

Abstract
Neurotropic RNA viruses continue to emerge and are increasingly linked to diseases of the central nervous system (CNS) despite viral clearance. Indeed, the overall mortality of viral encephalitis in immunocompetent individuals is low, suggesting efficient mechanisms of virologic control within the CNS. Both immune and neural cells participate in this process, which requires extensive innate immune signaling between resident and infiltrating cells, including microglia and monocytes, that regulate the effector functions of antiviral T and B cells as they gain access to CNS compartments. While these interactions promote viral clearance via mainly neuroprotective mechanisms, they may also promote neuropathology and, in some cases, induce persistent alterations in CNS physiology and function that manifest as neurologic and psychiatric diseases. This review discusses mechanisms of RNA virus clearance and neurotoxicity during viral encephalitis with a focus on the cytokines essential for immune and neural cell inflammatory responses and interactions. Understanding neuroimmune communications in the setting of viral infections is essential for the development of treatments that augment neuroprotective processes while limiting ongoing immunopathological processes that cause ongoing CNS disease.
Keywords: RNA virus, encephalitis, neuroinflammation, blood-brain barrier, neurovascular unit, microglia, brain Trm
INTRODUCTION
Neurotropic RNA viruses continue to emerge and spread worldwide, leading to increased cases of acute infectious and autoimmune diseases of the central nervous system (CNS), and postinfection neurologic sequelae. These viruses gain access to the CNS via multiple routes, replicating within various subtypes of neurons, glia and microglia, depending on the type of virus and, for some viruses, the route of entry (1). Antiviral immunity can effectively clear viral infections in most tissues, and the brain is no exception: Innate immune cytokines and surveying T cells can clear even neurons of replicating viruses—in some cases without significant cellular injury (1). The rules of engagement with regard to neuroimmune mechanisms of virologic control and leukocyte entry exhibit numerous specializations and checkpoints that serve to minimize viral spread, improve efficiency in targeting only virally infected cells, and limit the overall numbers and level of activation of infiltrating immune cells and inflammation-amplifying resident neural cells, such as microglia and astrocytes.
Still, CNS dysfunction and injury often occur during acute viral infections of the CNS, and animal models of viral encephalitis and recovery indicate multiple molecular mechanisms that may negatively impact the functions of both immune and neural cell types as they differentially respond to the same molecules. Thus, cytokines that are critical for effector T cell responses may limit neuronal repair, and chemokines that localize infiltrating immune cells may decrease the efficiency of antiviral immune responses (2, 3). In addition, certain neural cells exhibit individual and regionally heterogeneous roles that influence the extent of neuroinvasion and control of viruses that specifically target the CNS (4, 5). In this review, we discuss the immune and neural cell types that may exhibit neuroprotective or neurotoxic effects during CNS infections with RNA viruses (Figure 1), with a focus on cellular and molecular mechanisms that can promote both efficient viral clearance and long-term neurologic dysfunction.
Figure 1.
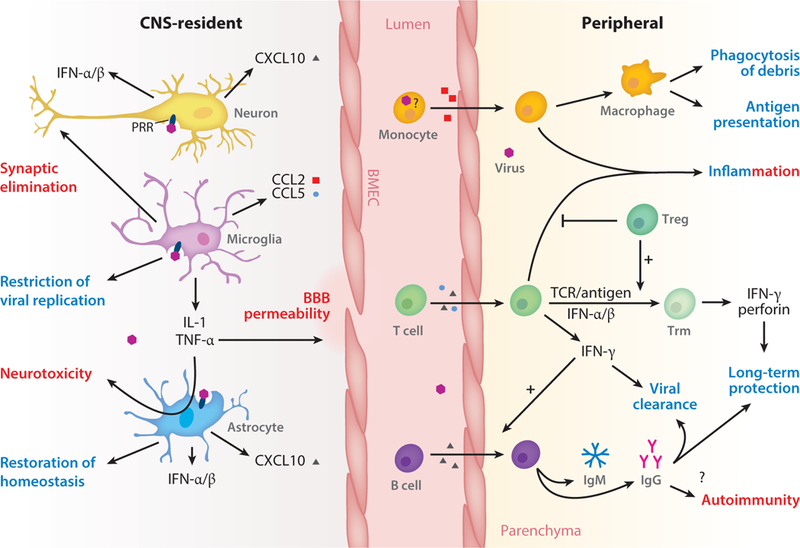
CNS-resident and peripheral immune responses promote antiviral and immunopathogenic outcomes. Activation of CNS-resident neurons and glia, either directly by viral-sensing PRRs or indirectly, promotes antiviral and inflammatory cytokine expression and leads to restriction and resolution of viral infection. Prolonged glia activation additionally may initiate neuropathogenic programs. The innate immune response of CNS-resident cells also shapes the recruitment and activation of peripheral immune cells through the expression of chemokines, modulation of the BBB, and cell-cell interaction (not shown). Infiltration of immune cells is critical for viral clearance and establishment of long-term protection in the CNS, but it may facilitate neuroinvasion or contribute to neuropathology both during and after infection. Antiviral and immunopathogenic outcomes are labeled blue and red, respectively. Abbreviations: BBB, blood-brain barrier; BMEC, brain microvascular endothelial cell; CNS, central nervous system; PRR, pattern recognition receptor; Treg, regulatory T cell; Trm, tissue-resident memory T cell.
VIRAL ACTION AT THE BLOOD-BRAIN BARRIER
Critical to protecting the CNS from peripheral infections is the blood-brain barrier (BBB). The BBB is a tightly regulated vascular interface that limits extravasation of macromolecules, peripheral cells, toxins, and invading pathogens, including viruses, from the blood to the parenchyma. The BBB is composed of highly specialized cell types, including brain microvascular endothelial cells (BMECs), pericytes and astrocyte end feet. Direct interactions or communications between these cell types govern various functions of the BBB, including its stability and transport of molecules and nutrients into and out of the CNS (6–8). The physical barrier of the BBB is formed by continuous tight junctions (TJs) and adherens junctions (AJs) between BMECs, which are linked to the actin cytoskeleton by scaffolding proteins—zonula occludens (ZO) 1–3 and catenins, respectively (6). There are two types of AJs with opposing effects. Linear AJs are connected to the circumferential actin bundles and enhance barrier integrity, whereas focal AJs are linked to radial actin bundles and their formation leads to increased barrier permeability (9). The small Rho GTPases, including Ras homolog gene family member A (RhoA), Ras-related C3 botulinum toxin substrate (Rac) 1, and cell division control protein 42 homolog (Cdc42), play a critical role in the formation and stabilization of both TJs and AJs between BMECs (10). While TJs and AJs can prevent paracellular movement of molecules and cells, they primarily maintain the polarized expression of molecules targeted to either the abluminal or luminal surfaces of the microvasculature.
In addition to the physical barrier, maintenance of the BBB is determined by several factors including limited expression of cellular adhesion molecules (CAMs) and reduced vesicular trafficking across the brain endothelium. Intercellular adhesion molecule (ICAM) 1 and vascular cell adhesion molecule (VCAM) 1 play a critical role in leukocyte interaction and extravasation at the BBB (11, 12). Under normal conditions, CAMs are minimally expressed on BMECs, limiting leukocyte infiltration inside the CNS (13). Another unique feature of BMECs is their uniquely low rate of transcytosis, which is critical for sustaining the restrictive nature of the BBB (14). Recently, major facilitator superfamily domain 2a (Mfsd2a) has been identified as a key regulator of vesicular trafficking at the BBB. Mfsd2a is selectively expressed on BMECs, and its genetic ablation results in vascular leakage, mainly by enhanced transcytosis rather than TJ disruption (7). Lipid flippase activity of Mfsd2a modulates the lipid composition of the brain endothelium in a way that prevents caveola formation and transport across BMECs (15). Several studies have reported that both RNA and DNA viruses exploit caveola-mediated transcytosis to cross the epithelial cell barriers of various organs (16–18). Hence, the low rate of transcytosis in BMECs may serve to limit neuroinvasion by impeding viral transport within vesicles across BMECs.
Viruses may be capable of disrupting the BBB to enhance viral neuroinvasion. For instance, tail vein injection of mice with HIV-1 Tat protein (to model Tat secretion from infected cells) promoted BBB opening through activation of cyclooxygenase 2 (COX-2), which in turn suppressed expression of TJs (19). Additionally, HIV-1 Tat protein stimulates MMP-9 in astrocytes, resulting in disruption of BBB integrity (20). However, some viruses can gain access to the CNS in the absence of BBB disruption (21, 22) via unknown mechanisms.
Innate Immune Sensing in the Neurovascular Unit
Once a virus has entered the CNS, the innate immune response is the first wave of host defense. While infiltrating immune cells are critical to the clearance of viral infection in the CNS, virtually all cells are capable of participating in innate immunity in some capacity. The innate immune response is initiated by cellular sensing of pathogen-associated molecular patterns (PAMPs) by pattern recognition receptors (PRRs). Activation of these receptors initiates signaling cascades that culminate in the expression of type I interferons and inflammatory cytokines by interferon regulatory transcription factor 3 (IRF3), IRF7, and NF-κB, respectively (reviewed in 23). These cytokines act by autocrine mechanisms to trigger interferon-stimulated genes and other antiviral factors. Additionally, they act by paracrine signaling, initiating antiviral innate immunity programs in neighboring cells and modulating the function of infiltrating immune cells and resident inflammation-amplifying cells.
For RNA viruses, PRRs primarily recognize PAMPs composed of single- or double-stranded RNA sequences present in the viral genome or generated during the course of infection. Specifically, these viral RNA PAMPs are recognized by multiple PRRs, including the endosomal Toll-like receptors (TLRs) TLR3 and TLR7, or the cytoplasmic retinoic acid–inducible gene I (RIG-I)-like receptors (RLRs), which include RIG-I and melanoma differentiation-associated protein 5 (MDA5). TLR3 and TLR7 recognize double-stranded and single-stranded RNA, respectively, while the RLRs are RNA helicases that recognize double-stranded RNA. TLR3, TLR7, RIG-I, and MDA5 are expressed and have demonstrable activity in the cells composing the neurovascular unit (NVU), including neurons, astrocytes, microglia, and BMECs (24–31).
The specific contribution of these RNA-sensing PRRs in the various cells of the NVU during viral infection has not been fully explored. However, in vitro studies of PRR activation in single-cell populations have demonstrated the capability of these signaling cascades to shape viral infection and the innate and adaptive immune responses. As expected, cytokine expression is a hallmark of TLR and RLR activation in NVU cells. Expression of type I interferons and antiviral interferon-stimulated genes is upregulated following activation of TLR3/7, RIG-I, or MDA5 in most NVU cell types in which they are expressed (24–27, 31–36). Conversely, inflammatory cytokines exhibit distinct but overlapping patterns of expression of tumor necrosis factor (TNF), IL-1, and IL-6 dependent on both the PRR and the cell type (25, 27, 37–43). In addition to cytokine expression, PRR signaling induces activation of microglia and astrocytes, suggesting that viral sensing initiates additional glial antiviral mechanisms that control CNS infection, as discussed below (44, 45). TLR 3/7 and RLR signaling in astrocytes and BMECs also upregulates expression of molecules that facilitate recruitment of immune cells, including chemokines (CCL2, CCL3, and CCL5) and CAMs (ICAM-1 and VCAM) (26, 32, 33, 43). Taken together these data demonstrate that viral sensing in the NVU not only acts to initialize antiviral signaling but also enhances the antiviral activity of both resident and infiltrating immune cells.
