

Abstract
A number of viruses and bacterial species have been implicated as contributors to atherosclerosis, potentially providing novel pathways for prevention. Epidemiological studies examining the association between Helicobacter pylori and cardiovascular disease have yielded variable results and no studies have been conducted in nonhuman primates. In this investigation, we examined the relationship between H. pylori infection and atherosclerosis development in socially housed, pre- and postmenopausal cynomolgus macaques consuming human-like diets. Ninety-four premenopausal cynomolgus monkeys (Macaca fascicularis) were fed for 36 months an atherogenic diet deriving its protein from either casein lactalbumin(CL) or high isoflavone soy (SOY). Animals were then ovariectomized and fed either the same or the alternate diet for an additional 36 months. Iliac artery biopsies were obtained at the time of ovariectomy and iliac and coronary artery sections were examined at the end of the study. Evidence of H. pylori infection was found in 64% of the monkeys and 46% of animals had live H. pylori within coronary atheromas as determined by mRNA-specific in situ hybridization. There was a significant linear relationship between the densities of gastric and atheroma organisms. Helicobactor pylori infection correlated with increased intimal plaque area and thickness at both the premenopausal and postmenopausal time points and regardless of diet (p< 0.01), although animals consuming the SOY diet throughout had the least amount of atherosclerosis. Additionally, plasma lipid profiles, intimal collagen accumulation, ICAM-1, and plaque macrophage densities were adversely affected by H. pylori infection among animals consuming the CL diet, while the SOY diet had the opposite effect. Plaque measurements were more highly associated with the densities of cagA-positive H. pylori within coronary atheromas than with the densities of gastric organisms, whereas plasma lipid changes were associated with H. pylori infection, but not cagA status. This study provides strong evidence that live H. pylori infects atheromas, exacerbates atherosclerotic plaque development, and alters plasma lipid profiles independently of diet or hormonal status. Finally, socially subordinate animals relative to their dominant counterparts had a greater prevalence of H. pylori, suggesting a stress effect. The results indicate that early H. pylori eradication could prevent or delay development of cardiovascular disease.
Introduction
Prevention of atherosclerosis–the pathobiological process that underlies cardiovascular disease (CVD)–has focused primarily on modifiable risk factors such as smoking, obesity, hypertension, and diet [1]. The hypothesis that chronic infections promote atherosclerosis is more controversial, even though acute infection has been recognized previously as a major contributor to atherosclerosis. For example, in the early 1900s, physicians noted that even young people who died from typhoid fever or other infections frequently had atherosclerotic plaques, whereas those dying from other causes did not [2]. More recently, correlative evidence between atherosclerosis and chronic bacterial and viral infections has been uncovered, or perhaps rediscovered. Potential contributors include periodontal disease, Herpes Simplex virus, Epstein-Barr virus, cytomegalovirus, Chlamydia pneumoniae, and H. pylori [3, 4].
H. pylori is a Gram negative, spiral or curved bacterium that colonizes the gastric mucosa of humans and other primates. Importantly, H. pylori is not an acidophile; it can only survive acidic conditions temporarily and must exit the acidic gastric lumen and enter the mucous layer, which has a neutral pH, in order to survive and grow [5–7]. Although H. pylori is best known for causing peptic ulcers and gastric cancer, it also has been associated with a number of extragastric disease processes, including atherosclerosis (reviewed in [8, 9]). This is not surprising in view of evidence that H. pylori colonizes extragastric tissues, as the bacteria have been cultured from the middle ear and liver and detected by immunohistochemistry in skin as well as nasal polyps [10–13]. However, elucidation of a role for H. pylori in extragastric diseases is complicated by the fact that H. pylori is genetically heterogeneous and some virulence factors are not universally present. Thus, only certain H. pylori strains may contribute to specific types of disease pathology. The major virulence factor of H. pylori is the CagA (Cytotoxin Associated Gene A) cytotoxin that is injected into host cells and alters host cell signaling pathways [14]. H. pylori strains that possess the cagA gene are associated with a greater risk of ulcers and gastric cancer relative to strains that do not [15, 16]; here we assess the degree to which CagA may also contribute to atherosclerosis and, by extension, CVD.
Early epidemiological studies that examined the potential for H. pylori to contribute to CVD yielded conflicting results [17–20]. However, recent studies increasingly suggest that H. pylori, and particularly cagA-positive H. pylori strains, may accelerate the development of atherosclerosis and the resulting CVD [21–25]. Finally, although H. pylori DNA has been detected in atherosclerotic plaques by numerous investigators using PCR[26–30], it remains uncertain whether the bacteria in these plaques are intact or viable.
CVD is the leading cause of death in women over 50 [31]. Improved prevention and treatment strategies have reduced CVD incidence in older individuals, but not among women under age 55 [32]. Premenopausal women have lower rates of CVD than similarly aged men do, yet the role of estrogen in cardioprotection remains unclear [31]. For these reasons, studies in women or suitable female animals are essential for clarifying CVD risk factors in young and older women.
Cynomolgus monkeys (Macaca fasciularis) have been used extensively to model the development of atherosclerosis in relation to diet, hormonal conditions, psychosocial factors, and pharmaceutical manipulations [33]. Atherosclerosis in this species closely mimics the pattern observed in human beings, including the relative inhibition of disease progression in reproductively mature females compared to males, the extensive involvement of the coronary and carotid arteries, and the exacerbating effect of traditional risk factors (e.g., hypertension and a diet high in cholesterol and saturated fat) as well as psychosocial stress [34–36]. Finally, responses to a wide variety of pharmaceutical interventions also resemble those observed in people [37].
The purpose of the present study was to determine in this well-established monkey model whether infectious agents–including H. pylori and a number of viruses–influence atherosclerosis and whether other risk factors, such as diet, reproductive hormones, and psychosocial stress modify any infection-associated effects. Hence, we measured atherosclerosis and plasma lipid composition in pre- and postmenopausal cynomologus macaques following consumption of diets that derived their protein from either animal (casein lactalbumin, CL) or high isoflavone soy isolate (SOY). Most notably we found that H. pylori, particularly cagA-positive H. pylori, positively correlates with atherosclerosis lesion size and dyslipidemia irrespective of diet treatment, whereas viral infections did not correlate with indices of atherosclerosis extent or severity. Psychosocial stress, as indicated by subordinate social status, was associated with an elevated prevalence of H. pylori.
Materials and methods
Animals and study design
The research described in this manuscript (including the acquisition of monkeys and all protocols for their use) was approved by the Wake Forest School of Medicine Institutional Animal Care and Use Committee (Protocols A97-157, A01-195, and A04-116, encompassing approximately eight years of in vivo investigation preceded by a one year run in and a one year close down). Wake Forest is an AALAC accredited institution and all animal care procedures followed the NIH guide and the Principles for the Ethical Treatment of Non-human Primates of the American Society of Primatologists. In addition to the foregoing, the research described here was conducted in accordance with the standard operating procedures of the Wake Forest Environmental Enrichment Program, which involves the provision of cage devices and structures that include at a minimum hanging devices, mirrors, and perches in each pen, and regular surveillance under a behavioral management program designed to identify and mitigate atypical behavior. Finally, all animals were housed continuously in stable social groups–a situation designed to maximize the opportunity to engage in normal, species typical behavior. Macaques were fed twice per day with sufficient food that some remained uneaten.
Ninety-four (94) monkeys were randomized into the treatment conditions for this study. Eighteen (18) died over the following seven years of in-vivo investigation and an additional 2 monkeys became diabetic and had to be removed from the study. This meant that seventy-four monkeys survived until experimental necropsy, which for all cases involved use of sodium pentobarbital to induce euthanasia.
Of the 18 in-study deaths, 2 occurred in year two, 5 in year three, 2 in year four, 6 in year five, 1 in year six, and 2 in year seven. A complete anatomic and histopathologic necropsy was performed on each dead animal and a specific cause of death could be determined in 15 of the 18 cases. Causes of death were varied and included gastric dilation (2 cases), bladder carcinoma, acute cardiac arrest, congestive heart failure, oligodendroglioma, hypoglycemia, hyperglycemia, arterial disease, right atrial thrombosis, and intraoperative hypoxia. There were no deaths due to trauma among the animals in this study. This number of deaths and the development of diabetes is not unusual in a study that persists for almost eight years in-vivo, involves consumption of a human-like diet, and includes a major surgery (oophorectomy) at the midpoint (years four and five).
All surgery and euthanasia was performed under aseptic conditions using sodium pentobarbital anesthesia to minimize suffering. Specifically, this investigation was categorized as USDA class D (animals upon which experiments, teaching, research, surgery, or tests are conducting involving accompanying pain or distress to the animals and for which appropriate anesthetic, analgesic, or tranquilizing drugs are provided). Survival surgery was initiated with ketamine HCl (10–15 mg/kg, IM) followed by butorphanol (0.25 mg/kg, IM). After surgery, all animals were returned to single cages and monitored at 30 minute intervals until they were sitting unaided. Butorphanol was administered for pain relief. Hunched posture, lack of appetite, or different from that typically observed after anesthesia for nonsurgical procedures was interpreted as signs of discomfort or pain. Nonsurgical procedures (including blood and fluid sampling, bone density scans) were conducted using ketamine HCl restraint. Animals were observed from the time of anesthesia until they were fully capable of sitting unaided. Procedures were generally conducted in the A.M. Further monitoring (after the animal was sitting unaided) was done on an hourly basis. Veterinary care was sought if any of the aforementioned signs of distress were observed following surgical or non-surgical procedures.
A crossover study was conducted as previously described (Fig 1) [38]. Briefly, adult female cynomolgus macaques (Macaca fascicularis) living in permanent social groups (n = 5 or 6 per group x 16 groups; N = 94) were randomized to receive either an atherogenic diet containing casein and lactalbumin (CL) as the protein source or an otherwise identical diet that substituted high-isoflavone soy protein isolate (SOY) for the animal protein. Both diets were formulated to be equivalent in caloric content of protein, fat, carbohydrates and cholesterol (0.28 mg cholesterol/Cal) and to mimic a typical diet consumed by women in the United States [39]. After 36 months, the animals were ovariectomized to induce menopause and a segment of the left common iliac (LCI) artery was removed to measure premenopausal atherosclerosis. Half of the social groups were then randomly selected to receive the alternate diet, while the remaining social groups continued with the original diet. The four resulting diet groups were designated CL-CL (casein/lactalbumin only), CL-SOY, (casein/lactalbumin followed by high isoflavone soy), SOY-CL (soy followed by casein/lactalbumin), or SOY-SOY (soy isolate only). The postmenopausal phase continued for an additional 36 months. Using procedures developed in numerous prior studies [40, 41], animals within each social group were categorized on the basis of competitive interactions as being socially dominant (n = 2 or 3/group) or subordinate (n = 3/group); thus approximately half of the animals were considered dominant and half subordinate.
Fig 1. Study design.

Cynomolgus macaques received either SOY or CL diets for 36 months, at which point animals were ovariectomized and a left common iliac segment was removed. Half of the animals received the alternate diet for an additional 36 months. The number (n) of surviving animals that could be classified as H. pylori-positive or–negative (70 out of 74) is given for each group.
Plasma lipid measurement
Total plasma cholesterol (TPC), high-density lipoprotein-associated cholesterol (HDLc), and plasma triglycerides (TGs) were determined on numerous occasions as well as at the end of the study by the Wake Forest Primate Center Clinical Chemistry Laboratory [42, 43], which is fully standardized with the Centers for Disease Control and Prevention-National Institutes of Health Lipid Standardization Program specifications for accuracy and precision. Non-HDLc, which approximates the sum of low-density lipoprotein (LDL) cholesterol and very-low-density lipoprotein cholesterol, was calculated by subtracting HDLc from TPC.
Screening for viral infections
Serum from each animal was submitted for viral detection. Antibodies against Herpes B virus, simian immunodeficiency virus (SIV), measles, and simian T cell leukemia virus (STLV-1 and -2) were assessed by ELISA. Rhesus cytomegalovirus (RhCMV), simian type D retrovirus (SRV/D), and STLV-1 and-2 were detected by immunofluorescence assay (CNPRC Pathogen Detection Laboratory, Davis, CA).
Necropsy procedures
At the conclusion of the study, surviving monkeys (n = 74) were euthanized using sodium pentobarbital (100mg/kg, IV; i.e. consistent with the recommendations of the panel on euthanasia of the American Veterinary Medical Association) and perfused with lactated Ringer’s solution. A complete necropsy was performed and all major organs, including the stomach, were collected and fixed in 10% neutral buffered formalin (NBF). The right common iliac artery (RCI) was removed for comparison with the left common iliac artery that was removed at the time of ovariectomy. Hearts were removed and coronary arteries were perfused-fixed with 10% NBF for 1h at 100 mm Hg pressure. The left anterior descending (LAD), left circumflex (LCX), and right coronary arteries (RCA) were then collected. Each artery was sectioned into five blocks (3 mm each in length), cut perpendicular to the long axis of the artery, processed routinely, and embedded in paraffin to generate formalin-fixed paraffin-embedded (FFPE) blocks. Whole sections of the left circumflex artery were also sent to the Uniformed Services University of the Health Sciences for detection of H. pylori. Due to postmortem degradation, usable gastric tissue could not be obtained from three animals. Coronary artery tissue from these animals was included in the analysis only if H. pylori was detected at that site, since lack of H. pylori in the artery does not guarantee that the animal was H. pylori-negative in the stomach.
