

Dynamics of a ground-state chemical reaction are revealed by ultrafast x-ray scattering.
Abstract
We explore the photo-induced kinetics of 1,3-cyclohexadiene upon excitation at 200 nm to the 3p state by ultrafast time-resolved, gas-phase x-ray scattering using the Linac Coherent Light Source. Analysis of the scattering anisotropy reveals that the excitation leads to the 3px and 3py Rydberg electronic states, which relax to the ground state with a time constant of 208 ± 11 fs. In contrast to the well-studied 266 nm excitation, at 200 nm the majority of the molecules (76 ± 3%) relax to vibrationally hot cyclohexadiene in the ground electronic state. A subsequent reaction on the ground electronic state surface leads from the hot cyclohexadiene to 1,3,5-hexatriene, with rates for the forward and backward reactions of 174 ± 13 and 355 ± 45 ps, respectively. The scattering pattern of the final hexatriene product reveals a thermal distribution of rotamers about the carbon-carbon single bonds.
INTRODUCTION
The ring-opening reaction of 1,3-cyclohexadiene (CHD) to 1,3,5-hexatriene (HT) is a prototypical photochemically induced electrocyclic reaction, and it is the reaction motif for the synthesis of vitamin D in the skin upon exposure to sunlight (1). Numerous experimental and computational studies have explored the dynamics of CHD upon excitation at 267 nm to the 1B valence state, which results in conrotatory ring opening according to the Woodward-Hoffmann rules (1). The wavepacket created by optical excitation evolves on the steeply sloped 1B surface, glancing by the conical intersection (CI) with the 2A state while staying on the adiabatic potential energy surface (2, 3). This passage propels the system into a symmetry-breaking direction and channels the wavepacket to the CI, where it transitions to the 1A surface in a region that leads to the HT product with about 50% yield (2, 4–8). Given the complexity of this intriguing mechanism, it stands to reason that different electronic excitations might lead to different outcomes. Yet the dynamics upon excitation to higher-lying states remains poorly understood. We report here on a remarkably different reaction outcome when the molecule is excited at 200 nm, which triggers the photochemical reaction on the ground electronic state.
Prior studies have established that the peaks in the ultraviolet (UV)–visible absorption spectrum near 200 nm (Fig. 1) are due to excitation to 3p Rydberg states. Merchán et al. (9) associated the two dominant peaks to a vibrational progression (≈1450 cm−1) in the 3p manifold belonging to the lowest adiabatic ionization potential at 8.12 eV. The Rydberg state character is borne out by photoionization-photoelectron studies, which show a dominant Δv = 0 vibronic transition (10). Ultrafast time-resolved spectra by Bühler et al. (11), excited at 207 nm, showed the Rydberg state to be stable but short lived (≈200 fs), in agreement with our own observations presented below. These authors also reported the first indication that at shorter wavelengths, CHD might not undergo the same ring-opening reaction as in the 1B electronic state, but instead return to the ground electronic state in the ring-closed form. However, two studies with two-photon excitation using 400-nm pulses by Sekikawa and co-workers (12, 13) challenged this finding. They interpreted photoelectron spectra taken with extreme UV high harmonics probe pulses as showing rapid conversion from the Rydberg state to 2A, followed by a dynamical evolution of the ring structure on the 2A surface, eventually causing ring opening on a 500- to 800-fs time scale via a CI, leading to HT with a minimum quantum yield of 0.46 (12). Subsequent experiments where the target CHD molecules themselves produce the high harmonics appeared supportive of the earlier findings (13). The dominance of ring opening in the high harmonics experiments is inconsistent with Bühler’s photoelectron spectra, yet the harmonics experiments are encumbered by substantial noise and the uncertainty generally associated with two-photon excitation. Consequently, there is no clarity regarding the reaction dynamics, the time scales, or the quantum yield of the photochemically induced dynamics of this important prototypical molecule at short wavelengths.
Fig. 1. UV absorption spectrum of CHD.
