

Abstract
Protein-catalyzed capture agents (PCCs) are synthetic and modular peptide-based affinity agents that are developed through the use of single generation in situ click chemistry screens against large peptide libraries. In such screens, the target protein, or a synthetic epitope fragment of that protein, provides a template for selectively promoting the non-copper catalyzed azide-alkyne dipolar cycloaddition click reaction between either a library peptide and a known ligand, or a library peptide and the synthetic epitope. The development of epitope-targeted PCCs was motivated by the desire to fully generalize pioneering work from the Sharpless and Finn groups in which in situ click screens were used to develop potent, divalent enzymatic inhibitors. In fact, a large degree of generality has now been achieved. Various PCCs have demonstrated utility for selective protein detection, as allosteric or direct inhibitors, as modulators of protein folding, and as tools for in vivo tumor imaging. We provide a historical context for PCCs, and place them within the broader scope of biological and synthetic aptamers. The development of PCCs is presented as: (i) Generation I PCCs, which are branched ligands engineered through an iterative, non-epitope targeted process, and (ii) Generation II PCCs, which are typically developed from macrocyclic peptide libraries and are precisely epitope targeted. We provide statistical comparisons of Generation II PCCs relative to monoclonal antibodies in which the protein target is the same. Finally, we discuss current challenges and future opportunities of PCCs.
Keywords: In situ click chemistry, affinity reagents, epitope-targeting, co-operative binding
Graphical Abstract

1. INTRODUCTION
In 2002, the Sharpless and Finn groups reported on target-guided in situ click chemistry.1 Over the past decade we have sought to broadly generalize that approach to create a class of synthetic ligands which we have called protein catalyzed capture agents (PCCs) (Figure 1), and which are the subject of this Chemical Review.
Figure 1.
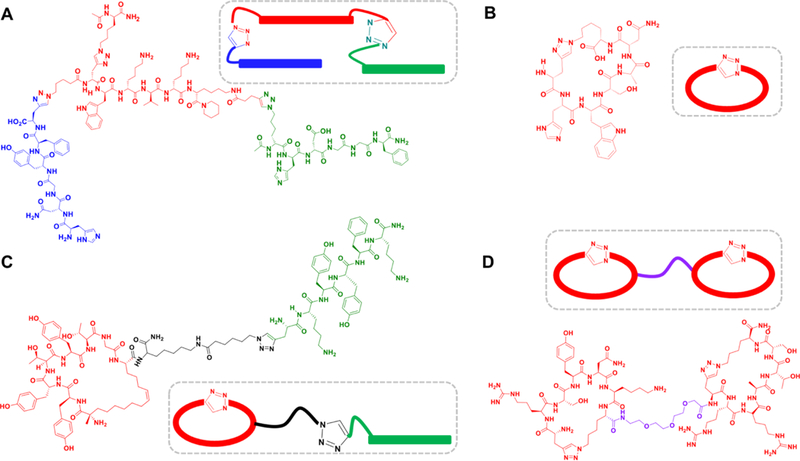
Examples of PCCs developed through chemical epitope targeting. The individual colored components of the PCCs are developed separately. Red: 1° (initial anchor) ligand, Green: 2° ligand, Blue: 3° ligand (A) Linear triligand PCC against the C-terminal S474 epitope of Akt2. The 1° ligand was developed through chemical epitope targeting, while the 2° and 3° components were identified through iterative in situ click screening steps. (B) Macrocyclic PCC developed against the lactate dehydrogenase biomarker for Plasmodium falciparum. (C) Biligand PCC against human Akt2 that contains a macrocyclic anchor ligand and a linear 2° ligand. (D) Biligand that cooperatively binds interleukin-17F comprised of two macrocyclic anchor peptides separately developed against adjacent epitopes, and then connected with a 3 unit polyethylene glycol linker to optimize cooperative binding interactions.171
The original in situ click approach used the substrate binding site of an enzyme target as a scaffold for the highly selective promotion of the Huisgen 1,3-dipolar cycloaddition ‘click’ reaction between specific elements of two small molecule libraries. The reaction, which resembles the (non-catalyzed) azide-alkyne cycloaddition reaction,2 is better viewed as a highly selective variant of the well-known Cu(I) catalyzed click reaction,3 with the key difference that the hydrophobic binding pocket of the acetylcholinesterase (AChE) enzyme was used in place of the Cu(I) catalyst. We review the Sharpless/Finn approach in some detail below, with just a brief description in this introduction.
AChE is a highly active enzyme that catalyzes the breakdown of certain choline esters that function as neurotransmitters. The active site of AChE comprises 2 subsites — an anionic site and an esteratic subsite. The basic in situ approach was to build small libraries of molecules that bind to either of these two subsites, and to modify those libraries so binders to subsite I present an acetylene group, and binders to subsite II present an azide group. The libraries are then incubated with AChE. If elements from each library sit within the AChE binding site in just the right orientation, then the click reaction is promoted, with reaction products detected using LC/MS.4 A powerful aspect of the method was that the resultant clicked inhibitor exhibited strong binding cooperativity, meaning that the binding energies of the two moieties were additive, and so the affinity of the biligand approached the full product of the affinities of the individual moieties.
Sharpless and Finn posited that any enzyme with an active site comprised of two subsites should be similarly targetable. In fact, they and their collaborators demonstrated a level of generality by similarly targeting several other proteins.5,6 In our labs, we have expanded on their original concept to demonstrate significantly broader levels of generality, with the goal of developing a rapid, all-synthetic approach towards producing protein capture agents that contain the best traits of biological capture agents, such as monoclonal antibodies (mAbs), while retaining advantages achievable through chemically synthesized small molecules. This work has included exploring the feasibility of the in situ click approach for site-selectively targeting virtually any exposed region of a protein, rather than just a hydrophobic, bifurcated binding pocket. A consequence of this effort is that the tertiary structure of the folded protein can now be viewed as a landscape that can be targeted in novel ways, with interesting parallels to biological agents. For example, the ability to target surface-exposed and continuous epitopes (i.e., contiguous polypeptide sequences) bears similarities to methods used in the development of mAbs.7–9 Further, continuous epitopes, as displayed on antigen presenting cells, are also the targets of T cell recognition.10,11 We have also shown that discontinuous epitopes (i.e., non-contiguous polypeptide sequences) can be targeted,12,13 and this draws parallels to many naturally occurring B cell epitopes.14
The original Sharpless approach required custom built molecular libraries, designed using knowledge of the protein target. This is an area that we have also sought to generalize. Specifically, we have explored the use of small linear and cyclic synthetic one-bead-one-compound (OBOC) comprehensive peptide libraries15 as easily prepared molecular basis sets from which the capture agents are selected, via a single generation in situ click screen. Further, by separately targeting epitopes that are proximal in the tertiary structure of the protein, the types of cooperative binding effects observed by Sharpless’ group may be achieved, but using a simplified and more general algorithm.13 The algorithm relies upon knowing which epitopes were targeted and the distance between those epitopes in the folded protein structure. That distance is then bridged using an appropriate length molecular linker. A net result of these and other developments is that it is now possible to identify and validate molecules that bind to specific regions of specific proteins, typically with low nanomolar-level affinities, all in about a period of a month. The extension to cooperative binding biligands to yield binders with picomolar-range affinities is a similarly rapid extension of the core PCC technology.
The ability to prepare epitope targeted PCCs has allowed us to target novel epitopes, such as the recognition sites of antibodies,16 phosphorylated epitopes,17 or oncogenic mutation sites,18 and to perturb protein functions ranging from enzymatic activity to protein folding and misfolding using novel allosteric19,20 and direct inhibition mechanisms.12 The chemical flexibility of small synthetic peptides has permitted strategies designed to promote cell penetration, or precisely tune for cooperative binding interactions. Finally, the focus on generalizing and standardizing the PCC production method has yielded methods for increasingly rapid production of high quality PCC ligands against specific targets. Figure 1 shows four PCCs that reflect conceptual and chemical advances over the past few years. The PCC of Fig 1A is the most labor intensive to develop, while the PCC of Fig 1D is the highest performing, and reflects the current state-of-the-art in rapid PCC production.
In this Chemical Review, we discuss these advances in PCC technology, broadly classifying early-stage PCCs, which were not epitope targeted, as Generation I PCCs, while classifying epitope targeted PCCs as Generation II. We first place PCCs within a historical context, and within the broader field of other aptamer approaches.
2. BIOLOGICAL AND SYNTHETIC APTAMERS
The term ‘aptamer’ refers to an oligonucleotide21,22 or peptide23 molecule or macromolecule that binds to a particular target molecule.24 PCCs comprise just one of a wide variety of aptamer technologies that span from biological reagents to all synthetic systems. Several motivators drive the development of non-antibody affinity reagents, including cost, increased stability,25–27 ease of preparation, reduced molecular weight, and tunable and potentially high avidity for the target. The ability to further engineer the capture agent for specific in vitro or in vivo tasks may be incorporated into certain aptamer platforms. Our goal in this section is to briefly cover representative peptide (or protein)-based biological and synthetic aptamer systems to provide context for PCCs. We focus on the chemistry and biochemistry of library construction, the basic scaffold structure of the different libraries, the screening methodologies, and some illustrative examples of how various aptamers have been used. We first discuss the biological aptamer classes known as DARPins28 and affimers,29 and then synthetic peptide ligands15 and, as an illustrative class of synthetic non-natural building blocks, peptoids. The importance of aptamers was highlighted by the recognition of George Smith, who shared the 2018 Nobel Prize in Chemistry for his work on phage display methods.30
2.1. DARPINs
DARPINs, which is an acronym for designed ankyrin repeat proteins,28,31 and affimers,29 are both genetically engineered antibody mimics. Repeat proteins occur naturally, with some serving as components of the adaptive immune system in jawless vertebrates.32 They are typically comprised of small, repeating structural motifs of 20–50 amino acids, often stacking together to form elongated structures that are stabilized by, for example, hydrogen bonds. A feature common to such repeat proteins and peptide macrocycles is that the structural constraints of the (macro)molecule reduce the entropy loss upon target binding, which is helpful for achieving high target binding affinities. Repeat proteins include the leucine-rich repeat family of proteins,33 ankyrin,33, and tetratricopeptide repeats.34 The development of protein libraries from these repeat proteins is a non-trivial exercise, as maintaining the overall protein folded structure and stability are a key consideration. Thus, most of the residues are conserved, while perhaps 6–10 are subjected to varying levels of mutational randomization (Figure 2A). For each randomized position, the side chain diversity is encoded to mimic the diversity seen in naturally occurring motifs. This assumes that the diversity arises from competition between maintaining the structural context of the protein, and maximizing the potential for protein-protein interactions.35 A typical library may contain 109 proteins, but each element ideally will have a similar folded structure, with similar solubility and stability characteristics. Non-natural or artificial amino acids are not easily incorporated into such libraries. While ribosome display28,36 was initially employed as a selection strategy for DARPins, the libraries were later adapted for generation using phage display methods.37 This enables individual screen hits to be sorted, and then identified via DNA sequencing. Multi-generation screens are typically carried out to find the best binders, using approaches similar to the SELEX method used for DNA aptamer development.38,39 DARPins have been developed against relatively large protein epitopes or domains. A recent illustrative example is the development of binders to the HIV-1 Gag polyprotein. The screened target was a 90-mer polypeptide target representing L343-F433 of the Pr55Gag sequence.40 DARPins typically have a molecular weight in the 15–20 kDa range, which is about 10-fold lighter than a typical IgG antibody, and about half the weight of a minibody.41 A specific DARPin can be co-expressed with its target within a cell, and in this way can be readily used for in-cell targeting.42 However, DARPins are, in general, not cell penetrant.
Figure 2.

Representative biological aptamers. (A) DARPin scaffold, with constant N- and C-terminal modules, and 3 copies of the ankyrin repeat module, with the sequence of that module shown below. The library is built through substitutions at the residues indicated by the red ‘x’. (B) The Adhiron affimer scaffold, which was derived from a large number of plant cystatin sequences. The library is built through substitutions at the purple-colored loops, which are expanded beyond the 7 indicated residues. The figures in A and B are from Tamaskovic, R. et al., Methods … 2012, 503, 101 (ref. 172) and Tiede, C. et al., Elife 2017, 6, e24903 (ref. 44), respectively).
2.2. AFFIMERS
Affimers are a second class of proteins that can be engineered into large libraries,43 and used for screening using phage display methods.44 Affimers are from a class of engineered proteins broadly referred to as monobodies. They are generated by altering solvent exposed, non-structured portions of a functionally inert protein (the scaffold) (Figure 2B). An example is the Adhiron scaffold,45 which was derived from the alignment of 57 plant cystatin sequences to yield a thermally stable construct. The scaffold contains two loops, originally with a total of 7 residues. These loops were targeted for engineering, with each expanded to 9 residues, yielding a library that displays 1010 clones. Like the case of DARPins, affimer scaffolds are adapted to phage display methods to permit multi-generation screens for affinity maturation. The development of affimer libraries is a matured process.
As with DARPins, affimers are not particularly suited to inhibiting intracellular targets. In-cell inhibition assays are often done by genetically encoding the affimer into the cells of interest.46 Further, it appears that DARPins and, to a lesser extent, affimers, exhibit preferential binding to convex regions of the protein target. This is likely related to the structural rigidity of the relevant scaffolds, and the nature of how the variable region of the aptamer is presented to the target.47 Several high performance affimers designed as immunohistochemical staining reagents, kinase inhibitors, and diagnostic capture agents have been reported.
Another similar development to affimers are Affibodies, which are small engineered proteins comprised of a parental 58 amino acids in a three-helix bundle, called the “Z domain.”48 Combinatorial libraries of Affibodies can be generated via phage and staphylococcal surface display.49 Chemically synthesized Affibodies are roughly 6 kDa in size and have been shown to fold rapidly into their correct structures.50 As alternatives to mAbs, Affibodies have demonstrated affinities in the subnanomolar range.51
Affibodies are an example of protein engineered affinity agents that have myriad applications. For example, Eigenbrot, et al. demonstrated one of the first examples of Affibody recognition of tumor associated targets through targeting HER2 (Kd = 22 pM).52 In 2006, Orlova, et al. reported on a HER2-specific Affibody that was successfully used to image tumor expression of HER2 in vivo.53 In 2007, the conjugation of a HER2-specific affibody to 177Lu was shown to have antitumor properties.54 Since their development, Affibody scaffolds have found utility as imaging (i.e., diagnostic) and therapeutic against with potential applications for clinical use.51
2.3. REPRESENTATIVE SYNTHETIC APTAMERS
Peptoids.
Peptoids, or N-substituted polyglycine polymers, are a class of synthetic polymer that was first disclosed in 1992.55,56 While similar to peptides in terms of having a polyamide backbone, the side chains migrated from the α-carbon to the amide nitrogen. As a result, the biomimetic polymer has an achiral backbone with tertiary amides that are completely resistant to proteolytic degradation. Peptoids are prepared via a modified solid-phase synthesis method called the submonomer method, which has been adapted to automated synthesis (Scheme 1).57 This iterative two-step process involves acetylation of the free amine group with a 2-halo acetic acid, typically 2-bromo acetic acid, followed by SN2 displacement of the halogen with a primary amine. The use of a primary amine to introduce functionality greatly expands the pool of potential side chains beyond the original twenty side chains found in α-amino acids. As a result, non-canonical side chains can be readily introduced. Most early reports of peptoids were linear, but recently cyclization methods have been developed for both solution phase and on-bead cyclization58 including head-to-tail amide formation,59–61 side-chain-tail cyclization,62 side-chain click reactions,63,64 side-chain ring-closing metathesis (RCM),65 and triazine thioether formation.66,67 Peptoid libraries are prepared using the aforementioned split-and-mix protocol to prepare OBOC libraries ranging in size from modest (~103−4) to large (~105−6) as the pool of viable amines increases. Unlike the biological aptamers, peptoids could easily be modified for use in PCC in situ click screens.
Scheme 1.