Histopathology and morphometry
For each animal, the first 5mm FFPE block of the LCX (n = 74) was serially sectioned at 5um thickness and sections were mounted on slides. The first five sections were stained with Verhoeff -van Gieson stain to highlight the internal elastic lamina, and plaque size was determined using computer assisted morphometric analysis (ImageProPlus 5.1); the cross-sectional area of plaque (defined as the area between the endothelial lining and internal elastic lamina) in each of the five sections was measured and plaque area for each animal was calculated as the mean of the five sections examined. Section 6 was stained with Hematoxylin-Eosin (H&E), examined by light microscopy for the presence of mineralization and/or necrosis within the tunica intima, and semi-quantitatively scored as follows: 0 = none, 1 = minimal, 2 = mild, 3 = moderate, 4 = marked, 5 = severe. Section seven was stained with Masson’s Trichrome to highlight areas of collagen deposition (fibrosis). The proportion of intima occupied by fibrosis was determined by overlay of a 25 x 25 pixel grid with evaluation of each crosshatch as positive or negative for collagen by a board-certified pathologist (JAC) blinded to treatment. The remaining LCX serial sections were utilized for immunohistochemistry as detailed below. Additional 5 μm sections of stomach tissue and LCX artery were generated, mounted on slides and stained with H&E and Genta-Robason (H&E, silver stain and Alcian blue), which is a conventional method for detecting the presence of H. pylori. Slides were examined by light microscopy for the presence/absence of argyrophilic curved or spiral-shaped bacteria. The remaining slides were used for in situ hybridization as detailed below.
Histochemistry and immunohistochemistry
For immunohistochemistry, the slides were incubated with mouse anti-human HAM56 (Dako Cytomation, Carpinteria, CA), mouse anti-human CD3 (BDBiosciences, San Jose, CA), mouse anti-human CD4 (Abcam, Cambridge, MA), and mouse anti-human CD8 (Abcam, Cambridge, MA) for 1.5 h at 25° C. Primary antibodies were localized with appropriate biotinylated secondary antibodies, streptavidin–alkaline phosphatase (Biogenex, San Ramon, CA) and Vector Red (Vector Labs, Burlingame, CA) substrate. Sections were counterstained with Mayer's Hematoxylin and examined by the study pathologist (JAC) using light microscopy. Thymus and lymph node were used as tissue controls; as an assay control, the primary antibodies were substituted with non-immune sera. Cell densities for macrophages, CD3+ cells, CD4+ cells, and CD8+ cells were determined by overlay of a 25 x 25 pixel grid with evaluation of each crosshatch for positive or negative staining by the pathologist (JAC), who was blinded to treatment. Cell densities are expressed as the percentage of intima occupied by positive staining.
RNA isolation and quantification
RNA was isolated from intima-media sections as described previously [44–46] using a Tri-Reagent protocol. Total RNA concentration and quality was assessed using an Agilent 2100 Bioanalyzer and aliquots were reverse transcribed using a high-capacity cDNA archive kit (Applied Biosystems). Quantitative real-time reverse transcriptase–polymerase chain reaction (PCR) was performed on an ABI Prism 7000 system using TaqMan FAM-MGB primer-probe assays (Applied Biosystems). Primers included cynomolgus macaque-specific primers (CCL2, encoding monocyte chemotactic protein-1 [MCP-1], VCAM1 [vascular cellular adhesion molecule-1], ICAM1 [intercellular adhesion molecule-1], CD3G, encoding the CD3 δ chain, CD4, ESR1, encoding estrogen receptor alpha [ER-α], and ESR2, encoding estrogen receptor beta [ER-β]) and human-specific primers (microsialin/CD68, T-cell receptor-β, and IL6 [interleukin-6]). Quantitative reverse transcriptase–PCR data were normalized to the geometric mean of the endogenous, constitutively expressed control genes glyceraldehyde-3-phosphate dehydrogenase, β-actin, and ribosomal protein L13a [47] using the 2–ΔCT procedure [48, 49]. Amplifications not reaching threshold by 40 cycles were designated as zero expression.
In situ hybridization
H. pylori was detected in coronary arteries (i.e., the LCX) and stomach tissue following euthanasia. All procedures were performed on paraffin sections according to previously described methodologies [Liu, 18648543; [50, 51]]. H. pylori-16S rRNA (antisense 5’-TAC CTC TCC CAC ACT CTA GAA TAG TAG TTT CAA ATGC-3’ and sense 5’ CTA TGA CGG GTA TCC GGC-3’) and H pylori cagA (antisense 5’CTG CAA AAG ATT GTT TGG CAG A-3’ and sense 5’-GAT AAC AGG CAA GCT TTT GAG G-3’) probes were used. The 5' ends of the oligonucleotides were labeled with biotin or digoxygenin (see below). All probes were synthesized in the Synthesis and Sequencing Facility, Biomedical Instrumentation Center (USUHS). Negative controls were routinely run as described previously [52]. These included using sense instead of antisense probes, hybridization buffer without probe, probe without biotin or digoxigenin label, pre-treatments with DNase I, RNase A or both, and incubation with an irrelevant probe, scorpion Buthus martensi Karsch neurotoxin (5’-GGC CAC GCG TCG ACT AGT AC-3’)[53].
In situ hybridization was performed as previously described [52, 54, 55]. Briefly, sections were deparaffinized in xylene, rehydrated, treated with proteinase K and washed with standard saline citrate solution (SSC). For pre-hybridization, sections were incubated with hybridization mixture (Denhardt’s solution, NaCl, formamide, dextran sulfate EDTA, bovine serum albumin) and hybridization solution (SSC and extracted salmon testis DNA). For hybridization, sections were incubated with the denatured primer overnight at 37˚C. For post-hybridization, unbound probe was removed by successive washes in decreasing concentrations of SSC. For visible light studies, biotin-labeled probes were detected using streptavidin-alkaline phosphatase and nitro blue tetrazolium/5-bromo-4-chloro-3-indolyl-phosphate (NBT/BCIP). Sections were counterstained with Nuclear Fast red. Serial sections were used to detect 16S rRNA and cagA mRNA. For fluorescence dual detection studies, 16S rRNA probes were labeled with biotin and detected using avidin-Texas red (red color), while cagA probes were labeled with digoxigenin-3-0-methylcarbonyl-ε-aminocaproic acid-N-hydroxy-succinimide ester (DIG-NHS ester, Roche Diagnostics, Indianapolis, IN) and detected using anti-digoxigenin-FITC (green color).
An Eclipse Nikon microscope was used to examine the section at X100, X400 and X1000 magnification for bright light and for fluorescence. Bacterial density was quantified as described previously [52]. Briefly, the number of positive-stained bacteria were quantified at 400X magnification in three randomly-selected fields according to a modification of the point-counting stereological method and using an intraocular reticle (27 mm diameter, covering 3,578 μm2, i.e. 17,892 μm3 for 5 μm-thick sections; No. KR-409 (Klarmann Rulings, Inc. Litchfield, NH), located in a Nikon Eclipse E 800. Tissues were analyzed randomly and blindly regardless of category. All data were expressed as mean ± standard error of the mean (SEM) number of bacteria per 106 μm3 (representing an imaginary cube with sides of 100 μm or 0.1 mm).
Statistical analysis
Descriptive statistics and graphical plots, such as means, standard deviations and standard errors, and comparative boxplots, were calculated and obtained for H. pylori densities, cagA densities, H. pylori infection status, and social status. Linear models and analysis of variance (ANOVA) were used to determine the influences of H. pylori infection, diet, and social status on severity of premenopausal and postmenopausal atherosclerosis, arterial inflammatory markers, and lipid profiles. Pairwise group comparisons were performed using Welch two-sample t-test and/or nonparametric rank tests (Kruskal-Wallis H test), the latter employed when assumptions for the validity of the t-test were not satisfied. Linear models and correlation analyses were used to compare the effect of H. pylori density in the artery and stomach on plaque area and thickness. Fisher’s exact test was used where indicated. The (effective) sample sizes in some of the calculations were smaller than the overall sample sizes due to missing values.
Results
Evidence of H. pylori infection in gastric and artery tissue
The study design is outlined in Fig 1. Curved or spiral organisms consistent with H. pylori were first detected in gastric mucosa using the Genta-Robason staining procedure. Subsequently live H. pylori were specifically detected in gastric mucosa and LCX coronary artery sections by in situ hybridization using RNA anti-sense probes specific for H. pylori 16S rRNA or cagA mRNA. These probes recognize RNA and the method has been previously validated [52]. The normal infection pattern for H. pylori is for the majority of organisms to reside in the mucus just above the epithelium, attached to epithelial cells, or within gastric crypts. H. pylori is occasionally intracellular or found within gastric capillaries[56]. We found similar results in our study. Within the stomach, the majority of H. pylori were located in the superficial gastric mucosa (Fig 2), but a few organisms were located in gastric blood vessels. In situ hybridization demonstrates co-expression of 16S rRNA and cagA in the same bacteria (Fig 3), further confirming that the organisms are H. pylori and demonstrating that some macaques were infected with cagA-positive strains, some were infected with cagA-negative strains, and some were infected with a mixture of strains. Infection with multiple H. pylori strains is not unusual.
Fig 2. Detection of H. pylori in gastric tissue.
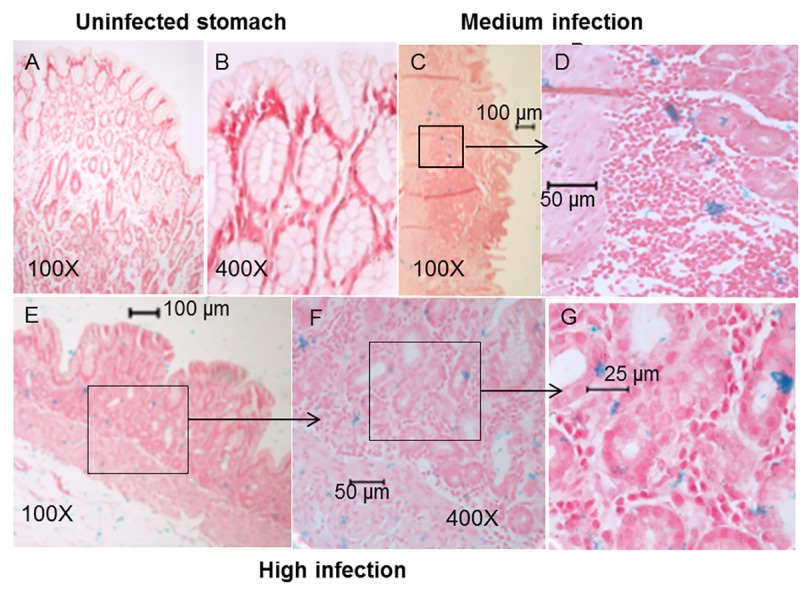
In situ hybridization using the H. pylori-specific 16S rRNA probe (blue staining) demonstrates absence and presence of H. pylori in gastric sections. Sections were counterstained with Nuclear Fast Red. A, B) Stomach from an uninfected animal (no blue staining). C, D) Stomach showing a medium infection density. E, F) Stomach showing a high density of H. pylori.
Fig 3. Identification of cagA-positive H. pylori in gastric tissue.

Fluorescence in situ hybridization of H. pylori 16S rRNA (red) and cagA (green) was done on tissue sections. H. pylori (circled) are seen attached to epithelial cells within a gastric gland. Colocalization of red and green staining indicates that the organisms are cagA-positive.
Of potentially greater interest, H. pylori was detected within coronary atherosclerotic plaques as isolated organisms, or small colonies, randomly distributed throughout the fibrofatty admixture of foam cells and extracellular matrix. Fig 4 shows examples of uninfected, moderately infected, and highly infected LCX arteries as measured using colorimetric ISH with a 16S probe. Tunica intima and media were often involved, but no colonization was observed in the tunica adventitia, suggesting that H. pylori originates from the vessel lumen and does not easily cross the external elastic membrane. Bacteria often appeared in clusters, suggesting limited replication (Fig 4D). Using serial sections, some H. pylori were found to express cagA. This latter observation was confirmed by dual fluorescence in which 16S rRNA was stained with Texas Red and cagA mRNA was stained with FITC (green) (Fig 5). Occasional individual bacteria were noted in collateral vessels (Fig 6), but there was no evidence of bacterial growth in non-atherosclerotic vessels. Animals were defined as H. pylori positive if H. pylori was visualized in either gastric or coronary artery sections following euthanasia. Iliac arteries were not assessed for H. pylori infection. Of the animals with evaluable data sets, 64% were H. pylori-positive. Thus, there were adequate numbers of infected and uninfected animals for statistical determination of the effects of H. pylori infection on atherosclerosis, arterial inflammation, and plasma lipids. All but seven of the 45 H. pylori-positive animals were infected with at least some cagA-positive strains, with the percentages of cagA-positive bacteria varying between 11% and 100% in a single artery section. Four animals were infected with only cagA-positive strains, while the remainder had mixed infections with both cagA-positive and cagA-negative strains, as seen in Fig 5. Of the H. pylori-positive animals, 64% had evidence of H. pylori infection in both the stomach and arteries and 29% had H. pylori only in the stomach. As expected, the density of H. pylori was highest in the gastric mucosa, but impressive numbers of H. pylori were found in the LCX arteries (up to 43 per 106 μm3). When the densities of H. pylori in the coronary arteries were plotted against gastric mucosal densities, a substantial linear correlation (coefficient of determination of R2 = 0.6747, p< 0.0001) was found (Fig 7). A correlation matrix of H. pylori 16S and cagA densities in the stomach and LCX coronary artery revealed correlations of r ≥0.73 for all pairings, further indicating that the densities of cagA-positive or–negative H. pylori in the artery are significantly associated (p< 0.0001) with densities in the stomach. In other words, live H. pylori were frequently present in coronary arteries of H. pylori-infected monkeys and the density of bacteria found in plaques was strongly related to colonization density in the stomach.
