined the possibility that reverse transport through the AMT mediates postsynaptic release of eCBs during retrograde signaling. AMT inhibitors (VDM11 and AM404) were loaded into the postsynaptic neuron via the patch pipette in an effort to prevent transport of eCBs out of this cell. Intracellular loading with VDM11 (10 μm) prevented the HFS-induced decrease in EPSC amplitude (115.6 ± 8.7% of baseline; n = 8) (Fig. 2a). Loading the cell with VDM11 had no effect on baseline EPSC amplitude in the absence of HFS (93.0 ± 3.0% of baseline; n = 5), demonstrating that intracellular loading of VDM11 alone does not alter EPSC amplitude during the course of the experiment. LTD was also blocked in cells loaded with AM404 (2 μm) (122 ± 18.7% of baseline; n = 5) (Fig. 2b). In cells loaded with vehicle alone (0.02% DMSO), LTD was readily induced (66.4 ± 3.7% of baseline; n = 8) (Fig. 2A). Extracellular application of VDM11 did not affect baseline EPSC amplitude in the striatum (112.0 ± 5.4%; n = 4). Pretreatment of slices extracellularly with VDM11 (10 μm) for at least 30 min before HFS neither blocked nor enhanced HFS-induced synaptic depression (71.0 ± 5.7% of baseline; n = 7) (Fig. 2c), demonstrating that release of eCBs is more sensitive to intracellular than extracellular AMT blockade. In addition, we wanted to determine whether intracellular application of VDM11 blocked expression of DSI, a CB1-dependent short-lasting plasticity, in hippocampal CA1 pyramidal neurons. We could evoke IPSCs of 0.7-2.4 nA amplitude in VDM11-loaded neurons. Robust DSI was observed in these cells (IPSC amplitude, 45.7 ± 8.1% of baseline; n = 6) and was completely reversible. The magnitude and time course of the inhibition were comparable with DSI described previously in the literature (Wilson and Nicoll, 2001; Chevaleyre and Castillo, 2003).
Figure 2.

Intracellular, but not extracellular, application of AMT inhibitors blocks striatal LTD. a, Including the AMT inhibitor VDM11 (10 μm) intracellularly via the patch pipette blocked LTD (•), whereas a similarly included vehicle (0.02% DMSO) had no effect (○). Top, Sample traces for VDM11 inside (top) and vehicle inside (bottom); traces are as in Figure 1. b, LTD was also blocked when AM404 (2 μm) was included intracellularly. Top, Traces are as in Figure 1. c, Slices pretreated extracellularly with VDM11 (10 μm) expressed LTD. Top, Traces are as in Figure 1. Calibration: 100 pA, 25 msec. d, PPR of EPSCs 20-30 min after HFS relative to baseline (percentage). Vehicle (Veh)-loaded cells and cells pretreated extracellularly with VDM11 (VDM11 out) exhibited enhanced PPR after HFS. Cells loaded intracellularly with either VDM11 (VDM11 in) or AM404 (AM404 in) did not exhibit this PPR increase.
In addition to EPSC amplitude, we examined the PPR of evoked EPSCs (eEPSCs) before and after HFS using paired stimuli separated by 50 msec. Changes in the PPR are often indicative of changes in the probability of release. Typically, the PPR increases after HFS, and we did indeed observe increases in the PPR after HFS in cells loaded with vehicle (120.6 ± 6.4% of baseline; p < 0.0001; n = 8) (Fig. 2d) or cells pretreated with extracellular 10 μm VDM11 (120.1 ± 10.3%; p < 0.0001; n = 7 of 8) (Fig. 2d). However, intracellular application of 10 μm VDM11 (99.4 ± 6.8%; p > 0.05; n = 8) or 2 μm AM404 (102.0 ± 4.0%; p > 0.05; n = 5) (Fig. 2d) prevented the HFS-induced increase in the PPR. There were no differences among conditions in the baseline PPR measured before HFS (see Fig. 5). We can rule out any changes in striatal fast GABAergic transmission (which also are also sensitive to inhibition via presynaptic CB1 receptors) (Szabo et al., 1997) because an antagonist for GABAA receptors (picrotoxin) was included in the perfusion solution. Taken together, these findings demonstrate that striatal LTD, and the decrease in presynaptic glutamate release accompanying LTD expression, are disrupted by postsynaptic loading of drugs that interfere with eCB transport.