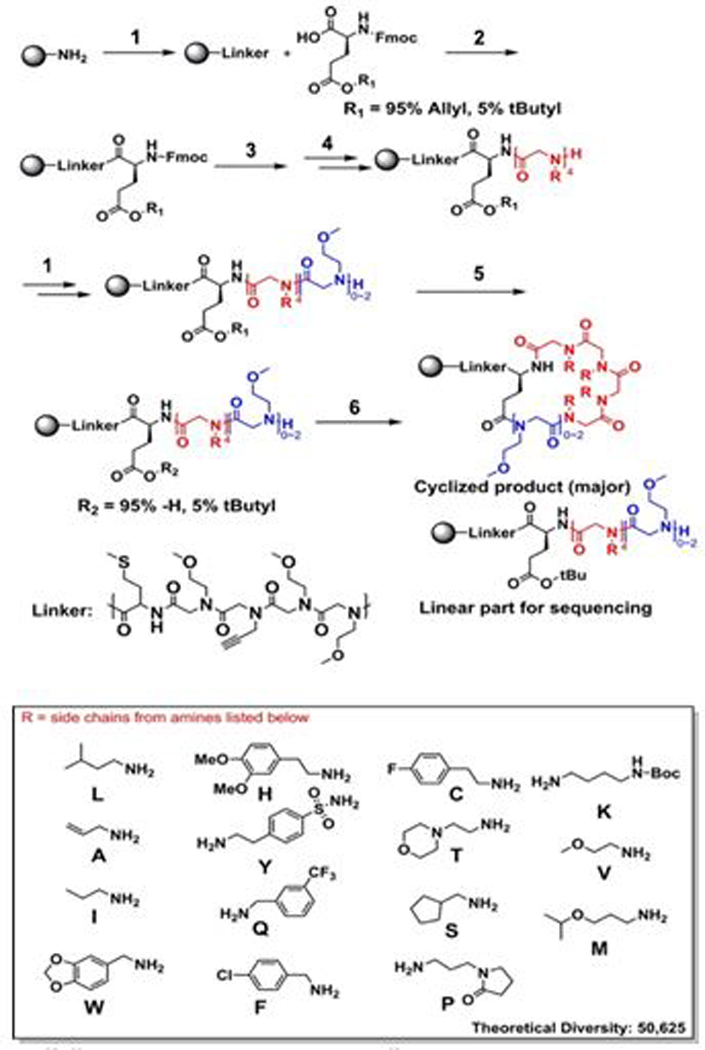
Preparation of linear and cyclic peptoid libraries for screening. The synthesis conditions for each reaction step are as follows: (1) Standard peptoid synthesis (see the Supporting Information of Ref. 73 for details). (2) 4:4:4 equiv of DIC/Oxyma/glutamic acid mixture (95% Fmoc-Glu(OAll)-OH, 5% Fmoc-Glu-(OtBu)-OH) in DMF, 2 h × 2. (3) 20% piperidine in DMF, 15 min × 2. (4) Split-pool synthesis of four peptoid units using 15 primary amines listed in the box. (5) Pd(0)(PPh3)4, 2 equiv in 37:2:1 CHCl3− AcOH-NMM under argon, 2 h. (6) PyBOP 4 equiv., DIEA 10 equiv. in DMF, 2h × 2 (Adapted from Gao & Kodadek, ACS Comb Sci 2015; Fig 1). 73
Peptoid libraries, DARPins, and affimers can be screened using methods that select for target binding, and with interferent screens to improve binding specificity. A classic OBOC library screen protocol for peptoid libraries was reported by Kodadek et al. that invokes a secondary screening step of hit peptoid beads to distinguish true hits from false positives.68 The same group also reported on a two-color cell-based procedure for identifying peptoids that bind to overexpressed membrane proteins,69 but not normally expressing control cells. Another screening protocol involves incubation of the peptoid library with a His6-tagged target protein that can be captured with anti-His6 on magnetic beads.44
The hits from a library screen are sequenced via Edman degradation on bead or tandem mass-spectrometry, after liberation of the molecules from beads.70 In the case of cyclic peptoids, a methionine residue is incorporated both into the macrocycle and adjacent to the resin linker to allow cleavage off the beads.68 Validation of the scaled-up hits involves measuring their desired performance off-bead and can occur after spotting the hits onto a spin-coated glass slide in a microarray format. Weidemann et al. disclosed a strategy where hits can be analyzed for binding affinity before scale-up.71 After resin cleavage, a fluorescent tag is conjugated to the peptoid hit to determine binding affinity with fluorescence polarization (FP). As a result, only the most promising hits are subject to further investigation.
Peptoids have been raised against a variety of protein targets72 and for functionally affecting the target proteins, making them amenable as potential therapeutics.69,73 While peptoid binders can be generated against a wide variety of proteins, their affinities are modest, perhaps due to their conformationally unrestricted linear nature. Cyclic peptoids would be expected to have enhanced affinity/functional effects as a result of their more constrained nature.
Epitope-targeted ligands.
A valuable feature of monoclonal antibodies (mAbs) is that they can be raised against specific sites (epitopes) of specific protein targets. This is similarly an advantage of Generation II PCCs, which are discussed in detail in section §4.3. Here, we provide a brief overview of literature precedents for epitope targeted ligands, which are tabulated in Table 2.
Table 2.
Non-PCC ligands developed with epitope targeted strategies.
|
Protein & Target epitope |
Selection criteria |
Best IC50 Kd/EC50 |
Screen strategy | Ref. | |||
|---|---|---|---|---|---|---|---|
| Dopamine D2 receptor SWYDDDLER (res 7–15) |
Extracellular |
--- | 1 nM | Magnetic sorting, OBOC library | 77 | ||
| H-Ras GSMSCKCVLS (res 180–189) |
Pharmacophore |
100 μM | Cu-assay, OBOC library | 78 | |||
| Interleukin-1β NEAYVHDGPVRSLN (res 110 – 123) |
Pharmacophore |
--- | 2 μM | λ repressor-reconstitution assay | 79 | ||
| Human insulin-like growth factor (IGFI) KPAKSARSVRAQR (res 113–125) |
NA |
--- | 2–20 μM | λ repressor-reconstitution assay | 79 | ||
The archetypical affinity agents, mAbs, can be raised against virtually any epitope by immunizing an organism with the target epitope to elicit an immune response. Specific B cells isolated from the organism are then used to produce more of these antibodies.74 As many epitopes are not inherently antigenic, target epitopes are often tethered to an immunogenic carrier protein (e.g., bovine serum albumin, keyhole limpet hemocyanin) before introduction into the host.75,76 In this way, specific B cells will produce mAbs against the tethered epitope, as well against the protein target.
Peptide ligands against epitopes in dopamine 2 receptor (D2R) were developed by Sasaki et al., who used magnetic beads to screen a pentapeptide library for binding to epitopes on the protein.77 They reasoned that peptides that bound particular epitopes held utility for selective capture of the target protein. They incubated a linear OBOC pentapeptide library with magnetic beads that were appended with a surface-exposed epitope of D2R that bore the sequence SWYDDDLER. OBOC beads that presented ligands with high affinity to this epitope bound the magnetic particles, and were thus separated. Their ligands exhibited Kd values of 1 to 560 nM to D2R.
A separate OBOC screening strategy for epitope-targeted ligand discovery was demonstrated by Dong et al. for the C-terminus on oncogenic Ras protein.78 The C-terminal epitope must be post-translationally farsenylated to support Ras function. 78 They posited that Ras function could be inhibited if small peptides bound to the C-terminal epitope to prevent farsenylation. To identify ligands, the group incubated a His6 tagged version of the C-terminal Ras epitope (Table 2) with an OBOC library of branched tetrapeptides. Beads that bound this epitope would subsequently bind copper ions, and then take on color through the copper-catalyzed reduction of benzidine. Ligands from this work inhibited Ras with an IC50 of 100 μM.
Zhang et al. identified epitope-targeted peptide ligands via a genetic selection strategy called a λ repressor-reconstitution assay.79 Briefly, this assay exploits strong interactions among a target epitope and a library-encoded peptide within an E. coli cell to impart resistance of the cell to phage lambda. E. coli cells that express ligands that interact strongly with the target peptide will survive challenge by phage lambda and generate resistant colonies, which can be sequenced to establish the identity of the library-encoded ligand. This method produced peptide ligands against epitopes on Interlekin-1β and Human Insulin-like Growth Factor with Kd values of 2 to 20 μM.
3. CLICK CHEMISTRY
3.1. CU-CATALYZED CLICK CHEMISTRY
The term ‘click chemistry’ was first coined by Sharpless, Kolb, and Finn to describe facile chemical reactions that created carbon-heteroatom bonds (C-X-C, where X = heteroatom). In general, click chemistry refers to high yielding chemical reactions that are straightforward to perform, generate inoffensive byproducts, are stereospecific, and are insensitive to the presence of water and oxygen.3 An ideal click reaction allows for the modular synthesis of physiologically stable molecules that can be readily isolated without chromatography.3,80 Whilst the term click chemistry encompasses several means of C-C and C-X bond formation, the most famous click reaction is the irreversible Huisgen 1,3-dipolar cycloaddition reaction between an azide (1,3-dipole) and an alkyne (dipolarophile) to generate 1,2,3-triazoles, which are five-membered heterocyclic rings (Figure 3A).2
Figure 3.
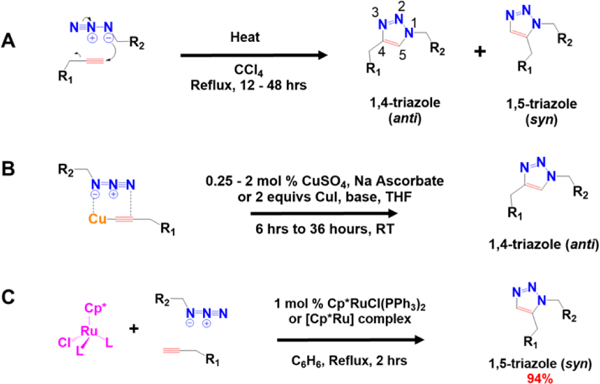
(A) The Huisgen 1,3-dipolar cycloaddition reaction between an azide/alkyne has an activation barrier of ~25 kcal/mol,2,82–84 requiring heating during implementation. It is non-regioselective and generates a mixture of 1,4- and 1,5-triazole products. (B) The Cu(I) catalyzed click reaction conducted in aqueous or organic media is regioselective for the 1,4-triazole product.84 (C) The 1,5-triazole regioisomer can be selectively synthesized using a [Cp*Ru] catalyst under reflux in organic solvent.
Azide and alkyne functional groups can be readily installed on molecular frameworks by nucleophilic and electrophilic reactions.81 For example, artificial amino acids that contain acetylene or azide bearing side chains bearing myriad chemical structures are commercially available (ChemPep). Azide-containing side chains, or azido amino acids, include 4-azidobutyric acid, azidolysine, and structures where the conventional amino group is replaced with an azido group. Likewise, commercially available acetylene-containing amino acids include propargylglycine and 2-(2’-propynyl) alanine. As amino acids, these click handles can be incorporated onto molecular scaffolds without extensive chemical modifications.
The azide and alkyne moieties are slow to react, rendering them kinetically stable and relatively inert in aqueous environments.6,80,81 The exergonic 1,3-dipolar cycloaddition reaction requires heating and refluxing to overcome the ~25 kcal/mol activation barrier.2,82–84 The reaction is nonregioselective and generates a mixture of 1,4- and 1,5-triazole isomers. In 2002, the use of copper(I) to catalyze 1,3-dipolar cycloaddition was reported concurrently by Tornøe82 and Fokin and Sharpless83 (Figure 3B). Fokin and Sharpless demonstrated the utility of copper(I) catalysts that are generated in situ by reduction of 0.25 – 2 mol% Cu(II) salts.83 Copper(I) salts such as CuI and CuOTf·6 H2O can also be used with a reducing reagent to catalyze the cycloaddition reaction. CuI can also be used in the presence of a nitrogen base such for the preparation of peptidotriazoles on solid phase.82,83 Fokin, Jia, and Sharpless also found that regioselective synthesis of the 1,5-triazole product could also be accomplished using the pentamethyl Ru(II) ([Cp*Ru]) complexes at 1 mol % catalyst loading (Figure 3C).84
Although the screen used to identify PCC candidate ligands is an in situ (non-catalyzed) click reaction, metal catalyzed click reactions using ruthenium in the form of Grubbs I catalyst (Figure 3C) or Cu(I) are used for the scaled-up production of PCCs, once those lead hits have been identified. For example, a common general scaffold for the synthesis of PCCs is a pentameric peptide that is appended on the N- and C-termini with propargylglycine and azidolysine. Cyclization is accomplished by the addition of Cu(I) catalyst in the presence of L-ascorbic acid as a reducing agent, and base such as piperidine. We have also generated i, i + 7 hydrocarbon stapled peptides85 where the PCC is appended on either end with olefin-bearing sidechains. The cycle, or the stapling of the linear peptide into a macrocycle, is generated by olefin metathesis catalyzed first generation Ru(II) Grubbs catalyst.86 These metal catalyzed reactions also hold utility within the Heath lab for the generation of macrocyclic peptide-based combinatorial libraries that are described in this review (see §4.1).
3.2. IN SITU CLICK CHEMISTRY
The kinetic inertness of azides and alkynes in aqueous media bodes well for their application towards addressing biological environments.81,87,88 Of particular interest is the application of combinatorial libraries towards generating multivalent molecules through covalently bridging two or more ligands that bind to distinct sites on target proteins. Such molecules can be applied as inhibitors, modulators of protein function, or affinity agents. Developing leads with combinatorial libraries typically requires iterative screening cycles (see §4.2), which is time consuming and costly. However, in situ click chemistry, which is a type of kinetic target-guided synthesis (TGS), relies on the structural landscape of the protein target to template the assembly of bivalent inhibitors from a pool of known site-specific binders. This methodology has yielded high affinity binders to enzyme/protein targets and accelerated the process of lead discovery.
Huisgen 1,3-dipolar cycloaddition is considered to be the “cream of the crop” of click reactions, particularly when applied to TGS.3,80 The cycloaddition is biorthogonal and the reactive moieties require no protecting groups, making them ideal for generating covalent bonds in native cellular systems. In 1983, Mock and coworkers reported on the addition of cucurbituril, an organic cage compound, into reactions with terminal azides and alkynes.89,90 Cucurbituril served an enzyme-like purpose by orienting the azide and alkyne containing reactants within its caged structure. The researchers observed that a 1,3-dipolar cycloaddition was accelerated by a factor of 104 to 105 when reactants were encapsulated within cucurbituril. The caged structure of cucurbituril facilitated the optimal orientation of azide and alkynes to catalyze the regioselective formation of the heterocyclic 1,3-triazole covalent product. Two causative mechanisms were proposed: (a) cucurbituril removed entropic constraints by serving an enzyme-like manner to optimally orient the reactant functionalities, and (b) both substrates within the catalyst cavity induced strain which would kinetically accelerate the reaction further than proximal and proper orientation alone. Taken together, these findings demonstrate that close proximity and proper orientation of terminal azides and alkynes were sufficient to induce 1,3-dipolar cycloaddition in the absence of a metal catalyst.89,90 Naturally, it is expected that an enzyme scaffold might serve the same function as cucurbituril to generate bivalent inhibitors using 1,3-dipolar cycloaddition for ligation of distinct ligands.
We return back to the 2002 Sharpless and Finn report of the in situ click synthesis of AChE bivalent inhibitor.1 The AChE active site is within a narrow 20 Å gorge lined with aromatic residues.91,92 This structure made AChE a model for demonstrating how one might screen for a bivalent inhibitor using TGS. A peripheral anionic binding site also exists on the rim of the aromatic gorge.
The screen for a bivalent inhibitor began with a library of possible lead compounds. Tacrine93 and phenanthridinium94 are known AChE inhibitors specific to the esteratic and anionic sites, respectively (Figure 4). A small building block library of tacrine (active site binder) and phenanthridinium (peripheral site binder) derivatives were functionalized with alkyl azides and alkyl acetylenes, respectively.1 These building blocks were incubated with AChE at ambient temperature and with no Cu(I) catalyst. Only 1 out of 49 potential reactions generated a detectable amount of the covalent triazole click product.1 Blocking of the AChE active site prevented formation of a triazole product. The bivalent inhibitor had Kd = 77 to 14000 fM to different species of AChE. Computational studies of the docked bivalent inhibitor showed that the triazole linker rested deep in the narrow gorge, reinforcing the concept that the gorge provided a reaction template. Additionally, the affinity of the biligand approached the product of the affinity of the individual components,95,94 strongly suggestive of cooperative binding.1
Figure 4.
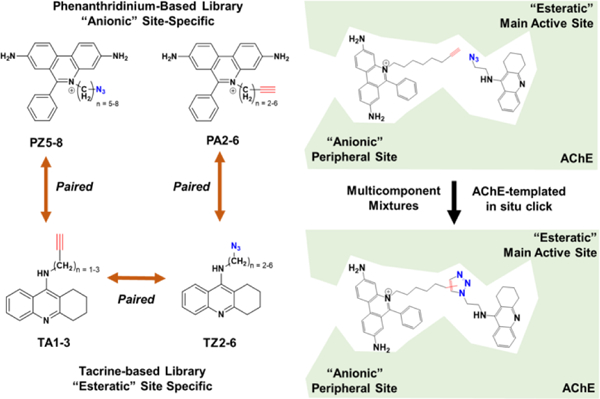
Proof-of-principle of in situ click chemistry for assembling a bi-specific inhibitor against acetylcholinesterase (AChE). The bi-specific active site of AChE comprises the esteratic site and a peripheral anionic site. Site-specific libraries were generated from known structures bearing variable linkers affixed with azide or acetylene click handles. The double-ended arrows represent pairings of complimentary azide and alkyne bearing libraries elements that were incubated in the presence of acetylcholinesterase. The enzyme templates the arrangement of the reactive click moieties and promotes in situ click chemistry between paired components. (Figure has been adapted and reprinted with permission from Lewis, et. al., JACS, 2004, 126 (40), 12809–12818 and Krasiński, et. al., JACS. 2005, 127 (18), 6686–6692. Copyright 2004 – 2005 ACS.)
The development of the biligand AChE inhibitor was a beautiful demonstration of a new concept in TGS. With respect to generality, there were some caveats. First, some prior knowledge of active site binders was required for library construction, and the library itself required significant chemistry. Second, the bi-specific active site was an important component of the process. However, the approach had some generality. In 2004 and 20054, additional novel AChE inhibitors were generated through targeting the bi-specific active site. These new inhibitors were derived from unique libraries of tacrine and phenylphenanthridinium functionalized with azides/alkynes.96 The introduction of analytical LC/MS-SIM for product identification greatly enhanced the sensitivity of in situ click lead discovery.