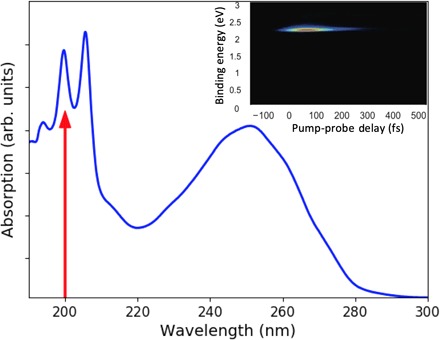
The broad band between 220 and 280 nm reflects the absorption to the 1B state. The sharper peaks at short wavelengths are due to the 3p Rydberg states. A red arrow indicates the 200-nm excitation wavelength used in the present experiment. The inset shows the corresponding time-resolved photoelectron spectrum for the short-lived 3p state.
Hot HT and CHD reaction products are difficult to distinguish and quantitatively analyze using spectroscopic methods because the intensity of spectroscopic signals depends on the transition dipole moments, which in turn may depend on the structure, and on the line shapes of the transition, which are broad in the hot reaction products. X-ray scattering signals depend on the number of electrons in a molecule and on the molecular structure and therefore are a more direct measure of chemical transformations. This makes ultrafast x-ray scattering an attractive alternative to spectroscopic experiments for determining reaction dynamics and quantum yields. We exploit the short pulse duration and brightness of the Linac Coherent Light Source (LCLS) X-ray Free-Electron Laser at the SLAC National Accelerator Laboratory to measure the ultrafast CHD dynamics in a dilute gas. A novel analysis technique that uses a large pool of instantaneous structures obtained from simulations aids the analysis of the experimental data.
MATERIALS AND METHODS
We performed time-resolved gas-phase x-ray scattering experiments at the Coherent x-ray Imaging instrument (14) of the LCLS at the SLAC National Accelerator Laboratory as previously described (15–17). CHD was introduced as a room temperature gas with a pressure of 6 torr and intersected by a 200-nm pump pulse. A Cornell-SLAC pixel array detector (18) recorded single-shot scattering patterns, which were binned by their time relative to the pump laser and averaged over many shots. The pump-probe signals are given as percent differences
| (1) |
where Ion(q, t) is the signal with the pump laser on, which depends on the magnitude of the scattering vector q and the delay time t, while Ioff(q) is the scattering signal with the pump laser off. Taking the difference signal is advantageous as many of the poorly characterized experimental parameters, such as background signals, and gas pressure fluctuations cancel out. The percent difference signal depends on the excitation fraction, which is kept low to minimize multiphoton processes.
We simulate the ground-state dynamics of hot HT using the dynamics code SHARC (19, 20) interfaced with the electronic structure package MOLPRO (21), with electronic structure calculations performed at the SS-CAS(2,2)/6-311+G(d) level of theory. The initial conditions for hot HT were sampled from the Wigner distributions for the cZc-HT and tZt-HT forms of the molecule. The excess kinetic energy in each case corresponded to the 200-nm photon energy randomly distributed among all degrees of freedom of the molecules, corrected for the energy relative to the CHD ground state (1.02 eV for cZc-HT and 0.69 eV for tZt-HT). For each form, 50 trajectories were propagated for 3 ps, allowing the molecule to traverse the entire ground-state potential energy surface. Last, 600,000 geometries were extracted for the hot HT conformers (covering all three possible hot HT conformers; see table S1).
RESULTS AND DISCUSSION
The linear polarization of the pump laser induces an alignment in the molecular sample that gives rise to an anisotropy of the time-dependent scattering patterns. As has been pointed out previously (22, 23), this anisotropy depends sensitively on the alignment of the molecule’s transition dipole moment with respect to the molecular axes. Figure 2 shows the experimental pump-probe scattering image at 1 ns, where the anisotropy is recovered because of rotational rephasing (see the Supplementary Materials) (24). A computational model of the scattering signals, described in detail previously (21), gives rise to the three computed scattering images shown in Fig. 2 (top). A comparison of the computed scattering image with the experimental ones gives an optimal fit with an admixture of 62 + 2% 3px and 38 ± 2% 3py, indicating that the excitation to the out-of-plane 3px Rydberg orbital is dominant. This is in close agreement with the photoelectron spectra by Bühler et al. (11) and the computed transition moments by Merchán et al (9). Both the 3px and the 3py orbitals are of B symmetry in the C2 point group of the CHD molecule.
Fig. 2. The anisotropic components of the pump-probe scattering patterns.