Figure 5.

Effects of intracellular loading with AEA or 2-AG are blocked with coloading of an AMT inhibitor. a, b, Including AEA or 2-AG in the postsynaptic neuron was associated with an increase in the PPR. This increase was blocked when VDM11 (10 μm) was also included in the pipette but not when VDM11 was applied extracellularly. Top, Ten trace average of EPSCs recorded when AEA (a) or 2-AG (b) was coloaded with VDM11 (left) or pretreated extracellularly with VDM11 (right). Calibration: 100 pA, 25 msec. c, Intracellular or extracellular application of VDM11 alone had no effect on the PPR relative to vehicle-loaded cells. This was also true for extracellular application of the VDM11 vehicle Tocrisolve100. d, When stimulus intensities were identical across experiments, the PPR was enhanced in cells loaded with 50 mm AEA, and this increase was blocked with extracellular application of SR141716A and cointracellular loading with AM404 (10 mm). The number of cells is indicated above each bar. *p < 0.05, relative to vehicle-loaded cells.
Synaptic depression produced by intracellular loading of eCBs: prevention by AMT inhibitors
Activation of presynaptic CB1 receptors, during direct agonist application or as a result of LTD induction, decreases neurotransmitter release probability and is associated with an increase in the PPR (Gerdeman and Lovinger, 2001; Gerdeman et al., 2002). Moreover, depression of synaptic transmission and increased PPR were produced when postsynaptic MSNs were loaded with the eCB AEA (Gerdeman et al., 2002), suggesting that eCBs can act as retrograde messengers when high concentrations are present in the postsynaptic neuron. In the present study, we wanted to determine whether 2-AG, in addition to AEA, could signal in this retrograde manner. We performed experiments in which either eCB was included in the postsynaptic neuron (Fig. 3). Response amplitude was set to ∼200 pA to normalize response amplitude and quantal content across experimental groups. When AEA (50 μm; PPR, 1.24 ± 0.06; n = 9) or 2-AG (50 μm; PPR, 1.23 ± 0.06; n = 11) was loaded into the patch pipette, there was a significant increase in the PPR relative to cells loaded with vehicle alone (0.2% DMSO; PPR, 0.93 ± 0.05; n = 12) (p < 0.001 and p < 0.01, respectively). Pretreatment (30-90 min) of slices with extracellular SR141716A (3 μm) blocked this increase in the PPR (AEA PPR: 0.98 ± 0.03 n = 6; 2-AG PPR, 0.92 ± 0.07, n = 10), demonstrating involvement of a CB receptor.
Figure 3.

PPR increases in cells loaded intracellularly with AEA or 2-AG. Vehicle-loaded cells (Veh; 0.2% DMSO) had a low PPR, consistent with PPR values obtained previously from MSNs at this age (Choi and Lovinger, 1997A). Intracellular loading with AEA or 2-AG (50 μm) was associated with a significant increase in the PPR, whereas similarly loaded cells in slices pretreated with the CB1 antagonist SR141716A (3 μm) did not exhibit this PPR increase. SR141716A alone did not significantly affect the PPR. The number of cells is indicated above each bar. *p < 0.05, relative to vehicle. Calibration: 100 pA, 25 msec.
We also wanted to determine whether CB1 receptor activation was sufficient to produce LTD. If LTD were produced by agonist application alone, then one would expect agonist-induced synaptic depression to be resistant to reversal by antagonist applied several minutes after the onset of the full antagonist effect (as was the case for HFS-induced LTD shown in Fig. 1). As can be seen in Figure 4a, application of SR141716A reversed depression of synaptic responses produced by extracellular application of the CB1 agonist WIN55,212-2 (WIN55,212-2, 56.9 ± 4.7%; SR141716A, 103.9 ± 18.2% baseline PS; n = 5). Also, when the eCB AEA was loaded into the postsynaptic cell, subsequent extracellular application of SR141716A produced an increase in EPSC amplitude (161.4 ± 13.8% baseline; n = 4) (Fig. 4b). In these experiments, the PPR also decreased in three of four neurons examined (85.2 ± 2.2% of baseline), and the one cell in which the PPR did not increase did not show a marked increase in EPSC amplitude. Application of SR141716A to neurons not loaded with AEA did not increase synaptic response amplitude (100.6 ± 4.9% of baseline; n = 4) (Fig. 4b). These findings indicate that agonist activation of CB1 receptors depresses transmission, and this decrease persists for as long as the receptor is occupied, but agonist treatment is not sufficient to induce LTD.
