

Abstract
Purpose
To determine the safety, pharmacokinetics, and recommended phase 2 dose of an antibody drug conjugate (ADC) targeting ectonucleotide phosphodiesterases-pyrophosphatase 3 (ENPP3) conjugated to monomethyl auristatin F (MMAF) in subjects with advanced metastatic renal cell carcinoma (mRCC).
Experimental Design
Two phase 1 studies were conducted sequentially with 2 ADCs considered equivalent, hybridoma derived AGS-16M8F and Chinese Hamster Ovary derived AGS-16C3F. AGS-16M8F was administered intravenously every 3 weeks at 5 dose levels ranging from 0.6 to 4.8 mg/kg until unacceptable toxicity or progression. The study was terminated before reaching the maximum tolerated dose (MTD). A second study with AGS-16C3F started with the AGS-16M8F bridging dose of 4.8 mg/kg given every 3 weeks.
Results
The AGS-16M8F study (n=26) closed before reaching the MTD. The median duration of treatment was 12 weeks (1.7 - 83 weeks). One subject had durable partial remission (PR) (83 weeks) and 1 subject had prolonged stable disease (48 weeks). In the AGS-16C3F study (n=34), the protocol defined MTD was 3.6 mg/kg but this was not tolerated in multiple doses. Reversible keratopathy was dose limiting and required multiple dose de-escalations. The 1.8 mg/kg dose was determined to be safe and was associated with clinically relevant signs of antitumor response. Three of 13 subjects at 1.8 mg/kg had durable PRs (range 100-143 weeks). Eight (8) subjects at 2.7 mg/kg and 1.8 mg/kg had disease control > 37 weeks (37.5 – 141 weeks).
Conclusion
AGS-16C3F was tolerated and had durable antitumor activity at 1.8 mg/kg every 3 weeks.
INTRODUCTION
The mechanisms of resistance to chemotherapy in renal cell carcinoma (RCC) are largely unknown (1). Several hypotheses have been put forward to explain this, including increased drug efflux due to overexpression of ATP-driven pumps such as PgP (MDR1) and modified expression of tubulin isotypes affecting sensitivity to taxanes (4) (5). ADCs represent a different modality of chemotherapy in which a potent payload is delivered specifically to target-positive tumor cells, sparing normal cells to a large degree (6) (7). Importantly, due to the long half-life of ADCs, the exposure to the active component is significantly increased from hours to days when compared to traditional chemotherapy (6). This increased exposure and higher potency of the active payload for an ADC compared with traditional chemotherapy may contribute to overcoming resistance mechanisms. Furthermore, ADCs containing the non-cleavable linker mcMMAF liberate the active moiety Cys-mcMMAF after processing in lysosomes (8). Cys-mcMMAF is not very membrane permeable because it is positively charged at a physiological pH (8), facilitating accumulation in target cells and, theoretically it is also a poor MDR1 substrate (6), reducing drug efflux. Altogether, these properties suggest that ADCs may represent a feasible treatment for RCC.
AGS-16M8F and AGS-16C3F are ADCs composed of fully human IgG2a antibodies conjugated to MMAF via a non-cleavable linker (9). The antibody components target ectonucleotide pyrophosphatase/phosphodiesterase 3 (ENPP3: CD203a), a member of the ENPP family. ENPP3 is expressed in a subset of renal tubules and on activated basophils/mast cells. Among cancers, ENPP3 is expressed by most RCC of clear cell histology (94%) and about 60% of those with papillary histology (9). Preclinical experiments confirmed that both ADCs internalize and induce cytotoxicity in both in vitro and in vivo models of RCC (9).
The first product tested in humans was the cell line-derived hybridoma designated AGS-16M8F(Hyb). While production of ADCs via hybridomas is cost and time effective for limited scale production, the method is not suitable for scaled up production. We planned to switch to a Chinese Hamster Ovary (CHO) cell line system, which is suitable for later stage development, after safety and biological activities were evaluated with AGS-16M8F(Hyb). The first study was terminated before reaching the MTD when the CHO-derived product, designated AGS-16C3F(CHO) became available. A new phase 1 study with AGS-16C3F(CHO) was implemented from where the first study left off to continue dose escalation and to determine the AGS-16C3F(CHO) dose suitable for late stage development.
MATERIALS AND METHODS
Objectives
For both studies in subjects with mRCC, the primary objective was to evaluate the safety and pharmacokinetics of AGS-16M8F(Hyb)/AGS-16C3F(CHO), and the secondary objective was to assess the immunogenicity and effectiveness of AGS-16M8F(Hyb)/AGS-16C3F(CHO).
Study Population
In both studies, eligibility criteria included a diagnosis of mRCC of all histologies, age ≥ 18 years, Eastern Cooperative Oncology Group (ECOG) performance status ≤ 1, adequate organ and bone marrow function; creatinine ≤ 1.5× upper limit of normal (ULN) or calculated creatinine clearance > 50 mL/min; total bilirubin ≤ 1.5× ULN; AST/ALT ≤ 2.5× ULN (or ≤ 5× ULN if known hepatic metastases); absolute neutrophil count ≥ 1,500/μL; and platelet count ≥ 100,000/μL. Exclusion criteria common to the two studies included: uncontrolled CNS metastases, significant underlying cardiac problems, investigational therapy within 4 weeks of enrollment, or thrombotic events in the prior 3 months. In addition, the AGS-16C3F(CHO) study required measurable disease as defined by RECIST version 1.1 (10), at least one prior anti-angiogenic therapy for subjects with clear cell histology, and ENPP3 positivity by immunohistochemistry for subjects with non-clear cell histology. Subjects who had recent cataract surgery or ocular disorders significantly affecting vision were excluded.
Study Design
The AGS-16M8F(Hyb) and AGS-16C3F(CHO) studies were phase 1, open-label, dose-escalation clinical trials. The institutional review boards at all participating institutions approved the study protocols and all subjects gave written informed consent. The studies were registered in ClinicalTrials.gov (identifier and , respectively) and were conducted in accordance with Good Clinical Practice guidelines, as provided by the International Council on Harmonisation and principles of the Declaration of Helsinki. The AGS-16M8F(Hyb) study was conducted in 3 study sites in the US from August 2010 to November 2012 and the AGS-16C3F(CHO) study in 9 study sites in US and Canada from July 2012 to February 2017.
