

The discovery of protein biomarkers is hampered by the limited availability of assays needed for the reproducible quantification of proteins in complex biological matrices such as blood plasma. Here, we describe a biomarker development strategy based on large scale targeted proteomics measurements including a rigorous experimental design and data analysis. Applied to epithelial ovarian cancer, we identified a five-protein biomarker signature that in combination with CA125 increased sensitivity in detecting epithelial ovarian cancer compared to CA125 alone.
Keywords: Cancer biomarker(s), ovarian cancer, selected reaction monitoring, targeted mass spectrometry, plasma or serum analysis, quantification, statistics
Graphical Abstract

Highlights
Rigorous experimental design and data analysis for large-scale SRM studies.
Plasma-based biomarker signature combined with CA125 for ovarian cancer detection.
Broadly applicable strategy for the development of diagnostic biomarker assays.
Abstract
Protein biomarkers for epithelial ovarian cancer are critical for the early detection of the cancer to improve patient prognosis and for the clinical management of the disease to monitor treatment response and to detect recurrences. Unfortunately, the discovery of protein biomarkers is hampered by the limited availability of reliable and sensitive assays needed for the reproducible quantification of proteins in complex biological matrices such as blood plasma. In recent years, targeted mass spectrometry, exemplified by selected reaction monitoring (SRM) has emerged as a method, capable of overcoming this limitation. Here, we present a comprehensive SRM-based strategy for developing plasma-based protein biomarkers for epithelial ovarian cancer and illustrate how the SRM platform, when combined with rigorous experimental design and statistical analysis, can result in detection of predictive analytes.
Our biomarker development strategy first involved a discovery-driven proteomic effort to derive potential N-glycoprotein biomarker candidates for plasma-based detection of human ovarian cancer from a genetically engineered mouse model of endometrioid ovarian cancer, which accurately recapitulates the human disease. Next, 65 candidate markers selected from proteins of different abundance in the discovery dataset were reproducibly quantified with SRM assays across a large cohort of over 200 plasma samples from ovarian cancer patients and healthy controls. Finally, these measurements were used to derive a 5-protein signature for distinguishing individuals with epithelial ovarian cancer from healthy controls. The sensitivity of the candidate biomarker signature in combination with CA125 ELISA-based measurements currently used in clinic, exceeded that of CA125 ELISA-based measurements alone. The SRM-based strategy in this study is broadly applicable. It can be used in any study that requires accurate and reproducible quantification of selected proteins in a high-throughput and multiplexed fashion.
Clinical management of aggressive tumors requires sensitive and specific protein biomarkers that can be monitored in a noninvasive way (1). To establish the clinical value of such biomarkers, it is imperative to reliably quantify proteins of interest in large subject cohorts (2). Blood plasma is the preferred source of protein biomarkers as blood collection is minimally invasive (3). However, the complex and large dynamic range of protein concentrations in plasma pose a technical challenge for the accurate, sensitive and reproducible quantification of biomarker candidates across hundreds of samples (4). Although affinity-based assays, such as enzyme-linked immunosorbent assay (ELISA), have traditionally been the method of choice, they are constrained by their limited availability for human proteins and time-consuming development of new and reliable assays.
Targeted mass spectrometry (MS) based on selected reaction monitoring (SRM)1 is a highly sensitive MS approach for accurate and reproducible protein quantification and for fast and cost-effective development of assays. It has been proposed several years ago as an alternative method to immune reagent-based measurements for developing biomarkers (2, 3). A requirement for successful application of SRM in this area is a rigorous study design, reproducible sample preparation, and appropriate statistical analysis (5). Importantly, the reliable detection of biomarkers requires studying large subject cohorts. Such large-scale studies face substantial challenges on many different levels. First, the investment of substantial resources for SRM-based quantification across large subject cohorts requires a careful selection of protein targets. Second, the collection of clinical samples often spans many years and the duration and condition of sample storage may confound bona fide biomarkers. Third, the concurrent processing of hundreds of samples is typically challenging, requiring the sample set to be processed in batches, thus potentially introducing batch effects. Finally, because the measurements will likely span a considerable time on the mass spectrometer, controls such as heavy labeled internal standards should be included to account for variability in instrument performance.
Even though SRM is now well established, few studies have successfully applied it to large subject cohorts (6–10) and there is currently no consistent way to deal with the aforementioned challenges of large-scale SRM-based studies. This manuscript demonstrates the importance of rigorous experimental design, combined with various controls to account for variabilities in SRM measurements, sample preparation and measurements across batches, when using a case study of developing proteomic biomarkers of epithelial ovarian cancer (EOC). We found that these controls were key for achieving optimal predictive performance of the biomarker signature for detecting EOC.
EOC is the fifth leading cause of cancer death in women and the leading cause of death from gynecological malignancies (11). The 5-year survival rate for EOC is low as most cases are diagnosed with advanced stage III-IV disease. If diagnosed at an early stage when the cancer remains confined to the ovaries, most patients can be cured by a combination of debulking surgery and platinum- and taxane-based chemotherapy (11). However, only 20% of the patients with EOC are currently diagnosed with early stage tumors. Therefore, much effort is invested in finding effective strategies for early diagnosis.
Cancer antigen 125 (CA125) (12) and human epididymis protein 4 (HE4) (13, 14) are currently approved by the Food and Drug Administration (FDA) as blood-based biomarkers for monitoring the disease and treatment response. These two markers are also used in the clinic in combination with transvaginal sonography or computer tomography to support the diagnosis of EOC in women with a pelvic mass. More recent in vitro diagnostic multivariate index assays have been cleared by the FDA for assessing the EOC risk in women diagnosed with an ovarian tumor before surgery, OVA1 and ROMA (14, 15). However, these markers lack the sensitivity and the specificity required for stand-alone diagnostic use. Multiple other biomarker panels have been discovered, initially showing promising results for the detection of EOC (16–20). However, follow-up studies in a large prospective cohort collected before clinical diagnosis of EOC demonstrated that the tested biomarkers combined with CA125 showed only little improvement compared with CA125 alone in the early detection of EOC using prediagnostic samples (21). The challenge of detecting reliable novel EOC plasma biomarkers is due in part to the molecular and cellular heterogeneity of EOC. EOC is characterized by disease heterogeneity as it relates to its multiple histotypes (serous, endometrioid, mucinous, and clear cell) and cellular grade (low and high grade for serous and endometrioid EOC) (22, 23). The heterogeneity of EOCs indicates that not a single biomarker, but a multivariate protein panel, is required for accurate tumor detection of multiple histotypes.
This manuscript presents a biomarker development strategy that consists of three phases. The first phase is the generation of a discovery list of EOC biomarker candidates. The second phase is their SRM-based quantification in blood plasma. The third phase is the development and validation of a protein biomarker signature for EOC in patient samples (Fig. 1) (6, 7, 24). The discovery list of EOC biomarker candidates was compiled using data from global quantitative MS measurements of tumors collected from a genetically engineered mouse model (GEMM) of endometrioid ovarian cancer (25). The list was further augmented with biomarker candidates, either previously discovered or from ongoing cancer biomarker studies (26–30). Next, the biomarker candidates were quantified using a multiplexed, targeted MS method in a cohort of more than 200 plasma samples from EOC patients and healthy controls. The SRM data was then used to develop and evaluate the performance of a 5-protein signature, consisting of IGHG2, LGALS3BP, DSG2, L1CAM, and THBS1. The 5-protein signature in combination with CA125 detected EOC with a sensitivity of 94%, which outperformed CA125 alone that had a sensitivity of 87%, albeit at a lower specificity (94% versus 97%). At a specificity of 97% the combined panel showed an improved sensitivity of 94%. Finally, we correlated the abundance differences observed for LGALS3BP in blood plasma samples to protein levels in patient tumors.
Fig. 1.

Study overview. A, In the discovery phase, epithelial ovarian cancer (EOC) biomarker candidates were discovered based on a proteomics-based discovery study using tissue samples from an EOC conditional GEMM. B, Biomarker candidates, i.e. the plasma-detectable, orthologous human proteins detected as differentially abundant in the discovery phase were subsequently quantified in plasma samples derived from a large cohort of EOC patients and healthy controls using selected reaction monitoring (SRM). C, Finally, the most predictive biomarker candidates for the detection of EOC were selected, combined in a protein biomarker signature, and further evaluated in an independent validation set.
Below we describe the step-by-step experimental design needed for large-scale SRM-based biomarker development studies and we focus specifically on EOC. However, these considerations are broadly applicable and can be used in large-scale studies of other diseases or biological systems.
EXPERIMENTAL PROCEDURES
Experimental Design and Statistical Rationale
For the discovery of biomarker candidates, we selected tissue samples from OC bearing mice (n = 5) and control samples (n = 4) each of them representing 8 ovaries pooled from nontumor bearing mice. All tissue samples were measured in technical triplicates on the mass spectrometer. For quantification of biomarker candidates, we obtained blood plasma from 124 patients with EOC and 110 healthy controls. Healthy controls were age and gender matched to EOC patients and had no previous cancer disease.
Collection and Preparation of Murine Tissue Samples
We chose a genetically engineered mouse model for endometrioid OC, which accurately recapitulates the human disease, to identify biomarker candidates for detection of OC in blood plasma. For the conditional endometrioid OC mouse model, the generation of LSL-K-RasG12D/+; PtenloxP/loxP mice and adenoviral induction of ovarian tumors was accomplished as previously described (25). Control mice were littermates that underwent the same surgical procedure but were injected with Adeno-empty virus instead of Adeno-Cre. At 12 weeks following Adeno-Cre induction, mice were sacrificed, and tumor-bearing mice showed large ovarian tumors that had metastasized to pelvic or peritoneal locations as previously described (25). Murine ovarian tumors and controls were dissected and snap frozen. To get enough starting material, 8 murine control ovaries were pooled into one control sample. Tumor-bearing ovaries contained enough material for individual analysis.