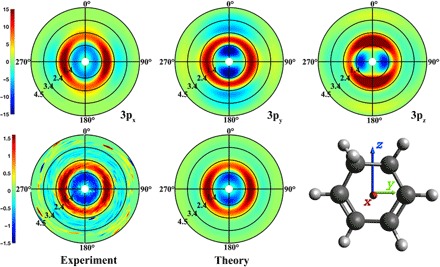
Top: Computed patterns for alignments of the transition dipole moments along the x, y, and z directions. Bottom left: Experimental pattern at 1 ns. Bottom center: Simulated pattern with the optimal admixture of transition dipole moments.
The isotropic component of the difference scattering signal (Fig. 3) reveals a time dependence that stretches from the femtosecond regime to the 1-ns maximum time of the experiment. On the femtosecond time scale, the molecules are excited to the 3p Rydberg states, where they persist for about 200 fs according to the photoelectron studies of Bühler et al. (11). The femtosecond time scale signal therefore shows the initially excited state and the products immediately following the electronic decay out of those states. The picosecond regime signal, i.e., the signal between 1 and 15 ps in Fig. 3, could show any dynamics or kinetics immediately following the electronic decay. Inspection of Fig. 3 suggests little activity during that time regime. At longer times, 15 ps to 1 ns, the difference signal increases markedly, suggesting a reaction on the hot ground state of the system.
Fig. 3. The isotropic component of the time-dependent experimental percent difference scattering signal of CHD as a function of time.
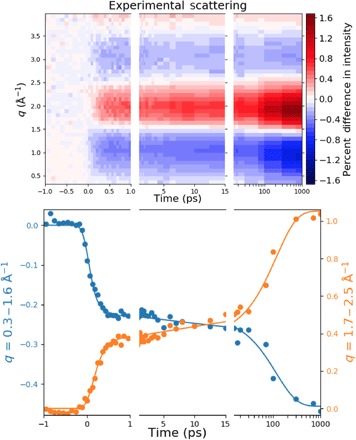
Plotted on top is the percent difference in scattering intensity (color bar) induced by the laser pulse as a function of the absolute value of the scattering vector, q, and the pump-probe time delay. The bottom panel shows averages over two q ranges (dots) and the kinetic fit as described in the text (lines). Because of the large range of experimental delay times the panels are divided into three time segments: one from −1 to 1 ps to show the ultrafast temporal response to the pump laser pulse; the times from 1ps to 15 ps showing the initial ground-state population; and the time range from 15 ps to 1 ns (on a log scale) giving the increase in HT population as the molecules equilibrate on the ground state potential energy surface.
We model these data according to the reaction sequence shown in Fig. 4 and in Eq. 2 (A to C). The pump photon excites the molecules to the electronically excited 3px/3py states, indicated as CHD*. The photoelectron spectra by Bühler et al. (11) showed that the 3px and 3py states have very similar rates; thus, the 3px and 3py states are given the same decay constant (k1) for the reaction to hot CHD and hot HT in the electronic ground state. Thermal ring opening and closing reactions between these hot isomers eventually lead to an equilibrium. We note that the direct photochemical electrocyclic ring-opening reaction is conrotatory, while the secondary ring-opening reaction on the electronic ground state should follow a thermal disrotatory mechanism. The lack of substituents in CHD does not permit the two stereospecific pathways to be distinguished, but their yields should depend on the quantum yield of the initial electronic decay.
Fig. 4. Reaction Pathway of Rydberg-Excited CHD.
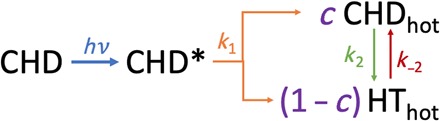
The reaction scheme gives rise to the following kinetic equations for the time-dependent populations of CHD*, CHDhot, and HThot
| (2A) |
| (2B) |
| (2C) |
with
Here, k1 is the rate constant for the decay of the electronically excited CHD, c is the fraction of excited CHD that initially decays to CHDhot, and k2 and k−2 are the rate constants for the ground-state hot CHD ring opening to form hot HT and its back reaction, respectively. The rate equations, when convoluted with a Gaussian instrument function, are fit to the experimental signals. The initial ground-state structure of CHD at room temperature is well known (25) and is used to calculate the Ioff(q) signal in Eq. 1.