The ADCs were given as monotherapy via intravenous infusion over 60 min every 3 weeks until unacceptable toxicity, progression, or investigator decision. Both studies used a 3+3 dose escalation design. The AGS-16C3F(CHO) study also included a dose expansion cohort for clear cell and papillary histologies after the dose escalation phase.
The AGS-16M8F(Hyb) study enrolled 26 subjects and tested 6 dose levels (0.6, 1.2, 1.8, 2.4, 3.6 and 4.8 mg/kg). The protocol was designed to explore doses up to 8 mg/kg. However, when the CHO-derived AGS-16C3F(CHO) became available, the AGS-16M8F(Hyb) study closed and a phase 1 study of AGS-16C3F(CHO) was opened at the highest dose (4.8 mg/kg) tested in the prior study. Toxicities at 4.8 and 3.6 mg/kg dose levels required amending the protocol to include lower dose levels. The amended protocol added planned dose levels of 2.7, 1.8, 1.2 and 0.6 mg/kg and dose finding was done through dose de-escalation.
Safety
Toxicities were graded according to the National Cancer Institute Common Toxicity Criteria (NCI-CTCAE) version 4.0. A dose-limiting toxicity (DLT) in the dose determining cohorts was defined as an adverse event occurring in Cycle 1 (Day 1 - 22) including any non-laboratory grade 3 or higher adverse event. The following were also DLTs in both studies: Grade 4 neutropenia lasting > 5 days, Grade 4 thrombocytopenia, Grade 3 thrombocytopenia with bleeding, any requirement for a platelet transfusion, and Grade 4 anemia unexplained by underlying disease. In the AGS-16C3F(CHO) study, DLTs also included Grade 3 infusion related reaction not resolving within 24 hours, febrile neutropenia, ≥ Grade 3 neutropenia with bacterial infection, Grade 4 non-hematological laboratory abnormalities, ≥ Grade 3 non-hematological laboratory abnormalities with clinical consequences not resolving within 24 hours, and ALT > 3xULN with bilirubin > 2xULN.
Clinical and laboratory assessments were similar in both studies, with the exception that ophthalmology exams were required at baseline, Cycle 3 and Cycle 5 in the AGS-16C3F(CHO) study.
Pharmacokinetics
In both AGS-16M8F(Hyb) and AGS-16C3F studies, blood samples were collected for pharmacokinetic (PK) analysis. PK analysis measured serum total antibody (serum free antibody + serum antibody drug conjugate), serum ADC, and serum Cys-mcMMAF. PK parameters assessed included maximum observed plasma concentration (Cmax) after the first and fourth dose, time of maximum observed plasma concentration (Tmax) for Cys-mcMMAF only, partial area under the concentration-time curve (AUC), and terminal elimination half-life (t1/2).
Immunogenicity
To assess immunogenicity, subject serum samples were collected prior to cycles 1 - 4 or 5 and every 12 weeks thereafter, and evaluated for anti-drug antibodies to AGS-16M8F(Hyb) or AGS-16C3F(CHO).
Antitumor Effects
Objective response rate (ORR) and disease control rate were assessed by RECIST 1.1 criteria. Disease evaluations were performed at baseline and every 12 weeks thereafter in the AGS-16M8F(Hyb) study and every 8 weeks in the AGS-16C3F(CHO) study.
ENPP3 Expression by Immunohistochemistry (IHC)
A mouse anti-ENPP3 monoclonal antibody (M16-48(4)29.1.1.1) was generated by immunizing Balb/c mice with the ENPP3 extracellular domain (ECD) (9). Sections were stained in a Bond Max IHC autostainer (Leica Biosystems, Buffalo Grove, IL). Antigen retrieval was carried out using proteinase K (Dako). M16-48(4)29.1.1.1 or MOPC21 (negative control), both at 6 μg/mL, were applied to the sections and incubated for 45 min at room temperature. ENPP3 was visualized using the Bond Refine Polymer kit DC9800 (Leica, Buffalo Grove, IL) with 3.3′-diaminobenzidine (DAB) as the chromogen. Positivity was defined as anything greater than an H-score of 0, with one exception where a sample was considered positive based on staining alone (fewer than 100 cells).
Tissue collection was optional for all subjects in the AGS-16M8F(Hyb) study; in the AGS-16C3F(CHO) study, this was required for all non-clear cell subjects and optional for clear cell subjects.
RESULTS
Subjects and Treatment
Subject characteristics in the 2 studies are listed in Table 1.
Table 1.
Summary of Subject Characteristics
| AGS-16M8F(Hyb) | AGS-16C3F(CHO) | ||
|---|---|---|---|
| Enrollment and Histology | 26 total, 19 clear, 7 non-clear | Dose Determining | 14 total, 10 clear, 4 non-clear cell |
| Dose Expansion | 20 total, 15 clear, 5 papillary | ||
| Gender | 19 male, 7 female | 27 male, 7 female | |
| Median Age | 65 (47-80) | 64 (46-84) | |
| ECOG | 0 (10, 38.5%), 1 (15, 57.7%), 1 missing | 0 (11, 32.4%), 1 (23, 67.6%) | |
| Median Prior Therapies | 3 (0-8) 24/26 (92.3% had at least 1 prior line of treatment) | 3 (0-7) 33/34 (97.1%) had at least 1 prior line of treatment | |
The AGS-16M8F(Hyb) study enrolled 26 subjects in 6 dose cohorts (6 at 0.6 mg/kg, 3 each at 1.2, 1.8, 2.4, and 3.6 mg/kg and 8 at 4.8 mg/kg).
The AGS-16C3F(CHO) study enrolled 34 subjects. Fourteen (14) subjects were treated in the dose-determining phase; 2 at 4.8 mg/kg, and 6 each at 3.6 and 2.7 mg/kg. Twenty (20) subjects were treated in the dose expansion cohort which included both 2.7 and 1.8 mg/kg dose levels. The expansion phase opened at 2.7 mg/kg and enrolled 7 subjects, but the dose was further reduced to 1.8 mg/kg due to toxicity where 3 subjects experienced an ocular AE, of which 2 discontinued treatment due to this AE. Thirteen (13) additional subjects were enrolled in the dose expansion phase for a total of 20 subjects. The decision to dose reduce to 1.8 mg/kg applied to all new subjects enrolled in the expansion phase and all subjects whose treatment was ongoing at that time. Five subjects’ treatment was ongoing at the time at 2.7 mg/kg, 2 from dose determining phase and 3 from the dose expansion phase. These 5 subjects received 1-3 doses at 2.7 mg/kg, but later continued treatment at 1.8 mg/kg (Table 2).