Protein Extraction and Glycopeptide Enrichment from Tissue
Tissue samples were homogenized in a Microdismembranator (Sartorious, Goettingen, Germany), subjected to protein extraction in lysis buffer (50% PBS liquid, pH7.4 (GIBCO, Invitrogen, Carlsbad, CA) and 50% 2,2,2-Trifluoroethanol (99.9% purity, Fluka, Buchs, Switzerland)) and solubilized with 1% Rapigest (Waters, Milford, MA) in 250 mm ammonium bicarbonate. Ultra sonication in a vial-tweeter ultrasonicator (Hielscher, Teltow, Germany) at 4 °C was used to further disintegrate the homogenized tissue. Proteins were denatured at 60 °C for 2 h, reduced with 5 mm dithiotreitol (DTT, Sigma-Aldrich, St. Louis, MS) at 60 °C for 30 min, and alkylated with 25 mm iodoacetamide (IAA, Sigma-Aldrich) at 25 °C for 45 min in the dark. Samples were diluted to 15% TFE in 100 mm ammonium bicarbonate and proteolyzed with sequencing grade porcine trypsin (Promega, Madison, WI) at a protease to substrate ratio of 1:100, at 37 °C for 1 5h. Peptide mixtures were desalted with Sep-Pak tC18 cartridges (Waters), eluted with 50% acetonitrile/0.1% formic acid, evaporated to dryness, and resolubilized in 100 μl 20 mm sodium acetate, 100 mm sodium chloride, pH 5. Glycopeptides were isolated as described previously (31). N-linked glycosylated peptides were released with N-glycosidase F (PNGase F; Roche and New England Biolabs). Formerly glycosylated peptides were desalted as above and resolubilized in 100 μl HPLC grade water/2% acetonitrile/0.1% formic acid.
Discovery-driven LC-MS/MS Acquisition and Analysis of Murine Tissue N-glycosites
LC-MS/MS analysis was carried out on a Thermo hybrid LTQ-FT-ICR mass spectrometer (Thermo Fisher Scientific, Waltham, MA) interfaced to a nanoelectrospray ion source (Thermo Fisher Scientific) and coupled online to a Tempo 1D-plus nanoLC (Sciex, Framingham, MA). 2 μl of N-glycosite samples were loaded from a cooled (4 °C) autosampler (Sciex) and separated on a 15 cm fused silica emitter, 75 μm diameter, packed in-house with Magic C18 AQ 3 μm resin (Michrom BioResources, Auburn, CA) using a linear gradient from 5% to 35% acetonitrile/0.1% formic acid over 60 min at a flow rate of 0.3 μl/min. To obtain good quantitative data for generating a biomarker candidate list, the MS instrument was operated to maximize the quality of LC-MS feature maps as opposed to maximizing the number of identifications. Therefore, for each peptide sample a standard data-dependent acquisition (DDA) of the three most intense ions per MS-scan was performed. Each survey scan acquired in the ICR cell at 100 000 FWHM was followed by MS/MS scans of the three most intense precursor ions in the linear ion trap, resulting in an overall cycle time of ∼1 s. Charge stage screening was employed, allowing fragmentation of doubly and higher charged ions, and rejecting ions of single or unknown charge state. A threshold of 200 ion counts was set to trigger an MS/MS attempt. Each sample was analyzed in triplicates. The raw data acquired by the LTQ-FT was converted to the centroid mzXML format using ReAdW (version 4.3.1) applying default parameters (32). MS/MS spectra were searched against the UniProt/TREMBL mouse database (release_2010–07, 65306 protein entries) using the SORCERER-SEQUEST™ v4.0.4 algorithm (33). The search criteria were set as follows: at least one tryptic terminus (cleavage after lysine or arginine residues, unless followed by proline); two missed cleavages were allowed; carbamidomethylation (C) was set as fixed modification; oxidation (M), deamidation (N; formerly N-glycosylated asparagines are converted to aspartic acid by PNGaseF release) were applied as variable modifications; monoisotopic parent and fragment masses and precursor ion mass tolerance of 50 ppm. The database search results were further processed through the mass spectrometry Trans-Proteomic-Pipeline 4.0.2 (TPP). In the TPP, the database search results were validated using the PeptideProphet software (34) and ProteinProphet software (35). The false positive error rate was set to 1% on the peptide and on the protein level as determined by PeptideProphet (34) and ProteinProphet (35). RAW data and search results have been deposited to the ProteomeXchange Consortium via the PRIDE partner repository with the dataset identifier PXD005665 (36, 37).
Relative Quantification and Statistical Testing of Murine OC Tissue N-glycosites
The acquired raw data were converted to the profile mzXML format using ReAdW (version 4.3.1) (32). These files were processed together with the SEQUEST search results by the software tool SuperHirn (version 3) into a MasterMap that includes the MS/MS-spectra assignments for label-free quantification as described previously (38). The data were filtered for fully tryptic peptides containing a deamidation at the NxST-sequence motif. A log2 transformation was applied to raw intensities and constant normalization was performed based on the median of all intensities per MS run to reduce the run-to-run variation (39). Features were removed that had missing intensities in more than 12 out of the 18 MS runs representing 3 biological replicates for both tumor and control samples which were measured in 3 technical replicates. For the remaining features with at least one missing intensity value, a G-test of independence was used to test for independence between missing values and conditions (tumor/control) (40). p values were adjusted to control for the false discovery rate (FDR) using the Benjamini-Hochberg method (41). A significant test result (i.e. adjusted p value < 0.01) for a feature indicates that the missing values appear more frequently in one of the conditions than it is expected by random chance. These features were assumed to be present in the samples, but below the detection limit of the instrument. Therefore, the missing values for these features were substituted by the estimated limit of detection. To estimate the limit of detection for this experiment, we calculated the minimum intensities across all features for each sample (total 18 samples) and then calculated the average of these 18 minimum intensities per sample. Statistical analyses of this data set was performed using a protein-level linear mixed effect model in the MSstats R package (v1.0) (42). Proteins with a fold change > 4 and an adjusted p value < 0.01 comparing tumor bearing to control ovarian tissue were selected as biomarker candidates.
Clinical Cohort
Patients with suspected adnexal tumor that underwent surgery at the gynecological department, Skåne University Hospital between 2004 and 2013 were included in the Skåne University Hospital ovarian tumor biobank. Until November 2011, when the current study started, 461 patients had been included in the biobank. The blood samples of healthy controls were collected between January 2004 and June 2008. Controls were age and gender matched to EOC cases and had no previous cancer disease. All individuals gave written informed consent for participation. Ethical permission for the biobank was obtained from the Lund University Ethics Committee. All blood samples were drawn into EDTA-tubes and centrifuged at 2000 × g for 10 min. The plasma samples were stored at −80 Celsius degrees within approximately two hours from sampling. The current study included blood plasma from patients in the biobank, 124 patients with epithelial ovarian cancer (EOC) and 110 healthy controls. Patients with borderline tumors were excluded from this study, because of the uncertain malignant potential of these benign tumors. The healthy controls were voluntary partners to cancer patients at Skåne University Hospital.
Glycoprotein Enrichment from Plasma
Formerly N-linked glycosylated peptides from each plasma sample were isolated using the N-linked glycopeptide capture procedure as described by Zhang et al. (31). Enrichment of N-glycosylated peptides is performed once for each subject except for the subjects that were included in both batches. Glycoproteins were first oxidized by adding sodium periodate (Perbio, Helsingborg, Sweden); thereafter, the sample was conjugated to the hydrazide resin. Nonglycoproteins were then extensively washed off the resin. Trypsin was added to digest the glycoproteins directly on the solid-phase resin. The trypsin-released peptides were removed by a second washing procedure. N-linked glycopeptides were released enzymatically using PNGase F (Roche, Basel, Switzerland, and New England Biolabs, Ipswich, MA). The whole protocol was adapted to be performed in 96-well plate format. Additionally, two bovine standard N-glycoproteins (Fetuin, Alpha-1-acid glycoprotein) were added before the glycopeptide enrichment to each plasma sample in the same amount (10pmol/protein) to follow the relative losses because of the solid phase extraction of glycopeptides procedure. Samples were prepared using a block randomized design according to their clinical features to prevent introduction of experimental bias. Because samples were prepared in 96-well plate format and a plate was treated as a block. To remove plate bias, we assigned a similar number of samples per condition in each plate and a similar distribution of ages in each condition. The location of samples was randomized within a plate.
Targeted LC-SRM Analysis of Plasma N-glycosites
SRM assays were retrieved from the N-glycoprotein SRM atlas (http://www.srmatlas.org/) (43), reanalyzed to select the best transitions for endogenous detection in plasma, split to multiple SRM methods, or used to optimize a single SRM method. Tier 2 SRM assays were used to quantify the N-glycosites employing internal standard peptides labeled with heavy isotopes at the C-terminal lysine or arginine, +8 or +10 Da, respectively (Thermo Fisher Scientific, Sigma-Aldrich, or JPT Peptide Technology, Berlin, Germany) to identity peptides based on analogy of chromatographic and fragmentation properties to the reference and quantify peptides.
SRM analyses for pilot batch 1 was performed on a 4000QTRAP and 5500QTRAP (Sciex) equipped with a nanoelectrospray ion source. All samples were analyzed on both instruments, biomarker candidates that were not detected on the 4000QTRAP were targeted on the 5500QTRAP. Chromatographic separations of peptides were performed by a Tempo nano LC system (Eksigent, Dublin, CA) coupled to a 15 cm fused silica emitter, 75 μm diameter, packed with a Magic C18 AQ 5 μm resin (Michrom BioResources). For each peptide, the best 3–4 transitions for the internal standard as well as the endogenous peptide were monitored in a scheduled fashion with a retention time window of 4 min and a cycle time fixed to 2.75 s (4000QTRAP) and 2 s (5500QTRAP). Peptides were loaded on the column from a cooled (4 °C) Tempo autosampler (Eksigent) and separated in 35 min by a linear gradient of acetonitrile (5–35%) and water, containing 0.1% formic acid at a flow rate of 300 nL/min. SRM acquisition was performed with Q1 and Q3 operated at unit resolution (0.7 m/z half maximum peak width).
