

Objectives:
IL-27 is known as an antiviral cytokine that inhibits HIV, hepatitis C virus, and other viruses. We have previously demonstrated that, IL-27 posttreatment after HIV-infection inhibits viral replication in primary CD4+ T cells.
Design:
Here, we evaluated the anti-HIV effect of IL-27 pretreatment in CD4+ T cells from healthy donors prior to HIV infection with HIVNL4.3 or vesicular stomatitis virus G glycoprotein (VSV-G)-pseudotyped HIV-luciferase virus (HIV-LUC-V).
Methods:
IL-27-treated CD4+ T cells were infected with HIVNL4.3 or HIV-LUC-V and assessed the anti-HIV effect. HIV infection was monitored by p24 antigen ELISA or luciferase assay. HIV fusion/entry and uncoating were determined by BlaM-Vpr assay and HIV fate of capsid and/or HIV Entry/Uncoating assay based on core-packaged RNA availability and Translation assay, respectively. HIV proviral copy number was determined by real-time PCR. Gene expression profile from IL-27-pretreated CD4+ T cells was determined using Genechip array. Posttranslational modification of global proteins from IL-27-pretreated CD4+ T cells was determined by a combination of 2-dimensional difference-in-gel-electrophoresis (2D-DIG), western-blot and protein mass spectrometry.
Results:
IL-27 pretreatment inhibited HIVNL4.3 and HIV-LUC-V infection in CD4+ T cells. HIV copy assay demonstrated that IL-27-treatment suppressed an early step of reverse transcription during HIV infection. A combination of 2D-DIG-electrophoresis and western blot assays demonstrated that IL-27-treatment induces a change in posttranslational modification of Y box binding protein-1 (YB-1). Overexpression of domain negative YB-1 mutants illustrated that a residue Lysine at 118 plays a key role in supporting HIV infection in CD4+ T cells.
Conclusion:
IL-27-pretreatment inhibits HIV-1 infection by suppressing an HIV-reverse transcription product formation/uncoating step by suppressing the acetylation of YB-1 in primary CD4+ T cells.
Keywords: 2-dimensional difference-in-gel-electrophoresis (2D-DIG), acetylation, cytokine, IL-27, posttranslational modification, primary T cells, Y box binding protein-1
Introduction
We have previously reported that the pleotropic cytokine IL-27 inhibits HIV replication in primary CD4+ T cells, monocyte-derived macrophages, and dendritic cells [1–4]. IL-27 is a heterodimeric cytokine composed of Epstein–Barr virus-induced gene 3 and IL-27p28 that belongs to the IL-6/IL-12 family of cytokines [5]. It is primarily secreted by macrophages and dendritic cells [6–8]. A recently published report indicated that plasma IL-27 levels in HIV patients were not significantly altered during HIV-1 infection [9], suggesting that IL-27 could potentially be used in anti-HIV therapy [10].
HIV utilizes host proteins to complete every step of its life cycle and replication process [11–13]. In addition, the host cells are equipped with proteins known as ‘host restriction factors’ that counter the HIV replication process. Cell survival and function are critically dependent on numerous cellular signals that are mostly mediated by posttranslational modifications (PTMs) of the proteins. Several studies report that PTMs of the host proteins either enhance or inhibit HIV replication [14–17], it is reported that Y-box binding protein-1 (YB-1), a DNA-binding and RNA-binding protein [18,19], serves as a host protein that stabilizes the genomic RNA of HIV-1 to promote HIV production in 293T cells [20]. It is also involved in HIV replication in Jurkat T cells [21]. In the current study, we investigated the anti-HIV mechanism of IL-27 in primary T cells. We now report, for the first time, that acetylated YB-1 is a key factor for HIV infection in primary T cells and that IL-27 regulates acetylation followed by HIV infection in T cells.
Material and methods
Participants were informed written consent prior to blood being drawn. All experimental procedures in these studies were approved by the National Cancer Institute at Frederick and National Institute of Allergy and Infectious Diseases (NIAID) Institutional Review Board and performed in accordance with the relevant guidelines and regulations.
Plasmids
pNL4.3 [22] and pNL4.3.Luc [23,24] were obtained from the AIDS Reagents Program of the NIAID/National Institutes of Health (NIH) YB-1 expression vectors were constructed as follows: YB-1 wild-type and YB-1 lysine-mutated genes were synthesized (Eurofins MWG Operon, Louisville Kentucky, USA) and cloned into pCMV5.1-Flag to obtain plasmid wild-type-YB-1-Flag and Mut-YB-1-Flag.
Cells and viruses
CD4+ T cells were isolated from peripheral blood mononuclear cells of healthy donors using CD4+ microbeads according to the manufacturer's instructions (Miltenyi Biotec, San Diego, California USA), as previously described [1]. Isolated CD4+ T cells were stimulated with phytohemagglutinin (PHA) (Sigma-Aldrich, St. Louis, Missouri, USA) at 5 μg/ml for 3 days in RPMI-1640 media supplemented with 10% fetal bovine serum (FBS, Hyclone; Thermo Fisher Scientific, Waltham, Massachusetts, USA), 10 mmol/l HEPES: 2-[4-(2-hydroxyethyl)piperazin-1-yl]ethanesulfonic acid (Quality Biological, Gaithersburg, Maryland, USA), and 5 μg/ml of gentamycin (Thermo Fisher Scientific) (RP-10) [1]. 293T cells were obtained from the American Type Culture Collection and maintained in D-MEM (Thermo Fisher Scientific) containing 10% FBS. HIV-1 stocks were prepared using plasmids encoding the full length of HIVNL4.3 [22] as previously mentioned [2,25]. VSV-G-pseudotyped HIV-luciferase virus (HIV-LUC-V) virus was prepared as previously mentioned [3]. HIV-1 virions containing BlaM-Vpr were produced by cotransfection of 293T cells (1.5 × 106 cells) with pNL4–3 proviral DNA (6 μg), pCMV-BlaM-Vpr (2 μg), and pAdVAntage vectors (1 μg) as described [26]. HIV Entry/Uncoating assay based on core-packaged RNA availability and Translation (EURT) virus was produced as previously described [27].
Pretreatment of CD4+ T cells with IL-27 and panabinostat
The PHA-stimulated CD4+ T cells were pretreated with IL-27 prior to HIV infection. Briefly, the cells were cultured with either 20 U/ml of recombinant IL-2 (Roche, Mannheim, Germany) alone (control cells) or IL-2 with 100 ng/ml of recombinant IL-27 (R&D Systems, Minneapolis, Minnesota, USA) (treated cells) for 3 days in RP-10 at 2 × 106 cells/ml. After incubation, the cells were washed with prewarmed RP-10 medium at 500 × g for 5 min for three times, and then maintained in RP-10 with 20 U/ml of IL-2. Panabinostat (Selleckche, Houston, Texas, USA) treatment was performed as follows: PHA-stimulated CD4+ T cells were pretreated with 35 nmol/l Panabinostat for 24 h. Following the pretreatment, CD4+ T cells were treated with IL-2 or IL-2 with IL-27 for 3 days and then cells were washed as described above.
