

Abstract
Neutrophils are the most abundant circulating leukocyte and the first point of contact between many drug delivery formulations and human cells. Despite their prevalence and implication in a range of immune functions, little is known about how human neutrophils respond to synthetic particulates. Here, we describe how ex vivo human neutrophils respond to particles which vary in both size (5 nm to 2 μm) and chemistry (lipids, poly(styrene), poly(lactic-co-glycolic acid), and gold). In particular, we show that (i) particle uptake is rapid, typically plateauing within 15 min; (ii) for a given particle chemistry, neutrophils preferentially take up larger particles at the nanoscale, up to 200 nm in size; (iii) uptake of nanoscale poly(styrene) and liposomal particles at concentrations of up to 5 μg/mL does not enhance apoptosis, activation, or cell death; (iv) particle-laden neutrophils retain the ability to degranulate normally in response to chemical stimulation; and (v) ingested particles reside in intracellular compartments that are retained during activation and degranulation. Aside from the implications for design of intravenously delivered particulate formulations in general, we expect these observations to be of particular use for targeting nanoparticles to circulating neutrophils, their clearance site (bone marrow), or distal sites of active inflammation.
Keywords: neutrophils, nanoparticles, nanomaterials, leukocytes, drug delivery
Graphical Abstract
The human neutrophil1 is uniquely poised at the locus of two pressing challenges in modern medicine: (i) targeted therapeutic delivery to diseased tissue or cellular subsets2 and (ii) precise, powerful immunomodulation.3 Generated and recycled in the bone marrow and abundant in both human blood (representig 50–70% of circulating leukocytes) and organs such as the liver, spleen,4 and lung,5 neutrophils are perhaps best known for their sentinel-like ability to home to sites of inflammation, attract adaptive immune cells, phagocytose foreign organisms, and “activate”, releasing granules containing a potent suite of immunomodulators, proteases, and biotoxins.6,7 Far from being blunt antibacterial instruments, neutrophil granules come in multiple subtypes (known as azurophil, specific, and gelatinase granules; secretory vesicles also play a role), with each subtype characterized by a different molecular “armory” and released in response to different stimuli. Engineering the release of these potent granules for nonendogenous purposes or delivering synthetic nanoparticles to granules for a hitchhiking-type release within inflammatory environments represent attractive targets in immune engineering. In addition, phenotypically distinct neutrophil subsets have also been implicated in a wide range of inflammatory disorders (e.g., cancer, atherosclerosis, chronic inflammation at biomaterial implants, rheumatoid arthritis, and other auto-immune diseases) and normal immune functions (e.g., immunoregulation, wound healing, and resolution of infection).8
In the context of drug delivery, this combination of high abundance in circulation, natural capacity for tissue homing, phenotypic plasticity, and potency represents an attractive opportunity to (i) facilitate cellular uptake of encapsulated therapeutic payloads that (ii) potentiate specific immune responses at (iii) precise locations within the body, whether distal sites of inflammation or difficult-to-reach anatomical locations like bone marrow. Nevertheless, the neutrophil has been largely ignored in the context of drug delivery, perhaps due to the challenges of working with short-lived (half-life 1–5 days),9,10 terminally differentiated and nonproliferating primary cells. Recent work has (a) utilized neutrophils to deliver therapeutic-bearing nanoparticles to tumors11 and extravascular sites of inflammation;12 (b) modulated the behavior of activated neutrophils with therapeutic nano-particles to resolve active inflammation;13 and (c) studied the influence of nanoscale particulate uptake on neutrophil behavior.14–24 Recent delivery-focused studies have primarily leveraged murine models 11–13,17,25–27; however, mouse neutrophils represent only 10–15% of circulating leukocytes and possess limited ability to accurately reflect neutrophil behavior in humans.28 The few studies which describe interactions of ex vivo human neutrophils and colloidal-scale particles emphasize toxicology and particulates found in environmental pollutants,14,20 assess isolated particulate formulations,29,19,21 or utilize parameters (concentration, exposure time, materials, etc.) that fall outside the bounds of clinical relevance.15,23,22,24 A systematic approach to studying particulate–neutrophil interactions in the context of drug delivery is lacking.
Here, we take an initial step toward understanding the nature of interactions between neutrophils and the nano- to micrometer-scale particles routinely used in drug delivery formulations. In particular, we examine the impact of incubation time, size, particle chemistry, mass concentration, and the presence/absence of serum protein on particle internalization and neutrophil phenotype. We find that particulates ranging in size from 20 nm to >1 μm and spanning a variety of chemistries at concentrations ranging up to 0.5 mg/mL are internalized rapidly (uptake plateaus within 2 h, in general) by ex vivo human neutrophils. The presence of human serum protein drastically inhibits the uptake of certain particles, like poly(styrene) (PS), while enhancing uptake of others, like poly(lactic-co-glycolic acid) (PLGA). Interestingly, passive adsorption of human serum albumin (HSA) to the surface of PLGA particles at all tested sizes was found to enhance internalization when compared to bare PLGA. At the concentrations tested, particle internalization had no observed impact on neutrophil viability, apoptosis, or activation. Importantly, particle-laden cells degranulate normally upon activation, and particles remain inside the cell afterward.
RESULTS AND DISCUSSION
Neutrophils Rapidly Internalize Nanomaterials in the Absence of Serum.
To assess the capability of nonactivated ex vivo neutrophils to ingest nanoscale particulates, we exposed cells to fluorescently labeled nanoparticles (NPs) for various lengths of time in serum-free cell culture media (Figure 1). Neutrophils were isolated from human blood according to a well-established protocol; activation (via CD62L shedding), apoptosis (via CD16 shedding), and viability (live/dead dye) were assessed via flow cytometry (Table S1). After isolation, >99% of neutrophils are alive, >90% are nonapoptotic, and >95% are nonactivated (Figure S1). This indicates that asisolated neutrophils exist in a relatively unperturbed state and are as representative of the in vivo environment as possible prior to encountering the PS, PLGA, and liposomal nano-particles used here. PS, with a tightly controllable size distribution, is widely used as a “generic” polymeric nano-particle formulation when assessing the impact of size on uptake. PLGA has attracted both preclinical and clinical interest for its modular character, ease of formulation, and capability for controlled release of various payloads. Liposomal formulations, in turn, are already in clinical use due to their ability to encapsulate and favorably modify the therapeutic index of highly toxic drugs (e.g., Doxil for ovarian cancer and multiple myeloma). After just a briefincubation (15 min), cells cultured with dilute (1–5 μg/mL) PS and liposomal nanoparticles exhibited 1–2 order of magnitude increases in fluorescence via flow cytometry (Figures 2a–c).
Figure 1.
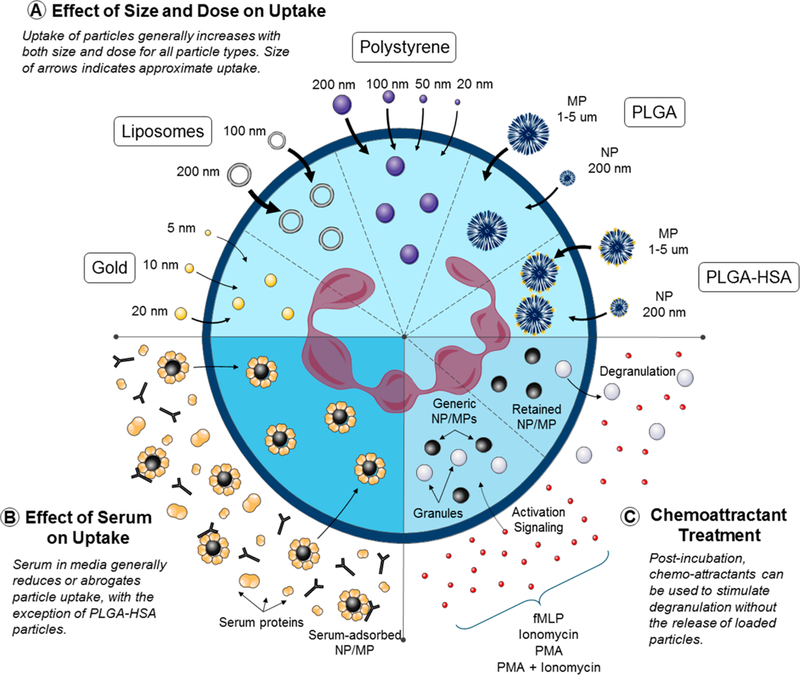
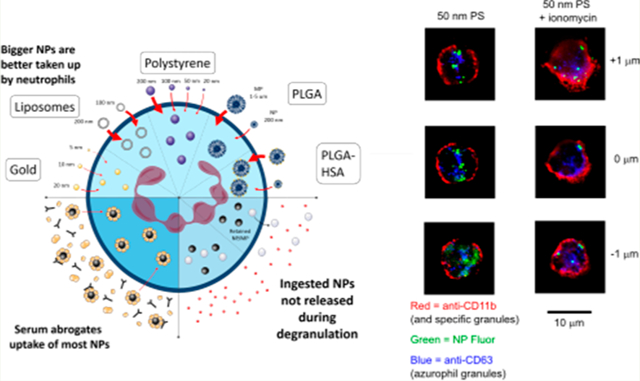
Schematic illustrating (a) the interaction of neutrophils with small (nano- to microscale), synthetic particles, (b) the impact of physiologically relevant quantities of serum on such interactions, and (c) how particle-loaded neutrophils respond to degranulation stimuli.