Fig 4. In situ hybridization using the H. pylori 16S rRNA probe (blue staining) demonstrating presence and absence of H. pylori in left circumflex artery sections.
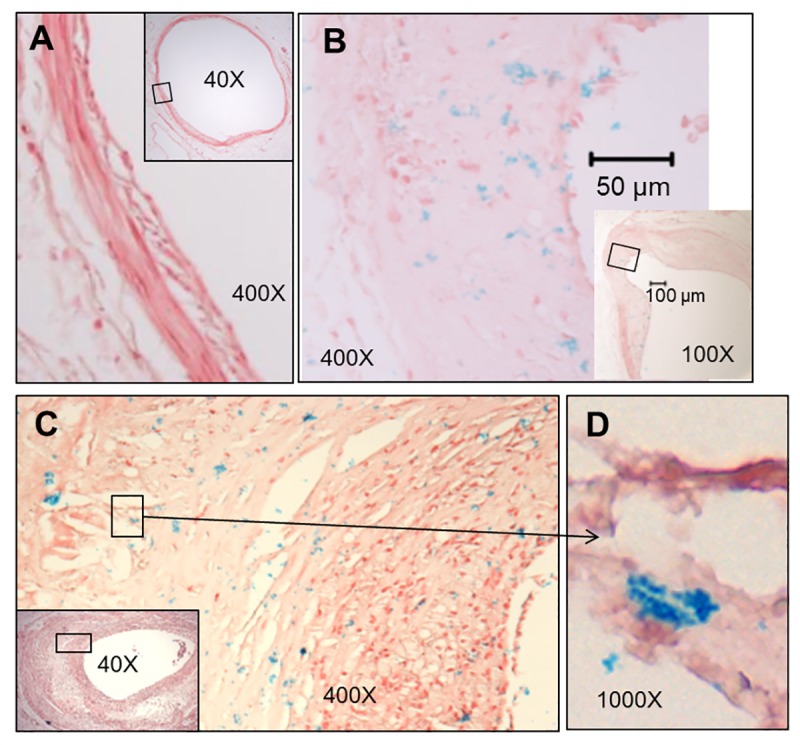
Sections were counterstained with Nuclear Fast Red and boxed regions of low magnification images indicate the locations of the high magnification images. A) Artery from an uninfected animal. B) Artery showing a medium infection density (blue dots). C) Artery showing a high density of bacteria. D) High magnification reveals a cluster of H. pylori suggestive of limited growth.
Fig 5. Fluorescence in situ hybridization on an artery section demonstrating mixed cagA-positive and cagA-negative H. pylori.
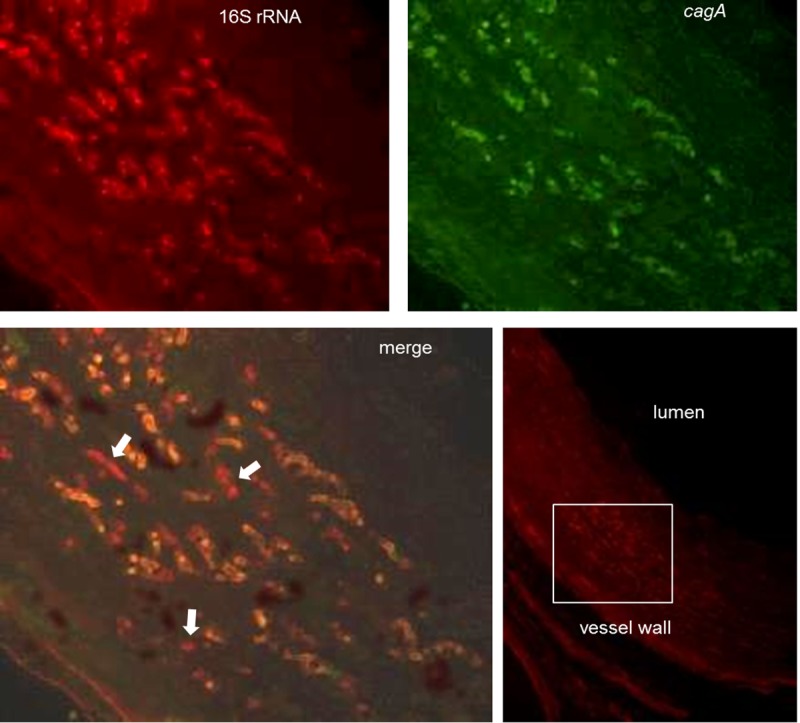
Fluorescence in situ hybridization of H. pylori 16S rRNA (red) and cagA (green) was conducted on tissue sections and imaged using confocal microscopy. White arrows indicate H. pylori lacking cagA expression. The bottom right panel shows the region of interest (white box) in a lower magnification image.
Fig 6. H. pylori attached to the endothelium of a collateral arteriole.
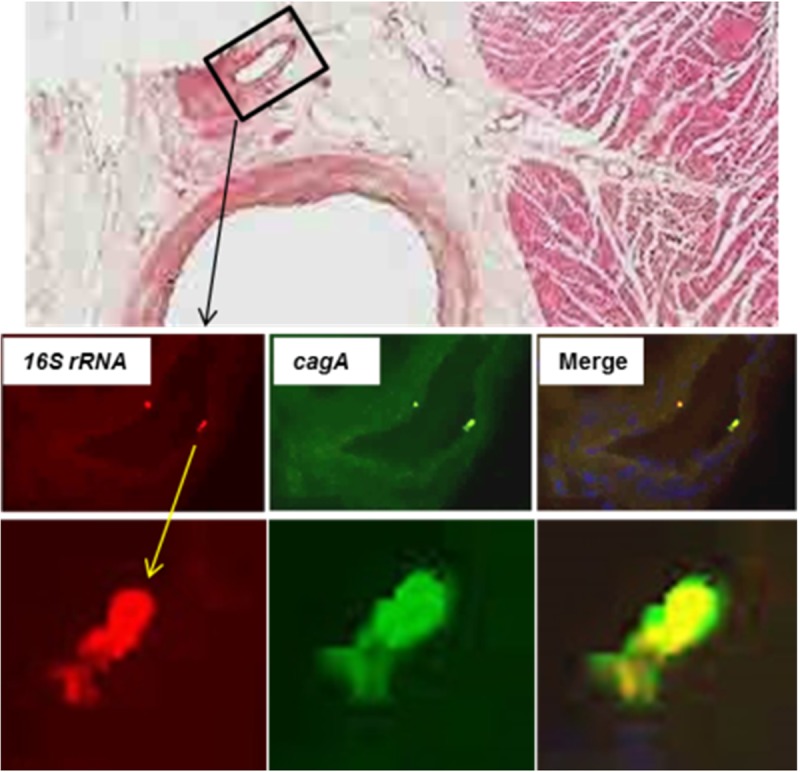
Fluorescence in situ hybridization of H. pylori 16S rRNA (red) and cagA (green) was carried out on tissue sections with DAPI staining of nuclei (blue). Top image, boxed area shows the arteriole shown in the lower images. Middle row, 400x; Bottom row, 1,000x.
Fig 7. The density of H. pylori in the stomach correlates with the density in the left circumflex coronary artery.
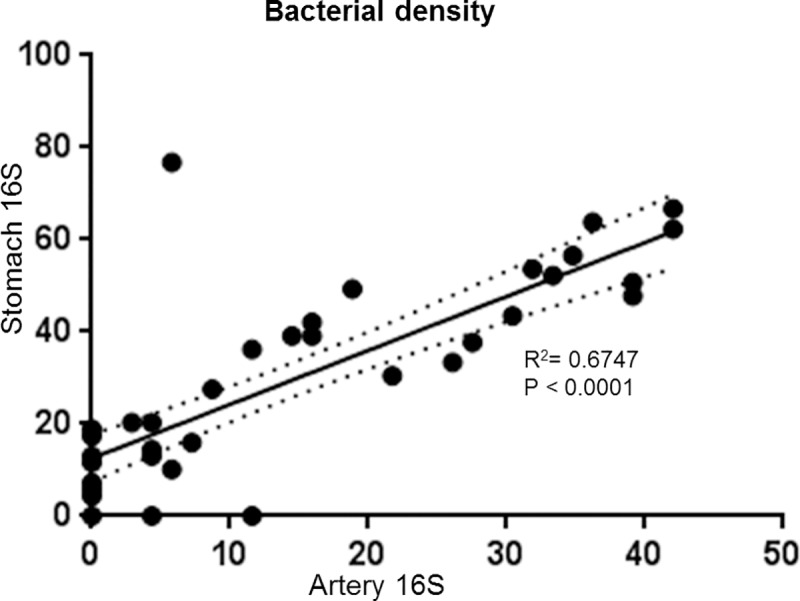
H. pylori density was determined by counting the number of bacteria per field (as determined by 16S rRNA in situ hybridization) and is expressed as bacteria per 106 μm3. Only H. pylori positive animals with both stomach and artery data are included in the analysis (n = 44). Solid line, linear regression best fit; dotted line, 95% confidence interval; R2, coefficient of determination.
H. pylori infection influences severity of premenopausal and postmenopausal atherosclerosis
The influence of H. pylori infection (as determined in postmenopausal gastric/coronary tissue) was evident at both premenopausal and postmenopausal time points. Premenopausal atherosclerosis was assessed in the LCI at the time of ovariectomy and postmenopausal atherosclerosis was assessed in both the RCI and coronary arteries (LCX, LAD, RCA). Both the intimal area (IA) and maximum intimal thickness (MXIT) were strongly associated with H. pylori infection (p<0.005) even when diet was included in the statistical model (Table 1). Few H. pylori-negative animals had plaques of appreciable size at the premenopausal time point. Only 16% of H. pylori-negative animals had intimal areas greater than 0.5 mm2, whereas 55% of infected animals had premenopausal plaque areas above that value (p< 0.001 by Fisher’s exact test). There was no significant association between H. pylori infection status and premenopausal estrogen receptor expression. Other premenopausal measurements shown in Table 1 will be discussed below.
Table 1. Premenopausal iliac artery plaque parameters and inflammatory biomarkers.
| H. pylori negative | H. pylori positive | Ratio of Means | ||||
|---|---|---|---|---|---|---|
| Mean (n)b | SEM | Mean (n) | SEM | Hp+/Hp- | P- Valuea, c | |
| Max plaque thickness | 0.1451 (25) | 0.0223 | 0.2555 (45) | 0.0207 | 1.7607 | 0.0003 ** |
| Plaque area | 0.2419 (25) | 0.0566 | 0.5361 (45) | 0.0614 | 2.2165 | 0.0004 ** |
| Mineralization | 1.4000 (25) | 0.3416 | 2.1591 (44) | 0.2640 | 1.5422 | 0.0423 * |
| Intimal collagen | 28.2063 (25) | 5.7756 | 41.6703 (42) | 3.1962 | 1.4773 | 0.0241 * |
| Intimal HAM56+ | 0.6943 (25) | 0.3766 | 2.0685 (44) | 0.3622 | 2.9791 | 0.0054 ** |
| CD68 | 0.0522 (23) | 0.0192 | 0.1332 (41) | 0.0224 | 2.5501 | 0.0040 ** |
| CD3 | 0.0017 (23) | 0.0004 | 0.0031 (41) | 0.0005 | 1.7664 | 0.0206 * |
| CD4 | 0.0135 (23) | 0.0032 | 0.0269 (41) | 0.0046 | 1.9907 | 0.0104 * |
| MCP-1 | 0.0126 (23) | 0.0040 | 0.0219 (41) | 0.0039 | 1.7387 | 0.0542ns |
| VCAM-1 | .0000355 (23) | .000000782 | .00000588 (41) | .00000131 | 1.6526 | 0.0661ns |
| ICAM-1 | 0.0080 (23) | 0.0014 | 0.0127 (41) | 0.0014 | 1.5923 | 0.0099 ** |
| ER-α | 0.0085 (23) | 0.0006 | 0.0074 (41) | 0.0005 | 0.8723 | 0.1875 ns (2-sided) |
| ER-β | 0.000125 (23) | 0.0000177 | 0.0000932 (41) | 0.00000786 | 0.7438 | 0.1079ns (2-sided) |
| IL-6 | 0.000442 (23) | 0.0000934 | 0.000864 (41) | 0.000152 | 1.9531 | 0.0108 * |
aBold text indicates statistical significance. Unless otherwise indicated, the p-value are based on a one-sided Welch Two-Sample T-Test with alternative hypothesis that the mean for the H. pylori negative is smaller than the mean for the H. pylori positive.
b(n) represents the effective sample size used in computing the mean and standard error of the mean (SEM).
c[ns = not significant
* = significant at 5% level
** = significant at 1% level.]
As shown in Fig 1, following ovariectomy and for an additional 36 months, animals were either fed the same diet they had consumed premenopausally or switched to the other diet. MXIT and IA measurements were taken on the right common iliac artery and three coronary arteries (LCX, LAD, and RCA). Postmenopausal results are summarized in Table 2. Fig 8B and 8D show measurements of iliac artery plaques at the postmenopausal time point as compared with premenopausal values. Progression of iliac plaque area, as determined by subtracting premenopausal MXIT and IA from postmenopausal values in each animal, trended higher in H. pylori-infected animals, but progression of plaque thickness was unaffected by infection status.