Figure 4.
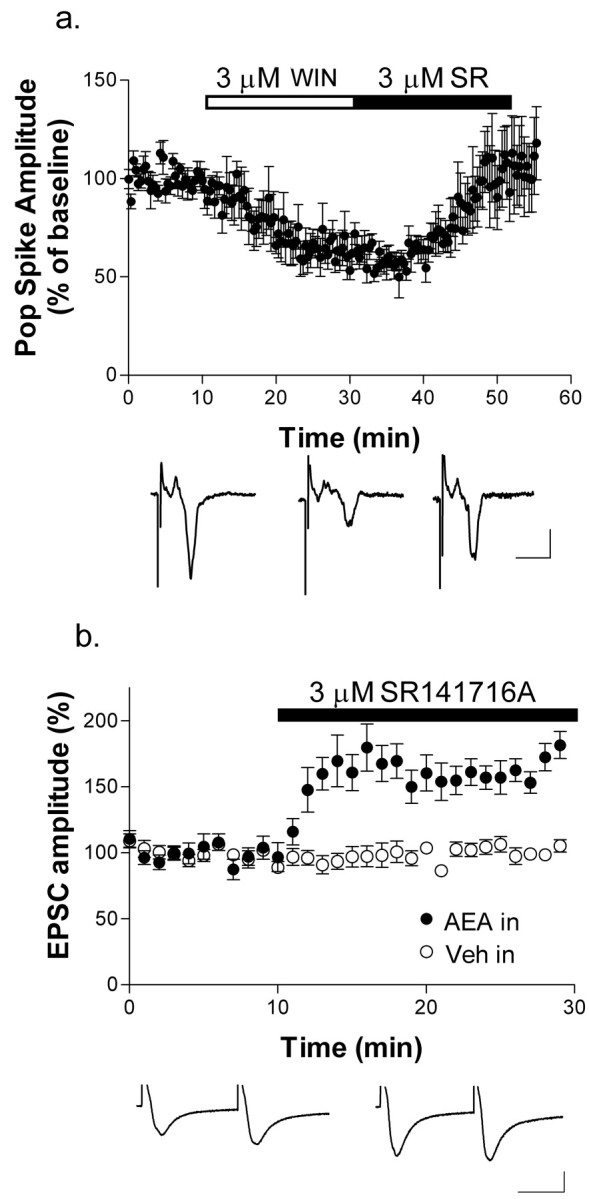
CB1 activation alone is not sufficient to induce striatal LTD. a, Extracellular application of the CB1 agonist WIN55,212-2 (WIN; 3 μm) caused a significant decrease in PS amplitude. Subsequent bath application of SR141716A (SR; 3 μm) reversed this inhibition. Calibration: 0.2 mV, 10 msec. b, Extracellular bath application of SR141716A subsequent to intracellular AEA (50 μm) loading caused an increase in EPSC amplitude (•). SR141716A alone had no effect on baseline EPSC amplitude (○). Calibration: 200 pA, 25 msec.