Table 2.
AGS-16C3F(CHO) study dose level de-escalation
| Dose level | Dose Determining | Expansion | Total |
|---|---|---|---|
| 4.8 mg/kg | 2 | 2 | |
| 3.6 mg/kg | 6 | 6 | |
| 2.7 mg/kg | 6 (2 reduced to 1.8 mg/kg) | 7 (3 reduced to 1.8 mg/kg) | 13 |
| 1.8 mg/kg | 13 | 13 |
Safety
In the AGS-16M8F(Hyb) study, all subjects had at least one adverse event (AE). The most common AEs were fatigue, thrombocytopenia, constipation, dyspnea, and nausea. There was only 1 DLT in the 0.6 mg/kg cohort, which was pulmonary embolism with dyspnea and chest pain. Dose escalation continued up to 3.6 mg/kg with 3 subjects in each cohort and without DLTs. At 4.8 mg/kg, a total of 8 subjects were enrolled. Two of the first 6 discontinued after the first dose, 1 due to progressive disease and another for metamorphosia (visual distortions). The decision was then made to enroll 2 additional subjects before closing the study and initiating the study with AGS-16C3F(CHO). There were no DLTs in the 8 subjects at 4.8 mg/kg and hence the MTD of AGS-16M8F(Hyb) was not established before study closure.
The AGS-16C3F(CHO) study started with 2 subjects at 4.8 mg/kg. Both had DLTs: grade 4 keratopathy in one (keratopathy is used as an umbrella term to include any pathology affecting the cornea, i.e., keratitis, microcystic epitheliopathy, superficial keratopathy, etc.) and posterior reversible encephalopathy in the other; the latter subject was on bevacizumab treatment until 10 weeks before the first dose of AGS-16C3F. The MTD was exceeded and the dose was de-escalated. Only 1 of 6 subjects treated at 3.6 mg/kg had a DLT (Gr 4 thrombocytopenia), but 4 of these subjects discontinued therapy after the second dose due to keratopathy (2 subjects) or progressive disease (2 subjects). The dose was again reduced to 2.7 mg/kg and 3 subjects were enrolled. While there were no DLTs in these 3 subjects, 2 subjects had delayed administration of the second dose at a reduced dose of 1.8 mg/kg, 1 subject due to Gr 3 fatigue based on pre-existing condition and another due to Grade 2 creatinine increase. A decision was therefore made to enroll 3 additional subjects at 2.7 mg/kg. No DLTs were reported from the additional 3 subjects and the study team then decided to start the expansion phase at 2.7 mg/kg. The dose initially appeared to be tolerated, but of the first 7 subjects treated, 3 subjects experienced grade 2-4 keratopathy after 1 or 2 doses, and 2 subjects discontinued treatment due to keratopathy, which was reversible. Consequently, the dose was further reduced to 1.8 mg/kg. The expansion phase continued at the 1.8 mg/kg dose level and the 5 subjects who were still being treated at 2.7 mg/kg from both the escalation and expansion cohorts were dose-reduced. The expansion phase enrolled another 13 subjects at 1.8 mg/kg. This dose level was tolerated in multiple doses. Of the 13 subjects enrolled at 1.8 mg/kg, 12 subjects reported changes in the eyes; ophthalmology examination revealed keratitis/keratopathy in 5 subjects (38%), and 7 subjects (54%) experienced ocular symptoms with no obvious keratopathy. Only 1/13 subjects discontinued treatment due to keratopathy. Ocular symptoms and keratopathy were all reversible with discontinuation of the treatment. Overall, all 34 subjects had at least one AE.
The most common (> 20%) AEs from the 2 studies are shown in Table 3. Gr 3-4 AEs are shown in Table 4.
Table 3.
Most common (>20%) AEs
| All Adverse Event | AGS-16M8F(Hyb) N=26 | AGS-16C3F(CHO) N=34 |
|---|---|---|
| Subjects with at least 1 event | 26 (100%) | 34 (100%) |
| Fatigue | 12 (46.2%) | 24 (70.6%) |
| Nausea | 7 (26.9%) | 19 (55.9%) |
| Dry eye | 17 (50.0%) | |
| Decreased appetite | 15 (44.1%) | |
| Vision blurred | 15 (44.1%) | |
| Vomiting | 13 (38.2%) | |
| Thrombocytopenia | 8 (30.8%) | 12 (35.3%) |
| Headache | 11 (32.4%) | |
| Keratitis | 11 (32.4%) | |
| Anaemia | 10 (29.4%) | |
| Constipation | 8 (30.8%) | 10 (29.4%) |
| Dyspnoea | 7 (26.9%) | 10 (29.4%) |
| Pyrexia | 8 (23.5%) | |
| Epistaxis | 7 (20.6%) | |
| Infusion related reaction | 7 (20.6%) | |
| Oedema peripheral | 7 (20.6%) |
Table 4.
Grade 3-4 AEs (> 1 subject)
| AGS-16M8F(Hyb) N=26 | AGS-16C3F(CHO) N=34 | |
|---|---|---|
| Subjects with at least 1 event | 16 (61.5%) | 25 (73.5%) |
| Adverse Events | ||
| Thrombocytopenia | 3 (11.5%) | 6 (17.6%) |
| Anaemia | 7 (20.6%) | |
| Keratitis | 6 (17.6%) | |
| Fatigue | 4 (11.8%) | |
| Asthenia | 2 (5.9%) | |
| Back pain | 2 (5.9%) | |
| Dry eye | 2 (5.9%) | |
| Hypertension | 2 (5.9%) | |
| Nausea | 2 (5.9%) | |
| Oedema | 2 (5.9%) | |
| Vision blurred | 2 (5.9%) | |
| Vomiting | 2 (5.9%) | |
| Dyspnoea | 2 (7.7%) | |
| Hypophosphataemia | 2 (7.7%) |
Ocular AEs
Ophthalmology exams were not required in the AGS-16M8F(Hyb) study but were required at baseline, week 7, and week 13 in the AGS-16C3F(CHO) study.