Figure 2.
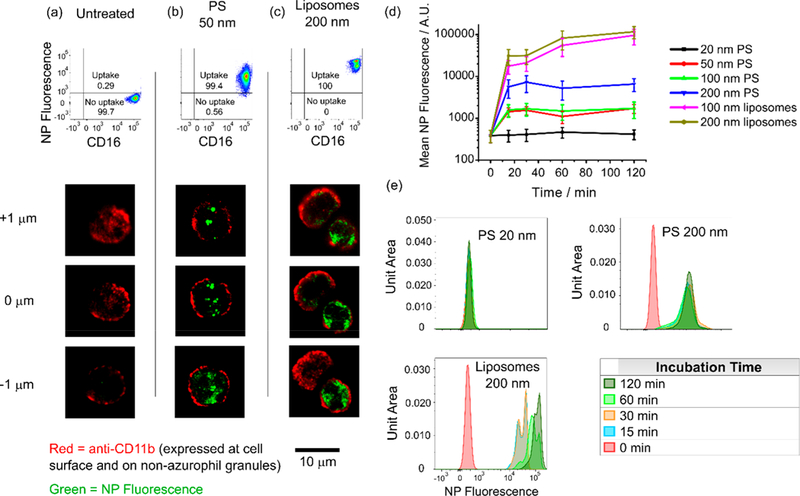
Nanoparticles are rapidly internalized by ex vivo human neutrophils in the absence of serum proteins. (a–c) Flow cytometry and confocal microscopy z-stacks indicate internalization of NPs by neutrophils incubated with NPs (b and c) for 15 min (FC) or 3 h (confocal) compared to untreated controls (a). (d) Mean fluorescence intensity and (e) histogram distributions for the NP fluorescence channel for neutrophils incubated with NPs for varying lengths of time. Uptake occurs rapidly and plateaus for all particle types showing uptake, with the majority of uptake occurring within 15 min. Note that the results here should not be interpreted as a comparison of uptake efficiencies between cell types due to differing fluorophores and fluorophore concentrations between certain formulations. All NP fluorescence data from flow cytometry was first gated on live, nonapoptotic, nonactivated neutrophils as described in Figure S1. Cells were incubated with P) nanoparticles at 1 μg/mL and liposomes at 10 μM.
Flow cytometry alone cannot readily distinguish between uptake and cell surface association. When studying neutrophils, it is of critical importance that surface association via neutrophil extracellular traps (NETs) be excluded. NETs are “sticky” complexes formed from nuclear DNA and a variety of proteins, exocytosed by dying neutrophils. Exclusion of NETosis as a mechanism for cell–NP association is possible if surface markers are carefully selected to ensure that cells are alive and nonactivated both prior to and post-uptake. A nominally lobular nuclear morphology, intact cell membrane, and lack of activation/apoptosis markers point to an absence of NETosis. If the possibility of NETosis is excluded, uptake and cell surface association can be determined by standardized methods. We sought to understand trends in NP uptake using the accepted combination of conventional flow cytometry for quantitative data on particle association and confocal laser microscopy to obtain qualitative data on internalization.30 To build a precise quantitative model of NP uptake or to verify intracellular location, imaging flow cytometry or the integration of quantitative confocal microscopy and flow cytometry data would be required, an aim beyond the scope of this work.30
Thus, to verify that NPs were actually internalized, we imaged NP-laden neutrophils via confocal microscopy. Neutrophils were identified by their characteristic polymer-phonuclear structure. Figures 2a–c show z-stacked images of control (Figure 2a) and NP-incubated (Figures 2b,c) neutrophils at 1 μm steps, stained for CD11b after a 2 h incubation. In human neutrophils, CD11b is expressed on both the exterior surface of the plasma membrane and in three of the four key granule subtypes.31 Compared to the control, bright punctate staining with z-continuity throughout the interior of the cell was observed for neutrophils incubated with 50 nm PS particles (Figure 2b). This indicates that particles are indeed internalized by neutrophils and are likely contained within membrane-bound intracellular compartments. Neutrophils incubated with 200 nm unilamellar liposomes showed similar intracellular dye localization but with diffuse cytoplasmic fluorescence that lacks the compartmentalization observed for poly(styrene) particulates (Figure 2c). The data are consistent with either (1) intact liposomes that escaped vesicles and uniformly spread throughout the intracellular space or (2) liposomal structures that degraded post-uptake, leaving BODIPY-modified cholesterol to be integrated into membranes uniformly throughout the intracellular space. In the context of previous literature, option 2 is more likely; phosphatidylcholine-containing liposomes have been shown to degrade in macrophages through several kinetically rapid processes, the fastest of which exhibited a half-life of 13 min.32 It is unlikely that liposomes degraded significantly prior to contact with cells, as fluorescent cholesterol would have been integrated to a greater extent into the plasma membrane than what is observed here.
We next assessed the uptake kinetics for PS particles ranging in size from 20 to 200 nm and unilamellar liposomes ranging in size from 100 to 200 nm (Figure 2d). Polymeric nanoparticles in general are attractive in both preclinical and clinical research for their modular character, ease of formulation, and capability for controlled release of various payloads. Liposomal formulations, in turn, are already in clinical use; their ability to encapsulate and favorably modify the therapeutic index of highly toxic drugs (e.g., Doxil for ovarian cancer and multiple myeloma). For PS nanoparticles, neither the mean fluorescence intensity in the NP channel nor the accompanying distribution histogram exhibited any change after 15 min (Figure 2d,e). Though liposomes exhibited additional uptake between 15 min and 2 h, a 100-fold increase occurred within the first 15 min; the mean intensity increased by only a factor of 2 in the subsequent 1 h and 45 min (Figures 2d and e). Figure 2 clearly indicates rapid, plateauing uptake of all NP types by neutrophils and is not intended to demonstrate a comparison of uptake efficiency between different types of particles. This is a crucial distinction considering the differences in NP formulations and selection of fluorophores.
That neutrophils ingest nanoscale particles is unsurprising, given their known phagocytic capability. However, the rapid plateauing of uptake (100 000 neutrophils per well, >29 000 particles per cell, representing 106% of the average neutrophil membrane surface area of 214 μm2 per cell calculated at 1 μg/ mL for 50 nm PS, for example) is somewhat unexpected. More unexpected still is the observation that larger particles (which have lower surface area per unit mass than smaller particles) are taken up preferentially on a per mass basis. That difference would become even more exaggerated if uptake were normalized to total particle surface area. Taken in the context of clinical drug delivery, such rapid uptake should be considered favorably. In a scenario wherein a nanoencapsulated therapeutic payload is injected into the bloodstream, each particle will have <1 min (1 min being the approximate amount of time it takes a RBC to complete a circulatory circuit) to become associated with neutrophils before encountering filtration organs like the kidneys or liver. That neutrophils in the bloodstream are present at an approximately 10-fold higher concentration than that used here (0.5 × 106 cells/mL) portends favorably for translation; if prolonged interaction driven by sluggish neutrophil uptake were required in vitro, a “stealth” formulation to boost circulation half-life,33 at minimum, would be required in vivo, substantially reducing any chance at successful neutrophil targeting. However, the in vivo environment is significantly more intricate and dynamic than that tested here; further experiments would be required to confirm the existence of similar nanoparticle-neutrophil interactions in vivo.