The generality of in situ click chemistry was further explored by Fokin, Elder, and Sharpless, by targeting HIV-1 protease (HIV-1-Pr)5. The structural landscape of HIV-1-Pr was used to template the formation of a known bivalent inhibitor of the enzyme with 18:1 enhancement in regioselectivity of the 1,4-triazole over the 1,5-triazole product. This work underscored the importance of enzymatic active sites for templating the in situ click reaction.
In 2005, Kolb, Sharpless and coworkers extended in situ click chemistry to develop inhibitors of carbonic anhydrase (CA) II, a metalloenzyme that catalyzes the reversible hydration of carbon dioxide to carbonic acid.6,97 Again, the results of this work demonstrated the requirement of an active site for in situ inhibitor assembly by triazole formation. A known inhibitor (Kd = 37 nM) bearing an acetylene was incubated with CA II and a complementary library of azide reagents of micromolar affinities. Several divalent inhibitors generated from this screen displayed sub-nanomolar affinities. Although impressive, these affinities were not the full products of individual ligands as observed for the AChE study.1,96 This work illustrated further generality of the basic concept, but also indicated that binding cooperativity is a subtle subject. Other examples of combinatorial screens involving TGS with in situ click chemistry have been reported (Table 1).
Table 1.
Examples of novel assembly or rate enhancement of bivalent molecules through in situ click chemistry methodology where two sites are targeted. The applications of this methodology include antibiotic synthesis, inhibitors, and stabilizers.
| Target | Affinity/Activity | Ref |
|---|---|---|
| AChE (multiple species) | Kd = 77 to 14000 fM | 1 |
| AChE (mouse) | Kd = 99 to 140000 fM | 96 |
| AChE (multiple species) | Kd = 0.36 to > 400 μM | 4 |
| Acetylcholine binding protein (AChBp, two subunit interface) | Kd = 0.6 to 1900 aM | 99 |
| b(h)CAII | Kd = 0.2 to 7.1 nM | 6 |
| Cyclooxygenase-2 | IC50 = 0.05 to 0.09 μMIC50 = 0.06 to 0.08 μM (Cell) | 100 |
| Transcriptional Repressor EthR | IC50 = 580 nM to 7.4 μM | 101 |
| HIV-1 Protease (18:1 rate enhancement) | IC50=6 nM | 5 |
| S. marcesans chitinases (SmChis A, B, and C) | IC50 = 0.061 to > 30 μM | 102 |
| 70s E. coli Ribosomes (Macrolide synthesis) | Kd = 0.6 to 5.0 nM | 103 |
| S. aureus biotin protein ligase (Rate enhancement) | Ki = 0.09 μM | 104 |
Our early development of PCCs was a collaboration with the Scripps groups19,98 through which we sought to address certain of the limitations associated with the in situ click TGS approach. Namely, we sought to avoid (i) the need for a bi-specific binding pocket, (ii) the need for knowledge of site-specific binding molecules, and (iii) the reliance on small libraries that required significant chemical expertise to construct. We stipulated these limitations could be overcome through the sampling of sufficiently large chemical space, and through strategies that could amplify the presence of in situ clicked products.
4. PCCs
All PCCs are developed via screens against OBOC libraries (§4.1), with libraries that have evolved over time from linear peptides to macrocycles. Generation I PCCs are developed by first identifying an anchor ligand, either through a binding assay, or from the literature. That anchor ligand is then matured into a biligand or triligand using iterative in situ click screens (§4.2).
Generation II PCCs are developed through an in situ click screen of an OBOC library against a chemically modified fragment of the target protein, using the method of all synthetic chemical epitope targeting (§4.3). Those PCCs may also be matured into biligands, but the most advanced of those biligands are developed by optimally linking two Generation II PCCs that were separately developed against strategically identified epitopes on the same target protein.
4.1. OBOC LIBRARIES
“One-bead, one-compound” (OBOC) libraries were first introduced by Kit Lam in 1991,105 and are prepared using the “split-and-mix” technique. The resin is divided into n reaction vessels, where n is the number (typically 18–20) of amino acid residues used to construct the library. In each vessel, a single amino acid is coupled onto the beads. The resins are pooled and then re-distributed for a second cycle of coupling, and so on, until the peptide library is completed. Using this approach, a linear pentapeptide library constructed from 18 natural amino acids (L-cysteine and methionine are often left out of the variable region) will contain nearly 2 million (=185) unique sequences. Individual beads may be sequenced using Edman degradation. Subsequent advances for determining sequences included N-terminal capping a small portion of the peptides at each coupling cycle (“ladder-synthesis”),106 MALDI-TOF/TOF algorithms,107 and DNA encoding.108
The OBOC strategy permits building blocks other than natural amino acids. For example, D-amino acids109 (used in most PCC screens) can impart resistance to protease digestion, as can peptoids (see Section 2.3)70,110 Stability can also be introduced by cyclizing these peptide ligands.111
In 2015, Das and co-workers112 reported on a 1,2,3-triazole linked macrocyclic library (Figure 5A), which has subsequently become the standard for Generation II PCC development. For this library, an L-azidolysine (Az4) was coupled onto TentaGel S NH2 resin (Rapp Polymere; Tübingen, Germany) using standard solid phase peptide synthesis (SPPS). The “split-mix” technique was then utilized to generate a linear library with a 5-residue variable region built from a basis set of 18 D-amino acids. After that, L-propargylglycine (Pra) was coupled to the sequence. The library elements were then cyclized through 1,3-dipolar cycloaddition between the azide and alkyne using Cu(I) as the catalyst. On-bead cyclization chemistry was confirmed through infrared spectroscopy where the azide IR stretch at ~2100 cm−1 was only observed with the uncyclized peptide. Finally, a second Pra or Az4 was coupled onto this 2-million element library as a click handle for in situ click screening.
Figure 5.

OBOC libraries used for PCC development (A) Cyclic library used for Chemical Epitope Targeting. (B) An Encoded Cyclic Peptide Library (ECPL) may display H2N-Pra-cyclo(Pra-X1X2X3X4X5-Az4) on 80% of the bead and H2N-Pra-Gly-X1X2X3X4X5-Az4, the corresponding linear peptide, on 20% of the bead.
Sequences of the macrocycles could be determined via Edman degradation. Following release of the appended click handle on the library, the second degradation cycle opens the peptide macrocycle, while the subsequent cycles release individual residues for analysis. This cyclic library was utilized to obtain epitope targeted binders against Plasmodium falciparum-specific proteins PfHRP2, PfLDH, and botulinum neurotoxin (BoNT).112
As a high-throughput alternative to Edman degradation, MALDI-TOF/TOF was developed for rapid de novo sequencing of beads from a cyclic peptide library. For this purpose, an encoded cyclic peptide library (ECPL) was created where 20% of the peptide on each bead is a linear tag for MALDI-TOF/TOF sequencing (Figure 5B), while 80% remains the cyclic peptide for ligand discovery. When synthesizing the ECPL, methionine is first coupled to the TentaGel beads as a CNBr-selective cleavage handle. Then, Az4 and the 5-residue variable region are coupled via the “split-mix” technique, respectively. A mixture of 80:20 Pra:Gly (mol/mol) is subsequently coupled onto the resins. Then, Cu(I) is added to cyclize the Pra-coupled peptide with the Az4, while the Gly-terminated peptides remain linear for MALDI-TOF/TOF sequencing. Finally, Pra is coupled onto the N-terminus of the library as a click handle in situ click screening. To confirm the success of library synthesis, a randomly selected sample of library beads is removed. The peptides are released from the bead upon microwave irradiation treatment with aqueous acidic cyanogen bromide (CNBr).113 Cyclic and linear peptides released from the ECPL differ in mass by 38 m/z in MS mode. The 20% linear peptide is further analyzed in MS/MS mode for sequence determination. Observation of an equal representation of constituent amino acids from these randomly selected library beads provides confirmation of sequencing accuracy. Of course, specific library elements can be synthesized and sequenced to provide additional validations.
Camperi and co-workers114 reported on an alternative approach towards a MS-sequence-able OBOC macrocycle library, in which glycine is first coupled onto the resin. Then, 20% of the peptide on each bead is coupled with alanine, while 80% was coupled with aspartic acid residue bearing a 2-phenylisopropylester side-chain protecting group (Asp(OPp)). The “split-mix” technique was then used to generate a heptapeptide library. This was followed by the selective cleavage of the OPp protecting group with mild acidic conditions. The deprotected carboxylic acid side chain of the aspartic acid was then coupled to the N-terminus of the peptide, thus generating a cyclic peptide. The Ala-Gly terminated peptide remains linear and is available for MALDI-TOF/TOF sequencing.
A second generation of the library was developed to be fully cyclic. In this version of the library, Asp(OPp) was first coupled, followed by the 5-residue variable region via the “split-and-mix” technique. Glycolic acid, followed by alanine, were then coupled to the N-terminus. Selective cleavage of the OPp protecting group and cyclization between the aspartic acid and the N-terminal Ala generated the cyclic peptide. To analyze the peptide sequence using MALDI-TOF/TOF, ring opening of the macrocycle was achieved by treating the glycoamidic ester with ammonium hydroxide.115
Topologically segregated bilayer beads can also be employed to decode libraries,116 where the library compounds are presented on the outer surface of the bead, while the sequencing tag is present in the inner core. An added advantage of this bead motif is the reduction of ligand presented on the bead surface, which decreases non-specific binding between the ligand and the target.117 Pei and co-workers have demonstrated cyclic peptide sequence determination using topologically segregated bilayer beads.118 Using the outer and inner core segregation technique,119 the cyclic peptides are presented on the outer core for lead discovery, while their corresponding linear peptides reside in the inner core for sequencing.
In principle, all of these various libraries could be used for PCC production.
4.2. IN SITU CLICK SCREENING METHODS
The technique of Iterative Peptide In Situ Click Chemistry (IPISC) draws from the methodologies established by Sharpless’ group described in §3.2 and is the major tool for engineering Generation I PCCs. Through IPISC, the target protein is used to template the covalent coupling of two binding peptides and proceeds stepwise. Using this approach, a known binder, developed from a traditional screen for target binding,98 or identified from the literature,16 is considered as the initial anchor, or 1° ligand. For example, for the development of a triligand capture agent against bovine carbonic anhydrase II (bCAII), the 1° ligand was selected for target binding from a linear OBOC pentapeptide library (X = 18 D-amino acids).98 In situ click screening of the acetylene-presenting OBOC peptide library with the target protein and biotin-labeled, azide-presenting 1° ligand was used to identify a biligand via screening for clicked product. The biligand was then modified to present an azide, and it served as the anchor in a similar triligand screen. Bi-, tri-, and tetraligands display improved affinity and/or selectivity for the target protein, relative to the individual component ligands, due to the avidity arising from the combination of multiple binders engaging with the target.120 A general schematic of IPISC is shown in Figure 6.
Figure 6.
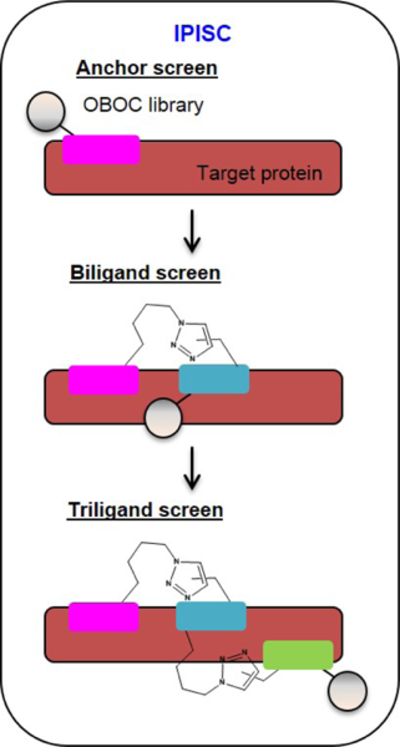
Iterative Peptide In Situ Click Chemistry (IPISC). An anchor ligand (pink) is selected for binding to a target protein (brown) from an OBOC peptide library (grey sphere). The anchor ligand is modified with an azide and incubated with the protein and an OBOC library of alkyne-modified peptides. Target-templated in situ click chemistry links the anchor ligand to a secondary peptide (teal) to form a biligand. The biligand is then modified with an azide and the in situ click screening is repeated to append a tertiary peptide (green) to form a triligand.
When IPISC is used to expand anchor ligands into branched triligands or tetraligands, those larger PCCs can present significant synthetic challenges. Nevertheless, the iterative approach does typically yield ligand performance improvements in both affinity and selectivity at each step. Examples of Generation I and Generation II PCCs developed through the use of IPISC are provided in §5.1 and §5.2.
4.3. CHEMICAL EPITOPE TARGETING
Generation II PCCs utilize the approach of chemical epitope targeting to identify the individual anchor ligands. Those ligands may be evolved into biligands using either IPISC, or via the optimal coupling of independently developed PCC anchor ligands. Early Generation II PCCs were developed from linear OBOC libraries, while more recent PCCs arose from cyclic OBOC libraries.
Chemical epitope targeting begins with the selection of a continuous 10- to 30-mer region of the target protein of interest. The properties of various screened epitopes are discussed in §4.5. The epitope of interest is first synthesized as a polypeptide, with a biotin detection label and a strategically substituted alkyne- (or azide-) presenting amino acid.112 This synthetic epitope (SynEp), which is typically 9–30 amino acids in length, acts as both a target and anchor ligand in the in situ click screening of linear or cyclic OBOC libraries appending azide- (or alkyne-) presenting peptides. During the screen, hit peptides are those that bind to the SynEp in a preferred orientation so as to template the click reaction. After thoroughly washing to remove non-covalently bound copies of the SynEp, treatment with alkaline phosphatase (AP)-conjugated streptavidin or anti-biotin antibody visualizes the biotin detection label coupled to the hit beads. Note that this is not a standard binding assay but instead is a reaction product screen, where hit beads are defined as those with covalently coupled SynEp. This screening concept is a major differentiator of PCCs from general affinity screens.
Beads are picked for sequencing, and hit peptides are then scaled up and tested for binding to the full-length target protein. A variation of the epitope targeting approach utilized a zinc chelator as the anchor ligand to engage a phosphopeptide epitope of Akt2.17 Promising binders have been utilized as anchor ligands in IPISC to develop a multiligand PCCs.17,18 Starting at the biligand screen, the full-length protein is used as the target instead of the SynEp to allow the library to sample the surface of the folded protein beyond the site of the targeted epitope.
An epitope targeted in situ click screen (Figure 7) is performed as a single generation screen, using multiple steps to filter the results. The cyclic OBOC library used for recent Generation II PCCs is a heptapeptide on TentaGel S NH2 resin (0.3 mmol/g amine loading) with a 5-residue variable region, cyclization of the flanking alkyne- and azide-modified amino acid side chains by copper-catalyzed azide-alkyne cycloaddition (CuAAC), and an N-terminal alkyne-modified amino acid handle for in situ click screening (Figure 7B). The OBOC library is first pre-cleared to eliminate beads that exhibit non-specific binding to scrambled epitopes, interferents, and/or reagents that may be used for detection in a later screening step. The remaining library is washed, re-equilibrated in the screening buffer, and subjected to the primary screen. This product screen is designed to detect those hit beads containing the triazole-linked SynEp labeled with biotin (Figure 7B)112. In many ways, this is a selectivity screen, since non-selective library elements are removed in the pre-screening steps, and the product screen only selects those peptides that interact with the SynEp in such an orientation to promote that azide and alkyne coupling.12 A typical screen against a single SynEp may will only yield about 5 or so hits from a 2 million element OBOC library, but multiple SynEps representing different epitopes of the same protein can be screened simultaneously. Hit beads are decolorized, washed, and re-equilibrated in the screening buffer. Prior to sequencing the hits, a secondary target screen is optionally performed to identify those hits that bind to the full-length target protein (Figure 7C–i). Binding of the target protein is visualized using an AP-conjugated antibody that turns over a colorimetric substrate. This last target screen is designed to capture any differences in the epitope presentation between SynEp and target protein.
Figure 7.

(A) Molecular structure of IL-17F with potential target epitopes colored in blue and green, with various structures of SynEps for the blue epitope in which click handles introduced either at the C-terminus, N-terminus, or near the center, and a biotin assay handle at the N-terminus. (B) Schematic diagram of a process flow to discover PCCs that bind an epitope on IL-17F. PDB ID: 1JPY. (C) Secondary screens are optionally performed against the purified target protein (i) and/or target in a cellular context (ii) to confirm target binding, and/or human serum (iii, anti-screen) to select for low non-specific binding. Peptides from hit beads can be re-synthesized after the primary screen (iv) or after one or more of the secondary screen(s).
Another secondary screen option that can be utilized for targets that are expressed on the cell surface incorporates the use of live cells. Hit beads are incubated with cells expressing the target of interest on the cell surface (Figure 7C–ii). Beads that show binding to cells will be homogenously coated with a monolayer of cells over the entire bead surface.121 In this cell-based screen, the cell surface target is presented in the context of the cell membrane, which may be different from presentation as a SynEp or purified target protein. For a cell surface protein, only the extracellular domain is exposed. Following the screen with target-positive cells, the hit beads should also be subjected to an anti-screen against target-negative cells to eliminate hits that display non-specific binding to cells.