In the AGS-16M8F(Hyb) study, ocular AEs were reported only at the 3 highest dose levels from 8 subjects (31%); 1 subject each (33%) at 2.7 and 3.6 mg/kg, and 6 subjects (75%) at 4.8 mg/kg. The 2 subjects at 2.7 and 3.6 mg/kg only reported Gr 1 dry eye. Of the 6 subjects at 4.8 mg/kg dose level with ocular AEs, 4 subjects reported Gr 2 blurred vision, 3 subjects had dry eye (2 Gr 2 and 1 Gr 3), 1 subject had Gr 1 eye pruritis, and 1 subject experienced Gr 1 metamorphosia.
In the AGS-16C3F(CHO) study, 29/34 subjects (85%) experienced ocular signs and symptoms. Keratopathy was diagnosed in 20/34 subjects (59%) and 9/34 subjects (26%) reported ocular symptoms without objective keratopathy. Five subjects (15%) did not report any ocular symptoms and showed no keratopathy; however, most only received 1 dose (2 subjects) or 2 doses (2 subjects) before discontinuing treatment; one subject received 12 doses before discontinuing treatment.
Approximately half of the subjects reported ocular symptoms such as dry eye (17/34, 50%) and blurred vision (15/34, 44%). The most common ophthalmological findings were corneal lesions described as microcysts. While subjects frequently reported ocular signs and symptoms, these were asynchronous with clinical findings.
Gr 3 corneal events were observed in 7/34 subjects (21%) and were reported more frequently at higher dose levels (1/2 at 4.8 mg/kg, 3/6 at 3.6 mg/kg, 2/13 at 2.7 mg/kg and 1/13 at 1.8 mg/kg). Of these 7 subjects, 1 subject at 2.7 mg/kg also experienced a Gr 4 corneal event. Six (6) subjects discontinued study drug due to keratopathy (1 subject (50%) at 4.8 mg/kg, 2 subjects each at 3.6 and 2.7 mg/kg (33% and 15% respectively), and 1 subject (8%) at 1.8 mg/kg); the best overall response for these 6 subjects was 3 PD, 2 SD, and 1NE.
Most changes in the eyes, including corneal changes, were observed early in treatment, within the first two cycles. Overall, while the frequency of ocular AEs was consistent across all dose levels, the severity was dose dependent. Severity decreased with lower doses and at 1.8 mg/kg; the eye symptoms were better tolerated and manageable. In both studies, ocular AE management method, mitigation or intervention, was not specified. The needed interventions for the management of ocular AEs were variable, determined by treating investigators and local ophthalmologists. Interventions included, but were not limited to, lubrication with artificial tears and steroid eye drops.
Keratopathy reported in both studies was reversible after study drug cessation. Time to resolution of keratopathy, both symptoms and objective findings, varied from a few weeks to several months.
Pre-clinical data suggest that these ocular AEs are mediated through macropinocytosis (a regulated form of endocytosis that mediates the non-selective uptake of solute molecules, nutrients and antigens) by corneal epithelial cells, which do not express ENPP3 (11).
Thrombocytopenia
A transient decrease in platelet number was observed in the first 7-15 days in 24/25 subjects (96%) (1 subject had no data) and 32/34 (94%) in subjects treated with AGS-16M8F(Hyb) and AGS-16C3F(CHO) respectively. Importantly, the target for this ADC, ENPP3, is not expressed in platelets or megakaryocytes (12) The median change in platelet count from baseline was −52% (min −6.9%, max −85%) for AGS-16M8F(Hyb) and for AGS-16C3F(CHO), this was −51% (min −8.4% and max −95.8%). The decreased platelet count was mostly Gr 1 and 2. In the AGS-16M8F(Hyb) study, 1 subject each at 1.2, 2.7, and 4.8 mg/kg had at least 1 Gr 3 platelet count decrease during the study. In the AGS-16C3F(CHO) study, 4 subjects (1 subject each at 1.8 and 2.7 mg/kg and 2 subjects at 3.6 mg/kg) experienced Gr 3 and 2 subjects (1 subject each at 2.7 and 3.6 mg/kg) had Gr 4 thrombocytopenia. The Gr 4 event at 3.6 mg/kg was a DLT.
In general, all subjects at all dose levels experienced an initial drop in platelet count after the first dose but there was sufficient recovery spontaneously by Day 22 to receive the next dose. The platelet count stabilized at a lower than baseline threshold during treatment and improved after cessation of treatment. Study subjects were not followed long enough to determine if full recovery of platelet count would occur as subjects continued on to another treatment or died. In the AGS-16C3F(CHO) study, Gr 3 and 4 thrombocytopenia were dose dependent and the incidence of thrombocytopenia decreased with decreasing dose level. At 1.8 mg/kg, 3/13 (23%) subjects reported thrombocytopenia, only one of which was Gr 3.
Bone marrow biopsies performed in 2 subjects at 1.8 mg/kg did not show myelosuppression. One result showed hypercellularity with increased normal megakaryocytes and the other showed normal bone marrow. In the case that showed normal bone marrow, a peripheral blood smear was performed and this result determined that platelet clumping caused the low platelet count. No bleeding events requiring transfusion, or interventions other than nasal tamponade, were observed during thrombocytopenia.
While the mechanism of thrombocytopenia induced by AGS-16C3F(CHO) is not fully understood, pre-clinical data suggest that this is not a direct effect on platelets, but rather through macropinocytosis-mediated uptake by developing megakaryocytes (12).