Flow cytometry analysis
Expression of CD4+, CXCR4, and CD3 on CD4+ T cells was analyzed by flow cytometry. Cells were stained for 15 min at room temperature in PBS containing 2% bovine serum albumin with the following antibodies: Pacific Blue-conjugated anti-CD3 (Becton Dickinson, Franklin Lakes, New Jersey, USA), APC-Cy7-conjugated anti-CD4+ (Becton Dickinson), and phycoerythrin (PE)-conjugated anti-CXCR4 (Becton Dickinson), all of which were performed following the vendor's protocol (Abcam, Cambridge, Massachusetts, USA). The cell preparations were analyzed with a FACSCalibur flow cytometer (Becton Dickinson). Positive and negative events were determined with matching isotype controls.
HIV-1 replication assay and HIV single-round infection
IL-2 or IL-2/IL27-pretreated CD4+ T cells were infected with HIVNL4.3 as previously described [2]. Half of the culture supernatants were replaced with fresh medium on day 3. Viral replication was gauged from p24 levels in culture supernatants using an HIV-1 p24 ELISA kit (PerkinElmer, Waltham, Massachusetts, USA). For single-round infection, CD4+ T cells were incubated with HIV-LUC-V (100 ng/ml p24) for 2 h. Cells were washed and then cultured for 2 days in the presence of IL-2. Luciferase activity was measured with a Bright-Glo Luciferase Assay System (Promega, Madison, Wisconsin, USA).
HIV binding assay
CD4+ T cells were incubated with virus at 4 °C for 80 min to allow binding but not membrane fusion, as previously described [28], and then washed to remove unbound virus. Cells were lysed using RIPA buffer (Boston BioProducts, Ashland, Massachusetts, USA) to obtain the protein, which was followed by a HIV-1 p24 ELISA to assess the level of bound HIV.
Quantitative reverse transcription-PCR
Cells were washed using cold PBS, and RNA was isolated from the cells using an RNeasy Isolation kit (Qiagen, Germantown, Maryland, USA). Total complementary DNA (cDNA) was synthesized using TaqMan reverse transcription reagents (Applied Biosystems, Foster City, California, USA). YB-1 and other gene expression levels were measured using quantitative real-time polymerase chain reaction (qRT-PCR) on a CFX96 Real-Time system (Bio-Rad Laboratories, Hercules, California, USA) and normalized by glyceraldehyde 3-phosphate dehydrogenase (GAPDH). Probes information is as follows: YB-1/YBX-1 (Hs00358903_g1), SUB1 (Hs00743451_s1), tryptophanyl-tRNA synthetase (WARS) (Hs00188259_m1), HnRNPH1 (Hs03044422_g1), CALM1 (Hs00300085_s1), MYL12B (Hs00853081_g1), PDCD6 (Hs00918238_m1), TPI1 (Hs03806547_s1), PGAM1 (Hs01652468_g1), PPIA (Hs04194521_s1), IMMT (Hs00986184_mH), LAP3 (Hs00429769_m1), CAPG (Hs00156249_m1), ACTG1 (Hs03044422_g1), STAT1 (Hs01014002_m1), MX1 (Hs00182073-ml), and GAPDH (Hs99999905_m1) (Applied Biosystems).
HIV reverse transcription product assay
Control and IL-27-pretreated CD4+ T cells were infected with 100 ng/ml of pseudotyped HIV (DNase I-pretreated) for 2 h. Twelve hours postinfection, genomic DNA was isolated using a QIAamp DNA Blood Mini Kit (Qiagen). A total of 500 ng of genomic DNA was used for each reaction of qRT-PCR. Absolute copy number per 1 × 106 cells were normalized by RNase P housekeeping gene (Applied Biosystems), as previously described [3].
SDS–PAGE and western blot analysis
CD4+ T cells were washed with ice-cold PBS and resuspended in RIPA buffer with a protease inhibitor cocktail (Sigma-Aldrich) and phosphatase inhibitors (Thermo Fisher Scientific) at 1 × 106 cells/100 μl at 4 °C for 10 min. The protein concentration was determined using a bicinchoninic acid (BCA) protein assay kit (Thermo Fisher Scientific). Using a total of 20-μg protein, western blot analysis was performed as previously described [29], and the intensity of the band was analyzed using NIH ImageJ (http://rsbweb.nih.gov/ij/). The western blot membrane was stripped (Restore western Blot Stripping Buffer, Thermo Fisher Scientific) and reprobed with another antibody or β-actin (Sigma-Aldrich) wherever necessary. The antibody information is as follows: PDCD6 (Proteintech, Rosemont, Illinois, USA), (Mitofilin, IMMT, WARS, Cyclophilin, HnRNPH1, MYL12B, PC4, TPI1, CALM1, LAP3, CAPG, PGAM, YBX-1/YB-1) (Abcam), β-actin (Sigma-Aldrich), STAT1 and phospho-S102-YB-1 (Cell Signaling Technology, Danvers, Massachusetts, USA).
2-Dimensional difference-in-gel-electrophoresis, two-dimensional gel electrophoresis, and western blot analysis
Whole-cell lysate protein samples from control and treated CD4+ T cells were supplied to Applied Biomics (Hayward, California, USA) for identification of regulated global proteins by 2-dimensional difference-in-gel-electrophoresis (2D-DIGE).
Whole-cell lysate proteins (100 μg) were subjected to 2D-PAGE analysis as described [30]. After separation by 2D-PAGE, the samples were transferred to a membrane for western blot analysis.
Transfection of small interfering RNA (siRNA) or DNA into CD4+ T cells
YB-1-specific siRNA (SR303243-C) and scrambled control siRNA (SR30004) were purchased from OriGene (Rockville, Maryland, USA). CD4+ T cells (7 × 106/cuvette) were transfected with siRNA (300 nmol/l) or plasmid DNA (2 μg) using a P3 Primary Cell 4D-Nucleofector Kit (V4XP-3024) using E0–115 pulse program code (Lonza, Basel, Switzerland) followed by warm RP-10 medium incubation overnight. The cells were washed and stimulated with PHA/IL-2 for 3 days. siRNA/DNA nucleofection efficiency was assessed by reverse transcription-PCR and western blot after 3 days of stimulation.
BlaM-Vpr assay for fusion and entry
The BlaM-Vpr entry/fusion assay was performed as previously described [26], and the fusion and entry activity in the samples were analyzed by flow cytometry using excitation at 410 nm and emission at 450/550 and 525/550 nm.
Immunoprecipitation and identification of posttranslational modification of Y box binding protein-1
Following nucleofection of CD4+ T cells with 2 μg of wild-type-YB-1-Flag plasmid, cells were stimulated with PHA, followed by IL-2 or IL-2/IL-27 treatment for 3 days. Cells were harvested and lysed in IP lysis buffer (Thermo Fisher Scientific). A total of 500 μg of protein was used for immunoprecipitation, following the manufacturer's protocol (Sigma-Aldrich) using glycine/Tris elution. The eluted protein was concentrated using methanol/chloroform precipitation, and samples were subjected to mass spectrometry (MC) analysis for identification of PTM.
Fate-of-capsid assay
The fate-of-capsid assay was performed as previously described [31], with 10 × 106 control or IL-27-treated CD4+ T cells infected with pseudotyped HIV-LUC-V virus (250 ng/ml) for 2 h, then washed and incubated for 12 h in RP-10 medium with IL-2. Infected CD4+ T cells were harvested and resuspended in 2.5 ml of hypotonic lysis buffer (10 mmol/l Tris–Cl pH 8.0, 10 mmol/l KCl, 1 mmol/l ethylenediaminetetraacetic acid (EDTA), complete protease inhibitors (Roche) and a phosphatase inhibitor). After ultracentrifugation as previously described [31], the top layer above the sucrose cushion was collected as the ‘soluble’ fraction (1 ml), and, once the remaining sucrose was removed, SDS–PAGE loading dye was added to the tube to resuspend the ‘pellet’ fraction. Fractions were analyzed for capsid protein content by immunoblotting using a mouse monoclonal anti-HIV-1 p24 antibody (Abcam).