Table 2. Postmenopausal plaque measurements and lipid profiles.
| H. pylori negative | H. pylori positive | ratio | ||||
|---|---|---|---|---|---|---|
| mean | SEM | mean | SEM | Hp+/Hp- | p value | |
| avg. coronary area | 0.331 | 0.076 | 0.961 | 0.236 | 2.90 | 0.0070 |
| avg. coronary thickness | 0.121 | 0.021 | 0.282 | 0.042 | 2.32 | 0.0006 |
| iliac area | 0.601 | 0.137 | 1.090 | 0.122 | 1.82 | 0.0048 |
| iliac area progression | 0.359 | 0.097 | 0.545 | 0.083 | 1.52 | 0.0750 |
| iliac thickness | 0.233 | 0.034 | 0.364 | 0.029 | 1.56 | 0.0024 |
| iliac thickness progression | 0.088 | 0.022 | 0.105 | 0.020 | 1.19 | 0.2898 |
| total cholesterol | 217.52 | 13.54 | 242.16 | 12.01 | 1.11 | 0.0894 |
| triglycerides | 34.52 | 3.95 | 35.39 | 6.47 | 1.03 | 0.4547 |
| HDL cholesterol | 66.63 | 4.84 | 49.57 | 2.92 | 0.74 | 0.0048 (2-sided) |
| LDL + VLDL cholesterol | 151.00 | 16.19 | 192.70 | 13.21 | 1.28 | 0.0255 |
| TPC/HDLc | 3.89 | 0.50 | 5.75 | 0.44 | 1.48 | 0.0036 |
Bold text indicates statistical significance. There were 25 observations for H. pylori-negative group and 44 observations for H. pylori -positive group. P-values are obtained from Welch two-sample t-test based on a one-sided alternative hypothesis (except for HDL cholesterol, which is two-sided).
Fig 8. Premenopausal and postmenopausal effects of H. pylori on atherosclerotic plaque thickness.
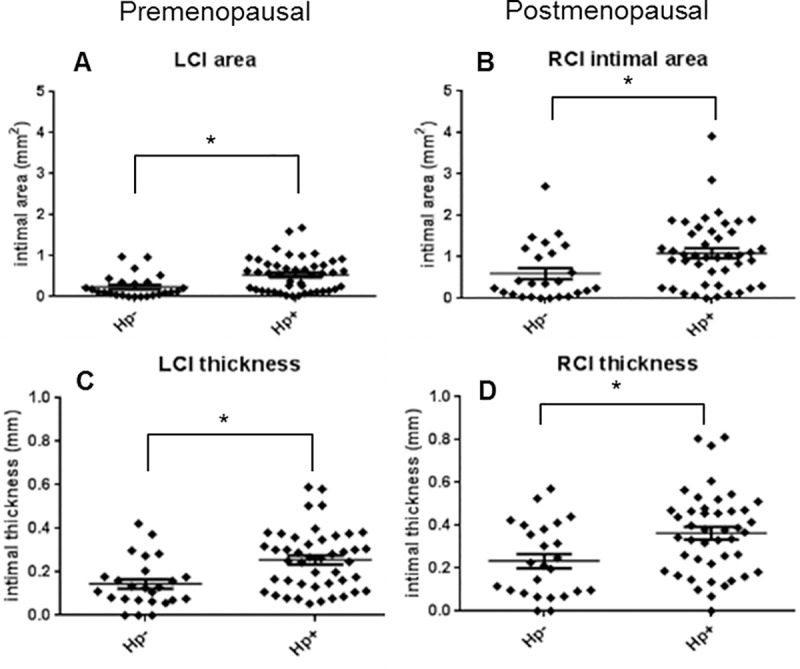
Common iliac artery plaque intimal areas and maximum intimal thicknesses were measured at premenopausal (A and C, n = 70) and postmenopausal time points (B and D, n = 69). Hp, H. pylori; LCI, left common iliac; RCI, right common iliac). Graphs show mean +/- standard error of the mean. *P< 0.05.
The micrographs in Fig 9A–9C represent examples of extreme LCX artery pathology, which was seen in animals infected with cagA-positive H. pylori. Large necrotic regions and bulging plaques in the LCX were found in six of the cagA-positive animals and none of the cagA-negative or uninfected animals (Fisher’s exact test p< 0.05). Plaques representative of median thickness values for each cagA-positive, cagA-negative, and uninfected macaques are shown in Fig 9D–9F. Note that these values represent the maximum thickness of the LCX slice in which H. pylori density was determined, not the maximum thickness of the entire artery section. Plaque measurements for the three coronary arteries were averaged and the differences between H. pylori-positive and–negative animals irrespective of diet group are shown in Fig 10A and 10B. H. pylori infection substantially increased intimal area and thickness (p = 0.0004 and p = 0.0003, respectively). Panels 10C and 10D demonstrate the effects of both H. pylori infection and diet group. H. pylori-negative animals fed a SOY-SOY diet had essentially no plaque accumulation, whereas several H. pylori-positive animals on the same diet had modest amounts of plaque. Almost all H. pylori-positive animals that received a casein/lactalbumin diet at any point had moderate to severe atherosclerosis. Analyses of the effects of diet on atherosclerosis in these animals have been previously published [38, 44], and therefore are only briefly mentioned in this report.
Fig 9. Coronary artery pathology seen in H. pylori-infected and uninfected macaques.
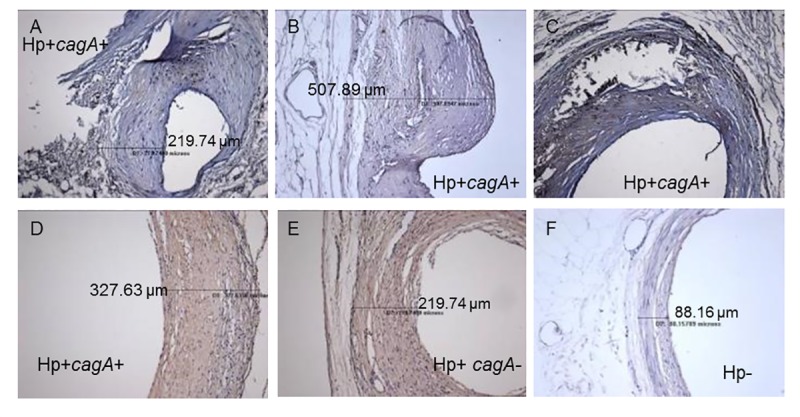
Panels A, B, C, and D show arteries from cagA positive (cagA+) animals. Panels A and C show severe necrosis. Panel B shows an atypical bulging plaque. Panels D, E, and F depict arteries that are near the median intimal thickness values for cagA-positive, cagA-negative (cagA-), and uninfected (Hp-) animals, respectively. Measurement lines shown in some images indicate the maximal intimal thickness for those sections.
Fig 10. Effects of H. pylori infection and diet on coronary artery plaque measurements.

The values shown represent the average plaque measurements of the left circumflex, left anterior descending, and right coronary arteries. Values are grouped according to H. pylori (Hp) status (A and B) or according to H. pylori status and pre/postmenopausal diet group (C and D). Graphs show mean +/- standard error of the mean. *P< 0.05.
The outlier seen in Fig 10 represents a cagA-positive animal receiving the casein-lactalbumin diet for the duration of the study. There is no a priori basis for invalidating the measurements from this animal. Moreover, the results of analyses were similar whether the outlier was included or not, and all statistical analyses discussed herein are inclusive of all animals.
Effects of H. pylori on plaque size are due to local rather than systemic effects
We next sought to determine whether the effects of H. pylori on plaque measurements were due to local effects of bacteria within the plaques or due to systemic effects stemming from gastric infection. To this end, we performed multiple regression analysis and correlation analysis. As shown in Table 3, the density of cagA-positive H. pylori in the LCX coronary artery showed a stronger correlation to plaque area and thickness than did the density of cagA-positive bacteria in the stomach. On the other hand, the overall densities of H. pylori, as measured by 16S in the stomach or artery, did not correlate with plaque measurements (Table 3). Taken together, these data suggest that the presence of cagA is critical for artery pathology and that bacteria are acting locally rather than systemically.
Table 3. Correlations between plaque measurements and H. pylori density in the artery or stomach.
| artery 16S | artery cagA | stomach 16S | stomach cagA | |||||
|---|---|---|---|---|---|---|---|---|
| P value | Corr. | P value | Corr. | P value | Corr. | P value | Corr. | |
| premenopausal iliac plaque area | .0698 | .2165 | .0046 | .3329 | .1683 | .1678 | .0446 | 0.2426 |
| premenopausal iliac plaque thickness | .0710 | .2156 | .0068 | .3182 | .18 | .1633 | .1015 | 0.1988 |
| LCX area | 0.5241 | 0.0769 | 0.0532 | .2305 | .2427 | 0.1425 | .0595 | 0.2281 |
| avg. cardiac plaque area | .1043 | .3866 | .0268 | .2628 | .1474 | 0.1763 | 0.0245 | 0.2707 |
| LCX plaque thickness | .2746 | .1314 | .0101 | .3034 | .1564 | 0.1725 | 0.0263 | 0.2674 |
| avg. cardiac plaque thickness | .1930 | .1563 | .0061 | .3222 | .0877 | 0.2071 | 0.0127 | 0.2985 |
| HDL cholesterol | .0178 | -0.2826 | 0.0851 | -0.2069 | 0.0032 | -0.3524 | 0.0079 | -0.3197 |
| LD+VLD cholesterol | 0.6492 | 0.0553 | 0.5853 | 0.0663 | 0.2939 | 0.1292 | 0.2963 | 0.1285 |
| TPC/HDLc | 0.1338 | 0.1810 | 0.2077 | 0.1524 | 0.0400 | 0.2497 | 0.0466 | 0.2422 |
Bold text indicates statistical significance. Estimates of the Pearson’s correlation coefficient together with their p-values for testing, based on a t-test, that the correlation coefficient is not equal to zero between bacterial densities in the artery or stomach and plaque or plasma lipid measurements.
H. pylori infection influences arterial inflammatory markers
Additional measurements of inflammation and plaque physiology were performed on sections of the LCI artery at the premenopausal time point. Intimal area containing collagen was significantly increased in H. pylori-infected animals (Fig 11A and Table 1), while mineralization was modestly increased (Fig 11B and Table 1). Macrophage populations were increased by more than two fold in H. pylori-infected monkeys as measured by both immunohistochemistry (HAM56; Table 1) and CD68 expression (Fig 11C and Table 1). ICAM1, IL-6 and expression of the T cell markers CD3 and CD4 were significantly increased and expression of the cytokine MCP-1 tended to be higher in infected animals (Fig 11 and Table 1). Our findings indicate an increased inflammatory response due to H. pylori infection. Overall, the effects of H. pylori infection were the opposite of the anti-inflammatory effects seen with exposure to the SOY treatment, which decreased macrophage and T cell infiltration, ICAM-1, MCP-1, and IL-6 in these animals[44].
Fig 11. Effect of H. pylori infection on molecular markers of plaque inflammation.