Pharmacokinetics and Immunogenicity
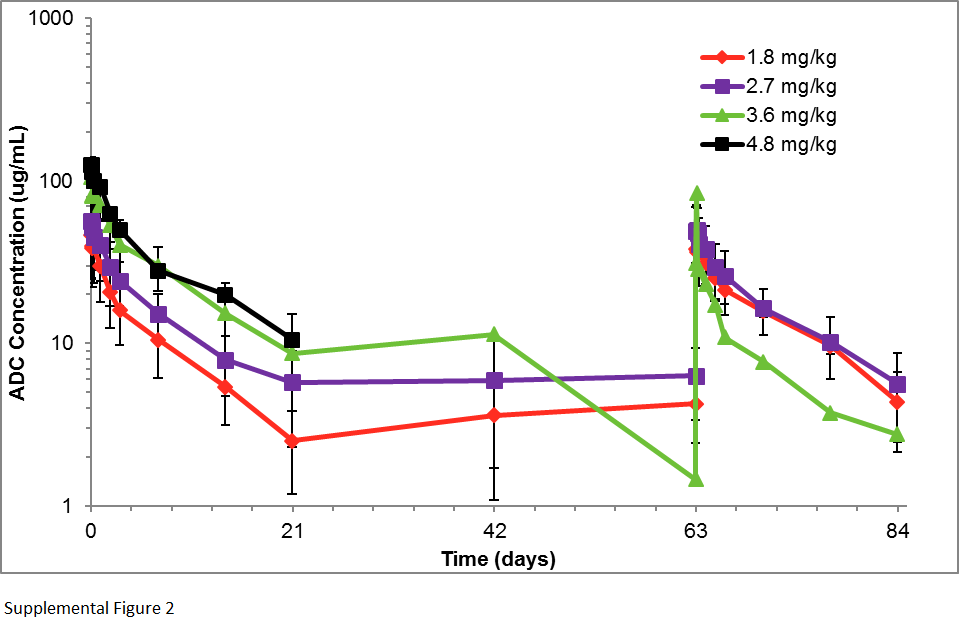
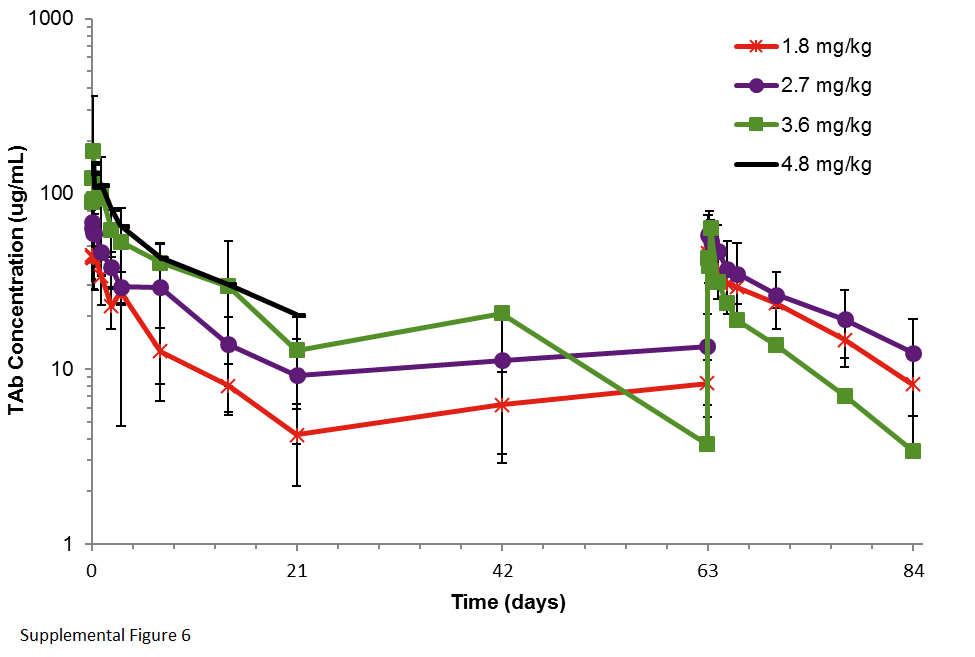
For every 3-week AGS-16M8F(Hyb) and AGS-16C3F(CHO) dosing, Cmax and AUC increased linearly with increasing doses and without significant ADC accumulation (Supplementary Figure 1, Supplementary Figure 2, Supplementary Table 1). The AUC and Cmax for free Cys-mcMMAF increased in line with increases in overall doses of AGS-16M8F(Hyb) and AGS-16C3F(CHO) and no accumulation was observed with repeat administration (Supplementary Figure 3, Supplementary Figure 4, Supplementary Table 2). The median Tmax for peak Cys-mcMMAF concentrations from AGS-16M8F(Hyb) and AGS-16C3F(CHO) was observed at 5 hours (range 3 - 8 hours) for both AGS-16M8F(Hyb) and AGS-16C3F(CHO). The total antibody concentration (TAb), comprising free antibody and ADC, was higher overall compared to ADC concentration alone, implying deconjugation of the ADC (Supplementary Figure 5, Supplementary Figure 6, Supplementary Table 3). Similar to the ADC pharmacokinetics, the AUC and Cmax of the TAb and Cys-mcMMAF increased proportionally to the dose given. As summarized in Supplementary Table 4, the overall pharmacokinetic properties of both ADCs were comparable.
No subject developed immunogenicity during the study (Supplementary Table 5).
Clinical Antitumor Effects
In the AGS-16M8F(Hyb) study, 1 clear cell subject treated at 2.7 mg/kg had a PR at Week 23 and response was confirmed at Week 29. This subject remained on study for 56 weeks. Additionally, 1 clear cell subject treated at 0.6 mg/kg had prolonged SD (48 weeks).
In the AGS-16C3F(CHO) study, three (3) subjects at 1.8 mg/kg achieved durable PR (3/13, 23%), 2 with clear cell (Fuhrman Gr 2 and 3) and 1 with type 2 papillary histology (Fuhrman Gr 3). Response was observed at week 8 (1 subject) and at week 16 (2 subjects). The 3 PR subjects were on treatment for 100, 116, and 143 weeks. The disease control rate at 1.8 mg/kg was 92% (12/13 subjects). The disease control rate for the entire study was 59% (20/34 subjects). The waterfall plot from the AGS-16C3F(CHO) study showing the maximum change from baseline in total tumor burden by best overall response and duration of treatment is shown in Figure 1.
Figure 1.