To further address the role of the AMT as a mediator of eCB efflux, we examined the effect of loading postsynaptic neurons with AMT inhibitors on the increased PPR produced by postsynaptically loaded AEA and 2-AG. When the AMT inhibitor VDM11 (10 μm) was included in the pipette with 50 μm AEA, there was no increase in the PPR, in contrast to what was observed with AEA alone (AEA plus VDM11 PPR, 1.0 ± 0.04; p > 0.05 relative to AEA alone; n = 10) (Fig. 5a).Similar results were obtained when cells were loaded with 50 μm 2-AG (2-AG plus VDM11 PPR, 0.90 ± 0.11; p > 0.05 relative to 2-AG alone; n = 7) (Fig. 5b). We also wanted to determine whether VDM11 blockade of eCB efflux was specific for intracellularly loaded AMT inhibitor, as was the case for LTD. Thus, in another experiment, we found that brain slices pretreated extracellularly with VDM11 (10 μm; 30-90 min) and cells loaded intracellularly with eCBs still exhibited an increase in the PPR relative to vehicle-loaded cells. PPRs averaged 1.26 ± 0.10 (p < 0.01 relative to the vehicle group; n = 7) for AEA plus extracellular VDM11 and 1.19 ± 0.03 (p < 0.01 relative to the vehicle group; n = 6) for 2-AG plus extracellular VDM11 (Fig. 5a,b). Cells with VDM11 only intracellularly or extracellularly showed no change in the PPR relative to vehicle controls [VDMin: PPR = 0.91 ± 05, p > 0.05; VDMout: PPR = 0.95 ± 0.08, p > 0.05; n = 10) (Fig. 5c). This finding indicates that blockade of AMT does not alter synaptic transmission in the absence of postsynaptic eCB.
In the previous experiments, stimulus intensities were adjusted such that the first of the paired EPSCs had a peak amplitude of ∼200 pA. However, when recordings were made at identical stimulus intensities, without controlling first EPSC amplitude, there was an increase in the PPR as well as an associated decrease in first EPSC peak amplitude in cells loaded with AEA. At this stimulus intensity (1.5 mA, 0.2 msec), AEA-loaded cells exhibited an increase in the PPR (1.2 ± 0.02; n = 14) (Fig. 5d) and a decrease in eEPSC amplitude (44 ± 15 pA; n = 14) (Fig. 5d) relative to vehicle-loaded cells (Tocrisolve100: PPR = 0.93 ± 0.06; 206 ± 58 pA; n = 4) (Fig. 5d). The effects of intracellular AEA under these conditions were prevented by bath application of the CB1 antagonist SR141716A (5 μm) (EPSC amplitude, 737+/-157 pA; PPR, 0.98 ± 0.01; n = 8) (Fig. 5d), as described previously (Gerdeman et al., 2002), and with intracellular loading of the AMT inhibitor AM404 (10 μm) (EPSC amplitude, 262 ± 23 pA; PPR, 0.86 ± 0.04; n = 5) (Fig. 5d). These findings further support the idea that the effects of postsynaptically loaded eCBs can be reversed when eCB transport is inhibited.
Discussion
Although striatal LTD requires postsynaptic activation, there is strong evidence for a presynaptic expression mechanism, demonstrating the necessity for a retrograde messenger. In this study, we have shown that two eCBs, AEA and 2-AG, are capable of being released from MSNs after intracellular loading via the patch pipette. Subsequent activation of CB1 receptors presynaptically reduces the probability of glutamate release, as evidenced by an increase in the PPR. These data support previous findings (Gerdeman et al., 2002) that eCBs can act as retrograde messengers at glutamatergic synapses in striatum.
After HFS of glutamatergic inputs to the striatum, there is a long-lasting decrease in synaptic efficacy that requires release of eCBs and activation of CB1. We have shown that CB1 activation is necessary for induction, but not maintenance, of striatal LTD. The effects of antagonists applied at different times before, during, and after HFS are consistent with the notion that eCB signaling occurs during a limited time window during and for a few minutes after HFS. Comparable findings were demonstrated for heterosynaptic LTD at GABAergic synapses in CA1 of hippocampus (Chevaleyre and Castillo, 2003) and spike-timing-dependent LTD in neocortical layer 5 pyramidal neuron pairs (Sjöström et al., 2003).
The observation that CB1 antagonists reverse agonist-induced synaptic depression but do not alter established LTD (i.e., when applied 10 min after HFS) indicates that CB1 receptor activation is not sufficient to induce LTD. It is possible that another presynaptic signal that coincides with CB1R activation is needed to produce the presynaptic changes necessary to establish LTD. This idea is consistent with the dual role of CB1 and NMDA receptors in visual cortex spike-timing LTD reported by Sjöström et al. (2003). However, NMDA receptors are not involved in striatal LTD (Calabresi et al., 1992; Partridge et al., 2000), and, thus, the coincident signal for LTD at corticostriatal synapses is likely different from the signal involved in visual cortical LTD.