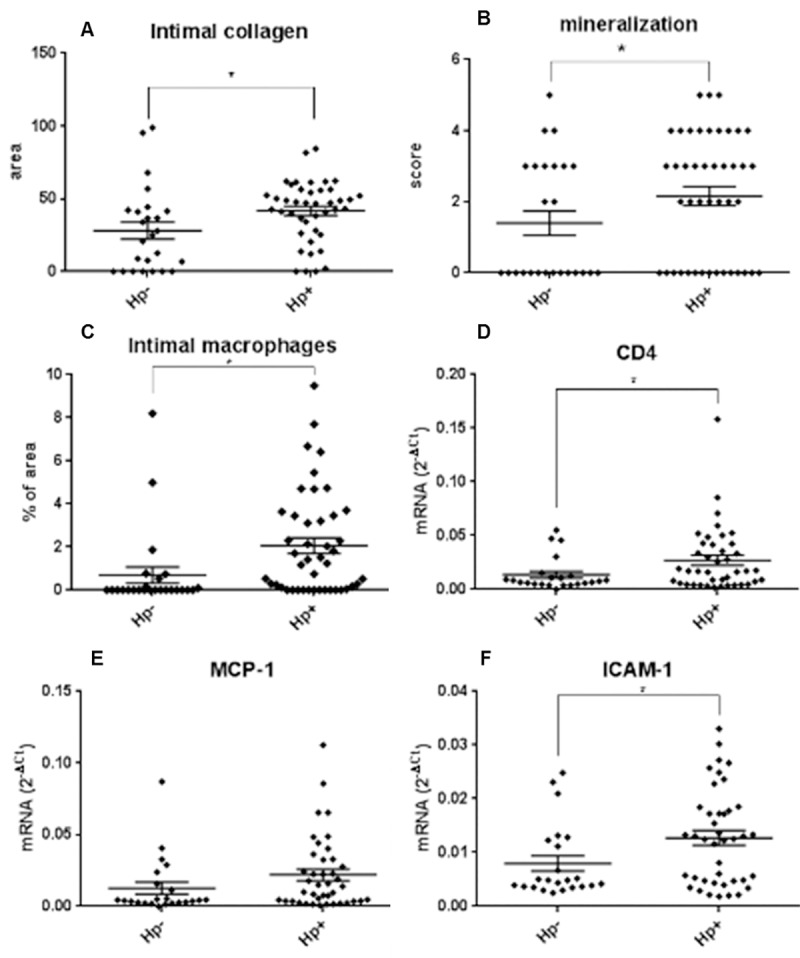
Intimal collagen area was determined by computer-assisted image analysis (A, C). Quantitative PCR was carried out using mRNA from intima-media sections of the left common iliac artery (D-F). Data were normalized to the geometric mean of endogenous control genes GAPDH, ACTB, and RPL0. *p< 0.05. (one-sided two-sample T-test) A, n = 67; B and C, n = 69; D-F, n = 64.
H. pylori alters plasma lipid profiles
H. pylori has been linked to dyslipidemia in humans, as indicated by epidemiological studies and following measurement of plasma lipids before and after H. pylori eradication [57, 58]. Similarly, we found that H. pylori infection in monkeys correlated with decreased HDLc, increased non-HDLc (low density and very low density lipoprotein-associated cholesterol), and increased TPC/HDLc (Castelli’s ratio) (all p<0.05; Table 2). In contrast with plaque measurements, the density of H. pylori in the gastric mucosa (16S and CagA) correlated better with plasma lipids than did H. pylori density in the artery. Also, the presence of cagA did not appear to be an important contributor to dyslipidemia, as indicated by the stronger correlations between plasma lipids and stomach H. pylori 16S than plasma lipids and artery or stomach cagA. This suggests that either local effects of H. pylori in the stomach or systemic effects of infection are responsible for plasma lipid alterations. Body weight, total cholesterol, and triglycerides were not associated with H. pylori infection.
Known viral infections do not influence atherosclerosis in these animals
Human epidemiological studies have noted a relationship between atherosclerosis and viral infections [59]. To explore this possibility, monkeys were tested for Herpes B virus, simian immunodeficiency virus (SIV), measles, Rhesus cytomegalovirus (RhCMV), simian T cell leukemia virus (STLV), types 1 and 2, and simian type D retrovirus (SRV/D). None of the viral infections correlated with the atherosclerosis biomarkers tested in this study.
Subordinate social status is associated with increased H. pylori burden
Premenopausal female cynomolgus macaques of low social status are typically at greater risk of atherosclerosis than their dominant counterparts [41]. Could H. pylori infection influence this risk? We found that subordinate status was significantly associated with increased risk for H. pylori infection and increased infection density (p< 0.05 using either Fisher’s exact test or Pearson’s Chi-squared test with Yates’ continuity correction). Only 48% of the dominant monkeys were infected with H. pylori as compared with 78% of the subordinate animals; however, rank did not influence either premenopausal or postmenopausal plaque measurements when H. pylori infection status was included in the predictive model. Therefore, H. pylori infection may be a more important contributor to atherosclerosis than social status or, alternatively, status may exert its effects in part through modified resistance to this bacterial infection.
Discussion
This work is the first study to examine the effects of H. pylori on development of atherosclerosis in non-human primates and the first to demonstrate that H. pylori within atherosclerotic plaques are alive. Natural infection with H. pylori has been documented in both rhesus (M. mulatta) and cynomolgus macaques [60–62], and rhesus macaques have long been used to study H. pylori gastric pathogenesis, evolution, and carcinogenesis [60, 63, 64]. However, the potential for natural infection by H. pylori to influence extragastric pathology in non-human primates had not been explored previously.
Detection of H. pylori DNA by PCR in human atherosclerotic plaques was first reported by Farsak et al. in 2000 and has since been confirmed by a number of other investigators [26–29]; nonetheless, PCR of DNA does not prove the presence of live H. pylori within plaques. In situ hybridization has been routinely used to detect mRNA in paraffin-embedded tissue and is a sensitive and specific method for detecting H. pylori in gastric biopsies [65, 66]. Our detection of 16S and cagA mRNA, combined with corresponding structures matching the size and morphology of H. pylori, confirm that live H. pylori inhabit atherosclerotic plaques. The presence of bacterial clusters further suggests that limited bacterial growth occurs within these same plaques. Seeding of heart valves by hematogenously spread oral flora is a well-documented risk and H. pylori within gastric and oral blood vessels has been previously documented [56, 67], so perhaps it is not surprising that H. pylori entering the bloodstream can reach the heart and adhere. H. pylori requires host cholesterol for optimal membrane integrity [68, 69] and the cholesterol-rich atherosclerotic plaques may encourage survival and growth of H. pylori. The distinction between living or dead H. pylori within plaques is critical because live H. pylori can inject bacterial components including the CagA toxin, peptidoglycan, DNA, and metabolites of lipopolysaccharide into the host cell cytoplasm via the type IV secretion apparatus [70, 71]; dead H. pylori cannot influence host cell signaling in this manner. Therefore, in terms of atherosclerosis H. pylori is likely to directly activate inflammatory pathways within the atheroma in addition to acting indirectly via distal activity at the site of gastric colonization.
H. pylori significantly worsened several plaque parameters, including common iliac artery intimal thickness and area in the same animals evaluated pre- and postmenopausally. Intimal collagen density and mineralization were also affected. Although the majority of macaques were infected with a mixture of cagA-positive and cagA-negative H. pylori strains, we were able to examine the contributions of cagA by determining whether LCX artery measurements were more greatly influenced by total bacterial density (as determined by 16S) or by cagA-positive bacterial density. That analysis revealed that cagA-positive strains were more atherogenic than cagA-negative strains. Moreover, H. pylori densities found within the arteries had a greater impact than bacterial density in the stomach. Though not conclusive, these data suggest that plaque development is due to local effects of cagA-positive H. pylori. Notably, these findings are consistent with epidemiological studies conducted in humans. While early epidemiological studies investigating the role of H. pylori in cardiovascular diseases yielded mixed results [17–20], recent studies that differentiate between cagA-positive vs. cagA-negative strains have yielded more consistent positive associations [24, 72–74]. Interestingly, Franceschi et al reported cross-reactivity between CagA-specific antibodies and artery tissue [75]. This immunoreactivity was assumed to be due to molecular mimicry, yet the authors did not determine whether antibodies specific for other H. pylori proteins reacted with the tissue or prove that the “cross-reactive” antigen was of human origin. In light of the evidence shown here, it is likely that CagA itself was being detected.
Another intriguing finding of this study was that the densities of H. pylori in the gastric mucosa were highly correlated with the densities in the coronary artery plaques. Gastric colonization density is known to vary among individuals and is believed to be related to both the H. pylori strain and the type of immune response generated by the host [76–78]. We could not determine whether colonization density varied over time, but the high correlation between postmenopausal bacterial density and both pre- and postmenopausal plaque measurements may suggest that colonization density is stable. Additional studies will be needed to determine how H. pylori densities in both locations change over time and whether gastric biopsies could be used to estimate the risk of severe atherosclerosis.
In addition to affecting plaque measurements, H. pylori infection was associated with increases in some arterial inflammatory markers, providing a possible mechanism for H. pylori-mediated atherosclerosis exacerbation. VCAM-1, ICAM-1, and MCP-1 all contribute to recruitment of macrophages to plaques [79, 80] and all tended to be increased in H. pylori-infected macaques. IL-6, which induces ICAM-1 [81], was also elevated by H. pylori. Our findings are consistent with in vitro induction of P-selectin, E-selectin, VCAM-1, and ICAM-1 and enhanced transendothelial migration of activated CD4+ and CD8+ T cells in human umbilical vein endothelial cell cultures following H. pylori exposure [82, 83]. We similarly found elevated plaque CD4+ T cells, which are pro-atherogenic [84], in H. pylori-positive animals. Others have reported that H. pylori induces MCP-1, IL-6, and ICAM-1 in various cell types [85–88] and endothelial ICAM1 expression strongly correlates with the presence of H. pylori DNA in human atherosclerotic plaques [89]. Furthermore, Figura et al. report that acute coronary artery disease patients infected with cagA-positive H. pylori have higher circulating IL-6 levels than uninfected patients [90]. Our results confirm and expand upon previous results, since we have established that live H. pylori within atherosclerotic lesions can directly interact with macrophages and/or endothelial cells to induce cytokine and adhesion molecule production.
Clinical studies have also reported decreased HDL and increased LDL in association with H. pylori, and some studies have found reversal of dyslipidemia following H. pylori eradication [57, 58, 91], although not all studies have found such a link [92]. One Japanese study found H. pylori-associated dyslipidemia in male, but not female subjects [93]; we clearly saw dyslipidemia in female, postmenopausal macaques. The varied findings in human studies likely result from the considerable differences among humans in terms of diet, lifestyle, genetics, and many other factors, many of which were controlled for in our monkey study. Some studies have linked dyslipidemia with atrophic gastritis in H. pylori-positive patients [94, 95]. This is consistent with our observation that plasma lipid changes were more closely related to gastric H. pylori densities than arterial densities.
Atherosclerosis development is delayed in women compared with men and estrogens likely mediate this effect based on studies showing that premenopausal oophorectomy or premature ovarian failure are associated with increased coronary heart disease risk, an effect that can be mitigated by treatment with exogenous estrogen in younger, but not older women [96]. Soy phytoestrogens improve cardiovascular disease biomarkers in some human studies involving men and post-menopausal women [97]. This may be in part due to the effects of soy on ER-α and ER-β, which are expressed in arteries [48]. H. pylori increased atheroma measurements both pre- and post-menopausally and generally counteracted the beneficial effects of SOY on inflammatory markers in cynomolgus macaques (reported by Walker et al [48]); however, unlike the SOY diet, H. pylori did not significantly alter expression of ER-α or ER-β. Therefore, the increased risk associated with H. pylori infection seems unaffected by estrogen.
The differing H. pylori burdens among dominant and subordinate animals have not been previously reported. Female subordinate macaques are under considerably more stress than dominant females and typically develop more atherosclerosis than their dominant counterparts when consuming an atherogenic diet [41], an effect that co-occurs with substantial ovarian disruption [96]. In humans, lower socioeconomic status positively correlates with H. pylori prevalence, presumably due to poor hygiene and overcrowding [98]. Although human socioeconomic status is not homologous to monkey dominance relationships [40], subordinate female macaques are directly stressed as a result of being physically harassed by their dominant counterparts during competitive interactions over food, space, and preferred grooming partners [99]. Moreover, in the small group context used in the study reported here, dominant animals demonstrably outcompete subordinates for fresh food and water and desirable perching locations, potentially exposing subordinates to more fecal contamination as they consume food and water that drop or spill to the ground. Such contamination, in turn, is one proposed transmission route for H. pylori [100]. Of note, several large epidemiological studies report an increased risk of cardiovascular disease in humans with low socioeconomic status. Although, again, human socioeconomic status differs conceptually from monkey dominance status [40], the increased H. pylori burden in humans with low socioeconomic status could contribute to higher atherosclerosis risk in this group.
The primary weakness of this study is that we are not certain when the animals became infected or whether bacterial prevalence changed over time. Humans most often become colonized during childhood, but transmission can occur later in life, particularly between spouses [101–103]. In rhesus macaques, H. pylori transmission occurs within 90 days via social contact, rather than fomites, but some animals remain uninfected, possibly due to host genetic factors [104, 105]. H. pylori infection persists for at least a decade in Rhesus macaques [63] indicating that infection is as persistent in non-human primates as it is in humans. If the animals in this study were not previously infected, then transmission likely occurred soon after assignment to social groups. The substantial correlations between postmenopausal H. pylori status and premenopausal plaque parameters further indicate that animals were infected before the study midpoint.