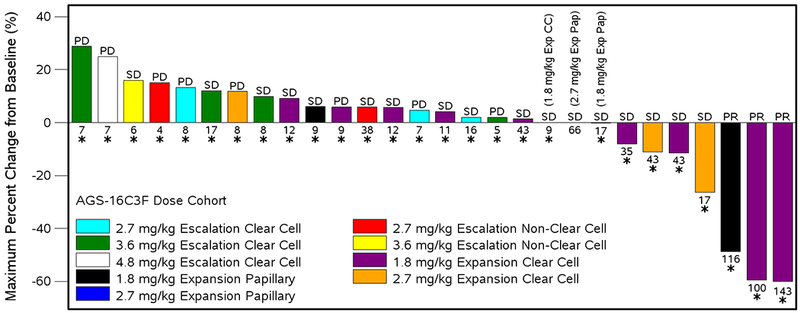
Waterfall Plot for AGS-16C3F(CHO): Maximum percent change from baseline in total tumor burden by best overall response and duration of treatment
Best overall response was determined according to RECIST: CR=complete response, PR=partial response, SD=stable disease, PD=progressive disease
The following subjects are not represented on the graph due to having a nonevaluable best overall response: 0001-0103 (2.7 mg/kg Escalation Non-Clear Cell), 0006-0007 (3.6 mg/kg Escalation Clear Cell), 0001-0002 (4.8 Escalation Non-Clear Cell mg/kg), 0003-1005 (2.7 mg/kg Expansion Clear Cell), 0008-1004 (2.7 mg/kg Epansion Clear Cell), 0003-1102 (2.7 mg/kg Expansion Papillary)
Numbers below the bars represent number of weeks between first dose and decision made to end treatment.
Bars with an asterisk (*) indicate a subject who received prior systemic therapy.
Note: Maximum change is defined as subject’s best response and calculated so that bars below 0 represent good outcomes.
Prog: csr\dev\programs\tfl\f-tumor-wtrfl-dur.sas, f14-3-2-f-tumor-wtrfl-dur.rtf (24May2017:16:41)
Immunohistochemistry
ENPP3 immunohistochemical staining was performed on tumor samples from 66 subjects. Only 63 were evaluable and were analyzed using the H-score system (13); 2 had an insufficient number of tumor cells and one sample was poorly processed, preventing accurate immunohistochemical staining. Of the tumors with clear cell histology, 93% (25/27) were positive, as were 73% (16/22) of the papillary carcinoma samples, although expression was somewhat lower overall in this group. Thirteen (13) of the remaining 14 were either of unclassified (12) or chromophobe type (1) and had mixed expression with 62% (8/13) being positive (Figure 2). The other remaining sample was a lung biopsy from a subject with RCC shown to be a primary squamous cell carcinoma rather than a renal cancer metastasis; this sample was excluded from the IHC analysis (Supplementary Table 6).
Figure 2.
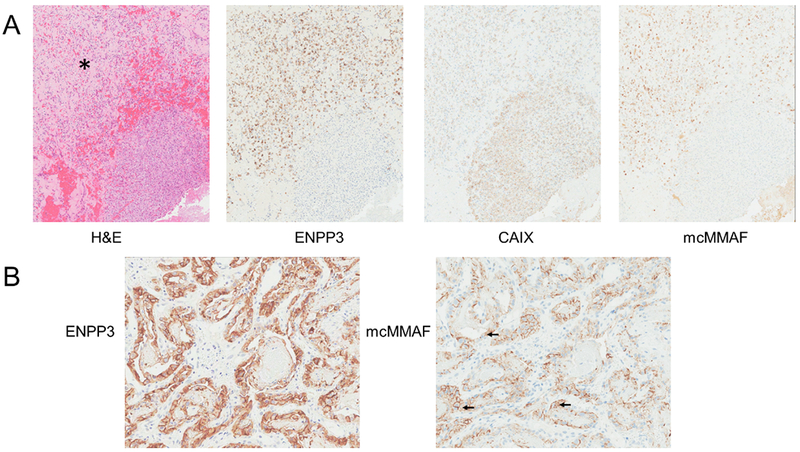
Immunohistochemical characterization of tumor metastases from 2 patients. (A) Sample of tumor from humerus from a patient treated with AGS-16M8F(Hyb) at 2.7 mg/kg who had a PR. From left to right: hematoxylin and eosin showing a fibrotic area (*); ENPP3 staining shows positivity in the area of fibrosis only; CAIX staining shows tumor cell positivity throughout the section; mcMMAF showing areas of positivity where AGS-16M8F(Hyb) may be bound. (B) Sample of tumor from bronchus from a patient treated with AGS-16M8F(Hyb) at 0.6 mg/kg who had an SD, From left to right: ENPP3 staining of the tumor cells; mcMMAF showing areas of positivity where AGS-16M8F(Hyb) may be bound or internalized (arrows).
Samples of tumor metastases were collected from 2 subjects after dosing with AGS-16M8F(Hyb). One of the samples was from a subject treated at 2.7 mg/kg who had a PR. The sample from the primary tumor for this subject had an H-score of 277. The metastatic sample acquired post treatment showed decreased ENPP3 positivity and contained 2 different areas. One area was comprised of tumor cells, as defined by CAIX staining, which were also ENPP3 positive and stained using an antibody that recognizes MMAF, demonstrating binding of AGS-16M8F(Hyb) (Figure 2A). This area includes fibrosis suggestive of cell death. The other area of the section contained tightly packed tumor cells with no fibrosis that were positive for CAIX but negative for ENPP3 and showed no binding of AGS-16M8F(Hyb) (Figure 2A). Lack of ENPP3 expression in these areas suggests a possible mechanism of escape from AGS-16M8F(Hyb) therapy. The second sample was from a subject who had a best overall response of SD. The sample showed high levels of ENPP3 (Figure 2B) expression as well as surface and intracellular staining for AGS-16M8F(Hyb) after dosing with 0.6 mg/kg, the lowest dosage investigated (Figure 2B). Altogether, these data demonstrated specific binding of AGS-16M8F(Hyb) to ENPP3 positive cells.
DISCUSSION
The AGS-16CF phase 1 study results demonstrated that an ADC targeting ENPP3 can be administered safely at 1.8 mg/kg every 3 weeks and has anti-tumor activity in a heavily-pretreated, refractory mRCC population, in both clear cell and papillary histologies. Three (3) of the 13 subjects treated at the recommended phase 2 dose experienced partial responses that lasted between 100 and 143 weeks (23.3 and 33.4 months). These durations of response are longer than the typical durations of response associated with second or later lines of therapy using mTOR inhibitors (9.2 months for everolimus) (Choueiri, personal communication and (14)) or tyrosine kinase inhibitors (13 months for lenvantanib combined with everolimus) (15). In addition, responses to AGS-16C3F(CHO) treatment lasted longer than the subject’s response to their most recent therapy in 2 of the 3 partial response cases, a finding that supports the conclusion that the responses were due to AGS-16C3F(CHO) and not to delayed effects from prior therapy (16).