Ex Vivo Human Neutrophils Preferentially Internalize Larger Particles.
Particle size and surface chemistry are key design parameters for drug delivery formulations. Particle size has been shown to affect interactions with cell membranes,34 the ability of a formulation to circumvent the various biological barriers to successful therapeutic delivery at a desired anatomical location,33 and particle surface chemistry itself via variations in the protein corona formed in the presence of serum.35 Chemistry of the particle surface, being the interface with which cells and blood proteins interact, is of obvious importance. For three different particle chemistries (gold, poly(styrene), and unilamellar liposomes), we evaluated the impact of size within a formulation and the impact of particle chemistry between formulations of identical size.
To make accurate cell internalization comparisons among particles of identical chemistry, we first needed to take into account the specific fluorescence of each formulation per unit mass. The results of this analysis are depicted in Figure 3a, and a more detailed description of the methodology is provided in the Materials and Methods section under Nanoparticle Synthesis. For instance, Alexa Fluor 488 is conjugated to the surface of gold nanoparticles; surface area decreases per unit mass with increasing particle size, and as such, the fluorescence per unit mass experiences a similar decrease. Poly(styrene) particles, which contain a proprietary fluorophore distributed throughout the bulk of the particle, exhibit more consistent fluorescence intensities per unit mass: the 50, 100, and 200 nm formulations exhibit roughly identical fluorescence per unit mass, as do liposomes. The 20 nm formulation, however, is significantly dimmer. In addition, we took into account the differences stemming from variations in (i) fluorophore chemistry between formulations and (ii) the cytometer laser/detector configuration and the optical configuration achievable on a fluorescence plate reader. Gold and liposomal configurations can thus be expected to show similar brightness per unit mass on the cytometer; 50–200 nm PS particles are slightly dimmer, and 20 nm PS particles are the dimmest per unit mass. It is important to note the limitations of this approach. Because different fluorophores with different amounts per unit mass of particle were used (albeit using the same excitation source/detector pair on the cytometer), we are only able to make limited comparisons. Normalization of cytometer intensities using precise excitation/emission measurements on a plate reader at equivalent unit mass, as we did, enable a “rough” comparison of radically different formulations. But this comparison must necessarily be “rough” and cannot be used to draw quantitative conclusions regarding uptake efficiency between formulations. Because of this the only comparison between formulations with specific intensity or fluorophore differences is shown in Figure 3c, where the data clearly indicate that gold NPs are clearly not taken up, small PS particles are taken up in roughly equivalent amounts, and large particles of all types are taken up in significantly greater amounts.
Figure 3.
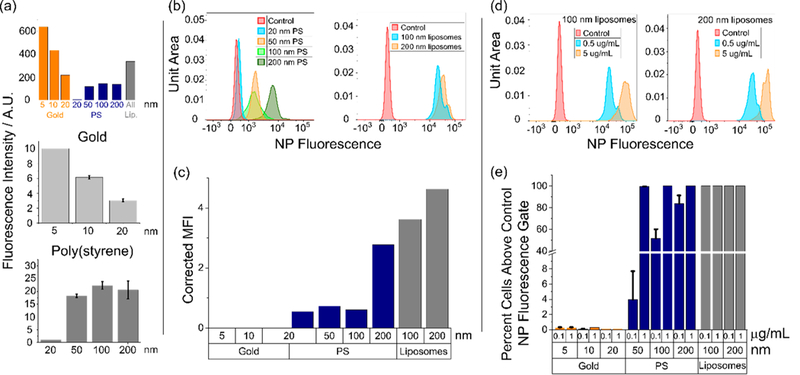
Neutrophil internalization of nanomaterials is size-dependent. All particle incubation times are 3 h unless otherwise noted. (a) Theoretical expected relative fluorescence intensities for different particle formulations as they would be observed by a flow cytometer, calculated for fixed particle concentrations with known detector gains, fluorophore properties, and cytometer excitation/emission wavelengths (top). Comparison of actual fluorescence emission intensities observed at 1.0 μg/mL for gold (middle) and poly(styrene) (bottom) particles on a fluorescence plate reader. (b) NP fluorescence intensity distribution histograms for poly(styrene) and liposomes taken up by nonapoptotic, nonactivated neutrophils as a function of particle size. PS particles incubated at 1 μg/mL. Liposomes incubated at 0.5 μg/mL. (c) Mean NP fluorescence intensities as observed by the flow cytometer for nonapoptotic, nonactivated neutrophils. Particle concentrations were 1.0 μg/mL. Fluorescence intensities as observed by the cytometer were adjusted by subtracting the untreated control fluorescence and applying the appropriate correction factor calculated in Figure 3a. (d) Dose response of NP fluorescence intensities for liposomes in nonapoptotic, nonactivated neutrophils. (e) Dose response for gold, PS, and liposomal formulations in nonapoptotic, nonactivated neutrophils. Percent cells with fluorescence exceeding the gate (maximum value of the control) calculated according to the gating shown in Figures 2a–c. Particle incubation time was 12 h.
When incubated with ex vivo human neutrophils, a significant increase in uptake was observed for 200 nm particles over that of particles <100 nm in diameter (Figure 3b). When interformulation brightness differences were taken into account, PS particles from 20 to 100 nm in diameter exhibited nearly identical mean fluorescence at the cytometer (Figure 3c). Two-hundred nanometer PS particles and both the 100 and 200 nm unilamellar liposome formulations were taken up to a similar extent (Figure 3c). Gold nanoparticles ranging from 5 to 20 nm were not taken up at all, consistent with previous literature (Figure 3c).18 The response to nanoparticles of varying size is also consistent with prior theory and observations that phagocytic activity increases with particle size for particulates with hydrophobic surfaces.23,24,34
A clear dose response was also observed for PS and liposomal particles. The mean fluorescence for liposomes exhibited significant increases in the distribution of fluorescence values observed by flow cytometry as the concentration increased from 0.5 to 5 μg/mL (Figure 3d). The fluorescence intensity of PS particles responded similarly as concentrations increased from 0.1–1 μg/mL. When gated as in Figures 2a–c, the number of cells incubated with PS 50 nm exhibiting fluorescence above the gated value increased from 4 to 100% as the concentration changed from 0.1–1 μg/mL, for instance (Figure 3e). Gold nanoparticles exhibited no appreciable uptake within the same range of mass concentrations.
These data, when taken together with the saturation-type kinetics observed in Figure 2, are consistent with two potential conclusions. The first is that at the low concentrations tested, the number of particles present is the limiting factor in neutrophil uptake. Alternatively, when exposed to large numbers of particulates, neutrophils undergo a rapid and transient burst of phagocytic activity. The second hypothesis is more easily reconciled with previous observations on particle uptake by neutrophils: similarly rapid uptake kinetics were observed at much higher PS particle concentrations (~1 mg/mL) for all sizes tested (0.1–5 μm).23 Whether or not this burst of phagocytic activity would occur in vivo in human blood requires further investigation.
Human Serum Albumin Reduces Uptake of Poly-(styrene) and Liposomal Nanoparticles and Enhances Uptake of PLGA Particles.
One of the first interactions that injectable drug delivery formulations have with the human body is the rapid adsorption of serum proteins (including opsonins) to the particle surface.36 To evaluate how this interaction might impact internalization of particulates, we incubated nanoparticles with neutrophils in culture medium containing 10% human serum, blood type AB. Under these conditions, poly(styrene) uptake was completely abrogated for all sizes (Figure 4a). Uptake of liposomes was reduced by approximately an order of magnitude (Figure 4a). Three potential explanations are consistent with this data: (i) exposure of nanoparticles to serum results in particle aggregation,37 (ii) modification of the particle surface with serum protein generates a barrier to the otherwise “sticky” interactions between hydrophobic elements of the particle surface and the neutrophil cell membrane, and (iii) exposure to the proteins and small particulates in serum makes neutrophils intrinsically less prone to take up particles.
Figure 4.