Effect of Inflammation on Blood-Brain Barrier Function
Innate cytokines such as TNF, IL-1β, and type I interferons have been shown to modulate BBB integrity via multiple mechanisms. TNF and IL-1β can activate matrix metalloproteinase 9 (MMP-9), which mediates degradation of claudin 5 and ZO-1 between BMECs (46). Additionally, TNF, IL-1β, and type I interferons modulate BBB integrity through differential regulation of Rho GTPases in BMECs (reviewed in 47, 48). RhoA triggers the formation of stress fibers and actomyosin contraction by activating its downstream effectors, such as mammalian diaphanous and Rho-associated protein kinase (ROCK) (49). Enhanced ROCK activity exerts a pulling force on AJs that dissociates their interaction with the actin cytoskeleton (50). Additionally, Rho kinase activity mediates phosphorylation and degradation of TJ protein claudin 5, leading to increased BBB permeability (51). Conversely, Cdc42 promotes BBB integrity by inducing the assembly and stabilization of linear AJs between BMECs (52, 53). Rac-1 can exert contrasting effects on the BBB integrity depending on the stimulant involved. Activation of Rac-1 by a synergic action of type I interferons with c-Mer proto-oncogene tyrosine kinase (Mertk), a TAM receptor tyrosine kinase, enhances TJ integrity (27, 54). Hence, cooperative signaling between Mertk and type I interferon receptor (IFNAR) signaling induces BBB tightening thus protecting against viral neuroinvasion. The mechanism of BBB tightening by type I interferons also depends on suppression of IL-1β expression, likely through inhibition of inflammasome activity that mediates caspase-1-dependent IL-1β maturation (55). Similar to type I interferons, IFN-λ (which belongs to the type III family of interferons and signals through distinct receptors) promotes BBB integrity during neurotropic viral infection (56). Nonetheless, encephalitic viruses have evolved multiple strategies to block type I interferon activities (57). Whether and how these directly impact on NVU functions and promote viral neuroinvasion is unclear (58).
Elevated CAM expression is reported to occur during infection of mice with multiple neurotropic viruses such as West Nile virus (WNV), Japanese encephalitis virus (JEV), and Venezuelan equine encephalitis virus (VEEV) (59–61). In addition to playing a critical role in leukocyte extravasation, elevated CAM expression may lead to further modification of the BBB. Leukocyte interaction with CAMs induces phosphorylation and internalization of vascular endothelial cadherins, which destabilizes AJs, resulting in BBB disruption (10). Additionally, leukocyte interaction with ICAM-1 on endothelial cells augments caveola-mediated transcytosis across the pulmonary endothelium, leading to increased vascular leakage (62). Consistently, pharmacological blockade ofVCAM-1 or its ligand ameliorated the severity of immunopathology associated with WNV infection (4). Further studies that more definitively delineate protective and pathological effects of leukocyte subsets with identification of biomarkers that could establish appropriate time points of intervention are needed to further develop biologic agents that limit leukocyte-mediated immunopathology.
ASTROCYTES AND NEURONS IN RNA VIRAL INFECTIONS
Astrocytes Are Activated During Viral Infections
Astrocytes are essential for brain homeostasis, performing a variety of neuroprotective functions. They provide metabolic support of neurons and synaptic function, regulate extracellular balance of ions and neurotransmitters (63), and maintain BBB integrity (64). In addition, these glial cells play a role in controlling immune cell trafficking and activation. Astrocytes can detect injury signals and respond by secreting cytokines and chemokines and activating immune defenses (65). They respond to CNS viral infections by increasing expression of many proteins, most notably GFAP (glial fibrillary acidic protein), and by proliferating in a process known as astrogliosis (66). Activation of astrocytes during viral infection may occur in multiple ways, including direct viral infection, release of viral particles, neuronal cell death, and via other immune mediators (67). A body of work suggests that microglia also play a role in inducing reactive astrocytes during viral infection as well as other neural injuries (4, 68, 69). Their production of proinflammatory cytokines such as IL-1, IL-6, TNF-α, and IFN-γ has been shown to favor astrogliosis (69, 70).
Astrocytes Participate in Innate and Adaptive Viral Immune Responses
Astrocytes play an important role in initiating immune responses during viral infection of the CNS. Infection of cultured astrocytes with different flaviviruses, including tick-borne encephalitis, JEV, WNV, and Zika virus (ZIKV), induces the production of type I interferons and restricts viral spread (71, 72). Type I interferons are critical to the inhibition of viral spread after initial infection. Global deletion of IFNAR, or downstream type I interferon signaling, leads to increased mortality in WNV-infected mice and an inability to clear virus in a model of lymphocytic choriomeningitis virus (LCMV) (73, 74). The role of astrocytes in regulating BBB integrity to limit inflammation has been shown to be regionally specific in vivo, as the loss of IFNAR signaling in astrocytes enhances BBB permeability in the hindbrain due to WNV infection (4). Throughout the CNS, astrocytes exhibit distinct and regional heterogeneity in gene expression, morphology, and signaling functions (75).
In addition to playing critical roles in initiating the innate immune response, activated astrocytes continue to regulate the adaptive immune response, albeit to a smaller extent than other cell types. Members of the Flaviviridae family induce the expression of MHC class I and II molecules involved in the activation of CD8+ T cells (76, 77). Astrocytes normally express low levels of MHC class I molecules, but in vivo infection of mouse hepatitis virus (MHV), in particular strain JHM (JHMV), upregulates expression, yet not to the level of expression by microglia. In contrast, MHC class II molecules remained relatively undetectable on the majority of astrocytes (78). In a more recent study, cultured astrocytes infected with St. Louis encephalitis virus responded by increasing expression of MHC class I molecules (79). Whether the increased cell surface expression on astrocytes is functional in recognizing T cells has yet to be studied. Christensen et al. (80) found that in a murine model of LCMV infection, astrocytes are the major producers of chemokine CXCL10 in the neural parenchyma. The same group showed that CXCL10 is a key ligand for CXCR3 on CD8+ T cells at sites of CNS viral inflammation (81). Further studies of this interplay between astrocytes and T cells may reveal mechanisms for long-term deficits after viral infection.
Beneficial and Pathogenic Impact of Astrocyte Activation
Activated astrocytes are generally accepted as beneficial because they play critical roles in removing toxic molecules and restoring the BBB. As described above, they are key initiators of the immune response needed to limit viral burden. These glial cells are also important for signaling growth factor that promotes axonal and neuronal growth after infection (66). On the other hand, activated astrocytes can be viewed as detrimental for neuronal function and regeneration. Studies looking at flavivirus-infected astrocytes have indicated the production of IL-1β, IL-6, IFN-α, TNF-α, CXCL10, and CCL4 (68, 82). Cytokines such as TNF-α and IL-1β have also been reported to regulate neural correlates of memory, including adult neurogenesis, synaptic plasticity, and modulation of long-term potentiation (83–85). Most recently, a study demonstrated that IL-1R1 signaling after WNV infection in the mouse leads to an increased production of proinflammatory, reactive astrocytes at the expense of adult neurogenesis. These newly generated astrocytes then become a source of antineurogenic cytokines (68). While the initial role of activated astrocytes is indisputably important for viral control, the persistence of these glial cells may lead to more long-term neurocognitive dysfunctions that need to be further studied, as discussed below.
Neurons Can Express Chemokines and Cytokines
Chemokine expression in the infected CNS is often regulated by cytokine production via infiltrating leukocytes. Infection by several different viruses has been correlated with a rapid induction of chemokines from astrocytes and microglia. However, whether neurons contribute to the recruitment of leukocytes to the CNS after viral infection is still under study. Neurons themselves are the major target in the CNS in a variety of viruses, including JEV, WNV, and LCMV (86–88). Given their susceptibility to these viruses, neurons are likely to be the first responders in initiating immune signaling. Early studies using a mouse model of measles virus showed an association between the neuronal expression of CCL5 and CXCL10 and T cell recruitment, suggesting that neurons might play a role in activating immune responses to viral infection (89). A further study, using a mouse model of WNV, describes the role of neuronal CXCL10 in recruiting CXCR3+ T cells to the CNS (88). Neutralization or genetic silencing of CXCL10 in mouse models of WNV reduced T cell migration to the brain, preventing efficient viral control and reducing survival (88). Like astrocytes, the regional heterogeneity of expression of CXCL10 leads to differences in inflammatory cell migration. In a WNV model of viral encephalitis, a subdued secretion of CXCL10 by cerebellar neurons alters migration of inflammatory cells (90). More recently, inoculation of the CNS with Theiler’s murine encephalomyelitis virus (TMEV) rapidly induced production of neuronal CCL2, a source necessary for infiltration of inflammatory monocytes into the hippocampus (91). Along similar lines, neurons have been shown to produce type I interferons and express MHC class I molecules, taking a further active role in the antiviral defense (92, 93). In murine models of TMEV and La Cross virus (LCAV), neurons can produce type I interferons, while Borna disease virus (BDV) infection of cultured neurons leads to MHC-I expression. Infection of BDV also increases the presentation of CD8+ T cells, leading to the secretion of cytokines, including IFN-γ and IL-10. Together, these findings suggest a role of neuronal production of chemokines and cytokines in the activation of leukocyte infiltration.
ACUTE VIROLOGIC CONTROL IN THE CENTRAL NERVOUS SYSTEM
Antiviral T Cell Responses Are Neuroprotective
T cells play a critical role during viral infections of the CNS, although the response must be carefully controlled to limit immunopathology in an organ with limited capacity for repair. T cell recruitment to the CNS occurs primarily via chemokine signaling pathways. During WNV infection, neuronal production of T cell chemoattractants, such as CXCL10, leads to the recruitment of CXCR3+ T cells to the CNS to control viral infection and promote survival (88). Reactivation of recruited T cells occurs via interaction with CD11c+ dendritic cells in an IL1-R1-dependent manner (94), and release of T cells from the perivascular space into the parenchyma is in part mediated via IL-1-mediated alterations in polarity of CXCL12 expression at the endothelium (2, 3). Recent work in a model of neurotropic coronavirus infection highlights the importance of CD8+ T cell reactivation in perivascular spaces of the CNS for viral control and survival. In this work, expression of CCL19 and 21 by stromal endothelial cells and fibroblast-like reticular cells in the perivascular spaces of the CNS was shown to be important for coordinating recruitment and reactivation of antiviral CD8+ T cells to the CNS via CCR7 signaling (95). Local restimulation of T cells critically limits immunopathology by limiting the number of T cells needed to improve virologic control, thus improving the efficiency of antiviral responses in the CNS (96).
