

Abstract
Multiple sclerosis (MS) is a neuroinflammatory disease with a relapsing-remitting disease course at early stages, distinct lesion characteristics in cortical gray versus subcortical white matter, and neurodegeneration at chronic stages. We assessed multilineage cell expression changes using single-nucleus RNA sequencing (snRNA-seq) and validated results using multiplex in situ hybridization in MS lesions. We found selective vulnerability and loss of excitatory CUX2-expressing projection neurons in upper cortical layers underlying meningeal inflammation; such MS neuron populations showed upregulation of stress pathway genes and long non-coding RNAs. Signatures of stressed oligodendrocytes, reactive astrocytes and activated phagocytosing cells mapped most strongly to the rim of MS plaques. Interestingly, snRNA-seq identified phagocytosing microglia and/or macrophages by their ingestion and perinuclear import of myelin transcripts, confirmed by functional mouse and human culture assays. Our findings indicate lineage- and region-specific transcriptomic changes associated with selective cortical neuron damage and glial activation contributing to MS lesion progression.
Multiple sclerosis (MS) is a progressive neuroinflammatory autoimmune disease affecting about 2.3 million people worldwide1. Immune-mediated cytotoxic damage to oligodendrocytes (OLs) causes demyelination and focal plaque formation2,3 accompanied by progressive axonal damage in white matter (WM)4,5, and active MS plaques typically show a rim of inflammation with myelin phagocytosis. MS lesion heterogeneity in WM versus gray matter (GM) compartments suggests that the underlying pathobiology and potential for repair is likely to vary in a region-restricted manner. Cortical GM pathologies include demyelination and damage to the axon, neurite and neuron cell body6, particularly in areas underlying meningeal inflammation with plasma cell infiltration7–9. However, whether this process affects all or a subset of cortical neurons is poorly understood10. Indeed, cell type-specific mechanisms of MS progression, including scar formation with slowly expanding WM lesions11 and cortical atrophy12 are unclear.
Single-cell transcriptomic techniques are well suited to identify cellular heterogeneity in the human brain; recently, they have been applied to individual glial lineages in MS13,14. Here, we took a multilineage approach to brain-resident populations (neurons, astrocytes, OLs, microglia) to better understand molecular, cellular and spatially-restricted substrates of progressive MS pathology. We used frozen human brain samples from MS cases and controls to perform unbiased isolation of nuclei from cortical and subcortical lesion and non-lesion areas followed by single-nucleus RNA-sequencing (snRNA-seq)13,15 and in situ validation of RNA gene expression across large anatomical areas. Our results indicate that genes most dysregulated in MS map spatially to vulnerable upper cortical layer neurons and reactive glia at the borders of subcortical MS lesions associated with progression in MS
Results
snRNA-seq using post-mortem frozen MS tissue reveals cell-type specific molecular changes associated with MS pathogenesis
We used snRNA-seq to profile cortical GM and adjacent subcortical WM MS lesion areas at various stages of inflammation and demyelination, and control tissue from unaffected individuals. We established a pipeline for serial sectioning of entire tissue blocks including lesion and non-lesion GM and WM areas plus meningeal tissue. Tissue sections were screened for RNA integrity number (RIN) of >6.5. Using this criterion, 12/19 MS tissue samples screened from 17 individuals and 9/16 samples screened from control individuals were further procesed (Fig. 1a; Supplementary Table 1). Confounding variables of age, sex, postmortem interval and RIN were not significantly different between control and MS subjects (p > 0.1, Mann-Whitney U test).
Fig. 1. Experimental approach and characteristics of snRNA-seq using frozen MS tissue.

(a) Cortical and subcortical control tissue and MS lesion types (DM = demyelination, NA = normal appearing). (b) Experimental approach for isolating nuclei from postmortem snap-frozen brain samples of MS and control patients. (c) Cell types from individual samples (left), cell-type specific clusters (center; ctrl, n= 9; MS, n= 12) and sample contribution to individual clusters (right). Note separation of EN-L2-3 and OL cells into MS-specific clusters EN-L2-3-A/B and OL-B/C. (d) tSNE plots highlight marker genes for neurons, astrocytes, OLs and microglia. (e) Bar chart shows contributions of normalized control and MS cell numbers to major cell-type clusters. Note that EN-L2-3-A cell enrichment and concomitant decrease in EN-L2-3-B in control samples over MS was not statistically significant (p = 0.165 and 0.082). (f) Specific loss of EN-L2-3 versus EN-L4, EN-L5-6 or IN-VIP neurons based on normalized cell numbers. (g) Differential gene expression (DGE) analysis showing highest number of dysregulated genes in EN-L2-3 followed by EN-L4 and OL cells; least differentially expressed genes were found in SST INs and OPCs. Box plots represent median and interquartile range (IQR) of differentially expressed gene number calculated after downsampling (100 DGE analyses per cell cluster; ctrl, n= 9; n= 12 MS). Wiskers extend to the largest values within 1.5 IQR from box boundaries, outliers shown as dots, notches represent a 95% confidence interval around the median. Two-tailed Mann-Whitney tests performed in e and f (ctrl, n= 9; MS, n= 12); *P ≤ 0.05. Data presented as mean ± SEM. For tSNE plots, data shown from a total of 48,919 nuclei (ctrl, n= 9; n= 12 MS).
We optimized and performed unbiased nuclei isolation using sucrose-gradient ultracentrifugation (Extended Data Fig. 1a), followed by snRNA-barcoding (10x Genomics) and cDNA sequencing. After quality control filtering, snRNA-seq yielded 48,919 single-nuclei profiles (Fig. 1b-c). We normalized data and applied several independent analysis techniques. As shown (Fig. 1c), unbiased clustering identified 22 cell clusters (n.b., none comprised nuclei captured from individual MS or control samples). We detected a median of 1,400 genes and 2,400 transcripts per nucleus with higher numbers detected in neuronal versus glial populations (Extended Data Fig. 1b, Supplementary Table 2).
Next, we annotated cell clusters based on expression of lineage marker genes for excitatory and inhibitory cortical neurons, astrocytes, OL lineage cells and microglia, as well as smaller cell populations (Fig. 1d, Extended Data Fig. 1e, Supplementary Table 3)16. Neuronal subtype markers included excitatory neuron marker SCL17A7, upper layer marker CUX2, layer 4 marker RORB, deep layer marker TLE4, as well as interneuron (IN) marker GAD2 and subtype markers PVALB, SST, VIP and SV2C. Comparing normalized nuclei numbers from MS and controls (Supplementary Table 4), we observed a selective reduction of upper-layer excitatory projection neuron (EN-L2-3A/B) numbers in MS samples with cortical demyelination (Fig. 1e-f). In contrast, numbers of intermediate (EN-L4) and deep-layer (EN-L5-6) excitatory neurons, THY1/NRGN-high-expressing pyramidal cells, VIP-expressing, somatostatin (SST) - and parvalbumin (PVALB)-expressing INs were similar between MS samples and controls (Fig. 1e-f). MS-associated genes showed greatest differential expression in EN-L2-3, followed by EN-L4 and myelinating OLs (Fig. 1g). Notably, for EN-L2-3 and OLs, transcriptomic changes distinguished subclusters from MS or control samples (Fig. 1c and 1e). In contrast to EN-L2-3 cells, gene dysregulation was less pronounced in upper layer VIP-expressing INs (Fig. 1g). These findings suggested cell-type vulnerability of layer 2/3 excitatory CUX2-expressing neurons.
Selective vulnerability of CUX2-expressing upper layer neurons in MS
We investigated changes in CUX2-expressing EN-L2-3 cells in MS lesion pathology using unsupervised pseudotime trajectory analysis to identify dynamic gene expression changes. As shown (Fig. 2a), cell distribution along the trajectory separated control from MS in EN-L2-3 cells. Interestingly, progression along the trajectory correlated with conventional inflammatory lesion staging and the degree of upper layer cortical demyelination (Fig. 2b, Extended Data Fig. 1c-d), e.g., CUX2-expressing neurons, which localized towards the trajectory end, derived mainly from samples harboring late chronic inactive lesions with extensive subpial demyelination versus lesions with less upper cortical demyelination (Fig. 2c).
Fig. 2. Pseudotime trajectory analysis of upper layer excitatory projection neurons.

(a) Trajectory analysis of CUX2-expressing EN-L2-3 cells (upper left). Unsupervised pseudotime trajectories within the EN-L2-3 (upper right) cluster reflected cellular origin from MS samples or controls (lower left) and inflammatory lesion stage (lower right). (b) EN-L2-3 pseudotime trajectories showed similar features as (a) and suggested loss of normalized EN-L2-3 numbers (lower left). Strongest association with EN-L2-3 trajectories found for upper cortical layer demyelination (upper right) versus deep cortical layer (center right) and subcortical demyelination (lower right). (c) Note selective enrichment of dysregulated genes in EN-L2-3 cells from samples with late chronic inactive lesions versus acute/chronic-active and control samples. (d) Visualization of GO terms (enrichment calculated using GSEA, FDR adjusted p ≤ 0.05, no terms significantly decreased) in genes significantly regulated in EN-L2-3 in a pseudotime-dependent manner (Moran’s I test, FDR adjusted p ≤ 0.0001). Note enrichment of severe cell stress processes. (e) Trajectory-dependent upregulated (f) and downregulated EN-L2-3 genes of interest. Grey shading represent 95% confidence interval based on gene expression in all (n= 5,938) sampled EN-L2-3 nuclei.
Trajectory analysis highlighted gene ontology (GO) terms and dynamic upregulation of oxidative stress, mitochondrial dysfunction and cell death pathways in EN-L2-3 cells, including FAIM2, ATF4, CLU, B2M (cell stress/death), HSPH1, HSP90AA1 (heat-shock response), APP, NEFL, UBB (protein accumulation, axon degradation), COX7C, PKM, PPIA (energy metabolism, oxidative stress) and long-noncoding (lnc) RNAs LINC00657 (NORAD) and BCYRN1 (BC200) (Fig. 2d-e, Extended Data Fig. 2a, Supplementary Table 5)17,18. Conversely, we noted dynamic downregulation of transcripts associated with mitochondrial energy consumption (FARS2), glutamate signaling (GRIA4, GRM5), potassium/cation homeostasis (KCNB2, KCNN2, SLC22A10), neuronal signaling (NELL1), axon plasticity (ROBO1) and lncRNA LINC01266 (Fig. 2f). Neurons from all cortical layers in MS showed enrichment of cell stress pathways compared to controls (Extended Data Fig. 2b, Supplementary Table 6); in contrast, PVALB- and VIP-expressing INs showed only one GO term (associated with protein folding) enriched for dysregulated genes. Together, these findings highlighted a selective transcriptomic damage signature for CUX2-expressing neurons in MS.
Loss of CUX2-expressing neurons in demyelinated cortical MS lesions in situ
We next used large area spatial transcriptomic (‘LaST’) mapping19 to validate cell type-specific gene expression changes. We optimized chromogenic and multiplex small molecule fluorescent in situ hybridization (smFISH) protocols to overcome high levels of background auto-fluorescence in WM and GM areas in frozen human brain samples. As shown (Fig. 3a), we achieved a favorable signal-to-noise ratio over tissue sections for neuronal markers CUX2 and SYT1 combined with immunohistochemistry for myelin oligodendrocyte glycoprotein (MOG), and we confirmed layer-associated expression of neuronal subtype markers RORB, THY1, TLE4, VIP and SST (Fig. 3a, Extended Data Fig. 3a).
Fig. 3. Cellular and molecular neuronal pathology in cortical MS lesions.