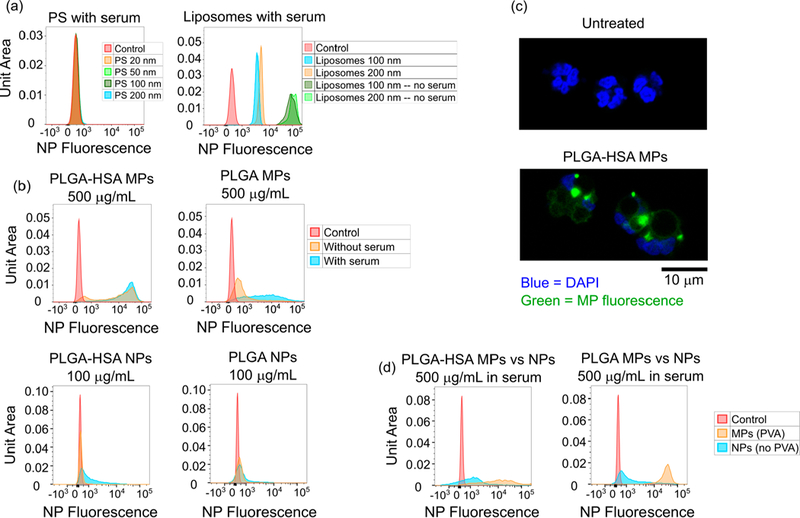
Serum dramatically reduces particle uptake for PS and liposomal nanoformulations but enhances uptake of PLGA microparticles and nanoparticles. (a) NP fluorescence after a 3 h incubation with PS particles of various sizes (at 1 μg/mL) is completely abrogated. Uptake of liposomes (at 5 μg/mL) is reduced by approximately an order of magnitude. Gating is for nonapoptotic, nonactivated neutrophils. (b) The presence of serum improves the uptake of PVA-stabilized PLGA microparticles. Particle preadsorption with human serum albumin even further improves uptake as assessed by NP fluorescence on flow cytometry, gated for nonapoptotic, nonactivated neutrophils. Incubation time was 2 h. (c) Confocal microscopy images of untreated neutrophils and neutrophils incubated with HSA-preadsorbed PLGA microparticles at 500 μg/mL for 2 h. (d) Uptake of PLGA particles is size-dependent. In formulations both with and without HSA, uptake of PLGA microparticles exceeds uptake of nanoparticles at a dose of 500 μg/mL. Incubation time was 2 h.
To differentiate between hypotheses (i)/(ii) and (iii), we investigated the impact of serum on uptake of PLGA microparticles. Micrometer-size particles have previously been shown to maximize uptake by human neutrophils, albeit under conditions less relevant to drug delivery.23 In addition, microparticles formed from PVA-stabilized PLGA may be intrinsically more stable due to the expected greater resilience of PVA to displacement by serum proteins compared to that of Tween-20 (used to stabilize the PS formulations). Intriguingly, PLGA microparticles showed enhanced uptake in the presence of serum compared to serum-free incubation conditions (Figure 4b). Preadsorption of PLGA microparticles with human serum albumin increased uptake by nearly 2 orders of magnitude when incubated with neutrophils in serum-free conditions; incubation of HSA-preadsorbed particles with neutrophils in the presence of serum generated little additional benefit (Figure 4b). PLGA-HSA microparticle-laden neutrophils were also imaged with confocal microscopy; uptake of multiple microparticles per cell was routinely observed (Figure 4c). This would seem to indicate that neutrophils do not experience any intrinsic reduction in phagocytic capability in the presence of serum.
It is important to note that a surfactant-free method was used to formulate PGLA nanoparticles, contrasting with the formulation of PLGA microparticles using poly(vinyl alcohol) (PVA). PVA, an emulsifier, is commonly used in the formulation of PLA and PLGA nanoparticles to increase uniformity and improve dispersity in an aqueous medium.38 During formulation, PVA forms an interconnected network with molecules at the particle surface, resulting in residual surface PVA despite washing.39 The amount of residual PVA influences particle size, zeta potential, polydispersity index, surface hydrophobility, protein loading, and in vitro protein release to varying degrees.39
In this study, size is measured postformulation and is thus accounted for in the comparison between surfactant-free NPs and MPs with residual PVA. However, the surface MP particles with residual PVA may be more hydrophilic, limiting a strict comparison of uptake between the smaller NPs and larger MPs.39 Nonetheless, the decreased hydrophobicity of particles with residual PVA generally decreases cellular uptake, depending on the percentage of surfactant used during formulation.39 Figure 4d compares PLGA-HSA MPs formulated with PVA and PLGA-HSA NPs formulated without PVA. Although the presence of PVA limits interpretation, the literature suggests that the uptake of MPs by neutrophils in this study would theoretically be the same or lower than MPs formulated without PVA. Thus, the increased uptake of PVA-positive MPs over PVA-negative NPs is a reasonable indication that uptake is size-dependent and may in fact understate the preferential uptake of larger polymeric particles by neutrophils.
The exceptional uptake of albumin-preadsorbed PLGA microparticles by neutrophils, even in the presence of serum, bodes well for future clinical applications. PLGA is a biodegradable polymer that is generally regarded as safe by multiple regulatory agencies and is currently in use as a component of clinical formulations. Drug-loaded PLGA microparticles, injected intravenously, would encounter approximately 5 × 106 neutrophils/mL, a 10-fold increase over the concentration used in this study. Rapid and substantial uptake could (i) reduce the need for highly engineered ligand-targeted coatings designed to improve targeting precision, (ii) reduce the size constraints enjoined upon formulations by filtration organs like the liver and spleen40 and (iii) eliminate the need for stealth coatings designed to improve circulation half-life of nanoscale formulations.
Particle Uptake Does Not Appreciably Alter Neutrophil Activation or Capacity for Degranulation.
Central to the strategy of using neutrophils as carriers for drug-loaded particulate formulations is the notion that particle uptake does not appreciably perturb degranulation, an important neutrophil function. To support this hypothesis, we assessed the impact of incubation with nanoparticles on neutrophil viability, apoptosis, and activation. At the concentrations used in this study, no significant impact on neutrophil viability was observed for any formulation of poly(styrene) or liposomes (Figure 5a). Particles similarly failed to impact CD16 and CD62L expression, markers of apoptosis and activation, respectively (Figures 5b and c).
Figure 5.
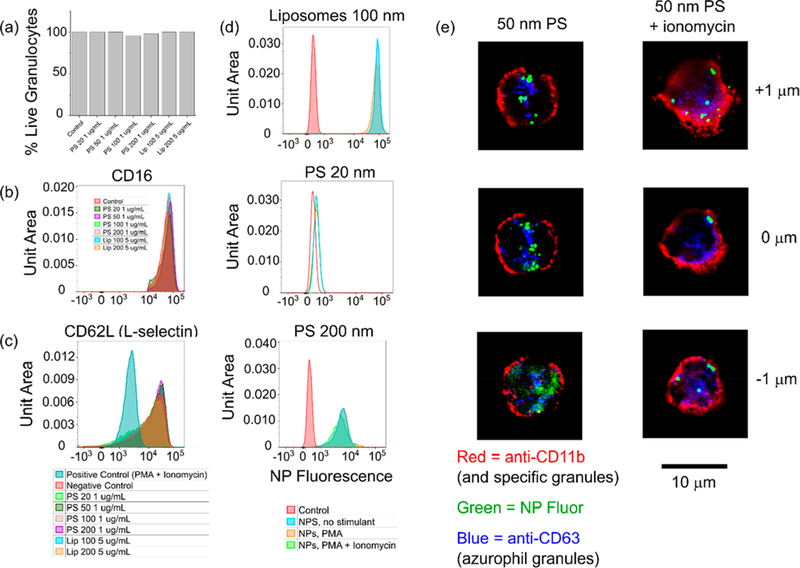
Critical aspects of neutrophil phenotype are not perturbed by particle uptake; particles remain inside the cell following degranulation. Incubation times were 3 h unless otherwise noted. (a) Cell viability is not affected by particle uptake. At least 40 000 cells from 2 separate experiments were used to generate the chart. (b) Neutrophil apoptosis as measured by CD16 shedding was not impacted by particle uptake. (c) Neutrophil activation as measured by CD62L shedding was not impacted by particle uptake. (d and e) Neutrophils incubated with 1 μg/mL PS or 5 μg/mL liposomes for 3 h and then treated with 50 nM PMA, 1 μM ionomycin, or 50 nM PMA + 1 μM ionomycin for 30 min at 37 °C do not exocytose particles previously internalized.