Regulatory T cells (Tregs) additionally limit immunopathology in the CNS by controlling antiviral responses during acute encephalitis. Transfer of virus-specific Tregs improved survival from MHV-induced encephalitis by limiting pathogenic Th1 cells, associated with decreases in microglial activation (97). In a clinical study of blood donors testing positive for WNV, Tregs were found to expand after infection, and asymptomatic patients had higher levels of Tregs than symptomatic patients, suggesting that Treg control of antiviral immune responses might influence the severity of clinical disease (98). Indeed, in the same study, animals depleted of Tregs showed more severe symptoms and had a significantly higher mortality rate than control animals following WNV infection (98). Loss of Tregs led to decreased expression of CD103 on T cells persisting in the brain after WNV infection (99), suggesting that Tregs also influence the development of immune memory in the CNS.
Cytokines, chemokines, and the strain of virus together influence T cell polarization and the resulting antiviral and/or immunopathological response. For example, loss of IL-10 signaling led to either Th17 - or Th1-mediated increases in morbidity and mortality following infection depending on the virulence of the strain of Sindbis virus (SINV) (100, 101). The specific chemokine milieu in the infected CNS appears to also influence T cell activity and antiviral response. During acute infection with LCMV, cytotoxic T lymphocytes express chemokines, which leads to peripheral myeloid cell recruitment, severe immunopathology, and death (102). However, during chronic LCMV infection, therapeutic administration of virus-specific T cells can lead to noncytopathic clearance of virus (96). Cytokines also exert different antiviral effects in the periphery and the CNS. IFN-γ critically controls infectious virus replication in the periphery but clears virus from the CNS without affecting acute viral replication (103, 104).
T cells can persist in the CNS for months to years following neurotropic infection with dengue virus (DENV) (105), vesicular stomatitis virus (106), WNV (99, 107), or influenza (108); however, the functional implications of this persistence are relatively unknown. Tissue-resident memory T cells (Trm cells) are a recently described class of long-lived memory T cells that reside in nonlymphoid tissues at sites of prior antigen exposure and coordinate a rapid recall response to rechallenge (109, 110). Trm cells are classically defined by the expression of the integrin αE (CD103) and the early activation marker CD69; however, expression of these two markers is not uniform across all circulation-independent Trm cells in nonlymphoid tissues. CD69 is rapidly up-regulated upon activation through TCR signaling and exposure to type I interferons and interferes with signaling through the sphingosine 1 phosphate 1 receptor S1P1, preventing T cell egress from the tissue (111, 112). In the CNS, generation of Trm cells appears to be antigen dependent (106, 113). However, in vivo infection models with vesicular stomatitis virus or neurotropic influenza suggest that persistent antigen exposure was not required for Trm persistence in the CNS (106, 114). CD69+ brain Trm cells also express high levels of the prosurvival molecule Bcl-2 (114), which may explain their long-term survival despite lack of continuous antigen exposure.
Consistent with studies of Trm cells in other tissues, brain Trm cells are poised to provide enhanced protection against returning pathogens. An early study identified CD69+CD8+ T cells persisting in the CNS to 320 days postinfection (the latest time point analyzed) that retained killing function when exposed to peptide- or virus-loaded cells ex vivo (108). More recent studies have demonstrated that brain Trm cells can provide enhanced protection and survival in mice upon rechallenge (114, 115). This protection could occur despite depletion of peripheral CD4+ and CD8+ T cells and was dependent on IFN-γ and perforin production by the brain Trm cells (114). While these studies demonstrate the existence of brain Trm cells, and the capacity to provide functional pathogen control upon reexposure, very little is known about how the presence and activity of persistent T cells might influence activity of resident CNS cells, including microglia, astrocytes, and neurons, in the absence of overt infection.
B Cells Protect the CNS During Acute Encephalitis
B cell functions, including antiviral humoral immune responses, are critical for control of viral dissemination in the periphery and neuroinvasion during neurotropic viral infections (reviewed in 116). Studies in murine models of neurotropic RNA viral infections, including those caused by flaviviruses and alphaviruses, indicate that both virus-specific and nonspecific B cells also enter the CNS during acute viral infection with CNS virus-specific, antibody-secreting cells (ASCs) increasing in the brain after their initial development within lymphoid tissues. Early studies in mice infected with SINV indicated that while most isotype switching from IgM to IgG and IgA occurs in peripheral lymphoid tissues, numbers of ASCs producing IgG1 continue to rise in the brain through the course of acute encephalitis. As described above, T cells are the first to enter the CNS during viral infection, with T cell–derived cytokines, such as IFN-γ, critically involved in viral clearance and the amplification of immune cell infiltration through upregulation of chemokines. Early infiltrating B cells express CXCR3, CXCR5, and CCR7, whose ligands, CXCL9, CXCL10, CXCL13, CCL19, and CCL21, are upregulated within the CNS during viral infections. Animals that lack T cell production of IFN-γ or ability to signal through its receptor, IFNγR, exhibit decreased CNS B cell recruitment with limited expression of SINV-specific IgM, IgG2a, and IgG2b (103). Detection of IgM within the cerebrospinal fluid of patients with neuroinvasive disease due to WNV suggests that IgM-expressing CD19+CD38+CD138−Blimp-1+ plasmablast cells might initially traffic into the brain as an early response to viral neuroinvasion. Indeed, studies in viral encephalitis due to SINV or WNV showed that IgM-expressing ASCs are among the earliest infiltrating immune cells and are required for virologic control within the CNS (117). These ASCs express IgG and IgA during the period of viral clearance, and both CD19+CD38+ memory B cells expressing surface immunoglobulin and CD38−CD138+Blimp-1+ plasma cells are retained in the CNS long after elimination of virus (107).
Studies examining survival and differentiation of CNS B cells indicate that acute infection with virus triggers long-term alterations in inflammatory cascades that support their persistence. IL-10 and IL-21, which promote B cell proliferation and differentiation, increase early during infection and remain elevated long after clearance of infectious virus (118). Proliferating ASCs that originate in cervical lymph nodes during acute infection and enter the CNS during neuroinvasion are retained within the CNS, where they continue to proliferate and differentiate. Elevated levels of B cell–activating factor (BAFF) in the CNS and BAFF receptor expression by B cells contribute to long-term maintenance of virus-specific ASCs in the brain (118). Given the recent identification of postinfection, antibody-based neurologic diseases diagnosed in patients after recovery from CNS infection with both DNA and RNA viruses (119, 120), it is possible that these persistent CNS B cells or plasmablasts contribute to the development of rare types of CNS autoimmunity in certain patients.
Several studies have also evaluated the role ofIL-10, which is expressed by a variety of cell types, including CNS-infiltrating regulatory B cells. IL-10 can limit the immunopathology induced by Th1/Th17 effector cells and inhibit cytokine expression by activated microglia (100, 121). Of interest, recent studies suggest that viral virulence also impacts on IL-10-mediated restriction of helper T cell responses. In a model using a recombinant SINV strain with intermediate virulence, IL-10 deficiency led to enhanced disease via increased differentiation of immunopathogenic Th1, but not Th17, cells (101).
MONOCYTES/MACROPHAGES
Monocyte Infiltration into the CNS
Neurotropic RNA viruses gain access to permissive cells via attachment to entry receptors (e.g., TLR3, glycosaminoglycans, RIG-1) expressed by neural cells including neurons, astrocytes, and microglia (reviewed in 122). As described above, PRR detection of viral nucleic acids induces expression of chemoattractants that promote the parenchymal entry of mononuclear cells, including peripheral blood monocytes, which differentiate into tissue macrophages at specific sites of injury. For viral CNS insults there is debate as to whether these cells aid in viral clearance and recovery or mediate continuing damaging inflammation.
Macrophages can produce anti-inflammatory mediators, scavenge the area and phagocytose foreign material and debris, and regulate the extracellular matrix and glial scar surrounding the damaged area (123). However, these cells have also been shown to have potent effector functions including antigen presentation, T cell stimulation, and production and secretion of numerous proinflammatory mediators as well as reactive oxygen species (124). More work must be done to elucidate the different mechanisms that induce different macrophage and monocyte phenotypes in disease processes.
Chemokines Are Critical for CNS Monocyte Recruitment
During CNS infections with RNA viruses, proinflammatory chemokines, such as CCL2 and CCL7, are crucial for the CNS recruitment of monocytes. In a WNV mouse model, deficiency of CCL7 resulted in increased viral burden and mortality. Subsequent restoration of CCL7 by exogenous administration increased monocyte and neutrophil recruitment to the CNS and improved survival (125). In this study, while CCL2 and CCL7 were shown to be required for efficient monocytosis, only CCL7 played a role in viral clearance. CCL2 aided in monocyte upregulation, while CCL7 was involved with increases of monocytes, neutrophils, and CD8+ T cells to the CNS (104). This suggests that both chemokines work differentially to assist in leukocyte upregulation and decrease viral burden.
The previous study also highlights the debate over the differential roles of chemokines and chemokine receptors in monocyte recruitment and viral clearance. In another study, CCR7 depletion led to a phenotype similar to that of CCL7 depletion. CCR7 is a chemokine receptor that binds CCL21 and CCL19. Mice deficient in this receptor and infected with WNV have higher viral loads and increased mortality rates as compared to wild-type infected mice (125). Importantly, this depletion leads to a panleukocytosis, with increases in the migration of CD4+ T cells, CD8+ T cells, inflammatory monocytes, and neutrophils to the CNS (125). In light of these findings, the authors suggest that CCR7 acts as a gatekeeper under physiological conditions and normally restricts leukocyte migration into the brain to limit neuroinflammation during viral infection.
Characterization of CNS-infiltrating leukocytes in JEV-infected CCL2−/− and CCR2−/− mice produced different phenotypes than those infected with WNV. CCL2 ablation led to increased susceptibility to JEV infection, while CCR2 deficiency enhanced resistance to the virus (126). Both groups of mice had equal viral burden in the periphery, but only CCL2-deficient mice exhibited increased viral burden and CD11b+Ly-6Chi monocytes in CNS tissue (126). One reason for this discrepancy could be that chemokine receptors, including CCR2, are able to bind multiple chemokines. Thus, when CCL2 binds CCR2, it recruits immune cells, which enhances viral clearance. However, competition for CCR2 binding by several chemokines might diminish the effective concentration of CCL2, making the CNS exhibit lack of virologic control.
Monocytes as Biomarkers in CNS Viral Infections
Although the role of monocytes mainly involves viral clearance, some viral pathogens hijack the CNS homing mechanisms to infiltrate the brain parenchyma and infect other CNS cells. Studies have shown that HIV uses this mechanism, and infection leads to increased CCR2 and CCL2 sensitivity in CD14+CD16+ monocytes (127). Furthermore, blocking CCR2 with targeted therapeutic agents reduces transmigration of HIV+CD14+CD16+ monocytes to the brain, helping to diminish viral entry (128). There has also been evidence of monocytes as useful biomarkers of disease progression in HIV/SIV (simian immunodeficiency virus) infection. In a cohort study, patients with HIV-associated neurocognitive dementia (HAND) had significantly increased CCR2 expression on CD14+CD16+ monocytes compared to HIV-infected patients with normal cognition (129). When paired with an in vitro model of the human BBB, this study showed the CD14+CD16+ monocytes taken from the HAND patients migrated across the BBB model in significantly higher numbers compared to controls. Together these studies reveal a promising role for the measurement and inhibition of monocytes and their respective chemokines in disease monitoring and modification.