HIV Entry/Uncoating assay based on core-packaged RNA availability and translation assay
The HIV EURT assay for HIV core protein uncoating was performed as previously described [27]. Briefly, control and treated CD4+ T cells were infected with 500 ng/ml p24 HIV EURT virus for 2 h. Following infection, the cells were incubated in RP-10 medium with IL-2 for 12 h. Luciferase activity was measured with a Bright-Glo Luciferase Assay System (Promega).
Statistical analysis
Statistical analyses were performed using GraphPad Prism 5 software (San Diego, California, USA). Error bars indicate SDs or standard errors (SE) from means as noted. An unpaired Student's t test was used, and P values lower than 0.05 were considered significant.
Results
Pretreatment with IL-27 confers CD4+ T cells with resistance to HIV-1 infection
PHA-stimulated IL-27-pretreated CD4+ T cells were infected with HIV-1NL4.3 or pseudotyped HIV-LUC-V, and HIV replication was then monitored. IL-27 pretreatment inhibited p24 in culture by 66 ± 10% (P < 0.01, n = 3) (Fig. 1a) and inhibited luciferase activity in CD4+ T cells by 68 ± 4.2% (P < 0.01, n = 7) (Fig. 1b). IL-27 did not alter the expression of CD4+ and CXCR4 on CD4+ T cells (Suppl. Fig. S1a) and showed no change in cell cycle progression (Suppl. Fig. S1b), indicating that IL-27 doesn’t change the replication status or cell growth of CD4+ T cells. The supernatants of control and treated CD4+ T cells showed no significant difference in cytokine release profiles, confirming that IL-27-mediated HIV resistance may be caused by a direct effect and not by other secreted cytokines (Suppl. Fig. S1c). These observations confirm that pretreatment with IL-27 directly mediates resistance to HIV-1 infection without compromising the normal functions of CD4+ T cells.
Fig. 1.
IL-27 exhibits anti-HIV effects in primary CD4+ T cells.
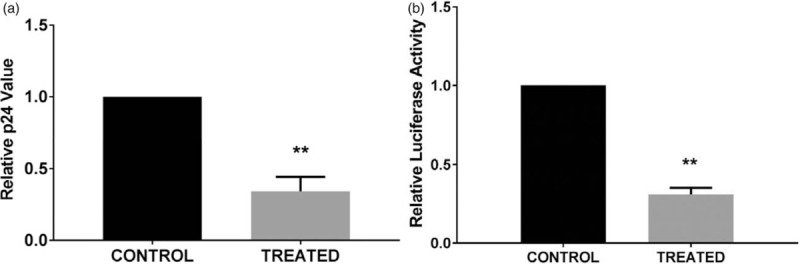
(a) IL-27 pretreatment inhibits HIV-1 replication in CD4+ T cells. Untreated (control) or IL-27-treated (treated) CD4+ T cells were infected with replication-competent HIV-1 and cultured for 7 days. A p24 ELISA was performed on the seventh day postinfection to quantitate the HIV p24 antigen in supernatants of HIV-infected control and treated cells. Relative HIV replication was compared between control and treated cells. Data show mean ± standard errors from three independent assays. (b) IL-27 pretreatment inhibits pseudo-typed HIV-1 (HIV-LUC-G) infection. Control and treated cells were infected with HIV-Luc-G virus and then cultured for 2 days. HIV infection was monitored by luciferase activity in cells. Relative HIV infection was compared between control and treated cells. Data show mean ± standard errors from seven independent assays.
IL-27 inhibits the early steps of HIV replication in CD4+ T cells
To determine which part of the HIV life cycle is suppressed in the IL-27-treated cells, we first compared HIV-1NL4.3 binding to CD4+ T cells. The amount of viral p24 on the cell surface was not altered by IL-27 pretreatment, indicating that HIV binds at the same intensity during infection in both control and treated CD4+ T cells (Fig. 2a). Next, we assessed the impact of IL-27 on HIV internalization or fusion/entry into the host cells using the BlaM-Vpr fusion/entry assay [26]. No significant differences were observed between the control and treated cells in terms of CCF2-AM dye cleavage, an indicator of viral fusion/entry. Thus, IL-27 doesn’t affect the fusion/entry step of HIV infection (Fig. 2b), suggesting that IL-27 treatment may affect the reverse transcription step.
Fig. 2.
IL-27 treatment inhibits the early steps of HIV replication in CD4+ T cells.
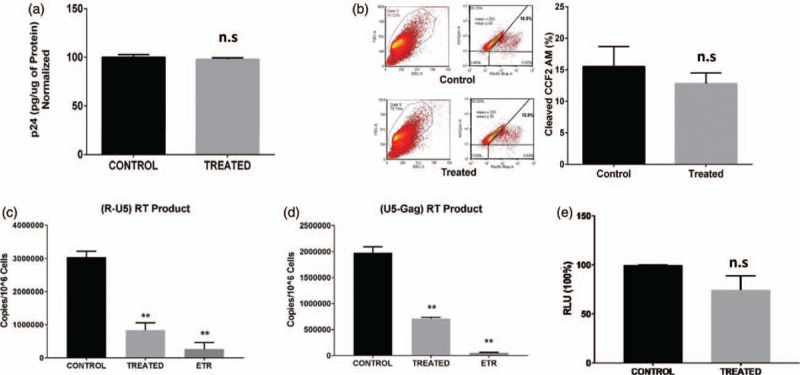
(a) IL-27 pretreatment had no effect on HIV-1 binding to the CD4+ T cells. Untreated (control) or IL-27-treated (treated) CD4+ T cells were incubated with replication-competent HIV-1 on ice for 80 min. The amount of bound HIV was monitored by p24 ELISA, and the total cell numbers were normalized by BCA protein assay. Relative HIV binding to the cells was compared between control and treated cells. Data show mean ± standard errors from three independent assays. (b) IL-27 pretreatment had no effect on HIV fusion/entry. Control and treated CD4+ T cells were infected with HIV-1 expressing BLaM-Vpr. The cleaved substrate of CCF2-AM by BLaM was measured by excitation of the substrate at 410 nm and emission shift from green (520 nm) to blue (450 nm) dye. Data show mean ± standard errors from three independent assays. (c and d) Control and treated cells were infected with a pseudotyped HIV-1 (HIV-LUC-G), and the copy number of HIV early (c) and late (d) reverse transcription products was determined by a real-time PCR. Data show mean ± standard errors from three independent assays. As a positive control of reverse transcription inhibition, control cells were treated with a reverse transcription inhibitor etravirine. (e) IL-27 treatment had no impact on HIV promoter activity. Control and treated cells were co-transfected with three constructs: a construct encoding HIV LTR with Firefly luciferase, a construct containing the HIV tat and a construct containing Renila luciferase [32] and then cultured for 48 h. Luciferase activity in the cells was monitored by Dual-Glo Luciferase Assay (Promega). Data show mean ± standard errors from three independent assays. BCA, bicinchoninic acid.