Samples from batch 2 were analyzed on TSQ Vantage (Thermo Fisher Scientific) equipped with a nanoelectrospray ion source. Chromatographic separation of peptides was carried out on a nano LC system (Eksigent). In each injection, peptides were loaded onto a 75 μm diameter and 15 cm long fused silica microcapillary reverse phase column, in house packed with Magic C18 AQ material (200 Å pore, 5 μm diameter; Michrom BioResources). For peptide separation, a linear 40 min gradient from 2 to 40% solvent B (solvent A: 98% water, 2% acetonitrile, 0.1% formic acid; solvent B: 98% acetonitrile, 2% water, 0.1% formic acid) at a 300 nl/min flow rate was applied. The mass spectrometer was operated in the positive ion mode using ESI with a capillary temperature of 270 C°, a spray voltage of 1,350 V, and a collision gas pressure of 1.5 mTorr. SRM transitions were monitored with a mass window of 0.7 half maximum peak width (unit resolution) in Q1 and Q3. All the measurements were performed in scheduled mode, applying a retention time window of 3 min and a cycle time of 2c. To prevent bias during data collection we measured the samples in the same randomized order which was used for the sample preparation (see above).
Data Processing and Normalization for Targeted LC-SRM of Plasma N-glycosites
Raw data files from both data sets were uploaded to Skyline (v1.3) to perform automatic SRM peak integration, detect interferences and extract single transition intensities (44). SRM data can be accessed, queried, and downloaded via Panorama (https://panoramaweb.org/ovarian_cancer_biomarker.url) (45). Peak intensity data sets were exported from Skyline into the MSstats report format. All the transition intensities were log2 transformed to more closely conform to Normal distribution and better satisfy the statistical modeling assumptions for downstream analysis (5). The normalization consisted of three steps, as follows (Fig. 3A, supplemental Fig. S1). The first and second step of the normalization were conducted, separately by batch 1 and batch 2, to remove systematic variation because of the instrument performance and losses in the sample preparation process. The first normalization step (supplemental Fig. S1A) was based on the isotopically labeled synthetic peptides (heavy peptides) with sequence matching the targeted endogenous peptides, spiked in each sample to account for systemic shifts in peak intensities between mass spectrometric runs. The normalization was performed on the log2 transformed intensities using the following steps: (1) The median of the log2 transformed heavy reference intensities for each run were calculated; (2) an overall median was calculated of the medians of each run; (3) the difference between the median for each run and the median of the median was calculated; (4) then, these differences were added to log2-intensities from heavy and endogenous peptides in the corresponding run. The second normalization (supplemental Fig. S1B) used two bovine (standard) proteins, AIAG and FETUA, spiked with the same amount in each sample, to account for other potential artifacts that could occurred during sample preparation, before data acquisition. The endogenous peak intensities of two standard proteins in each MS run were summarized in a single value per run per protein using MSstats (v3.6.0) (42). The means of two summarized peak intensities from two standard proteins per MS run were equalized across the runs, and endogenous peak intensities of the other proteins in the corresponding run were shifted by the same amount. The third normalization step (supplemental Fig. S1C) was conducted to account for batch effects. With the assumption that subjects included in multiple batches have the same protein abundance, batch effects could be removed by equalizing their relative abundance of peptides and transitions. Samples from 38 patients, which we refer to as 'repeated samples', were measured twice (once in batch 1 and once in batch 2). The repeated samples were used to account for differences in scale between the datasets. The difference between the median of peak intensities among the repeated samples for each feature (peptides × transitions) in batch 2 and that in batch 1 was added to the corresponding feature in the batch 1, separately for reference and endogenous intensities. The repeated samples were removed from batch 1 after normalization, and batch 1 and batch 2 were merged into one dataset.
Fig. 3.

Experimental design for SRM-based evaluation of biomarker candidates in a large EOC patient cohort. A, Overview of the experimental design. The repeated samples of 38 subjects (green tubes), two standard proteins, and heavy reference peptides were used as controls in multiple steps of experiments. The subject samples were processed and measured in two batches. Three normalization steps were applied to combine the datasets derived from two batches, thereby accounting for variability in instrument performance, variability in sample preparation, and other batch effects. The merged normalized dataset from both batches was divided into a training set and a validation set. Feature intensities were summarized into protein abundances for each subject, separately for the training set and the validation set. B, Distribution of subjects across the training and the validation sets based on confounding factors, age and stage of EOC. C, Heatmap of standardized protein abundances of 65 proteins across all the subjects in the training set. The subjects are arranged in columns, and the proteins in rows. For the purposes of visual display, the summarized protein abundances were standardized by protein (i.e. by row) on a quantile scale. Red shades indicate increased protein abundance, and blue shades indicate reduced protein abundance. Hierarchical clustering with Euclidian distance and Ward linkage was employed to cluster subjects by similarity of standardized protein abundance. The disease status of subjects is labeled with green for healthy cases, yellow for EOC cases at the top of heatmap.
Division Into Training Set and Validation Set and Relative Quantification
Two hundred thirty-four unique subjects were divided randomly into training set with 173 subjects and validation set with 61 subjects, while keeping a similar distribution of age and EOC stage (early or late stage) between the sets. We first determined the required number of subjects per subgroup (combination of stage and age group) for either training set or validation set. We then selected the corresponding number of subjects randomly within each subgroup. The normalized transition peak intensities for each subject were summarized using MSstats (v3.6.0, without the internal decision for the censored missing values by the option, maxQuantileforCensored = NULL) on a relative scale into a single number per subject, separately in the training and validation set (42). The summarized protein abundance per subject was used for the predictive analysis.
CA125 Measurements in Human Plasma Samples
For batch 1, CA125 plasma concentration was measured by sandwich ELISA following the manufacturer's instructions for the CA125 (Human) ELISA Kit (Abnova Corporation, Walnut, CA). Samples were diluted 1/3 in sample buffer (1/1 LowCross Buffer (Candor Bioscience, Wangen, Germany), 1% BSA in PBS). The CA125 standards and the samples were incubated with Enzyme Conjugate Reagent in the already coated 96-well plate for 90 min at 37 °C. After washing the wells with wash buffer, they were incubated with TMB Reagent for 20 min at room temperature in the dark. Stop Solution was added to stop the reaction and optical density was read at 450 nm. The lowest standard used for the ELISA was 5 U/ml and used as minimal value for the dataset. CA125 plasma levels were measured from 20 μl EDTA-plasma using a fully automated electrochemiluminescence immunoassay (ECLIA) on a Cobas E 601 system (Roche Diagnostics, Basel, Switzerland). Thirty-eight plasma samples included in both batches were used to calibrate ELISA and ECLIA CA125 concentration levels. Plasma samples in batch 1 with minimal CA125 concentration of 5 U/ml, were removed for calibration. Calibration between ELISA and ECLIA for CA125 was conducted by fitting the linear regression model between log2 transformed ELISA and ECLIA. ECLIA CA125 concentrations for the rest of batch 1 (45 samples) were estimated with the coefficients from the fitted linear regression model and measured ELISA CA125. ECLIA CA125 concentrations (original value in batch 2 and estimated value in batch 1) were used for further predictive analysis.
Predictive Analysis
Eight-fold cross-validation was used to find the most predictive proteins for biomarker signature between EOC patients and healthy controls in the training set. Within each fold, proteins were tested for differential abundance between EOC patients and healthy controls using seven-eighth of the subjects by MSstats (v3.6.0) (42). The relative abundances of the significant proteins (adjusted p value < 0.05 and fold change cutoff ≥1.1) were used as input to a logistic regression model. The subset of significant proteins was selected by stepwise method (i.e. by repetitively adding or dropping variables in the model until minimizing the Akaike information criterion (AIC)). The predictive accuracy of the logistic regression model with selected proteins was then evaluated on the remaining one-eighth of the subjects in the training set. The procedure was repeated eight times by rotating the one-eighth of the left-out subjects in the training set. The final consensus logistic regression model with five protein abundances (IGHG2, LGALS3BP, DSG2, L1CAM, THBS1), which were selected more than six times among eight folds, and log2 transformed CA125 as predictors was fit to the training set. Probability cutoff for classification was determined to maximize the predictive accuracy, the fraction of all cases and controls who were correctly classified, on the training set. The predictive ability of the fitted model was then evaluated on the independent validation set, in terms of the area under the ROC curve, accuracy, sensitivity, specificity, negative predictive value and positive predictive value. The performance of CA125 alone was evaluated with the cutoff = 35 U/ml (as is standard in the clinical practice) on the same independent validation set. The ROC curves, as well as other measures of performances (i.e. accuracy, sensitivity, specificity, negative predictive value and positive predictive value) were obtained with the R package pROC. 95% confidence intervals for the area under the ROC curve, accuracy, sensitivity, specificity, negative predictive value and positive predictive value were derived from 2000 bootstrap replicates.
Tissue Microarray Analysis (TMA) of Ovarian Tissue
The aim of this analysis was to measure tissue protein abundance of LGALS3BP by immunohistochemistry and correlate it with clinicopathologic variables. The study protocol was approved by the local ethics committee (KEK Zurich, KEK-ZH-No. 2014–0604). The cohort consisted of primary EOC tissues (n = 150) and borderline tumors of the ovary (n = 34) which were diagnosed at the Institute of Surgical Pathology, University Hospital Zurich (Switzerland) between 1995 and 2005. Normal tube- and surface epithelium of ovaries from patients without a history of ovarian cancer (diagnosed with e.g. endometriosis) was used as control (n = 14). EOC specimen, borderline tumors and the normal tube- and surface epithelial tissues were immediately formalin fixed overnight and completely paraffin embedded. Further workup comprised 2 μm serial sectioning of selected tumor blocks and construction of a tissue microarray as described previously (46). Immunohistochemical staining was performed using the Ventana Benchmark (Roche Ventana Medical Systems, Inc., Tucson, AZ) automated staining system with the following primary antibody anti-LGALS3BP (rabbit polyclonal, Atlas Antibodies AB; dilution 1:1000). Detection was performed with ultraView DAB-kit (Ventana) using the heat-induced epitope retrieval CC1 solution. Slides were counterstained with hematoxylin (Ventana), dehydrated and mounted. A surgical pathologist (P.J.W.) performed a blinded expression analysis. A semiquantitative scoring system was applied to quantify the cytoplasmic expression of LGALS3BP: score 0, negative; score 1+, weak; score 2+, moderate; score 3+, strong. Samples were excluded if they showed uninterpretable results caused by lack of target tissue, presence of necrosis, or crush artifacts. Statistical analysis was performed using the software IBM SPSS Statistics Version 22 (SPSS Inc., Chicago, IL). To study associations between clinicopathologic and immunohistochemical data, the two-sided Fisher's exact test was used, the standard procedure in biometrics to test for homogeneity/nonhomogeneity in cross tables. All correlations were significant with p values below 0.05.