MICROGLIA DURING ACUTE RNA VIRAL INFECTION
Microglia Origin, Differentiation, and Distribution
Microglia are the resident mononuclear phagocytes of the CNS, accounting for 5–12% of cells in the adult mouse brain (130) and 0.5–16.6% of cells in the adult human brain (131) with regional variations in density in both mice and humans. Although microglia were initially thought to originate from the neuroectoderm, it is now generally accepted that microglia are of myeloid origin (132); however, the nature of microglial precursors and progenitors is still debated. Only recently has it been convincingly demonstrated that microglia originate from embryonic yolk sac precursors rather than the fetal liver (133). Under certain conditions, monocytes are recruited to the CNS and can differentiate into microglia-like cells, such as in transcription factor PU.1 knockout mice when the endogenous microglial niche is vacant (134), or during infection (135); however, this does not seem to be a significant source for maintaining the microglial population under homeostatic conditions. Rather, microglia appear to be long-lived cells that are self-renewing without contribution from circulating progenitors (136).
Colony-stimulating factor 1 receptor (CSF1R) signaling is essential for the development of many mononuclear phagocytes, including microglia. Tissue-resident macrophages including microglia fail to develop in Csf1r−/− mice (133), but in contrast to many tissue macrophages, adult microglia can still form in Csf1op/op mice, which carry a natural null mutation in the Csf1 gene (137). This is due to an alternative CSF1R ligand, IL34, that is highly expressed by neurons (138, 139). Similar to Csf1op/op mice, 1L34−/− mice exhibit a reduction in microglial development that varies by brain region. While the cortex exhibits approximately 65% decrease in microglia, the cerebellum has fewer microglia at baseline and no significant decrease with IL34 elimination (140).
Microglia Restrict Viral Replication via Innate Immune Mechanisms
Microglia are thought to protect the CNS from viral infection through several mechanisms, including the production of antiviral cytokines, phagocytosis of virus-infected and dying neurons, and the induction of neuronal repair and homeostasis (141). However, assessing specific immune contributions of microglia during infection has been challenging. Although powerful for mechanistic studies, in vitro microglial culture systems may not faithfully model in vivo environmental cues, which may alter both homeostatic and reactive gene expression patterns (142). Furthermore, many viruses of importance to human health infect neurons and astrocytes rather than microglia (143), thus requiring a more complex mixed cell culture system for analysis of microglial response to and interaction with infected cells. In vivo, it is difficult to distinguish CNS resident microglia from infiltrating peripheral monocytes and macrophages; thus, assigning specific immune contributions to each cell type is challenging.
Researchers attempting to determine specific contributions of microglia during infection have used several strategies to isolate the role of microglia. Using an ex vivo spinal cord slice culture system that isolates microglial response from peripheral immune responses, Quick et al. (144) showed that WNV-infected tissues upregulate inflammatory cytokines including CXCL10, CXCL1, CCL5, CCL2, CCL3, TNF-α, TNF-related apoptosis-inducing ligand (TRAIL), and IL-6. Inhibition of microglial activation with minocycline reduced expression of CCL5, CCL2, and IL-6, indicating that microglia are responsible for expression of these proinflammatory cytokines (144). Several strategies have also been used to reduce microglial numbers. IL34−/− mice were more susceptible to infection after intracranial inoculation with attenuated WNV (140). This was not due to increased viral burden but rather increased neuronal death, suggesting that microglia might provide neurotrophic support and protection rather than limit viral replication (140). However, Wheeler et al. (145) demonstrated that microglia restrict replication of neuroattenuated murine coronavirus, MHV, early in the course of infection using CSF1R antagonist PLX5622 to deplete microglia. Similarly, depletion of microglia using liposome-encapsulated clodronate causes increased viral replication in a neonatal model of DENV encephalitis (146). Impacts of pharmacological agent on peripheral monocytes and macrophages may account for these differences between genetic and pharmacological depletion of microglia.
Microglia Orchestrate Peripheral Immune Cell Interaction
In addition to restricting viral replication, microglia orchestrate the peripheral immune response to invading pathogens. In a healthy CNS, microglial cells do not express MHCs; however, when activated by pathological conditions, they upregulate these molecules (146). The extent to which microglia express costimulatory molecules that define professional antigen-presenting cells and their impact on infiltrating immune cells are less clear (147). Depletion of microglia by CSF1R antagonist PLX5622 suggests that microglia are not necessary to prime a virus-specific T cell response, but their depletion diminishes aspects of the CD4+ T cell response to MHV (145). This may be due to loss of the primary MHC-II-expressing cell type in the brain as well as decreased MHC-II expression on infiltrating monocytes/macrophages. Whereas PLX5622-treated mice infected with MHV showed a modest increase in CD8+ T cell recruitment to the CNS (145), intracerebral infection with DENV following depletion of microglia with clodronate-containing liposomes in neonatal mice resulted in diminished CD8+ T cell recruitment to the brain (146). Although both studies implicate microglia in efficient T cell response to viral encephalitis, discrepancies may reflect differences in the virus, methods of microglial depletion, and/or age of mice being infected.
Pathogenic Impacts of Microglia
A growing body of literature illustrates fundamental roles for microglia beyond immune functions, in controlling neuronal proliferation and differentiation as well as maintaining synaptic connections. During homeostatic conditions, microglia survey the microenvironment and modify or eliminate synaptic structures (148); however, during infection, prolonged microglia activation may contribute to astrocyte-mediated neurotoxicity (68, 69) and excessive complement-mediated synapse elimination (149). On the other hand, Wheeler et al. (145) found that the absence of microglia diminished the Treg response, which is important for modulating the inflammatory response and limiting host damage. Interestingly, recent data indicate that microglia may exhibit epigenetic-mediated immune memory in the forms of both immune training and immune tolerance, which impacts the neuropathology of neurodegenerative mouse models (150). Thus, microglial immune memory may affect the severity of postinfection neurologic sequelae.
VIRAL PERSISTENCE AND POSTINFECTION SYNDROMES
Many autoantibody and demyelinating diseases have been hypothesized to be related to postinfection etiologies, particularly in the absence of a detectable oncologic process. Even in cases where virus is undetectable in the serum or cerebrospinal fluid at the onset of CNS disease, a patient history of preceding infection can often be identified. While an antibody-mediated mechanism is noted in many cases, viral persistence has also been demonstrated to cause CNS disease well after the acute phase of infection. Here, we discuss the postinfection neurologic sequelae that may occur after recovery from infections with RNA viruses (Table 1).
Table 1.
Specific RNA viruses and associated neurologic defects
| Virus | Acute defects | Subacute or chronic defects |
|---|---|---|
| Measlesvirus (154–158) | Encephalitis, blindness | Subacute sclerosing panencephalitis, cognitive defects, seizures, motor dysfunction, coma, cerebellar ataxia |
| West Nile virus (159–162) | Meningoencephalitis, acute flaccid paralysis | Cognitive defects |
| Zikavirus (163–168) | Meningoencephalitis, Guillain-Barré syndrome | Birth defects (microcephaly, developmental delay, motor dysfunction), myelopathy, possible cognitive defects |
| Japanese encephalitis virus (169–170) | Encephalitis, acute flaccid paralysis | Motor dysfunction, ataxia, tremor |
| Venezuelan equine encephalitis virus (171) | Encephalitis | Seizures, cognitive defects, psychiatric illness |
| Western equine encephalitis virus (171) | Encephalitis | Seizures, cognitive defects, psychiatric illness, motor dysfunction |
| Eastern equine encephalitis virus (171) | Encephalitis | Seizures, cognitive defects, psychiatric illness, motor dysfunction |
| Influenza virus (172–175) | Encephalitis | Not reported to date |
Although measles virus infection outbreaks have significantly decreased since the advent and administration of the vaccine, recent outbreaks of acute and chronic infections in the United States and Europe have occurred in unvaccinated populations (151–153). Acute measles infection causes fever, cough, runny nose, and conjunctivitis (154). However, it is the persistent CNS infection, subacute sclerosing panencephalitis (SSPE), that causes significant morbidity and mortality. Studies have shown tropism for neurons and glial cells in CNS measles virus infection, and a robust infiltration of innate immune cells also occurs (155). SSPE symptoms include cognitive defects, seizures and motor dysfunction, and coma, and these symptoms are thought to result from a combination of the infection of CNS-resident cells and the immune response to infection. Rare, disease-associated disease foci also include the eye, with presentations of chorioretinitis and macular scarring with pigmentation (156), and the hindbrain, with acute cerebellar ataxia as a presenting symptom (157). SSPE cases occur at a high rate in unvaccinated children, especially when the infection is acquired during infancy (158).
WNV infection can also cause neuroinvasive disease with both acute and postinfection syndromes. Acute WNV neuroinvasive disease (WNND) can manifest as encephalitis, meningitis or acute flaccid paralysis (159, 160). Although virus is typically cleared within 1–2 weeks, with many recovering from acute WNND, up to 50% of patients, many of whom did not have overt acute WNND, have persistent neurocognitive dysfunction after infection (161, 162). Murine models of WNND demonstrate that there is no change in hippocampal volume, so the neurocognitive dysfunction might be related to more subtle changes in synapse physiology and repair (68, 149). While a recent animal study demonstrated that administration of anti-IL-1R1 agents might prevent the development of cognitive dysfunction after recovery from WNV (68), prognostic biomarkers are needed to aid in risk-benefit analyses that might support immunosuppressant therapies in selected patients.
ZIKV outbreaks in tropical climates in the last five years have brought interest and attention to the neurologic sequelae of ZIKV neuroinvasive disease. Most notably, a clear correlation between maternal ZIKV infection and teratogenic effects on the growing fetus, including microcephaly and developmental delay, has been observed (163). In addition, there has been a surge of Guillain-Barré syndrome cases associated with outbreaks of human ZIKV infection (164). Recently, ZIKV infections of adults were associated with transverse myelitis (165), encephalopathy, and meningomyelitis or encephalomyelitis, that latter of which may be associated with defects in memory and cognition with unknown long-term outcomes for survivors (164–168). An appreciation of the full spectrum of ZIKV-mediated diseases and studies providing mechanistic insights and potential therapeutic targets are forthcoming.
JEV occurs mostly in Southeast Asia and the Western Pacific. Acute infection symptoms include fever, headache, vomiting, encephalopathy, and seizures. Acute flaccid paralysis is also common. Approximately half of surviving patients will have neurologic sequelae, including persistent mixed upper and lower motor neuron disease as well as extrapyramidal symptoms and cerebellar signs (169). Although many of these symptoms may be related to viral destruction of neurons, JEV-specific antibodies and viral antigens have both been detected in previously infected patients 50–180 days after symptom onset (170), suggesting that postinfection sequelae might be due to ongoing virologic or immunologic processes.
Infections of alphaviruses such as VEEV, Western equine encephalitis virus, and Eastern equine encephalitis virus in humans also produce neurologic sequelae beyond acute encephalitis, including cognitive defects, seizures, psychiatric illness, and motor dysfunction. Similar to those of JEV, neurologic sequelae of alphaviruses are likely related to a combination of viral persistence and the host immune response (171).