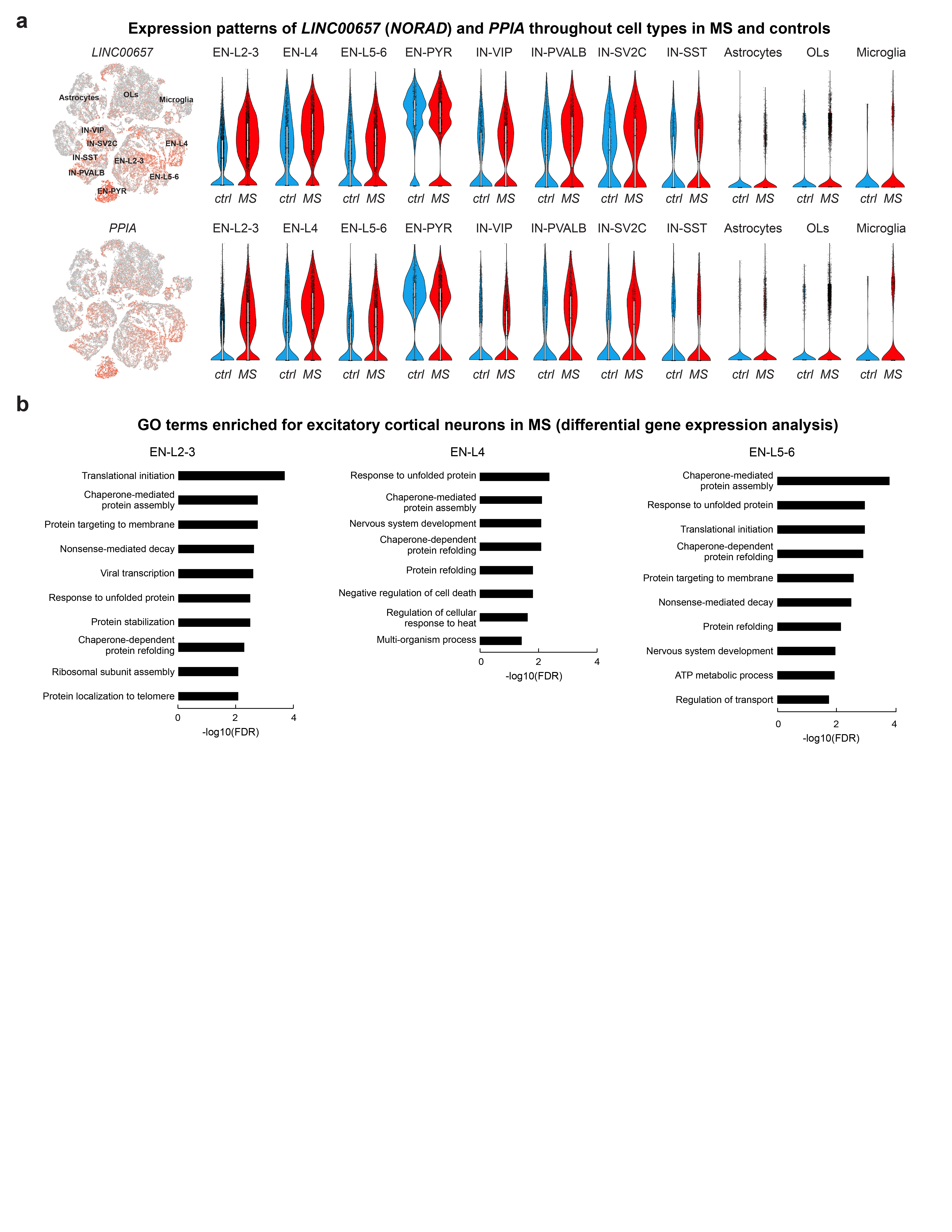
(a) tSNE plots CUX2, VIP and TLE4-expressing neurons (left). Spatial transcriptomics showing layer-specific expression of CUX2 in lesion (indicated by loss of MOG) versus non-lesion areas (center left). Schematic illustrates layer-specific neuron subtype diversity (center). Note CUX2 and VIP expression in upper and TLE4 in deep cortical layers by smFISH (center right; ctrl, n=5), and validation of neuronal expression by SYT1 ISH (black arrowheads; ctrl, n=5). (b) CUX2 and VIP smFISH demonstrate reduction of CUX2- but not VIP-expressing upper layer neurons in DMGM underlying meningeal inflammation (upper left and right) versus incomplete demyelinated (IDMGM), NAGM and control cortical GM (bottom left). ANOVA with Kruskal Wallis multiple comparison tests were performed (ctrl, n=5 (CUX2), n=4 (VIP); MS, n=8; *P ≤ 0.05; different samples with NAWM, IDMGM and DMGM MS lesion areas from same sections; representative images). (c) Upregulation of neuronal PPIA in DMGM and NAGM versus control GM (left, white circles indicate perinuclear areas of PPIA quantification). Neuronal upregulation and cytoplasmic accumulation of LINC00657 (NORAD) in DMGM versus NAGM and control areas (right, black arrowheads). ANOVA with Tukey’s multiple comparison tests were performed (ctrl, n=3; MS, n=4; **p ≤ 0.01, ***p ≤ 0.001, ****p ≤ 0.0001; different samples with NAWM and DMGM areas from same sections; representative images). Data presented as mean ± SEM. For tSNE plots, data shown from a total of 48,919 nuclei (ctrl, n = 9; n = 12 MS). Violin plots represent DGE (normalized log transformed UMIs) in EN-L2-3 (EN-L2-3-A and EN-L2-3-B) nuclei (ctrl, n = 3,481; n = 2,639 MS); box plots represent median and standard deviation of gene expression.
Given snRNA-seq findings above, we investigated expression of co-located upper layer CUX2- and VIP-expressing populations by smFISH in MS and control sections (Fig. 3b). We found a significant reduction of CUX2-expressing neurons in completely and incompletely demyelinated cortical areas; in contrast, numbers of abutting VIP-expressing INs were maintained. Of note, meningeal infiltration of IGHG1/ MZB1-expressing plasma cells (that predominated over SKAP1+ T cells) was a common finding in sulci with underlying upper cortical layer demyelination and loss of CUX2-expressing neurons (Extended Data Fig. 3b)7,20.
We next used smFISH to validate upregulation of the cell stress markers, including PPIA (encoding prolyl isomerase cyclophilin A, Extended Data Fig. 2a) in MS EN-L2-3 and EN-L4 neurons. As shown (Fig. 3c), PPIA transcripts were increased in neurons from demyelinated and adjacent normal-appearing cortical lesion areas21. We confirmed upregulated NORAD in EN-L2-3 and EN-L4 neurons (Extended Data Fig. 2a) by chromogenic and fluorescent smFISH, and observed cytoplasmic NORAD accumulation in MS lesions as compared to normal-appearing areas with intact myelin (Fig. 3c). Together, these findings confirm degeneration and selective loss of CUX2-expressing upper layer excitatory neurons in cortical MS lesions, while co-located inhibitory and other cortical excitatory neuron subtypes were relatively preserved.
Distinct spatial macroglial signatures in cortical and subcortical MS lesions
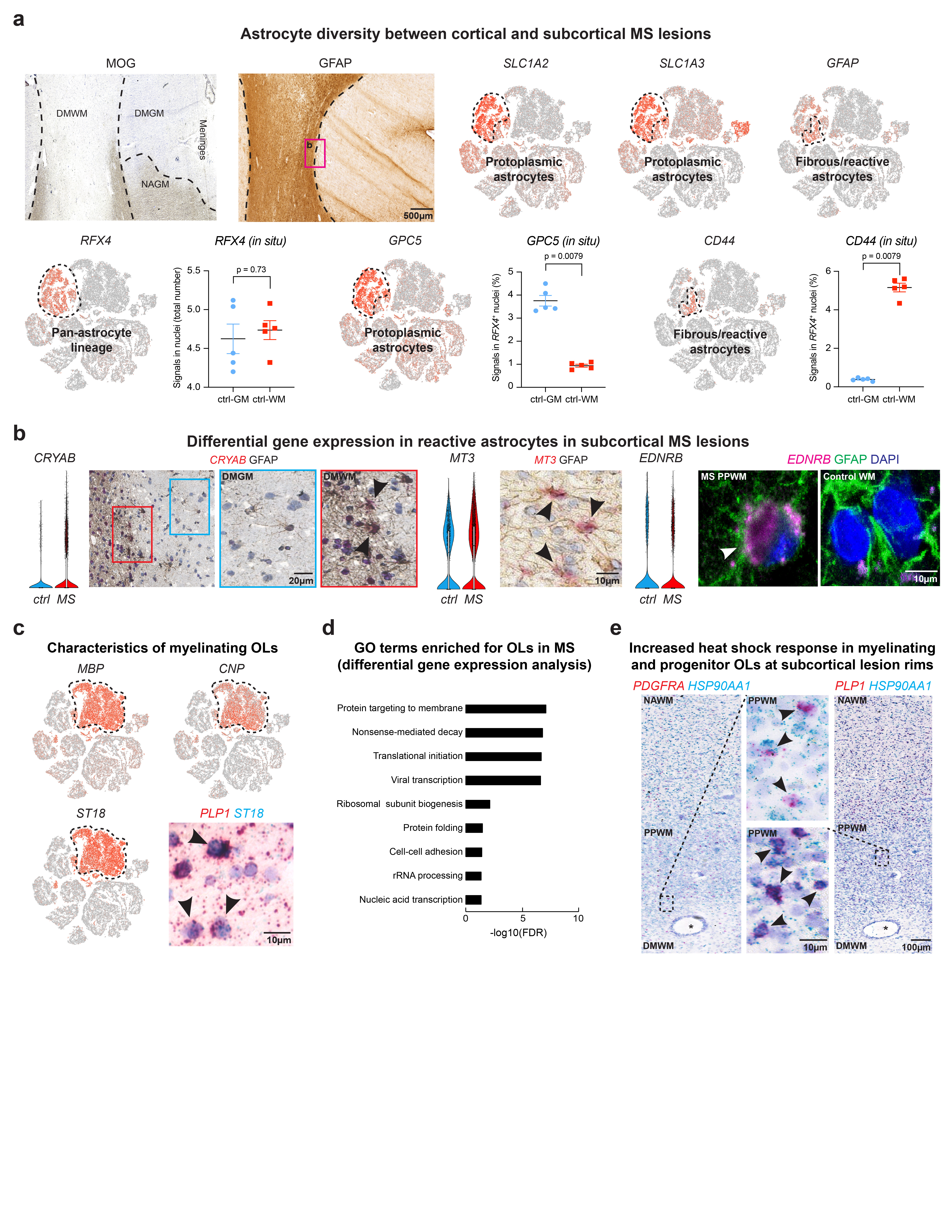
Prior studies have indicated differential gene expression and functionally diverse properties of reactive astrocytes that can be antagonistic or beneficial to repair after injury22,23. We identified astrogliosis by enhanced immunoreactivity for glial fibrillary acidic protein (GFAP) in regions of subcortical demyelinated WM that did not cross into the demyelinated cortex in MS lesions (Extended Data Fig. 4a). The GFAP signature in demyelinated WM overlapped with CD44-expressing reactive astrocytes24; CD44 showed upregulation at the lesion rim in astrocytes that co-expressed CRYAB and MT3 (Extended Data Fig. 4a-b)25. As shown (Fig. 4a, Extended Data Fig. 4a), RFX4 expression was specific to the astrocyte lineage and captured all SLC1A2-positive GM and CD44-expressing WM astrocytes (Supplementary Table 3, Extended Data Fig. 4a). We observed downregulation of genes for glutamate (SLC1A2, GLUL) and potassium homeostasis (KCNJ10)26 in cortical GM astrocytes and confirmed expression of GPC5, a marker that co-localizes with RFX4-expressing GM astrocytes, in lesion and non-lesion cortical areas in situ (Fig. 4a-b, Extended Data Fig. 4a). Reactive astrocytes at inflammatory chronic active lesion rims showed strong expression of the transcription factors BCL6, FOS (encoding c-FOS) – associated with astrocyte endothelin receptor type B (EDNRB) upregulation – and LINC01088 (Fig. 4b, Extended Data Fig. 4b)27. Thus, spatial transcriptomics revealed distinct expression patterns for cortical versus subcortical reactive astrocytes in the MS lesion microenvironment.