The CHD*, the hot CHD, and the hot HT each was associated with a time-independent scattering pattern with adjustable q dependence Sx(q), with x being CHD*, CHDhot, or HThot. The time dependence arises from the populations in the respective states [Eq. 2 (A to C)]. Therefore, the two-dimensional fit to the isotropic scattering Piso(q, t) (Fig. 3, top) takes the following form
| (3) |
Fitting the results of the scattering experiment (Fig. 3) to the analytical form of Eq. 2, with the scattering signals of HThot, CHDhot, and CHD* as adjustable parameters (Eq. 3), results in an excellent fit (Fig. 3, bottom). The experimentally derived hot scattering pattern for HT, SHThot(q), is shown in Fig. 5 (uncertainties are given in 1σ).
Fig. 5. Percent difference scattering signal, scaled for 100% excitation probability, for the hot (6.20 eV) molecules relative to the cold CHD (room temperature) molecules.
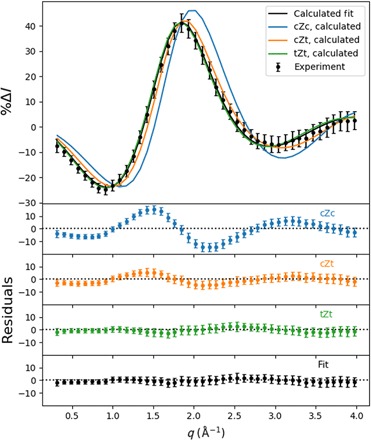
(Top) The experimental scattering signal, represented by black dots with error bars (given in 1σ), and the theoretical patterns to the three conformers, given in blue, orange, and green. A fit to a mixture of the three theoretical signals (20% cZt, 80% tZt) is given as a black line. (Bottom) The separate residuals of the experimental percent difference scattering signals with respect to each of the HT theoretical patterns and the optimal admixture.
The conventional approach to account for vibrational excitation in the calculation of scattering signals involves temperature-dependent amplitude terms (26). Our experiment inserts 6.2 eV of the pump photon energy into vibrations, and assuming harmonic vibrations as reported (27), the vibrational temperature of CHD in the electronic ground state would be 2870 K. It is questionable whether the conventional approach works under these extreme conditions. Instead, we use a large pool of hot CHD and HT structures, as described in Materials and Methods. The HT conformers are divided into cZc, cZt, and tZt rotamers based on the criteria indicated in table S1, adapted from Wolf et al. (5). This results in the scattering patterns shown in Fig. 5 (solid lines), where only the isotropic components are shown. The cZc pattern is distinguishable, while the cZt and tZt patterns are quite similar. The hot HT pattern was fit to an admixture of the three calculated hot HT patterns. The best fit gave an HT conformational distribution of 20 ± 15% cZt and 80 ± 10% tZt. The conformational distribution was averaged over all time points since the signal did not indicate any coherent dynamics between hot HT rotamers, as had been observed previously with 1B excitation (5). The fit also yields the overall percent excitation as 6.0 ± 0.2%.
The CHD* decays in 208 ± 11 fs out of the originally excited electronic state, in excellent agreement with prior lifetime measurements (11, 13). The decay leads back to the ring-closed ground-state CHD with a quantum yield of 76 ± 3%. The dominant relaxation pathway is, therefore, not to the ring-open HT, as suggested in (12, 13), but rather to the ring-closed CHD as postulated by Bühler et al. (11). Once on the electronic ground state, the ring opening proceeds with a time constant of 174 ± 13 ps, while the reverse ring closing has a time constant of 355 ± 45 ps. These kinetic reaction processes are represented in Fig. 6 by the relative populations of the three main components of the reaction. The interconversion of CHDhot to HThot proceeds on a time scale of hundreds of picoseconds, eventually leading to an equilibrium with 33% CHDhot and 67% HThot, respectively.
Fig. 6. Relative populations of the three components of the photoreaction (Eq. 2) on a logarithmic timescale: Electronically excited CHD*, the hot ground-state CHDhot, and the hot ground-state HThot.
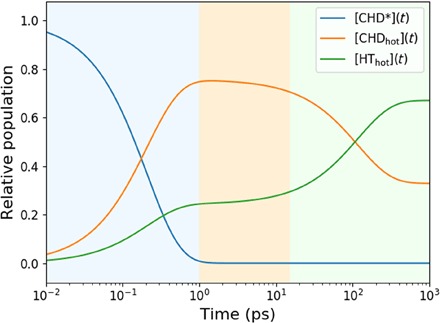
The background colors indicate the different time regimes of the kinetics: Blue is dominated by the decay of CHD*, orange is dominated by the hot CHD, and green indicates the equilibration to 67% HThot and 33% CHDhot.