Neutrophils that have taken up nanoparticles must also retain their ability to carry out normal functions if the possibility of “hijacking” the cells for in vivo cell therapy is to be realized. As a first step, we investigated the ability of neutrophils to degranulate following particle uptake. Degranulation is a critical neutrophil function;1 although a reduction in the cells’ ability to do so might be considered useful in certain therapeutic contexts,13 such a reduction would appreciably diminish the potency available for immunomodulatory cell therapy. Neutrophils incubated with nanoparticles were then stimulated to degranulate by exposure to fMLP, ionomycin, PMA or PMA and ionomycin. Neutrophil gelatinase-associated lipocalin (NGAL) was used as a marker for degranulation, as it is present in three of the four granule subsets exocytosed upon activation.41 For 1 μg/mL PS and 5 μg/mL liposomes, no significant differences in the cells’ response to any of these stimulant cocktails was observed, indicating retention of normal activity (Figure 6).
Figure 6.
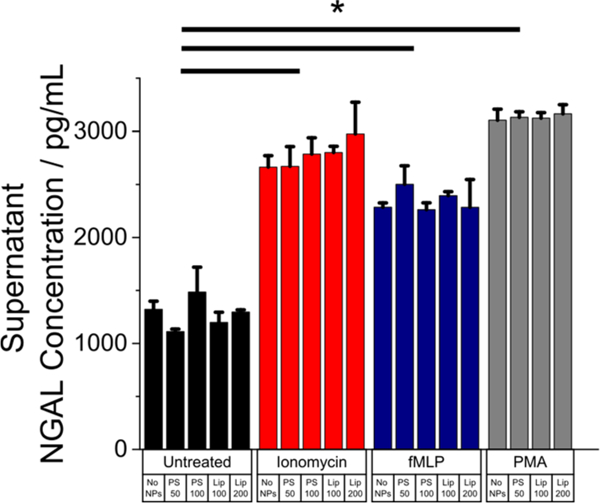
Release of NGAL by neutrophils during degranulation is unchanged by prior uptake of NPs. Neutrophils previously incubated with nanoparticles for 2 h at 1 μg/mL (PS) or 5 μg/mL (liposomes) exhibit no change in their ability to release NGAL, a critical degranulation marker, upon stimulation with 1 μM fMLP, 50 nM PMA, or 1 μM ionomycin for 30 min at 37 °C. n = 2. * = p < 0.05.
Finally, we assessed the fate of internalized particles after degranulation. In neutrophils, as in other granulocytes, phagosomes containing ingested foreign bodies may often fuse with granules, depending on the uptake mechanism.42 In this study, however, degranulating neutrophils did not release particles (Figures 5d and e). No decreases in NP fluorescence were observed after treatment with neutrophil stimulants (Figure 5d). Confocal microscopy shows neutrophils with typical signs of degranulation after treatment with ionomycin, including a more diffuse-appearing membrane and mobilization of CD63-stained azurophil granules to the cell surface (Figure 5e). While some nanoparticle-bearing compartments do appear to be in very close proximity to the cell membrane, they are definitively not released (Figure 5d).
Although we studied degranulation due to its potential to force the release of internalized particles, subsequent work examining the effect of internalized NPs on a full array of neutrophil functions would be helpful. Specifically, a follow-up study could examine changes to degranulation and the impact of uptake on the migratory capacity of neutrophils.
CONCLUSION
Ex vivo human neutrophils are shown to internalize polymeric and liposomal particles ranging in size from 0.02 to 5 μm. Uptake occurs rapidly, typically plateauing within 15 min to 2 h. Within the range of submicrometer formulations tested, a size-dependent response was observed: neutrophils preferentially internalized larger particles. Including serum in the neutrophil culture media completely abrogated uptake for nanoscale poly(styrene) and reduced uptake by of unilamellar liposomes by an order of magnitude. However, PLGA microparticles exhibited significant increases in uptake when incubation occurred in the presence of serum. When PLGA microparticles were preadsorbed with human serum albumin, the impact on uptake was dramatic, increasing by two orders of magnitude over non-preadsorbed PLGA. No impact of particle uptake on neutrophil viability, apoptosis or activation was observed; particleladen cells retained the ability to degranulate in response to the potent secretagogues fMLP, ionomycin, and PMA. When degranulation was induced post-uptake, internalized particles were retained within the cell.
By using ex vivo human neutrophils and common materials like PS, PLGA, gold, and lipid-cholesterol liposomes, we take an initial step toward clinically oriented formulation designs that take neutrophil behavior into account. With this information, drug delivery formulations could be designed to better avoid uptake by circulating neutrophils. We also expect this data set to have implications for the design of cell-based immunomodulatory therapeutics for disorders like cancer, autoimmune diseases, and atherosclerosis that do not require removing cells from the body; rather, a formulation may be designed for rapid uptake by the blood neutrophils which initially encounter injected particles. Subsequent to uptake, existing delivery motifs43 (i.e., delayed release, triggered release, and targeting of specific subcellular compartments) may be readily leveraged to modulate the function of the neutrophil itself or “hijack” the neutrophil to reach either distal sites of inflammation or the otherwise cloistered bone marrow compartment, where neutrophils are ultimately recycled.
This study of NP uptake by neutrophils complements the growing body of literature on nanomaterial interactions with monocytes and macrophages, two other phagocytes with critical roles in the inflammatory response. Whereas neutrophils are generally involved in the acute response, monocytes and macrophages play a large role in chronic inflammation. Similar work linking nanomaterial size, chemistry, and serum adsorption to trends in uptake by macrophages and monocytes can be interpreted alongside our results.44–50 Uptake by macrophages is charge- and size- dependent, as well as serum-dependent depending on NP surface functionalization or PEG incorporation. Further work comparing nanomaterial interactions across all phagocytes (i.e., neutrophils vs macrophages/monocytes) would be valuable in developing improved clinical approaches for treating inflammatory disorders. Other possibilities for future work include: (l) assessing the impact of other physicochemical parameters like charge or hydrophobicity and (2) studying the mechanisms by which particle-laden neutrophils can migrate to and deliver particles to various sites in the body, even without exocytosing the particles. For instance, a study that examines whether or not particle-laden neutrophils are recycled in the bone marrow, as with unladen cells, could shed light on the neutrophils’ utility in delivering drugs to immature blood cells in the hard-to-reach marrow.
In summary, we anticipate these results will serve as a promising foundation to (i) better design drug delivery formulations that either avoid or target neutrophil uptake, (ii) chart a path toward in vivo cell therapies enabling delivery of therapeutic payloads to bone marrow (the recycling site for most neutrophils) or distal sites of inflammation, and (iii) begin solving the delivery challenge required to selectively target neutrophils for immunomodulation in vivo.
MATERIALS AND METHODS
Materials.
Tables 1 and 2 list materials and formulations used.
Table 1.
Materials Used
| NP/MP type | sizes | description |
|---|---|---|
| gold | 5, 10, 20 nm | Nanocs (New York, NY) fluorescently labeled gold NPs fluorophore (ex/em): Alexa Fluor 488 (491/515 nm) |
| polystyrene | 20, 50, 100, 200 nm | Phosphorex (Hopkinton, MA) fluorescent poly(styrene) NPs size: 20 (–COOH modified), 50, 100, 200 nm stabilized with Tween-20 fluorophore (ex/em): proprietary (460/500 nm); spectrum available via www.degradex.com |
| liposomes | 100, 200 nm | unilamellar liposomes synthesized from egg L-α-lysophosphatidyl-choline (Egg PC), egg sphingomyelin (Egg SM) and ovine wool cholesterol (Chol) (all from Avanti Polar Lipids [Alabaster, AL]) using a previously established protocol51 fluorophore (ex/em): TOPFLUOR BODIPY-conjugated cholesterol (495/507 nm) at 2.5% by mass |
| PLGA | 1–3 μm | PLGA particles were synthesized using a single-emulsion evaporation technique according to a previously published protocol52 |
| PLGA-HSA | 1–3 μm | stabilized with poly(vinyl alcohol) HSA was passively adsorbed onto PLGA-HSA MPs fluorophore (ex/em): FITC (491/515 nm) |
Table 2.