Additional anti-screen steps can be conducted to remove beads that exhibit binding to off-target proteins found in human serum (Figure 7C–iii). These anti-screens are particularly useful for PCC agents designed to detect analytes from blood. For example, in the development of capture agents against the Bacillus anthracis PA27 or against anti-HIV1 (gp41-specific16) antibodies, anti-screens in the presence of 1% human serum were performed to remove hits that exhibited binding to off-target proteins present in human serum. Beads exhibiting off-target binding were detected using a pan anti-human serum antibody, and in the case of ligands against Bacillus anthracis PA, the anti-screen eliminated about 75% of beads from the pool of hits that were further tested in the product screen.27
4.4. EPITOPE TARGETED ANTI-IL-17F PCCS
Biligands developed against IL-17F (Fig 1D) represent the current state-of-the-art in PCC development,13 and so a brief description of the strategy used to develop those biligands is provided here.
The IL-17 family is comprised of 6 proteins, IL-17A-F. These proteins are associated with various immune and autoimmune diseases and are both diagnostic and therapeutic targets. Of these, IL-17A and IL-17F are the most closely related, with high sequence homology (Figure 8A), and bind as both homodimers and heterodimers. However, they exhibit very different functional differences and are associated with different disease conditions, ranging from psoriasis to adverse responses in CAR-T cell therapies. The goal was to develop PCCs that were selective capture agents for IL-17A or IL-17F, but not both. The red and green highlighted sequences of Figure 8A are regions that exhibit the greatest differences. These two regions are shown in Figure 8B for IL-17F, and labeled epitope #1 and #2. These sequences informed the construction of SynEps, and SynEp 1 (represented as epitope #1) is shown in Figure 8C, with an L-azidolysine substituted for Cys48. The SynEps were subjected to in situ click screens to yield two consensus ligands, L1 (Figure 8D) and L2 (not shown), which exhibited Kd values of 50±15 and 28±8 nM, respectively. The distance between the two protein-bound SynEps was calculated to be around 18Å, based upon the crystal structure of IL-17F. Thus, a series of biligands, linked by polyethylene glycol (PEG) oligomers of varying lengths, were prepared (Figure 8E), and tested for binding affinity against IL-17F (Figure 8E). The optimal biligand was estimated to be PEG3 by distance and, in fact, enhanced binding cooperativity was observed for the biligand with the PEG3 linker (Figure 8F), which exhibited a Kd of around 200 pM. The selectivity of this cooperative biligand was determined by titrating IL-17F and IL-17A proteins against surface-immobilized biligand. These experiments showed 4:1 selectivity for the IL-17F isoform (Figure 8F). An analogous set of ligands were prepared against IL-17A, with similar performance and differential selectivity. Of course, PCCs developed against other protein targets may exhibit difficulties that were not observed for the IL-17 PCCs, but the process illustrated in Figure 8 has been demonstrated for other PCCs against very different targets.
Figure 8.

(A) Primary sequences of IL-17F and IL-17A with target epitopes #1 (IL-17F residues 40–54) and #2 (IL-17F residues 60–69) colored in red and green, respectively. Schematic process flow for anti-IL-17F PCC development, which included (B) epitope selection based on epitope proximity, (C) synthesis of SynEp(s), and (D) in situ click screen to identify high-affinity PCC ligands. (E) Subsequent design of an IL-17F biligand involved linking top-performing PCCs that target epitope #1 and #2 with PEG linkers of various lengths. (F) Biligands conjoined by a PEG3 linker exhibited the strongest affinity, suggesting co-operative mode of binding, and also (G) selectively bound IL-17F over IL-17A.
4.5. COMPARISON OF EPITOPES TARGETED BY BOTH PCCS AND ANTIBODIES
The epitope targeting strategy of Figure 7 appears to have a good deal of generality. However, how it depends upon properties such as secondary or tertiary protein structure, surface exposure, hydrophilicity, etc., is unclear. Insights into the versatility of the epitope targeting strategy can be gained by a comparative analysis of PCC- and antibodies-epitope binding, which can be done by tabulating the properties for epitopes targeted by PCCs against which epitope-specific antibodies also exist. Principal component analyses (PCA) can then provide the comparison of PCCs and antibodies across this high dimensional chemical and structural space. Properties selected for this PCA analysis included residue hydrophobicity and charge, as well as secondary structure, surface exposure, and B cell antigenicity, as predicted from the protein sequence by using informatics tools.122,123 Each epitope was assigned a representative value for each property, which was the average of the property value over all residues in the epitope. Epitopes included those targeted by PCCs in 14 different proteins (some targeted at multiple sites) and from antibody antigens of these same proteins that were obtained from the Immune Epitope DataBase.124 PCA yields orthogonal vectors, or Principal Components (PCs), which capture the variability in the data, so that the first PC captures the most variability, and so on. For structural analysis of targeted epitopes, the top 3 PCs captured >75% of the variance.
The results of this calculation are provided in Figure 9. These plots reveal differences between antibodies and PCCs, as well as highlight epitope factors that are important for both. With a limited number of Generation II PCCs, the PCC epitope list may be influenced by researcher bias with respect to how the epitopes were selected, although typically the epitopes are selected for practical reasons (§4.4) and because the epitope is surface-exposed, rather than for the additional properties that go into the two calculations. In Figure 9, PC1 correlates positively with alpha helical content, and is anticorrelated antigenicity and random coil character. PC2 is dominated by the antigenicity and alpha helix character of the epitope, while PC3 is dominated by beta-sheet character. Both PCA plots in Figure 9 reveal clustering of PCC epitope data points (Figure 9A, blue circles) at negative PC1 values. As PC1 is negatively correlated with antigenicity and random coil content, the data implies that PCCs readily bind antigenic and unstructured epitopes. Surface exposure is also negatively correlated with PC1, suggesting that epitopes targeted by PCCs tend to be surface exposed, as expected. The proclivity of PCCs to bind antigenic sites is borne out by the fact that most epitopes targeted by PCCs have positive PC2 values, which has positive contributions from antigenicity. However, PC2 also has a strong positive correlation with alpha helix content, suggesting that PCCs can be targeted against epitopes with alpha helical contents. Antibody antigens have a weaker tendency towards negative PC1, implying a broader class of epitopes are available for targeting. PCC epitopes have little amplitude for PC3, relative to antibody epitopes.
Figure 9.
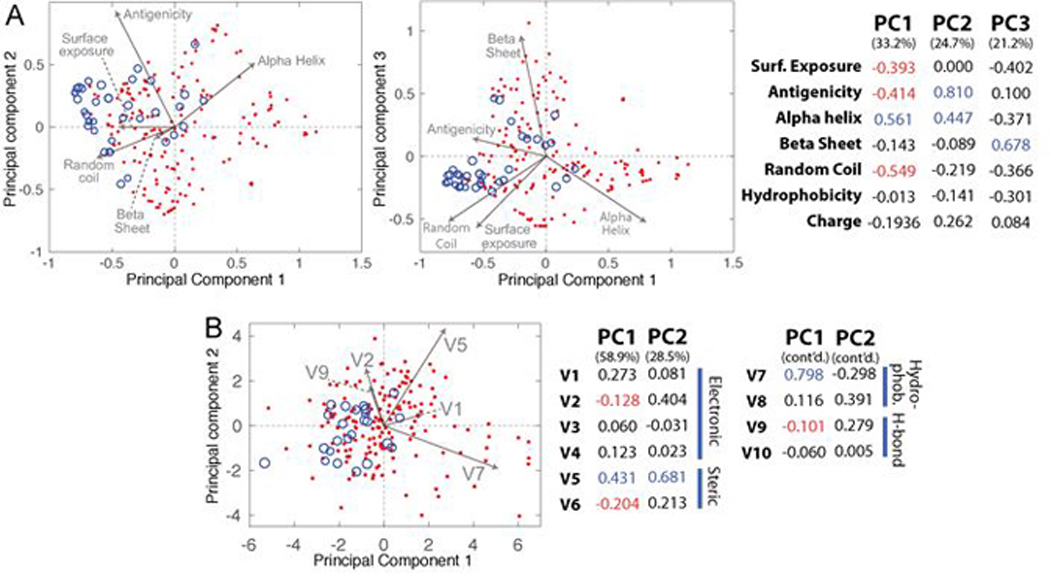
PCA analyses of epitopes based on (A,B) structural properties and (C) divided physicochemical property scores (DPPS). The blue circles represent data from PCC-targeted epitopes on 14 different proteins, and the size of the data point corresponds to the relative affinity (IC50 or KD) of the PCC to the full-length protein, with the smallest having greatest affinity. The red dots are associated with antibody antigens on the same proteins, as obtained from the IEDB free antigen database. Structural properties include surface exposure, B-cell antigenicity, secondary structure, hydropathy, and charge. Each epitope was assigned a value for each property, which was the average of the property value over all residues in the epitope or antigen. Grey vectors represent the direction and relative magnitude of the PCA components of a particular variable in the PC space.
PCC epitopes were also analyzed by their physicochemical properties to gain insight into the intermolecular interactions responsible for PCC-epitope binding. Namely, we exploit divided physicochemical property scores (DPPS) by Tian et al125 to define physicochemical properties of epitopes. These researchers derived the DPPS were derived by performing PCA on literature values of 119 different physicochemical properties for each natural amino acid. The resulting ten PCs (V1-V10) classify amino acids based on their physicochemical attributes, which included electronic, steric, hydrophobic and hydrogen-bonding (see key to right of plot). Tian et al identified that a subset of these DPPS eigenvectors most clearly delineated physical properties, namely: V1 (electrostatic) which increases with positive charge, V2 (electrostatic) increases with polarity, greater V5 (steric) values correlated with larger (>80A3) residues, V7 (hydrophobic) increases with residue hydrophobicity, and V9 (hydrogen-bonding) associates with ability to form hydrogen bonds.125 We confine our analyses to these DPPS eigenvectors, as the others are not readily associated with a physical property. PCC- and antibody-targeted epitope residue compositions were transformed based on the DPPS and averaged, and subsequent PCA analyses produced two PCs that captured >85% of the variance.
A plot of the PC1 and PC2 of the DPPS-based PCA is shown in Figure 9B. This analysis underscores specific physicochemical attributes of epitopes targeted by PCCs and highlights specific interactions that account for binding. A clear trend is that data points for PCC epitopes cluster at negative PC1 values. Interestingly, however, the two DPPS eigenvectors with the largest magnitude contributions to PC1, V5 (steric) and V7 (hydrophobic), contribute positively to PC1. As increasing V5 and V7 values reflect increased bulkiness and hydrophobicity,125 clustering of PCC-targeted epitopes at negative PC1 values establishes that these epitopes typically lack bulky or hydrophobic residues. This implies that PCC-epitope binding doesn’t rely strongly on hydrophobicity. In addition to low bulkiness and hydrophobicity, another possible reason for the large negative PC1 values for epitopes targeted by PCCs are contributions from V2 (electrostatic) and V9 (hydrogen-bonding) DPPS eigenvectors, both of which both anti-correlate with PC1. The values of V2 and V9 increase with polarity and capability to form hydrogen bonds. This supports that PCC-epitope binding is driven mainly by electrostatic and hydrogen-bonding interactions.
5. PROPERTIES OF DEVELOPED PCCs
5.1. GENERATION I PCCS
Here we provide a brief summary of several individual PCCs made in our respective labs. These PCCs represent scientific demonstrations of diagnostic reagents16, allosteric19 and direct12 inhibitors, in-cell protein inhibitors via degradation tags126, and in vivo imaging.26 Certain metrics of these PCCs are listed in Table 3.
Table 3.
Summary of targets and performances of Generation I and II PCCs
| Purpose of PCC | Site / Epitope Sequence and Residue #’s |
EC50 or IC50 (bold) | KD | Ref |
|---|---|---|---|---|
| Detection of b(h)CAII | Not active site | 45 nM (hCAII) 64 nM bCAII | 98 | |
| Allosteric inh. of Akt1 | kinase domain | Tri.: 200 nM | 19 | |
| Detection of gp41-specific anti-HIV IgGs | IgG Variable region | from 1 – 50 nM (various IgG clones) | 16 | |
| In vivo imaging of VEGF | VEGF receptor binding interface | Mono: 240(21) nM Tri: 2.6(0.5) μM Tetra: 740(50) nM | 26,69 | |
| Detection of Anthrax PA | unknown | Bi: 190 nM | Bi.: 216(7) nM | 26,27 |
| Allosteric Inh. of Akt2 | Res: 450–581 (Bt–PEG5–ITPPDRYDSLGLLEL QRTHFPQF(pS+click))YSASIRE) | Mono: 3 μM | 17 | |
| Bi: 1 μM | ||||
| Tri: (N-tL): 19 nM | ||||
| Tri: (C-tL): 120 nM Tri. (C-tL) 4 μM | ||||
| Selective allosteric inh. of Akt2E17K relative to WT | Res 1–32 (Bt–PEG5–PEVAIVKEGWLKKRGK Y[I→Pra]KTWRPRYFLLKNDG) | selective inh. of mutant protein; IC50 not quantified |
Mono.: 54(7) nM Tri.: 115(9) nM Mono.: 78(13) nM |
18 |
| Cell penetrant direct inh. of BoNT | Res: 166–177 Az4-SFGHEVLNLTRN –PEG4–Bt | Bi: 165(15) pM | 12 | |
| Detection of PfLDH | Res: 218–229 LISDAELEAIFD–Az4–PEG5 -Bt | 23.4 nM | 40.6 nM | 112 |
| Detection of Plasmodium LDH (non P. falciparum isotypes) | Res: 297–308 Bt–PEG5–GVEQV–Pra–ELQLN | 1.7 μm | 112 | |
| Detection of PfHRP2 | C-term. AHHATDAHHAAAHHEAATHC–Pra–PEG5–Bt | 20 nM | 54.2 nM | 112 |
| Repeat sequence Bt–PEG5–AHHAADAHHA–Pra | 538 pM | |||
| Repeat sequence Bt–PEG5–AHHAHHAAD–Pra | 218 nM | |||
| N-term. Bt–PEG5–LHETQAHVDD–Pra | 4 nM | |||
| Detection of L1R myristyl protein | Res: 161–185 Bt–PEG5–KALMQLTTKATQIA–Pra–PKQVAGTGVQ | 875 nM | 112 | |
| Selective detection of Interleukin IL-17F and not IL-17A | 66(9) nM | 50(15)nM | 112 | |
| Res: 40–54 Bt-PEG3-FFQKPES[C→Az4]PPVPGGS |
||||
| Res: 60–69 Bt-PEG3-GI[I →Az4]NENQRVS | Mono: 15(5) nM; Bi: 246 pM | Mono: 28(9)nM Bi: (252(13)pM) | 112 | |
| Promote folding of SOD1 | Res: 121–144 EKADD[L→Pra]GKGGNEESTKTGNAG-PEG1-Propionic Acid-Bt | 8 μM (holo SOD1) 0.94 μM (apo SOD1) | 20 | |
| Selective detection of IL-17A over IL-17F | Res: 33–49 Biotin–PEG3–PNSEDKNFPRTVNL-Az4 | 4.5(0.4) nM | 13 | |
| Allosteric Inh. of KRas | Switch II Res: 25–40 FVDEYD[P→Az4 ]TIEDSY | 3.3(0.6) μM 24 (1.2) μM | 139 |
Linear Triligand against bovine (b) and human (h) CAII
The original Generation I PCC was a branched triligand against bCAII and hCAII. It was comprised of three linear peptides developed using the IPISC process, and was developed in collaboration with Fokin and Sharpless.98 The OBOC libraries were built from penta- to heptameric peptides comprised of D-stereoisomers to ensure proteolytic resistance. The development process required no prior knowledge of the protein target. This PCC provided a successful test of the hypothesis that a protein structure, not just a hydrophobic binding pocket, could provide a template for target guided in situ click chemistry. The anchor and biligand and triligand exhibited affinities of 500 μM, 3 μM, and 64 (and 45 nM) binders to bCAII (and hCAII). Single generation screens yielded a high sequence homology implying selectivity. This was shown with the triligand, which could selectively detect bCAII from 10% porcine serum with a limit of detection at 20 ng. Importantly the PCC did not interfere with the catalytic activity of bCAII, implying that it bound away from the hydrophobic binding pocket.
Branched Triligand allosteric inhibitor against Akt1
Millward et al.19 used IPISC to develop a branched triligand PCC composed of D-peptides as an allosteric inhibitor against the Akt1 isoform of protein kinase B. The triligand bound to the kinase domain of the protein. Similar to the case of the anti-bCAII PCC, the triligand Kd (200 nM) was orders of magnitude improved over the anchor (> 25 μM). This work demonstrated that PCCs might provide a synthetic alternative to inhibitory mAbs, but for challenging intracellular targets, although cell penetration was not demonstrated. The inhibitory potency of the PCC was not high, but Akt1 itself is known to be an extremely challenging drug target, regardless of the strategy for drugging.127–129
Biligand PCC cocktail for detecting polyclonal IgGs against the gp41 antigen of HIV
Pfeilsticker et al16 used IPISC to develop a cocktail of PCCS designed to sensitively measure the (polyclonal) immune response of patients during specific stages of HIV infection.130 The PCCs utilized chemically modified variants of the gp41 HIV antigen (IWCGSGKLICTTA),131,132 which is a component of the HIV envelope protein) as the anchor peptide. These were expanded into biligands by in situ click screens against a polyclonal cocktail of anti-gp41 antibodies. The goal was to develop a cocktail of PCCs that could more effectively detect IgGs against gp41 across patient populations.16 The anchor ligands had affinities in the range of Kd = 1 – 50 nM, while the developed biligand cocktail performed 2.5 times better than the commercially available affinity agent133 for detecting anti-gp41 antibodies from the blood of a cohort of HIV+ patients. The cocktail of developed PCCs exhibited no degradation when stored at temperatures near 60 °C for 2 months.