It has been reported that upon excitation at 212 nm, CHD may also decompose into benzene and hydrogen molecules (28). We tested for the presence of that reaction channel in a separate fit but found that adding the possibility of dissociation did not yield a better fit. The scattering experiment is very sensitive to dissociation reactions (29), and so we conclude that the dissociation channel is not active on the 1-ns time scale of the experiment.
The discovery that the 200-nm photoinduced ring-opening reaction proceeds mostly thermally and on the ground-state surface stands in contrast to the conrotatory photochemical mechanism observed at longer wavelengths and merits further discussion. While the lower-energy excitation to the 1B state leads to HT with a 50% quantum yield, we find the yield of immediate HT formation to be substantially lower for 200-nm excitation (24%). This suggests that the dynamics through the CI between the Rydberg states and the steep 2A state does not favor ring opening.
The experiment determines that the 3px and 3py Rydberg states are excited upon 200-nm excitation. Both of them are of B symmetry, as is the 1B state that is reached with the longer wavelength photons. As illustrated in Fig. 7, it is likely that the doubly excited 2A state, which is the ring-opening reaction coordinate, crosses the Rydberg states at higher energies. The intersection with the 3s state, of A symmetry, leads to an avoided crossing, consistent with the observation by Garavelli et al. (3) that in the Franck-Condon region the 3s Rydberg state is mixed with the 2A surface. The crossings with the 3px and 3py states give rise to CIs. This picture is consistent with the 221-fs lifetime of the 3px and 3py Rydberg states, and a lifetime of the 3s state that is so short that it cannot be identified in the broad absorption spectrum (Fig. 1).
Fig. 7. The potential energy surfaces of CHD allow for reversion to the ground state of the reactant molecule, favored when the wavepacket is close to the plane of symmetry, or ring opening to the HT product molecule, favored when the wavepacket is off symmetry.
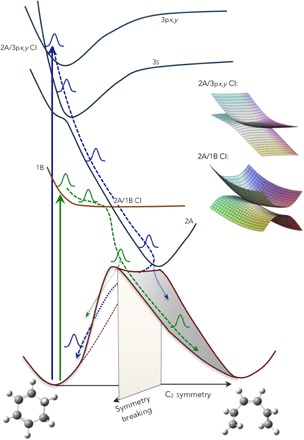
The path through the 2A/1B CIs can either deflect the wavepacket away from the symmetry plane (1B excitation, leading preferentially to the HT product) or focus it onto the symmetry plane (3px,y excitation, leading preferentially to the hot CHD).
The CIs between the 2A surface and the 1B and 3px,y surfaces, respectively, are therefore of similar topology. However, the coupling between the Rydberg state and the doubly excited 2A valence state is likely much smaller since the electron density of the Rydberg state in the overlap region is small compared with that of the 1B valence state. Consequently, the Rydberg state retains much of the shape of its potential energy surface, consistent with the photoelectron spectrum (Fig. 1) that shows no Franck-Condon progression in the photoionization.
An important aspect of the ring-opening reaction is the approach to the strongly coupled 1B/2A CI. With 200-nm excitation, there is no pronounced cusp in the 3p/2A CI because of the smaller coupling, and the vibrational wave function in the 3px,y state probably looks much like an unperturbed vibrational wave function within the Rydberg potential. As the amplitude crosses into 2A, it does, therefore, not scatter into an out-of-symmetry direction. The curvature of the 2A state is, moreover, such that the energy in the plane of symmetry is lowest, implying a funneling of the traveling wavepacket toward the CI on the C2 symmetry plane. Both of these effects focus the wavepacket toward the symmetry plane when it reaches the bottom of the 2A well, which is less favorable for an exit toward the HT product. With the 1B excitation at 266 nm, however, the wavepacket approaches the CI from the lower surface, causing it to be deflected away from the maximum at the CI into an out-of-symmetry direction, aiming it directly toward the region where the transition to the ground state occurs (1, 3, 30, 31).