Fig. 4. Transcriptomic changes in astrocytes and myelinating oligodendrocytes in cortical and subcortical MS lesions.

(a) Downregulation of SLC1A2 and GPC5 and upregulation of GFAP and CD44 in MS astrocytes (upper left). LaST ISH experiments confirm SLC1A2 downregulation in DMGM underlying meningeal inflammation, whereas CD44 shows ubiquitous expression in NAWM and PPWM (periplaque white matter, center left) and upregulation in reactive astrocytes at lesion rims in b1 (center right). Note CD44 and GPC5 co-expression with pan-astrocyte marker RFX4 (white/black arrowheads, lower left and right) and association of CD44 with fibrous/reactive WM astrocytes and GPC5 with protoplasmic cortical GM astrocytes (black arrowheads; right; white star indicates blood vessel). (b) Downregulation of GLUL and KCNJ10 in MS astrocytes (left). Note differential upregulation of BCL6 and FOS in reactive astrocytes at PPWM (center, black arrowheads) and LINC01088 in fibrous/reactive WM astrocytes (right, black arrowhead). (c) Violin plots for selected genes linked to cell stress (upregulated, top), myelin biosynthesis and axon maintenance (downregulated, bottom) in MS OLs. (d) FTL and FTH1 upregulation in PLP1-expressing OLs at iron-laden lesions rims (left, black arrowheads). Note differential upregulation of B2M and HLA-C in PLP1-expressing OLs at PPWM (right; yellow arrowheads [white arrowheads mark OLs without B2M ISH signals in NAWM]). For ISH, representative images shown (ctrl, n = 3; n = 4 MS). For tSNE plots, data shown from a total of 48,919 nuclei (ctrl, n = 9; n = 12 MS). Violin plots represent DGE (normalized log transformed UMIs) in nuclei (astrocytes: ctrl, n = 1,571; n = 3,810 MS; OLs [OL-A, OL-B and OL-C]: ctrl, n = 3,070; n = 9,324 MS;); box plots represent median and standard deviation of gene expression.
Myelinating OLs characterized by myelin gene expression and the transcription factor ST18 (Fig. 1d, Extended Data Fig. 4c) exhibited the third highest number of differentially expressed genes (Fig. 1g) consistent with enriched stress pathways (Extended Data Fig. 4d) and known cell loss in MS. Differential gene expression analysis indicated upregulation of genes for heat shock response (HSP90AA1) (Extended Data Fig. 4e)34, cell stress (FAIM2, ATF4), MHC class I upregulation (B2M, HLA-C), iron accumulation (FTL, FTH1)28, ubiquitin-mediated protein degradation (UBB) and LINC00657 (NORAD) and LINC00844 (Fig. 4c-d, Extended Data Fig. 2a). Conversely, we observed downregulation of markers for OL differentiation and myelin synthesis (BCAS1, SGMS1)29, potassium/cation homeostasis (KCNJ10)26, cell-cell-interaction (SEMA6A) and formation of the node of Ranvier (GLDN) in MS OLs at lesion borders (Fig. 4c). Our findings indicate severe cell stress in MS OLs that can be mapped back to periplaque rim areas of subcortical lesions.
Activated phagocytosing microglial cells can be identified by snRNA-seq and mapped to chronic-active MS lesion rims
Given dramatic expansion of microglia in MS samples (Fig. 1e), we performed hierarchical clustering (Extended Data Fig. 5) and observed microglial cells with a homeostatic gene expression signature (P2RY12, RUNX1, CSF1R) in MS and control samples as well as MS-specific cells with enrichment for transcripts encoding activation markers, complement factors and MHC-II associated proteins14, and lipid degradation (ASAH1, ACSL1, DPYD) (Fig. 5a-b, Extended Data Fig. 5, Supplementary Table S5). Downregulated genes in MS microglia included synapse remodeling transcript SYNDIG1 and potassium channel KCNQ3. As shown (Fig. 5a-b), marker genes for microglia reactivity (CD68, CD74, FTL, MSR1) colocalized with the lineage microglia marker RUNX1, and mapped such activated cells to chronic active boundaries of subcortical MS lesions.
Fig. 5. Transcriptomic changes in activated and phagocytosing microglia subsets.

(a) Violin and tSNE plots for upregulated genes in MS microglia linked to myelin phagocytosis/breakdown (left), microglia activation and iron handling (center); note downregulation of genes encoding for synapse function (SYNDIG1) and potassium homeostasis (KCNQ3) (right). (b) Pseudo low resolution 3D rendering of confocal images showing subcortical WM lesions of different inflammatory stages by MBP smFISH and CD68 IHC; white arrowheads indicate CD68+ cells with MBP+ ISH signals; note colocalization of MBP, CD74 and RUNX1 in CD68-positive cells (center left, white arrowheads). CD68 IHC identifies WM lesion (blood vessel, black star; upper right) with upregulation of MSR1 at lesion rims, co-expressed with RUNX1 (lower right) and FTL (upper right, black arrowheads); representative images from different tissue sections (ctrl, n=3; MS, n=4). (c) Human (upper left; n=3 individual biopsies) and mouse (upper center right; n=4 independent cultures) myelin-microglia engulfment assays confirming ingestion of MBP and PLP1 transcripts derived from rat myelin. Note localization to nuclear/perinuclear spaces (white arrowheads). Microglia labeled by pHrodo (human) and Iba1/CD68 (mouse) with LMNA/C and DAPI nuclear counterstain. Schematic illustrates myelin phagocytosis and uptake into microglial (peri-)nuclear spaces (upper right). MBP persistence up to 4 days after ingestion in mouse microglia as shown by smFISH (4 independent cultures; lower left); note upregulation of Cd163 and downregulation of P2ry12 in phagocytosing mouse (6 independent cultures) and human MS microglia (lower right). Two-tailed Mann-Whitney tests performed. Data presented as mean ± SEM. For tSNE plots, data shown from a total of 48,919 nuclei (ctrl, n = 9; n = 12 MS). Violin plots represent DGE (normalized log transformed UMIs) in microglia nuclei (ctrl, n = 159; n = 1,524 MS [microglial and phagocytosing cells]); box plots represent median and standard deviation of gene expression.
Interestingly, we found a cluster of microglial cells characterized by phagocytosis and enrichment for OL-specific markers PLP1, MBP and ST18 (Fig. 1c-d, Extended Data Fig. 4c and 5, Supplementary Table 7) suggesting the possibility that ingested myelin transcripts co-purified with nuclei of phagocytosing cells in MS. To provide functional evidence for putative myelin RNA microglial phagocytosis, we cultured human and mouse microglia exposed to purified myelin from rat brain (Fig. 5c), which contains myelin transcripts (Extended Data Fig. 6)30. As shown (Fig. 5c), PLP1 and MBP transcripts were observed in intracellular, perinuclear and nuclear compartments of cultured human or mouse microglia at 1-day post exposure to labeled (pHrodo) myelin; ingested MBP mRNA was observed in mouse microglia up to 4-days post-feeding. In parallel, we observed morphological changes in phagocytosing mouse microglia, differential upregulation of the activation marker Cd163 and downregulation of the homeostatic microglia marker P2ry1231. Such changes in mouse microglia showed parallel gene expression changes in human MS microglia by snRNA-seq (Fig. 5c).
Interactive single-cell web browser to visualize snRNA-seq data
We created an interactive web browser to analyze cell-type specific expression levels of genes and transcriptomic changes in MS versus control tissue (https://ms.cells.ucsc.edu).
Discussion
MS lesions are heterogeneous in cortical and subcortical areas with distinct patterns of inflammatory demyelination10,32,33. We found cell type-specific gene expression changes in regions of cortical neurodegeneration and at the rim of chronic active subcortical lesions involved in progression and cortical atrophy. Our technical finding of snRNA-seq feasibility in MS is consistent with recent observations13–15. We used high-quality archival samples from patients, who did not receive modern immunomodulatory therapies; thus, they represent the endpoint of the natural disease course with relatively early death of patients (30-50 yrs). However, the number of MS samples studied could have resulted in under-reporting of certain lineages.
Computational analysis of differential gene expression and trajectory analysis of a total of 12 MS and 9 control samples pointed most strongly to the neuronal compartment and indicated dramatic cellular stress and loss of CUX2-expressing upper layer excitatory projection neurons in demyelinated and partially remyelinated cortical MS lesions. As such lesions underlie meningeal inflammation with pronounced plasma B cell infiltration, these findings suggest the importance of B cells in progressive MS7,8 and that damaged cortical neuron populations potentially benefited from B cell depleting therapies34.
We validated candidate gene expression using spatial transcriptomics of human MS brain. Markers of stressed CUX2-expressing neurons included PPIA (cyclophilin A) and NORAD, a neuronal lncRNA that helps stabilize DNA upon genomic stress by binding to PUMILIO and RBMX proteins 17,35, as well as other pathways for protein degradation, heat shock response and metabolic exhaustion36,37. Whereas most transcriptional changes and neuronal cell loss occurred in demyelinated regions, we also observed abnormal gene expression features (e.g., PPIA) in normal-appearing cortical areas suggesting a gradient of pathology38. While it is possible that CUX2-expressing projection neurons are damaged by both sustained meningeal inflammation and retrograde axon pathology from juxtacortical WM lesions in MS33, additional intrinsic factors might account for their lack of resilience, especially considering that neighboring inhibitory and excitatory neurons of the cortex showed relatively little cell loss.
Recent studies used MS WM lesion single-nuclei and single-cell RNA-seq to study the OL13 and microglia14 lineages and reported subsets linked to MS pathobiology. Here, we used spatial transcriptomics to map dysregulated glial gene expression in cortical and subcortical lesion and non-lesion areas. Transcriptomic changes associated with OL, microglia and astrocyte activation mapped predominantly to the rim areas of chronic active subcortical lesions11,39. In particular, lesion rim OLs28 showed molecular changes indicating cellular degeneration and iron overload. Notably, both stressed myelinating OLs and upper layer cortical projection neurons upregulated genes for self-antigen presentation to immune cells (B2M, HLA-C) suggesting processes perpetuate degeneration and inflammation40,41.
In another example of spatial diversity in MS, we detected distinct transcripts for cortical versus subcortical lesion astrocytes, indicating molecular differences in the tissue microenvironment. Further, we found that snRNA-seq can distinguish phagocytosing cells in MS based on their transport of ingested myelin transcripts into peri-nuclear structures or the nucleus itself. Future work is needed to determine whether this biology is beneficial or detrimental in disease course, e.g., by exacerbating inflammation. In summary, multilineage and spatial gene expression analysis indicates cell type-specific neuron vulnerability and glial activation patterns relevant to neurodegeneration and MS lesion progression.