Influenza-associated encephalitis/encephalopathy (IAE) is a rare but serious complication of influenza infection. In a report of the Australian Childhood Encephalitis Study, 13 patients with IAE were identified. Neuroinvasive complications included encephalopathy and seizures that were often associated with imaging findings on brain magnetic resonance imaging to suggest alteration in the structural integrity of brain tissue. Seven of the 13 children in that study died or had significant neurology morbidity as a result of IAE (172). Prior vaccination for influenza and antiviral therapy were infrequent in patients with significant morbidity and mortality. Similar findings were reported for a pediatric population in Japan (173) as well as adults (174, 175). Postinfection syndromes such as transverse myelitis have also been reported and, of note, associated with myelin-oligodendrocyte glycoprotein antibodies in one case (176). In summary, postinfection neurologic consequences of viral infections, even among nonneurotropic viruses, are an important clinical entity whose study might identify generalizable mechanisms for related seizure, CNS autoimmune, and memory disorders.
CONCLUDING REMARKS
Immune and neural cell types can exhibit neuroprotective and neurotoxic effects during CNS viral infections depending on the context. At the BBB, endothelial cells and astrocytes control viral entry into the CNS and are strongly influenced by TLR and PRR activation. Endothelial cells respond to these cues and modulate barrier integrity through altered expression of CAMs and Rho GTPases, while astrocytes can be activated to produce proinflammatory cytokines such as IL-1, IL-6, TNF-α, and IFN-γ. These proinflammatory cytokines aid in viral clearance through recruitment of mononuclear cells but may also be detrimental long-term to neuronal function and regeneration. While T cells that infiltrate from the blood and perivascular spaces participate in direct control of acute viral infection through cytokine release, they are also capable of remaining in the CNS for months or years as Trm cells. The long-term neurologic consequences of these Trm cells and antibody-secreting B cells within the CNS are still unclear. Monocyte recruitment to the CNS meant to decrease viral load may be hijacked by viral machinery and lead to increased CNS viral burden in specific instances. Furthermore, the CNS has intrinsic microglia that orchestrate immune cell activation and viral clearance via MHC upregulation. However, prolonged microglial activation after infection may lead to astrocyte-mediated neuronal damage and excessive synaptic pruning. Taken together, these data suggest the importance of understanding the immune system’s role in clearing CNS infection but also of examining its impact on long-term neuronal and immunologic function. Studying the differential impacts of neuroimmune responses may help us better prevent and treat neurologic sequelae that often follow infection.
Many questions still remain unanswered as we aim to develop effective therapeutics. For one, the mechanisms underlying the contribution of persistent neuroinflammation to CNS pathophysiology are unclear. Secondly, although this review was divided into sections by cell type, neural and immune cells function in conversational groups, whose molecular contributions to these interactions are incompletely understood. For example, we know glial cells actively participate in infection control, but we are still examining the exact cues they provide to and receive from T cells. Finally, there is evidence suggesting the ability of peripheral infection alone to influence long-term CNS function and outcomes (150), but a lack of details regarding the processes.
The study of neural and immune cell interactions during and after viral infections may apply to a much broader context as well. Recent research has shown important roles for immune cells in almost all neurologic systems, including neurodevelopmental, neurodegenerative, and aging-related processes. Thus, the induction of inflammatory events after viral entry may prime the CNS to be more or less susceptible to the development of other diseases. Examining consequences of neurotropic viral infections may provide insight into the development of neurodegenerative processes as well.
ACKNOWLEDGMENTS
S.A.’s current research is funded by an NIH-T32 postdoctoral training grant. Additional funding support is provided by NIH grants U19AI083019, R01NS052632, R01AI101400, and HDTRA1-15-1-0032 to R.S.K.
Footnotes
DISCLOSURE STATEMENT
S.A. holds patents previously licensed to Elucid Bioimaging.
LITERATURE CITED
- 1.Manglani M, McGavern DB. 2018. New advances in CNS immunity against viral infection. Curr. Opin. Virol. 28:116–26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Durrant DM, Daniels BP, Klein RS. 2014. IL-1R1 signaling regulates CXCL12-mediated T cell localization and fate within the central nervous system during West Nile Virus encephalitis. J. Immunol. 193:4095–106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.McCandless EE, Zhang B, Diamond MS, Klein RS. 2008. CXCR4 antagonism increases T cell trafficking in the central nervous system and improves survival from West Nile virus encephalitis. PNAS 105:11270–75 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Daniels BP, Jujjavarapu H, Durrant DM, Williams JL, Green RR, et al. 2017. Regional astrocyte IFN signaling restricts pathogenesis during neurotropic viral infection. J. Clin. Investig. 127:843–56 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cho H, Proll SC, Szretter KJ, Katze MG, Gale M Jr., Diamond MS. 2013. Differential innate immune response programs in neuronal subtypes determine susceptibility to infection in the brain by positive-stranded RNA viruses. Nat. Med. 19:458–64 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Abbott NJ, Patabendige AA, Dolman DE, Yusof SR, Begley DJ. 2010. Structure and function of the blood-brain barrier. Neurobiol. Dis. 37:13–25 [DOI] [PubMed] [Google Scholar]
- 7.Ben-Zvi A, Lacoste B, Kur E, Andreone BJ, Mayshar Y, et al. 2014. Mfsd2a is critical for the formation and function of the blood-brain barrier. Nature 509:507–11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Daneman R, Zhou L, Kebede AA, Barres BA. 2010. Pericytes are required for blood-brain barrier integrity during embryogenesis. Nature 468:562–66 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Millan J, Cain RJ, Reglero-Real N, Bigarella C, Marcos-Ramiro B, et al. 2010. Adherens junctions connect stress fibres between adjacent endothelial cells. BMC Biol. 8:11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Rho SS, Ando K, Fukuhara S. 2017. Dynamic regulation of vascular permeability by vascular endothelial cadherin-mediated endothelial cell-cell junctions. J. Nippon Med. Sch. 84:148–59 [DOI] [PubMed] [Google Scholar]
- 11.Lopes Pinheiro MA, Kooij G,Mizee MR, Kamermans A, Enzmann G, et al. 2016. Immune cell trafficking across the barriers of the central nervous system in multiple sclerosis and stroke. Biochim. Biophys. Acta 1862:461–71 [DOI] [PubMed] [Google Scholar]
- 12.Takeshita Y, Ransohoff RM. 2012. Inflammatory cell trafficking across the blood-brain barrier: chemokine regulation and in vitro models. Immunol. Rev. 248:228–39 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Schafer A, Whitmore AC, Konopka JL, Johnston RE. 2009. Replicon particles of Venezuelan equine encephalitis virus as a reductionist murine model for encephalitis. J. Virol. 83:4275–86 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Siegenthaler JA, Sohet F, Daneman R. 2013. ‘Sealing off the CNS’: cellular and molecular regulation of blood–brain barriergenesis. Curr. Opin. Neurobiol. 23:1057–64 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Andreone BJ, Chow BW, Tata A, Lacoste B, Ben-Zvi A, et al. 2017. Blood-brain barrier permeability is regulated by lipid transport-dependent suppression of caveolae-mediated transcytosis. Neuron 94:581–94.e5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bobardt MD, Chatterji U, Selvarajah S, Van der Schueren B, David G, et al. 2007. Cell-free human immunodeficiency virus type 1 transcytosis through primary genital epithelial cells. J. Virol. 81:395–405 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tugizov SM, Herrera R, Palefsky JM. 2013. Epstein-Barr virus transcytosis through polarized oral epithelial cells. J. Virol. 87:8179–94 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yu C, Achazi K, Moller L, Schulzke JD, Niedrig M, Bucker R. 2014. Tick-borne encephalitis virus replication, intracellular trafficking, and pathogenicity in human intestinal Caco-2 cell monolayers. PLOS ONE 9:e96957 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Pu H, Hayashi K, Andras IE, Eum SY, Hennig B, Toborek M. 2007. Limited role of COX-2 in HIV Tat-induced alterations of tight junction protein expression and disruption of the blood-brain barrier. Brain Res. 1184:333–44 [DOI] [PubMed] [Google Scholar]
- 20.Ju SM, Song HY, Lee JA, Lee SJ, Choi SY, Park J. 2009. Extracellular HIV-1 Tat up-regulates expression of matrix metalloproteinase-9 via a MAPK-NF-κB dependent pathway in human astrocytes. Exp. Mol. Med. 41:86–93 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cain MD, Salimi H, Gong Y, Yang L, Hamilton SL, et al. 2017. Virus entry and replication in the brain precedes blood-brain barrier disruption during intranasal alphavirus infection. J. Neuroimmunol. 308:118–30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Schafer A, Brooke CB, Whitmore AC, Johnston RE. 2011. The role of the blood-brain barrier during Venezuelan equine encephalitis virus infection. J. Virol. 85:10682–90 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kawai T, Akira S. 2009. The roles of TLRs, RLRs and NLRs in pathogen recognition. Int. Immunol. 21:317–37 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Butchi NB, Du M, Peterson KE. 2010. Interactions between TLR7 and TLR9 agonists and receptors regulate innate immune responses by astrocytes and microglia. Glia 58:650–64 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Crill EK, Furr-Rogers SR, Marriott I. 2015. RIG-I is required for VSV-induced cytokine production by murine glia and acts in combination with DAI to initiate responses to HSV-1. Glia 63:2168–80 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.da Conceicao TM, Rust NM, Berbel AC, Martins NB, do Nascimento Santos CA, et al. 2013. Essential role of RIG-I in the activation of endothelial cells by dengue virus. Virology 435:281–92 [DOI] [PubMed] [Google Scholar]