To define IL-27's role, an early reverse transcription product (R-U5) and a late reverse transcription product (U5-Gag) were semi-quantitated using qPCR. IL-27 inhibited the early reverse transcription product by 72 ± 7.0% and inhibited the late reverse transcription product by 64 ± 1.0% (n = 3) (Fig. 2c and d). In its natural state, HIV-1 provirus utilizes long terminal repeat (LTR) and Tat to mediate viral gene transcription. By cotransfecting the plasmid-encoding LTR-Luc and Tat [3], we were able to evaluate HIV promoter activity in control and IL-27-pretreated CD4+ T cells. Pretreating CD4+ T cells with IL-27 had no significant effect on viral transcription (n = 3, P = 0.15) (Fig. 2e). Thus, IL-27 pretreatment inhibits the reverse transcription step in the HIV-1 life cycle.
IL-27 regulates the posttranslational modification
HIV-1 utilizes host cell proteins as host-required factors to facilitate and complete its replication cycle. We hypothesized that IL-27 changes the expression of host factors in CD4+ T cells. To identify the potential host factor involved in IL-27-mediated HIV-1 inhibition, we compared gene expression between control and IL-27-treated T cells. A total of 25 000 genes were analyzed (Suppl. Fig. S2a), and only Radical S-Adenosyl Methionine Domain Containing Protein-2 (RSAD2) and Interferon-Induced Protein with Tetratricopeptide Repeats 3 (IFIT3) were upregulated more than four-fold in IL-27-treated cells. None of the genes were downregulated by more than four-fold. Real-time PCR confirmed increased gene expression in IL-27-treated CD4+ T cells (Suppl. Fig. S2b). The knockdown of either RSAD2 or IFIT3 by siRNA had no effect on the HIV-LUC-V infection (Suppl. Fig. S2c and d), suggesting that IL-27 may regulate an unknown host factor by PTM.
Since PTM regulation is an essential factor in cellular responses to stimuli and infection, IL-27 may mediate HIV-1 inhibition through PTM. We utilized a 2D-DIGE approach to determine the global protein modifications (Fig. 3a). A total of 17 proteins were either downregulated or upregulated in the 2D-DIGE analysis (Suppl. Table 1). No significant differences in the total protein expression of each selected proteins were found in 1D-gel western blot (Suppl. Fig. S3a). However, a 2D-western blot analysis revealed that IL-27 treatment significantly changed in PTM of Signal Transducer and Activator of Transcription-1, Myxovirus Resistance Protein-1, Tryptophanyl-TRNA Synthetase (WARS), and YB-1 (Fig. 3b, Suppl. Fig. S3b). In 2D gel-western blot YB-1 expression from control CD4+ T cells shifted the pI toward pH 4 from a pI of pH 9.87, indicating an additional negative charge on YB-1. In the IL-27-treated CD4+ T cells, YB-1 expression shifted from pH 4 through pH 7, with another smear of band around pH 9.5 (Fig. 3b). Quantitative analysis using ImageJ (NIH) demonstrated that unmodified YB-1 in control and treated cells represented 20 and 60%, respectively, of total YB-1 (n = 4) (Suppl. Fig. S3c).
Fig. 3.
Identification of host factors regulated by IL-27.
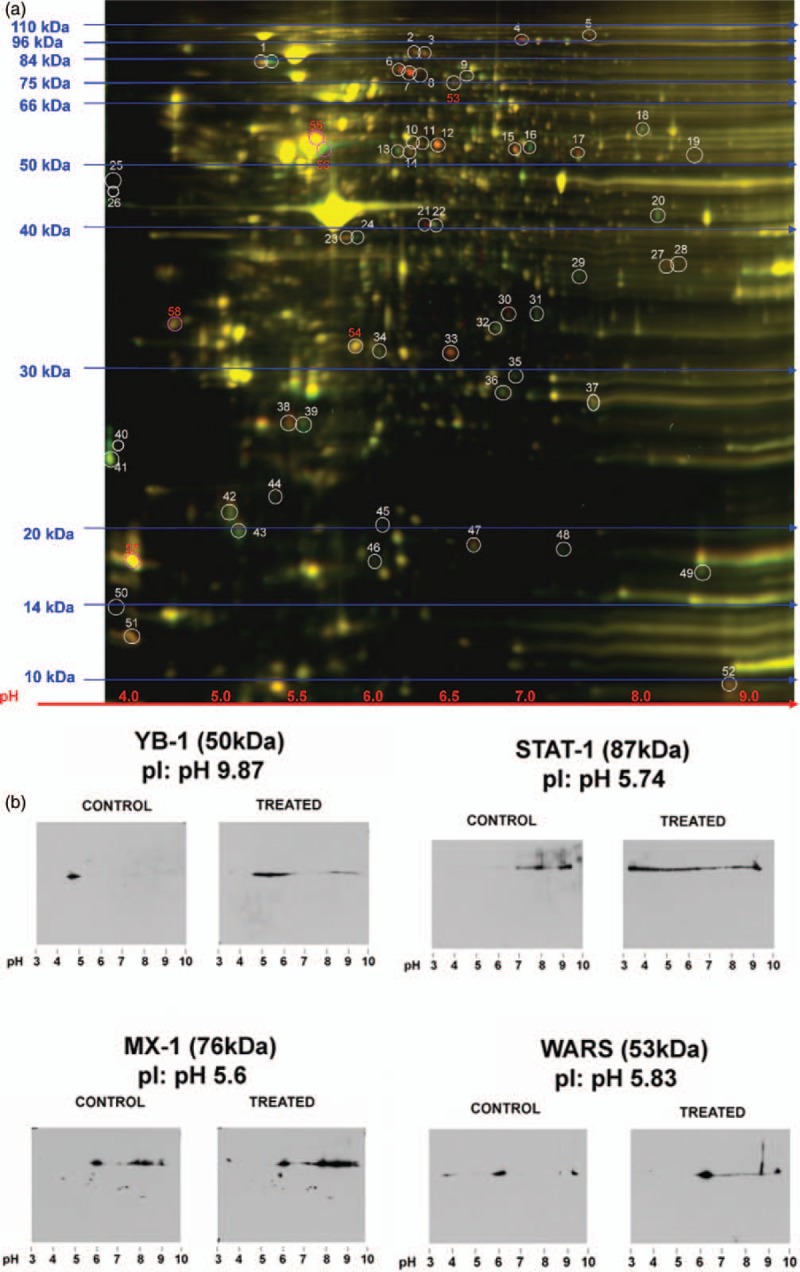
(a) 2-Dimensional difference-in-gel-electrophoresis gel showing control (green) and treated (red) samples. In total, 52 different spots were differentially regulated by IL-27 pretreatment (n = 3) (b) Western blot of 2-dimensional gel electrophoresis of differentially regulated proteins. Whole-cell lysate of untreated (control) and IL-27-treated (treated) CD4+ T cells were subjected to 2-dimensional gels followed by western blot using anti-Y box binding protein-1, Signal Transducer and Activator of Transcription-1 (STAT-1), Myxovirus Resistance Protein-1 (MX-1), or tryptophanyl-tRNA synthetase (WARS) antibodies. The isoelectric focusing (pI) points of Signal Transducer and Activator of Transcription-1, Myxovirus Resistance Protein-1, WARS, and Y box binding protein-1 are pH 5.74, pH 5.6, pH 5.8, and pH 9.87, respectively.
Y box binding protein-1 is an essential host-required factor for HIV infection
We sought the anti-HIV effects of the four posttranslationally regulated proteins using siRNA. Of the four, only siRNA-knockdown of YB-1 expression significantly inhibited the HIV-LUC-V infection (Fig. 4a and b).