Formulations Used and Size Measurement Techniques
| Formulation | Size, Distribution |
Size Measurement Technique |
|---|---|---|
| Gold 5–20 nm | 5–20 nm | • TEM as provided by manufacturer. |
| Polystyrene 20 nm | 24 nm +/− 7 nm | • DLS as provided by manufacturer. |
| Polystyrene 50 nm | 50 nm +/−12 nm | • DLS as provided by manufacturer. |
| Polystyrene 100 nm | 124 nm +/− 17 nm | • DLS as provided by manufacturer |
| Polystyrene 200 nm | 210 nm +/− 38 nm | • DLS as provided by manufacturer |
| Liposomes 100 nm | 95 nm, PDI 0.071 | • DLS on as-prepared samples. |
| Liposomes 200 nm | 178 nm, PDI 0.242 | • DLS on as-prepared samples. |
| PLGA Microparticles | 1–3 μm | • SEM
|
The following flow cytometry antibodies and their isotype controls were purchased from eBioscience (Thermo Fisher, Carlsbad, CA): anti-CD16 PE-Cy7 (eBioCB16, mouse IgG1, kappa); anti-CD62L APC (DREG56, mouse IgG1, kappa); anti-CD45 APC-eFluor780 (HI30, mouse IgG1). Anti-CD11b Alexa Fluor 647 (EPR1344, rabbit), Anti-CD63 (MEM-259, mouse IgG1), human and rat preadsorbed rabbit antimouse IgG Alexa Fluor 597, their isotype controls and normal goat serum for immunofluorescence microscopy were purchased from Abcam (Cambridge, MA). N-formyl-Met-Leu-Phe [fMLP], phorbol 12-myristate 13-acetate [PMA], dextran (MW 450–650 kDa) and human serum albumin (HSA) were purchased from Sigma-Aldrich (St. Louis, MO – now Merck KGaA, Darmstadt Germany). Ionomycin, RPMI 1640 cell culture medium and the LIVE/DEAD Aqua fixable dead cell stain were purchased from Life Technologies (Thermo Fisher, Carlsbad, CA). The neutrophil gelatinase-associated lipocalin (NGAL) DuoSet for ELISA, HRP and ELISA visualization reagents were purchased from R&D Systems (Minneapolis, MN). Lymphoprep was purchased from StemCell Technologies (Vancouver, British Columbia, Canada). Human AB serum was purchased from Fisher Scientific (Thermo Fisher, Carlsbad, CA).
Nanoparticle Synthesis.
The liposomes, composed of egg L-α-lysophosphatidylcholine (Egg PC), egg sphingomyelin (Egg SM) and ovine wool cholesterol (Chol) and TOPFLUOR cholesterol (CholF), at weight ratios 60%:30%:7.5%2.5%: (Egg PC/Egg SM/Chol/CholF) in a total lipid concentration of 20 mM, were prepared using a thin lipid film method.51 Briefly, stock solutions of all lipids were prepared by dissolving powdered lipids in chloroform and appropriate volumes of the lipids were taken from the stock solution to make lipids with above concentrations in a glass tube and gently dried under nitrogen. To ensure complete removal of chloroform, the lipids were left under vacuum for an additional 12 h. The lipid film was hydrated with a liposome buffer composed of 150 mM NaCl, 10 mM Hepes and 1 mM MgCl2 dissolved in nuclease-free water to create multilamellar liposomes. The resulting multilamellar liposomes were sized by repeated thawing and freezing and then subjected to 15 extrusion cycles at 60 °C through different pore size polycarbonate membranes to produce unilamellar liposomes. Sizing was performed using a Malvern Instruments (Malvern, United Kingdom) ZetaSizer ZS dynamic light scattering instrument.
Fluorescent PLGA microparticles were synthesized using a single-emulsion evaporation technique according to a previously established protocol.52 FITC was used as the fluorophore. PLGA-HSA particles were generated by allowing dissolved HSA to adsorb to the surface of as-synthesized PLGA particles. PLGA nanoparticles were prepared with a PVA-free formulation method. PLGA and fluorescent compound were dissolved in acetonitrile and the mixture was then added dropwise to 2–4 volumes of stirring water giving a final polymer concentration of 2 mg/mL. The mixture was stirred for 2 h under reduced pressure to allow the nanoparticles to form by self-assembly and the organic solvent was removed. The PLGA nanoparticles were then concentrated by centrifugation at 3,000xg for 10 min using an Amicon filter (MWCO 20KDa), washed twice, and reconstituted in PBS for physicochemical characterization.
To conduct a comparison of flow cytometry results for different nanoparticle formulations, which in general utilize fluorophores with distinct spectra, extinction coefficients, and specific particle loading (per unit mass), we measured fluorescence intensities of each particle formulation across a range of mass concentrations using a Tecan Safire plate reader. At fixed particle concentration, detector gain, and excitation/emission wavelengths, the fluorescence intensities observed can be used in conjunction with the fluorophore’s excitation and emission spectrum and knowledge of the cytometer excitation/emission wavelengths to calculate the expected relative fluorescence intensities, as observed by the cytometer for a fixed mass of particles. Importantly, this approach is semiquantitative because of a few simplifying assumptions. The first is that the filter bandwidths on both the plate reader and the cytometer have an ideal square shape; i.e., a sensitivity of 1 within the passband and 0 outside. The second is that the spectrum of the fluorophores inside the cell is identical to that in an unbuffered aqueous environment. When correcting the mean fluorescence intensities of cytometer-based uptake results, we subtracted the control MFI in the particle fluorescence channel and then applied the correction factor gleaned from the plate reader-based specific fluorescence analysis.
Neutrophil Isolation.
Fresh buffy coats obtained from whole blood of anonymous healthy donors with sodium citrate as an anticoagulant were obtained from Research Blood Components (Brighton, MA). Samples were collected under Institutional Review Board approval in place through Research Blood Components. Buffy coats were briefly stored at room temperature (<1 h) before further processing. To isolate neutrophils, buffy coats were mixed 1:1 (v/v) with a solution of 2% dextran in normal saline and incubated at room temperature for 30 min to selectively sediment red blood cells. The cloudy yellow supernatant was then centrifuged at 200g for 6 min at 4 °C with a low brake to remove platelets. All subsequent steps take place at 4 °C. The pellet was gently resuspended in cold 0.9% NaCl solution, layered on top of a Lymphoprep solution, and centrifuged at 400g for 30 min at 4 °C with no brake. The pellet, containing residual red blood cells and granulocytes, was resuspended in cold distilled water to remove red blood cells via hypotonic lysis. Addition of a 1.8% sodium chloride solution in equal volume restored isotonicity. This protocol yields neutrophils with >98% viability, verified by Trypan blue exclusion, and >95% purity, verified by flow cytometry. Remaining impurities include eosinophil granulocytes and residual monocytes/lymphocytes.
Nanoparticle Incubation and Neutrophil Activation.
Isolated neutrophils were resuspended in RPMI-1640 media at 500 000 cells/mL in 96-well plates coated overnight in 1% BSA to prevent neutrophil adhesion. Cells were incubated with fluorescently labeled NPs in a 37 °C incubator with 5% CO2 for varying lengths of time, up to 12 h. For experiments performed in the absence of serum, RPMI-1640 was used as a culture medium. For experiments performed in the presence of serum, RPMI-1640 + 10% human AB serum was used.
Exogenous neutrophil activation was performed by incubating ~15 000 neutrophils/well in RPMI-1640 for 30 min at 37 °C in the presence of one of the following stimulants: 1 μM fMLP, 50 nM PMA, 1 μM ionomycin, or 1 μM ionomycin + 50 nM PMA. Where applicable, NPs were first removed from media by washing with RPMI-1640 prior to activation. NGAL is present in three of the four neutrophil granule subsets (secretory vesicles, secondary granules, and gelatinase granules but not azurophil granules) and hence was used as a marker for the degranulation, which occurs when neutrophils are activated. NGAL concentration in cell supernatants following activation was assessed via ELISA.
Assessing Nanoparticle Internalization.
To assess NP internalization and NP release following chemoattractant treatments, flow cytometry was used. All flow cytometry measurements were taken using a Becton Dickinson BD FACS Canto II with a high-throughput sampling apparatus and running BD FACSDIVA software. The analyzer was equipped with lasers with the following properties (laser wavelength [detector center wavelength/filter bandwidth]): 405 nm [450/50 nm; 510/50 nm], 488 nm [530/30 nm; 585/42 nm; 670 nm/long-pass; 780/60 nm] and 640 nm [660/20 nm; 780/60 nm]. Analysis was carried out using the FlowJo software (FlowJo LLC, Version 10). For all FC analysis graphs shown, at least 20 000 cells comprising at least 2 separate experiments were acquired unless otherwise noted.