We observed unexpected differences in the safety profile of AGS-16M8F(Hyb) and AGS-16C3F(CHO), 2 ADCs that contained the same antibody derived from different cell lines and carried the same payload. The two ADCs were deemed comparable based on tests of critical quality attributes that included analytical and preclinical biological characterization (9). Comparability shown by these test results allowed the AGS-16C3F(CHO) study to start with the bridging dose from the AGS-16M8F(Hyb) study.
The human pharmacokinetic properties of the 2 ADCs were comparable as well. Serum concentrations decreased multi-exponentially and exposure was dose-proportional. Repeated administration of AGS-16M8F(Hyb) and AGS-16C3F(CHO) over a 3-week period did not demonstrate a cumulative increase at trough levels to suggest drug accumulation over cycles. In both clinical studies, the mean terminal half-life was approximately 7 - 8 days for the intact drug and approximately 4 days for Cys-MMAF. Despite the fact that the pharmacokinetic profiles of the 2 ADCs were almost superimposable, the bridging dose of 4.8 mg/kg that was tolerable in the AGS-16M8F(Hyb) study was not tolerated in the AGS-16C3F(CHO) study, most notably for ocular AEs. As a result, the AGS-16C3F(CHO) study was modified to become a reverse dose finding study from the starting dose of 4.8 mg/kg. Multiple dose reductions were required to define the MTD and the recommended phase 2 dose of 1.8 mg/kg for AGS-16C3F(CHO).
The immunohistochemical analysis of the metastatic tumors of two patients post AGS-16C3F(CHO) treatment showed the specific targeting of this ADC to ENPP3 positive tumor cells, either membrane bound or internalized in the cytoplasm, supporting the proposed mechanism of action. Importantly, in one of the samples from a patient who experienced a PR, areas of the metastatic tumor collected after clinical progression were ENPP3 negative and AGS-16C3F(CHO) was not found in those cells. Furthermore, Cys-mcMMAF, the active metabolite, has no bystander effect due to its poor membrane permeability. Thus, disappearance of the target, ENPP3, may contribute to resistance to AGS-16C3F(CHO) therapy as it has been suggested for other ADCs (17) (18). Combination of AGS-16C3F(CHO) with other active agents may reduce the impact of the lack of bystander effect and possible loss of ENPP3. Given the small number of subjects treated in each study and 4 objective responses (PRs) in the 2 studies combined, response correlation to target expression, positivity or positivity strength, cannot be made. This is being investigated further in a randomized Phase 2 study. Other potential uses of ADCs targeting ENPP3 include combination use in earlier lines of treatment of mRCC and in mastocytosis.
While ocular signs and symptoms were commonly reported, they were asynchronous with clinical findings. Objective ocular findings were limited to the cornea and were reversible with treatment cessation. ADC-induced keratopathy has been previously described and appears to be an off-target effect. (19) (2) (3)
The most objective way to grade ocular AEs is to use visual acuity of each eye, as grading according to symptoms by definition is subjective. CTCAE v4.0 has vision demarcation at 20/40 (Gr 2 for vision equal to or better than 20/40, Gr 3 for 20/50 to better than 20/200, and Gr 4 for 20/200 and worse). However, the limitation of CTCAE ocular AE vision demarcations is the assumption that a patient has baseline vision of 20/20, which is often not the case. For example, when a patient at baseline with a vision of 20/30 experiences an ocular AE where the vision declines to 20/50, according to CTCAE v4, it would assign the AE as Gr 3 simply because the vision is 20/50. The decline in two lines of visual acuity, however, should be assigned as Gr 2 as is the case for a patient whose vision of 20/20 declines two lines to 20/30. Grading ocular AE by visual acuity demarcations erroneously assigns a higher grade AE, which may be reflected in this study. Future studies using the change in visual acuity (delta vision), accounting for various baseline visions to grade ocular AE will more accurately capture the severity of keratopathy. As such, a modified CTCAE ocular AE grading scale may be beneficial to reach consistency among participating sites.
Systemic therapies for mRCC that are in broad use include the tyrosine kinase inhibitors targeting VEGF +/− MET, mTOR inhibitors, and the immune checkpoint inhibitor nivolumab. Many patients receive serial lines of therapy with tyrosine kinase inhibitors that have similar mechanisms of action, thus limiting the potential benefit of successor therapy on progression-free survival. The results of the AGS-16C3F(CHO) phase 1 study support further development of AGS-16C3F(CHO) or other next generation anti-ENPP3 ADCs as potential new treatments of mRCC.
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
TRANSLATIONAL RELEVANCE.
ADCs are designed to deliver a cytotoxic payload specifically to tumor cells expressing the target, while mostly sparing normal tissue. ENPP3 is a novel target specific to RCC with minimal expression in normal tissue. An ADC composed of an antibody against this target conjugated to the microtubule-disrupting agent MMAF via the linker maleimidocaproyl (mc) was developed (1). This report describes the first in human experience with this ADC. ENPP3 was detected at high levels in archived tumor samples in agreement with preclinical data. Despite historical failures of cytotoxic agents in RCC, AGS-16C3F had encouraging clinical activity with 3 PRs and tolerable toxicity. As reported with other ADCs containing mcMMAF, reversible corneal toxicity was dose limiting and independent of antibody target (2) (3). These findings warrant further clinical investigation of AGS-16C3F for the treatment of RCC.
Acknowledgments
Financial support:
- Agensys, Inc.,
- Patients treated at Memorial Sloan Kettering Cancer Center were supported in part by Memorial Sloan Kettering Cancer Center Support Grant/Core Grant (P30 CA008748)
Footnotes
Disclosure of Potential Conflicts of Interest: Authors include study investigators and employees of Agensys, Inc during the period of the studies.