RESULTS
Generating a Discovery List of EOC Biomarker Candidates and Testing Their Detectability in Plasma
The first critical step for a biomarker study is the generation of a biomarker candidate list for subsequent quantification in large subject cohorts. Because large scale studies require time and resources, it is essential to start with a high confidence candidate list specific to the clinical question. To optimize the discovery of EOC-specific candidates for noninvasive detection, we performed a global, quantitative proteomic study in tumors collected from a GEMM for endometrioid OC (Fig. 1A). Although this model accurately recapitulates the clinical disease, a potential caveat is that most EOCs are serous histology and biomarker candidates derived from it could be specific for endometrioid OC. However, a previous paper using the same K-ras/Pten model for biomarker discovery showed that upregulated secreted proteins identified in murine plasma during tumor development, including HE4, were more broadly representative of multiple ovarian cancer histological subtypes (30). The conditional mouse model was generated by Adeno-Cre induction of oncogenic K-rasG12D/+ and suppression of Pten (Pten−/−) in the ovarian surface epithelium, which led to the development of widespread, metastatic endometrioid OC (25). To identify biomarker candidates secreted or shed by tumors into circulation, we specifically focused on glycoproteins, which are representative of most currently approved biomarkers (24). However, by restricting our discovery to glycoproteins we are aware that we exclude other potential promising biomarker candidates. N-linked glycopeptides were isolated from tryptic digests of protein extracts using an approach based on hydrazide chemistry and solid phase extraction of glycopeptides (31). The protein extracts were collected from murine endometrioid ovarian tumors and control ovaries at 12 weeks following Adeno-Cre and Adeno-empty induction, respectively. The N-glycosites (de-glycosylated forms of peptides that are glycosylated in the native protein) were then analyzed by high-resolution liquid chromatography coupled to MS (LC-MS). Label-free quantification was performed (38) to determine N-glycoproteins that are differentially abundant between tumor bearing and control ovarian tissue and the results were subjected to statistical analysis using MS stats (42). Using this method we identified 906 glycoproteins in murine ovarian tumors (supplemental Table S1, supplemental Table S2), of which 275 glycoproteins were either significantly up- (124) or downregulated (151) in endometrioid OC compared with control ovarian tissue (adjusted p value < 0.01, fold change > 4) (supplemental Table S3). We then determined the human orthologues of the candidate murine proteins identified. The mouse model-derived list was further augmented with 110 additional human N-glycoproteins, which were either previously reported as candidates in other EOC biomarker studies performed in human tissue, ascites fluid, or blood sera (26–30) or represented candidates from ongoing cancer biomarker studies (6, 7) (supplemental Table S4). Overall, the biomarker candidates in the discovery list represented diverse biological processes, such as cell adhesion, extracellular matrix organization, proteolysis, angiogenesis, cell migration, and others.
The human blood plasma proteome is highly complex; it contains subsets of tissue proteomes and spans more than 10 orders of magnitude in concentration range (4). Therefore, we first verified the detectability of our biomarker candidates in N-glycosites enriched from a pooled plasma sample. N-glycosites representing the biomarker candidates and their optimized SRM assays were extracted from the human N-glycoprotein SRM Atlas (43) (supplemental Table S5). Heavy labeled reference peptides were synthesized for all N-glycosites and added to the samples to guide peptide identification in the complex sample matrix. Out of 376 biomarker candidates, 65 proteins were detectable in N-glycosites enriched from plasma (supplemental Table S6). The 65 detected biomarker candidates spanned a concentration range of 5 orders of magnitude with the majority of undetected candidates residing in sub-nanogram per milliliter concentration range in plasma (Fig. 2A). The candidates represented most of the biological processes that were present in the initial discovery list (Fig. 2B). In the next phase, the detectable fraction of our EOC biomarker candidates in human plasma was selected for quantification in a large subject cohort containing plasma samples from EOC patients and healthy controls.
Fig. 2.
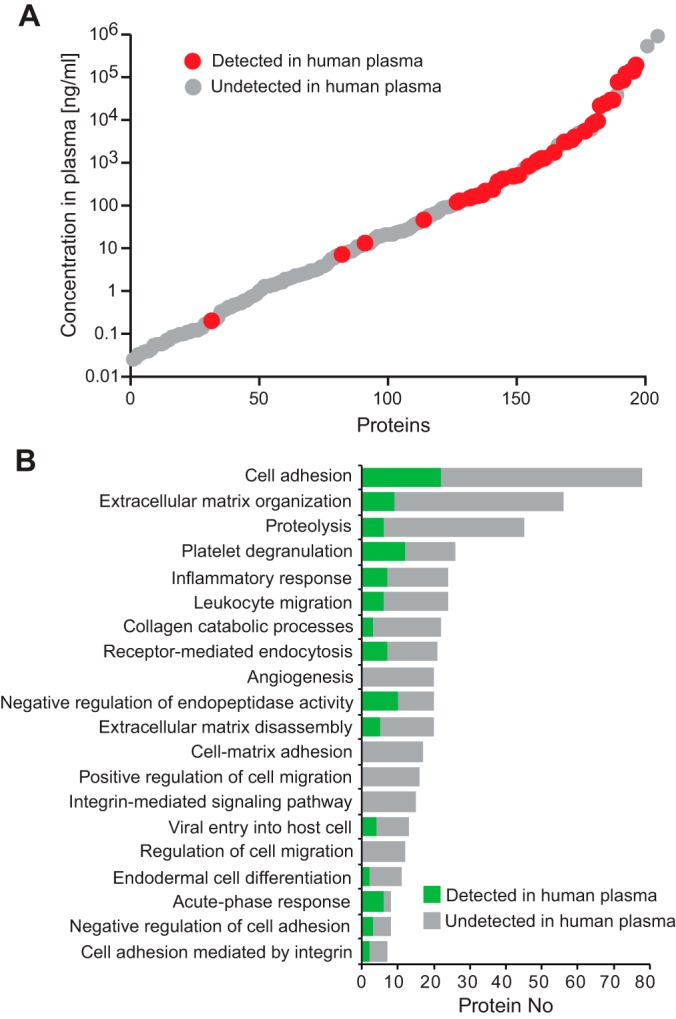
Characteristics of biomarker candidates. A, Estimated concentrations of biomarker candidates in human blood plasma. The candidates that were detectable by SRM in human plasma are indicated in red. B, Gene ontology biological processes enriched among all biomarker candidates. For each biological process the fraction of detected proteins in plasma is indicated in green.
Experimental Design for SRM-based Quantification of Biomarker Candidates in Large EOC Cohort
The second phase of a biomarker investigation involves the quantification of biomarker candidates in human blood across the large cohort of subjects. Typically, such large-scale studies cannot process and acquire data from all the samples in parallel and may even involve different instruments to increase sample throughput. A rigorous study design requires experimental controls, which account for variation in sample preparation, within and between different batches, as well as for variation introduced by MS instruments and their performance over time. The experimental controls help normalize the data sets, thereby reducing the experimental artifacts and producing more accurate quantification of protein abundance.
In our investigation, we aimed at quantifying detectable biomarker candidates from the discovery phase in a subject cohort consisting of 234 blood plasma derived from 110 healthy women and 124 EOC patients, to identify a subset of biomarker candidates that can discriminate between EOC and healthy subjects (Fig. 1B). The cohort reflected the underlying population with EOC in terms of histological subtype and stage (Table I, supplemental Table S7). Healthy controls were age and gender matched to EOC patients and had no previous cancer disease. We first performed measurements on a smaller pilot batch of 66 plasma samples to evaluate our experimental setup, before scaling up to the larger batch containing 206 plasma samples (supplemental Table S7). The two batches were prepared at different times and samples were measured on different instruments. Therefore, our study design included controls that accounted for variation introduced by the batch effects, sample preparation, as well as MS performance (Fig. 3A). For the purpose of batch effect normalization, 38 plasma samples from the first batch were also included in the second batch. To reduce variability in sample preparation, all plasma samples were subjected to N-glycosite enrichment in a 96-well plate format. To account for the variability in sample preparation within and across plates, two bovine N-glycoproteins (AIAG, FETUA) were added in equal amounts as internal standards to each sample before enrichment (Fig. 3A). The SRM measurements quantified each protein with between one and three peptides, and each peptide with three transitions. Heavy labeled internal standard peptides for each N-glycosite were spiked in equal amounts into each sample to account for variability in MS performance, and to allow for accurate identification and quantification of N-glycosites (Fig. 3A). The resulting data were analyzed using Skyline for identification and extraction of SRM feature intensities and log-transformed (44). Using this targeted proteomic approach, we were able to obtain a highly consistent dataset quantifying 65 biomarker candidates across 234 human blood plasma samples.