Fig. 4.
Y box binding protein-1 is an essential host-required factor.
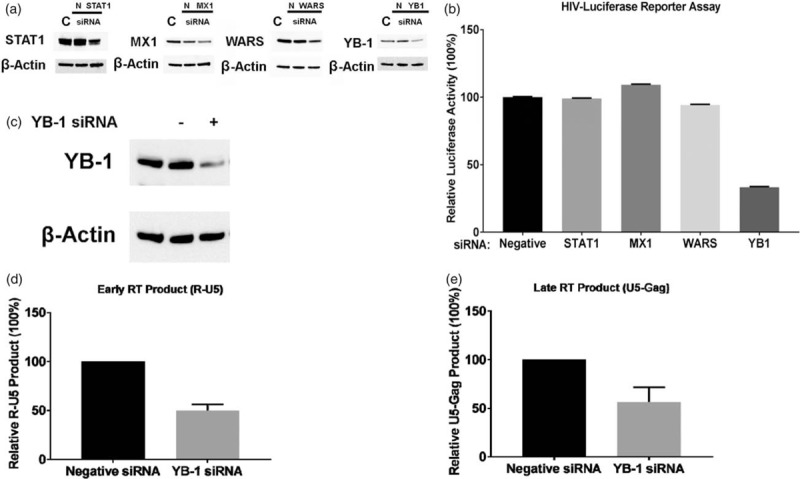
(a and b) CD4+ T cells were transfected with 300 nmol/l of a negative control small interfering RNA (siRNA), siRNA against Signal Transducer and Activator of Transcription-1 (STAT-1), Myxovirus Resistance Protein-1 (MX-1), WARS, or Y box binding protein-1 (YB-1), and then infected with pseudotyped HIV (HIV-LUC-V). The infected cells were cultured for 48 h, and then luciferase activity was measured. The results show means ± standard errors from three independent assay. (c) CD4+ T cells were transfected with a negative control siRNA or a siRNA against Y box binding protein-1, and the expression of Y box binding protein-1 protein in each cell was compared by western blot. (d and e) The siRNA-transfected CD4+ T cells were infected with VSV-G-pseudotyped HIV-luciferase virus. HIV reverse transcription products [the early reverse transcription product (R-U5) and the late reverse transcription product (U5-Gag)] in each cell were quantitated by real-time PCR.
We further analyzed the effect of knocking down YB-1 in reverse transcription product formation following HIV-LUC-V infection. Figure 4c shows the knockdown efficiency of YB-1 siRNA in CD4+ T cells (69% inhibition of YB-1 expression). This led to downregulation of the early and late reverse transcription product by 50 and 46%, respectively, compared with negative-control siRNA-transfected CD4+ T cells (Fig. 4d and e). This result suggested that YB-1 is a host requirement factor that supports the HIV reverse transcription reaction in CD4+ T cells during HIV replication.
Acetylated Y box binding protein-1 at lysine 118 residue is an essential host-required factor
To identify the PTM of YB-1 responsible for HIV replication, we overexpressed Flag-tagged wild-type-YB-1 in CD4+ T cells and cultured with IL-2 either alone or with IL-27. A 2D-gel western blot using an anti-Flag antibody demonstrated that overexpressed Flag-YB-1 exhibits a PTM pattern much like endogenous YB-1 (Fig. 5a and b). Therefore, we used the cell lysate of the YB-1-Flag-overexpressed CD4+ T cells for MC to determine PTM as described in the materials and methods.
Fig. 5.
Acetylated Y box binding protein-1 at lysine 118 residue is an essential host factor.
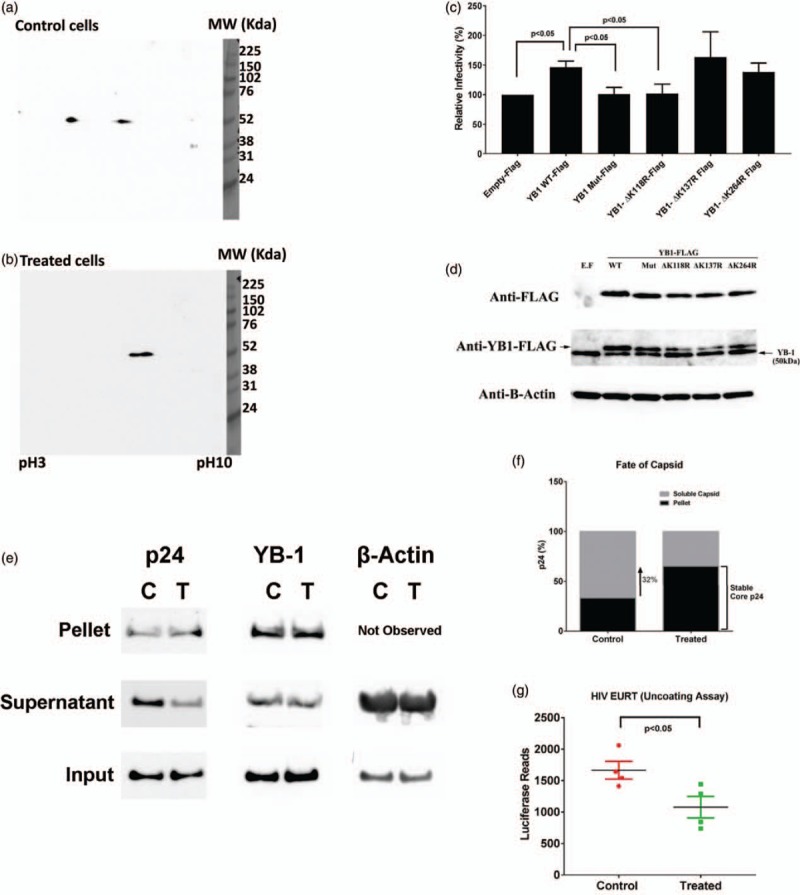
(a and b) wild-type-Y box binding protein-1-Flag-overexpressed CD4+ T cells were treated without (control) or with (treated) IL-27. We then performed 2-dimensional-gel electrophoresis followed by western blot using anti-Flag antibody. (c) CD4+ T cells were transfected with a series of expression vectors [wild-type-Y box binding protein-1-Flag, Y box binding protein-1-Flag lacking three acetylation sites (Y box binding protein-1-mutated-Flag), or Y box binding protein-1-Flag lacking a single acetylation site (Y box binding protein-1-ΔK118R-Flag, Y box binding protein-1-ΔK137R-Flag, Y box binding protein-1-ΔK264R)], and then infected with HIV-LUC-V. HIV infection activity was monitored by luciferase assay. Results show means ± SD from three independent assays. (d) Overexpression of Flag-Tag Y box binding protein-1 was detected by western blot using anti-Flag antibody. (e) Fate-of-capsid assay was performed as described in the materials and methods. Levels of HIV p24, Y box binding protein-1, and β-actin in pellet and supernatant fractions of control (C) and IL-27-treated CD4+ T cells (T) were detected by western blot using antip24, Y box binding protein-1, or β-actin antibody, respectively. Input is the sample obtained before the separation of soluble and stable core proteins using sucrose separation. (f) ImageJ analysis of soluble and stable core p24 protein amounts in C and T was performed and normalized to the total input protein amount. The stack bars indicate percentage of stable core form of p24 (black) in a total p24 in control cells (C) and treated cells (T). (g) HIV Entry/Uncoating assay based on core-packaged RNA availability and Translation assay was performed as described in the materials and methods. Data show mean ± standard errors from four independent assays.