Tetraligand for detection of VEGF via in vivo Positron Emission Tomography (PET) molecular imaging
Coppock et al.,26 utilized IPISC to develop triligand and tetraligand PCCs against Vascular Epithelial Growth Factor (VEGF) protein, which plays a role in tumor angiogenesis. Certain of the PCCs were advanced as PET probes for in vivo imaging.134 An initial peptide macrocycle anchor peptide was derived by modification of an existent anti-VEGF cyclic peptide derived from phage display technologies.135 Unlike later PCC macrocycles, this anchor was cyclized by a disulfide bridge between two cysteine residues. It was matured into a tetraligand PCC (EC50 = 6.1 + 0.2 nM) which approached the affinity of Bevacizumab Fab, a known therapeutic anti-VEGF antibody. The tetraligand also inhibited VEGF and VEGFR2 binding (IC50 = 0.74 + 0.05 μM). A triligand (IC50 = 2.6 + 0.5 μM) conjugated to DOTA for 64Cu chelation was shown to bind to VEGF in mouse models of cancer using in vivo PET imaging.
PCCs designed for the Sensitive Electrochemical Detection of Anthrax Protective Antigen (PA)
Farrow and Hong et al.27 developed a biligand against anthrax protective antigen through improvement of an anchor peptide previously developed from bacterial display screening.136 That anchor was fitted with a click handle for IPISC screening against a linear OBOC D-peptide library in the presence of PA. The resultant biligand from this screen exhibited an IC50 = 190 nM. It was coupled with an electrochemical ELISA, which afforded a limit-of-detection of 170 pg/mL or 2.1 pM. Extensive thermal stability studies showed that the electrochemical ELISA biosensor retained over 81% of its binding activity after heating to 90 °C in buffer solution.26
5.2. GENERATION II PCCS
Allosteric Inhibitors Targeted to the Phosphorylated C-terminus of Akt2
The first epitope-targeted PCC was developed by Nag et al, and was directed against the “hydrophobic motif” (HM) epitope (residues 450–481) of the Akt2 isoform of protein kinase B.17 The activity of Akt2 increases approximately 10-fold upon phosphorylation of serine 474 of the HM epitope.137 This allosteric activation effect provided the rationale for this epitope as a site for allosteric inhibition. PCCs that bound specifically to the HM were discovered by screening an alkyne-bearing OBOC library of PCCs against a synthesized version of the HM epitope (the SynEp) of Akt2, in which an azide click handle was introduced through a Zn-chelator138 of the phospho-group on Ser474. The in situ click screen yielded a single linear PCC that bound Akt2 with a Kd of 3 μM. Using this monoligand as a lead compound, the application of iterative generation I in situ click screens produced triligands with improved affinities (EC50 down to 19 nM) to Akt2, and with exhibited selectivities of 5:1 and 10:1 relative to the close kinase analogues Akt1 and Akt3, respectively. Linear (N-tL; Table 3) and branched (C-tL) triligands were shown to activate or inhibit Akt2 enzymatic function, respectively. This indicated that the p-Ser474 site is a sort of enzymatic saddle point, where distortion in one direction or another can have an activating or repressing influence over protein function. This, in turn, indicated that epitope-targeted PCCs could provide valuable probes for interrogating of protein structure/function relationships.
The N-tL PCC was further modified for cellular penetration by appending a cell penetrant (HIV TAT) peptide.140 This PCC was shown to selectively activate Akt2 in OVCAR3 and SKOV3 cancer cells.141 The PCC was further modified with a Hif-1α protein degradation tag.142–144 The modified PCC was shown to rapidly promote the in-cell degradation of Akt2 in OVCAR3 cells in a dose-dependent manner.141 This demonstrated the potential of epitope targeted PCCs as highly specific inhibitors of intracellular protein function.
PCCs Targeted to the oncogenic E17K point mutation of Akt1
Deyle et al. demonstrated that epitope-targeted PCCs could selectively target an epitope with a single point mutation versus the wild type epitope sequence.18 The E17K point mutation in Akt1 deregulates the recruitment of Akt1 to cellular membranes and promotes the constitutive activation of the PI3K pathway,145,146 which leads to leukemia in mice. This oncogenic role of AktE17K makes it an important therapeutic target. A PCC anchor ligand was developed against the N-terminus (residues 1–32) of Akt1E17K, and was shown to bind the Akt1E17K protein with a Kd of 54 nM and an ~18:1 selectivity over the wild type protein. Using this PCC as an anchor ligand, IPISC screens yielded bi- and tri-ligands that inhibited Akt1 activity. The tri-ligand in particular exhibited ~10:1 selectivity for the Akt1E17K mutant versus the wild type protein. Epitope-selective binding was demonstrated by modifying the PCC anchor ligand (sequence: yleaf) at the N terminus to contain a tosylate linker attached to a Cy5 dye molecule. Using this chemistry, the dye payload is transferred onto the protein through an SN2 reaction with proximal nucleophilic amino-acid side chains. Thus, regions next to the binding spot of the PCC were labeled and identified via sequencing. This work showed an impressive level of binding specificity could be achieved for epitope-targeted PCCs.
A Trojan Horse-type inhibitor of Botulinum neurotoxin light chain (BoNT A)
A study by Farrow et al. reported on two major advances for epitope-targeted PCCs, namely a macrocyclic PCC direct inhibitor, and an in situ click screen to discover linkers for multi-valent ligands.12 BoNT is the most lethal known neurotoxin and doubles as both a novel cosmetic drug and a biological threat agent. Existing BoNT inhibitors exhibit high cytotoxicities and require high dosages for effectiveness.147–149 BoNT is not toxic outside of a cell but once it enters the cell, a fragment of the protein (the ‘belt’) is cleaved, unveiling a highly potent enzymatically active site. That site is blocked for drugging prior to cell entry. Farrow et al. developed BoNT inhibitor by optimally linking two separately developed epitope targeted PCC macrocycles. PCC #1 is bound to the active site, while PCC #2 binds to a region adjacent to the active site (residues 166–179 of BoNT A). The individual PCCs exhibited sub 100 nM affinities, but, when optimally linked, yielded a 165 pM inhibitor. Outside of neurons, the PCC is bound to BoNT only via the interaction of PCC #2. The BoNT protein itself transports the PCC into the cell, where the active site is unmasked, and PCC #1 binds. The optimized linker between PCC #1 and #2 was identified through an in situ click screen.
Epitope Targeted PCCs against Malarial Proteins
Das et al. demonstrated the epitope-targeted in situ click strategy as a general procedure to develop PCC ligands against epitopes with a broad range of chemistries.112 This work identified PCC ligands that exhibited high affinities (EC50s down to 538 pM) to epitopes on several proteins that are diagnostic and/or therapeutic in humans, bacteria, and viruses. Several of these PCCs are against malaria-pathogen specific proteins Lactate dehydrogenase (LDH) and Histidine-Rich Protein 2 (HRP2) (Table 3), and implied that epitope targeted PCCs could be developed against proteins that ranged from well-folded structures to random coils.
Foldase-like PCCs against Superoxidase Dismutase 1 (SOD1)
Bunck et al.20 showed that PCCs could also modify the energetics and kinetics of protein folding in superoxidase dismutase I (SOD1),150 the misfolded and aggregated forms of which lead to amyotrophic lateral sclerosis. PCCs targeted to electrostatic loop region of SOD1 (residues 121–144), as guided by literature studies of the biophysics of this protein,151,152 stabilized the protein in a native conformation and accelerated the rate of SOD1 folding. Solution-state NMR analyses of the top-performing monoligand PCC was shown to interact with residues in the electrostatic loop. This work demonstrate that a foldase-like153 (large enzyme) allosteric interaction could be achieved by appropriately targeted small molecule PCCs.
PCC Agents that selectively bind to one member (Interleukins IL-17A or IL-17F) of a high homology protein family (17A-F). See §4.4.
An allosteric inhibitor against the ‘undruggable’ Kirsten Rat Sarcoma (KRAS) protein
McCarthy et al.139 showed that PCCs could serve as allosteric inhibitors of the notoriously undruggable protein Kirsten Rat Sarcoma (KRas).154 Mutated KRas is the most dominant oncogene in all of cancer, with KRasG12D driving most pancreatic cancers, amongst others.155 The KRas protein contains two “Switch” regions (residues 25–40 & 57–75) that are known to allosterically influence KRas function.156 Macrocyclic PCCs targeted to “Switch” regions (residues 25–40 & 57–75) of KRas were shown to inhibit KRAS activity at an IC50 of ~24 μM. Further, this paper reported on a high throughput microchip platform for rapidly assessing the performance of PCC candidate hits from an in situ click screen.
6. DISCUSSION
Within the past decade, we have made strides in developing all-synthetic peptide-based molecules as protein-catalyzed capture agents, or PCCs, that address many target applications. Our PCCs have demonstrated utility as affinity agents that can outperform antibodies if sufficiently engineered. They can serve as modulators of protein folding, as allosteric inhibitors, as cell penetrant direct inhibitors, and they can be used for in vivo tumor imaging. PCCs are engineered through a generalization of the in situ click chemistry methodology developed by Sharpless et al. through an approach that has been engineered to be increasingly high-throughput. Currently, the PCCs can site-specifically target both structured and unstructured proteins, yielding high affinity binders within a single generation screen.
Looking forward, we envision expanding the PCC technology to address current challenges and applications. PCCs against gp41-specific IgGs, PLDH, PfLDH, and PfHRP2 were originally developed for use in in vitro diagnostics within standard laboratory settings. A future challenge will be to integrate these synthetic affinity agents into formats suitable for lateral flow diagnostic assays157 or similar robust, fieldable platforms.
A second challenge involves designing PCCs for cell penetration. PCCs, like most peptides, are not generally cell penetrant. Specifically, can one use the synthetic flexibility and epitope targeting precision of PCCs to elucidate the molecular engineering rules for promoting trafficking across cell membranes?158,159
A third area involves developing PCCs for theranostic applications,160,161 which are rapidly coming of age in the clinic for treating drug-refractory solid tumors. Theranostic probes can be peptide based.162,163 When equipped with one radiolabel, they are an in vivo positron emission tomography (PET) imaging probe.164–166 Substitution of a second radiolabel doesn’t influence the targeting, but converts the same targeting molecule into a potent vehicle for site-directed radiotherapy.163,167 This is an application space that can draw from many of the unique advantages of PCCs, including the modularity of the platform, the low molecular weight (for rapid in vivo clearance), and precise epitope targeting for binding to unique disease-specific targets.
Inhibiting protein-protein interactions (PPIs) represents a key opportunity.168 Many proteins interact over relatively large areas through which multiple weak interactions are formed. This makes such interfaces hard to drug with conventional small-molecules. By comparison, PCCs can be developed to target epitopes of arbitrary hydropathy (Figure 9). However, the disruption of protein-protein interactions is still very much an art. A related possibility is that the combined targeting flexibility of PCCs, coupled with increasingly rapid production, might enable their use as molecular tools for extracting some of the basic rules for blocking protein-protein interactions.
Ultimately, PCC production can be automated, analogous to monoclonal antibody development. Recent improvements include the use of automated bead sorting to accelerate screening steps, as well as our recent report of a barcoded rapid assay microchip platform designed to generate full binding curves for many candidate PCCs, using a fraction of the biological reagents required for typical ELISA assays.139 Going forward, a key challenge is to speed up hit identification, perhaps by screening PCCs on high-density peptide microarrays in which peptide sequences are encoded spatially.169,170 The time for developing a high quality PCC ligand can likely be reduced to about a week.
7. CONCLUSIONS
PCCs are emerging as versatile synthetic affinity agents for a various biotools, diagnostic, and therapeutic applications. The PCC technology has been developed by focusing on ways to generalize the (non-catalyzed) in situ click reaction originally pioneered by the Sharpless and Finn groups. The development of all synthetic, epitope-targeted PCCs has, in fact, brought a great deal of generality to the original concept.
The past decade has witnessed several exciting landmarks towards the practical applications of PCCs. PCCs have been demonstrated to perform a host of important functions beyond simple binding, including inhibiting protein function, promotion of protein degradation, allosteric modulation, and reporting on biological processes in vivo. Moreover, the generality and efficacy of the PCC approach has been borne out by the development of PCCs against challenging protein targets, ranging from unstructured phospho-epitopes to random coil structures to oncogenic mutations. The ability of PCCs to target such traditionally difficult biomolecules marks a key milestone for synthetic ligands and is a unique capability of PCCs.
ACKNOWLEDGMENT
We thank Drs. Max Robinson and Michael Strasser for input on the principle component analysis.
Funding Sources
Funding for this work was provided by the National Cancer Institute U54 grant (grant no.: CA199090–01; JRH PI), the Jean Perkins Foundation, and the Ben and Kathy Ivy Foundation. This work was also supported by the Institute for Collaborative Biotechnologies through grant W911NF-09-D-0001 from the U.S. Army Research Office.
ABBREVIATIONS
- PCC
Protein Catalyzed Capture Agent
- OBOC
one-bead one-compound
- Akt
Protein Kinase B
- pAkt2
protein kinase B2
- AChE
Acetylcholinesterase
- D2R
Dopamine Receptor D2
- pfHRP2
Plasmodium falciparum Histidine Rich Protein 2
- pfLDH
Plasmodium falciparum Lactate Dehydrogenase
- BoNT
Botulinum Neurotoxin
- SOD1
Superoxidase Dismutase 1
- CNBr
Cyanogen Bromide
- ECPL
encoded cyclic peptide library
- VEGF
Vascular Epithelial Growth Factor
- PA
Protective Antigen
- SynEp
Synthetic Epitope
- IPISC
Iterative Peptide In Situ Click
- FP
Fluorescence Polarization
- TGS
Target Guided Synthesis
- HIV-1-Pr
Human Immunodeficiency Virus 1 Protease
- CA
Carbonic Anhydrase
- SPPS
Solid Phase Peptide Synthesis
- AP
Alkaline Phosphatase
- IL
Interleukin
- CuAAC
Copper-catalyzed azide-alkyne cycloaddition
- RCM
ring-closing metathesis
- PC
Principal Component
- bCA
bovine Carbonic Anhydrase
- hCA
human Carbonic Anhydrase
- Az4
Azidolysine
- Pra
Propargylglycine
- CAR-T
Chimeric Antigen Receptor T-Cell
- MALDI
Matrix Assisted Laser Desorption/Ionization
- TOF
Time of flight
- LC
Liquid Chromatography
- MS
Mass Spectroscopy
- HM
hydrophobic motif
- BBB
blood-brain barrier
- DMF
dimethylformamide
- DIC
diisopropylcarbodiimide
- Fmoc
fluorenylmethyloxycarbonyl
- Fmoc-Glu(OAll)-OH
Fmoc-L-glutamic acid 5-allyl ester
- Fmoc-Glu(OtBu)-OH
Fmoc-L-glutamic acid 5-tert-burtyl ester
- Pd
Palladium
- PyBOP
benzotriazole-1-yl-oxytripyrrolidinophosphonium hexafluorophosphate
- DIEA
diethylisopropylamine
Biographies
Heather D. Agnew attended Penn State University, where she received a B.S. in Chemistry and a B.S. in Biochemistry & Molecular Biology. As a Gates Cambridge Scholar, she then obtained an M. Phil in Chemistry from University of Cambridge. Under the guidance of Professor James R. Heath, she then earned a Ph.D. in Chemistry from California Institute of Technology, where she developed techniques to create synthetic, site-specific binding molecules as protein-catalyzed capture agents (PCCs) that address many target applications. Heather is currently Vice President R&D Operations at Indi Molecular, an emerging life sciences company that is focused on commercialization of PCCs. She also holds an adjunct appointment in the David Geffen School of Medicine at UCLA.
Dr. Matthew B. Coppock obtained his B.S. in chemistry from the University of Richmond and received his Ph.D. degree in chemistry from Penn State University under the guidance of Dr. Mary Beth Williams. He joined the U.S. Army Research Laboratory as an Oak Ridge Associated Universities (ORAU) postdoctoral fellow before becoming a civilian employee. His research interests include synthetic peptide chemistry, proteomics, spectroscopy, and assay development. He is currently the technical lead for the development and integration of stable, high affinity and highly selective peptide-based receptors for ruggedized biological assays.
Matthew Idso attended the University of Washington at Seattle, where he received a B.S. in Chemical Engineering. He then pursued a PhD in Chemical Engineering at the University of California, Santa Barbara. Working with Prof. Bradley Chmelka, Matthew studied the incorporation of photoactive molecular guests into nanostructured materials. Matthew then joined Heath lab as a post-doctoral scholar and is exploring new applications for protein-catalyzed capture agents in treating cancer and infectious diseases.