Methods
Human tissue samples, ethical compliance and clinical information
All tissue included in this study was provided by the UK Multiple Sclerosis Tissue Bank at Imperial College, London, UK and the University of Maryland Brain Bank through the NIH NeuroBioBank. Human MS and control tissues were obtained via a prospective donor scheme following ethical approval by the National Research Ethics Committee in the UK (08/MRE09/31). We have complied with all relevant ethical regulations regarding the use of human postmortem tissue samples. We examined a total of 35 (19 MS and 16 controls) snap-frozen brain tissue blocks obtained at autopsies from 17 MS patients and 16 controls.
RNA extraction and integrity measurements
Frozen brain tissue samples encompassing the entire span of cortical GM with attached meninges and underlying subcortical WM were sectioned on a CM3050S cryostat (Leica Microsystems) to collect 100 μm-thick sections for total RNA and nuclei isolation. Total RNA from 10 mg tissue was isolated using Trizol (Invitrogen) and purified using the RNAeasy Kit (Qiagen) according to manufacturer’s instructions. Next, we performed RNA integrity analysis on the Agilent 2100 Bioanalyzer using the RNA 6000 Pico Kit (Agilent, 5067-1514). Only samples with an RNA integrity number (RIN) ≥ 6.5 were used to perform nuclei isolation followed by snRNA-seq as samples with lower RIN generated low quality data. As the result, we excluded 14 samples (7 MS and 7 control samples) and performed snRNA-seq on total of 12 snap-frozen brain tissue blocks obtained at autopsies from 7 female and 3 male MS patients (1 primary progressive MS, 9 secondary progressive MS; Supplementary table 1). The age of the MS patients ranged from 34 to 55 years (median 46 years), and the disease duration from 5 to 43 years (median 18 years). For control tissue, we included a total 9 snap-frozen brain tissue blocks obtained at autopsies from 4 female and 5 male individuals. The age of control patients ranged from 35 to 82 years (median 54 years; Supplementary table 1).
Nuclei isolation and snRNA-seq on the 10X Genomics platform
Control and MS samples were processed in the same nuclei isolation batch to minimize potential batch effects. 40 mg of sectioned brain tissue was homogenized in 5 mL of RNAse-free lysis buffer (0.32M sucrose, 5 mM CaCl2, 3 mM MgAc2, 0.1 mM EDTA, 10 mM Tris-HCl pH 8, 1 mM DTT, 0.1% Triton X-100 in DEPC-treated water) using a glass Dounce homogenizer (Thomas Scientific) on ice42. The homogenate was loaded into a 30 ml thick polycarbonate ultracentrifuge tube (Beckman Coulter). 9 ml of sucrose solution (1.8 M sucrose, 3 mM MgAc2, 1 mM DTT, 10 mM Tris-HCl in DEPC-treated water) was added to the bottom of the tube under the homogenate and centrifuged at 107,000 g for 2.5 hours at 4°C. Supernatant was aspirated, and nuclei pellet was incubated in 250 μL of DEPC-treated water-based PBS for 20 min on ice before resuspending the pellet. Nuclei suspension were analyzed for the presence of debris, nuclei were counted using a hemocytometer and diluted to 2,000 nuclei/μL before performing single-nuclei capture using the 10X Genomics Single-Cell 3’ system (Extended Data Fig. 1a). Target capture of 4,000 nuclei per sample was used. Control and MS samples were loaded on the same 10X chip to minimize potential batch effects. Single-nuclei libraries from individual samples were pulled and sequenced on the Illumina HiSeq 2500 machine. 10X nuclei capture and library preparation protocol was carried out according to the manufacturer’s recommendation without modification.
snRNA-seq data processing with 10X Genomics CellRanger software and data filtering
For library demultiplexing, fastq file generation, read alignment and unique molecular identifier (UMI) quantification, CellRanger software v 1.3.1 was used. CellRanger was used with default parameters, except for using pre-mRNA reference file (ENSEMBL GRCh38) to insure capturing intronic reads originating from pre-mRNA transcripts abundant in the nuclear fraction.
Individual expression matrices containing numbers of UMIs per gene in each nucleus were filtered to retain nuclei with at least 500 genes and 1000 transcripts expressed. Genes expressed in less than three nuclei were filtered out. Mitochondrial RNA genes were filtered out as well to exclude transcripts originating from outside the nucleus and avoid biases introduced by nuclei isolation and ultracentrifugation. Individual matrices were combined, UMIs were normalized to the total UMIs per nucleus and log-transformed.
Dimensionality reduction, clustering and t-SNE visualization
A filtered log-transformed UMI matrix containing genes expressed in more than five cells was used to perform truncated singular value decomposition (SVD) with k=50. A screen plot was generated to select the numbers of significant principle components (PCs) by localizing the last PC before the explained variance reaches plateau. This resulted in selection of 11 PCs. The significant PCs were used to calculate Jaccard distance-weighted nearest neighbor distances; number of nearest neighbors was assigned to root square of number of nuclei. The resulting graph with Jaccard-weighted edges was used to perform Louvain clustering43. To visualize nuclei transcriptomic profiles in two-dimensional space, t-distributed stochastic neighbor embedding (t-SNE) was performed44. Several original clusters expressed a combination of cell type markers, including interneuron subtypes, T cells, B cells, stromal cells and endothelial cells. These clusters were further subclustered by repeating PCA analysis of selected cell populations and performing partitioning around medoids (PAM) bi-clustering (Supplementary table 2).
Cell type annotation
Cell types were annotated based on the expression of known marker genes visualized by t-SNE plot, as well as by performing unbiased gene marker analysis (Supplementary table 3). For the latter, MAST was used to perform differential gene expression analysis by comparing nuclei in each cluster to the rest of nuclei profiles. Genes with a false discovery rate (FDR) <0.05 and at least two-fold gene expression upregulation were selected as cell type markers. Subtypes of projection neurons and interneurons were annotated based on combinatorial expression of inhibitory and excitatory markers and projection neurons and interneuron subtype markers.
Quantification of number of cell for cell types in MS and control samples
To get insight into enrichment or depletion of cell types in MS, numbers of nuclei in each cluster and individual were normalized to the total number of nuclei captured from each individual. The following formula was used:
Then, normalized cell numbers in each sample and cell type were compared between MS and control groups using Mann-Whitney test (Supplementary Table 4).
Differential gene expression analysis based on repeated down sampling
To estimate the degree of disease affection for different CNS cell types, the number of differentially expressed genes (DEG) between MS patients and controls was used as a surrogate parameter. We reasoned that the power to identify DEG is partially dependent on the number of cells detected in each cluster. Thus, we devised an analytical approach that corrects for cell count based on repeated down sampling to identical cell numbers for each donor-cluster combination. Specifically, 100 iterations of down sampling were performed, where 20 cells were randomly drawn from each donor for each cluster and combined into synthetic bulk samples as input for a differential gene expression analysis using DESeq2 version 1.20.045. In this case we favored a computationally less intensive analysis using DESeq2 without covariates on synthetic bulk samples over MAST to facilitate the execution of a sufficient number of iterations. The results of this screening approach were plotted as notched box plots and relevant differences between clusters were assumed where notches did not overlap (Fig. 1g). Notably, very small clusters which had less than 4 samples with a minimum of 20 cells available, were excluded from the analysis.
Trajectory pseudotime analysis
A single cell trajectory for excitatory cortical layer 2-3 neurons was determined and analyzed using the Monocle package version 3 alpha46. FDR-corrected p values were calculated using the Monocle 3alpha R package using 5,938 EN-L2-3 nuclei (Supplementary Table 5). Briefly, single cell transcriptomes of all CUX2-expressing cells were dimensionally reduced by principal component analysis (PCA) followed by uniform manifold approximation and projection (UMAP). Next, an unsupervised trajectory through the reduced space was identified using the SimplePPT algorithm. The root of the resulting tree was set to where most cells of the control samples clustered. Pseudotime values were then automatically assigned to each cell depending on its distance on the trajectory relative to the root node. Moran’s I test as implemented in Monocle 3 alpha was used to identify genes significantly regulated over pseudotime. For each gene the adjusted p value was signed by the direction of regulation determined by comparing expression in the first 5% of cells in pseudotime with the last 5%. The resulting gene list, ordered by signed adjusted p value, was the input for Gene Set Enrichment Analysis (GSEA) to test for enriched gene ontology (GO) terms using the clusterProfiler package version 3.10.147. Gene sets with a FDR < 0.05 were considered as significantly enriched. The results of the analysis were plotted as a GO term map using the emapplot() function of the clusterProfiler package to cluster terms based on their gene set relation. Clusters of gene sets were annotated with representative labels.
To analyze enrichment of individual gene sets on a single cell level, we employed the AUCell algorithm48. The algorithm measures geneset enrichment towards the top of an expression ranked gene list for each cell. The resulting area under the curve values (AUC) were plotted for all cells along pseudotime.
Differential gene expression analysis using linear mixed model regression
To identify genes differentially expressed in MS compared to control samples per cell type. P values were calculated and FDR-corrected using MAST R package. All nuclei from 9 control and 12 MS samples for corresponding cell types were used (Supplementary Table 6). MAST was used to perform zero-inflated regression analysis by fitting a linear mixed model. To exclude gene expression changes stemming from confounders, such as age, sex, RIN, cortical region, fractions of ribosomal and mitochondrial transcripts, 10X capture batch and sequencing batch, the following model was fit with MAST:
zlm(~diagnosis + sequencer + (1|ind) + cngeneson + age + sex + RIN + region + Capbatch + Seqbatch + ribo_perc + mito_perc, sca, method = "glmer", ebayes = F, silent=T)
Where cngeneson is gene detection rate (factor recommended in MAST tutorial), Capbatch is 10X capture batch, Seqbatch is sequencing batch, ind is individual label, ribo_perc is ribosomal RNA fraction and mito_perc is mitochondrial RNA fraction.
To identify genes differentially expressed due to the disease effect, likelihood ratio test (LRT) was performed by comparing the model with and without the diagnosis factor. Genes with at least 10% increase or decrease in expression in MS vs control and an FDR <0.05 were selected as differentially expressed. In addition, we calculated raw fold changes of gene expression by repeating MAST analysis with only the diagnosis factor in the model and filtered out genes with raw fold change of expression less than 7%. The latter filtering step allowed removing genes, whose fold change of expression was heavily dependent on the confounding factors, rather than clinical diagnosis.
Gene Ontology (GO) analysis for differentially expressed genes
PANTHER software (Broad Institute, https://software.broadinstitute.org/panther) was used to perform statistical overrepresentation tests for DEGs with respect to individual clusters. All genes expressed in a given cluster were used as a background list, and GO term analysis for enriched biological processes was performed. Processes with an FDR < 0.05 were considered and sorted by FDR.
Heatmap data presentation and hierarchical cluster analysis
Hierarchical clustering was performed with the online Morpheus software (Broad Institute, https://software.broadinstitute.org/morpheus) using 1-Pearson correlation as distance metric and complete clustering of rows (genes) and columns (cells or samples). To perform hierarchical clustering, we used single-nuclei gene expression matrix of cell type markers for lymphocytes, microglia and phagocytes (Extended Data Fig. 5, Supplementary Table 7).