Uptake of eCBs across the cellular membrane occurs via a putative transporter (AMT) by facilitated diffusion and, by this definition, should be reversible. Our observation that AMT inhibitors prevent LTD as well as the effects of postsynaptically loaded eCBs provides evidence consistent with the idea that the AMT is a key player in mediating retrograde signaling in striatal MSNs. Interestingly, we have shown that DSI, another form of CB1-dependent plasticity, is not inhibited with intracellular blockade of the AMT. This suggests that although the reversal of the AMT is a primary process mediating retrograde transport of eCBs for striatal LTD, it does not seem to be so for hippocampal DSI. Other fundamental differences between expression mechanisms of hippocampal inhibitory LTD, another type of LTD, and DSI were described and discussed previously (Chevaleyre and Castillo, 2003).
The magnitude of the changes in the PPR during LTD expression seem to be smaller than the corresponding changes in EPSC amplitude. This is most likely attributable to a nonlinear relationship between the PPR and release probability. One elegant demonstration of this nonlinear relationship was made by Atzori et al. (2001) in pairs of connected principal neurons in layer II/III of the A1 auditory cortex. When probability of release was determined using the method of failures, paired-pulse facilitation was not seen unless release probability was rather low (i.e., ∼0.3). Therefore, it is not surprising that seemingly large changes in EPSC amplitude are associated with relatively small changes in the PPR, because small changes in the probability of release might not be detected as a change in the PPR if initial probability is sufficiently high at particular synapses within the sampled population.
AEA and 2-AG are both AMT substrates, and, thus, it is not surprising that effects of both eCBs are prevented by transport inhibitors. Nonetheless, our findings should be interpreted with due caution. The AMT is not characterized or cloned molecularly, and there is some controversy regarding its existence. It has been argued that AEA uptake by the AMT is actually a function of the activity of fatty acid amide hydrolase (FAAH) (the enzyme responsible for AEA hydrolysis) (Glaser et al., 2003). It was found, at least in certain cell types, that FAAH activity promotes AEA uptake by maintaining the driving force necessary for uptake. In addition, Glaser et al. (2003) showed that certain AMT inhibitors also decrease FAAH activity. However, our findings are not consistent with a role for FAAH inhibition in the actions of intracellularly loaded VDM11 or AM404. These compounds would be expected to inhibit FAAH activity in the postsynaptic neuron, leading to decreased AEA uptake (Glaser et al., 2003) that would, if anything, enhance LTD and the eCB actions. The fact that LTD is prevented by these compounds indicates that FAAH inhibition is not involved. In addition, the observation that actions of postsynaptically loaded 2-AG are prevented by intracellular VDM11 application is inconsistent with a role for FAAH inhibition, given that 2-AG is not thought to be a FAAH substrate in vivo (Dinh et al., 2002). Overall, our data are inconsistent with an inhibition of FAAH by postsynaptic loading of VDM11. Similarly, in primary cortical neuronal cultures, selective inhibitors of FAAH blocked AEA breakdown without affecting uptake via the AMT (Kathuria et al., 2003). This uptake was blocked with extracellular application of AM404, demonstrating a distinct mechanism of action for FAAH inhibitors and AMT inhibitors. Our findings are most consistent with the existence of the AMT and a role for this transporter in retrograde eCB signaling during LTD induction.