In summary, we find that photoexcitation of CHD at 200 nm leads to rapid dynamics through a series of CIs and avoided crossings but dominantly (76%) recovers the CHD molecule in its ground state. The internal energy remains high, however, triggering a thermal reaction to the ring-open HT product on a 174-ps time scale under collision-free conditions. While the lack of substituents on CHD renders it impossible to distinguish conrotatory and disrotatory reaction products, it seems rational to postulate a disrotatory path for this reaction of the hot molecule on the ground electronic state. The final product distribution for this photothermal reaction is therefore dominantly disrotatory, contrary to the traditional Woodward-Hoffmann rules that propose a conrotatory reaction outcome for the photochemical reaction. Further investigations in the deep-UV range of the spectrum, including on systems with less symmetry, may be rewarding.
Supplementary Material
Acknowledgments
Funding: This work was supported by the U.S. Department of Energy, Office of Science, Basic Energy Sciences under award DE-SC0017995. Use of the Linac Coherent Light Source (LCLS), SLAC National Accelerator Laboratory, is supported by the U.S. Department of Energy, Office of Science, Office of Basic Energy Sciences under contract no. DE-AC02-76SF00515. A.K. acknowledges support from the Royal Society of Edinburgh Sabbatical Fellowship (58507) and a research grant from the Carnegie Trust for the Universities of Scotland (CRG050414), N.Z. a Carnegie Ph.D. scholarship, and D.B. an EPSRC Ph.D. studentship from the University of Edinburgh. Author contributions: P.M.W., A.K., and M.P.M. directed the project. M.L. and S.B. performed x-ray alignment and data collection. S.C., J.S.R., and M.P.M. performed laser alignment. J.K. provided software support during the experiment. W.D. and Y.C. performed record keeping during the experiment. J.M.R. and H.Y. performed the analysis on the experimental data. H.Y., N.Z., A.M.C., and D.B. performed ab initio calculations and scattering pattern simulations. J.M.R., H.Y., B.S., A.M.C., W.D., N.G., Y.C., A.O., D.B., A.K., and P.M.W. interpreted the results. J.R., H.Y., A.K., and P.M.W. wrote the manuscript in consultation with all the other authors. Competing interests: The authors declare that they have no conflicts of interest. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/5/9/eaax6625/DC1
Section S1. Anisotropic and isotropic scattering decomposition
Section S2. Kinetics fit
Section S3. Separating HT rotamers
Fig. S1. The time-resolved anisotropic scattering signal for CHD excited at 200 nm.
Fig. S2. The residuals of the kinetics fit, divided into temporal regions as shown in Fig. 3 of the main text.
Table S1. Criteria for fitting the HT rotamers.
REFERENCES AND NOTES
- 1.Deb S., Weber P. M., The ultrafast pathway of photon-induced electrocyclic ring-opening reactions: The case of 1, 3-cyclohexadiene. Ann. Rev. Phys. Chem. 62, 19–39 (2011). [DOI] [PubMed] [Google Scholar]
- 2.Celani P., Ottani S., Olivucci M., Bernardi F., Robb M. A., What happens during the picosecond lifetime of 2A1 cyclohexa-1,3-diene? A CAS-SCF study of the cyclohexadiene/hexatriene photochemical interconversion. J. Am. Chem. Soc. 116, 10141–10151 (1994). [Google Scholar]
- 3.Garavelli M., Page C. S., Celani P., Olivucci M., Scmid W. E., Trushin S. A., Fuss W., Reaction path of a sub-200 fs photochemical electrocyclic reaction. J. Phys. Chem. A 105, 4458–4469 (2001). [Google Scholar]
- 4.Anderson N. A., Pullen S. H., Walker L. A., Shiang J. J., Sension R. J., Ultrafast polyene dynamics in solution: The conformational relaxation and thermalization of highly excited cis-1,3,5-hexatriene as a function of initial conformation and solvent. J. Phys. Chem. A 102, 10588–10598 (1998). [Google Scholar]