Immunohistochemistry
16 μm-cryosections were collected on superfrost slides (VWR) using a CM3050S cryostat (Leica Microsystems) and fixed in either 4% PFA at room temperature (RT) or ice-cold methanol. Next, sections were blocked in 0.1M PBS/0.1% Triton X-100/ 10% goat/horse/donkey sera for 1 hour at RT. Primary antibody incubations were carried out overnight at 4°C. The following antibodies were used for immunohistochemistry: mouse anti-MOG (clone 8-18C5, 1:1,000 [1:200 after in situ hybridization], Millipore Sigma), rat anti-GFAP (clone 2.2B10, 13-0300, Thermo Fisher, 1:1,000 [1:200 after in situ hybridization]), rat anti-CD3 (clone CD3-12, Bio-Rad, 1:100), rabbit anti-MZB1 (polyclonal, Thermo Fisher, 1:1,000), rabbit anti-SKAP1 (polyclonal, Sigma Aldrich, 1:100), mouse anti-CD138 (clone DL-101, Biolegend, 1:100), mouse anti-CD68 (clone 514H12, Bio-Rad, 1:100), mouse anti-Neurofilament H (NF-H), nonphosphorylated (clone SMI32, 801701, Biolegend, 1:10,000), mouse anti-NeuN (clone MAB377, Sigma Aldrich, 1:1,000). After washing in 0.1M PBS, cryosections were incubated with secondary antibodies diluted in 0.1M PSB/ 0.1% Triton X-100 for 2 hours, RT. For chromogenic assays, sections were incubated with biotinylated secondary IgG antibodies (1:500, Thermo Fisher) followed by avidin-biotin complex for 1-hour incubation (1:500, Vector) and subsequent color revelation using diaminobenzidine according to the manufacturer’s recommendations (DAB, Dako). For immunofluorescence, Alexa fluochrome-tagged secondary IgG antibodies (1:500, Thermo Fisher) were used for primary antibody detection. Slides with fluorescent antibodies were mounted with DAPI Fluoromount-G (SouthernBiotech). Negative control sections without primary antibodies were processed in parallel. For diagnostic purposes, hematoxylin and eosin (HE) and Luxol fast blue (LFB) staining was carried out.
Iron staining
Tissue non-heme iron was stained according to previously published protocols49. Sections of fixed, frozen human tissue was allowed to warm to room temperature and dried for 15 minutes in a laminar flow hood. Endogenous peroxidase activity was quenched by immersion in a solution of 0.3% H2O2 (v/v) in methanol for 20 minutes and washed three times in deionized water (dH2O). Sections were then placed in a solution of fresh 1% (w/v) potassium ferrocyanide (Sigma-Aldrich, UK), pH 1 with HCL for 40 minutes, followed by three washes in dH2O. Sections were then placed in 0.01M NaN3, 0.3% H2O2 in methanol for 60 minutes, followed by three washes in PBS. Iron staining was intensified using 3’-diaminobenzidine (DAB) (10% v/v) solution from Pierce DAB substrate kit (Thermo Fisher) in PBS with 0.005% H2O2 (v/v) for five hours. DAB reaction was halted with three washes in PBS, 1 wash in 100% methanol and a further three washes in Bond Wash solution (Leica Biosystems).
Chromogenic single/duplex in situ RNA hybridization
Single molecule in situ hybridization was performed according to the manufacturer’s recommendations (RNAscope 2.5 brown, red and duplex chromogenic manual assay kits, Biotechne). Sequences of target probes, preamplifier, amplifier, and label probes are proprietary and commercially available (Advanced Cell Diagnostics). Typically, target probes contain 20 ZZ probe pairs (approx. 50 bp/pair) covering 1,000 bp. The following human manual RNAscope assay probes were used: CUX2, RORB, TLE4, THY1, VIP, SST, HSP90AA1, LINC00657, PPIA, FTL, B2M, PIEZO2, IGHG1, MSR1, LINC01088, GPC5, CD44, BCL6, FOS, EDNRB, ST18, RUNX1-C2, SLC1A2-C2, CD44-C2, RFX4-C2, PDGFRA-C2, SYT1-C2, PLP1-C2. Following red chromogenic single-molecule in situ hybridization, we performed immunohistochemistry using either chromogenic or fluorescence assays (see above). After duplex single molecule in situ hybridization we performed hematoxylin staining of nuclei.
Fluorescence multiplex in situ RNA hybridization and human brain tissue optimization
For small molecule fluorescence in situ RNA hybridization (smFISH) on human brain cryosections performed on an automated BOND RX robotic stainer (Leica), the following procedure was used. Fresh snap frozen human brain tissue was cryosectioned and slides immediately stored at -80°C. Because human brain tissue often showed high levels of autofluorescence, several treatments were needed to minimize interference with FISH signals. Using spectral analysis, we identified sources of autofluorescence to be mainly lipofuscin in grey matter (emission wavelengths ~450-650nm) and collagen/elastin in white matter (emission wavelengths ~470-520nm). Initial experiments in fixed tissue also displayed low levels of formaldehyde-induced fluorescence in the green yellow spectra (~420-470nm); thus, sections of human brain tissue were not formaldehyde-fixed before storage. On the day of the experiment, with minimal exposure to (RT) air to keep oxidation of endogenous fluorescent proteins low, slides where directly transferred from -80°C into pre-chilled PFA 4% (methanol-free). Following 45 minutes incubation sections where immediately submerged in boiling citrate buffer (pH 3.0, Sigma) for 15 minutes to loosen up the recent crosslinking. Slides where then rinsed twice in PBS and dehydrated. To avoid interference of background fluorescence, experiments were designed so that low expressing probes were detected using fluorophores with low background, i.e., Opal 570, Opal 650. All samples in this study were treated in the same way regardless of disease/stage and age. The assay was then performed for 2-3 genes by FISH using the RNAScope LS Multiplex Assay (Biotechne).
Samples were initially permeabilized with heat and protease treatment to improve probe penetration and hybridization. For heat treatment, samples were incubated in BOND ER2 buffer (pH 9.0, Leica) at 95°C for 10 minutes. For protease treatment, samples were incubated in ACD protease reagent at 42°C for 10 minutes. Prior to probe hybridization, samples were incubated in hydrogen peroxide for 10 minutes to inactivate endogenous peroxidases and ACD protease. Subsequently, samples were incubated in target z-probe mixtures (C1-C4) for 2 hours at 42°C. Each slide wash flushed three times in order to obtain optimal hybridization to transcripts. The following human RNAScope LS assay probes were used: SYT1, CUX2, LINC00657, B2M, CD74, RUNX1, RFX4, SLC1A2, CD44, PLP1, MBP.
Following hybridization, branched DNA amplification trees were built through sequential incubations in AMP1, AMP2 and AMP3 reagents for 15-30 minutes each at 42°C with LS Rinse buffer (Leica) high stringency washes between incubation steps. After amplification, probe channels were detected sequentially via HRP-TSA labeling. Here, samples were incubated in channel-specific HRP reagents for 15 minutes at 42°C, TSA fluorophores for 30 minutes and HRP blocking reagent for 15 minutes at 42°C. Probes were labeled using Opal 520, 570 and 650 TSA fluorophores (Perkin Elmer, 1:300). Directly following FISH assay, localization of MOG myelin protein was performed by BOND RX assisted IHC, where samples were incubated with anti-MOG antibody in blocking solution for 1 hour (1:200). To develop the antibody signal, samples were incubated in donkey anti-mouse HRP (Abcam, ab205719, 1:500) for 1 hour, TSA-biotin (PE, NEL700A001KT, 1:200) for 10 minutes and streptavidin-conjugated Alexa 700 (Sigma, 1:200) for 30 minutes.
PCR for myelin and neuron transcripts from rat myelin preparations
RNA from myelin was purified using phenol-chloroform extraction by adding 100 μl of chloroform (Sigma Aldrich) to 500 μl of Tri-Reagent containing 50 μl of enriched rat CNS myelin. Samples were vortexed and centrifuged at 12,000 gmax for 15 minutes at 4°C. The upper aqueous phase was collected and an equal volume of 70% ethanol added and vortexed. RNA was purified using PureLink RNA Mini-Kit (Thermo Fisher) according to manufactures instructions. RNA was eluted with 30 μl of RNAse free water and concentration determined using a SPECTROStar Nano. cDNA was synthesized from 0.3 μg of RNA using SuperScript III (Thermo Fisher) according to manufactures instructions with or without inclusion of RT enzyme. PCRs for rat Mbp and synaptophysin (Syp) were performed using 20 μl of PCR MegaMix Blue (Client Life Science), 1 μl of cDNA and 0.5 μl of 10 μM forward (F) and reverse (R) primers in an ABI Veriti 96 Well thermal cycler (Thermo-Fisher) for 30 cycles at 950C, 720C and 540C:
Mbp-F: GTGGTATGTGAGCACAGGCT
Mbp-R: TAAAAGCACCTGCTCTGGGG
Syp-F: TGCCATCTTCGCCTTTGCTA
Syp-R: GCCTGTCTCCTTGAACACGA
Amplified products were loaded onto 1% E-Gel (Thermo-Fisher) according to manufactures instructions and imaged using E-Gel imager (Thermo-Fisher).
Western blot and Coomassie staining for myelin and neuron protein from rat myelin preparations
15-20 μg of protein were separated on 4-12% Bis-Tris NuPAGE gels (Thermo-Fisher) according to manufactures instructions. Gels were either stained for total protein using 0.3% w/v brilliant blue-G (Sigma) in 40% v/v methanol and 7% v/v glacial acetic acid overnight. Destaining was done with several washes in 40% v/v methanol and 7% v/v glacial acetic. For Western blotting, proteins in gels were transferred onto PVDF membranes (Millipore) using Bolt transfer buffer (Thermo-Fisher) for 1 hour at 15 V constant voltage. Membranes were blocked with Li-Cor Blocking Buffer (Li-Cor) for 1 hour at room temperature on a platform shaker. Membranes were incubated overnight in primary antibodies (1:2000 dilution) rabbit-anti-Mbp or mouse-anti-Mog (Cell Signaling), rabbit-anti Neurofilament heavy or mouse anti-synaptophysin (Sigma) in 50% Li-Cor Blocking buffer in tris buffered saline with tween-20 (0.001% v/v) (TBS-T). Membranes were washed three times with TBS-T and Li-Cor 680-RD secondaries (1:5000) (Li-Cor) applied in 50% v/v Li-Cor Blocking Buffer in TBS-T for 1 hour at room temperature. Membranes were three times in TBS-T and imaged on a Li-Cor Odyssey (Li-Cor).
Myelin Enrichment assay and polymerase chain reaction
Myelin enrichment of adult rat CNS was performed according to Jahn et al.50. Unless otherwise stated all buffers were prepared in DEPC treated water and all procedures carried out at 4°C. Briefly, adult rats were perfused with saline and the brains rapidly dissected, olfactory bulbs removed and kept on ice. Brains were cut into hemispheres, and one hemisphere was used for each preparation. Hemispheres were homogenized using a glass Dounce in 6 ml of 0.32 M sucrose prepared in DEPC treated water with HALT protease inhibitor cocktail without EDTA (Thermo Fisher). 1ml of homogenate was retained for further biochemical analysis and 6ml of homogenate loaded on top of 6ml of 0.85 M sucrose treated with DEPC with HALT protease inhibitors in 14ml thin walled centrifuge tubes (Beckman Coulter, UK). Samples were centrifuged at 75,000 gmax for 35 minutes at 4°C. The interface between 0.85 and 0.32 M sucrose was collected, resuspended in water and centrifuged at 75,000 gmax for 15 minutes at 4°C. The pellet was subjected to two rounds of osmotic shock by resuspension in water, left on ice for 10 min and centrifuged at 12,500 gmax for 15 minutes at 4°C. The pellet was resuspended in 6 ml of 0.32M sucrose and overlaid on a bed of 0.85 M sucrose and centrifuged at 75,000 gmax for 35 minutes at 4°C. The purified myelin was collected from the 0.32M and 0.85M sucrose interface. The myelin was then washed in 10 ml of water and centrifuged at 75,000 gmax for 35 minutes at 4°C and resuspended in either 0.25 M bicarbonate pH 8.3 or Tris Buffered Saline (TBS) pH 7.4 to final volume of 400 μl. A 50 μl was retained and 500 μl of Tri-Reagent (Thermo Fisher) was added to preserve the RNA. Protein concentration was measured using Pierce-BCA Protein Assay (Thermo Fisher) according to manufactures instructions.