Table I. Summary characteristics of EOC patients and healthy controls included in the study.
| Variable | Ovarian cancer (OC) | Controls |
|---|---|---|
| Total # patients | 124 | 110 |
| Age [y] | 66 (27–88) | 60 (24–77) |
| Endometrioid | 14 | |
| Mucinous | 11 | |
| Serous | 89 | |
| Clear cell | 3 | |
| Other | 7 | |
| Stage 1/2 | 35 | |
| Stage 3/4 | 89 |
Data derived from the two batches were integrated using three steps of normalization (Fig. 3A, supplemental Fig. S1). The first normalization step accounted for variability in MS data acquisition, separately for each batch. It equalized the median log-intensities of the heavy reference peptides across the samples in a batch (Fig. 3A, supplemental Fig. S1A). The second normalization step accounted for variability in sample preparation, separately for each batch. It equalized the log-intensities of the two bovine glycoproteins, AIAG and FETUA, across the samples in a batch (Fig. 3A, supplemental Fig. S1B). The last normalization step accounted for the variability between the batches. Plasma samples quantified in both batches were used to equalize median log-intensities for each transition between both batches, reference and endogenous separately, thereby keeping the relative abundance changes across samples unchanged after combining the two batches. (Fig. 3A, supplemental Fig. S1C).
The combined dataset was divided into a training (173 subjects) and validation (61 subjects) set with a ratio of three to one (Fig. 3B). To avoid systematic differences between the training and validation set, subjects were equally distributed based on their age as well as cancer stage (Fig. 3B). Quantification of protein abundances across subjects, separately by training and validation set, was performed using MSstats (42). Hierarchical clustering of subjects in training set according to abundances of all 65 biomarker candidates showed coclustering of EOC patients and healthy controls to some extent but could not completely separate the two groups (Fig. 3C).
Development of a Protein Biomarker Signature for Detecting EOC
The final phase of a biomarker study is aimed at defining the most predictive analytes from the large-scale SRM dataset to develop a multivariate biomarker signature. At present, no single diagnostic marker or procedure has adequate sensitivity and specificity for EOC diagnostic (47). Although CA125 is the most widely used plasma-based biomarker for ovarian cancer, it has only 50–60% sensitivity for early stage EOC and 90% for late stage disease (47). Therefore, our goal was to develop a multivariate biomarker signature that, when combined with CA125, improves the sensitivity and specificity for detecting EOC (Fig. 1C). To select predictive protein components of the signature we performed an 8-fold cross validation on the training set (Fig. 4A). Within each fold of the training set, we tested proteins for differential abundance between EOC patients and healthy controls (Adjusted p value ≤ 0.05, FC cutoff ± 1.1) (Fig. 4A, supplemental Table S8) (42). The differentially abundant proteins in each fold were used as input to a logistic regression that classified EOC patients versus healthy controls. Within each fold, stepwise variable selection identified the most discriminative subset of the biomarker candidates (Fig. 4A). After the procedure was repeated for all eight folds, biomarker candidates selected in six or more folds were retained as predictors in a consensus logistic regression model fit on the entire training set (Fig. 4A). Finally, one more logistic regression combined the retained proteins, with CA125 as an additional predictor, and fit on the entire training set.
Fig. 4.

Protein biomarker signature development. A, The training set was used to determine a predictive biomarker signature using multivariate logistic regression and 8-fold cross-validation. The subjects were systematically rotated between eight folds. Within each fold, differentially abundant proteins were determined comparing EOC patients to healthy controls. The plot indicates the results per fold. Red squares indicate significantly up-regulated proteins, and blue squares indicate significantly down-regulated significant protein. Gray color indicates no statistically significant changes (fold change cutoff ±1.1, adjusted p value < 0.05). Next, a logistic regression model was fit with stepwise method for selecting predictive proteins (black squares show the selected proteins for each fold) and the disease status was predicted for the 'left-out' subjects in each fold (AUC sub-validation). The final consensus logistic regression model was generated using the biomarker candidates selected more than six times among eight folds in the cross validation (selected proteins are indicated with a red dot). B, The consensus logistic regression model was fit in the training set, combining the selected biomarker candidates with CA125. The probability cutoff was selected to maximize the predictive accuracy on the training set. C, The validation set was used to evaluate the performance of the final consensus logistic regression model. Detection of disease status of subjects with EOC and healthy controls was summarized in an ROC curve comparing CA125 to the novel five-protein signature and its combination with CA125. Summary statistics are listed for five-protein signature plus CA125 and CA125 alone.
The retained proteins were Ig gamma-2 chain C region (IGHG2), Galectin-3-binding protein (LGALS3BP), Desmoglein-2 (DSG2), Neural cell adhesion molecule L1 (L1CAM), and Thrombospondin-1 (THBS1) (Fig. 4B). For LGAL3BP, DSG2, and THBS1 increased abundance in plasma was associated with EOC patients. In contrast, for IGHG2, and L1CAM decreased abundance was associated with EOC (supplemental Fig. S2). The in-sample predictive ability of the consensus logistic regression with five retained proteins and CA125 was evaluated on the complete training set, resulting in area under the ROC curve of 0.973 (Fig. 4B).
We also evaluated the predictive ability on the independent validation set, which was never used for the training of the signature. We further compared the predictive ability of the biomarker signature to that of CA125 alone, quantified by ELISA and using an established cutoff of 35, as well as to the signature combining CA125 with SRM-based measurements of IGHG2, LGALS3BP, DSG2, L1CAM, and THBS1 (Fig. 4C). Although CA125 performed well on the validation set (with specificity 97% and sensitivity 87%), its combination with the 5-protein signature maximized the area under the ROC curve (0.99 compared with 0.96) on the validation set, with a higher sensitivity of 94%, but a slightly lower specificity of 93% (Fig. 4C). However, when we compared the performance of CA125 alone with its combination with the 5-protein signature at the same specificity of 97%, the combination still outperformed CA125 alone in terms of sensitivity (supplemental Fig. S3). These results demonstrated that, although CA125 alone had a high specificity in the validation cohort, the addition of the multivariate protein signature increased the sensitivity of detecting EOC.
Validating LGALS3BP Blood Plasma Results in Human EOC Tissue from an Independent Subject Cohort
Many cancer biomarker studies hypothesize that the development of a solid tumor gives rise to protein abundance changes that can be measured noninvasively in blood plasma. To test this hypothesis, we sought to validate, in EOC tissue, protein abundance differences found in blood plasma of EOC patients compared with healthy individuals. To evaluate protein abundances in tissue samples, we used a tissue microarray (TMA) containing formalin-fixed tissues from 145 EOC patients, 30 benign ovarian tumor patients and 13 healthy controls (46). Tissue samples included on the TMA were derived from an independent subject cohort collected at a different hospital. Based on the availability of antibodies for immunohistochemistry, we stained the TMA for LGALS3BP. Staining intensities were graded by a pathologist as exemplified in Fig. 5A. LGALS3BP had shown significantly higher protein abundance in plasma samples from EOC patients compared with healthy individuals (supplemental Fig. S2) and the TMA results were in concordance with the plasma results (Fig. 5B). Specifically, comparing the staining intensities for LGALS3BP in EOC tissue compared with benign ovarian tumor tissue as well as ovarian tissue from healthy controls we detected a significantly stronger intensity for LGALS3BP in EOC tissue (p value < 0.001, Fisher's exact test) (Fig. 5B). The TMA results suggested that it is indeed possible to detect protein abundance changes caused by a solid tumor remotely in blood plasma.
Fig. 5.

Connecting LGALS3BP blood plasma results to protein abundances in human EOC tissue. A, Grading scheme for immunohistochemistry staining of LGALS3BP in human ovarian tissue. B, Tissue microarray staining results for ovarian cancer, borderline tumors, and control epithelial ovarian tissue.
DISCUSSION
In this study, we derived an accurate 5-protein signature for distinguishing individuals with EOC from healthy controls, the sensitivity of the signature in combination with CA125 measurements exceeding that of CA125 ELISA-based measurements alone. Aside from the actual biomarker signature, the clinical importance and novelty of this paper lies in the large-scale application of SRM measurements with a rigorous experimental design and statistical analysis. Our SRM-based strategy is broadly applicable and can be used in any disease entity for the development of diagnostic biomarker assays.
Even though SRM has shown great promise as a tool for biomarker studies, it has only been applied for a few large-scale studies to date. This is in part because of the requirement of a rigorous study design, reproducible sample preparation, and appropriate statistical analysis for performing SRM measurements across large subject cohorts and developing a biomarker signature. In this study, we implemented a rigorous experimental design and data analysis strategy for protein biomarker development using SRM-based targeted MS. This strategy was applied to discover and validate novel biomarker candidates for EOC in human blood plasma. A high-quality list of biomarker candidates was compiled from a quantitative proteomic study using a GEMM representative of endometrioid OC. The list was augmented with candidates, which were published or from ongoing cancer biomarker studies (Fig. 2). To quantify biomarker candidates in a large subject cohort, we designed an experimental and data analysis strategy, which would account for experimental variability in instrument performance, sample preparation and sample batch effects (Fig. 3). In the case of EOC, we demonstrated the effectiveness of this strategy to identify a signature of selected biomarker candidates, which in combination with CA125 allowed the detection of EOC with a higher sensitivity than CA125 alone (Fig. 4).
To assemble a biomarker candidate list, we used a conditional GEMM of EOC generated by Adeno-Cre induction of oncogenic K-ras and Pten suppression specifically in the ovarian surface epithelium, which led to widespread, metastatic ovarian endometrioid OC (25). Ideally, biomarker discovery should include multiple animal models or patient samples recapitulating all histological subtypes. However, despite endometrioid OC being only the second most common histological EOC after high-grade serous OC (48, 49), the chosen GEMM is still highly relevant for biomarker discovery as it accurately recapitulates the clinical disease. In addition, the RAS and PI3K/PTEN signaling pathways are altered in various histological subtypes of EOC, such as serous, endometrioid and mucinous OC (48, 49). Further, CA125, the biomarker currently most widely used for OC diagnosis, has shown a better performance for detecting high grade serous OC (HGSC) but revealed a reduced sensitivity in detecting early tumors (27, 50). Therefore, identifying biomarker candidates that are specific for each histological EOC subtype and combining them with CA125 might increase the sensitivity necessary for early detection of multiple EOC histotypes. We augmented the biomarker candidate list with potential markers for EOC that have been proposed previously, but their performance in detecting EOC was to our knowledge never evaluated systematically in human blood plasma. The relevance of including biomarker candidates from various sources is underscored by the fact that the final biomarker candidate signature combining five novel proteins with CA125 consists of proteins selected from all sources.