It is possible that we are blocking AEA and 2-AG synthesis by intracellular loading of VDM11 or AM404. However, this is unlikely because there is no increase in the PPR in cells loaded intracellularly with both eCB and AMT inhibitor. In this case, impairment of synthesis would not matter because we are supplying a large reserve of eCB via the patch pipette. Additionally, there is no evidence to date that demonstrates any decrease in eCB levels after application of these drugs (and, in fact, there is consistently an increase in extracellular eCB levels) (Giuffrida et al., 2000; Kathuria et al., 2003; Ligresti et al., 2003). In addition to being an inhibitor of the AMT, AM404 is an agonist at the capsaicin-sensitive transient receptor potential vanilloid 1 (TRPV1) (De Petrocellis et al., 2000). That is one reason why we undertook experiments using the more selective AMT inhibitor VDM11, an AMT inhibitor that exhibits lower potency for inhibition of FAAH (IC50, >50 μm) or activation of TRPV1 (De Petrocellis et al., 2000) relative to AM404. The observation that VDM11 has actions similar to AM404 indicates that TRPV1 activation is not involved in actions of these drugs that block LTD and eCB-induced synaptic inhibition. The lack of any role for TRPV1 in LTD is supported by our observations that TRPV1 agonists do not alter synaptic transmission and the TRPV1 antagonist capsazepine does not alter LTD induction (J. Ronesi and D. M. Lovinger, unpublished observations). VDM11 applied extracellularly has no effect on striatal LTD, and, interestingly, bath-applied AM404 actually rescues LTD in cells loaded with the calcium chelator EGTA (Gerdeman et al., 2002). Furthermore, extracellular application of AM404 in the hippocampus inhibits transmission and prevents DSI (Wilson and Nicoll, 2001), consistent with an increase in eCB levels produced by AMT inhibition. The difference in effects of intracellularly and extracellularly applied AMT inhibitors suggests that there may be an intracellular binding site for inhibitors on the transporter that is distinct from the extracellular site. It seems that the direction of the AEA/2-AG concentration gradient determines whether the blocker works from the inside or the outside. If internal concentration is high, then an internal blocker is more effective by competing for the internal AMT binding site, therein blocking binding and release of eCBs. Perhaps there is no effect with extracellular application of AMT inhibitors because there is no appreciable driving force (i.e., no extracellular eCB) to promote accumulation of these molecules intracellularly. Given the limited time frame of our experiments, there may not be enough time for these extracellularly applied inhibitors to accumulate intracellularly and then subsequently block release during and immediately after HFS.
Although our findings are consistent with a role for the AMT in eCB postsynaptic release, it is possible, albeit unlikely, that AM404 and VDM11 inhibit LTD induction through an as yet uncharacterized mechanism. Even if this proves to be the case, the fact that postsynaptic application of these compounds prevents LTD induction further strengthens the model that eCBs released postsynaptically act as retrograde messengers at corticostriatal synapses.
Our findings indicate that eCBs are released postsynaptically, most likely via an AMT-like eCB transporter, after high-frequency activation of corticostriatal synapses. The eCBs seem to have a transient action that interacts with another putative presynaptic signal to induce LTD. Cannabinoid-dependent LTD has now been observed in a number of brain regions and has been linked to fear extinction in the amygdala (Marsicano et al., 2002). This widespread mechanism of synaptic efficacy may well play key roles in refinement of information storage throughout the brain.
Footnotes
This work was supported by the intramural program of the National Institute on Alcohol Abuse and Alcoholism.
Correspondence should be addressed to Dr. David M. Lovinger, Laboratory for Integrative Neuroscience, National Institute on Alcohol Abuse and Alcoholism, 158H Park 5 Building, 12420 Parklawn Drive, Rockville, MD 20852-0615. E-mail: lovindav@mail.nih.gov.