Animals used for myelin-microglia engulfment assays
Wildtype C57Bl/6J mice (stock #000664) were obtained from Jackson Laboratories (Bar Harbor, ME). All animal experiments were carried out at the animal facility at the University of Massachusetts Medical School and approved (#A-2496-17) by Animal Care and Use Committees (IACUC) and performed under NIH guidelines for proper animal welfare.
Purification and treatment of primary mouse microglia
Purified primary brain-derived microglia were obtained from mixed glial cultures by modified standard protocols as described before51. Briefly, cerebral cortices from male and female postnatal day 0.5 C57Bl/6J wildtype mice were dissected free of meninges, chopped into small pieces and mechanically dissociated until a single cell suspension was obtained. Cells were then seeded in 10 ml DMEM (Thermo Fisher) supplemented with 10% FBS (Life Technologies) and 1% penicillin-streptomycin (Life Technologies) at a density of one brain per 75cm2 flask, and cultured for 7 days at 37°C in humidified 5% CO2/95% air. By shaking the culture flasks for 3 hours at 180 rpm, loosely adhering microglia were detached. The suspended microglial cells were seeded onto glass coverslips at a density of 80.000 cells/well in a 24-well plate and cultured overnight. 24 h prior to treatment cell culture medium was changed to neurobasal medium supplemented with 1x sodium pyruvate, 1x B27 (all from Thermo Fisher), 1x GlutaMAX, 1x penicillin-streptomycin (both from Life Technologies), 5 μg/ml insulin, 1x SATO, 5 μg/ml N-acetyl-L-cysteine, 40 ng/ml T3 (all from Sigma), and 10 ng/ml mouse macrophage colony stimulating factor (Shenandoah). Finally, microglia were treated with 6.45 μg purified myelin fraction from rat brain for 4 hours, before myelin was removed and cells were fixed or harvested for analysis at the indicated time points. To visualize engulfment of myelin proteins into microglial lysosomes, myelin protein was labeled with pHrodo (Thermo Fisher) under RNase free conditions according to the manufacture’s recommendations prior to treatment.
Mbp RNA hybridization on primary mouse microglia
RNA in situ hybridization was performed according to the manufacturer’s recommendations (ACDBio). Briefly, after treatment, cells were fixed with 4% PFA, dehydrated and stored at -20°C for up to 7 days before further use. Prior to RNA hybridization, cells were rehydrated, rinsed in PBS and treated with 1:15 diluted “Protease III” for 15 minutes at 40°C. Then, probes against Mbp (ACDBio) were added and incubated for 2 hours at 40°C. Subsequent amplification steps were performed according to the manufacturer’s instructions. To confirm specificity of RNA signals, some samples were treated with 10 mg/ml RNaseA (Thermo Fisher Scientific) for 1h at 37°C prior to incubation with probes. To immunostain samples following Mbp RNA hybridization, cells were washed in PBS, blocked in 2% normal goat serum supplemented with 0.01% TritonX-100 for 30 minutes and incubated with the following primary antibodies: rabbit polyclonal anti-Iba1 (Wako Chemicals) and rat monoclonal anti-CD68 (clone FA-11, AbD Serotec, MCA1957) (both 1:100). The following day, cells were incubated with appropriate Alexa-fluorophore-conjugated secondary antibodies (Thermo Fisher Scientific) and mounted with vectashield containing DAPI (Vector laboratories). Random 63x fields of all cultures were imaged using identical settings on a Zeiss Observer Spinning Disk Confocal microscope equipped with diode lasers (405nm, 488nm, 594nm, 647nm) and Zen Blue acquisition software (Zeiss). For unbiased quantification of Mbp puncta, signals were co-localized to Iba1+ microglia blind to treatment of the samples and the number of total puncta as well as signals associated with microglia nuclei (0.5μm distance from the nucleus) were determined using ImageJ (NIH). Moreover, 630x z-stacks were acquired with 35-50 steps at 0.22 μm spacing and processed in Imaris (Bitplane, Switzerland) to 3D surface render engulfed signals as previously described.
RNA isolation and quantitative reverse transcriptase polymerase chain reaction
Total RNA from microglia was extracted using TRIzol (Life Technologies) acco52rding to manufacturer’s recommendations. 500 ng total RNA samples were transcribed into cDNA using Power SYBR™ Green Cells-to-CT Kit (Thermo Fisher Technologies) according to manufacturer’s instructions. Relative Cd163 and P2ry12 expression was determined by quantitative polymerase chain reaction (qPCR) in relation to Gapdh housekeeping gene expression using the following forward (F) and reverse (R) primers:
Cd163-F: GGGTCATTCAGAGGCACACTG
Cd163-R: CTGGCTGTCCTGTCAAGGCT
P2ry12-F: GTTCTACGTGAAGGAGAGCA
P2ry12-R: CTACATTGGGGTCTCTTCGC
Gapdh-F: TGTCCGTCGTGGATCTGAC
Gapdh-R: CCTGCTTCACCACCTTCTTG
Human tissue sampling for primary human microglia assays
Human brain tissue was obtained with informed consent under protocol REC 16/LO/2168 approved by the NHS Health Research Authority. Adult human brain tissue was obtained from three biopsies (age 17, male, diffuse axonal injury, right frontal lobe; age 61, male, unruptured cerebral aneurysm, right gyrus rectus; age 70, male, normal pressure hydrocephalus, right parietal lobe) taken from the site of neurosurgery resection for the original clinical indication. Tissue was transferred to Hibernate A low fluorescence (HALF) supplemented with 1x SOS (Cell Guidance Systems), 2% Glutamax (Life Technologies), 1% P/S (Sigma), 0.1% BSA (Sigma), insulin (4g/ml, Sigma), pyruvate (220 g/ml, Gibco) and DNase 1 Type IV (40 g/ml, Sigma) on ice and transported to a dedicated BCL 2 laboratory.
Dissociation of human brain tissue and purification of human microglia
Brain tissue was mechanically digested in fresh ice-cold HALF supplemented with 1x SOS (Cell Guidance Systems), 2% Glutamax (Life Technologies), 1% P/S (Sigma), 0.1% BSA (Sigma), insulin (4g/ml, Sigma), pyruvate (220 g/ml, Gibco) and DNase 1 Type IV (40 g/ml, Sigma). The prepared mix was spun in HBSS+ (Life Technologies) at 300g for 5 mins and supernatant discarded. The digested tissue was rigorously triturated at 4°C and filtered through a 70μm nylon cell strainer (Falcon) to remove large cell debris and undigested tissue. Filtrate was spun in a 22% Percoll (Sigma) gradient with DMEM F12 (Sigma) at 800g for 20 minutes. Supernatant was discarded and the pellet was re-suspended in ice cold supplemented HALF. The isolated cell suspension was incubated with anti-CD11b conjugated magnetic beads (Miltenyi) for 15 minutes at 4°C. Cells were washed twice with supplemented HALF and passed through an MS column (Miltenyi). Each sample was washed three time in the column and then extracted. Cells were plated in DMEM F12 with 10% foetal bovine serum and 0.1% Macrophage colony-stimulating factor (M-CSF). Note that incubators for all cell culture incubators are regularly tested for mycoplasma contamination.
Mbp and Plp1 RNA hybridization on primary mouse microglia
Purified rat brain myelin extracts from three biological replicates were diluted to 1 mg/mL (total protein) in 0.1 M sodium bicarbonate buffer, pH 8.3 in a volume of 100 μl. The pH sensitive fluorescent dye succinimidyl ester known as pHrodoRed (Thermo Fisher) was added from a 10 mM stock in DMSO to a final of 100 μM to each myelin extract for 45 minutes at room temperature. Samples were centrifuged for 30 minutes at 17,000x g at 4°C and the supernatant discarded. The labelled myelin pellets were resuspended in 100 μl of 0.1 M sodium bicarbonate buffer, pH 8.3 to a final of 1mg/ml of protein and 1.5 μl added to wells of human microglia in 24 well glass bottom plate (Cellvis) for phagocytosis over 18 hours. The next day, the cells were washed twice with PBS before fixation with 4% PFA at room temperature for 10 minutes and washing with PBS.
Cells were manually stained for RNA using RNAScope using a modified automated procedure for the Leica BOND RX (Leica). Fixed cells were washed twice with BOND wash solution (Leica) before antigen retrieval with BOND Epitope Retrieval Solution 2 (Leica) at 95°C and allowed to cool to room temperature, and followed by three washes with BOND wash. Cells were permeabilized with 0.5x RNAScope 2.5 LS Protease III (Biotechne) in PBS at 37°C for 5 min, followed by cold BOND wash (40°C) and then two more BOND washes at room temperature. Endogenous peroxidase activity was quenched with RNAScope 2.5 LS Hydrogen Peroxide (Biotechne) for 10 minutes and followed by two more BOND washes. RNAScope probes for mouse Mbp (Biotechne) and Plp-1 (Biotechne) were diluted 1:50 in C1 probe. Mbp and Plp-1 probes were amplified using sequential treatments with RNAScope LS Multiplex AMP 1, 2 and 3 (Biotechne) for 30 minutes at 42°C with BOND washing and RNAScope 2.5 LS Rinse Reagent (Biotechne) for 5 minutes each between each amplification step. Probe channel C2 for Mbp was fluorescently developed using RNAScope Multiplex HRP-C2 (Biotechne) for 15 minutes at 42°C, followed by BOND washes and incubation with tyramide-conjugated Opal 520 dye at 1: 2,500 (Perkin Elmer) for 30 minutes followed with two more BOND washes. Residual HRP activity was quenched with RNAScope LS Multiplex HRP Blocker (Biotechne) for 15 minutes at 42°C, followed by BOND washes. Probe channel C3 for Plp-1 was developed as for C2 but using RNAScope LS Multiplex HRP-C3 (Biotechne) and Opal 650 dye at 1:2,500 (Perkin Elmer, UK) and followed by RNAScope LS Multiplex HRP Blocker (Biotechne) with BOND washing. Staining of LMNA/C (Laminutes A/C) was done after RNAScope development by incubating cells with mouse anti-porcine Laminutes A/C antibody (Insight Biotechnology, UK) at 1:200 and rabbit anti-Iba1 biotin conjugated antibody at 1:200 for 60 minutes at RT. Excess primary antibodies were washed away with BOND wash and cells incubated goat anti-mouse IgG2B AlexaFlour 350 at 1:500 and streptavidin-conjugated AlexaFlour 700 at 1:1,000 for 60 minutes at RT. Cells were wash three times in BOND wash and twice in PBS before imaging on an Operetta CLS (Perkin-Elmer) spinning disk confocal microscope.
Image acquisition and analysis of human IHC and ISH experiments
Bright field images were acquired on Zeiss Axio Imager 2 and Leica DMi8 microscopes equipped with Zeiss Axiocam 512 color and Leica DMC5400 cameras. Fluorescent images were taken using Leica TCS SP8 and TCS SPE laser confocal and DMi8 widefield (equipped with Leica DFC7000 GT camera) microscopes with either 10x, 20x, 40x or 63x objectives; all fluorescent confocal pictures are Z-stack images, unless stated otherwise. High-resolution FISH images of human tissue sections were acquired on a spinning disk Operetta CLS (Perkin Elmer) in confocal mode using a sCMOS camera and a 40X NA 1.1 automated-water dispensing objective. The field-of-view was 320 x 320 μm and voxel size 0.3 x 0.3 x 1 μm. Each field was imaged as a z-stack consisting of 20 to 30 planes with a 1 μm step size. z-heights of tissue sections were manually identified by imaging DAPI on sample fields prior to tissue-wide scans. Each z-plane was imaged across 5 channels depending on the experiment with exposure between 60 and 120 ms at 90% LED power. 3D projections were generated using raw imaging data in Volocity 6.3 software (Perkin Elmer). Images were processed using Fiji ImageJ or Photoshop software (Adobe) and exported to Illustrator vector-based software (Adobe) for figure generation.