Bert T. Lai obtained his undergraduate degree in Chemistry at the Johns Hopkins University. He then received his Ph.D. in Chemistry from California Institute of Technology, where he characterized membrane bound structures of α-synuclein under the direction of Professor Harry Gray. Bert is currently the Senior Director of Chemistry at Indi Molecular, where he leads the chemistry effort in developing PCCs against targets of interest.
JingXin Liang obtained her Bachelor of Science in Chemistry at Temple University. She then obtained her Ph.D. in Chemistry from California Institute of Technology, in the lab of Professor James R. Heath, where she developed peptide-based capture agents for infectious disease diagnostics. JingXin is currently a postdoctoral fellow at Institute for Systems Biology, where she is studying mechanisms of drug resistance in cancer.
Amy M. McCarthy-Torrens attended the University of California Berkeley, where she received a B.S. in Chemistry. She then pursued a Ph.D. in Chemistry under the guidance of Professor James Heath at the California Institute of Technology. While at Caltech Amy developed a rapid assay platform for efficiently and simultaneously measuring the binding affinity of multiple protein-catalyzed capture (PCC) agents, and also identified PCCs that inhibit the intrinsic GTPase activity of KRas protein. Now graduated, Amy intends to pursue a chemical biology postdoc in London.
Carmen M. Warren received a B.S. in Biochemistry & Cell Biology and an M.S. in Biology from the University of California, San Diego. She obtained her Ph.D. in Biological Chemistry from the University of California, Los Angeles, where she studied the post-translational control of the ERBB family of receptor tyrosine kinases under the guidance of Dr. Ralf Landraf. Carmen then completed a postdoctoral fellowship in the laboratory of Dr. Luisa Iruela-Arispe at the University of California, Los Angeles, where she examined mechanisms of pathological angiogenesis and VEGF signaling in diabetes and in the tumor microenvironment. She is currently a Senior Scientist in Translational Biology at Indi Molecular, where protein catalyzed capture agents are being developed for therapeutics, in vivo imaging, and in vitro diagnostics.
Jim Heath is the President of the Institute for Systems Biology, and holds faculty appointments at the University of Washington (Biomolecular Engineering) and UCLA Geffen School of Medicine (Dept of Molecular and Medical Pharmacology). From 2003–2018 he served as the Elizabeth Gilloon Professor of Chemistry at Caltech, after starting his academic career at UCLA. Heath’s research is multidisciplinary in nature, focusing on problems at the interface of chemistry, physics, bioengineering, and oncology. He has founded several companies, including Isoplexis, Sofie Biosciences, Indi Molecular, and PACT Pharma.
Footnotes
ASSOCIATED CONTENT
Conflict of Interest Statement
JRH is a founder and board member of Indi Molecular, Inc., which is a company seeking to commercialize PCC technology.
REFERENCES
- (1).Lewis WG; Green LG; Grynszpan F; Radić Z; Carlier PR; Taylor P; Finn MG; Sharpless KB Click Chemistry in Situ: Acetylcholinesterase as a Reaction Vessel for the Selective Assembly of a Femtomolar Inhibitor from an Array of Building Blocks. Angew. Chemie - Int. Ed. 2002, 114, 1095–1099. [DOI] [PubMed] [Google Scholar]
- (2).Huisgen R Proceedings of the Chemical Society. October 1961; 1961; pp 357–396. [Google Scholar]
- (3).Kolb HC; Finn MG; Sharpless KB Click Chemistry: Diverse Chemical Function from a Few Good Reactions. Angew. Chemie - Int. Ed. 2001, 40, 2004–2021. [DOI] [PubMed] [Google Scholar]
- (4).Krasiński A; Radić Z; Manetsch R; Raushel J; Taylor P; Sharpless KB; Kolb HC In Situ Selection of Lead Compounds by Click Chemistry: Target-Guided Optimization of Acetylcholinesterase Inhibitors. J. Am. Chem. Soc. 2005, 127 (3), 6686–6692. [DOI] [PubMed] [Google Scholar]
- (5).Whiting M; Muldoon J; Lin YC; Silverman SM; Lindstrom W; Olson AJ; Kolb HC; Finn MG; Sharpless KB; Elder JH; et al. Inhibitors of HIV-1 Protease by Using in Situ Click Chemistry. Angew. Chemie - Int. Ed. 2006, 45 (9), 1435–1439. [DOI] [PubMed] [Google Scholar]
- (6).Mocharla VP; Colasson B; Lee LV; Röper S; Sharpless KB; Wong CH; Kolb HC In Situ Click Chemistry: Enzyme-Generated Inhibitors of Carbonic Anhydrase II. Angew. Chemie - Int. Ed. 2005, 44 (1), 116–120. [DOI] [PubMed] [Google Scholar]
- (7).Köhler G; Milstein C Continuous Cultures of Fused Cells Secreting Antibody of Predefined Specificity. Nature 1975, 256, 495–497. [DOI] [PubMed] [Google Scholar]
- (8).Little M; Kipriyanov SM; Le Gall F; Moldenhauer G Of Mice and Men: Hybridoma and Recombinant Antibodies. Immunol. Today 2000, 21, 364–370. [DOI] [PubMed] [Google Scholar]
- (9).Liu JKHH The History of Monoclonal Antibody Development - Progress, Remaining Challenges and Future Innovations. Ann. Med. Surg. 2014, 3 (4), 113–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Davis MM; Bjorkman PJ T-Cell Antigen Receptor Genes and T-Cell Recognition. Nature 1988, 334, 395–402. [DOI] [PubMed] [Google Scholar]
- (11).Guermonperez P; Valladeau J; Zitvogel L; Théry C; Amigorena S Antigen Presentation and T Cell Stimulation by Dendritic Cells. Annu. Rev. Immunol. 2002, 20, 621–667. [DOI] [PubMed] [Google Scholar]
- (12).Farrow B; Wong M; Malette J; Lai B; Deyle KM; Das S; Nag A; Agnew HD; Heath JR Epitope-Targeting of Tertiary Protein Structure Enables Target-Guided Synthesis of a Potent in Cell Inhibitor of Botulinum Neurotoxin. Angew. Chem. Int. Ed. Engl. 2015, 54 (24), 7114–7119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Lai BT; Wilson JA; Loredo JM; Pitram SM; LaBerge NA; Heath JR; Agnew HD Epitope Targeted Macrocyclic Peptide Ligand with Picomolar Cooperative Binding to Interleukin-17F. Chem. - A Eur. J 2018, 24 (15), 3760–3767. [DOI] [PubMed] [Google Scholar]
- (14).Barlow DJ; Edwards MS; Thornton JM Continuous and Discontinuous Protein Antigenic Determinants. Nature 1986, 322, 747–748. [DOI] [PubMed] [Google Scholar]
- (15).Lam KS; Lebl M; Krchňák V The “One-Bead-One-Compound” Combinatorial Library Method. Chem. Rev. 1997, 97, 411–448. [DOI] [PubMed] [Google Scholar]
- (16).Pfeilsticker JA; Umeda A; Farrow B; Hsueh CL; Deyle KM; Kim JT; Lai BT; Heath JR A Cocktail of Thermally Stable, Chemically Synthesized Capture Agents for the Efficient Detection of Anti-Gp41 Antibodies from Human Sera. PLoS One 2013, 8 (10), 125–130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Nag A; Das S; Yu MB; Deyle KM; Millward SW; Heath JR A Chemical Epitope-Targeting Strategy for Protein Capture Agents: The Serine 474 Epitope of the Kinase Akt2. Angew. Chemie Int. Ed. 2013, 52 (52), 13975–13979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Deyle KM; Farrow B; Hee YQ; Work J; Wong M; Lai B; Umeda A; Millward SW; Nag A; Das S; et al. A Protein-Targeting Strategy Used to Develop a Selective Inhibitor of the E17K Point Mutation in the PH Domain of Akt1. Nat. Chem. 2015, 7, 455–462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Millward SW; Henning RK; Kwong GA; Pitram S; Agnew HD; Deyle KM; Nag A; Hein J; Lee SS; Lim J; et al. Iterative in Situ Click Chemistry Assembles a Branched Capture Agent and Allosteric Inhibitor for Akt1. J. Am. Chem. Soc. 2011, 133, 18280–18288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Bunck DN; Atsavapranee B; Museth AK; Vandervelde D; Heath JR Modulating the Folding Landscape of Superoxide Dismutase 1 with Targeted Molecular Binders. Angew. Chemie 2018, 130 (2), 6320–6323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Ellington AD; Szostak JW In Vitro Selection of RNA Molecules That Bind Specific Ligands. Nature 1990, 346, 818–822. [DOI] [PubMed] [Google Scholar]
- (22).Kang K-N; Lee Y-S RNA Aptamers: A Review of Recent Trends and Applications In Future Trends in Biotechnology. Advances in Biochemical Engineering/Biotechnology, vol 131; Springer, Berlin, Heidelberg, 2012; pp 153–169. [DOI] [PubMed] [Google Scholar]
- (23).Reverdatto S; Burz D; Shekhtman A Peptide Aptamers: Development and Applications. Curr. Top. Med. Chem. 2015, 15, 153–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Mascini M; Palchetti I; Tombelli S Nucleic Acid and Peptide Aptamers: Fundamentals and Bioanalytical Aspects. Angew. Chemie - Int. Ed 2012, 51, 1316–1332. [DOI] [PubMed] [Google Scholar]
- (25).Wetzel SK; Settanni G; Kenig M; Binz HK; Plückthun A Folding and Unfolding Mechanism of Highly Stable Full-Consensus Ankyrin Repeat Proteins. J. Mol. Biol. 2008, 376, 241–257. [DOI] [PubMed] [Google Scholar]
- (26).Coppock MB; Warner CR; Dorsey B; Orlicki JA; Sarkes DA; Lai BT; Pitram SM; Rohde RD; Malette J; Wilson JA; et al. Protein Catalyzed Capture Agents with Tailored Performance for in Vitro and in Vivo Applications. Pept. Sci. 2016, 108 (2), e22934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Farrow B; Hong SA; Romero E; Lai B; Coppock MB; Deyle KM; Finch AS; Stratis-Cullum DN; Agnew H; Yang S; et al. A Chemically Synthesized Capture Agent Enables the Selective, Sensitive, and Robust Electrochemical Detection of Anthrax Protective Antigen. ACS Nano 2013, 7 (10), 9452–9460. [DOI] [PubMed] [Google Scholar]
- (28).Binz HK; Amstutz P; Kohl A; Stumpp MT; Briand C; Forrer P; Grütter MG; Plückthun A High-Affinity Binders Selected from Designed Ankyrin Repeat Protein Libraries. Nat. Biotechnol. 2004, 22, 575. [DOI] [PubMed] [Google Scholar]
- (29).Colas P; Cohen B; Jessen T; Grishina I; McCoy J; Brent R Genetic Selection of Peptide Aptamers That Recognize and Inhibit Cyclin-Dependent Kinase 2. Nature 1996, 380, 548–550. [DOI] [PubMed] [Google Scholar]
- (30).Smith GP; Petrenko VA Phage Display. Chem. Rev. 1997, 97, 491–410. [DOI] [PubMed] [Google Scholar]
- (31).Boersma YL; Plückthun A DARPins and Other Repeat Protein Scaffolds: Advances in Engineering and Applications. Curr. Opin. Biotechnol. 2011, 22, 849–857. [DOI] [PubMed] [Google Scholar]
- (32).Pancer Z; Cooper MD The Evolution of Adaptive Immunity. Annu. Rev. Immunol. 2006, 24, 497–518. [DOI] [PubMed] [Google Scholar]
- (33).Stumpp MT; Forrer P; Binz HK; Plückthun A Designing Repeat Proteins: Modular Leucine-Rich Repeat Protein Libraries Based on the Mammalian Ribonuclease Inhibitor Family. J. Mol. Biol. 2003, 332, 471–487. [DOI] [PubMed] [Google Scholar]
- (34).Main ERG; Lowe AR; Mochrie SGJ; Jackson SE; Regan L A Recurring Theme in Protein Engineering: The Design, Stability and Folding of Repeat Proteins. Curr. Opin. Struct. Biol. 2005, 15, 464–471. [DOI] [PubMed] [Google Scholar]
- (35).Guellouz A; Valerio-Lepiniec M; Urvoas A; Chevrel A; Graille M; Fourati-Kammoun Z; Desmadril M; van Tilbeurgh H; Minard P Selection of Specific Protein Binders for Pre-Defined Targets from an Optimized Library of Artificial Helicoidal Repeat Proteins (AlphaRep). PLoS One 2013, 8, e71512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Hanes J; Plückthun A In Vitro Selection and Evolution of Functional Proteins by Using Ribosome Display. Proc. Natl. Acad. Sci. 1997, 94 (10), 4937–4942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Steiner D; Forrer P; Stumpp MT; Plückthun A Signal Sequences Directing Cotranslational Translocation Expand the Range of Proteins Amenable to Phage Display. Nat. Biotechnol. 2006, 24, 823–831. [DOI] [PubMed] [Google Scholar]
- (38).Blind M; Blank M Aptamer Selection Technology and Recent Advances. Mol. Ther. - Nucleic Acids 2015, 4, e223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Daniels DA; Chen H; Hicke BJ; Swiderek KM; Gold L A Tenascin-C Aptamer Identified by Tumor Cell SELEX: Systematic Evolution of Ligands by Exponential Enrichment. Proc. Natl. Acad. Sci. 2003, 100, 15416–15421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Hadpech S; Nangola S; Chupradit K; Fanhchaksai K; Furnon W; Urvoas A; Valerio-Lepiniec M; Minard P; Boulanger P; Hong SS; et al. Alpha-Helicoidal HEAT-like Repeat Proteins (ΑRep) Selected as Interactors of HIV-1 Nucleocapsid Negatively Interfere with Viral Genome Packaging and Virus Maturation. Sci. Rep. 2017, 7, 16335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Holliger P; Hudson PJ Engineered Antibody Fragments and the Rise of Single Domains. Nat. Biotechnol. 2005, 23, 1126–1136. [DOI] [PubMed] [Google Scholar]
- (42).Wyler E; Kaminska M; Coïc Y-M; Baleux F; Véron M; Agou F Inhibition of NF-ΚB Activation with Designed Ankyrin-Repeat Proteins Targeting the Ubiquitin-Binding/Oligomerization Domain of NEMO. Protein Sci. 2007, 16, 2013–2022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).Nord K; Gunneriusson E; Ringdahl J; Ståhl S; Uhlén M; Nygren P-Å Binding Proteins Selected from Combinatorial Libraries of an α-Helical Bacterial Receptor Domain. Nat. Biotechnol. 1997, 15, 772–777. [DOI] [PubMed] [Google Scholar]
- (44).Tiede C; Bedford R; Heseltine SJ; Smith G; Wijetunga I; Ross R; Alqallaf D; Roberts APE; Balls A; Curd A; et al. Affimer Proteins Are Versatile and Renewable Affinity Reagents. Elife 2017, 6, e24903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (45).Tiede C; Tang AAS; Deacon SE; Mandal U; Nettleship JE; Owen RL; George SE; Harrison DJ; Owens RJ; Tomlinson DC; et al. Adhiron: A Stable and Versatile Peptide Display Scaffold for Molecular Recognition Applications. Protein Eng. Des. Sel. 2014, 27, 145–155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (46).Spencer-Smith R; Koide A; Zhou Y; Eguchi RR; Sha F; Gajwani P; Santana D; Gupta A; Jacobs M; Herrero-Garcia E; et al. Inhibition of RAS Function through Targeting an Allosteric Regulatory Site. Nat. Chem. Biol. 2017, 13, 62–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (47).Sha F; Salzman G; Gupta A; Koide S Monobodies and Other Synthetic Binding Proteins for Expanding Protein Science. Protein Sci. 2017, 26, 910–924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (48).Nilsson B; Moks T; Jansson B; Abrahmsen L; Elmblad A; Holmgren E; Henrichson C; Jones TA A Synthetic IgG-Binding Domain Based on Staphylococcal Protein A. Protein Eng. 1987, 1 (2), 107–113. [DOI] [PubMed] [Google Scholar]
- (49).Kronqvist N; Malm M; Göstring L; Gunneriusson E; Nilsson M; Höidén Guthenberg I; Gedda L; Frejd FY; Ståhl S; Löfblom J Combining Phage and Staphylococcal Surface Display for Generation of ErbB3-Specific Affibody Molecules. Protein Eng. Des. Sel. 2011, 24 (4), 385–396. [DOI] [PubMed] [Google Scholar]