- 27.Daniels BP, Holman DW, Cruz-Orengo L, Jujjavarapu H, Durrant DM, Klein RS. 2014. Viral pathogen-associated molecular patterns regulate blood-brain barrier integrity via competing innate cytokine signals. mBio 5:e01476–14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Li J, Wang Y, Wang X, Ye L, Zhou Y, et al. 2013. Immune activation of human brain microvascular endothelial cells inhibits HIV replication in macrophages. Blood 121:2934–42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.McCarthy GM, Bridges CR, Blednov YA, Harris RA. 2017. CNS cell-type localization and LPS response of TLR signaling pathways. F1000Res 6:1144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Nazmi A, Dutta K, Basu A. 2011. RIG-I mediates innate immune response in mouse neurons following Japanese encephalitis virus infection. PLOS ONE 6:e21761 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Peltier DC, Simms A, Farmer JR, Miller DJ. 2010. Human neuronal cells possess functional cytoplasmic and TLR-mediated innate immune pathways influenced by phosphatidylinositol-3 kinase signaling. J. Immunol. 184:7010–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Carpentier PA, Begolka WS, Olson JK, Elhofy A, Karpus WJ, Miller SD. 2005. Differential activation of astrocytes by innate and adaptive immune stimuli. Glia 49:360–74 [DOI] [PubMed] [Google Scholar]
- 33.De Miranda J, Yaddanapudi K, Hornig M, Lipkin WI. 2009. Astrocytes recognize intracellular polyinosinic-polycytidylic acid via MDA-5. FASEB J. 23:1064–71 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Roth-Cross JK, Bender SJ, Weiss SR. 2008. Murine coronavirus mouse hepatitis virus is recognized by MDA5 and induces type I interferon in brain macrophages/microglia. J. Virol. 82:9829–38 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Town T, Jeng D, Alexopoulou L, Tan J, Flavell RA. 2006. Microglia recognize double-stranded RNA viaTLR3. J. Immunol. 176:3804–12 [DOI] [PubMed] [Google Scholar]
- 36.Zhou Y, Ye L, Wan Q, Zhou L, Wang X, et al. 2009. Activation of Toll-like receptors inhibits herpes simplex virus-1 infection of human neuronal cells. J. Neurosci. Res. 87:2916–25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bhargavan B, Kanmogne GD. 2018. Toll-like receptor-3 mediates HIV-1-induced interleukin-6 expression in the human brain endothelium via TAK1 and JNK pathways: implications for viral neuropathogenesis. Mol. Neurobiol. 55:5976–92 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Borysiewicz E, Fil D, Konat GW. 2009. Rho proteins are negative regulators of TLR2, TLR3, and TLR4 signaling in astrocytes. J. Neurosci. Res. 87:1565–72 [DOI] [PubMed] [Google Scholar]
- 39.Das A, Chai JC, Kim SH, Lee YS, Park KS, et al. 2015. Transcriptome sequencing of microglial cells stimulated with TLR3 and TLR4 ligands. BMC Genom. 16:517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Jiang R, Ye J, Zhu B, Song Y, Chen H, Cao S. 2014. Roles of TLR3 and RIG-I in mediating the inflammatory response in mouse microglia following Japanese encephalitis virus infection. J. Immunol. Res. 2014:787023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lawrimore CJ, Crews FT. 2017. Ethanol, TLR3, and TLR4 agonists have unique innate immune responses in neuron-like SH-SY5Y and microglia-like BV2. Alcohol Clin. Exp. Res. 41:939–54 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Liu HY, Hong YF, Huang CM, Chen CY, Huang TN, Hsueh YP. 2013. TLR7 negatively regulates dendrite outgrowth through the Myd88-c-Fos-IL-6 pathway. J. Neurosci. 33:11479–93 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Madeddu S, Woods TA, Mukherjee P, Sturdevant D, Butchi NB, Peterson KE. 2015. Identification of glial activation markers by comparison of transcriptome changes between astrocytes and microglia following innate immune stimulation. PLOS ONE 10:e0127336 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Nerurkar L, McColl A, Graham G, Cavanagh J. 2017. The systemic response to topical Aldara treatment is mediated through direct TLR7 stimulation as imiquimod enters the circulation. Sci. Rep. 7:16570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Tarassishin L, Suh HS, Lee SC. 2014. LPS and IL-1 differentially activate mouse and human astrocytes: role of CD14. Glia 62:999–1013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zhou L, Yan C, Gieling RG, Kida Y, Garner W, et al. 2009. Tumor necrosis factor-alpha induced expression of matrix metalloproteinase-9 through p21-activated kinase-1. BMC Immunol. 10:15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Al-Obaidi MMJ, Bahadoran A, Wang SM, Manikam R, Raju CS, Sekaran SD. 2018. Disruption of the blood brain barrier is vital property of neurotropic viral infection of the central nervous system. Acta Virol. 62:16–27 [DOI] [PubMed] [Google Scholar]
- 48.Klein RS, Hunter CA. 2017. Protective and pathological immunity during central nervous system infections. Immunity 46:891–909 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ando K, Fukuhara S, Moriya T, Obara Y, Nakahata N, Mochizuki N. 2013. Rap1 potentiates endothelial cell junctions by spatially controlling myosin II activity and actin organization. J. Cell Biol. 202:901–16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Huveneers S, Oldenburg J, Spanjaard E, van der Krogt G, Grigoriev I, et al. 2012. Vinculin associates with endothelial VE-cadherin junctions to control force-dependent remodeling. J. Cell Biol. 196:641–52 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Yamamoto M, Ramirez SH, Sato S, Kiyota T, Cerny RL, et al. 2008. Phosphorylation of claudin-5 and occludin by rho kinase in brain endothelial cells. Am. J. Pathol. 172:521–33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Amado-Azevedo J, Reinhard NR, van Bezu J, de Menezes RX, van Beusechem VW, et al. 2017. A CDC42-centered signaling unit is a dominant positive regulator of endothelial integrity. Sci. Rep. 7:10132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Broman MT, Kouklis P, Gao X, Ramchandran R, Neamu RF, et al. 2006. Cdc42 regulates adherens junction stability and endothelial permeability by inducing alpha-catenin interaction with the vascular endothelial cadherin complex. Circ. Res. 98:73–80 [DOI] [PubMed] [Google Scholar]
- 54.Miner JJ, Daniels BP, Shrestha B, Proenca-Modena JL, Lew ED, et al. 2015. The TAM receptor Mertk protects against neuroinvasive viral infection by maintaining blood-brain barrier integrity. Nat. Med. 21:1464–72 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Guarda G, Braun M, Staehli F, Tardivel A, Mattmann C, et al. 2011. Type I interferon inhibits interleukin-1 production and inflammasome activation. Immunity 34:213–23 [DOI] [PubMed] [Google Scholar]
- 56.Lazear HM, Daniels BP, Pinto AK, Huang AC, Vick SC, et al. 2015. Interferon-lambda restricts West Nile virus neuroinvasion by tightening the blood-brain barrier. Sci. Transl. Med. 7:284ra59 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Schulz KS, Mossman KL. 2016. Viral evasion strategies in type IIFN signaling—a summary of recent developments. Front. Immunol. 7:498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Bleau C, Filliol A, Samson M, Lamontagne L. 2015. Brain invasion by mouse hepatitis virus depends on impairment of tight junctions and beta interferon production in brain microvascular endothelial cells. J. Virol. 89:9896–908 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Sharma A, Bhomia M, Honnold SP,Maheshwari RK. 2011. Role of adhesion molecules and inflammation in Venezuelan equine encephalitis virus infected mouse brain. Virol. J. 8:197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Cook-Mills JM, Marchese ME, Abdala-Valencia H. 2011. Vascular cell adhesion molecule-1 expression and signaling during disease: regulation by reactive oxygen species and antioxidants. Antioxid. Redox Signal. 15:1607–38 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Li F, Wang Y, Yu L, Cao S, Wang K, et al. 2015. Viral infection of the central nervous system and neuroinflammation precede blood-brain barrier disruption during Japanese encephalitis virus infection. J. Virol. 89:5602–14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Hu G, Vogel SM, Schwartz DE, Malik AB, Minshall RD. 2008. Intercellular adhesion molecule-1-dependent neutrophil adhesion to endothelial cells induces caveolae-mediated pulmonary vascular hyperpermeability. Circ. Res. 102:e120–31 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Clarke LE, Barres BA. 2013. Emerging roles of astrocytes in neural circuit development. Nat. Rev. Neurosci. 14:311–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Abbott NJ, Ronnback L, Hansson E. 2006. Astrocyte-endothelial interactions at the blood-brain barrier. Nat. Rev. Neurosci. 7:41–53 [DOI] [PubMed] [Google Scholar]
- 65.Dong Y, Benveniste EN. 2001. Immune function of astrocytes. Glia 36:180–90 [DOI] [PubMed] [Google Scholar]
- 66.Sofroniew MV, Vinters HV. 2010. Astrocytes: biology and pathology. Acta Neuropathol. 119:7–35 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Chen CJ, Ou YC, Lin SY, Raung SL, Liao SL, et al. 2010. Glial activation involvement in neuronal death by Japanese encephalitis virus infection. J. Gen. Virol. 91:1028–37 [DOI] [PubMed] [Google Scholar]
- 68.Garber C, Vasek MJ, Vollmer LL, Sun T, Jiang X, Klein RS. 2018. Astrocytes decrease adult neurogenesis during virus-induced memory dysfunction via interleukin-1. Nat. Immunol. 19:151–61 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Liddelow SA, Guttenplan KA, Clarke LE, Bennett FC, Bohlen CJ, et al. 2017. Neurotoxic reactive astrocytes are induced by activated microglia. Nature 541:481–87 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Rohl C, Lucius R, Sievers J. 2007. The effect of activated microglia on astrogliosis parameters in astrocyte cultures. Brain Res. 1129:43–52 [DOI] [PubMed] [Google Scholar]
- 71.Pfefferkorn C, Kallfass C, Lienenklaus S, Spanier J, Kalinke U, et al. 2016. Abortively infected astrocytes appear to represent the main source of interferon beta in the virus-infected brain. J. Virol. 90:2031–38 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Lindqvist R, Mundt F, Gilthorpe JD, Wolfel S, Gekara NO, et al. 2016. Fast type I interferon response protects astrocytes from flavivirus infection and virus-induced cytopathic effects. J. Neuroinflammation 13:277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Samuel MA, Diamond MS. 2005. Alpha/beta interferon protects against lethal West Nile virus infection by restricting cellular tropism and enhancing neuronal survival. J. Virol. 79:13350–61 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Zhou S, Cerny AM, Fitzgerald KA, Kurt-Jones EA, Finberg RW. 2012. Role of interferon regulatory factor 7 in T cell responses during acute lymphocytic choriomeningitis virus infection. J. Virol. 86:11254–65 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Zhang Y, Barres BA. 2010. Astrocyte heterogeneity: an underappreciated topic in neurobiology. Curr. Opin. Neurobiol. 20:588–94 [DOI] [PubMed] [Google Scholar]
- 76.Cheng Y, King NJ, Kesson AM. 2004. Major histocompatibility complex class I (MHC-I) induction by West Nile virus: involvement of 2 signaling pathways in MHC-I up-regulation. J. Infect. Dis. 189:658–68 [DOI] [PubMed] [Google Scholar]
- 77.Kesson AM, Cheng Y, King NJ. 2002. Regulation of immune recognition molecules by flavivirus, West Nile. Viral 1mmunol. 15:273–83 [DOI] [PubMed] [Google Scholar]
- 78.Hamo L, Stohlman SA, Otto-Duessel M, Bergmann CC. 2007. Distinct regulation of MHC molecule expression on astrocytes and microglia during viral encephalomyelitis. Glia 55:1169–77 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Zuza AL, Barros HL, de Mattos Silva Oliveira TF, Chavez-Pavoni JH, RG Zanon. 2016. Astrocyte response to St. Louis encephalitis virus. Virus Res. 217:92–100 [DOI] [PubMed] [Google Scholar]