Statistical Analysis
Data are presented as mean ± standard error of mean (SEM). Analyses were performed using two-tailed parametric or non-parametric (Mann-Whitney, Kruskal-Wallis) t-tests for two groups and if applicable, one-way ANOVA with corresponding post-hoc tests for multiple group comparisons. P values were designated as follows: *p ≤ 0.05, **p ≤ 0.01, *** p ≤ 0.001, **** p ≤ 0.0001. Analyses were performed using GraphPad Prism (GraphPad Software).
Extended Data
Extended Data Fig. 1. Sample and disease contribution of cell types captured by snRNA-seq.

(a) Representative images selected from nuclei suspensions (ctrl, n=9; MS, n=12) after ultracentrifugation and before capturing by 10X Genomics confirming DAPI nuclear counterstaining with presence of smaller and larger DAPI+ nuclei. Note that larger nuclei are co-stained with anti-NeuN antibody confirming neuronal origin (white arrowheads). (b) Colored t-SNE plots showing numbers of genes (left) and UMIs (right) per captured nuclei from control and MS samples. (c) Colored t-SNE plot visualizing nuclei from different lesion stages based on classic pathological MS lesion staging. (d) Colored t-SNE plots visualizing nuclei from samples with different levels of upper and deep layer cortical demyelination as well as subcortical demyelination. (e) Representative tSNE plots with cell-type specific marker genes for OL progenitor cells, stromal cells including pericytes, endothelial cells, and leukocytes. For tSNE plots, data shown from 9 control and 12 MS samples and a total of 48,919 nuclei.
Extended Data Fig. 2. Molecular changes in cortical neuron subtypes in MS lesions.

(a) NORAD and PPIA expression patterns in cortical neurons and selected glial subtypes. Note baseline expression of NORAD and PPIA in neuronal versus glial subtypes and preferential upregulation of both NORAD and PPIA in upper cortical layer excitatory neurons (EN-L2-3 and EN-L4) in MS lesion tissue versus deep cortical layer excitatory and inhibitory neurons (EN-L5-6 and IN-SST). For all tSNE and violin plots, data are shown from 9 control and 12 MS samples. For tSNE plots, data from 48,919 nuclei are shown. For EN-L2-3, EN-L4 and EN-L5-6 violin plots, data shown from 6,120, 3,125 and 3,058 nuclei. Box plots inside violin plots represent median and standard deviation of gene expression. (b) Visualization of enriched GO terms in EN-L2-3, EN-L4 and EN-L5-6 cells based on differential gene expression analysis (linear mixed model regression). Binomial test with FDR correction was utilized to calculate FDR-corrected p values using genes differentially expressed in EN-L2-3, EN-L4 and EN-L5-6 nuclei (n= 428, 364 and 327).
Extended Data Fig. 3. Cortical neuron and lymphocyte subtype analysis in MS lesions.

(a) tSNE plots for neuron subtype specific expression of RORB, THY1, NRGN, SST, SV2C and PVALB (left). LaST (ctrl, n= 5) showing layer-specific expression of neuronal RORB in intermediate cortical layer 4 and widespread expression of pyramidal neuron marker THY1 with enrichment in layer 5; note that SST-expressing interneurons preferentially map to deep cortical layers. Co-expression studies (ctrl, n= 5) with SYT1 confirm neuronal expression of RORB, THY1 and SST (black arrowheads). (b) Heatmap with hierarchical clustering of lymphocyte-associated transcripts allowing sub clustering of lymphocytes in T cells, B cells and plasma cells based on marker gene expression (upper left). tSNE plots for typical B/plasma cell and T cell marker genes enriched in lymphocyte clusters (upper right). IHC for T cell marker SKAP1 (black arrowheads mark SKAP1+ T cells) together with spatial transcriptomics for B cell-associated IGHG1 encoding immunoglobulin G1 (IgG1) (magenta-colored arrowheads; lower left); note preferential clustering of plasma cell-associated MZB1+ and IGHG1-expressing B cells (white arrowheads, lower right) in inflamed meningeal tissue versus mixed T and B cell infiltration in perivascular cuffs of subcortical lesions (lower panels). One caveat to these findings is the relatively small number of MS cases samples, which limited our ability to cluster T cell populations. For tSNE plots (a, b) and hierarchical clustering (b), data shown from 9 control and 12 MS samples. For tSNE plots, data shown for all 48,919 nuclei; for hierarchical clustering, data shown from 53 nuclei in the B cell cluster. For ISH and IHC experiments in b, representative images shown from individual tissue sections (ctrl, n= 4; MS, n= 7).
Extended Data Fig. 4. Astrocyte and oligodendrocyte cluster analysis and spatial transcriptomics in MS lesions.

(a) Differential spatial expression patterns of astroglial GFAP in subcortical versus cortical demyelination by IHC (left); tSNE plots visualizing astrocyte specific genes corresponding to all (RFX4) protoplasmic (SLC1A2, GPC5) and fibrous/reactive astrocytes (GFAP, CD44). Quantification of RFX4+ ISH signals per nuclei in GM and WM of control samples validates RFX4 as a canonical astrocyte marker (ctrl, n= 5); quantification of GPC5+ and CD44+ ISH signals per RFX4+ astrocytes confirms validates GPC5 as protoplasmic GM and CD44 as fibrous WM marker. Two-tailed Mann-Whitney tests were performed. Data presented as mean ± SEM. (b) Upregulation of astroglial CRYAB, MT3 (black arrowheads) and endothelin type B receptor transcript EDNRB (white arrowhead) in reactive astrocytes in subcortical lesions. (c) tSNE plots showing OL-specific expression of myelin genes MBP, CNP and transcription factor ST18; note co-expression of ST18 with PLP in control WM by ISH. (d) Visualization of enriched GO terms in myelinating OLs based on differential gene expression analysis. Binomial test with FDR correction was utilized to calculate FDR-corrected p values using 151 genes differentially expressed in OLs. (e) Co-expression spatial transcriptomic studies confirming upregulation of heat shock protein 90 transcript HSP90AA1 in both progenitor (PDGFRA-expressing) and myelinating (PLP1-expressing) OLs at lesion rims (PPWM, black arrowheads). For tSNE and violin plots, data shown from 9 control and 12 MS samples. For astrocyte violin plots, 1,571 control and 3,810 MS nuclei are shown. Box plots inside violin plots represent median and standard deviation of gene expression. For ISH and IHC experiments, representative images from from 3 control and 4 MS individual tissue sections are shown.
Extended Data Fig. 5. Cluster analysis of activated and phagocytosing microglia subtypes.

Hierarchical cluster analysis identifies several homeostatic and activated MS-specific microglia subtypes according to inflammatory lesion stages allowing transcriptomic staging of microglia subtypes. Clusters with enriched genes are marked and annotated a-f (see Supplementary Table 8 for gene list). Note that phagocytosing cells are identified by presence of OL/myelin genes (cluster “f” on bottom of heatmap).
Extended Data Fig. 6. PCR for rat Mbp from myelin preparation.

(a) Representative Coomassie stain of brain homogenate (Hom.) and purified myelin (P.M.) from adult rat brain (left). Western blots for myelin basic protein (Mbp), myelin oligodendrocyte glycoprotein (Mog), synaptophysin (Syp) and neurofilament heavy molecular weight (NF-H) (center). PCRs of myelin basic protein (Mbp) and synaptophysin (Syp) transcripts in brain homogenate and purified myelin fractions (right). (b) Densitometric quantification of myelin and homogenates prepared from n= 4 independent rat hemispheres for Coomassie (total protein), Western blot proteins and PCRs shown in (a) of purified myelin fractions normalized to their respective homogenates. Data is shown as median and error bars ± standard error of the mean of the 4 biological replicates. Similar results were obtained with Hom. and P.M. fractions not used in this study. P values calculated from Students’s two tailed t-test with Welch's correction and p values less than 0.05 considered significant.
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Acknowledgements
We thank Jason Cyster (UCSF, San Francisco, USA), Daniel Reich (NIH, Bethesda, USA) and Sarah Teichmann (Wellcome Sanger Institute, Hinxton, UK) for advice and comments on the manuscript, Irina Pshenichnaya for technical assistance and Anna Hupalowska for figure illustrations. Djordje Gveric and Alexandra LeFevre provided human brain samples from the UK Multiple Sclerosis Tissue Bank, funded by the Multiple Sclerosis Society of Great Britain and Northern Ireland, and the National Institutes of Health (NIH) NeuroBioBank at the University of Maryland, respectively. L.S. was supported by postdoctoral fellowships from the German Research Foundation (DFG, SCHI 1330/1-1) and the National Multiple Sclerosis Society (NMSS) funded in part by the Dave Tomlinson Research Fund (FG-1607-25111). D.V. was supported by a BOLD & BASIC fellowship from the UCSF Quantitative Biosciences Institute. S.W. was supported by a postdoctoral fellowship from the DFG (WE 6170/1-1), and S.M. was supported by EMBO (ALTF_393-2015) and DFG (MA 7374/1-1). A.B. acknowledges an NIH postdoctoral fellowship (F32NS103266). D.H.R. is a Paul G. Allen Frontiers Group Distinguished Investigator. This work was funded by the Dr. Miriam and Sheldon G. Adelson Medical Research Foundation (D.H.R., D.P.S., R.J.M.F.), the Hertie Foundation (medMS-MyLab program; P1180016 to L.S.), the National Human Genome Research Institute (4U41HG002371 to M.H.), the California Institute for Regenerative Medicine (GC1R-06673-C to M.H.), the Silicon Valley Community Foundation (2018-182809 to M.H.), the NMSS (PP-1609-25953, D.H.R.), the NIHR Cambridge Biomedical Research Center (D.H.R.), and grants from the NIH/NINDS (NS040511 to D.H.R., R35NS097305 to A.R.K.), European Research Council and the Wellcome Trust (to D.H.R).
Footnotes
Data Availability
All raw snRNA-seq data (fastq files) were deposited to the Sequence Read Archive (SRA), accession number PRJNA544731 (NCBI Bioproject ID: 544731).
Author contributions
L.S., D.V., A.R.K. and D.H.R. designed, coordinated and interpreted all studies and wrote the manuscript. L.S. and R.R. selected control and MS samples. L.S., D.V. and D.J. performed snRNA-Seq assisted by B.T. and N.G. D.V. and M.K. performed regression and trajectory analysis of single cell data, assisted by A.B. and J.B.E., who modified analytical scripts with oversight from M.F., A.R.K. and D.H.R.. S.H., L.S., D.J., S.V. and S.M performed smFISH with oversight from O.A.B.. S.W., J.H.S., A.Y. and M.S. conducted mouse and human myelin-microglia engulfment assays and analysis, supervised by D.P.S. and R.J.M.F.. L.R.S. analyzed findings related to immune cells. M.H. generated the single-cell web browser to visualize control and MS sequencing data. All coauthors read, revised and approved the manuscript. D.H.R. and A.R.K. supervised all experiments.