A major limitation for protein biomarker quantification in blood plasma is the complex nature of the body fluid. Its dynamic protein concentration range spans 12 orders of magnitude, with few highly abundant proteins making up 90% of the plasma proteome (3, 4). Even though we focused on the N-glycosylated proteome, thereby reducing the complexity of the plasma samples, we were able to detect and consistently quantify 65 out of 376 biomarker candidates across hundreds of plasma samples (i.e. the detection rate was 17%). Despite this fact, proteins for which detectability in plasma by SRM was established, could be detected in the sample cohort with a high degree of consistency (only 0.3% missing values across 234 samples). The majority of the undetected biomarker candidates resided in the sub-nanogramm per milliliter concentration range in plasma (Fig. 2A) and were only accessible by MS after extensive sample fractionation and workup. Additionally, biomarker candidates initially discovered in murine tissue had to be translated into their human orthologues, which resulted in some biomarker candidates lacking N-glycosites, and therefore intractable with our strategy. However, the alternative option for systematically quantifying biomarker candidates relies on immunoassays, which require the availability of specific antibodies for each protein, are only available for a small subset of the human proteome and are usually biased for frequently studied proteins (51).
Accurate and reproducible quantification of proteins across a large subject cohort is crucial for biomarker development. We implemented experimental controls in our study and demonstrated a multistep normalization approach, which accounted for variability on all experimental levels: SRM measurements, sample preparation, batch effects and usage of different MS instruments (Fig. 3A). For the enrichment of N-glycosites from plasma we added two N-glycosylated bovine standard proteins in equal amounts to each sample. Because the sample preparation for this study was performed in a 96-well plate format, variability in sample preparation was limited and two standard proteins appeared to be sufficient. However, for future applications it might be beneficial to include a higher number of standard proteins to better estimate the variability in sample preparation. Additionally, unused standard proteins could be used to assess the quantitative accuracy of the SRM assay. Unlike ELISA-based measurements, MS-based quantification is performed on a relative scale. Therefore, the quantitative measurements from different MS batches may not be comparable directly. To remove batch effects between samples profiled on different MS instrument platforms, we included a subset of subjects in all the batches. With the assumption that subjects included in multiple batches have the same protein abundance, batch effects could be removed by equalizing their relative abundance of peptides and transitions. The experimental design and data analysis strategy presented here is broadly applicable to other biomarker studies including large cohorts measured in multiple batches, as well as studies requiring the accurate and reproducible MS-based quantification of proteins across many subjects.
Finally, to develop a biomarker signature for the detection of EOC, we selected proteins with high predictive ability that fit a logistic regression model on the training set (Fig. 4A). The best performing signature included 5 proteins, namely IGHG2, LGALS3BP, DSG2, L1CAM, and THBS1, which were combined with the ELISA measurement of CA125, the current clinical standard, into a final signature (Fig. 4B). Based on the independent validation set, the 5-protein signature combined with CA125 detected EOC with a higher sensitivity than CA125 alone (Fig. 4C).
Among the proteins in the signature, LGALS3BP and THBS1 were initially discovered in the endometrioid OC GEMM, DSG2 was derived from previously published OC biomarker studies, and L1CAM and IGHG2 represented candidates from ongoing cancer biomarker studies. LGALS3BP is a member of the scavenger receptor cysteine-rich domain family of proteins (52), which has not only been previously associated with various malignant tumors but also suggested as a potential biomarker (53–55). DSG2, a desmosomal cadherin expressed in epithelial derived tissues (56), is also overexpressed in various malignancies, such as nonsmall cell lung cancer and melanoma (57, 58). Knockout of DSG2 was reported to suppress colon and nonsmall cell lung cancer cell proliferation (57, 59). THBS1 is an endogenous angiogenesis inhibitor which has previously been associated with the development of tumor microenvironment and angiogenesis and has been shown to promote migration of cancer cells (60–62). L1CAM is a cell adhesion molecule, which was originally identified as a neural cell adhesion molecule in the central nervous system (63), but L1CAM expression has been identified in a variety of tumor types (64) and has been recently reported the be involved in the progression of endometrial cancer (65). Lastly, IGHG2 forms the constant region of immunoglobulin heavy chains. It represents a high abundance plasma protein and has to our knowledge not been linked to cancer. Both, IGHG2 and L1CAM, showed a decreased level in plasma of cancer patients, that cannot be biologically explained at this point. Changes in high abundant plasma proteins, such as IGHG2, could result from secondary effects at late stages of EOC and in fact most patients included in our study have late stage EOC. Therefore, we currently do not have evidence that the protein signature reported here could be applied for early stage EOC detection.
LGALS3BP, DSG2 and L1CAM have been associated with other malignancies indicating that these proteins might not be specific biomarker candidates for EOC. To ensure the specificity of the signature, only a subset of the biomarker candidates in the signature needs to be specifically elevated or downregulated in EOC. The combination with additional general cancer markers can help to increase sensitivity in detecting EOC. For example, a recent study assessed blood-based N-glycoproteins across five solid carcinomas and found significantly different expression levels for THBS1 in four out of five carcinomas compared with controls suggesting THBS1 as a general cancer marker (66). To evaluate the specificity of the suggested biomarker signature for detection of EOC and its application for early detection of EOC, a follow-up study using an independent patient cohort should be designed to include other malignancies and benign conditions of the ovaries, which are known to result in elevated CA125 levels, as well as pre- and/or early EOC.
Targeted proteomics is a promising tool for biomarker development and the quantification of biomarker candidates in complex sample matrices without the necessity of a lengthy and expensive development of specific antibodies against the proteins of interest. However, to date only a few large-scale biomarker studies have been conducted using targeted proteomics for systematic quantification, which mostly focused on a few candidates for which antibody-based assays are available (51). The experimental design and data analysis considerations detailed in this study as it pertains to EOC will contribute to a broader applicability of targeted proteomics in large-scale studies.
Data Availability
For DDA experiments, RAW data and search results have been deposited to the ProteomeXchange Consortium via the PRIDE partner repository with the dataset identifier PXD005665 (https://www.ebi.ac.uk/pride/archive/projects/PXD005665) (36, 37). For SRM experiments, SRM data can be accessed, queried, and downloaded via Panorama (https://panoramaweb.org/ovarian_cancer_biomarker.url) (45).
Supplementary Material
Acknowledgments
We would like to thank Ralph Schiess and Bernd Wollscheid for helpful discussions and enthusiasm throughout the project.
Footnotes
* The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institute of Health. E.N. was supported by the Swedish Breast Cancer Association (BRO), Region Skåne, Governmental Funding of Research within the Swedish National Health Service (ALF), Mrs Berta Kamprad Foundation, BioCARE, Marcus and Marianne Wallenberg Foundation. R.A. was supported by the Swiss National Science Foundation (Grant no. 3100A0-688 107679). This work is supported by grants awarded to D.M.D. by the DOD OCRP (W81XWH-15-1-0089), American Cancer Society (RSG-13-083-01-TBG), Ovarian Cancer Research Fund Liz Tilberis award, and the Burroughs-Wellcome Fund Career Award in the Biomedical Sciences 1005320.01. P.J.W. work was funded in part by an Oncosuisse grant.
 This article contains supplemental Figures and Tables.
This article contains supplemental Figures and Tables.
1 The abbreviations used are:
- SRM
- selected reaction monitoring
- ELISA
- enzyme-linked immunosorbent assay
- EOC
- epithelial ovarian cancer
- FDA
- Food and Drug Administration
- GEMM
- genetically engineered mouse model
- LC-MS
- liquid chromatography coupled to mass spectrometry
- TMA
- tissue microarray
- HGSC
- high grade serous ovarian cancer
- DDA
- data-dependent acquisition
- FDR
- false discovery range.