Liquid chromatography–MC (LC/MS) revealed that lysine (K) residues K118, K137, and K264 in YB-1 were acetylated in control versus treated samples (Suppl. Fig. S4a–c) indicating that the acetylated YB-1 plays a key role in HIV reverse transcription step and IL-27-mediated anti-HIV effect is led by inhibition of the YB-1 acetylation, which may be caused by either suppression of acetylation or enhancement of deacetylation of YB-1 by IL-27. To further confirm that posttranslational regulation of YB-1 (inhibition of acetylation) acetylation is a key event in IL-27-mediated anti-HIV effect in CD4+ T cells, we pretreated the PHA-stimulated CD4+ T cells with a pan-histone deacetylase inhibitor, Panabinostat (35 nmol/l for 24 h at 37 °C), followed by IL-27 pretreatment, and infected the cells with HIV-LUC-V. Without Panabinostat treatment, IL-27 suppressed HIV replication to 47 ± 8.5% (n = 3) and the inhibitor treatment restored the HIV replication to 84.5 ± 2.7% (n = 3) (Suppl. Fig. S4d). This result illustrated that IL-27-mediated anti-HIV effect is led by induction of deacetylation of YB-1.
To identify the K residues responsible for HIV replication in YB-1, we designed YB-1 mutants with all three K residues mutated and three-mutants with a single K residue mutated to arginine (R). Following overexpression of wild-type-YB-1 and mutant-YB-1, CD4+ T cells were infected with HIV-LUC-V. If YB-1 played a key role in HIV infection in T cells, it was assumed that overexpression of YB-1 would increase HIV infection. As expected, the overexpression of YB-1-wild-type-Flag increased HIV infection (Fig. 5c). Overexpression of YB-1 mutants lacking K137 (YB-1-ΔK137R-Flag) or K264 (YB-1-ΔK264R-Flag) also increased HIV infection. In contrast, overexpression of the YB-1 mutant lacking K118 (YB-1-ΔK118R-Flag) and YB-1-Mut-Flag (all three mutations: K118R/K137R/K264R) had no impact on HIV infection (n = 3) (Fig. 5c). Overexpression of each YB-1 protein in CD4+ T cells was confirmed by western blot (Fig. 5d). Acetylation of lysine-118 residue in YB-1 is therefore key in HIV replication. When this site was mutated to disable acetylation, HIV replication was set to the basal level in CD4+ T cells.
IL-27 inhibits the uncoating of the HIV capsid core
After infection, the HIV core proteins must be disassembled in the host cell cytoplasm to release the reverse-transcribed viral genome. Therefore, the reverse transcription process is closely tied to the shedding of the viral capsid core proteins [33]. Since IL-27 inhibits the reverse transcription process in CD4+ T cells, we further investigated the role of IL-27 during the uncoating process. If IL-27 inhibited the uncoating steps, the amount of intact core protein (stable core) in IL-27-treated cells would be higher. The fate-of-capsid assay (Fig. 5e and f, Suppl. Fig. S5a–d) revealed that 34 ± 0.4% (n = 3) and 59 ± 5.3% (n = 3) of a total p24 proteins were in intact capsid core of control and IL-27-treated cells, respectively. This indicates that the capsid core shedding is 1.8-fold (P < 0.05) lesser in IL-27-treated CD4+ T cells. In the pelleted fraction (capsid core fraction), YB-1 protein was also detected, indicating either a potential interaction between capsid p24 and YB-1 protein during the viral capsid uncoating process or that YB-1 binds to HIV genomic RNA and gets pulled down in the pelleted fraction [20].
A recently established EURT using a reporter mRNA-coding luciferase [27] was applied to the control and IL-27-treated CD4+ T cells. Then, we measured the firefly luciferase activity and observed a 35% reduction in the treated CD4+ T cells (Fig. 5g, n = 4), indicating that control CD4+ T cells exhibit optimal uncoating and IL-27 treatment inhibits the uncoating process. We have confirmed that IL-27-pretreated CD4+ T cells inhibit the uncoating process of the HIV capsid core protein, thereby inhibiting the reverse transcription product formation and, ultimately, suppressing HIV-1 replication.
Discussion
Our study has demonstrated that IL-27 promotes PHA-activated T cells into HIV-1-resistant T cells. IL-27 blocks HIV-1 infection post entry through a PTM of YB-1 that suppresses HIV core protein uncoating and inhibits the reverse transcription process. Silencing YB-1 in T cells strongly inhibits HIV-1 infection. Conversely, overexpression of YB-1 in T cells markedly increases the susceptibility of T cells to infection. Thus, our results suggest that modulating YB-1 by PTM or expression is an effective way to render human T cells resistant to HIV-1 infection (all assays and results are summarized in Suppl. Table 2).
Several reports have indicated that capsid uncoating is required for reverse transcription complex (RTC) formation/activation [34,35], which collectively supports the idea that HIV core uncoating precedes the reverse transcription process. In our study, the PTM of YB-1 by IL-27-pretreatment profoundly delayed capsid core shedding, thereby inhibiting proper RTC activation (which is observed by the diminished reverse transcription products followed by inhibition of HIV-1 replication). This supports the finding that uncoating precedes the reverse transcription process. The capsid uncoating was only 40% lesser in IL-27-treated CD4+ T cells compared with control cells; however, the inhibition of HIV-1 replication in IL-27-treated CD4+ T cells was 65–70% (Fig. 1). This difference could be attributed to the fact that uncoating occurs early in the HIV-1 replication process, which eventually influences the reverse transcription, nuclear translocation, and integration. That has an additive effect on overall HIV-1 replication, which cumulatively amplifies the inhibitory effects. Therefore, IL-27-pretreatment inhibits HIV capsid core uncoating (Fig. 5e–g) and overall HIV-1 replication. In addition, when using HIV EURT virus, luciferase activity fell to 65% in IL-27-treated cells (n = 4) (Fig. 5g). Since we had observed low reverse transcription product (Fig. 4d and e) and inhibition of HIV-1 replication (Fig. 4b) in YB-1-silenced CD4+ T cells, we hypothesized that this leads to delayed HIV core uncoating in YB-1 k/d CD4+ T cells. In YB-1-silenced CD4+ T cells, increased accumulation of the pelleted p24 protein (1.42-fold) was observed compared with the negative siRNA control CD4+ T cells (Suppl Fig. S5e) which is an indication that in the absence of YB-1, there is suppressed or delayed uncoating of HIV p24 capsid core. This finding further supports our hypothesis that YB-1 supports HIV replication by facilitating uncoating of HIV p24 core protein and in the absence of YB-1, uncoating is delayed or suppressed in infected CD4+ T cells. To precisely define mechanism of acetylated YB-1 and role of residue of K118 on uncoating of core protein, we need further study.