- 80.Christensen JE, Simonsen S, Fenger C, Sorensen MR, Moos T, et al. 2009. Fulminant lymphocytic choriomeningitis virus-induced inflammation of the CNS involves a cytokine-chemokine-cytokine-chemokine cascade. J. 1mmunol. 182:1079–87 [DOI] [PubMed] [Google Scholar]
- 81.Christensen JE, de Lemos C,Moos T, Christensen JP, Thomsen AR. 2006. CXCL10is the key ligand for CXCR3 on CD8+ effector T cells involved in immune surveillance of the lymphocytic choriomeningitis virus-infected central nervous system. J. 1mmunol. 176:4235–43 [DOI] [PubMed] [Google Scholar]
- 82.Palus M, Bily T, Elsterova J, Langhansova H, Salat J, et al. 2014. Infection and injury of human astrocytes by tick-borne encephalitis virus. J. Gen. Virol. 95:2411–26 [DOI] [PubMed] [Google Scholar]
- 83.Riazi K, Galic MA, Kentner AC, Reid AY, Sharkey KA, Pittman QJ. 2015. Microglia-dependent alteration of glutamatergic synaptic transmission and plasticity in the hippocampus during peripheral inflammation. J. Neurosci. 35:4942–52 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.delRey A, Balschun D, Wetzel W, Randolf A, Besedovsky HO. 2013. A cytokine network involving brain-borne IL-1β, IL-1ra, IL-18, IL-6, and TNFα operates during long-term potentiation and learning. Brain Behav. 1mmun. 33:15–23 [DOI] [PubMed] [Google Scholar]
- 85.Wu MD, Montgomery SL, Rivera-Escalera F, Olschowka JA, O’Banion MK. 2013. Sustained IL-1β expression impairs adult hippocampal neurogenesis independent of IL-1 signaling in nestin+ neural precursor cells. Brain Behav. 1mmun. 32:9–18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Bonthius DJ, Mahoney J, Buchmeier MJ, Karacay B, Taggard D. 2002. Critical role for glial cells in the propagation and spread of lymphocytic choriomeningitis virus in the developing rat brain. J. Virol. 76:6618–35 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Kalia M, Khasa R, Sharma M, Nain M, Vrati S. 2013. Japanese encephalitis virus infects neuronal cells through a clathrin-independent endocytic mechanism. J. Virol. 87:148–62 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Klein RS, Lin E, Zhang B, Luster AD, Tollett J, et al. 2005. Neuronal CXCL10 directs CD8+ T-cell recruitment and control of West Nile virus encephalitis. J. Virol. 79:11457–66 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Patterson CE, Daley JK, Echols LA, Lane TE, Rall GF. 2003. Measles virus infection induces chemokine synthesis by neurons. J. Immunol. 171:3102–9 [DOI] [PubMed] [Google Scholar]
- 90.Zhang B, Chan YK, Lu B, Diamond MS, Klein RS. 2008. CXCR3 mediates region-specific antiviral T cell trafficking within the central nervous system during West Nile virus encephalitis. J. Immunol. 180:2641–49 [DOI] [PubMed] [Google Scholar]
- 91.Howe CL, LaFrance-Corey RG, Goddery EN, Johnson RK, Mirchia K. 2017. Neuronal CCL2 expression drives inflammatory monocyte infiltration into the brain during acute virus infection. J. Neuroinflammation 14:238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Delhaye S, Paul S, Blakqori G, Minet M, Weber F, et al. 2006. Neurons produce type I interferon during viral encephalitis. PNAS 103:7835–40 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Chevalier G, Suberbielle E, Monnet C, Duplan V, Martin-Blondel G, et al. 2011. Neurons are MHC class I-dependent targets for CD8 T cells upon neurotropic viral infection. PLOS Pathog. 7:e1002393 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Durrant DM, Robinette ML, Klein RS. 2013. IL-1R1 is required for dendritic cell-mediated T cell reactivation within the CNS during West Nile virus encephalitis. J. Exp. Med. 210:503–16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Cupovic J, Onder L, Gil-Cruz C, Weiler E, Caviezel-Firner S, et al. 2016. Central nervous system stromal cells control local CD8+ T cell responses during virus-induced neuroinflammation. Immunity 44:622–33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Herz J, Johnson KR, McGavern DB. 2015. Therapeutic antiviral T cells noncytopathically clear persistently infected microglia after conversion into antigen-presenting cells. J. Exp. Med. 212:1153–69 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Zhao J, Zhao J, Perlman S. 2014. Virus-specific regulatory T cells ameliorate encephalitis by repressing effector T cell functions from priming to effector stages. PLOS Pathog. 10:e1004279 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Lanteri MC, O’Brien KM, Purtha WE, Cameron MJ, Lund JM, et al. 2009. Tregs control the development of symptomatic West Nile virus infection in humans and mice. J. Clin. Investig. 119:3266–77 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Graham JB, Da Costa A, Lund JM. 2014. Regulatory T cells shape the resident memory T cell response to virus infection in the tissues. J. Immunol. 192:683–90 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Kulcsar KA, Baxter VK, Greene IP, Griffin DE. 2014. Interleukin 10 modulation of pathogenic Th17 cells during fatal alphavirus encephalomyelitis. PNAS 111:16053–58 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Martin NM, Griffin DE. 2018. Interleukin-10 modulation of virus clearance and disease in mice with alphaviral encephalomyelitis. J. Virol. 92:e01517–17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Kim JV, Kang SS, Dustin ML, McGavern DB. 2009. Myelomonocytic cell recruitment causes fatal CNS vascular injury during acute viral meningitis. Nature 457:191–95 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Baxter VK, Griffin DE. 2016. Interferon gamma modulation of disease manifestation and the local antibody response to alphavirus encephalomyelitis. J. Gen. Virol. 97:2908–25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Shrestha B, Wang T, Samuel MA, Whitby K, Craft J, et al. 2006. Gamma interferon plays a crucial early antiviral role in protection against West Nile virus infection. J. Virol. 80:5338–48 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.van der Most RG, Murali-Krishna K, Ahmed R. 2003. Prolonged presence of effector-memory CD8 T cells in the central nervous system after dengue virus encephalitis. Int. Immunol. 15:119–25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Wakim LM, Woodward-Davis A, Bevan MJ. 2010. Memory T cells persisting within the brain after local infection show functional adaptations to their tissue of residence. PNAS 107:17872–79 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Stewart BS, Demarest VL, Wong SJ, Green S, Bernard KA. 2011. Persistence of virus-specific immune responses in the central nervous system of mice after West Nile virus infection. BMC Immunol. 12:6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Hawke S, Stevenson PG, Freeman S, Bangham CR. 1998. Long-term persistence of activated cytotoxic T lymphocytes after viral infection of the central nervous system. J. Exp. Med. 187:1575–82 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Mueller SN, Mackay LK. 2016. Tissue-resident memory T cells: local specialists in immune defence. Nat. Rev. Immunol. 16:79–89 [DOI] [PubMed] [Google Scholar]
- 110.Schenkel JM, Fraser KA, Beura LK, Pauken KE, Vezys V, Masopust D. 2014. Resident memory CD8 T cells trigger protective innate and adaptive immune responses. Science 346:98–101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Shiow LR, Rosen DB, Brdickova N, Xu Y, An J, et al. 2006. CD69 acts downstream of interferon-alpha/beta to inhibit S1P1 and lymphocyte egress from lymphoid organs. Nature 440:540–44 [DOI] [PubMed] [Google Scholar]
- 112.Mackay LK, Braun A, Macleod BL, Collins N, Tebartz C, et al. 2015. Cutting edge: CD69 interference with sphingosine-1-phosphate receptor function regulates peripheral T cell retention. J. Immunol. 194:2059–63 [DOI] [PubMed] [Google Scholar]
- 113.Casey KA, Fraser KA, Schenkel JM, Moran A, Abt MC,et al. 2012. Antigen-independent differentiation and maintenance of effector-like resident memory T cells in tissues. J. Immunol. 188:4866–75 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Steinbach K, Vincenti I, Kreutzfeldt M, Page N, Muschaweckh A, et al. 2016. Brain-resident memory T cells represent an autonomous cytotoxic barrier to viral infection. J. Exp. Med. 213:1571–87 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Wakim LM, Woodward-Davis A, Liu R, Hu Y, Villadangos J, et al. 2012. The molecular signature of tissue resident memory CD8 T cells isolated from the brain. J. Immunol. 189:3462–71 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Austin SK, Dowd KA. 2014. B cell response and mechanisms of antibody protection to West Nile virus. Viruses 6:1015–36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Metcalf TU, Griffin DE. 2011. Alphavirus-induced encephalomyelitis: antibody-secreting cells and viral clearance from the nervous system. J. Virol. 85:11490–501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Metcalf TU, Baxter VK, Nilaratanakul V, Griffin DE. 2013. Recruitment and retention of B cells in the central nervous system in response to alphavirus encephalomyelitis. J. Virol. 87:2420–29 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Acosta-Ampudia Y, Monsalve DM, Castillo-Medina LF, Rodriguez Y, Pacheco Y, et al. 2018. Autoimmune neurological conditions associated with Zika virus infection. Front. Mol. Neurosci. 11:116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Nosadini M, Mohammad SS, Corazza F, Ruga EM, Kothur K, et al. 2017. Herpes simplex virus-induced anti-N-methyl-D-aspartate receptor encephalitis: a systematic literature review with analysis of 43 cases. Dev. Med. Child Neurol. 59:796–805 [DOI] [PubMed] [Google Scholar]
- 121.Mutnal MB, Hu S, Schachtele SJ, Lokensgard JR. Infiltrating regulatory B cells control neuroinflammation following viral brain infection. J. Immunol. 193:6070–80 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Neal JW. 2014. Flaviviruses are neurotropic, but how do they invade the CNS? J. Infect. 69:203–15 [DOI] [PubMed] [Google Scholar]
- 123.London A, Cohen M, Schwartz M. 2013. Microglia and monocyte-derived macrophages: functionally distinct populations that act in concert in CNS plasticity and repair. Front. Cell Neurosci. 7:34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Terry RL, Getts DR, Deffrasnes C,vanVreden C, Campbell IL, King NJ. 2012. Inflammatory monocytes and the pathogenesis of viral encephalitis. J. Neuroinflamm. 9:270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Bardina SV, Michlmayr D, Hoffman KW, Obara CJ, Sum J, et al. 2015. Differential roles of chemokines CCL2 and CCL7 in monocytosis and leukocyte migration during West Nile virus infection. J. Immunol. 195:4306–18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Kim JH, Patil AM, Choi JY, Kim SB, Uyangaa E, et al. 2016. CCL2, but not its receptor, is essential to restrict immune privileged central nervous system-invasion of Japanese encephalitis virus via regulating accumulation of CD11b+ Ly-6Chi monocytes. Immunology 149:186–203 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Williams DW, Calderon TM, Lopez L, Carvallo-Torres L, Gaskill PJ, et al. 2013. Mechanisms of HIV entry into the CNS: increased sensitivity of HIV infected CD14+CD16+ monocytes to CCL2 and key roles ofCCR2, JAM-A, and ALCAM in diapedesis. PLOS ONE 8:e69270 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Veenstra M, Leon-Rivera R, Li M, Gama L, Clements JE, Berman JW. 2017. Mechanisms of CNS viral seeding by HIV+ CD14+ CD16+ monocytes: establishment and reseeding of viral reservoirs contributing to HIV-associated neurocognitive disorders. mBio 8:e01280–17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Williams DW, Byrd D, Rubin LH, Anastos K,Morgello S, Berman JW. 2014. CCR2 on CD14+CD16+ monocytes is a biomarker of HIV-associated neurocognitive disorders. Neurol. Neuroimmunol. Neuroinflamm. 1:e36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Lawson LJ, Perry VH, Dri P, Gordon S. 1990. Heterogeneity in the distribution and morphology of microglia in the normal adult mouse brain. Neuroscience 39:151–70 [DOI] [PubMed] [Google Scholar]