REFERENCES
- 1. Ludwig J. A., and Weinstein J. N. (2005) Biomarkers in cancer staging, prognosis and treatment selection. Nat. Rev. Cancer 5, 845–856 [DOI] [PubMed] [Google Scholar]
- 2. Huttenhain R., Malmström J., Picotti P., and Aebersold R. (2009) Perspectives of targeted mass spectrometry for protein biomarker verification. Curr. Opin. Chem. Biol. 13, 518–525 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Surinova S., Schiess R., Huttenhain R., Cerciello F., Wollscheid B., and Aebersold R. (2011) On the development of plasma protein biomarkers. J. Proteome Res. 10, 5–16 [DOI] [PubMed] [Google Scholar]
- 4. Anderson N. L., and Anderson N. G. (2002) The human plasma proteome: history, character, and diagnostic prospects. Mol. Cell. Proteomics 1, 845–867 [DOI] [PubMed] [Google Scholar]
- 5. Oberg A. L., and Vitek O. (2009) Statistical design of quantitative mass spectrometry-based proteomic experiments. J. Proteome Res. 8, 2144–2156 [DOI] [PubMed] [Google Scholar]
- 6. Cima I., Schiess R., Wild P., Kaelin M., Schüffler P., Lange V., Picotti P., Ossola R., Templeton A., Schubert O., Fuchs T., Leippold T., Wyler S., Zehetner J., Jochum W., Buhmann J., Cerny T., Moch H., Gillessen S., Aebersold R., and Krek W. (2011) Cancer genetics-guided discovery of serum biomarker signatures for diagnosis and prognosis of prostate cancer. Proc. Natl. Acad. Sci. U.S.A. 108, 3342–3347 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Surinova S., Choi M., Tao S., Schüffler P. J., Chang C.-Y., Clough T., Vysloužil K., Khoylou M., Srovnal J., Liu Y., Matondo M., Huttenhain R., Weisser H., Buhmann J. M., Hajdúch M., Brenner H., Vitek O., and Aebersold R. (2015) Prediction of colorectal cancer diagnosis based on circulating plasma proteins. EMBO Mol. Med. 7, 1166–1178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Drabovich A. P., Dimitromanolakis A., Saraon P., Soosaipillai A., Batruch I., Mullen B., Jarvi K., and Diamandis E. P. (2013) Differential diagnosis of azoospermia with proteomic biomarkers ECM1 and TEX101 quantified in seminal plasma. Sci. Transl. Med. 5, 212ra160–212ra160 [DOI] [PubMed] [Google Scholar]
- 9. Surinova S., Radová L., Choi M., Srovnal J., Brenner H., Vitek O., Hajdúch M., and Aebersold R. (2015) Non-invasive prognostic protein biomarker signatures associated with colorectal cancer. EMBO Mol. Med. 7, 1153–1165 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Duriez E., Masselon C. D., Mesmin C., Court M., Demeure K., Allory Y., Malats N., Matondo M., Radvanyi F., Garin J., and Domon B. (2017) Large-scale SRM screen of urothelial bladder cancer candidate biomarkers in urine. J. Proteome Res. 16, 1617–1631 [DOI] [PubMed] [Google Scholar]
- 11. Yap T. A., Carden C. P., and Kaye S. B. (2009) Beyond chemotherapy: targeted therapies in ovarian cancer. Nat. Rev. Cancer 9, 167–181 [DOI] [PubMed] [Google Scholar]
- 12. Bast R. C., Klug T. L., St John E., Jenison E., Niloff J. M., Lazarus H., Berkowitz R. S., Leavitt T., Griffiths C. T., Parker L., Zurawski V. R., and Knapp R. C. (1983) A radioimmunoassay using a monoclonal antibody to monitor the course of epithelial ovarian cancer. N. Engl. J. Med. 309, 883–887 [DOI] [PubMed] [Google Scholar]
- 13. Drapkin R., Horsten von, Lin H. H. Y., Mok S. C., Crum C. P., Welch W. R., and Hecht J. L. (2005) Human epididymis protein 4 (HE4) is a secreted glycoprotein that is overexpressed by serous and endometrioid ovarian carcinomas. Cancer Res. 65, 2162–2169 [DOI] [PubMed] [Google Scholar]
- 14. Moore R. G., McMeekin D. S., Brown A. K., Disilvestro P., Miller M. C., Allard W. J., Gajewski W., Kurman R., Bast R. C., and Skates S. J. (2009) A novel multiple marker bioassay utilizing HE4 and CA125 for the prediction of ovarian cancer in patients with a pelvic mass. Gynecol. Oncol. 112, 40–46 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Zhang Z., and Chan D. W. (2010) The road from discovery to clinical diagnostics: lessons learned from the first FDA-cleared in vitro diagnostic multivariate index assay of proteomic biomarkers. Cancer Epidemiol. Biomarkers Prev. 19, 2995–2999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Petricoin E. F., Ardekani A. M., Hitt B. A., Levine P. J., Fusaro V. A., Steinberg S. M., Mills G. B., Simone C., Fishman D. A., Kohn E. C., and Liotta L. A. (2002) Use of proteomic patterns in serum to identify ovarian cancer. Lancet 359, 572–577 [DOI] [PubMed] [Google Scholar]
- 17. Gorelik E., Landsittel D. P., Marrangoni A. M., Modugno F., Velikokhatnaya L., Winans M. T., Bigbee W. L., Herberman R. B., and Lokshin A. E. (2005) Multiplexed immunobead-based cytokine profiling for early detection of ovarian cancer. Cancer Epidemiol. Biomarkers Prev. 14, 981–987 [DOI] [PubMed] [Google Scholar]
- 18. Zhang Z., Bast R. C., Yu Y., Li J., Sokoll L. J., Rai A. J., Rosenzweig J. M., Cameron B., Wang Y. Y., Meng X.-Y., Berchuck A., Van Haaften-Day C., Hacker N. F., de Bruijn H. W. A., van der Zee A. G. J., Jacobs I. J., Fung E. T., and Chan D. W. (2004) Three biomarkers identified from serum proteomic analysis for the detection of early stage ovarian cancer. Cancer Res. 64, 5882–5890 [DOI] [PubMed] [Google Scholar]
- 19. Mor G., Visintin I., Lai Y., Zhao H., Schwartz P., Rutherford T., Yue L., Bray-Ward P., and Ward D. C. (2005) Serum protein markers for early detection of ovarian cancer. Proc. Natl. Acad. Sci. U.S.A. 102, 7677–7682 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Visintin I., Feng Z., Longton G., Ward D. C., Alvero A. B., Lai Y., Tenthorey J., Leiser A., Flores-Saaib R., Yu H., Azori M., Rutherford T., Schwartz P. E., and Mor G. (2008) Diagnostic markers for early detection of ovarian cancer. Clin. Cancer Res. 14, 1065–1072 [DOI] [PubMed] [Google Scholar]
- 21. Zhu C. S., Pinsky P. F., Cramer D. W., Ransohoff D. F., Hartge P., Pfeiffer R. M., Urban N., Mor G., Bast R. C., Moore L. E., Lokshin A. E., McIntosh M. W., Skates S. J., Vitonis A., Zhang Z., Ward D. C., Symanowski J. T., Lomakin A., Fung E. T., Sluss P. M., Scholler N., Lu K. H., Marrangoni A. M., Patriotis C., Srivastava S., Buys S. S., and Berg C. D.; PLCOProject Team. (2011) A framework for evaluating biomarkers for early detection: validation of biomarker panels for ovarian cancer. Cancer Prev. Res. 4, 375–383 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Kurman R. J., and Shih I.-M. (2008) Pathogenesis of ovarian cancer: lessons from morphology and molecular biology and their clinical implications. Int. J. Gynecol. Pathol. 27, 151–160 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Kurman R. J., and Shih I.-M. (2016) The dualistic model of ovarian carcinogenesis: revisited, revised, and expanded. Am. J. Pathol. 186, 733–747 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Schiess R., Wollscheid B., and Aebersold R. (2009) Targeted proteomic strategy for clinical biomarker discovery. Mol. Oncol. 3, 33–44 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Dinulescu D. M., Ince T. A., Quade B. J., Shafer S. A., Crowley D., and Jacks T. (2005) Role of K-ras and Pten in the development of mouse models of endometriosis and endometrioid ovarian cancer. Nat. Med. 11, 63–70 [DOI] [PubMed] [Google Scholar]
- 26. Bengtsson S., Krogh M., Szigyarto C. A.-K., Uhlen M., Schedvins K., Silfverswärd C., Linder S., Auer G., Alaiya A., and James P. (2007) Large-scale proteomics analysis of human ovarian cancer for biomarkers. J. Proteome Res. 6, 1440–1450 [DOI] [PubMed] [Google Scholar]
- 27. Köbel M., Kalloger S. E., Boyd N., McKinney S., Mehl E., Palmer C., Leung S., Bowen N. J., Ionescu D. N., Rajput A., Prentice L. M., Miller D., Santos J., Swenerton K., Gilks C. B., and Huntsman D. (2008) Ovarian carcinoma subtypes are different diseases: implications for biomarker studies. PLoS Med. 5, e232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Kuk C., Kulasingam V., Gunawardana C. G., Smith C. R., Batruch I., and Diamandis E. P. (2009) Mining the ovarian cancer ascites proteome for potential ovarian cancer biomarkers. Mol. Cell. Proteomics 8, 661–669 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Hudson M. E., Pozdnyakova I., Haines K., Mor G., and Snyder M. (2007) Identification of differentially expressed proteins in ovarian cancer using high-density protein microarrays. Proc. Natl. Acad. Sci. U.S.A. 104, 17494–17499 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Pitteri S. J., JeBailey L., Faça V. M., Thorpe J. D., Silva M. A., Ireton R. C., Horton M. B., Wang H., Pruitt L. C., Zhang Q., Cheng K. H., Urban N., Hanash S. M., and Dinulescu D. M. (2009) Integrated proteomic analysis of human cancer cells and plasma from tumor bearing mice for ovarian cancer biomarker discovery. PLoS ONE 4, e7916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Zhang H., Li X.-J., Martin D. B., and Aebersold R. (2003) Identification and quantification of N-linked glycoproteins using hydrazide chemistry, stable isotope labeling and mass spectrometry. Nat. Biotechnol. 21, 660–666 [DOI] [PubMed] [Google Scholar]
- 32. Pedrioli P. G. A., Eng J. K., Hubley R., Vogelzang M., Deutsch E. W., Raught B., Pratt B., Nilsson E., Angeletti R. H., Apweiler R., Cheung K., Costello C. E., Hermjakob H., Huang S., Julian R. K., Kapp E., McComb M. E., Oliver S. G., Omenn G., Paton N. W., Simpson R., Smith R., Taylor C. F., Zhu W., and Aebersold R. (2004) A common open representation of mass spectrometry data and its application to proteomics research. Nat. Biotechnol. 22, 1459–1466 [DOI] [PubMed] [Google Scholar]
- 33. Yates J. R., Eng J. K., McCormack A. L., and Schieltz D. (1995) Method to correlate tandem mass spectra of modified peptides to amino acid sequences in the protein database. Anal. Chem. 67, 1426–1436 [DOI] [PubMed] [Google Scholar]