Cells were washed with RPMI-1640 media, resuspended in Krebs-Ringer phosphate buffer with 0.1% BSA, and stained for CD45 (pan-leukocyte marker), CD16 (neutrophil marker in lymphocyte-depleted populations; shedding also indicates apoptosis), CD62-L (L-selectin, shed upon neutrophil activation), and Aqua fluorescent dead cell stain. Any fluorescent NP signals were evaluated on the FITC channel (488 nm excitation, 510/50 nm detection). Unless otherwise noted, all uptake experiments used only cells gated using the following hierarchical strategy: (1) FSC/SSC gating on the granulocyte population, (2) live cells (no aqua fluorescence), (3) nonapoptotic neutrophils (CD45+, CD16hi) and (4) nonactivated neutrophils (CD62Lhi).
Confocal microscopy was also used to image NP-loaded cells. Cells were fixed with 4% paraformaldehyde, permeabilized with 0.1% Triton X-100, and stained for CD11b (specific granules, gelatinase granules, and secretory vesicles marker), CD63 (azurophil granule marker), and DAPI (nucleus marker). A Cytospin cytocentrifuge was used to load fixed and stained neutrophils onto glass slides. Imaging was performed with RPI spinning disk confocal microscopy. MetaMorph Microscopy Automation and Image Analysis Software was used to collect and view images. ImageJ was used to process images and stacks for publication.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by the Bridge Project, a partnership between the Koch Institute for Integrative Cancer Research at MIT and the Dana-Farber/Harvard Cancer Center (to R.L. and M.J.M.). This work was supported in part by a Cancer Center Support (core) Grant P30-CA14051 from the National Cancer Institute and a grant from the Koch Institute’s Marble Center for Cancer Nanomedicine (to R.L.). M.J.M. was supported by a Burroughs Wellcome Fund Career Award at the Scientific Interface, a National Institutes of Health (NIH) Director’s New Innovator Award (DP2TR002776), and a grant from the American Cancer Society (129784-IRG-16–188-38-IRG). P.P.G.G. was supported by Leslie Misrock Cancer Nanotechnology Postdoctoral Fellowship and Fundação Estudar. S.G. was supported by a Fulbright Canada Killam Fellowship.
Footnotes
ASSOCIATED CONTENT
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsbiomaterials.8b01062.
Evaluation of neutrophil phenotype and purity by flow cytometry (PDF)
The authors declare no competing financial interest.
REFERENCES
- (1).Borregaard N Neutrophils, from Marrow to Microbes. Immunity 2010, 33 (5), 657–670. [DOI] [PubMed] [Google Scholar]
- (2).Farokhzad O; Langer R Impact of Nanotechnology on Drug Delivery. ACS Nano 2009, 3 (1), 16–20. [DOI] [PubMed] [Google Scholar]
- (3).Zolnik BS; González-Fernández Á; Sadrieh N; Dobrovolskaia MA Minireview: Nanoparticles and the Immune System. Endocrinology 2010, 151 (2), 458–465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Puga I; Cols M; Barra CM; He B; Cassis L; Gentile M; Comerma L; Chorny A; Shan M; Xu W; et al. B Cell–Helper Neutrophils Stimulate the Diversification and Production of Immunoglobulin in the Marginal Zone of the Spleen. Nat. Immunol. 2012, 13 (2), 170–180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Peters AM Just How Big Is the Pulmonary Granulocyte Pool? Clin. Sci. 1998, 94 (1), 7–19. [DOI] [PubMed] [Google Scholar]
- (6).Borregaard N; Sørensen OE; Theilgaard-Mönch K Neutrophil Granules: A Library of Innate Immunity Proteins. Trends Immunol. 2007, 28 (8), 340–345. [DOI] [PubMed] [Google Scholar]
- (7).Lim K; Hyun Y-M; Lambert-Emo K; Capece T; Bae S; Miller R; Topham DJ; Kim M Neutrophil Trails Guide Influenza-Specific CD8+ T Cells in the Airways. Science (Washington, DCC, U. S.) 2015, 349 (6252), aaa4352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Kolaczkowska E; Kubes P Neutrophil Recruitment and Function in Health and Inflammation. Nat. Rev. Immunol. 2013, 13 (3), 159–175. [DOI] [PubMed] [Google Scholar]
- (9).Summers C; Rankin SM; Condliffe AM; Singh N; Peters AM; Chilvers ER Neutrophil Kinetics in Health and Disease. Trends Immunol. 2010, 31 (8), 318–324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Pillay J; den Braber I; Vrisekoop N; Kwast LM; de Boer RJ; Borghans JAM; Tesselaar K; Koenderman L In Vivo Labeling with 2H2O Reveals a Human Neutrophil Lifespan of 5.4 Days. Blood 2010, 116 (4), 625–627. [DOI] [PubMed] [Google Scholar]
- (11).Chu D; Zhao Q; Yu J; Zhang F; Zhang H; Wang Z Nanoparticle Targeting of Neutrophils for Improved Cancer Immunotherapy. Adv. Healthcare Mater. 2016, 5 (9), 1088–1093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Chu D; Gao J; Wang Z Neutrophil-Mediated Delivery of Therapeutic Nanoparticles across Blood Vessel Barrier for Treatment of Inflammation and Infection. ACS Nano 2015, 9 (12), 11800–11811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Wang Z; Li J; Cho J; Malik AB Prevention of Vascular Inflammation by Nanoparticle Targeting of Adherent Neutrophils. Nat. Nanotechnol 2014, 9 (3), 204–210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Hedenborg M Titanium Dioxide Induced Chemiluminescence of Human Polymorphonuclear Leukocytes. Int. Arch. Occup. Environ. Health 1988, 61 (1–2), 1–6. [DOI] [PubMed] [Google Scholar]
- (15).Papatheofanis FJ; Barmada R Polymorphonuclear Leukocyte Degranulation with Exposure to Polymethylmethacrylate Nanoparticles. J. Biomed. Mater. Res. 1991, 25 (6), 761–771. [DOI] [PubMed] [Google Scholar]
- (16).Fromen CA; Kelley WJ; Fish MB; Adili R; Noble J; Hoenerhoff MJ; Holinstat M; Eniola-Adefeso O Neutrophil–Particle Interactions in Blood Circulation Drive Particle Clearance and Alter Neutrophil Responses in Acute Inflammation. ACS Nano 2017, 11 (11), 10797–10807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Mainardes RM; Gremião MPD; Brunetti IL; da Fonseca LM; Khalil NM Zidovudine-Loaded PLA and PLA–PEG Blend Nanoparticles: Influence of Polymer Type on Phagocytic Uptake by Polymorphonuclear Cells. J. Pharm. Sci. 2009, 98 (1), 257–267. [DOI] [PubMed] [Google Scholar]
- (18).Bartneck M; Keul HA; Zwadlo-Klarwasser G; Groll J Phagocytosis Independent Extracellular Nanoparticle Clearance by Human Immune Cells. Nano Lett. 2010, 10 (1), 59–63. [DOI] [PubMed] [Google Scholar]
- (19).Dianzani C; Cavalli R; Zara GP; Gallicchio M; Lombardi G; Gasco MR; Panzanelli P; Fantozzi R Cholesteryl Butyrate Solid Lipid Nanoparticles Inhibit Adhesion of Human Neutrophils to Endothelial Cells. Br. J. Pharmacol. 2006, 148 (5), 648–656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Girard D Focussing on Neutrophils for Evaluating In Vitro and In Vivo Inflammatory Activities of Nanoparticles. Nanomaterials - Toxicity and Risk Assessment 2015, 1 DOI: 10.5772/60703. [DOI] [Google Scholar]
- (21).Moeller S; Kegler R; Sternberg K; Mundkowski RG Influence of Sirolimus-Loaded Nanoparticles on Physiological Functions of Native Human Polymorphonuclear Neutrophils. Nanomedicine (N. Y, NY, U. S.) 2012, 8 (8), 1293–1300. [DOI] [PubMed] [Google Scholar]
- (22).Rudt S; Muller RH In Vitro Phagocytosis Assay of Nano- and Microparticles by Chemiluminescence. III. Uptake of Differently Sized Surface-Modified Particles, and Its Correlation to Particle Properties and in Vivo Distribution. Eur. J. Pharm. Sci. 1993, 1 (1), 31–39. [Google Scholar]