HIV EURT assay detects an entered/uncoated HIV-1 that is based on the direct translation of a viral genomic RNA mimic coding a luciferase reporter that is delivered in target cells after viral-to-cellular membrane fusion in viral cores [27]. The HIV-1 entry/uncoating RNA genome coding Firefly luciferase (EU-repRNA-Luc) is added with the HIV-1 packaging sequence (Ψ) upstream of Firefly luciferase followed by a poly A signal so that it behaves as a genomic viral RNA and gets encapsidated into virion particles. Upon virion maturation and Gag processing, this genome is contained within the viral core. Once the virion is delivered via infection into target cells, it is based on the two states of the capsid core (opened/partially opened and closed). Where opened/partially opened, it allows the cellular ribosome machinery to translate luciferase protein, reflecting the uncoating process. If the core is closed, the cellular translation machinery, lacking access to EU-repRNA-Luc, will not translate luciferase protein. Thus, the luciferase assay reflects the uncoating process (the fate and status of viral core) in the HIV EURT assay. Using both the fate-of-capsid assay and HIV EURT assay [27], we have confirmed that IL-27 pretreatment inhibits the HIV capsid core uncoating process; consequently, this could be the reason for reverse transcription product inhibition and, ultimately, HIV replication inhibition.
Recent data indicate that nucleotides required for reverse transcription are channeled into the core through electrostatic channels present in the capsid hexamer [36]. Thus, in control CD4+ T cells, the dynamic capsid pores (with optimal uncoating process) may support nucleotide import and enhance the encapsidated DNA synthesis. Conversely, in treated CD4+ T cells, the delayed uncoating process may disrupt the dynamics of nucleotide import, thereby inhibiting the reverse transcription products. However, this is just speculation, and a measure of incoming nucleotides into the capsid core is necessary to provide a stronger argument.
Rankovic et al. provided a strong argument that, during the reverse transcription process, the stiffness of the core increases immensely, which mechanically triggers capsid disassembly by stressing the capsid lattice, leading to core breakage [33]. Furthermore, they supported the idea that HIV uncoating may occur only after the core encounters a cytoplasmic environment favorable for completing reverse transcription [33]. Overall, there are several theories of HIV uncoating, and it remains poorly understood.
The YB-1 protein consists of 324 amino acids composed of three domains: the N-terminal Alanine/proline-rich domain (amino acids 1–54), Cold Shock Domain (CSD) (amino acids 55–128), and C-terminal domain (amino acids 129–324) [37]. From LC/MS, we found that lysine K-118, K-137, and K-264 residues are acetylated in CD4+ T cells permissive to HIV-1 replication and that this acetylation is inhibited in IL-27-treated CD4+ T cells resistant to HIV-1 replication. K-118 is a key residue for HIV-1 replication, which shows the importance of YB-1's CSD domain. Although the role of PTM of YB-1 has been well studied in cancer research [38] PTM of YB-1 in HIV-1 replication has not been reported. Our report provides the first evidence that acetylated YB-1 supports HIV-1 replication in human primary CD4+ T cells.
In conclusion, our data indicate that YB-1 is posttranslationally regulated by IL-27 in activated CD4+ T cells to inhibit HIV-1 replication. IL-27-mediated posttranslational regulation of YB-1 has a profound effect on the early stages of HIV replication inhibition. It is known that YB-1 is a multifunctional protein that plays important roles in transcriptional and translational regulation, DNA repair, drug resistance, and stress responses to extracellular signals [39]. A recent study using T-cell lines reported that YB-1 helps support HIV replication in cell line [40], as well, indicating that YB-1 supports HIV replication not only in primary T cells but also in the cell line. Whether or not the anti-HIV effect may have clinical activity, especially for the perspective of the persistent reservoir, requires further study. However, our study provides a potent novel target of posttranslational regulation of YB-1 in CD4+ T cells for anti-HIV therapy. It is possible that IL-27 could be used as a therapeutic intervention, especially in patients infected with multidrug resistant virus.
Acknowledgements
The authors thank Dr H. Clifford Lane for supporting and discussing this project; B. Sherman, X. Hu, and H. Sui for discussing the data and providing critical review of the article; and L. Fuentes and M. Bosche for running assays. The authors also thank Dr Andrea Cimarelli for kindly providing us the pEU-repRNA plasmid for the HIV EURT assay.
Author contributions: D.P. and T.I. designed the study. D.P., Q.C., S.G., J.W.A., A.H., S.D., and T.I. performed research and analyzed data. J.Y., R.L.H., and T.A. analyzed the data. All authors approved the final version of the article.
The project has been funded in whole or in part with federal funds from the National Cancer Institute, National Institutes of Health, under Contract No. HHSN261200800001E. The content of this publication does not necessarily reflect the views or policies of the Department of Health and Human Services, nor does mention of trade names, commercial products, or organizations imply endorsement by the US Government. This research was supported in part by the National Institute of Allergy and Infectious Disease.
Conflicts of interest
There are no conflicts of interest.
Supplementary Material
References
- 1.Fakruddin JM, Lempicki RA, Gorelick RJ, Yang J, Adelsberger JW, Garcia-Pineres AJ, et al. Noninfectious papilloma virus-like particles inhibit HIV-1 replication: implications for immune control of HIV-1 infection by IL-27. Blood 2007; 109:1841–1849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Imamichi T, Yang J, Huang DW, Brann TW, Fullmer BA, Adelsberger JW, et al. IL-27, a novel anti-HIV cytokine, activates multiple interferon-inducible genes in macrophages. AIDS 2008; 22:39–45. [DOI] [PubMed] [Google Scholar]
- 3.Dai L, Lidie KB, Chen Q, Adelsberger JW, Zheng X, Huang D, et al. IL-27 inhibits HIV-1 infection in human macrophages by down-regulating host factor SPTBN1 during monocyte to macrophage differentiation. J Exp Med 2013; 210:517–534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chen Q, Swaminathan S, Yang D, Dai L, Sui H, Yang J, et al. Interleukin-27 is a potent inhibitor of cis HIV-1 replication in monocyte-derived dendritic cells via a type I interferon-independent pathway. PLoS One 2013; 8:e59194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Pflanz S, Timans JC, Cheung J, Rosales R, Kanzler H, Gilbert J, et al. IL-27, a heterodimeric cytokine composed of EBI3 and p28 protein, induces proliferation of naive CD4+ T cells. Immunity 2002; 16:779–790. [DOI] [PubMed] [Google Scholar]
- 6.Pirhonen J, Siren J, Julkunen I, Matikainen S. IFN-alpha regulates Toll-like receptor-mediated IL-27 gene expression in human macrophages. J Leukoc Biol 2007; 82:1185–1192. [DOI] [PubMed] [Google Scholar]