- 34. Keller A., Nesvizhskii A. I., Kolker E., and Aebersold R. (2002) Empirical statistical model to estimate the accuracy of peptide identifications made by MS/MS and database search. Anal. Chem. 74, 5383–5392 [DOI] [PubMed] [Google Scholar]
- 35. Nesvizhskii A. I., Keller A., Kolker E., and Aebersold R. (2003) A statistical model for identifying proteins by tandem mass spectrometry. Anal. Chem. 75, 4646–4658 [DOI] [PubMed] [Google Scholar]
- 36. Jones P., Côté R. G., Martens L., Quinn A. F., Taylor C. F., Derache W., Hermjakob H., and Apweiler R. (2006) PRIDE: a public repository of protein and peptide identifications for the proteomics community. Nucleic Acids Res. 34, D659–D663 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Vizcaíno J. A., Deutsch E. W., Wang R., Csordas A., Reisinger F., Ríos D., Dianes J. A., Sun Z., Farrah T., Bandeira N., Binz P.-A., Xenarios I., Eisenacher M., Mayer G., Gatto L., Campos A., Chalkley R. J., Kraus H.-J., Albar J. P., Martinez-Bartolomé S., Apweiler R., Omenn G. S., Martens L., Jones A. R., and Hermjakob H. (2014) ProteomeXchange provides globally coordinated proteomics data submission and dissemination. Nat. Biotechnol. 32, 223–226 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Mueller L. N., Rinner O., Schmidt A., Letarte S., Bodenmiller B., Brusniak M.-Y., Vitek O., Aebersold R., and Müller M. (2007) SuperHirn - a novel tool for high resolution LC-MS-based peptide/protein profiling. Proteomics 7, 3470–3480 [DOI] [PubMed] [Google Scholar]
- 39. Smyth G. K. (2004) Linear models and empirical bayes methods for assessing differential expression in microarray experiments. Stat. Appl. Genet. Mol. Biol. 3, Article 3 [DOI] [PubMed] [Google Scholar]
- 40. Webb-Robertson B.-J. M., McCue L. A., Waters K. M., Matzke M. M., Jacobs J. M., Metz T. O., Varnum S. M., and Pounds J. G. (2010) Combined statistical analyses of peptide intensities and peptide occurrences improves identification of significant peptides from MS-based proteomics data. J. Proteome Res. 9, 5748–5756 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Benjamini Y., and Hochberg Y. (1995) Controlling the false discovery rate: a practical and powerful approach to multiple testing. J. Roy. Statistical Soc. 57, 289–300 [Google Scholar]
- 42. Choi M., Chang C.-Y., Clough T., Broudy D., Killeen T., MacLean B., and Vitek O. (2014) MSstats: an R package for statistical analysis of quantitative mass spectrometry-based proteomic experiments. Bioinformatics 30, 2524–2526 [DOI] [PubMed] [Google Scholar]
- 43. Huttenhain R., Surinova S., Ossola R., Sun Z., Campbell D., Cerciello F., Schiess R., Bausch-Fluck D., Rosenberger G., Chen J., Rinner O., Kusebauch U., Hajdúch M., Moritz R. L., Wollscheid B., and Aebersold R. (2013) N-glycoprotein SRMAtlas: a resource of mass spectrometric assays for N-glycosites enabling consistent and multiplexed protein quantification for clinical applications. Mol. Cell. Proteomics 12, 1005–1016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. MacLean B., Tomazela D. M., Shulman N., Chambers M., Finney G. L., Frewen B., Kern R., Tabb D. L., Liebler D. C., and MacCoss M. J. (2010) Skyline: an open source document editor for creating and analyzing targeted proteomics experiments. Bioinformatics 26, 966–968 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Sharma V., Eckels J., Taylor G. K., Shulman N. J., Stergachis A. B., Joyner S. A., Yan P., Whiteaker J. R., Halusa G. N., Schilling B., Gibson B. W., Colangelo C. M., Paulovich A. G., Carr S. A., Jaffe J. D., MacCoss M. J., and MacLean B. (2014) Panorama: a targeted proteomics knowledge base. J. Proteome Res. 13, 4205–4210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Rechsteiner M., Zimmermann A.-K., Wild P. J., Caduff R., Teichman von, Fink A. D., Moch H., and Noske A. (2013) TP53 mutations are common in all subtypes of epithelial ovarian cancer and occur concomitantly with KRAS mutations in the mucinous type. Exp. Mol. Pathol. 95, 235–241 [DOI] [PubMed] [Google Scholar]
- 47. Yang W. L., Lu Z., and Bast R. C. (2017) The role of biomarkers in the management of epithelial ovarian cancer. Expert Rev. Mol. Diagn. 17, 577–591 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Rojas V., Hirshfield K. M., Ganesan S., and Rodriguez-Rodriguez L. (2016) Molecular characterization of epithelial ovarian cancer: implications for diagnosis and treatment. Int. J. Mol. Sci. 17, 2113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Cancer Genome Atlas Research Network. (2011) Integrated genomic analyses of ovarian carcinoma. Nature 474, 609–615 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Buys S. S., Partridge E., Black A., Johnson C. C., Lamerato L., Isaacs C., Reding D. J., Greenlee R. T., Yokochi L. A., Kessel B., Crawford E. D., Church T. R., Andriole G. L., Weissfeld J. L., Fouad M. N., Chia D., O'Brien B., Ragard L. R., Clapp J. D., Rathmell J. M., Riley T. L., Hartge P., Pinsky P. F., Zhu C. S., Izmirlian G., Kramer B. S., Miller A. B., Xu J.-L., Prorok P. C., Gohagan J. K., Berg C. D., PLCOProject Team. (2011) Effect of screening on ovarian cancer mortality: the Prostate, Lung, Colorectal and Ovarian (PLCO) Cancer Screening Randomized Controlled Trial. JAMA 305, 2295–2303 [DOI] [PubMed] [Google Scholar]
- 51. Edwards A. M., Isserlin R., Bader G. D., Frye S. V., Willson T. M., and Yu F. H. (2011) Too many roads not taken. Nature 470, 163–165 [DOI] [PubMed] [Google Scholar]
- 52. Resnick D., Pearson A., and Krieger M. (1994) The SRCR superfamily: a family reminiscent of the Ig superfamily. Trends Biochem. Sci. 19, 5–8 [DOI] [PubMed] [Google Scholar]
- 53. Qu H., Chen Y., Cao G., Liu C., Xu J., Deng H., and Zhang Z. (2016) Identification and validation of differentially expressed proteins in epithelial ovarian cancers using quantitative proteomics. Oncotarget 7, 83187–83199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Piccolo E., Tinari N., D'Addario D., Rossi C., Iacobelli V., La Sorda R., Lattanzio R., D'Egidio M., Di Risio A., Piantelli M., Natali P. G., and Iacobelli S. (2015) Prognostic relevance of LGALS3BP in human colorectal carcinoma. J. Transl. Med. 13, 248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Park J., Yun H. S., Lee K. H., Lee K. T., Lee J. K., and Lee S.-Y. (2015) Discovery and validation of biomarkers that distinguish mucinous and nonmucinous pancreatic cysts. Cancer Res. 75, 3227–3235 [DOI] [PubMed] [Google Scholar]
- 56. Schäfer S., Koch P. J., and Franke W. W. (1994) Identification of the ubiquitous human desmoglein, Dsg2, and the expression catalogue of the desmoglein subfamily of desmosomal cadherins. Exp. Cell Res. 211, 391–399 [DOI] [PubMed] [Google Scholar]
- 57. Cai F., Zhu Q., Miao Y., Shen S., Su X., and Shi Y. (2017) Desmoglein-2 is overexpressed in non-small cell lung cancer tissues and its knockdown suppresses NSCLC growth by regulation of p27 and CDK2. J. Cancer Res. Clin. Oncol. 143, 59–69 [DOI] [PubMed] [Google Scholar]
- 58. Tan L. Y., Mintoff C., Johan M. Z., Ebert B. W., Fedele C., Zhang Y. F., Szeto P., Sheppard K. E., McArthur G. A., Foster-Smith E., Ruszkiewicz A., Brown M. P., Bonder C. S., Shackleton M., and Ebert L. M. (2016) Desmoglein 2 promotes vasculogenic mimicry in melanoma and is associated with poor clinical outcome. Oncotarget 7, 46492–46508 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Kamekura R., Kolegraff K. N., Nava P., Hilgarth R. S., Feng M., Parkos C. A., and Nusrat A. (2014) Loss of the desmosomal cadherin desmoglein-2 suppresses colon cancer cell proliferation through EGFR signaling. Oncogene 33, 4531–4536 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Pellatt A. J., Mullany L. E., Herrick J. S., Sakoda L. C., Wolff R. K., Samowitz W. S., and Slattery M. L. (2018) The TGFβ-signaling pathway and colorectal cancer: associations between dysregulated genes and miRNAs. J. Transl. Med. 16, 191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Roberts D. D. (2008) Thrombospondins: from structure to therapeutics. Cell. Mol. Life Sci. 65, 669–671 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Pal S. K., Nguyen C. T. K., Morita K.-I., Miki Y., Kayamori K., Yamaguchi A., and Sakamoto K. (2016) THBS1 is induced by TGFB1 in the cancer stroma and promotes invasion of oral squamous cell carcinoma. J. Oral Pathol. Med. 45, 730–739 [DOI] [PubMed] [Google Scholar]
- 63. Rathjen F. G., and Schachner M. (1984) Immunocytological and biochemical characterization of a new neuronal cell surface component (L1 antigen) which is involved in cell adhesion. EMBO J. 3, 1–10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Altevogt P., Doberstein K., and Fogel M. (2016) L1CAM in human cancer. Int. J. Cancer 138, 1565–1576 [DOI] [PubMed] [Google Scholar]
- 65. Notaro S., Reimer D., Duggan-Peer M., Fiegl H., Wiedermair A., Rössler J., Altevogt P., Marth C., and Zeimet A. G. (2016) Evaluating L1CAM expression in human endometrial cancer using qRT-PCR. Oncotarget 7, 40221–40232 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Sajic T., Liu Y., Arvaniti E., Surinova S., Williams E. G., Schiess R., Huttenhain R., Sethi A., Pan S., Brentnall T. A., Chen R., Blattmann P., Friedrich B., Niméus E., Malander S., Omlin A., Gillessen S., Claassen M., and Aebersold R. (2018) Similarities and differences of blood N-glycoproteins in five solid carcinomas at localized clinical stage analyzed by sWATH-MS. Cell Rep. 23, 2819–2831.e5 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
For DDA experiments, RAW data and search results have been deposited to the ProteomeXchange Consortium via the PRIDE partner repository with the dataset identifier PXD005665 (https://www.ebi.ac.uk/pride/archive/projects/PXD005665) (36, 37). For SRM experiments, SRM data can be accessed, queried, and downloaded via Panorama (https://panoramaweb.org/ovarian_cancer_biomarker.url) (45).