REFERENCES
- 1.Buti S, Bersanelli M, Sikokis A, Maines F, Facchinetti F, Bria E, et al. Chemotherapy in metastatic renal cell carcinoma today? A systematic review. Anti-Cancer Drugs 2013;24(6):535–54 doi 10.1097/CAD.0b013e3283609ec1. [DOI] [PubMed] [Google Scholar]
- 2.Tannir NM, Forero-Torres A, Ramchandren R, Pal SK, Ansell SM, Infante JR, et al. Phase I dose-escalation study of SGN-75 in patients with CD70-positive relapsed/refractory non-Hodgkin lymphoma or metastatic renal cell carcinoma. Investigational New Drugs 2014;32(6):1246–57 doi 10.1007/s10637-014-0151-0. [DOI] [PubMed] [Google Scholar]
- 3.Eaton JS, Miller PE, Mannis MJ, Murphy CJ. Ocular Adverse Events Associated with Antibody-Drug Conjugates in Human Clinical Trials. J Ocul Pharmacol Ther 2015;31(10):589–604 doi 10.1089/jop.2015.0064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Diamond E, Molina AM, Carbonaro M, Akhtar NH, Giannakakou P, Tagawa ST, et al. Cytotoxic chemotherapy in the treatment of advanced renal cell carcinoma in the era of targeted therapy. Critical Reviews in Oncology/Hematology 2015;96(3):518–26 doi 10.1016/j.critrevonc.2015.08.007. [DOI] [PubMed] [Google Scholar]
- 5.Hartmann JT, Bokemeyer C. Chemotherapy for renal cell carcinoma. Anticancer research 1999;19(2c):1541–3. [PubMed] [Google Scholar]
- 6.Beck A, Goetsch L, Dumontet C, Corvaia N. Strategies and challenges for the next generation of antibody-drug conjugates. Nature reviews Drug discovery 2017;16(5):315–37 doi 10.1038/nrd.2016.268. [DOI] [PubMed] [Google Scholar]
- 7.de Goeij BE, Lambert JM. New developments for antibody-drug conjugate-based therapeutic approaches. Current opinion in immunology 2016;40:14–23 doi 10.1016/j.coi.2016.02.008. [DOI] [PubMed] [Google Scholar]
- 8.Doronina SO, Mendelsohn BA, Bovee TD, Cerveny CG, Alley SC, Meyer DL, et al. Enhanced activity of monomethylauristatin F through monoclonal antibody delivery: effects of linker technology on efficacy and toxicity. Bioconjugate chemistry 2006;17(1):114–24 doi 10.1021/bc0502917. [DOI] [PubMed] [Google Scholar]
- 9.Donate F, Raitano A, Morrison K, An Z, Capo L, Avina H, et al. AGS16F Is a Novel Antibody Drug Conjugate Directed against ENPP3 for the Treatment of Renal Cell Carcinoma. Clin Cancer Res 2016;22(8):1989–99 doi 10.1158/1078-0432.CCR-15-1542. [DOI] [PubMed] [Google Scholar]
- 10.Eisenhauer EA, Therasse P, Bogaerts J, Schwartz LH, Sargent D, Ford R, et al. New response evaluation criteria in solid tumours: revised RECIST guideline (version 1.1). European journal of cancer (Oxford, England : 1990) 2009;45(2):228–47 doi 10.1016/j.ejca.2008.10.026. [DOI] [PubMed] [Google Scholar]
- 11.Zhao H, Atkinson J, Gulesserian S, Zeng Z, Nater J, Ou J, et al. Modulation of Macropinocytosis-Mediated Internalization Decreases Ocular Toxicity of Antibody-Drug Conjugates. Cancer research 2018;78(8):2115–26 doi 10.1158/0008-5472.CAN-17-3202. [DOI] [PubMed] [Google Scholar]
- 12.Zhao H, Gulesserian S, Ganesan SK, Ou J, Morrison K, Zeng Z, et al. Inhibition of Megakaryocyte Differentiation by Antibody-Drug Conjugates (ADCs) is Mediated by Macropinocytosis: Implications for ADC-induced Thrombocytopenia. Mol Cancer Ther 2017;16(9):1877–86 doi 10.1158/1535-7163.MCT-16-0710. [DOI] [PubMed] [Google Scholar]
- 13.McCarty KS Jr., Szabo E, Flowers JL, Cox EB, Leight GS, Miller L, et al. Use of a monoclonal anti-estrogen receptor antibody in the immunohistochemical evaluation of human tumors. Cancer research 1986;46(8 Suppl):4244s–8s. [PubMed] [Google Scholar]
- 14.Motzer RJ, Escudier B, McDermott DF, George S, Hammers HJ, Srinivas S, et al. Nivolumab versus Everolimus in Advanced Renal-Cell Carcinoma. New England Journal of Medicine 2015;373(19):1803–13 doi 10.1056/NEJMoa1510665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Motzer RJ, Hutson TE, Glen H, Michaelson MD, Molina A, Eisen T, et al. Lenvatinib, everolimus, and the combination in patients with metastatic renal cell carcinoma: a randomised, phase 2, open-label, multicentre trial. The Lancet Oncology;16(15):1473–82 doi 10.1016/S1470-2045(15)00290-9. [DOI] [PubMed] [Google Scholar]
- 16.Von Hoff DD, Stephenson JJ, Rosen P, Loesch DM, Borad MJ, Anthony S, et al. Pilot Study Using Molecular Profiling of Patients’ Tumors to Find Potential Targets and Select Treatments for Their Refractory Cancers. Journal of Clinical Oncology 2010;28(33):4877–83 doi 10.1200/JCO.2009.26.5983. [DOI] [PubMed] [Google Scholar]
- 17.Al-Rohil RN, Torres-Cabala CA, Patel A, Tetzlaff MT, Ivan D, Nagarajan P, et al. Loss of CD30 expression after treatment with brentuximab vedotin in a patient with anaplastic large cell lymphoma: a novel finding. J Cutan Pathol 2016;43(12):1161–6 doi 10.1111/cup.12797. [DOI] [PubMed] [Google Scholar]
- 18.Loganzo F, Sung M, Gerber HP. Mechanisms of Resistance to Antibody-Drug Conjugates. Mol Cancer Ther 2016;15(12):2825–34 doi 10.1158/1535-7163.mct-16-0408. [DOI] [PubMed] [Google Scholar]
- 19.Younes A, Kim S, Romaguera J, Copeland A, Farial Sde C, Kwak LW, et al. Phase I multidose-escalation study of the anti-CD19 maytansinoid immunoconjugate SAR3419 administered by intravenous infusion every 3 weeks to patients with relapsed/refractory B-cell lymphoma. Journal of clinical oncology : official journal of the American Society of Clinical Oncology 2012;30(22):2776–82 doi 10.1200/jco.2011.39.4403. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.