Author information
The authors state no relevant competing interests or disclosures.
References
- 1.Collaborators, G. D. a. I. I. a. P. Global, regional, and national incidence, prevalence, and years lived with disability for 310 diseases and injuries, 1990-2015: a systematic analysis for the Global Burden of Disease Study 2015. Lancet. 2016;388:1545–1602. doi: 10.1016/S0140-6736(16)31678-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Reich DS, Lucchinetti CF, Calabresi PA. Multiple Sclerosis. New England Journal of Medicine. 2018;378:169–180. doi: 10.1056/NEJMra1401483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lassmann H. Multiple Sclerosis Pathology. Cold Spring Harbor perspectives in medicine. 2018;8 doi: 10.1101/cshperspect.a028936. a028936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Trapp BD, et al. Axonal transection in the lesions of multiple sclerosis. The New England journal of medicine. 1998;338:278–285. doi: 10.1056/NEJM199801293380502. [DOI] [PubMed] [Google Scholar]
- 5.Schirmer L, Antel JP, Brück W, Stadelmann C. Axonal loss and neurofilament phosphorylation changes accompany lesion development and clinical progression in multiple sclerosis. Brain pathology (Zurich, Switzerland) 2011;21:428–440. doi: 10.1111/j.1750-3639.2010.00466.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Peterson JW, Bo L, Mork S, Chang A, Trapp BD. Transected neurites, apoptotic neurons, and reduced inflammation in cortical multiple sclerosis lesions. Annals of neurology. 2001;50:389–400. doi: 10.1002/ana.1123. [DOI] [PubMed] [Google Scholar]
- 7.Magliozzi R, et al. Meningeal B-cell follicles in secondary progressive multiple sclerosis associate with early onset of disease and severe cortical pathology. Brain : a journal of neurology. 2007;130:1089–1104. doi: 10.1093/brain/awm038. [DOI] [PubMed] [Google Scholar]
- 8.Magliozzi R, et al. A Gradient of neuronal loss and meningeal inflammation in multiple sclerosis. Annals of neurology. 2010;68:477–493. doi: 10.1002/ana.22230. [DOI] [PubMed] [Google Scholar]
- 9.Lucchinetti CF, et al. Inflammatory cortical demyelination in early multiple sclerosis. The New England journal of medicine. 2011;365:2188–2197. doi: 10.1056/NEJMoa1100648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Trapp BD, et al. Cortical neuronal densities and cerebral white matter demyelination in multiple sclerosis: a retrospective study. Lancet neurology. 2018;17:870–884. doi: 10.1016/S1474-4422(18)30245-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dal-Bianco A, et al. Slow expansion of multiple sclerosis iron rim lesions: pathology and 7 T magnetic resonance imaging. Acta neuropathologica. 2017;133:25–42. doi: 10.1007/s00401-016-1636-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mainero C, et al. A gradient in cortical pathology in multiple sclerosis by in vivo quantitative 7 T imaging. Brain : a journal of neurology. 2015;138:932–945. doi: 10.1093/brain/awv011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Jäkel S, et al. Altered human oligodendrocyte heterogeneity in multiple sclerosis. Nature. 2019;566:543–547. doi: 10.1038/s41586-019-0903-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Masuda T, et al. Spatial and temporal heterogeneity of mouse and human microglia at single-cell resolution. Nature. 2019;566:388–392. doi: 10.1038/s41586-019-0924-x. [DOI] [PubMed] [Google Scholar]
- 15.Lake BB, et al. Neuronal subtypes and diversity revealed by single-nucleus RNA sequencing of the human brain. Science (New York, N.Y.) 2016;352:1586–1590. doi: 10.1126/science.aaf1204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lodato S, Arlotta P. Generating neuronal diversity in the mammalian cerebral cortex. Annual review of cell and developmental biology. 2015;31:699–720. doi: 10.1146/annurev-cellbio-100814-125353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lee S, et al. Noncoding RNA NORAD Regulates Genomic Stability by Sequestering PUMILIO Proteins. Cell. 2016;164:69–80. doi: 10.1016/j.cell.2015.12.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mus E, Hof PR, Tiedge H. Dendritic BC200 RNA in aging and in Alzheimer's disease. Proc Natl Acad Sci U S A. 2007;104:10679–10684. doi: 10.1073/pnas.0701532104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bayraktar OA, et al. Single-cell in situ transcriptomic map of astrocyte cortical layer diversity. bioRxiv. 2018 doi: 10.1101/432104. 432104. [DOI] [Google Scholar]
- 20.Machado-Santos J, et al. The compartmentalized inflammatory response in the multiple sclerosis brain is composed of tissue-resident CD8+ T lymphocytes and B cells. Brain : a journal of neurology. 2018;141:2066–2082. doi: 10.1093/brain/awy151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Pasetto L, et al. Targeting Extracellular Cyclophilin A Reduces Neuroinflammation and Extends Survival in a Mouse Model of Amyotrophic Lateral Sclerosis. Journal of Neuroscience. 2017;37:1413–1427. doi: 10.1523/JNEUROSCI.2462-16.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Liddelow SA, et al. Neurotoxic reactive astrocytes are induced by activated microglia. Nature. 2017;541:481–487. doi: 10.1038/nature21029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Anderson MA, et al. Astrocyte scar formation aids central nervous system axon regeneration. Nature. 2016 doi: 10.1038/nature17623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chang A, et al. Cortical remyelination: a new target for repair therapies in multiple sclerosis. Annals of neurology. 2012;72:918–926. doi: 10.1002/ana.23693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ousman SS, et al. Protective and therapeutic role for alphaB-crystallin in autoimmune demyelination. Nature. 2007;448:474–479. doi: 10.1038/nature05935. [DOI] [PubMed] [Google Scholar]
- 26.Schirmer L, et al. Differential loss of KIR4.1 immunoreactivity in multiple sclerosis lesions. Annals of neurology. 2014;75:810–828. doi: 10.1002/ana.24168. [DOI] [PubMed] [Google Scholar]
- 27.Gadea A, Schinelli S, Gallo V. Endothelin-1 regulates astrocyte proliferation and reactive gliosis via a JNK/c-Jun signaling pathway. Journal of Neuroscience. 2008;28:2394–2408. doi: 10.1523/JNEUROSCI.5652-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hametner S, et al. Iron and neurodegeneration in the multiple sclerosis brain. Annals of neurology. 2013;74:848–861. doi: 10.1002/ana.23974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Fard MK, et al. BCAS1 expression defines a population of early myelinating oligodendrocytes in multiple sclerosis lesions. Science translational medicine. 2017;9 doi: 10.1126/scitranslmed.aam7816. eaam7816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Brophy PJ, Boccaccio GL, Colman DR. The distribution of myelin basic protein mRNAs within myelinating oligodendrocytes. Trends Neurosci. 1993;16:515–521. doi: 10.1016/0166-2236(93)90196-s. [DOI] [PubMed] [Google Scholar]
- 31.Zrzavy T, et al. Loss of ‘homeostatic’ microglia and patterns of their activation in active multiple sclerosis. Brain : a journal of neurology. 2017;140:1900–1913. doi: 10.1093/brain/awx113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lucchinetti C, et al. Heterogeneity of multiple sclerosis lesions: implications for the pathogenesis of demyelination. Annals of neurology. 2000;47:707–717. doi: 10.1002/1531-8249(200006)47:6<707::aid-ana3>3.0.co;2-q. [DOI] [PubMed] [Google Scholar]
- 33.Haider L, et al. The topograpy of demyelination and neurodegeneration in the multiple sclerosis brain. Brain : a journal of neurology. 2016;139:807–815. doi: 10.1093/brain/awv398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hauser SL, et al. B-cell depletion with rituximab in relapsing-remitting multiple sclerosis. The New England journal of medicine. 2008;356:676–688. doi: 10.1056/NEJMoa0706383. [DOI] [PubMed] [Google Scholar]
- 35.Munschauer M, et al. The NORAD lncRNA assembles a topoisomerase complex critical for genome stability. Nature. 2018;56:132–136. doi: 10.1038/s41586-018-0453-z. [DOI] [PubMed] [Google Scholar]
- 36.Campbell GR, et al. Mitochondrial DNA deletions and neurodegeneration in multiple sclerosis. Annals of neurology. 2011;69:481–492. doi: 10.1002/ana.22109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Fischer MT, et al. Disease-specific molecular events in cortical multiple sclerosis lesions. Brain : a journal of neurology. 2013;136:1799–1815. doi: 10.1093/brain/awt110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Carassiti D, et al. Neuronal loss, demyelination and volume change in the multiple sclerosis neocortex. Neuropathol Appl Neurobiol. 2018;44:377–390. doi: 10.1111/nan.12405. [DOI] [PubMed] [Google Scholar]
- 39.Absinta M, et al. Persistent 7-tesla phase rim predicts poor outcome in new multiple sclerosis patient lesions. J Clin Invest. 2016;126:2597–2609. doi: 10.1172/JCI86198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Falcão AM, et al. Disease-specific oligodendrocyte lineage cells arise in multiple sclerosis. Nat Med. 2018;24:1837–1844. doi: 10.1038/s41591-018-0236-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kirby L, et al. Oligodendrocyte Precursor Cells Are Co-Opted by the Immune System to Cross-Present Antigen and Mediate Cytotoxicity. bioRxiv. 2018 doi: 10.1101/461434. 461434. [DOI] [Google Scholar]
- 42.Matevossian A, Akbarian S. Neuronal nuclei isolation from human postmortem brain tissue. Journal of visualized experiments : JoVE. 2008 doi: 10.3791/914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Shekhar K, et al. Comprehensive Classification of Retinal Bipolar Neurons by Single-Cell Transcriptomics. Cell. 2016;166:1308–132.e1330. doi: 10.1016/j.cell.2016.07.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.v d Maaten L, Hinton G. Visualizing Data using t-SNE. Journal of Machine Learning Research. 2008;9:2579–2605. [Google Scholar]
- 45.Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biology. 2014;15:550. doi: 10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Trapnell C, et al. The dynamics and regulators of cell fate decisions are revealed by pseudotemporal ordering of single cells. Nature biotechnology. 2014;32:381–386. doi: 10.1038/nbt.2859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Yu G, Wang L-G, Han Y, He Q-Y. clusterProfiler: an R Package for Comparing Biological Themes Among Gene Clusters. 2012;16:284–287. doi: 10.1089/omi.2011.0118. home.liebertpub.com. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Aibar S, et al. SCENIC: single-cell regulatory network inference and clustering. Nature Methods. 2017;14:1083–1086. doi: 10.1038/nmeth.4463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Meguro R, et al. Nonheme-iron histochemistry for light and electron microscopy: a historical, theoretical and technical review. Archives of Histology and Cytology. 2007;70:1–19. doi: 10.1679/aohc.70.1. [DOI] [PubMed] [Google Scholar]
- 50.Jahn O, Tenzer S, Bartsch N, Patzig J, Werner HB. Vol. 79. Humana Press; Totowa, NJ: 2013. pp. 335–353. [Google Scholar]
- 51.Werneburg S, Mühlenhoff M, Stangel M, Hildebrandt H. Polysialic acid on SynCAM 1 in NG2 cells and on neuropilin-2 in microglia is confined to intracellular pools that are rapidly depleted upon stimulation. Glia. 2015;63:1240–1255. doi: 10.1002/glia.22815. [DOI] [PubMed] [Google Scholar]
- 52.Schafer DP, et al. Microglia Sculpt Postnatal Neural Circuits in an Activity and Complement-Dependent Manner. Neuron. 2012;74:691–705. doi: 10.1016/j.neuron.2012.03.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.