- 7.Liu J, Guan X, Ma X. Regulation of IL-27 p28 gene expression in macrophages through MyD88- and interferon-gamma-mediated pathways. J Exp Med 2007; 204:141–152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zhang J, Qian X, Ning H, Yang J, Xiong H, Liu J. Activation of IL-27 p28 gene transcription by interferon regulatory factor 8 in cooperation with interferon regulatory factor 1. J Biol Chem 2010; 285:21269–21281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Swaminathan S, Hu Z, Rupert AW, Higgins JM, Dewar RL, Stevens R, et al. Plasma interleukin-27 (IL-27) levels are not modulated in patients with chronic HIV-1 infection. PLoS One 2014; 9:e98989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Swaminathan S, Dai L, Lane HC, Imamichi T. Evaluating the potential of IL-27 as a novel therapeutic agent in HIV-1 infection. Cytokine Growth Factor Rev 2013; 24:571–577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zhou H, Xu M, Huang Q, Gates AT, Zhang XD, Castle JC, et al. Genome-scale RNAi screen for host factors required for HIV replication. Cell Host Microbe 2008; 4:495–504. [DOI] [PubMed] [Google Scholar]
- 12.Brass AL, Dykxhoorn DM, Benita Y, Yan N, Engelman A, Xavier RJ, et al. Identification of host proteins required for HIV infection through a functional genomic screen. Science 2008; 319:921–926. [DOI] [PubMed] [Google Scholar]
- 13.Konig R, Zhou Y, Elleder D, Diamond TL, Bonamy GM, Irelan JT, et al. Global analysis of host-pathogen interactions that regulate early-stage HIV-1 replication. Cell 2008; 135:49–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Shirakawa K, Takaori-Kondo A, Yokoyama M, Izumi T, Matsui M, Io K, et al. Phosphorylation of APOBEC3G by protein kinase A regulates its interaction with HIV-1 Vif. Nat Struct Mol Biol 2008; 15:1184–1191. [DOI] [PubMed] [Google Scholar]
- 15.Cribier A, Descours B, Valadao AL, Laguette N, Benkirane M. Phosphorylation of SAMHD1 by cyclin A2/CDK1 regulates its restriction activity toward HIV-1. Cell Rep 2013; 3:1036–1043. [DOI] [PubMed] [Google Scholar]
- 16.Farzan M, Babcock GJ, Vasilieva N, Wright PL, Kiprilov E, Mirzabekov T, et al. The role of posttranslational modifications of the CXCR4 amino terminus in stromal-derived factor 1 alpha association and HIV-1 entry. J Biol Chem 2002; 277:29484–29489. [DOI] [PubMed] [Google Scholar]
- 17.Yu X, Yu Y, Liu B, Luo K, Kong W, Mao P, et al. Induction of APOBEC3G ubiquitination and degradation by an HIV-1 Vif-Cul5-SCF complex. Science 2003; 302:1056–1060. [DOI] [PubMed] [Google Scholar]
- 18.Didier DK, Schiffenbauer J, Woulfe SL, Zacheis M, Schwartz BD. Characterization of the cDNA encoding a protein binding to the major histocompatibility complex class II Y box. Proc Natl Acad Sci U S A 1988; 85:7322–7326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Evdokimova VM, Wei CL, Sitikov AS, Simonenko PN, Lazarev OA, Vasilenko KS, et al. The major protein of messenger ribonucleoprotein particles in somatic cells is a member of the Y-box binding transcription factor family. J Biol Chem 1995; 270:3186–3192. [DOI] [PubMed] [Google Scholar]
- 20.Mu X, Li W, Wang X, Gao G. YB-1 stabilizes HIV-1 genomic RNA and enhances viral production. Protein Cell 2013; 4:591–597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Li Y, Frederick KM, Haverland NA, Ciborowski P, Belshan M. Investigation of the HIV-1 matrix interactome during virus replication. Proteomics Clin Appl 2016; 10:156–163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Adachi A, Gendelman HE, Koenig S, Folks T, Willey R, Rabson A, et al. Production of acquired immunodeficiency syndrome-associated retrovirus in human and nonhuman cells transfected with an infectious molecular clone. J Virol 1986; 59:284–291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Connor RI, Chen BK, Choe S, Landau NR. Vpr is required for efficient replication of human immunodeficiency virus type-1 in mononuclear phagocytes. Virology 1995; 206:935–944. [DOI] [PubMed] [Google Scholar]
- 24.He J, Choe S, Walker R, Di Marzio P, Morgan DO, Landau NR. Human immunodeficiency virus type 1 viral protein R (Vpr) arrests cells in the G2 phase of the cell cycle by inhibiting p34cdc2 activity. J Virol 1995; 69:6705–6711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Imamichi T, Berg SC, Imamichi H, Lopez JC, Metcalf JA, Falloon J, et al. Relative replication fitness of a high-level 3′-azido-3′-deoxythymidine-resistant variant of human immunodeficiency virus type 1 possessing an amino acid deletion at codon 67 and a novel substitution (Thr→Gly) at codon 69. J Virol 2000; 74:10958–10964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cavrois M, Neidleman J, Yonemoto W, Fenard D, Greene WC. HIV-1 virion fusion assay: uncoating not required and no effect of Nef on fusion. Virology 2004; 328:36–44. [DOI] [PubMed] [Google Scholar]
- 27.Da Silva Santos C, Tartour K, Cimarelli A. A novel entry/uncoating assay reveals the presence of at least two species of viral capsids during synchronized HIV-1 infection. PLoS Pathog 2016; 12:e1005897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.McClure MO, Moore JP, Blanc DF, Scotting P, Cook GM, Keynes RJ, et al. Investigations into the mechanism by which sulfated polysaccharides inhibit HIV infection in vitro. AIDS Res Hum Retroviruses 1992; 8:19–26. [DOI] [PubMed] [Google Scholar]
- 29.Poudyal D, Herman A, Adelsberger JW, Yang J, Hu X, Chen Q, et al. A novel microRNA, hsa-miR-6852 differentially regulated by interleukin-27 induces necrosis in cervical cancer cells by downregulating the FoxM1 expression. Sci Rep 2018; 8:900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sowrirajan B, Saito Y, Poudyal D, Chen Q, Sui H, DeRavin SS, et al. Interleukin-27 enhances the potential of reactive oxygen species generation from monocyte-derived macrophages and dendritic cells by induction of p47 (phox). Sci Rep 2017; 7:43441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yang Y, Luban J, Diaz-Griffero F. The fate of HIV-1 capsid: a biochemical assay for HIV-1 uncoating. Methods Mol Biol 2014; 1087:29–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Oguariri RM, Dai L, Adelsberger JW, Rupert A, Stevens R, Yang J, et al. Interleukin-2 inhibits HIV-1 replication in some human T cell lymphotrophic virus-1-infected cell lines via the induction and incorporation of APOBEC3G into the virion. J Biol Chem 2013; 288:17812–17822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Rankovic S, Varadarajan J, Ramalho R, Aiken C, Rousso I. Reverse transcription mechanically initiates HIV-1 capsid disassembly. J Virol 2017; 91:e00289-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Auewarakul P, Wacharapornin P, Srichatrapimuk S, Chutipongtanate S, Puthavathana P. Uncoating of HIV-1 requires cellular activation. Virology 2005; 337:93–101. [DOI] [PubMed] [Google Scholar]
- 35.Mamede JI, Cianci GC, Anderson MR, Hope TJ. Early cytoplasmic uncoating is associated with infectivity of HIV-1. Proc Natl Acad Sci U S A 2017; 114:E7169–E7178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Jacques DA, McEwan WA, Hilditch L, Price AJ, Towers GJ, James LC. HIV-1 uses dynamic capsid pores to import nucleotides and fuel encapsidated DNA synthesis. Nature 2016; 536:349–353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Matsumoto K, Wolffe AP. Gene regulation by Y-box proteins: coupling control of transcription and translation. Trends Cell Biol 1998; 8:318–323. [DOI] [PubMed] [Google Scholar]
- 38.Prabhu L, Hartley A-V, Martin M, Warsame F, Sun E, Lu T. Role of posttranslational modification of the Y box binding protein 1 in human cancers. Genes Dis 2015; 2:240–246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kohno K, Izumi H, Uchiumi T, Ashizuka M, Kuwano M. The pleiotropic functions of the Y-box-binding protein, YB-1. BioEssays 2003; 25:691–698. [DOI] [PubMed] [Google Scholar]
- 40.Weydert C, van Heertum B, Dirix L, De Houwer S, De Wit F, Mast J, et al. Y-box-binding protein 1 supports the early and late steps of HIV replication. PLoS One 2018; 13:e0200080. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.