- 5.Wolf T. J. A., Sanchez D. M., Yang J., Parrish R. M., Nunes J. P. F., Centurion M., Coffee R., Cryan J. P., Gühr M., Hegazy K., Kirrander A., Li R. K., Ruddock J., Shen X., Veccione T., Weathersby S. P., Weber P. M., Wilkin K., Yong H., Zheng Q., Wang X. J., Minitti M. P., Martinez T. J., The photochemical ring-opening of 1,3-cyclohexadiene imaged by ultrafast electron diffraction. Nat. Chem. 11, 504–509 (2019). [DOI] [PubMed] [Google Scholar]
- 6.J. M. Budarz, “Making a molecular movie: Imaging ultrafast reaction dynamics with gas-phase x-ray Scattering,” thesis, Brown University (2015). [Google Scholar]
- 7.Kim J., Tao H., Martinez T. J., Bucksbaum P., Ab initio multiple spawning on laser-dressed states: A study of 1,3-cyclohexadiene photoisomerization via light-induced conical intersections. J. Phys. B 48, 164003 (2015). [DOI] [PubMed] [Google Scholar]
- 8.Lochbrunner S., Fuss W., Schmid W. E., Kompa K. L., Electronic relaxation and ground-state dynamics of 1,3-cyclohexadiene and cis-hexatriene in ethanol. J. Phys. Chem. A 102, 9334–9344 (1998). [Google Scholar]
- 9.Merchán M., Serrano-Andrés L., Slater L. S., Roos B. O., McDiarmid R., Xing, Electronic spectra of 1,4-cyclohexadiene and 1,3-cyclohexadiene: A combined experimental and theoretical investigation. J. Phys. Chem. A 103, 5468–5476 (1999). [Google Scholar]
- 10.Kuthirummal N., Rudakov F. M., Evans C. L., Weber P. M., Spectroscopy and femtosecond dynamics of the ring opening reaction of 1,3-cyclohexadiene. J. Chem. Phys. 125, 133307 (2006). [DOI] [PubMed] [Google Scholar]
- 11.Bühler C. C., Minitti M. P., Deb S., Bao J., Weber P. M., Ultrafast dynamics of 1,3-cyclohexadiene in highly excited states. J. Atom.Molec.Opt. Phys. 2011, 637593 (2011). [Google Scholar]
- 12.Iikubo R., Sekikawa T., Harabuchi Y., Taketsugu T., Structural dynamics of photochemical reactions probed by time-resolved photoelectron spectroscopy using high harmonic pulses. Faraday Disc. 194, 147–160 (2016). [DOI] [PubMed] [Google Scholar]
- 13.Kaneshima K., Ninota Y., Sekikawa T., Time-resolved high-harmonic spectroscopy of ultrafast photoisomerization dynamics. Opt. Express 26, 31039–31054 (2018). [DOI] [PubMed] [Google Scholar]
- 14.Liang M., Williams G. J., Messerschmidt M., Seibert M. M., Montanez P. A., Hayes M., Milathianaki D., Aquila A., Hunter M. S., Koglin J. E., Schafer D. W., Guillet S., Busse A., Bergan R., Olson W., Fox K., Stewart N., Curtis R., Miahnahri A. A., Boutet S., The Coherent X-ray Imaging instrument at the Linac Coherent Light Source. J. Synchrotron Radiat. 22, 514–519 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Stankus B., Yong H., Zotev N., Ruddock J. M., Bellshaw D., Lane T. J., Liang M., Boutet S., Carbajo S., Robinson J. S., Du W., Goff N., Chang Y., Koglin J. E., Minitti M. P., Kirrander A., Weber P. M., Ultrafast X-Ray scattering reveals vibrational coherence following rydberg excitation. Nat. Chem. 11, 716–721 (2019). [DOI] [PubMed] [Google Scholar]
- 16.Budarz J. M., Minitti M. P., Cofer-Shabica D. V., Stankus B., Kirrander A., Hastings J. B., Weber P. M., Observation of femtosecond molecular dynamics via pump-probe gas phase X-ray scattering. J. Phys. B Atom. Molec. Opt. Phys. 49, 034001 (2016). [Google Scholar]
- 17.Minitti M. P., Budarz J. M., Kirrander A., Robinson J. S., Ratner D., Lane T. J., Zhu D., Glownia J. M., Kozina M., Feng Y., Nelson S., Saita K., Stankus B., Northey T., Hastings J. B., Weber P. M., Imaging molecular motion: Femtosecond X-ray scattering of an electrocyclic chemical reaction. Phys. Rev. Lett. 114, 255501 (2015). [DOI] [PubMed] [Google Scholar]
- 18.P. Hart, S. Boutet, G. Carini, M. Dubrovin, B. Duda, D. Fritz, G. Haller, R. Herbst, S. Herrmann, C. Kenney, N. Kurita, H. Lemke, M. Messerschmidt, M. Nordby, J. Pines, D. Schafer, M. Swift, M. Weaver, G. Williams, D. Zhu, N. Van Bakel, J. Morse, The CSPAD megapixel x-ray camera at LCLS, in Proceedings Volume 8504, X-Ray Free-Electron Lasers: Beam Diagnostics, Beamline Instrumentation, and Applications, 15 October 2012, San Diego, CA.