Copyright © 2004 Society for Neuroscience 0270-6474/04/241673-07$15.00/0
References
- Alger B (2002) Retrograde signaling in the regulation of synaptic transmission: focus on endocannabinoids. Prog Neurobiol 68: 247-286. [DOI] [PubMed] [Google Scholar]
- Atzori M, Lei S, Evans DI, Kanold PO, Phillips-Tansey E, McIntyre O, McBain CJ (2001) Differential synaptic processing separates stationary from transient inputs to the auditory cortex. J Neurosci 4: 1230-1237. [DOI] [PubMed] [Google Scholar]
- Beltramo M, Stella N, Calignano A, Lin SY, Makriyannis A, Piomelli D (1997) Functional role of high-affinity anandamide transport, as revealed by selective inhibition. Science 277: 1094-1097. [DOI] [PubMed] [Google Scholar]
- Bisogno T, MacCarrone M, DePetrocellis L, Jarrahian A, Finazziagro A, Hillard C, DiMarzo V (2001) The uptake of cells of 2-arachidonylglycerol, an endogenous agonist of cannabinoid receptors. Eur J Biochem 268: 1982-1989. [DOI] [PubMed] [Google Scholar]
- Calabresi P, Maj R, Pisani A, Mercuri NB, Bernardi G (1992) Long-term synaptic depression in the striatum: physiological and pharmacological characterization. J Neurosci 12: 4224-4233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calabresi P, Pisani A, Mercuri NB, Bernardi G (1996) The corticostriatal projection: from synaptic plasticity to dysfunctions of the basal ganglia. Trends Neurosci 19: 19-24. [DOI] [PubMed] [Google Scholar]
- Calabresi P, Saiardi A, Pisani A, Baik J-H, Centonze D, Mercuri N, Bernardi G, Borrelli E (1997) Abnormal synaptic plasticity in the striatum of mice lacking dopamine D2 receptors. J Neurosci 17: 4536-4544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chevaleyre V, Castillo PE (2003) Heterosynaptic LTD of hippocampal GABAergic synapses. A novel role of endocannabinoids in regulating excitability. Neuron 38: 461-472. [DOI] [PubMed] [Google Scholar]
- Choi S, Lovinger DM (1997a) Decreased probability of neurotransmitter release underlies striatal long-term depression and postnatal development of corticostriatal synapses. Proc Natl Acad Sci USA 94: 2665-2670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi S, Lovinger DM (1997b) Decreased frequency but not amplitude of quantal synaptic responses associated with expression of corticostriatal long-term depression. J Neurosci 17: 8613-8620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Petrocellis L, Bisogno T, Davis JB, Pertwee RG, DiMarzo V (2000) Overlap between the ligand recognition properties of the anandamide transport and the VR1 vanilloid receptor: inhibitors of anandamide uptake with negligible capsaicin-like activity. FEBS Lett 483: 52-56. [DOI] [PubMed] [Google Scholar]
- Dinh TP, Carpenter D, Leslie FM, Freund TF, Katona I, Sensi SL, Kathuria S, Piomelli D (2002) Brain monoglyceride lipase participating in endocannabinoid inactivation. Proc Natl Acad Sci USA 99: 10819-10824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freund TF, Katona I, Piomelli D (2003) Role of endogenous cannabinoids in synaptic signaling. Physiol Rev 83: 1017-1066. [DOI] [PubMed] [Google Scholar]
- Gerdeman GL, Lovinger DM (2001) CB1 cannabinoid receptor inhibits synaptic release of glutamate in rat dorsolateral striatum. J Neurophysiol 85: 468-471. [DOI] [PubMed] [Google Scholar]
- Gerdeman GL, Lovinger DM (2003) Emerging roles for enodcannabinoids in long-term synaptic depression. Br J Pharmacol 140: 781-789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerdeman GL, Ronesi J, Lovinger DM (2002) Postsynaptic endocannabinoids release is critical to long-term depression in the striatum. Nat Neurosci 5: 446-451. [DOI] [PubMed] [Google Scholar]
- Gerdeman GL, Partridge JG, Lupica CR, Lovinger DM (2003) It could be habit forming: drugs of abuse and striatal synaptic plasticity. Trends Neurosci 26: 184-192. [DOI] [PubMed] [Google Scholar]
- Giuffrida A, Parsons LH, Kerr TM, Rodriguez de Fonseca F, Navarro M, Piomelli D (1999) Dopamine activation of endogenous cannabinoids signaling in dorsal striatum. Nat Neurosci 2: 358-363. [DOI] [PubMed] [Google Scholar]