- 131.Mittelbronn M, Dietz K, Schluesener HJ, Meyermann R. 2001. Local distribution of microglia in the normal adult human central nervous system differs by up to one order of magnitude. Acta Neuropathol. 101:249–55 [DOI] [PubMed] [Google Scholar]
- 132.Walton MR, Gibbons H, MacGibbon GA, Sirimanne E, Saura J, et al. 2000. PU.1 expression in microglia. J. Neuroimmunol. 104:109–15 [DOI] [PubMed] [Google Scholar]
- 133.Ginhoux F, Greter M, Leboeuf M, Nandi S, See P, et al. 2010. Fate mapping analysis reveals that adult microglia derive from primitive macrophages. Science 330:841–45 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Beers DR, Henkel JS, Xiao Q, Zhao W, Wang J, et al. 2006. Wild-type microglia extend survival in PU.1 knockout mice with familial amyotrophic lateral sclerosis. PNAS 103:16021–26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Djukic M, Mildner A, Schmidt H, Czesnik D, Bruck W, et al. 2006. Circulating monocytes engraft in the brain, differentiate into microglia and contribute to the pathology following meningitis in mice. Brain 129:2394–403 [DOI] [PubMed] [Google Scholar]
- 136.Askew K, Li K, Olmos-Alonso A, Garcia-Moreno F, Liang Y, et al. 2017. Coupled proliferation and apoptosis maintain the rapid turnover of microglia in the adult brain. Cell Rep. 18:391–405 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Cecchini MG, Dominguez MG, Mocci S, Wetterwald A, Felix R, et al. 1994. Role of colony stimulating factor-1 in the establishment and regulation of tissue macrophages during postnatal development of the mouse. Development 120:1357–72 [DOI] [PubMed] [Google Scholar]
- 138.Chitu V, Gokhan S, Nandi S, Mehler MF, Stanley ER. 2016. Emerging roles for CSF-1 receptor and its ligands in the nervous system. Trends Neurosci. 39:378–93 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Nandi S, Gokhan S, Dai XM, Wei S, Enikolopov G, et al. 2012. The CSF-1 receptor ligands IL-34 and CSF-1 exhibit distinct developmental brain expression patterns and regulate neural progenitor cell maintenance and maturation. Dev. Biol. 367:100–13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Wang Y, Szretter KJ, Vermi W, Gilfillan S, Rossini C, et al. 2012. IL-34 is a tissue-restricted ligand of CSF1R required for the development of Langerhans cells and microglia. Nat. Immunol. 13:753–60 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Ransohoff RM, Cardona AE. 2010. The myeloid cells of the central nervous system parenchyma. Nature 468:253–62 [DOI] [PubMed] [Google Scholar]
- 142.Gosselin D, Skola D, Coufal NG, Holtman IR, Schlachetzki JCM, et al. 2017. An environment-dependent transcriptional network specifies human microglia identity. Science 356:eaal3222 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Ludlow M, Kortekaas J, Herden C, Hoffmann B, Tappe D, et al. 2016. Neurotropic virus infections as the cause of immediate and delayed neuropathology. Acta Neuropathol. 131:159–84 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Quick ED, Leser JS, Clarke P, Tyler KL. 2014. Activation of intrinsic immune responses and microglial phagocytosis in an ex vivo spinal cord slice culture model of West Nile virus infection. J. Virol. 88:13005–14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Wheeler DL, Sariol A, Meyerholz DK, Perlman S. 2018. Microglia are required for protection against lethal coronavirus encephalitis in mice. J. Clin. Investig. 128:931–43 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Tsai TT, Chen CL, Lin YS, Chang CP, Tsai CC, et al. 2016. Microglia retard dengue virus-induced acute viral encephalitis. Sci. Rep. 6:27670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Almolda B, de Labra C, Barrera I, Gruart A, Delgado-Garcia JM, et al. 2015. Alterations in microglial phenotype and hippocampal neuronal function in transgenic mice with astrocyte-targeted production of interleukin-10. Brain Behav. Immun. 45:80–97 [DOI] [PubMed] [Google Scholar]
- 148.Tremblay ME, Stevens B, Sierra A, Wake H, Bessis A, Nimmerjahn A. 2011. The role of microglia in the healthy brain. J. Neurosci. 31:16064–69 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Vasek MJ, Garber C, Dorsey D, Durrant DM, Bollman B, et al. 2016. A complement-microglial axis drives synapse loss during virus-induced memory impairment. Nature 534:538–43 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Wendeln AC, Degenhardt K, Kaurani L, Gertig M, Ulas T, et al. 2018. Innate immune memory in the brain shapes neurological disease hallmarks. Nature 556:332–38 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Bellini WJ Rota JS, Lowe LE, Katz RS, Dyken PR, et al. 2005. Subacute sclerosing panencephalitis: More cases of this fatal disease are prevented by measles immunization than was previously recognized. J. Infect. Dis. 192:1686–93 [DOI] [PubMed] [Google Scholar]
- 152.Schonberger K, Ludwig MS, Wildner M, Weissbrich B. 2013. Epidemiology of subacute sclerosing panencephalitis (SSPE) in Germany from 2003 to 2009: a risk estimation. PLOS ONE 8:e68909 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Campbell H, Andrews N, Brown KE, Miller E. 2007. Review of the effect of measles vaccination on the epidemiology of SSPE. Int. J. Epidemiol. 36:1334–48 [DOI] [PubMed] [Google Scholar]
- 154.Garg RK. 2008. Subacute sclerosing panencephalitis. J. Neurol. 255:1861–71 [DOI] [PubMed] [Google Scholar]
- 155.Esiri MM, Oppenheimer DR, Brownell B, Haire M. 1982. Distribution of measles antigen and immunoglobulin-containing cells in the CNS in subacute sclerosing panencephalitis (SSPE) and atypical measles encephalitis. J. Neurol. Sci. 53:29–43 [DOI] [PubMed] [Google Scholar]
- 156.Tripathy K, Chawla R, Mittal K, Farmania R, Venkatesh P, Gulati S. 2017. Ophthalmic examination as a means to diagnose subacute sclerosing panencephalitis: an optical coherence tomography and ultrawide field imaging evaluation. Eye Vis. 4:1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Saini AG, Sankhyan N, Padmanabh H, Sahu JK, Vyas S, Singhi P. 2016. Subacute sclerosing panencephalitis presenting as acute cerebellar ataxia and brain stem hyperintensities. Eur. J. Paediatr. Neurol. 20:435–38 [DOI] [PubMed] [Google Scholar]
- 158.Wendorf KA, Winter K, Zipprich J, Schechter R, Hacker JK, et al. 2017. Subacute sclerosing panencephalitis: the devastating measles complication that might be more common than previously estimated. Clin. Infect. Dis. 65:226–32 [DOI] [PubMed] [Google Scholar]
- 159.Campbell GL, Marfin AA, Lanciotti RS, Gubler DJ. 2002. West Nile virus. Lancet Infect. Dis. 2:519–29 [DOI] [PubMed] [Google Scholar]
- 160.Davis LE, DeBiasi R, Goade DE, Haaland KY, Harrington JA, et al. 2006. West Nile virus neuroinvasive disease. Ann. Neurol. 60:286–300 [DOI] [PubMed] [Google Scholar]
- 161.Athar P, Hasbun R, Nolan MS, Salazar L, Woods SP, et al. 2018. Long-term neuromuscular outcomes of West Nile virus infection: a clinical and electromyographic evaluation of patients with a history of infection. Muscle Nerve 57:77–82 [DOI] [PubMed] [Google Scholar]
- 162.Samaan Z, McDermid Vaz S, Bawor M, Potter TH, Eskandarian S, Loeb M. 2016. Neuropsychological impact of West Nile virus infection: an extensive neuropsychiatric assessment of 49 cases in Canada. PLOS ONE 11:e0158364 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Soares de Oliveira-Szejnfeld P, Levine D, Melo AS, Amorim MM, Batista AG, et al. 2016. Congenital brain abnormalities and Zika virus: what the radiologist can expect to see prenatally and postnatally. Radiology 281:203–18 [DOI] [PubMed] [Google Scholar]
- 164.Munoz LS, Parra B, Pardo CA, Neuroviruses Emerg. Am. Study. 2017. Neurological implications of Zika virus infection in adults. J. Infect. Dis. 216:S897–905 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Roth W, Tyshkov C, Thakur K, Vargas W. 2017. Encephalomyelitis following definitive Zika virus infection. Neurol. Neuroimmunol. Neuroinflamm. 4:e349 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Carteaux G, Maquart M, Bedet A, Contou D, Brugieres P, et al. 2016. Zika virus associated with meningoencephalitis. N. Engl. J. Med. 374:1595–96 [DOI] [PubMed] [Google Scholar]
- 167.Mehta R, Soares CN, Medialdea-Carrera R, Ellul M, da Silva MTT, et al. 2018. The spectrum of neurological disease associated with Zika and chikungunya viruses in adults in Rio de Janeiro, Brazil: a case series. PLOS Negl. Trop. Dis. 12:e0006212 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Soares CN, Brasil P, Carrera RM, Sequeira P, de Filippis AB, et al. 2016. Fatal encephalitis associated with Zika virus infection in an adult. J. Clin. Virol. 83:63–65 [DOI] [PubMed] [Google Scholar]
- 169.Solomon T, Dung NM, Kneen R, Gainsborough M, Vaughn DW, Khanh VT. 2000. Japanese encephalitis. J. Neurol. Neurosurg. Psychiatry 68:405–15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Ravi V, Desai AS, Shenoy PK, Satishchandra P, Chandramuki A, Gourie-Devi M. 1993. Persistence of Japanese encephalitis virus in the human nervous system. J. Med. Virol. 40:326–29 [DOI] [PubMed] [Google Scholar]
- 171.Ronca SE, Dineley KT, Paessler S. 2016. Neurological sequelae resulting from encephalitic alphavirus infection. Front. Microbiol. 7:959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Britton PN, Dale RC, Blyth CC, Macartney K, Crawford NW, et al. 2017. Influenza-associated encephalitis/encephalopathy identified by the Australian Childhood Encephalitis Study 2013–2015. Pediatr. Infect. Dis. J. 36(11):1021–26 [DOI] [PubMed] [Google Scholar]
- 173.Yoshikawa H, Yamazaki S, Watanabe T, Abe T. 2001. Study of influenza-associated encephalitis/encephalopathy in children during the 1997 to 2001 influenza seasons. J. Child Neurol. 16:885–90 [DOI] [PubMed] [Google Scholar]
- 174.Morishima T, Togashi T, Yokota S, Okuno Y, Miyazaki C, et al. 2002. Encephalitis and encephalopathy associated with an influenza epidemic in Japan. Clin. Infect. Dis. 35:512–17 [DOI] [PubMed] [Google Scholar]
- 175.Meijer WJ, Linn FH, Wensing AM, Leavis HL, van Riel D,et al. 2016. Acute influenza virus-associated encephalitis and encephalopathy in adults: a challenging diagnosis. JMM Case Rep. 3:e005076 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Amano H, Miyamoto N, Shimura H, Sato DK, Fujihara K, et al. 2014. Influenza-associated MOG antibody-positive longitudinally extensive transverse myelitis: a case report. BMC Neurol. 14:224. [DOI] [PMC free article] [PubMed] [Google Scholar]