- (23).Rudt S; Muller RH In Vitro Phagocytosis Assay of Nano- and Microparticles by Chemiluminescence. I. Effect of Analytical Parameters, Particle Size and Particle Concentration. J. Controlled Release 1992, 22 (3), 263–271. [Google Scholar]
- (24).Simon SI; Schmid-Schonbein GW Biophysical Aspects of Microsphere Engulfment by Human Neutrophils. Biophys. J. 1988, 53 (2), 163–173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Yan C; Li S; Li Z; Peng H; Yuan X; Jiang L; Zhang Y; Fan D; Hu X; Yang M; et al. Human Umbilical Cord Mesenchymal Stem Cells as Vehicles of CD20-Specific TRAIL Fusion Protein Delivery: A Double-Target Therapy against Non-Hodgkin’s Lymphoma. Mol. Pharmaceutics 2013, 10 (1), 142–151. [DOI] [PubMed] [Google Scholar]
- (26).Lin MH; Lin CF; Yang SC; Hung CF; Fang JY The Interplay between Nanoparticles and Neutrophils. J. Biomed. Nano- technol. 2018, 14, 66–85. [DOI] [PubMed] [Google Scholar]
- (27).Chu D; Dong X; Shi X; Zhang C; Wang Z Neutrophil- Based Drug Delivery Systems. Adv. Mater. (Weinheim, Ger.) 2018, 30, 1706245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Mestas J; Hughes CCW Of Mice and Not Men: Differences between Mouse and Human Immunology. J. Immunol. 2004, 172 (5), 2731–2738. [DOI] [PubMed] [Google Scholar]
- (29).Bartneck M; Keul HA; Singh S; Czaja K; Bornemann J; Bockstaller M; Moeller M; Zwadlo-Klarwasser G; Groll J Rapid Uptake of Gold Nanorods by Primary Human Blood Phagocytes and Immunomodulatory Effects of Surface Chemistry. ACS Nano 2010, 4 (6), 3073–3086. [DOI] [PubMed] [Google Scholar]
- (30).Gottstein C; Wu G; Wong BJ; Zasadzinski JA Precise Quantification of Nanoparticle Internalization. ACS Nano 2013, 7, 4933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Kjeldsen L; Sengeløv H; Borregaard. Subcellular Fractionation of Human Neutrophils on Percoll Density Gradients. J. Immunol. Methods 1999, 232 (1–2), 131–143. [DOI] [PubMed] [Google Scholar]
- (32).Harashima H; Hirai N; Kiwada H Kinetic Modelling of Liposome Degradation in Peritoneal Macrophages. Biopharm. Drug Dispos. 1995, 16 (2), 113–123. [DOI] [PubMed] [Google Scholar]
- (33).Blanco E; Shen H; Ferrari M Principles of Nanoparticle Design for Overcoming Biological Barriers to Drug Delivery. Nat. Biotechnol. 2015, 33 (9), 941–951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Champion JA; Walker A; Mitragotri S Role of Particle Size in Phagocytosis of Polymeric Microspheres. Pharm. Res. 2008, 25 (8), 1815–1821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Salvati A; Pitek AS; Monopoli MP; Prapainop K; Bombelli FB; Hristov DR; Kelly PM; Aberg C; Mahon E; Dawson KA Transferrin-Functionalized Nanoparticles Lose Their Targeting Capabilities When a Biomolecule Corona Adsorbs on the Surface. Nat. Nanotechnol. 2013, 8 (2), 137–143. [DOI] [PubMed] [Google Scholar]
- (36).Casals E; Pfaller T; Duschl A; Oostingh GJ; Puntes V Time Evolution of the Nanoparticle Protein Corona. ACS Nano 2010, 4 (7), 3623–3632. [DOI] [PubMed] [Google Scholar]
- (37).Guarnieri D; Guaccio A; Fusco S; Netti PA Effect of Serum Proteins on Polystyrene Nanoparticle Uptake and Intracellular Trafficking in Endothelial Cells. J. Nanopart. Res. 2011, 13 (9), 4295–4309. [Google Scholar]
- (38).Panyam J; Labhasetwar V Biodegradable Nanoparticles for Drug and Gene Delivery to Cells and Tissue. Adv. Drug Delivery Rev. 2012, 64, 61–71. [DOI] [PubMed] [Google Scholar]
- (39).Sahoo SK; Panyam J; Prabha S; Labhasetwar V Residual Polyvinyl Alcohol Associated with Poly (D,L-Lactide-Co-Glycolide) Nanoparticles Affects Their Physical Properties and Cellular Uptake. J. Controlled Release 2002, 82 (1), 105–114. [DOI] [PubMed] [Google Scholar]
- (40).Bertrand N; Leroux J-C The Journey of a Drug-Carrier in the Body: An Anatomo-Physiological Perspective. J. Controlled Release 2012, 161 (2), 152–163. [DOI] [PubMed] [Google Scholar]
- (41).Udby L; Borregaard N Subcellular Fractionation of Human Neutrophils and Analysis of Subcellular Markers. In Neutrophil Methods and Protocols’, Methods in Molecular Biology; Quinn, DeLeo MT, Bokoch FR, M. G, Eds.; Humana Press, 2007; Vol 412. [DOI] [PubMed] [Google Scholar]
- (42).Joiner KA The Opsonizing Ligand on Salmonella Typhimurium Influences Incorporation of Specific, but Not Azurophil, Granule Constituents into Neutrophil Phagosomes. J. Cell Biol. 1989, 109 (6), 2771–2782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).Kamaly N; Yameen B; Wu J; Farokhzad OC Degradable Controlled-Release Polymers and Polymeric Nanoparticles: Mechanisms of Controlling Drug Release. Chem. Rev. (Washington, DC, U. S.) 2016, 116 (4), 2602–2663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (44).Walkey CD; Olsen JB; Guo H; Emili A; Chan WCW Nanoparticle Size and Surface Chemistry Determine Serum Protein Adsorption and Macrophage Uptake. J. Am. Chem. Soc. 2012, 134, 2139. [DOI] [PubMed] [Google Scholar]
- (45).Lunov O; Syrovets T; Loos C; Beil J; Delacher M; Tron K; Nienhaus GU; Musyanovych A; Mailänder V; Landfester K; et al. Differential Uptake of Functionalized Polystyrene Nanoparticles by Human Macrophages and a Monocytic Cell Line. ACS Nano 2011, 5, 1657. [DOI] [PubMed] [Google Scholar]
- (46).Müller K; Skepper JN; Posfai M; Trivedi R; Howarth S; Corot C; Lancelot E; Thompson PW; Brown AP; Gillard JH Effect of Ultrasmall Superparamagnetic Iron Oxide Nanoparticles (Ferumoxtran-10) on Human Monocyte-Macrophages in Vitro. Biomaterials 2007, 28, 1629. [DOI] [PubMed] [Google Scholar]
- (47).Yu SS; Lau CM; Thomas SN; Gray Jerome W; Maron DJ; Dickerson JH; Hubbell JA; Giorgio TD Size- and Charge-Dependent Non-Specific Uptake of PEGylated Nanoparticles by Macrophages. Int. J. Nanomed. 2012, 799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (48).Arnida; Janát-Amsbury MM; Ray A; Peterson CM; Ghandehari H Geometry and Surface Characteristics of Gold Nanoparticles Influence Their Biodistribution and Uptake by Macrophages. Eur. J. Pharm. Biopharm. 2011, 77, 417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (49).Smith BR; Ghosn EEB; Rallapalli H; Prescher JA; Larson T; Herzenberg LA; Gambhir SS Selective Uptake of Single-Walled Carbon Nanotubes by Circulating Monocytes for Enhanced Tumour Delivery. Nat. Nanotechnol. 2014, 9, 481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (50).MacParland SA; Tsoi KM; Ouyang B; Ma XZ; Manuel J; Fawaz A; Ostrowski MA; Alman BA; Zilman A; Chan WCW; et al. Phenotype Determines Nanoparticle Uptake by Human Macrophages from Liver and Blood. ACS Nano 2017, 11, 2428. [DOI] [PubMed] [Google Scholar]
- (51).Mitchell MJ; Wayne E; Rana K; Schaffer CB; King MR TRAIL-Coated Leukocytes That Kill Cancer Cells in the Circulation. Proc. Natl. Acad. Sci. U. S. A. 2014, 111 (3), 930–935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (52).Ankrum JA; Miranda OR; Ng KS; Sarkar D; Xu C; Karp JM Engineering Cells with Intracellular Agent–Loaded Microparticles to Control Cell Phenotype. Nat. Protoc. 2014, 9 (2), 233–245. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.