For the first time, this study conclusively demonstrated that live H. pylori can reside within atherosclerotic plaques and that H. pylori is likely to have local effects on plaque size. Furthermore, H. pylori influenced macrophage infiltration, collagen deposition, ICAM1 expression, and plasma lipids. The effects of H. pylori on plaque parameters appeared to be primarily due to local effects of cagA-positive H. pylori, whereas changes in plasma lipids related to gastric infection by either cagA-positive or–negative strains. H. pylori status was relevant during both the premenopausal and postmenopausal phases as demonstrated by correlations between H. pylori infection and iliac artery plaque parameters measured at both ovariectomy and necropsy. Given the many similarities in atherogenesis between humans and non-human primates, future inclusion of cagA-positive H. pylori status in human epidemiological and clinical atherosclerosis studies is warranted.
Supporting information
(XLSX)
Acknowledgments
Drs. Andre Dubois and Cristina Semino-Mora unfortunately passed away prior to the writing of this manuscript. Their wisdom and expertise are sorely missed by their colleagues. The contents of this article are solely the responsibility of the authors and do not necessarily represent the official views of the Department of Defense, the Uniformed Services University, or the NIH.
Data Availability
All relevant data are within the manuscript and its Supporting Information files.
Funding Statement
This work was supported by the National Institutes of Health (https://www.nih.gov/) [R01 CA082312 to AD and DSM, R01HLO79421 to JRK, and P30GM103336-01A1 to EP and BQ]. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1.deGoma EM, Knowles JW, Angeli F, Budoff MJ, Rader DJ. The evolution and refinement of traditional risk factors for cardiovascular disease. Cardiol Rev. 2012;20(3):118–29. Epub 2011/12/21. 10.1097/CRD.0b013e318239b924 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ravnskov U, McCully KS. Infections may be causal in the pathogenesis of atherosclerosis. Am J Med Sci. 2012;344(5):391–4. Epub 2012/04/13. 10.1097/MAJ.0b013e31824ba6e0 . [DOI] [PubMed] [Google Scholar]
- 3.Chiu CH, Lee YT, Wang YC, Yin T, Kuo SC, Yang YS, et al. A retrospective study of the incidence, clinical characteristics, identification, and antimicrobial susceptibility of bacteremic isolates of Acinetobacter ursingii. BMC Infect Dis. 2015;15(1):400 Epub 2015/10/02. 10.1186/s12879-015-1145-z [pii]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Epstein SE, Zhou YF, Zhu J. Infection and atherosclerosis: emerging mechanistic paradigms. Circulation. 1999;100(4):e20–8. Epub 1999/07/27. 10.1161/01.cir.100.4.e20 . [DOI] [PubMed] [Google Scholar]
- 5.Rektorschek M, Weeks D, Sachs G, Melchers K. Influence of pH on metabolism and urease activity of Helicobacter pylori. Gastroenterology. 1998;115(3):628–41. Epub 1998/08/28. S0016508598002728 [pii]. 10.1016/s0016-5085(98)70142-8 . [DOI] [PubMed] [Google Scholar]
- 6.Morgan DR, Freedman R, Depew CE, Kraft WG. Growth of Campylobacter pylori in liquid media. J Clin Microbiol. 1987;25(11):2123–5. Epub 1987/11/01. ; Central PMCID: PMC269424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Phillipson M, Atuma C, Henriksnas J, Holm L. The importance of mucus layers and bicarbonate transport in preservation of gastric juxtamucosal pH. Am J Physiol Gastrointest Liver Physiol. 2002;282(2):G211–9. Epub 2002/01/24. 10.1152/ajpgi.00223.2001 . [DOI] [PubMed] [Google Scholar]
- 8.Testerman TL, Morris J. Beyond the stomach: an updated view of Helicobacter pylori pathogenesis, diagnosis, and treatment. World J Gastroenterol. 2014;20(36):12781–808. Epub 2014/10/04. 10.3748/wjg.v20.i36.12781 ; Central PMCID: PMC4177463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Pellicano R, Franceschi F, Saracco G, Fagoonee S, Roccarina D, Gasbarrini A. Helicobacters and extragastric diseases. Helicobacter. 2009;14 Suppl 1:58–68. 10.1111/j.1523-5378.2009.00699.x . [DOI] [PubMed] [Google Scholar]
- 10.Melake NA, Shaker GH, Salama MA. Incidence of Helicobacter pylori infection and their clarithromycin-resistant strains in otitis media with effusion regarding phenotypic and genotypic studies. Saudi Pharm J. 2012;20(4):345–53. Epub 2013/08/21. 10.1016/j.jsps.2012.02.004 SPJ138 [pii]. ; Central PMCID: PMC3745192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Silva LD, Rocha AM, Rocha GA, de Moura SB, Rocha MM, Dani R, et al. The presence of Helicobacter pylori in the liver depends on the Th1, Th17 and Treg cytokine profile of the patient. Mem Inst Oswaldo Cruz. 2011;106(6):748–54. Epub 2011/10/21. S0074-02762011000600016 [pii]. 10.1590/s0074-02762011000600016 . [DOI] [PubMed] [Google Scholar]
- 12.Missall TA, Pruden S, Nelson C, Fohn L, Vidal CI, Hurley MY. Identification of Helicobacter pylori in Skin Biopsy of Prurigo Pigmentosa. Am J Dermatopathol. 2012;34(4):446–8. Epub 2011/12/27. 10.1097/DAD.0b013e318231ae4a . [DOI] [PubMed] [Google Scholar]
- 13.Bansal D, Sharma S, Agarwal S, Saha R, Gupta N. Detection of Helicobacter pylori in Nasal Polyps. Head Neck Pathol. 2016;10(3):306–13. Epub 2016/02/03. 10.1007/s12105-016-0699-4 [pii]. ; Central PMCID: PMC4972759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Backert S, Tegtmeyer N. Type IV Secretion and Signal Transduction of Helicobacter pylori CagA through Interactions with Host Cell Receptors. Toxins (Basel). 2017;9(4). Epub 2017/03/25. E115 [pii] doi: 10.3390/toxins9040115 toxins9040115 [pii]. ; Central PMCID: PMC5408189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Axon AT. Are all helicobacters equal? Mechanisms of gastroduodenal pathology and their clinical implications. Gut. 1999;45 Suppl 1:I1–4. Epub 1999/08/24. 10.1136/gut.45.2008.i1 ; Central PMCID: PMC1766659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hatakeyama M. Oncogenic mechanisms of the Helicobacter pylori CagA protein. Nat Rev Cancer. 2004;4(9):688–94. Epub 2004/09/03. 10.1038/nrc1433 nrc1433 [pii]. . [DOI] [PubMed] [Google Scholar]
- 17.Danesh J. Is there a link between chronic Helicobacter pylori infection and coronary heart disease? Eur J Surg Suppl. 1998;(582):27–31. Epub 1999/02/24. . [DOI] [PubMed] [Google Scholar]
- 18.Danesh J, Wong Y, Ward M, Muir J. Chronic infection with Helicobacter pylori, Chlamydia pneumoniae, or cytomegalovirus: population based study of coronary heart disease. Heart. 1999;81(3):245–7. Epub 1999/02/23. 10.1136/hrt.81.3.245 ; Central PMCID: PMC1728960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Folsom AR, Nieto FJ, Sorlie P, Chambless LE, Graham DY. Helicobacter pylori seropositivity and coronary heart disease incidence. Atherosclerosis Risk In Communities (ARIC) Study Investigators. Circulation. 1998;98(9):845–50. Epub 1998/09/17. 10.1161/01.cir.98.9.845 . [DOI] [PubMed] [Google Scholar]
- 20.Pilotto A, Rumor F, Franceschi M, Leandro G, Novello R, Soffiati G, et al. Lack of association between Helicobacter pylori infection and extracardiac atherosclerosis in dyspeptic elderly subjects. Age Ageing. 1999;28(4):367–71. Epub 1999/08/25. 10.1093/ageing/28.4.367 . [DOI] [PubMed] [Google Scholar]
- 21.Niccoli G, Roberto M, D'Amario D, Scalone G, Fracassi F, Cosentino N, et al. Cytotoxin-associated gene antigen-positive strains of Helicobacter pylori and recurring acute coronary syndromes. Eur Heart J Acute Cardiovasc Care. 2016. Epub 2016/01/23. 2048872615627708 [pii] 10.1177/2048872615627708 . [DOI] [PubMed] [Google Scholar]
- 22.Franceschi F, Niccoli G, Ferrante G, Gasbarrini A, Baldi A, Candelli M, et al. CagA antigen of Helicobacter pylori and coronary instability: insight from a clinico-pathological study and a meta-analysis of 4241 cases. Atherosclerosis. 2009;202(2):535–42. Epub 2008/07/05. S0021-9150(08)00348-1 [pii] 10.1016/j.atherosclerosis.2008.04.051 . [DOI] [PubMed] [Google Scholar]
- 23.Wu Y, Tao Z, Song C, Jia Q, Bai J, Zhi K, et al. Overexpression of YKL-40 predicts plaque instability in carotid atherosclerosis with CagA-positive Helicobacter pylori infection. PLoS One. 2013;8(4):e59996 Epub 2013/04/11. 10.1371/journal.pone.0059996 PONE-D-12-28018 [pii]. ; Central PMCID: PMC3616092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mayr M, Kiechl S, Mendall MA, Willeit J, Wick G, Xu Q. Increased risk of atherosclerosis is confined to CagA-positive Helicobacter pylori strains: prospective results from the Bruneck study. Stroke. 2003;34(3):610–5. Epub 2003/03/08. 10.1161/01.STR.0000058481.82639.EF 01.STR.0000058481.82639.EF [pii]. . [DOI] [PubMed] [Google Scholar]
- 25.Xu Y, Wang Q, Liu Y, Cui R, Lu K, Zhao Y. Association between Helicobacter pylori infection and carotid atherosclerosis in patients with vascular dementia. J Neurol Sci. 2016;362:73–7. Epub 2016/03/06. 10.1016/j.jns.2016.01.025 S0022-510X(16)30025-9 [pii]. . [DOI] [PubMed] [Google Scholar]
- 26.Farsak B, Yildirir A, Akyon Y, Pinar A, Oc M, Boke E, et al. Detection of Chlamydia pneumoniae and Helicobacter pylori DNA in human atherosclerotic plaques by PCR. J Clin Microbiol. 2000;38(12):4408–11. Epub 2000/12/02. ; Central PMCID: PMC87613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kowalski M. Helicobacter pylori (H. pylori) infection in coronary artery disease: influence of H. pylori eradication on coronary artery lumen after percutaneous transluminal coronary angioplasty. The detection of H. pylori specific DNA in human coronary atherosclerotic plaque. J Physiol Pharmacol. 2001;52(1 Suppl 1):3–31. Epub 2002/01/25. . [PubMed] [Google Scholar]
- 28.Adiloglu AK, Ocal A, Can R, Duver H, Yavuz T, Aridogan BC. Detection of Helicobacter pylori and Chlamydia pneumoniae DNA in human coronary arteries and evaluation of the results with serologic evidence of inflammation. Saudi Med J. 2005;26(7):1068–74. Epub 2005/07/28. 20050051' [pii]. . [PubMed] [Google Scholar]
- 29.Kaplan M, Yavuz SS, Cinar B, Koksal V, Kut MS, Yapici F, et al. Detection of Chlamydia pneumoniae and Helicobacter pylori in atherosclerotic plaques of carotid artery by polymerase chain reaction. Int J Infect Dis. 2006;10(2):116–23. Epub 2005/09/27. S1201-9712(05)00096-2 [pii] 10.1016/j.ijid.2004.10.008 . [DOI] [PubMed] [Google Scholar]
- 30.Iriz E, Cirak MY, Engin ED, Zor MH, Erer D, Ozdogan ME, et al. Detection of Helicobacter pylori DNA in aortic and left internal mammary artery biopsies. Tex Heart Inst J. 2008;35(2):130–5. Epub 2008/07/10. ; Central PMCID: PMC2435450. [PMC free article] [PubMed] [Google Scholar]
- 31.Garcia M, Mulvagh SL, Merz CN, Buring JE, Manson JE. Cardiovascular Disease in Women: Clinical Perspectives. Circ Res. 2016;118(8):1273–93. Epub 2016/04/16. 10.1161/CIRCRESAHA.116.307547 CIRCRESAHA.116.307547 [pii]. ; Central PMCID: PMC4834856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wilmot KA, O'Flaherty M, Capewell S, Ford ES, Vaccarino V. Coronary Heart Disease Mortality Declines in the United States From 1979 Through 2011: Evidence for Stagnation in Young Adults, Especially Women. Circulation. 2015;132(11):997–1002. Epub 2015/08/26. 10.1161/CIRCULATIONAHA.115.015293 CIRCULATIONAHA.115.015293 [pii]. ; Central PMCID: PMC4828724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kaplan JR, Adams MR, Clarkson TB, Manuck SB, Shively CA. Social behavior and gender in biomedical investigations using monkeys: studies in atherogenesis. Lab Anim Sci. 1991;41(4):334–43. Epub 1991/08/01. . [PubMed] [Google Scholar]