- Giuffrida A, Rodriguez de Fonseca F, Nava F, Loubet-Lescoulie P, Piomelli D (2000) Elevated circulating levels of anandamide after administration of the transport inhibitor, AM404. Eur J Pharmacol 408: 161-168. [DOI] [PubMed] [Google Scholar]
- Glaser ST, Abumrad NA, Fatade F, Kaczocha M, Studholme KM, Deutsch DG (2003) Evidence against the presence of an anandamide transporter. Proc Natl Acad Sci USA 100: 4269-4274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graybiel AM (1990) Neurotransmitters and neuromodulators in the basal ganglia. Trends Neurosci 13: 244-253. [DOI] [PubMed] [Google Scholar]
- Gubellini P, Saulle E, Centzone D, Bonsi P, Pisani A, Bernardi G, Conquet F, Calabresi P (2001) Selective involvement of mGlu1 receptors in corticostriatal LTD. Neuropharmacology 40: 836-846. [DOI] [PubMed] [Google Scholar]
- Hillard CJ, Edgemond WS, Jarrahian A, Campbell WB (1997) Accumulation of N-arachidonoylethanolamine (anandamide) into cerebellar granule cells occurs via facilitated diffusion. J Neurochem 69: 631-638. [DOI] [PubMed] [Google Scholar]
- Jog MS, Kubota Y, Connolly CI, Hillegaart V, Graybiel AM (1999) Building neural representations of habits. Science 286: 1745-1749. [DOI] [PubMed] [Google Scholar]
- Kathuria S, Gaetani S, Fegley D, Valino F, Duranti A, Tontini A, Mor M, Tarzia G, La Rana G, Calignano A, Giustino A, Tattoli M, Palmery M, Cuomo V, Piomelli D (2003) Modulation of anxiety through blockade of anandamide hydrolysis. Nat Med 9: 76-81. [DOI] [PubMed] [Google Scholar]
- Kreitzer AC, Regehr WG (2001) Retrograde inhibition of presynaptic calcium influx by endogenous cannabinoids at excitatory synapses onto Purkinje cells. Neuron 29: 717-727. [DOI] [PubMed] [Google Scholar]
- Ligresti A, Bisogno T, Matias I, De Petrocellis L, Cascio MG, Cosenza V, D'argenio G, Scaglione G, Bifulco M, Sorrentini I, Di Marzo V (2003) Possible endocannabinoid control of colorectal cancer growth. Gastroenterology 125: 677-687. [DOI] [PubMed] [Google Scholar]
- Lovinger DM, Tyler EC, Merritt A (1993) Short and long-term synaptic plasticity in the neostriatum. J Neurophysiol 70: 1937-1949. [DOI] [PubMed] [Google Scholar]
- Marsicano G, Wotjak CT, Azad SC, Bisogno T, Rammes G, Cascio MG, Hermann H, Tang J, Hofmann C, Zieglgansberger W, Di Marzo V, Lutz B (2002) The endogenous cannabinoid system controls extinction of aversive memories. Nature 418: 530-534. [DOI] [PubMed] [Google Scholar]
- Partridge JG, Tang K-C, Lovinger DM (2000) Regional and postnatal heterogeneity of activity-dependent long-term changes in synaptic efficacy in the dorsal striatum. J Neurophysiol 84: 1422-1429. [DOI] [PubMed] [Google Scholar]
- Piomelli D, Beltramo M, Glasnapp S, Lin SY, Goutopoulos A, Xie WQ, Makriyannis A (1999) Structural determinants for recognition and translocation by the anandamide transporter. Proc Natl Acad Sci USA 96: 5802-5807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robbe D, Kopf M, Remaury A, Bockaert J, Manzoni OJ (2002) Endogenous cannabinoids mediate long-term synaptic depression in the nucleus accumbens. Proc Natl Acad Sci USA 99: 8384-8388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sjöström PJ, Turrigiano GG, Nelson CS (2003) Neocortical LTD via coincident activation of presynaptic NMDA and cannabinoid receptors. Neuron 39: 641-654. [DOI] [PubMed] [Google Scholar]
- Sung KW, Choi S, Lovinger DM (2001) Activation of group I mGluRs is necessary for induction of long-term depression at striatal synapses. J Neurophysiol 86: 2405-2412. [DOI] [PubMed] [Google Scholar]
- Szabo B, Dorner L, Pfreundtner C, Norenberg W, Starke K (1997) Inhibition of GABAergic inhibitory postsynaptic currents by cannabinoids in rat corpus striatum. Neuroscience 85: 395-403. [DOI] [PubMed] [Google Scholar]
- Tang K, Low MJ, Grandy DK, Lovinger DM (2001) Dopamine-dependent synaptic plasticity in striatum during in vivo development. Proc Natl Acad Sci USA 98: 1255-1260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson RI, Nicoll RA (2001) Endogenous cannabinoids mediate retrograde signaling at hippocampal synapses. Nature 410: 588-592. [DOI] [PubMed] [Google Scholar]