- 19.Mai S., Marquetand P., González L., Nonadiabatic dynamics: The SHARC approach. Wiley Interdiscip. Rev. Comput. Mol. Sci. 8, e1370 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Richter M., Marquetand P., González-Vázquez J., Sola I., González L., SHARC: Ab Initio molecular dynamics with surface hopping in the adiabatic representation including arbitrary couplings. J. Chem. Theory Comput. 7, 1253–1258 (2011). [DOI] [PubMed] [Google Scholar]
- 21.Werner H.-J., Knowles P. J., Knizia G., Manby F. R., Schütz M., Molpro: A general-purpose quantum chemistry program package. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2, 242–253 (2012). [Google Scholar]
- 22.Yong H., Zotev N., Stankus B., Ruddock J. M., Bellsahw D., Boutet S., Lane T. J., Liang M., Carbajo S., Robinson J. S., Du W., Goff N., Chang Y., Koglin J. E., Waters M. D. J., Sølling T. I., Minitti M. P., Kirrander A., Weber P. M., Determining orientations of optical transition dipole moments using ultrafast X-ray scattering. J. Phys. Chem. Lett. 9, 6556–6562 (2018). [DOI] [PubMed] [Google Scholar]
- 23.Lorenz U., Møller K. B., Henriksen N. E., On the interpretation of time-resolved anisotropic diffraction patterns. New J. Phys. 12, 113022 (2010). [Google Scholar]
- 24.Baskin J. S., Zewail A. H., Ultrafast electron diffraction: Oriented molecular structures in space and time. ChemPhysChem 6, 2261–2276 (2005). [DOI] [PubMed] [Google Scholar]
- 25.Cyvin S. J., Gebhardt O., Mean amplitudes of vibration for 1,3-cyclohexadiene from spectroscopic data. J. Mol. Struct. 27, 435–437 (1975). [Google Scholar]
- 26.I. Hargittai, M. Hargittai, Stereochemical Applications of Gas-Phase Electron Diffraction (VCH, 1988).
- 27.Autrey D., Choo J., Laane J., Spectroscopic Determination of the Ring-Twisting Potential Energy Function of 1,3-Cyclohexadiene and comparison with ab initio calculations. J. Phys. Chem. A 105, 10230–10236 (2001). [Google Scholar]
- 28.Cromwell E. F., Liu D. J., Vrakking M. J. J., Kung A. H., Lee Y. T., Dynamics of H2 elimination from cyclohexadiene. J. Chem. Phys. 95, 297–307 (1991). [Google Scholar]
- 29.Ruddock J. M., Zotev N., Stankus B., Yong H., Bellshaw D., Boutet S., Lane T. J., Liang M., Carbajo S., Du W., Kirrander A., Minitti M., Weber P. M., Simplicity beneath complexity: Counting molecular electrons reveals transients and kinetics of photodissociation reactions. Angew. Chem. Int. Ed. 58, 6371–6375 (2019). [DOI] [PubMed] [Google Scholar]
- 30.Kosma K., Trushin S. A., Fuß W., Schmid W. E., Cyclohexadiene ring opening observed with 13 fs resolution: Coherent oscillations confirm the reaction path. Phys. Chem. Chem. Phys. 11, 172–181 (2009). [DOI] [PubMed] [Google Scholar]
- 31.Schönborn J. B., Sielk J., Hartke B., Photochemical ring-opening of cyclohexadiene: Quantum wavepacket dynamics on a global ab initio potential energy surface. J. Phys. Chem. A 114, 4036–4044 (2010). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/5/9/eaax6625/DC1
Section S1. Anisotropic and isotropic scattering decomposition
Section S2. Kinetics fit
Section S3. Separating HT rotamers
Fig. S1. The time-resolved anisotropic scattering signal for CHD excited at 200 nm.
Fig. S2. The residuals of the kinetics fit, divided into temporal regions as shown in Fig. 3 of the main text.
Table S1. Criteria for fitting the HT rotamers.