- 34.Rudel LL. Genetic factors influence the atherogenic response of lipoproteins to dietary fat and cholesterol in nonhuman primates. J Am Coll Nutr. 1997;16(4):306–12. Epub 1997/08/01. 9263179. [DOI] [PubMed] [Google Scholar]
- 35.Weingand KW. Atherosclerosis research in cynomolgus monkeys (Macaca fascicularis). Exp Mol Pathol. 1989;50(1):1–15. Epub 1989/02/01. 0014-4800(89)90052-X [pii]. . [DOI] [PubMed] [Google Scholar]
- 36.Bond MG, Bullock BC, Bellinger DA, Hamm TE. Myocardial infarction in a large colony of nonhuman primates with coronary artery atherosclerosis. Am J Pathol. 1980;101(3):675–92. Epub 1980/12/01. ; Central PMCID: PMC1903666. [PMC free article] [PubMed] [Google Scholar]
- 37.Kaplan JR, Manuck SB. Antiatherogenic effects of beta-adrenergic blocking agents: theoretical, experimental, and epidemiologic considerations. Am Heart J. 1994;128(6 Pt 2):1316–28. Epub 1994/12/01. 0002-8703(94)90254-2 [pii]. 10.1016/0002-8703(94)90254-2 . [DOI] [PubMed] [Google Scholar]
- 38.Melendez GC, Register TC, Appt SE, Clarkson TB, Franke AA, Kaplan JR. Beneficial effects of soy supplementation on postmenopausal atherosclerosis are dependent on pretreatment stage of plaque progression. Menopause. 2015;22(3):289–96. Epub 2014/07/30. 10.1097/GME.0000000000000307 ; Central PMCID: PMC4309745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kaplan JR, Chen H, Appt SE, Lees CJ, Franke AA, Berga SL, et al. Impairment of ovarian function and associated health-related abnormalities are attributable to low social status in premenopausal monkeys and not mitigated by a high-isoflavone soy diet. Hum Reprod. 2010;25(12):3083–94. Epub 2010/10/20. 10.1093/humrep/deq288 deq288 [pii]. ; Central PMCID: PMC2989874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kaplan JR, Manuck SB. Status, stress, and atherosclerosis: the role of environment and individual behavior. Ann N Y Acad Sci. 1999;896:145–61. Epub 2000/02/22. 10.1111/j.1749-6632.1999.tb08112.x . [DOI] [PubMed] [Google Scholar]
- 41.Kaplan JR, Chen H, Manuck SB. The relationship between social status and atherosclerosis in male and female monkeys as revealed by meta-analysis. Am J Primatol. 2009;71(9):732–41. Epub 2009/05/20. 10.1002/ajp.20707 ; Central PMCID: PMC3366586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Adams MR, Golden DL, Williams JK, Franke AA, Register TC, Kaplan JR. Soy protein containing isoflavones reduces the size of atherosclerotic plaques without affecting coronary artery reactivity in adult male monkeys. J Nutr. 2005;135(12):2852–6. Epub 2005/12/01. 135/12/2852 [pii]. 10.1093/jn/135.12.2852 . [DOI] [PubMed] [Google Scholar]
- 43.Register TC, Cann JA, Kaplan JR, Williams JK, Adams MR, Morgan TM, et al. Effects of soy isoflavones and conjugated equine estrogens on inflammatory markers in atherosclerotic, ovariectomized monkeys. J Clin Endocrinol Metab. 2005;90(3):1734–40. Epub 2004/12/09. jc.2004-0939 [pii] 10.1210/jc.2004-0939 . [DOI] [PubMed] [Google Scholar]
- 44.Walker SE, Register TC, Appt SE, Adams MR, Clarkson TB, Chen H, et al. Plasma lipid-dependent and -independent effects of dietary soy protein and social status on atherogenesis in premenopausal monkeys: implications for postmenopausal atherosclerosis burden. Menopause. 2008;15(5):950–7. Epub 2008/04/23. 10.1097/gme.0b013e3181612cef ; Central PMCID: PMC3417141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Register TC, Adams MR. Coronary artery and cultured aortic smooth muscle cells express mRNA for both the classical estrogen receptor and the newly described estrogen receptor beta. J Steroid Biochem Mol Biol. 1998;64(3–4):187–91. Epub 1998/05/30. S0960-0760(97)00155-6 [pii]. . [DOI] [PubMed] [Google Scholar]
- 46.Chomczynski P. A reagent for the single-step simultaneous isolation of RNA, DNA and proteins from cell and tissue samples. Biotechniques. 1993;15(3):532–4, 6–7. Epub 1993/09/01. . [PubMed] [Google Scholar]
- 47.Vandesompele J, De Preter K, Pattyn F, Poppe B, Van Roy N, De Paepe A, et al. Accurate normalization of real-time quantitative RT-PCR data by geometric averaging of multiple internal control genes. Genome Biol. 2002;3(7):RESEARCH0034. Epub 2002/08/20. 10.1186/gb-2002-3-7-research0034 ; Central PMCID: PMC126239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Walker SE, Adams MR, Franke AA, Register TC. Effects of dietary soy protein on iliac and carotid artery atherosclerosis and gene expression in male monkeys. Atherosclerosis. 2008;196(1):106–13. Epub 2007/03/21. S0021-9150(07)00113-X [pii] 10.1016/j.atherosclerosis.2007.02.007 ; Central PMCID: PMC2657082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods. 2001;25(4):402–8. Epub 2002/02/16. 10.1006/meth.2001.1262 S1046-2023(01)91262-9 [pii]. . [DOI] [PubMed] [Google Scholar]
- 50.Semino-Mora C, Doi SQ, Marty A, Simko V, Carlstedt I, Dubois A. Intracellular and interstitial expression of Helicobacter pylori virulence genes in gastric precancerous intestinal metaplasia and adenocarcinoma. J Infect Dis. 2003;187(8):1165–77. 10.1086/368133 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Semino-Mora C, Testerman TL, Liu H, Whitmire JM, Studeman K, Jia Y, et al. Antibiotic treatment decreases microbial burden associated with pseudomyxoma peritonei and affects beta-catenin distribution. Clin Cancer Res. 2013;19(14):3966–76. Epub 2013/06/08. 10.1158/1078-0432.CCR-13-0616 1078-0432.CCR-13-0616 [pii]. . [DOI] [PubMed] [Google Scholar]
- 52.Semino-Mora C, Doi S, Marty A, Simko V, Carlstedt I, Dubois A. Intracellular and intersticial expression of Helicobacter pylori virulence genes in gastric precancerous intestinal metaplasia and adenocarcinoma. J Infect Diseases. 2003;187:1165–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Lan ZD, Dai L, Zhuo XL, Feng JC, Xu K, Chi CW. Gene cloning and sequencing of BmK AS and BmK AS-1, two novel neurotoxins from the scorpion Buthus martensi Karsch. Toxicon. 1999;37(5):815–23. Epub 1999/04/29. S0041-0101(98)00221-9 [pii]. 10.1016/s0041-0101(98)00221-9 . [DOI] [PubMed] [Google Scholar]
- 54.Semino-Mora C, Liu H, McAvoy T, Nieroda C, Studeman K, Sardi A, et al. Pseudomyxoma peritonei: is disease progression related to microbial agents? A study of bacteria, MUC2 AND MUC5AC expression in disseminated peritoneal adenomucinosis and peritoneal mucinous carcinomatosis. Ann Surg Oncol. 2008;15(5):1414–23. Epub 2008/02/27. 10.1245/s10434-007-9778-9 ; Central PMCID: PMC2570966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Liu H, Rahman A, Semino-Mora C, Doi SQ, Dubois A. Specific and sensitive detection of H. pylori in biological specimens by real-time RT-PCR and in situ hybridization. PLoS One. 2008;3(7):e2689 10.1371/journal.pone.0002689 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Aspholm M, Olfat FO, Norden J, Sonden B, Lundberg C, Sjostrom R, et al. SabA is the H. pylori hemagglutinin and is polymorphic in binding to sialylated glycans. PLoS Pathog. 2006;2(10):e110 10.1371/journal.ppat.0020110 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Nam SY, Ryu KH, Park BJ, Park S. Effects of Helicobacter pylori Infection and Its Eradication on Lipid Profiles and Cardiovascular Diseases. Helicobacter. 2015;20(2):125–32. Epub 2014/11/11. 10.1111/hel.12182 . [DOI] [PubMed] [Google Scholar]
- 58.Gen R, Demir M, Ataseven H. Effect of Helicobacter pylori eradication on insulin resistance, serum lipids and low-grade inflammation. South Med J. 2010;103(3):190–6. Epub 2010/02/06. 10.1097/SMJ.0b013e3181cf373f . [DOI] [PubMed] [Google Scholar]
- 59.Rosenfeld ME, Campbell LA. Pathogens and atherosclerosis: update on the potential contribution of multiple infectious organisms to the pathogenesis of atherosclerosis. Thromb Haemost. 2011;106(5):858–67. Epub 2011/10/21. 11-06-0392 [pii] 10.1160/TH11-06-0392 . [DOI] [PubMed] [Google Scholar]
- 60.Skoog EC, Deck SL, Entwistle HD, Hansen LM, Solnick JV. Characterization of the Cag pathogenicity island in Helicobacter pylori from naturally infected rhesus macaques. FEMS Microbiol Lett. 2016;363(24). Epub 2016/12/13. fnw275 [pii] 10.1093/femsle/fnw275 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Drevon-Gaillot E, Perron-Lepage MF, Clement C, Burnett R. A review of background findings in cynomolgus monkeys (Macaca fascicularis) from three different geographical origins. Exp Toxicol Pathol. 2006;58(2–3):77–88. Epub 2006/09/21. S0940-2993(06)00091-1 [pii] 10.1016/j.etp.2006.07.003 . [DOI] [PubMed] [Google Scholar]
- 62.Doi SQ, Kimbason T, Reindel J, Dubois A. Molecular characterization of Helicobacter pylori strains isolated from cynomolgus monkeys (M. fascicularis). Vet Microbiol. 2005;108(1–2):133–9. Epub 2005/05/12. S0378-1135(05)00135-5 [pii] 10.1016/j.vetmic.2005.04.003 ; Central PMCID: PMC2596641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Liu H, Fero JB, Mendez M, Carpenter BM, Servetas SL, Rahman A, et al. Analysis of a single Helicobacter pylori strain over a 10-year period in a primate model. Int J Med Microbiol. 2015;305(3):392–403. Epub 2015/03/26. 10.1016/j.ijmm.2015.03.002 S1438-4221(15)00021-1 [pii]. ; Central PMCID: PMC4376324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Liu H, Merrell DS, Semino-Mora C, Goldman M, Rahman A, Mog S, et al. Diet synergistically affects helicobacter pylori-induced gastric carcinogenesis in nonhuman primates. Gastroenterology. 2009;137(4):1367–79 e1-6. Epub 2009/07/23. S0016-5085(09)01161-5 [pii] 10.1053/j.gastro.2009.07.041 ; Central PMCID: PMC2774828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Hougaard DM, Hansen H, Larsson LI. Non-radioactive in situ hybridization for mRNA with emphasis on the use of oligodeoxynucleotide probes. Histochem Cell Biol. 1997;108(4–5):335–44. Epub 1997/12/05. . [DOI] [PubMed] [Google Scholar]
- 66.Tajbakhsh S, Samarbaf-Zadeh AR, Moosavian M. Comparison of fluorescent in situ hybridization and histological method for the diagnosis of Helicobacter pylori in gastric biopsy samples. Med Sci Monit. 2008;14(9):BR183–7. Epub 2008/09/02. 867956 [pii]. . [PubMed] [Google Scholar]