- (50).Nord K; Nord O; Uhlén M; Kelley B; Ljungqvist C; Nygren P-Å Recombinant Human Factor VIII-Specific Affinity Ligands Selected from Phage-Displayed Combinatorial Libraries of Protein A. Eur. J. Biochem. 2001, 268 (15), 4269–4277. [DOI] [PubMed] [Google Scholar]
- (51).Frejd FY; Kim K-T Affibody Molecules as Engineered Protein Drugs. Exp. Mol. Med. 2017, 49, e306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (52).Eigenbrot C; Ultsch M; Dubnovitsky A; Abrahmsén L; Bax A; Eigenbrot C; Ultsch M; Dubnovitskyylars A; Torleif H Structural Basis for High-Affinity HER2 Receptor Binding by an Engineered Protein. Proc. Natl. Acad. Sci. U. S. A. 2019, 107 (34), 15039–15044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (53).Orlova A; Magnusson M; Eriksson TLJ; Nilsson M; Larsson B; Höidén-Guthenberg I; Widström C; Carlsson J; Tolmachev V; Ståhl S; et al. Tumor Imaging Using a Picomolar Affinity HER2 Binding Affibody Molecule. Cancer Res. 2006, 66 (8), 4339–4348. [DOI] [PubMed] [Google Scholar]
- (54).Tolmachev V; Orlova A; Pehrson R; Galli J; Baastrup B; Andersson K; Sandstro M; Rosik D; Lundqvist H; Wennborg A; et al. Radionuclide Therapy of HER2-Positive Microxenografts Using a Lu-Labeled HER2-Specific Affibody Molecule. Cancer Res. 2007, 67 (6), 2773–2783. [DOI] [PubMed] [Google Scholar]
- (55).Simon RJ; Kania RS; Zuckermann RN; Huebner VD; Jewell DA; Banville S; Ng S; Wang L; Rosenberg S; Marlowe CK Peptoids: A Modular Approach to Drug Discovery. Proc. Natl. Acad. Sci. 1992, 89, 9367–9371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (56).Zuckermann RN Peptoid Origins. Pept. Sci. 2011, 96, 545–555. [DOI] [PubMed] [Google Scholar]
- (57).Zuckermann RN; Kerr JM; Moos WH; Kent SBH Efficient Method for the Preparation of Peptoids [Oligo(N-Substituted Glycines)] by Submonomer Solid-Phase Synthesis. J. Am. Chem. Soc. 1992, 114, 10646–10647. [Google Scholar]
- (58).Shin SBY; Yoo B; Todaro LJ; Kirshenbaum K Cyclic Peptoids. J. Am. Chem. Soc. 2007, 129, 3218–3225. [DOI] [PubMed] [Google Scholar]
- (59).D’Amato A; Schettini R; Della Sala G; Costabile C; Tedesco C; Izzo I; De Riccardis F Conformational Isomerism in Cyclic Peptoids and Its Specification. Org. Biomol. Chem. 2017, 15, 9932–9942. [DOI] [PubMed] [Google Scholar]
- (60).Andreev K; Martynowycz MW; Ivankin A; Huang ML; Kuzmenko I; Meron M; Lin B; Kirshenbaum K; Gidalevitz D Cyclization Improves Membrane Permeation by Antimicrobial Peptoids. Langmuir 2016, 32, 12905–12913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (61).Culf AS; Čuperlović-Culf M; Léger DA; Decken A Small Head-to-Tail Macrocyclic α-Peptoids. Org. Lett. 2014, 16, 2780–2783. [DOI] [PubMed] [Google Scholar]
- (62).Kaniraj PJ; Maayan G A Facile Strategy for the Construction of Cyclic Peptoids under Microwave Irradiation through a Simple Substitution Reaction. Org. Lett. 2015, 17, 2110–2113. [DOI] [PubMed] [Google Scholar]
- (63).Salvador CEM; Pieber B; Neu PM; Torvisco A; Kleber Z Andrade C; Kappe CO A Sequential Ugi Multicomponent/Cu-Catalyzed Azide-Alkyne Cycloaddition Approach for the Continuous Flow Generation of Cyclic Peptoids. J. Org. Chem. 2015, 80 (9), 4590–4602. [DOI] [PubMed] [Google Scholar]
- (64).Chirayil S; Luebke KJ Cyclization of Peptoids by Formation of Boronate Esters. Tetrahedron Lett. 2012, 53, 726–729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (65).Khan SN; Kim A; Grubbs RH; Kwon YU Ring-Closing Metathesis Approaches for the Solid-Phase Synthesis of Cyclic Peptoids. Org. Lett. 2011, 13, 1582–1585. [DOI] [PubMed] [Google Scholar]
- (66).Jang H; Fafarman A; Holub JM; Kirshenbaum K Click to Fit: Versatile Polyvalent Display on a Peptidomimetic Scaffold. Org. Lett. 2005, 7, 1951–1954. [DOI] [PubMed] [Google Scholar]
- (67).Lee JH; Kim HS; Lim HS Design and Facile Solid-Phase Synthesis of Conformationally Constrained Bicyclic Peptoids. Org. Lett. 2011, 13, 5012–5015. [DOI] [PubMed] [Google Scholar]
- (68).Simpson LS; Kodadek T A Cleavable Scaffold Strategy for the Synthesis of One-Bead One-Compound Cyclic Peptoid Libraries That Can Be Sequenced by Tandem Mass Spectrometry. Tetrahedron Lett. 2012, 53, 2341–2344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (69).Udugamasooriya DG; Dineen SP; Brekken RA; Kodadek T A Peptoid “Antibody Surrogate” That Antagonizes VEGF Receptor 2 Activity. J. Am. Chem. Soc. 2008, 130 (17), 5744–5752. [DOI] [PubMed] [Google Scholar]
- (70).Alluri PG; Reddy MM; Bachhawat-sikder K; Olivos HJ; Kodadek T Isolation of Protein Ligands from Large Peptoid Libraries. J. Am. Chem. Soc. 2003, 125, 13995–14004. [DOI] [PubMed] [Google Scholar]
- (71).Hintersteiner M; Kimmerlin T; Kalthoff F; Stoeckli M; Garavel G; Seifert JM; Meisner NC; Uhl V; Buehler C; Weidemann T; et al. Single Bead Labeling Method for Combining Confocal Fluorescence On-Bead Screening and Solution Validation of Tagged One-Bead One-Compound Libraries. Chem. Biol. 2009, 16, 724–735. [DOI] [PubMed] [Google Scholar]
- (72).Raveendra BL; Wu H; Baccala R; Reddy MM; Schilke J; Bennett JL; Theofilopoulos AN; Kodadek T Discovery of Peptoid Ligands for Anti-Aquaporin 4 Antibodies. Chem. Biol. 2013, 20, 351–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (73).Gao Y; Kodadek T Direct Comparison of Linear and Macrocyclic Compound Libraries as a Source of Protein Ligands. ACS Comb. Sci. 2015, 17, 190–195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (74).Liu JKH The History of Monoclonal Antibody Development - Progress, Remaining Challenges and Future Innovations. Ann. Med. Surg. 2014, 3 (4), 113–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (75).Harlow E; Lane D Antibodies: A Laboratory Manual; Cold Spring Harbor Laborary: New York, 1988. [Google Scholar]
- (76).Goding JW Monoclonal Antibodies: Priniples and Practice, Third; Harcourt Brace and Company, 1996. [Google Scholar]
- (77).Sasaki S; Takagi M; Tanaka Y; Maeda M A New Application of a Peptide Library to Identify Selective Interaction between Small Peptides in an Attempt to Develop Recognition Molecules toward Protein Surfaces. Tetrahedron Lett. 1996, 37, 85–88. [Google Scholar]
- (78).Dong DL; Liu R; Sherlock R; Wigler MH; Nestler HP Molecular Forceps from Combinatorial Libraries Prevent Farnesylation of Ras by Binding to Its Carboxyl Terminus. Chem. Biol. 1999, 6, 133–141. [DOI] [PubMed] [Google Scholar]
- (79).Zhang Z; Zhu W; Kodadek T Selection and Application of Peptide-Binding Peptides. Nat. Biotechnol. 2000, 18, 71–74. [DOI] [PubMed] [Google Scholar]
- (80).Kolb HC; Sharpless KB The Growing Impact of Click Chemistry on Drug Discovery. Drug Discov. Today 2003, 8, 1128–1137. [DOI] [PubMed] [Google Scholar]
- (81).Wang Q; Chan TR; Hilgraf R; Fokin VV; Sharpless KB; Finn MG Bioconjugation by Copper(I)-Catalyzed Azide-Alkyne [3 + 2] Cycloaddition. J. Am. Chem. Soc. 2003, 125, 3192–3193. [DOI] [PubMed] [Google Scholar]
- (82).Tornøe CW; Christensen C; Meldal M Peptidotriazoles on Solid Phase: [1,2,3]-Triazoles by Regiospecific Copper(I)-Catalyzed 1,3-Dipolar Cycloadditions of Terminal Alkynes to Azides. J. Org. Chem. 2002, 67, 3057–3064. [DOI] [PubMed] [Google Scholar]
- (83).Rostovtsev VV; Green LG; Fokin VV; Sharpless KB A Stepwise Huisgen Cycloaddition Process: Copper(I)-Catalyzed Regioselective “Ligation” of Azides and Terminal Alkynes. Angew. Chemie - Int. Ed. 2002, 41, 2596–2599. [DOI] [PubMed] [Google Scholar]
- (84).Zhang L; Chen X; Xue P; Sun HHY; Williams ID; Sharpless KB; Fokin VV; Jia G Ruthenium-Catalyzed Cycloaddition of Aryl Azides and Alkynes. J. Am. Chem. Soc. 2005, 127, 15998–15999. [DOI] [PubMed] [Google Scholar]
- (85).Verdine GL; Hilinski GJ Methods in Enzymology, Vol. 503; Wittrup KD, L VG, Eds.; Academic Press, 2012. [Google Scholar]
- (86).Zhang L; Chen X; Xue P; Sun HHY; Williams ID; Sharpless KB; Fokin VV; Jia G Ruthenium-Catalyzed Cycloaddition of Alkynes and Organic Azides. J. Am. Chem. Soc. 2005, 127, 15998–15999. [DOI] [PubMed] [Google Scholar]
- (87).Kiick KL; Saxon E; Tirrell DA; Bertozzi CR Incorporation of Azides into Recombinant Proteins for Chemoselective Modification by the Staudinger Ligation. Proc. Natl. Acad. Sci. 2002, 99, 19–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (88).Saxon E; Bertozzi CR Cell Surface Engineering by a Modified Staudinger Reaction. Science 2000, 287, 2007–2010. [DOI] [PubMed] [Google Scholar]
- (89).Mock WL; Irra TA; Wepsiec JP; Adhya M Catalysis by Cucurbituril. The Significance of Bound-Substrate Destabilization for Induced Triazole Formation. J. Org. Chem. 1989, 54 (22), 5302–5308. [Google Scholar]
- (90).Mock WL; Irra TA; Wepsiec JP; Manimaran TL Cycloaddition Induced by Cucurbituril. A Case of Pauling Principle Catalysis. J. Org. Chem. 1983, 48 (20), 3619–3620. [Google Scholar]
- (91).Harel M; Schalk I; Ehret-Sabatier L; Bouet F; Goeldner M; Hirth C; Axelsen PH; Silman I; Sussman JL Quaternary Ligand Binding to Aromatic Residues in the Active-Site Gorge of Acetylcholinesterase. Proc. Natl. Acad. Sci. 1993, 90 (19), 9031–9035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (92).Sussman J; Harel M; Frolow F; Oefner C; Goldman A; Toker L; Silman I Atomic Structure of Acetylcholinesterase from Torpedo Californica: A Prototypic Acetylcholine-Binding Protein. Science 1991, 253 (5022), 872–879. [DOI] [PubMed] [Google Scholar]
- (93).Radić Z; Taylor P Interaction Kinetics of Reversible Inhibitors and Substrates with Acetylcholinesterase and Its Fasciculin 2 Complex. J. Biol. Chem. 2001, 276 (7), 4622–4633. [DOI] [PubMed] [Google Scholar]
- (94).Berman HA; Decker MM; Nowak MW; Leonard KJ; McCauley M; Baker WM; Taylor P Site Selectivity of Fluorescent Bisquaternary Phenanthridinium Ligands for Acetylcholinesterase. Mol. Pharmacol. 1987, 31 (6), 610–616. [PubMed] [Google Scholar]
- (95).Carlier PR; Han YF; Chow ESH; Li CPL; Wang H; Xuan Lieu T; Sum Wong H; Pang YP Evaluation of Short-Tether Bis-THA AChE Inhibitors. A Further Test of the Dual Binding Site Hypothesis. Bioorganic Med. Chem. 1999, 7, 351–357. [DOI] [PubMed] [Google Scholar]
- (96).Manetsch R; Krasin A; Radic Z; Raushel J; Taylor P; Sharpless KB; Kolb HC In Situ Click Chemistry: Enzyme Inhibitors Made to Their Own Specifications. J. Am. Chem. Soc. 2004, 126 (18), 12809–12818. [DOI] [PubMed] [Google Scholar]
- (97).Lindskog S Structure and Mechanism of Carbonic Anhydrase. Pharmacol. Ther. 1997, 74, 1–20. [DOI] [PubMed] [Google Scholar]
- (98).Agnew HD; Rohde RD; Millward SW; Nag A; Yeo W-S; Hein JE; Pitram SM; Tariq AA; Burns VM; Krom RJ; et al. Iterative In Situ Click Chemistry Creates Antibody-like Protein-Capture Agents. Angew. Chem. Int. Ed. Engl. 2009, 48 (27), 4944–4948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (99).Grimster NP; Stump B; Fotsing JR; Weide T; Talley TT; Yamauchi JG; Nemecz Á; Kim C; Ho K-Y; Sharpless KB; et al. Generation of Candidate Ligands for Nicotinic Acetylcholine Receptors via in Situ Click Chemistry with a Soluble Acetylcholine Binding Protein Template. J. Am. Chem. Soc. 2012, 134 (15), 6732–6740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (100).Bhardwaj A; Kaur J; Wuest M; Wuest F In Situ Click Chemistry Generation of Cyclooxygenase-2 Inhibitors. Nat. Commun. 2017, 8 (1), 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (101).Willand N; Desroses M; Toto P; Dirié B; Lens Z; Villeret V; Rucktooa P; Locht C; Baulard A; Deprez B Exploring Drug Target Flexibility Using in Situ Click Chemistry: Application to a Mycobacterial Transcriptional Regulator. ACS Chem. Biol. 2010, 5 (11), 1007–1013. [DOI] [PubMed] [Google Scholar]
- (102).Hirose T; Sunazuka T; Ōmura S Rapid Identification via In Situ Click Chemistry of a Novel Chitinase Inhibitor. J. Synth. Org. Chem. Japan 2016, 74 (11), 1090–1097. [Google Scholar]
- (103).Glassford I; Teijaro CN; Daher SS; Weil A; Small MC; Redhu SK; Colussi DJ; Jacobson MA; Childers WE; Buttaro B; et al. Ribosome-Templated Azide–Alkyne Cycloadditions: Synthesis of Potent Macrolide Antibiotics by In Situ Click Chemistry. J. Am. Chem. Soc. 2016, 138 (9), 3136–3144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (104).Tieu W; Soares da Costa TP; Yap MY; Keeling KL; Wilce MCJ; Wallace JC; Booker GW; Polyak SW; Abell AD Optimising in Situ Click Chemistry: The Screening and Identification of Biotin Protein Ligase Inhibitors. Chem. Sci. 2013, 4 (9), 3533–3537. [Google Scholar]
- (105).Lam KS; Salmon SE; Hersh EM; Hruby VJ; Kazmierskit WM; Knappt RJ A New Type of Synthetic Peptide Library for Identifying Ligand-Binding Activity. Nature 1991, 354, 82–84. [DOI] [PubMed] [Google Scholar]
- (106).Youngquist RS; Fuentes GR; Lacey MP; Keough T Generation and Screening of Combinatorial Peptide Libraries Designed for Rapid Sequencing by Mass Spectrometry. J. Am. Chem. Soc. 1995, 117, 3900–3906. [Google Scholar]
- (107).Lee SS; Lim J; Tan S; Cha J; Yeo SY; Agnew HD; Heath JR Accurate MALDI-TOF/TOF Sequencing of One-Bead-One-Compound Peptide Libraries with Application to the Identification of Multiligand Protein Affinity Agents Using in Situ Click Chemistry Screening. Anal. Chem. 2010, 82, 672–679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (108).MacConnell AB; McEnaney PJ; Cavett VJ; Paegel BM DNA-Encoded Solid-Phase Synthesis: Encoding Language Design and Complex Oligomer Library Synthesis. ACS Comb. Sci. 2015, 17, 518–534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (109).Astle JM; Simpson LS; Huang Y; Reddy MM; Wilson R; Connell S; Wilson J; Kodadek T Seamless Bead to Microarray Screening : Rapid Identification of the Highest Affinity Protein Ligands from Large Combinatorial Libraries. Chem. Biol. 2010, 17 (1), 38–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (110).Kwon Y; Kodadek T Quantitative Evaluation of the Relative Cell Permeability of Peptoids and Peptides. J. Am. Chem. Soc. 2007, 127, 1508–1509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (111).Satoh T; Li S; Friedman TM; Wiaderkiewicz R; Korngold R; Huang Z Synthetic Peptides Derived from the Fourth Domain of CD4 Antagonize CD4 Function and Inhibit T Cell Activation. Biochem. Biophys. Res. Commun. 1996, 224, 438–443. [DOI] [PubMed] [Google Scholar]
- (112).Das S; Nag A; Liang J; Bunck DN; Umeda A; Farrow B; Coppock MB; Sarkes DA; Finch AS; Agnew HD; et al. A General Synthetic Approach for Designing Epitope Targeted Macrocyclic Peptide Ligands. Angew. Chemie Int. Ed. 2015, 54, 13219–13224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (113).Lee SS; Lim J; Cha J; Tan S; Heath JR Rapid Microwave-Assisted Cnbr Cleavage of Bead-Bound Peptides. J. Comb. Chem. 2008, 10, 807–809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (114).Giudicessi SL; Gurevich-Messina JM; Martínez-Ceron MC; Erra-Balsells R; Albericio F; Cascone O; Camperi SA Friendly Strategy to Prepare Encoded One Bead-One Compound Cyclic Peptide Library. ACS Comb. Sci. 2013, 15, 525–529. [DOI] [PubMed] [Google Scholar]
- (115).Gurevich-Messina JM; Giudicessi SL; Martínez-Ceron MC; Acosta G; Erra-Balsells R; Cascone O; Albericio F; Camperi SA A Simple Protocol for Combinatorial Cyclic Depsipeptide Libraries Sequencing by Matrix-Assisted Laser Desorption/Ionisation Mass Spectrometry. J. Pept. Sci. 2015, 21, 40–45. [DOI] [PubMed] [Google Scholar]
- (116).Wang X; Peng L; Liu R; Xu B; Lam KS Applicaitons of Topologically Segregated Bilayar Beads in “one-Bead One-Compound” Combinatorial Libraries. J. Pept. Sci. 2005, 65, 130–138. [DOI] [PubMed] [Google Scholar]
- (117).Chen X; Tan PH; Zhang Y; Pei D On-Bead Screening of Combinatorial Libraries: Reduction of Nonspecific Binding by Decreasing Surface Ligand Density. J. Comb. Chem. 2009, 11, 604–611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (118).Joo SH; Xiao Q; Ling Y; Gopishetty B; Pei D High-Throughput Sequence Determination of Cyclic Peptide Library Members by Partial Edman Degradation/Mass Spectrometry. J. Am. Chem. Soc. 2006, 128, 13000–13009. [DOI] [PubMed] [Google Scholar]
- (119).Liu R; Marik J; Lam KS A Novel Peptide-Based Encoding System for “One-Bead One-Compound” Peptidomimetic and Small Molecule Combinatorial Libraries. J. Am. Chem. Soc. 2002, 124, 7678–7680. [DOI] [PubMed] [Google Scholar]
- (120).Vauquelin G; Charlton SJ Exploring Avidity: Understanding the Potential Gains in Functional Affinity and Target Residence Time of Bivalent and Heterobivalent Ligands. Br. J. Pharmacol. 2013, 168, 1771–1785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (121).Peng L; Liu R; Marik J; Wang X; Takada Y; Lam KS Combinatorial Chemistry Identifies High-Affinity Peptidomimetics against Α4β1integrin for in Vivo Tumor Imaging. Nat. Chem. Biol. 2006, 2, 381–389. [DOI] [PubMed] [Google Scholar]
- (122).Klausen MS; Jespersen MC; Nielsen H; Jensen KK; Jurtz VI; Sønderby CK; Sommer MOA; Winther O; Nielsen M; Petersen B; et al. NetSurfP-2.0: Improved Prediction of Protein Structural Features by Integrated Deep Learning. BioRxiv 2018, 311209. [DOI] [PubMed] [Google Scholar]
- (123).Jespersen MC; Peters B; Nielsen M; Marcatili P BepiPred-2.0: Improving Sequence-Based B-Cell Epitope Prediction Using Conformational Epitopes. Nucleic Acids Res. 2017, 45 (W1), W24–W29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (124).Vita R; Overton JA; Greenbaum JA; Ponomarenko J; Clark JD; Cantrell JR; Wheeler DK; Gabbard JL; Hix D; Sette A; et al. The Immune Epitope Database (IEDB) 3.0. Nucleic Acids Res. 2015, 43 (D1), D405–D412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (125).Tian F; Yang L; Lv F; Yang Q; Zhou P In Silico Quantitative Prediction of Peptides Binding Affinity to Human MHC Molecule : An Intuitive Quantitative Structure – Activity Relationship Approach. Amino Acids 2009, 36, 535–554. [DOI] [PubMed] [Google Scholar]
- (126).Henning RK; Varghese JO; Das S; Nag A; Tang G; Tang K; Sutherland AM; Heath JR Degradation of Akt Using Protein-Catalyzed Capture Agents. J. Pept. Sci. 2016, 22 (4), 196–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (127).Luo Y; Shoemaker AR; Liu X; Woods KW; Thomas SA; de Jong R; Han EK; Li T; Stoll VS; Powlas JA; et al. Potent and Selective Inhibitors of Akt Kinases Slow the Progress of Tumors in Vivo. Mol . Cancer Ther. 2005, 4, 977–986. [DOI] [PubMed] [Google Scholar]
- (128).Okuzumi T; Fiedler D; Zhang C; Gray DC; Aizenstein B; Hoffman R; Shokat KM Inhibitor Hijacking of Akt Activation. Nat. Chem. Biol. 2009, 5, 484–493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (129).Testa JR; Tsichlis PN AKT Signaling in Normal and Malignant Cells. Oncogene 2005, 24, 7391–7393. [DOI] [PubMed] [Google Scholar]
- (130).Daskalakis D HIV Diagnostic Testing: Evolving Technology and Testing Strategies. Top. Antivir. Med. 2011, 19 (1), 18–22. [PMC free article] [PubMed] [Google Scholar]
- (131).Allen WJ; Rizzo RC Computer-Aided Approaches for Targeting HIVgp41. Biology. 2012, 1 (2), 311–338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (132).Xu JY; Gorny MK; Palker T; Karwowska S; Zolla-Pazner S Epitope Mapping of Two Immunodominant Domains of Gp41, the Transmembrane Protein of Human Immunodeficiency Virus Type 1, Using Ten Human Monoclonal Antibodies. J. Virol. 1991, 65 (9), 4832–4838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (133).Chin CD; Laksanasopin T; Cheung YK; Steinmiller D; Linder V; Parsa H; Wang J; Moore H; Rouse R; Umviligihozo G; et al. Microfluidics-Based Diagnostics of Infectious Diseases in the Developing World. Nat. Med. 2011, 17, 1015–1019. [DOI] [PubMed] [Google Scholar]
- (134).Phelps ME Positron Emission Tomography Provides Molecular Imaging of Biological Processes. Proc. Natl. Acad. Sci. 2000, 97, 9226–9233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (135).Fairbrother WJ; Christinger HW; Cochran AG; Fuh G; Keenan CJ; Quan C; Shriver SK; Tom JYKK; Wells JA; Cunningham BC Novel Peptides Selected to Bind Vascular Endothelial Growth Factor Target the Receptor-Binding Site. Biochemistry 1998, 37 (51), 17754–17764. [DOI] [PubMed] [Google Scholar]
- (136).Kogot JM; Zhang Y; Moore SJ; Pagano P; Stratis-Cullum DN; Chang-Yen D; Turewicz M; Pellegrino PM; de Fusco A; Soh HT; et al. Screening of Peptide Libraries against Protective Antigen of Bacillus Anthracis in a Disposable Microfluidic Cartridge. PLoS One 2011, 6 (11), e26925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (137).Yang J; Cron P; Good VM; Thompson V; Hemmings BA; Barford D Crystal Structure of an Activated Akt/Protein Kinase B Ternary Complex with GSK3-Peptide and AMP-PNP. Nat. Struct. Biol. 2002, 9 (12), 940–944. [DOI] [PubMed] [Google Scholar]
- (138).Sakamoto T; Ojida A; Hamachi I Molecular Recognition, Fluorescence Sensing, and Biological Assay of Phosphate Anion Derivatives Using Artificial Zn(II)-Dpa Complexes. Chem. Commun. 2009, No. 2, 141–152. [DOI] [PubMed] [Google Scholar]
- (139).McCarthy AM; Kim J; Museth AK; Henning RK; Heath JE; Winson E; Oh JJ; Liang J; Hong S; Heath JR Allosteric Inhibitor of KRas Identified Using a Barcoded Assay Microchip Platform. Anal. Chem. 2018, 90, 8824–8830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (140).Deshayes S; Morris MC; Divita G; Heitz F Cell-Penetrating Peptides: Tools for Intracellular Delivery of Therapeutics. Cell. Mol. Life Sci. 2005, 62, 1839–1849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (141).Henning RK; Varghese JO; Das S; Nag A; Tang G; Tang K; Sutherland AM; Heath JR Degradation of Akt Using Protein Catalyzed Capture Agents. J. Pept. Sci. 2016, 22 (4), 196–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (142).Hines J; Gough JD; Corson TW; Crews CM Posttranslational Protein Knockdown Coupled to Receptor Tyrosine Kinase Activation with PhosphoPROTACs. Proc. Natl. Acad. Sci. 2013, 110, 8942–8947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (143).Raina K; Crews CM Chemical Inducers of Targeted Protein Degradation. J. Biol. Chem. 2010, 285, 1057–11060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (144).Sakamoto KM; Kim KB; Kumagai A; Mercurio F; Crews CM; Deshaies RJ Protacs: Chimeric Molecules That Target Proteins to the Skp1-Cullin-F Box Complex for Ubiquitination and Degradation. Proc. Natl. Acad. Sci. 2001, 98, 8554–8559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (145).Carpten JD; Faber AL; Horn C; Donoho GP; Briggs SL; Robbins CM; Hostetter G; Boguslawski S; Moses TY; Savage S; et al. A Transforming Mutation in the Pleckstrin Homology Domain of AKT1 in Cancer. Nature 2007, 448 (7152), 439–444. [DOI] [PubMed] [Google Scholar]
- (146).Davies MA; Stemke-Hale K; Tellez C; Calderone TL; Deng W; Prieto VG; Lazar AJF; Gershenwald JE; Mills GB A Novel AKT3 Mutation in Melanoma Tumours and Cell Lines. Br. J. Cancer 2008, 99, 1265–1268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (147).Eubanks LM; Hixon MS; Jin W; Hong S; Clancy CM; Tepp WH; Baldwin MR; Malizio CJ; Goodnough MC; Barbieri JT; et al. An in Vitro and in Vivo Disconnect Uncovered through High-Throughput Identification of Botulinum Neurotoxin A Antagonists. Proc. Natl. Acad. Sci. U. S. A. 2007, 104 (15), 2602–2607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (148).Caglič D; Krutein MC; Bompiani KM; Barlow DJ; Benoni G; Pelletier JC; Reitz AB; Lairson LL; Houseknecht KL; Smith GR; et al. Identification of Clinically Viable Quinolinol Inhibitors of Botulinum Neurotoxin A Light Chain. J. Med. Chem. 2014, 57 (3), 669–676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (149).Burnett JC; Ruthel G; Stegmann CM; Panchal RG; Nguyen TL; Hermone AR; Stafford RG; Lane DJ; Kenny TA; McGrath CF; et al. Inhibition of Metalloprotease Botulinum Serotype A from a Pseudo-Peptide Binding Mode to a Small Molecule That Is Active in Primary Neurons. J. Biol. Chem. 2007, 282 (7), 5004–5014. [DOI] [PubMed] [Google Scholar]
- (150).Rotunno MS; Bosco DA An Emerging Role for Misfolded Wild-Type SOD1 in Sporadic ALS Pathogenesis. Front. Cell. Neurosci. 2013, 7, 253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (151).Antonyuk S; Elam JS; Hough MA; Strange RW; Doucette PA; Rodriguez JA; Hayward LJ; Valentine JS; Hart PJ; Hasnain SS Structural Consequences of the Familial Amyotrophic Lateral Sclerosis SOD1 Mutant His46Arg. Protein Sci. 2005, 14, 1201–1213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (152).Bosco DA; Morfini G; Karabacak NM; Song Y; Gros-Louis F; Pasinelli P; Goolsby H; Fontaine BA; Lemay N; McKenna-Yasek D; et al. Wild-Type and Mutant SOD1 Share an Aberrant Conformation and a Common Pathogenic Pathway in ALS. Nat. Neurosci. 2010, 13, 1396–1403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (153).Hartl FU; Bracher A; Hayer-Hartl M Molecular Chaperones in Protein Folding and Proteostasis. Nature 2011, 475, 324–332. [DOI] [PubMed] [Google Scholar]
- (154).Cox AD; Fesik SW; Kimmelman AC; Luo J; Der CJ Drugging the Undruggable Ras: Mission Possible? Nat. Rev. Drug Discov. 2014, 13 (11), 828–851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (155).Ying H; Kimmelman AC; Lyssiotis CA; Hua S; Chu GC; Fletcher-Sananikone E; Locasale JW; Son J; Zhang H; Coloff JL; et al. Oncogenic Kras Maintains Pancreatic Tumors through Regulation of Anabolic Glucose Metabolism. Cell 2013, 149, 656–670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (156).Hall BE; Yang SS; Boriack-Sjodin PA; Kuriyan J; Bar-Sagi D Structure-Based Mutagenesis Reveals Distinct Functions for Ras Switch 1 and Switch 2 in Sos-Catalyzed Guanine Nucleotide Exchange. J. Biol. Chem. 2001, 276 (29), 27629–27637. [DOI] [PubMed] [Google Scholar]
- (157).Gong MM; Sinton D Turning the Page: Advancing Paper-Based Microfluidics for Broad Diagnostic Application. Chem. Rev. 2017, 117 (12), 8447–8480. [DOI] [PubMed] [Google Scholar]
- (158).Gautam A; Chaudhary K; Kumar R; Sharma A; Kapoor P; Tyagi A; Consortium OSDD; Raghava GPS In Silico Approaches for Designing Highly Effective Cell Penetrating Peptides. J. Transl. Med. 2013, 11 (1), 74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (159).Chu Q; Moellering RE; Hilinski GJ; Kim Y-W; Grossmann TN; Yeh JT-H; Verdine GL Towards Understanding Cell Penetration by Stapled Peptides. Medchemcomm 2015, 6 (1), 111–119. [Google Scholar]
- (160).Ferdinandus J; Violet J; Sandhu S; Hofman MS Prostate-Specific Membrane Antigen Theranostics: Therapy with Lutetium-177. Curr. Opin. Urol. 2018, 28 (2), 197–204. [DOI] [PubMed] [Google Scholar]
- (161).Buck AK; Stolzenburg A; Hänscheid H; Schirbel A; Lückerath K; Schottelius M; Wester H-J; Lapa C Chemokine Receptor – Directed Imaging and Therapy. Methods 2017, 130, 63–71. [DOI] [PubMed] [Google Scholar]
- (162).Rizvi SFA; Naqvi SAR; Roohi S; Sherazi TA; Rasheed R 177Lu-DOTA-Coupled Minigastrin Peptides: Promising Theranostic Agents in Neuroendocrine Cancers. Mol. Biol. Rep. 2018, 1–9. [DOI] [PubMed] [Google Scholar]
- (163).van Essen M; Krenning EP; Kam BLR; de Jong M; Valkema R; Kwekkeboom DJ Peptide-Receptor Radionuclide Therapy for Endocrine Tumors. Nat. Rev. Endocrinol. 2009, 5, 382–393. [DOI] [PubMed] [Google Scholar]
- (164).Breeman WAP; de Blois E; Sze Chan H; Konijnenberg M; Kwekkeboom DJ; Krenning EP 68Ga-Labeled DOTA-Peptides and 68Ga-Labeled Radiopharmaceuticals for Positron Emission Tomography: Current Status of Research, Clinical Applications, and Future Perspectives. Semin. Nucl. Med. 2011, 41 (4), 314–321. [DOI] [PubMed] [Google Scholar]
- (165).Reubi JC; Maecke HR Peptide-Based Probes for Cancer Imaging. J. Nucl. Med. 2008, 49 (11), 1735–1738. [DOI] [PubMed] [Google Scholar]
- (166).Chakravarty R; Dash SC and Molecular A Imaging of Breast Cancer: Role of RGD Peptides. Mini-Reviews in Medicinal Chemistry. 2015, pp 1073–1094. [DOI] [PubMed] [Google Scholar]
- (167).De Pas T; Bodei L; Pelosi G; De Braud F; Villa G; Capanna R; Paganelli G; Group IS Peptide Receptor Radiotherapy: A New Option for the Management of Aggressive Fibromatosis on Behalf of the Italian Sarcoma Group. Br. J. Cancer 2003, 88 (5), 645–647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (168).Corbi-Verge C; Kim PM Motif Mediated Protein-Protein Interactions as Drug Targets. Cell Commun. Signal. 2016, 14 (1), 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (169).Hansen LB; Buus S; Schafer-Nielsen C Identification and Mapping of Linear Antibody Epitopes in Human Serum Albumin Using High-Density Peptide Arrays. PLoS One 2013, 8 (7), e68902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (170).Carmona SJ; Nielsen M; Schafer-Nielsen C; Mucci J; Altcheh J; Balouz V; Tekiel V; Frasch AC; Campetella O; Buscaglia CA; et al. Towards High-Throughput Immunomics for Infectious Diseases : Use of Next-Generation Peptide Microarrays for Rapid Discovery and Mapping of Antigenic Determinants. Mol. Cell. Proteomics 2015, 14, 1871–1884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (171).Ul-Ain Q; Kandler R; Gillespie D; Nag A Chemical Epitope Targeting: Review of a Novel Screening Technology. J. Med. Chem. Drug Des. 2018, 1 (2), 1–7. [Google Scholar]
- (172).Tamaskovic R; Simon M; Stefan N Designed Ankyrin Repeat Proteins (DARPins) from Research to Therapy. Methods Enzymol. 2012, 503, 101–134. [DOI] [PubMed] [Google Scholar]