- 67.Irani S, Monsef Esfahani A, Bidari Zerehpoush F. Detection of Helicobacter pylori in Oral Lesions. J Dent Res Dent Clin Dent Prospects. 2013;7(4):230–7. Epub 2014/03/01. 10.5681/joddd.2013.037 ; Central PMCID: PMC3935555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Shimomura H, Hosoda K, McGee DJ, Hayashi S, Yokota K, Hirai Y. Detoxification of 7-dehydrocholesterol fatal to Helicobacter pylori is a novel role of cholesterol glucosylation. J Bacteriol. 2013;195(2):359–67. Epub 2012/11/13. 10.1128/JB.01495-12 JB.01495-12 [pii]. ; Central PMCID: PMC3553845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Trainor EA, Horton KE, Savage PB, Testerman TL, McGee DJ. Role of the HefC efflux pump in Helicobacter pylori cholesterol-dependent resistance to ceragenins and bile salts. Infect Immun. 2011;79(1):88–97. Epub 2010/10/27. 10.1128/IAI.00974-09 IAI.00974-09 [pii]. ; Central PMCID: PMC3019907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Tegtmeyer N, Wessler S, Backert S. Role of the cag-pathogenicity island encoded type IV secretion system in Helicobacter pylori pathogenesis. FEBS J. 2011;278(8):1190–202. Epub 2011/03/01. 10.1111/j.1742-4658.2011.08035.x ; Central PMCID: PMC3070773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Varga MG, Shaffer CL, Sierra JC, Suarez G, Piazuelo MB, Whitaker ME, et al. Pathogenic Helicobacter pylori strains translocate DNA and activate TLR9 via the cancer-associated cag type IV secretion system. Oncogene. 2016;35(48):6262–9. Epub 2016/05/10. 10.1038/onc.2016.158 [pii]. ; Central PMCID: PMC5102820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Pietroiusti A, Diomedi M, Silvestrini M, Cupini LM, Luzzi I, Gomez-Miguel MJ, et al. Cytotoxin-associated gene-A—positive Helicobacter pylori strains are associated with atherosclerotic stroke. Circulation. 2002;106(5):580–4. Epub 2002/07/31. 10.1161/01.cir.0000023894.10871.2f . [DOI] [PubMed] [Google Scholar]
- 73.Gabrielli M, Santoliquido A, Cremonini F, Cicconi V, Candelli M, Serricchio M, et al. CagA-positive cytotoxic H. pylori strains as a link between plaque instability and atherosclerotic stroke. Eur Heart J. 2004;25(1):64–8. Epub 2003/12/20. 10.1016/j.ehj.2003.10.004 [pii]. . [DOI] [PubMed] [Google Scholar]
- 74.Niccoli G, Franceschi F, Cosentino N, Giupponi B, De Marco G, Merra G, et al. Coronary atherosclerotic burden in patients with infection by CagA-positive strains of Helicobacter pylori. Coron Artery Dis. 2010;21(4):217–21. Epub 2010/04/15. 10.1097/MCA.0b013e3283399f36 . [DOI] [PubMed] [Google Scholar]
- 75.Franceschi F, Sepulveda AR, Gasbarrini A, Pola P, Silveri NG, Gasbarrini G, et al. Cross-reactivity of anti-CagA antibodies with vascular wall antigens: possible pathogenic link between Helicobacter pylori infection and atherosclerosis. Circulation. 2002;106(4):430–4. Epub 2002/07/24. 10.1161/01.cir.0000024100.90140.19 . [DOI] [PubMed] [Google Scholar]
- 76.Michalkiewicz J, Helmin-Basa A, Grzywa R, Czerwionka-Szaflarska M, Szaflarska-Poplawska A, Mierzwa G, et al. Innate immunity components and cytokines in gastric mucosa in children with Helicobacter pylori infection. Mediators Inflamm. 2015;2015:176726 Epub 2015/05/08. 10.1155/2015/176726 ; Central PMCID: PMC4407632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Yamaoka Y, Kita M, Kodama T, Imamura S, Ohno T, Sawai N, et al. Helicobacter pylori infection in mice: Role of outer membrane proteins in colonization and inflammation. Gastroenterology. 2002;123(6):1992–2004. Epub 2002/11/28. 10.1053/gast.2002.37074 [pii]. . [DOI] [PubMed] [Google Scholar]
- 78.Sheu BS, Odenbreit S, Hung KH, Liu CP, Sheu SM, Yang HB, et al. Interaction between host gastric Sialyl-Lewis X and H. pylori SabA enhances H. pylori density in patients lacking gastric Lewis B antigen. Am J Gastroenterol. 2006;101(1):36–44. 10.1111/j.1572-0241.2006.00358.x . [DOI] [PubMed] [Google Scholar]
- 79.Drechsler M, Soehnlein O. The complexity of arterial classical monocyte recruitment. J Innate Immun. 2013;5(4):358–66. Epub 2013/04/11. 10.1159/000348795 000348795 [pii]. . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Lin J, Kakkar V, Lu X. Impact of MCP-1 in atherosclerosis. Curr Pharm Des. 2014;20(28):4580–8. Epub 2014/05/28. CPD-EPUB-60635 [pii]. 10.2174/1381612820666140522115801 . [DOI] [PubMed] [Google Scholar]
- 81.Wung BS, Ni CW, Wang DL. ICAM-1 induction by TNFalpha and IL-6 is mediated by distinct pathways via Rac in endothelial cells. J Biomed Sci. 2005;12(1):91–101. Epub 2005/05/03. 10.1007/s11373-004-8170-z . [DOI] [PubMed] [Google Scholar]
- 82.Byrne MF, Corcoran PA, Atherton JC, Sheehan KM, Murray FE, Fitzgerald DJ, et al. Stimulation of adhesion molecule expression by Helicobacter pylori and increased neutrophil adhesion to human umbilical vein endothelial cells. FEBS Lett. 2002;532(3):411–4. Epub 2002/12/17. S0014579302037286 [pii]. 10.1016/s0014-5793(02)03728-6 . [DOI] [PubMed] [Google Scholar]
- 83.Enarsson K, Brisslert M, Backert S, Quiding-Jarbrink M. Helicobacter pylori induces transendothelial migration of activated memory T cells. Infect Immun. 2005;73(2):761–9. Epub 2005/01/25. 73/2/761 [pii] 10.1128/IAI.73.2.761-769.2005 ; Central PMCID: PMC546998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Zhou X, Nicoletti A, Elhage R, Hansson GK. Transfer of CD4(+) T cells aggravates atherosclerosis in immunodeficient apolipoprotein E knockout mice. Circulation. 2000;102(24):2919–22. Epub 2000/01/11. 10.1161/01.cir.102.24.2919 . [DOI] [PubMed] [Google Scholar]
- 85.Gobert AP, Bambou JC, Werts C, Balloy V, Chignard M, Moran AP, et al. Helicobacter pylori heat shock protein 60 mediates interleukin-6 production by macrophages via a toll-like receptor (TLR)-2-, TLR-4-, and myeloid differentiation factor 88-independent mechanism. J Biol Chem. 2004;279(1):245–50. Epub 2003/10/24. 10.1074/jbc.M307858200 M307858200 [pii]. . [DOI] [PubMed] [Google Scholar]
- 86.Innocenti M, Thoreson AC, Ferrero RL, Stromberg E, Bolin I, Eriksson L, et al. Helicobacter pylori-induced activation of human endothelial cells. Infect Immun. 2002;70(8):4581–90. Epub 2002/07/16. 10.1128/IAI.70.8.4581-4590.2002 ; Central PMCID: PMC128191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Lu H, Wu JY, Kudo T, Ohno T, Graham DY, Yamaoka Y. Regulation of interleukin-6 promoter activation in gastric epithelial cells infected with Helicobacter pylori. Mol Biol Cell. 2005;16(10):4954–66. Epub 2005/07/21. E05-05-0426 [pii] 10.1091/mbc.E05-05-0426 ; Central PMCID: PMC1237095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Futagami S, Hiratsuka T, Tatsuguchi A, Suzuki K, Kusunoki M, Shinji Y, et al. Monocyte chemoattractant protein 1 (MCP-1) released from Helicobacter pylori stimulated gastric epithelial cells induces cyclooxygenase 2 expression and activation in T cells. Gut. 2003;52(9):1257–64. Epub 2003/08/13. 10.1136/gut.52.9.1257 ; Central PMCID: PMC1773780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Ameriso SF, Fridman EA, Leiguarda RC, Sevlever GE. Detection of Helicobacter pylori in human carotid atherosclerotic plaques. Stroke. 2001;32(2):385–91. Epub 2001/02/07. 10.1161/01.str.32.2.385 . [DOI] [PubMed] [Google Scholar]
- 90.Figura N, Palazzuoli A, Vaira D, Campagna M, Moretti E, Iacoponi F, et al. Cross-sectional study: CagA-positive Helicobacter pylori infection, acute coronary artery disease and systemic levels of B-type natriuretic peptide. J Clin Pathol. 2014;67(3):251–7. Epub 2013/12/18. 10.1136/jclinpath-2013-201743 [pii]. . [DOI] [PubMed] [Google Scholar]
- 91.Kim TJ, Lee H, Kang M, Kim JE, Choi YH, Min YW, et al. Helicobacter pylori is associated with dyslipidemia but not with other risk factors of cardiovascular disease. Sci Rep. 2016;6:38015 Epub 2016/11/29. 10.1038/srep38015 [pii]. ; Central PMCID: PMC5125092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Elizalde JI, Pique JM, Moreno V, Morillas JD, Elizalde I, Bujanda L, et al. Influence of Helicobacter pylori infection and eradication on blood lipids and fibrinogen. Aliment Pharmacol Ther. 2002;16(3):577–86. Epub 2002/03/06. 1202 [pii]. . [DOI] [PubMed] [Google Scholar]
- 93.Satoh H, Saijo Y, Yoshioka E, Tsutsui H. Helicobacter Pylori infection is a significant risk for modified lipid profile in Japanese male subjects. J Atheroscler Thromb. 2010;17(10):1041–8. Epub 2010/07/09. JST.JSTAGE/jat/5157 [pii]. 10.5551/jat.5157 . [DOI] [PubMed] [Google Scholar]
- 94.Bahadoran Z, Mirmiran P, Zarif-Yeaganeh M, Zojaji H, Azizi F. Helicobacter pylori Stool Antigen Levels and Serological Biomarkers of Gastric Inflammation are Associated with Cardio-Metabolic Risk Factors in Type 2 Diabetic Patients. Endocrinol Metab (Seoul). 2015;30(3):280–7. Epub 2015/10/06. 10.3803/EnM.2015.30.3.280 [pii]. ; Central PMCID: PMC4595352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Torisu T, Takata Y, Ansai T, Matsumoto T, Sonoki K, Soh I, et al. Possible association of atrophic gastritis and arterial stiffness in healthy middle-aged Japanese. J Atheroscler Thromb. 2009;16(5):691–7. Epub 2009/09/05. JST.JSTAGE/jat/943 [pii]. 10.5551/jat.943 . [DOI] [PubMed] [Google Scholar]
- 96.Kaplan JR, Manuck SB. Premenopausal Reproductive Health Modulates Future Cardiovascular Risk—Comparative Evidence from Monkeys and Women. Yale J Biol Med. 2017;90(3):499–507. Epub 2017/09/29. ; Central PMCID: PMC5612192. [PMC free article] [PubMed] [Google Scholar]
- 97.Gencel VB, Benjamin MM, Bahou SN, Khalil RA. Vascular effects of phytoestrogens and alternative menopausal hormone therapy in cardiovascular disease. Mini Rev Med Chem. 2012;12(2):149–74. Epub 2011/11/11. BSP/MRMC/E-Pub/270 [pii]. ; Central PMCID: PMC3288319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Eusebi LH, Zagari RM, Bazzoli F. Epidemiology of Helicobacter pylori infection. Helicobacter. 2014;19 Suppl 1:1–5. Epub 2014/08/30. 10.1111/hel.12165 . [DOI] [PubMed] [Google Scholar]
- 99.Silk JB. Practice random acts of aggression and senseless acts of intimidation: The logic of status contests in social groups. Evolutionary Anthropology. 2002;11(6):221–5. 10.1002/evan.10038 [DOI] [Google Scholar]
- 100.Goh KL, Chan WK, Shiota S, Yamaoka Y. Epidemiology of Helicobacter pylori infection and public health implications. Helicobacter. 2011;16 Suppl 1:1–9. Epub 2011/09/16. 10.1111/j.1523-5378.2011.00874.x ; Central PMCID: PMC3719046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Weyermann M, Rothenbacher D, Brenner H. Acquisition of Helicobacter pylori infection in early childhood: independent contributions of infected mothers, fathers, and siblings. Am J Gastroenterol. 2009;104(1):182–9. Epub 2008/12/23. 10.1038/ajg.2008.61 [pii]. . [DOI] [PubMed] [Google Scholar]
- 102.Georgopoulos SD, Mentis AF, Spiliadis CA, Tzouvelekis LS, Tzelepi E, Moshopoulos A, et al. Helicobacter pylori infection in spouses of patients with duodenal ulcers and comparison of ribosomal RNA gene patterns. Gut. 1996;39(5):634–8. Epub 1996/11/01. 10.1136/gut.39.5.634 ; Central PMCID: PMC1383384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Sari YS, Can D, Tunali V, Sahin O, Koc O, Bender O. H pylori: Treatment for the patient only or the whole family? World J Gastroenterol. 2008;14(8):1244–7. Epub 2008/02/27. 10.3748/wjg.14.1244 ; Central PMCID: PMC2690673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Solnick JV, Fong J, Hansen LM, Chang K, Canfield DR, Parsonnet J. Acquisition of Helicobacter pylori infection in rhesus macaques is most consistent with oral-oral transmission. J Clin Microbiol. 2006;44(10):3799–803. Epub 2006/10/06. 44/10/3799 [pii] 10.1128/JCM.01482-06 ; Central PMCID: PMC1594807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Linden S, Mahdavi J, Semino-Mora C, Olsen C, Carlstedt I, Boren T, et al. Role of ABO secretor status in mucosal innate immunity and H. pylori infection. PLoS Pathog. 2008;4(1):e2 Epub 2008/01/09. 10.1371/journal.ppat.0040002 [pii]. ; Central PMCID: PMC2174967. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
(XLSX)
Data Availability Statement
All relevant data are within the manuscript and its Supporting Information files.